 Ornella la fortune Tchoupou Saha1,2
Ornella la fortune Tchoupou Saha1,2 Grégory Dubourg1,2*
Grégory Dubourg1,2* Abdourahamane Yacouba1,2
Abdourahamane Yacouba1,2 Vincent Bossi2
Vincent Bossi2 Didier Raoult1,2
Didier Raoult1,2 Jean-Christophe Lagier1,2*
Jean-Christophe Lagier1,2*- 1Aix-Marseille Université, Institut de Recherche pour le Développement (IRD), Assistance Publique - Hôpitaux de Marseille (AP-HM), Microbes Evolution Phylogeny and Infections (MEPHI), Marseille, France
- 2IHU Méditerranée Infection, Marseille, France
While populations at risk for severe SARS-CoV-2 infections have been clearly identified, susceptibility to the infection and its clinical course remain unpredictable. As the nasopharyngeal microbiota may promote the acquisition of several respiratory infections and have an impact on the evolution of their outcome, we studied the nasopharyngeal microbiota of COVID-19 patients in association with baseline disease-related clinical features compared to that of patients tested negative. We retrospectively analyzed 120 nasopharyngeal pseudonymized samples, obtained for diagnosis, divided into groups (infected patients with a favorable outcome, asymptomatic, and deceased patients) and patients tested negative for SARS-CoV-2, by using Illumina-16S ribosomal ribonucleic acid (rRNA) sequencing and specific polymerase chain reaction (PCR) targeting pathogens. We first found a depletion of anaerobes among COVID-19 patients, irrespective of the clinical presentation of the infection (p < 0.029). We detected 9 taxa discriminating patients tested positive for SARS-CoV-2 from those that were negative including Corynebacterium propinquum/pseudodiphtericum (p ≤ 0.05), Moraxella catarrhalis (p ≤ 0.05), Bacillus massiliamazoniensis (p ≤ 0.01), Anaerobacillus alkalidiazotrophicus (p ≤ 0.05), Staphylococcus capitis subsp. capitis (p ≤ 0.001), and Afipia birgiae (p ≤ 0.001) with 16S rRNA sequencing, and Streptococcus pneumoniae (p ≤ 0.01), Klebsiella pneumoniae (p ≤ 0.01), and Enterococcus faecalis (p ≤ 0.05) using real-time PCR. By designing a specific real-time PCR, we also demonstrated that C. propinquum is decreased in asymptomatic individuals compared to other SARS-CoV 2 positive patients. These findings indicate that the nasopharyngeal microbiota as in any respiratory infection plays a role in the clinical course of the disease. Further studies are needed to elucidate the potential role in the clinical course of the disease of M. catarrhalis, Corynebacterium accolens, and more specifically Corynebacterium propinquum/diphteriticum in order to include them as predictors of the severity of COVID-19.
Introduction
The emerging viral pathology due to the new coronavirus that appeared in Wuhan in China in December 2019 is the cause of a public health crisis that threatens humanity (Na et al., 2020; Dahyot et al., 2021). It presents a very significant clinical symptomatology; that of a clinical course characterized by pneumonia of variable severity associated with age and underlying medical conditions (cancer, diabetes, immunosuppression, obesity, cardiac, renal, respiratory, hepatic, and neurological problems) (Allameh et al., 2020; Ding et al., 2020). While populations at risk of severe SARS-CoV-2 infection have been clearly identified, the susceptibility to infection and clinical course of the disease remain unpredictable.
Many mechanisms have been associated with disease progression, from a virological, immunological, and hemostatic point of view (Gautret et al., 2020). Since the SARS-CoV-2 primary site of infection is nasopharyngeal, upon entry into the host cell it binds primarily to angiotensin converting enzyme 2 (ACE2) receptors expressed on alveolar epithelial cells of the lung and on enterocytes of the gut (Hoffmann et al., 2020; Lundstrom et al., 2021), thus communicating with the nasopharyngeal and gut microbiota (Dhar and Mohanty, 2020). At the same time, it is currently known that the nasopharyngeal microbiota may promote the acquisition of several respiratory infections that have an impact on disease progression (Dubourg et al., 2019). Research in this area has grown exponentially with the advent of high-throughput sequencing approaches (Weinstock, 2012) that allow comprehensive exploration of the nasopharyngeal microbiota in a large population of individuals (Bogaert et al., 2011; Brugger et al., 2012; Edouard et al., 2018; Man et al., 2019). Indeed, associations between changes in the nasopharyngeal microbiota and bronchiolitis, influenza virus, or human rhinovirus have been reported (Allen et al., 2014; Hasegawa et al., 2016; Wen et al., 2018). Currently, studies conducted on the nasopharyngeal microbiota of COVID-19 patients show conflicting data, and very few studies have investigated the nasopharyngeal microbiota in these COVID-19 patients, mainly using 16S ribosomal RNA sequencing (De Maio et al., 2020; Mostafa et al., 2020; Zhang et al., 2020; Lloréns-Rico et al., 2021; Rosas-Salazar et al., 2021; Shilts et al., 2021). However, no previous studies have evaluated the association between disease severity and changes in the nasopharyngeal microbiota at different disease phases.
In this context, we therefore proposed to compare the nasopharyngeal microbiota of COVID-19 patients with that of uninfected individuals and to explore the taxonomic differences among COVID-19 patients according to the disease outcome (i.e., death or survival).
Materials and Methods
Patients and Materials
During of COVID-19 outbreak, we tested people presenting in IHU Méditerranée Infection for a diagnosis of COVID-19 (patients with symptoms or contact of COVID-19 infected patient) and/or respiratory infection using nasopharyngeal samples. Indeed, this study was conducted on a remaining material sampled for diagnosis, and patients were not specifically sampled for this investigation.
Samples were pseudonymized and we categorized two groups: COVID-19 negative and COVID-19 positive groups, confirmed by qualitative RT-PCR assay for the qualitative detection of SARS-CoV-2 nucleic acid in upper respiratory tract samples from COVID-19 suspects (Han et al., 2021). The distribution of individuals by group was done by taking into account the basic clinical characteristics related to COVID-19 disease (fever, dry cough, dyspnea, dysgeusia, anosmia, and hyposmia) (Allameh et al., 2020). The COVID-19 group was further divided into three subgroups; asymptomatic (patients without clinical symptoms), mild (patients who had positive responses to treatment after hospitalization), and death (patients who died because of severe and critical clinical symptoms). For this study, we used the remaining amount of nasopharyngeal samples obtained following COVID-19 testing by qPCR in our institute. We performed nucleic acid extraction using the EZ1 Virus Mini Kit v2. 0 on an EZ1 Advanced XL instrument (Qiagen, Courtaboeuf, France) or the KingFisher Flex system (Thermo Fisher Scientific, Waltham, MA, United States)® and for viral RNA amplification was carried out using the Master LightCycler 480 Probes kit (Roche Diagnostics, Meylan, France), as previously described (Colson et al., 2020). No additional specimen was collected as a part of this study. The study was validated by the Ethics Committee of the Méditerranée Infection Institute, under agreement number No. 2022-011. Patients were pseudonymized at the beginning of the study.
DNA Extraction and 16S rRNA Sequencing
Samples were extracted using two methods, as previously described (Angelakis et al., 2016). Briefly, a mechanical treatment was performed with powder glass beads, acid-washed (G4649-500g Sigma-Aldrich, St. Louis, United States) and 0.5 mm glass beads cell disruption media (Scientific Industries, Inc., New York, United States) using a FastPrep-24™ 5G Grinder (MP Biomedicals LLC, Corp., Irvine, CA, United States) at maximum speed (6.5 m/sec) for 90 s. Then the samples were treated with two kinds of lysis methods: method 1, with standard lysis and protease step, followed by purification on E.Z.N.A Tissue DNA Kit (Omega Bio-Tek, Norcross, GA, United States), and method 5, using a deglycosylation step and purification on the EZ1 Advanced XL device (Qiagen, Courtaboeuf, France) (Edouard et al., 2018).
Samples were then amplified, pooled, barcoded, and sequenced using Illumina MiSeq technology (Illumina, Inc., San Diego, CA, United States) with paired-end strategy, constructed according to the 16S Metagenomic Sequencing Library Preparation (Illumina).
For each protocol extraction, metagenomic DNA was amplified for the 16S “V3–V4” regions by polymerase chain reaction (PCR) for 45 cycles, using the Kapa HiFi Hotstart ReadyMix 2x (Kapa Biosystems Inc., Wilmington, MA, United States), and the surrounding conserved region V3_V4 primers with overhang adapters FwOvAd_341F
TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTA CGGGNGGCWGCAG; RevOvAd_785R,
GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGAC TACHVGGGTATCTAATCC. After purification on AMPure beads (Beckman Coulter Inc., Fullerton, CA, United States), the concentration was measured using high sensitivity Qubit technology (Beckman Coulter Inc., Fullerton, CA, United States) and dilution to 3.5 ng/μL was performed. At this step, the library of protocol 1 was pooled volume-to-volume to the library for protocol 5, and Illumina sequencing adapters and dual-index barcodes were added to the amplicon. After purification on AMPure beads (Beckman Coulter Inc., Fullerton, CA, United States), the first library was pooled with 95 multiplexed samples and the second library with 41 multiplexed samples. The global concentration was quantified by a Qubit assay with the high sensitivity kit (Life Technologies, Carlsbad, CA, United States).
Before loading for sequencing on MiSeq (Illumina Inc., San Diego, CA, United States) the pool was diluted at 8 pM. Automated cluster generation and paired-end sequencing with dual index reads were performed in a single 39-h run in a 2 bp × 250 bp.
The paired reads were filtered according to the read qualities. The raw data were configured in fastq files for R1 and R2 reads (Edouard et al., 2018).
Identification of Respiratory Bacteria
Real Time PCR
Real-time PCR amplification was performed using the Master LightCycler® 480 Probes kit (Roche Diagnostics, France) according to the manufacturer’s recommendations (Hoang et al., 2019). We amplified DNA of the following bacterial species: Staphylococcus aureus, Streptococcus pneumoniae, Streptococcus pyogenes, Klebsiella pneumoniae, Moraxella catarrhalis, Enterococcus faecalis, and Haemophilus influenzae based on their respective genes: LPSau, lytA, hpt, phoE, CopB, recN, and OMPp1, as previously described (Hoang et al., 2019). We developed a PCR in this study for the specific detection of C. propinquum targeting the DNA polymerase III subunit epsilon, and the primers used were Cprop_Dna3_MBF
TCACACTCACTGGCGAGTTC, Cprop_Dna3_MBR GACT ACCAGCACGGTGGTTT,
and Cprop_Dna3_MBP 6FAM- CCGGTTGTGGTCCGATA TCGC (Supplementary Table 1).
Routine Microbiological Analysis
Microbiological investigations included standard blood cultures (Dubourg et al., 2018), standard cultures of respiratory specimens (i.e., sputa, pleural fluids, bronchoalveolar lavages, bronchial aspirates, and pulmonary biopsies) (Dubourg et al., 2015), pneumococcal antigenic detection from urine specimens (Edelstein et al., 2020), and S. aureus PCR from bronchoalveolar lavage (BAL) (Oh et al., 2013). All positive results detected within 30 days following SARS-CoV-2 diagnosis were considered.
Statistical Analysis
Baseline characteristics between COVID-19 positive and negative groups were compared using the Fisher Exact test (for categorical variables) and the Kruskal–Wallis test (for continuous variables). Data were analyzed using SPSS software (IBM, New York, United States). All p-values < 0.05 were considered significant. MicrobiomeAnalyst software (Chong et al., 2020) was used to analyze 16S ribosomal ribonucleic acid (rRNA) sequencing data, as previously described (Amrane et al., 2019). Linear discriminant analysis (LDA) of effect size (LEfSe) was performed using read counts (Edouard et al., 2018) and presence/absence to determine taxa differentially represented between COVID-19 positive (patients with a favorable outcome, asymptomatic, and deceased patients) and negative groups. Principal component analysis was used to visualize taxa profile and bacterial diversity between different groups (Amrane et al., 2019).
Results
Patients
A total of 120 pseudonymized samples were enrolled in this study, including COVID-19 negative subjects and 90 COVID-19 positive patients (Table 1). According to their clinical outcome, the COVID-19 group was divided into three subgroups of 30 patients each, including patients with a favorable outcome, asymptomatic, and deceased patients. Our study population was predominantly female, with a sex ratio (F/M) of 1.1 in favor of females. The oldest population were the deceased patients, with an overall mean age of 59.03 years.
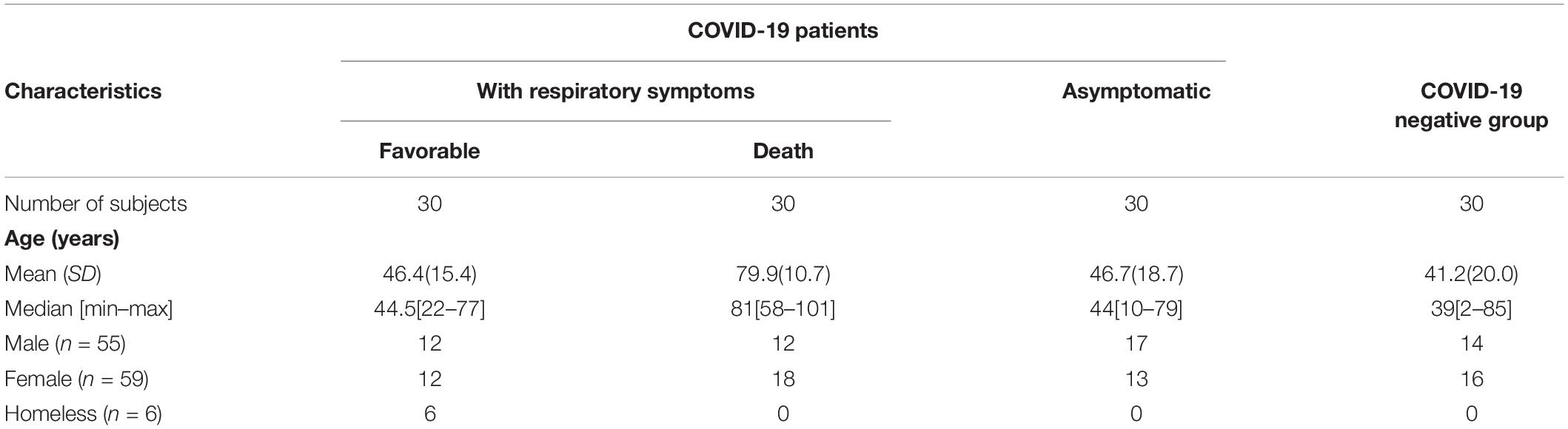
Table 1. Clinical characteristics of patients with COVID-19 (n = 90) and COVID-19 negative group.
General Composition of the Nasopharyngeal Microbiota
Among the 120 pseudonymized patients, we identified overall 1136 distinct OTUs (operational taxonomic unit) by 16S rRNA sequencing. Of these 1136 OTU, 214 were common to all groups, while 69 and 339 OTUs were only found in COVID-19 negative and COVID-19 positive subjects, respectively (Supplementary Figure 1). Proteobacteria, Firmicutes, and Actinobacteria were the phyla most identified, representing 64, 22, and 12%, respectively, of the reads generated among all groups. In asymptomatic COVID-19 patients, we observed an increase of Proteobacteria when compared to COVID-19 negative subjects (p ≤ 0.006), and a decrease of Firmicutes when compared to both deceased COVID-19 patients (p ≤ 0.04) and COVID-19 patients with a favorable outcome (p ≤ 0.004) (Supplementary Figure 2).
The principal component analysis performed on the raw data generated in this study allowed us to observe that the nasopharyngeal microbiota composition was different according to the group studied (Figure 1).
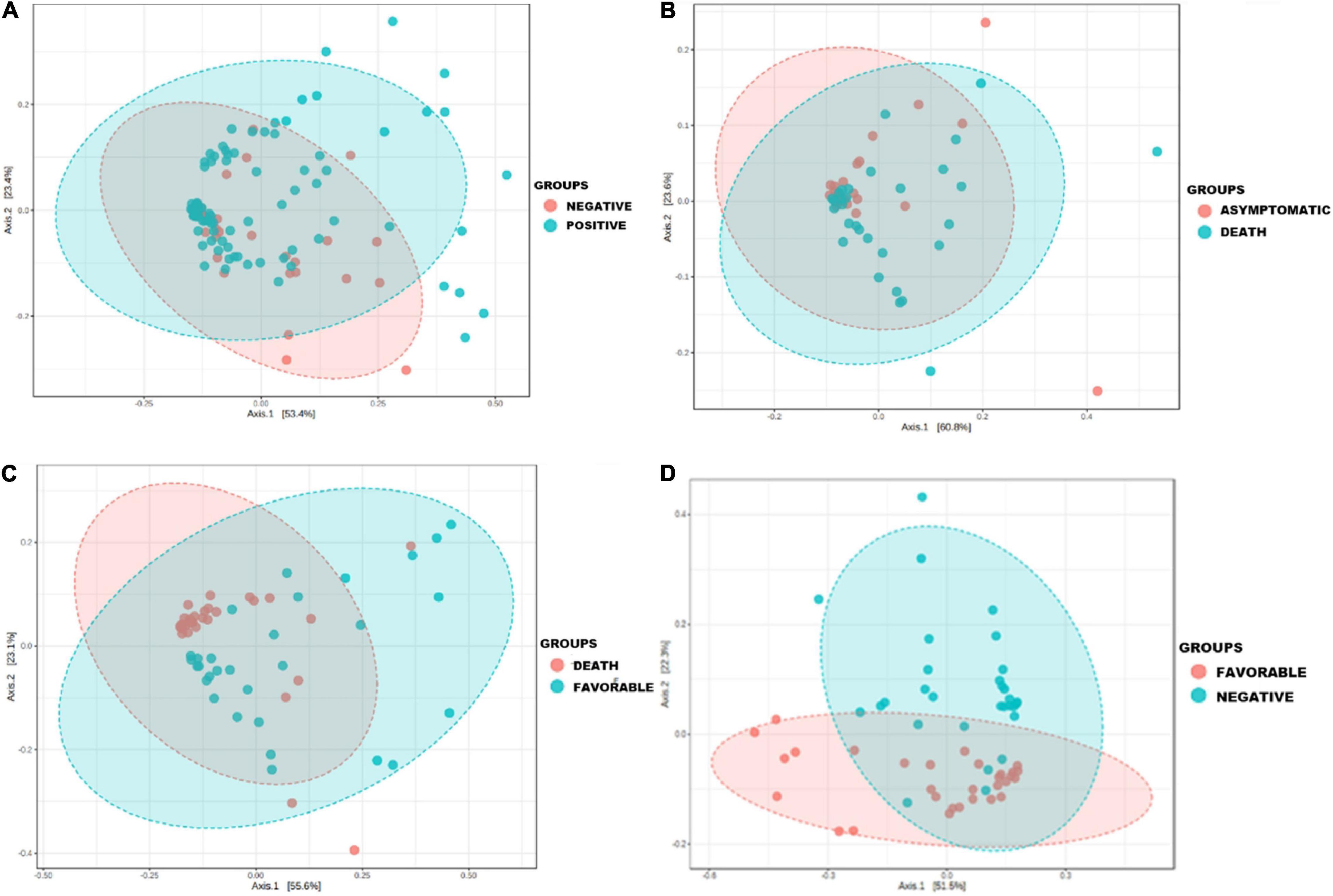
Figure 1. Principal coordinate analysis showing the change in the nasopharyngeal microbiota profile during COVID-19 airway infection in clinical patient groups versus COVID-19 negative group. Principal component analysis (PCoA) was performed on species from raw data (number of reads) using the Jensen Shannon divergence distance matrix and the Multivariate Analysis of Variance (MANOVA) statistical test. Each sample is represented by a point: (A) the microbiota profile of COVID-19 negative patients and cases (p ≤ 0.006); (B) the microbiota profile in asymptomatic patients and deceased patients (p ≤ 0.001); (C) the microbiota profile in patients with a favorable outcome and deceased patients (p ≤ 0.001); (D) the microbiota profile in patients with a favorable outcome and COVID-19 negative individuals (p ≤ 0.001).
The microbiota composition of most samples belonging to the positive group was far different than that belonging to the COVID-19 negative group (p ≤ 0.006) (Figure 1A), with a clear difference in the microbiota profile of deceased patients compared with patients in the favorable outcome group (p ≤ 0.001) (Figure 1C) and an approximation of the microbiota composition of some samples belonging to the favorable outcome group with that of other members of the negative group (p ≤ 0.001) (Figure 1D).
The number of reads per sample was significantly lower in COVID-19 negative subjects compared to COVID-19 patients (p ≤ 0.011), whether compared to that of the COVID-19 patients with a favorable outcome (p ≤ 0.0032) or deceased COVID-19 patients (p ≤ 0.063) subgroups (Supplementary Figure 4).
Specific Airway Microbiota of COVID-19 Patients When Compared to That of Healthy Individuals
First, we found a significant decrease of anaerobic bacteria among COVID-19 positive patients when compared to the COVID-19 negative group (p < 0.029). Indeed, 25% (72/283) of the bacterial species enriched in the latter group were anaerobic, while 20% (66/323) were anaerobic in the asymptomatic subgroup, 19% (60/316) in the deceased group, and 13% (28/219) in patients in the favorable outcome subgroup. Using linear discriminant analysis (LDA), we observed six discriminating OTUs among COVID-19 positive patients, including C. propinquum/pseudodiphtericum (p ≤ 0.05), M. catarrhalis (p ≤ 0.05), B. massiliamazoniensis (p ≤ 0.01), A. alkalidiazotrophicus (p ≤ 0.05), S. capitis subsp. capitis (p ≤ 0.001), and A. birgiae (p ≤ 0.001) (Figure 2 and Supplementary Figure 3A).
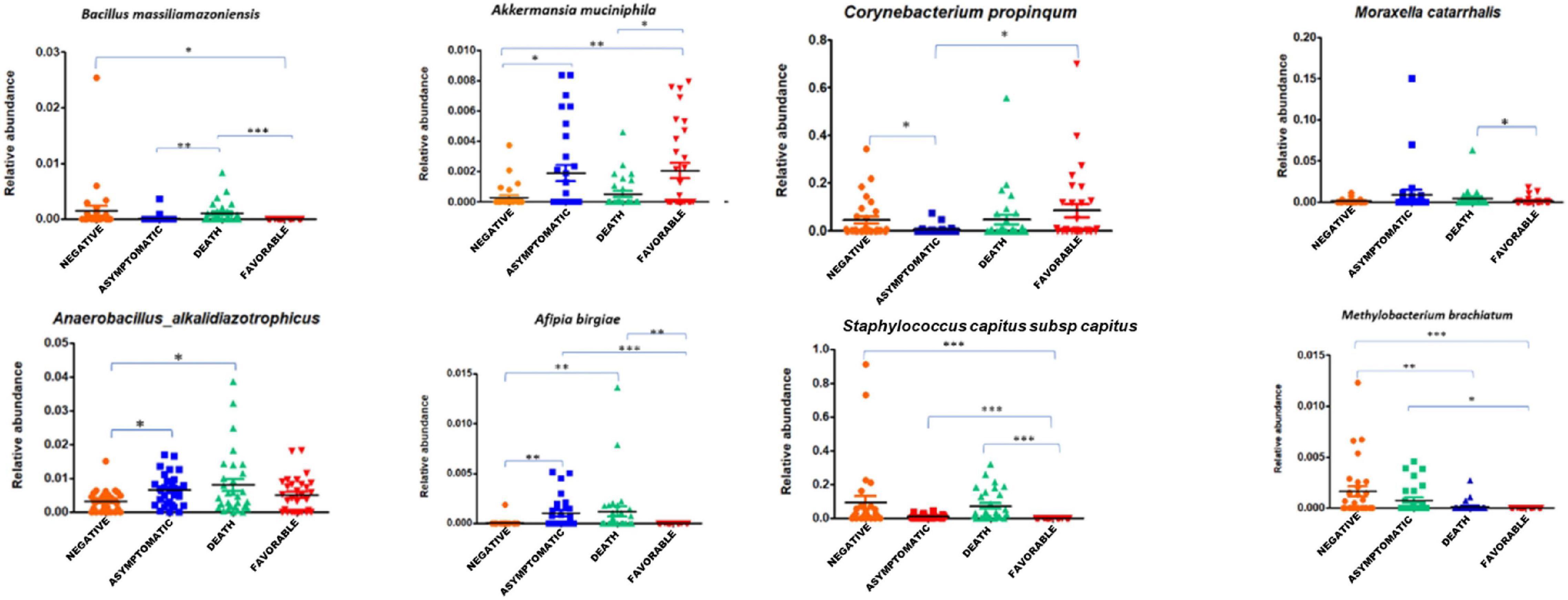
Figure 2. Association of specific taxa in the airway microbiota of COVID-19 patients. The relative abundance of differentially enriched taxa in the four clinical groups of COVID-19 patients (negative, asymptomatic, deceased patients, and patients with a favorable outcome). Five pathogens enrichment in groups of SARS-CoV-2 positive subjects: Corynebacterium propinquum/pseudodiphtericum (p ≤ 0.05), Moraxella catarrhalis (p ≤ 0.05), Bacillus massiliamazoniensis (p ≤ 0.01), Anaerobacillus alkalidiazotrophicus (p ≤ 0.05), and Staphylococcus capitis subsp. capitis (p ≤ 0.001). One-way ANOVA (Kruskal–Wallis statistic). *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001.
In the COVID-19 negative group, Methylobacterium brachiatum (p ≤ 0.001), Peptoniphilus gorbachi (p ≤ 0.01), Finegoldia magna (p ≤ 0.05), and Gemella haemolysans/sanguinis (p ≤ 0.05) were increased when compared to cases (patients with a favorable outcome, asymptomatic, and deceased patients) (Supplementary Figure 3A).
Correlation Between Indicator Taxa and the Clinical Course of the Disease
Subgroup analysis of taxa showed that the species S. capitis subsp. capitis (p ≤ 0.001), C. propinquum (p ≤ 0.01), C. accolens (p ≤ 0.01), and B. massiliamazoniensis (p ≤ 0.05) were enriched among deceased patients when compared to the asymptomatic subgroup (Figure 3A). Staphylococcus argenteus (p ≤ 0.001), Thermoleophilum minutum (p ≤ 0.01), Akkermansia muciniphila (p ≤ 0.01), Schlegelella aquatica (p ≤ 0.01) were enriched in patients with a favorable outcome compared to deceased patients (Figure 3B).
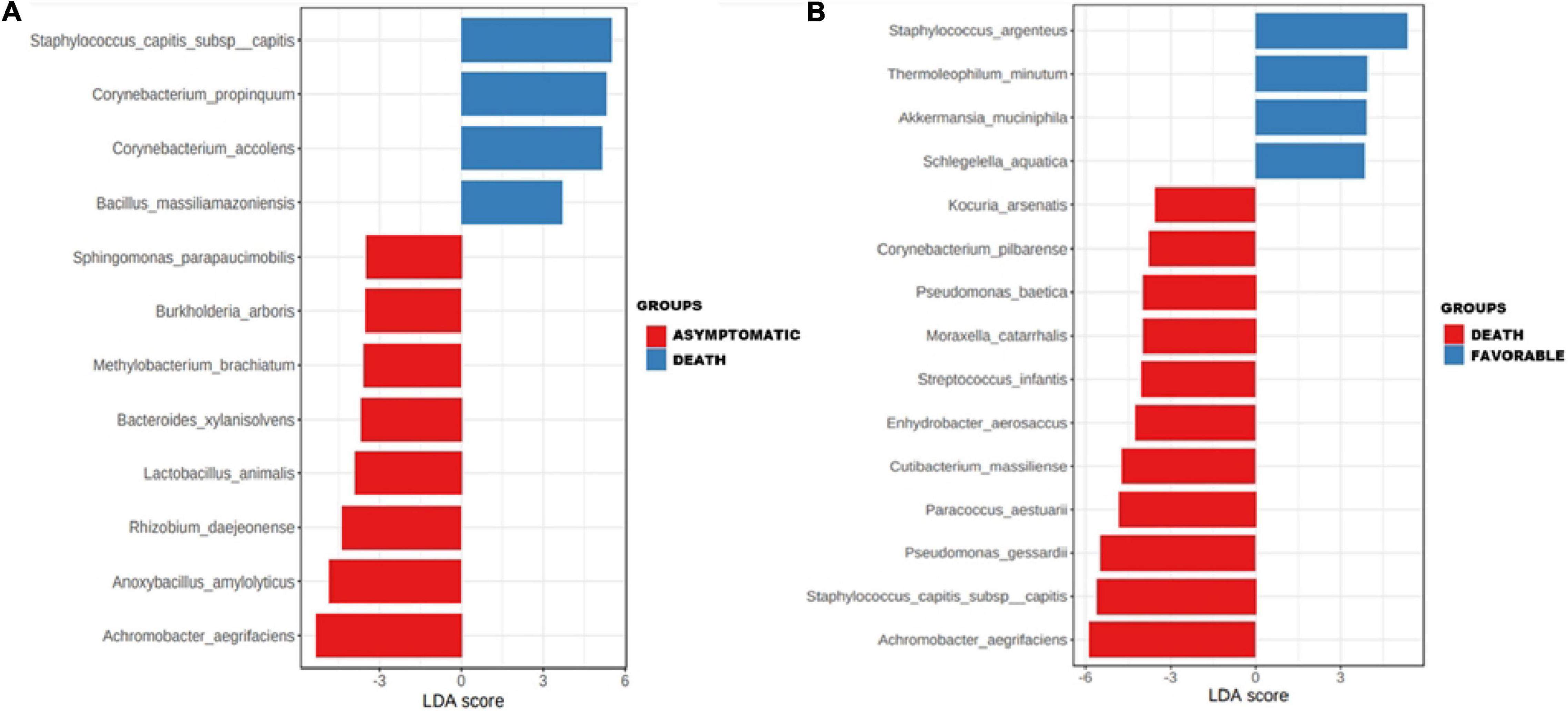
Figure 3. Association of specific taxa in the airway microbiota of COVID-19 patients. Enrichments of specific taxa in the respiratory microbiota of COVID-19 patients in asymptomatic and deceased patients (A) and in deceased patients and patients with a favorable outcome (B) by linear discriminant analysis (LDA) effect size (LEfSe).
Pathogenic Bacteria Amplified by Real-Time PCR and Routine Clinical Microbiology
The PCR tests performed on nasopharyngeal swabs from each group revealed a significantly higher frequency of S. pneumoniae (p ≤ 0.01) in the COVID-19 group when compared to COVID-19 negative subjects, while K. pneumoniae were more frequent in deceased patients (p ≤ 0.01) and patients in the favorable outcome group (p ≤ 0.01) compared to negative individuals and E. faecalis (p ≤ 0.05) in deceased patients compared to the COVID-19 negative group (Figure 4). C. propinquum was more frequently detected among cases (97.5%) than among negative patients (2.5%) (p ≤ 0.000006). However, the detection rate of C. propinquum was lower among asymptomatic patients (10%) than among deceased patients (42.5%) or patients with a favorable outcome (45%) (Figure 4).
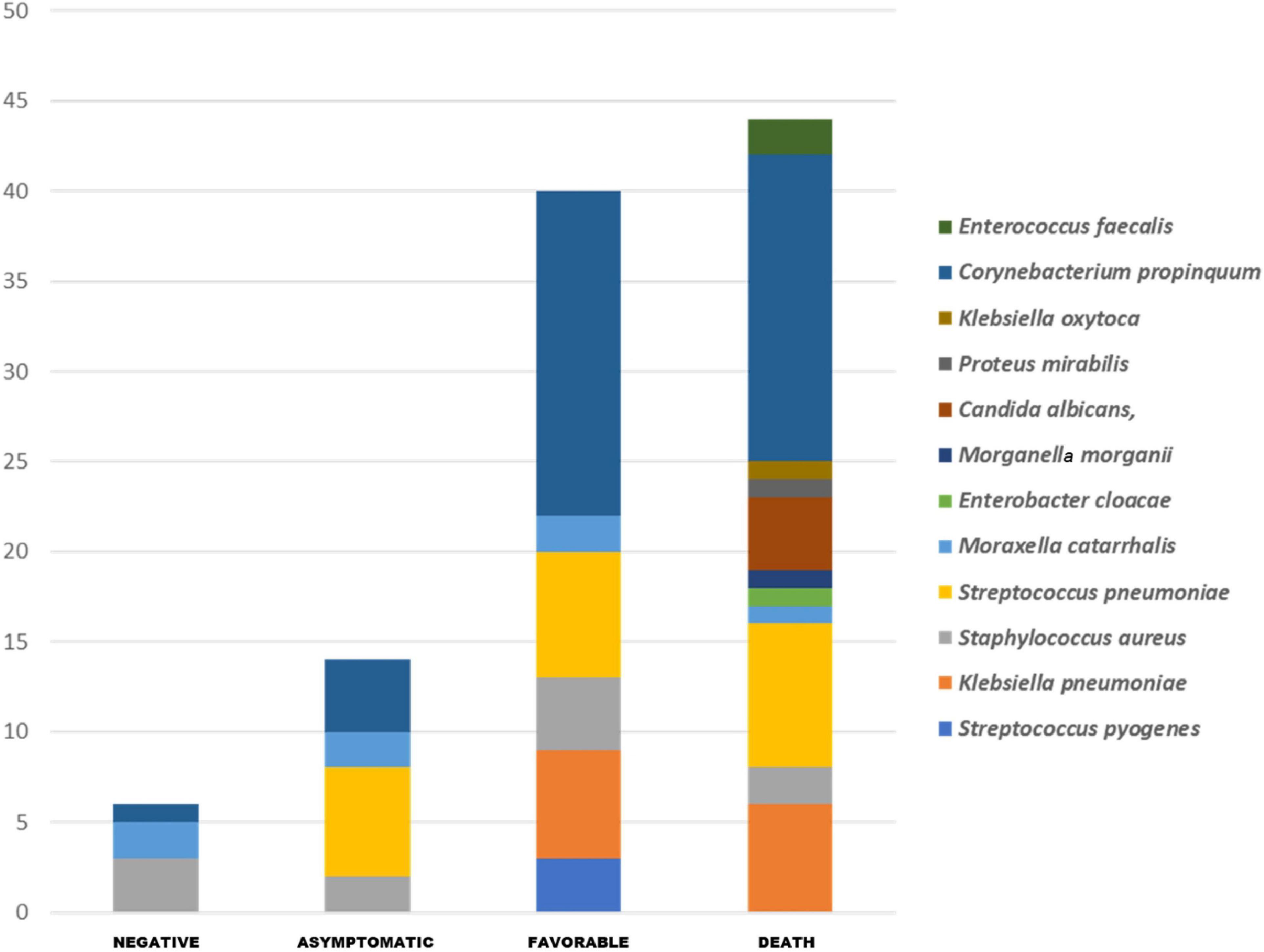
Figure 4. All respiratory pathogens routinely detected in our COVID-19 patients and by specific PCR. The pathogenic bacteria PCR shows the significant enrichment of Streptococcus pneumoniae (p ≤ 0.01) and Corynebacterium propinquum (p ≤ 0.000006) in the group of COVID-19 positive patients, and Klebsiella pneumoniae (p ≤ 0.01) in deceased patients and patients with a favorable outcome group compared to COVID-19 negative subjects, and Enterococcus faecalis (p ≤ 0.05) in deceased patients compared to negative individuals.
Discussion
In the present study, we were able to show that SARS-CoV-2 infection is associated with a modification of the nasopharyngeal microbiota according to different stages of the disease, thus conferring on the microbiota a microbiological signature during the clinical evolution of the infection. First, we found a depletion of anaerobes among COVID-19 patients, irrespective of the clinical presentation of the infection. These results are in line with the study of Edouard et al. (2018) that included patients with respiratory infections involving various viruses, but not SARS-CoV-2. These data suggest that the loss of anaerobes could be a specific consequence of respiratory viral infections. We also highlighted an increase of Proteobacteria in asymptomatic COVID-19 patients in contrast to COVID-negative individuals and a considerable decrease of Firmicutes. These differences corroborate those observed by De Maio et al. (2020) who despite their analysis did not find statistically significant differences, which could be explained by their relatively smaller sample size (Table 2).
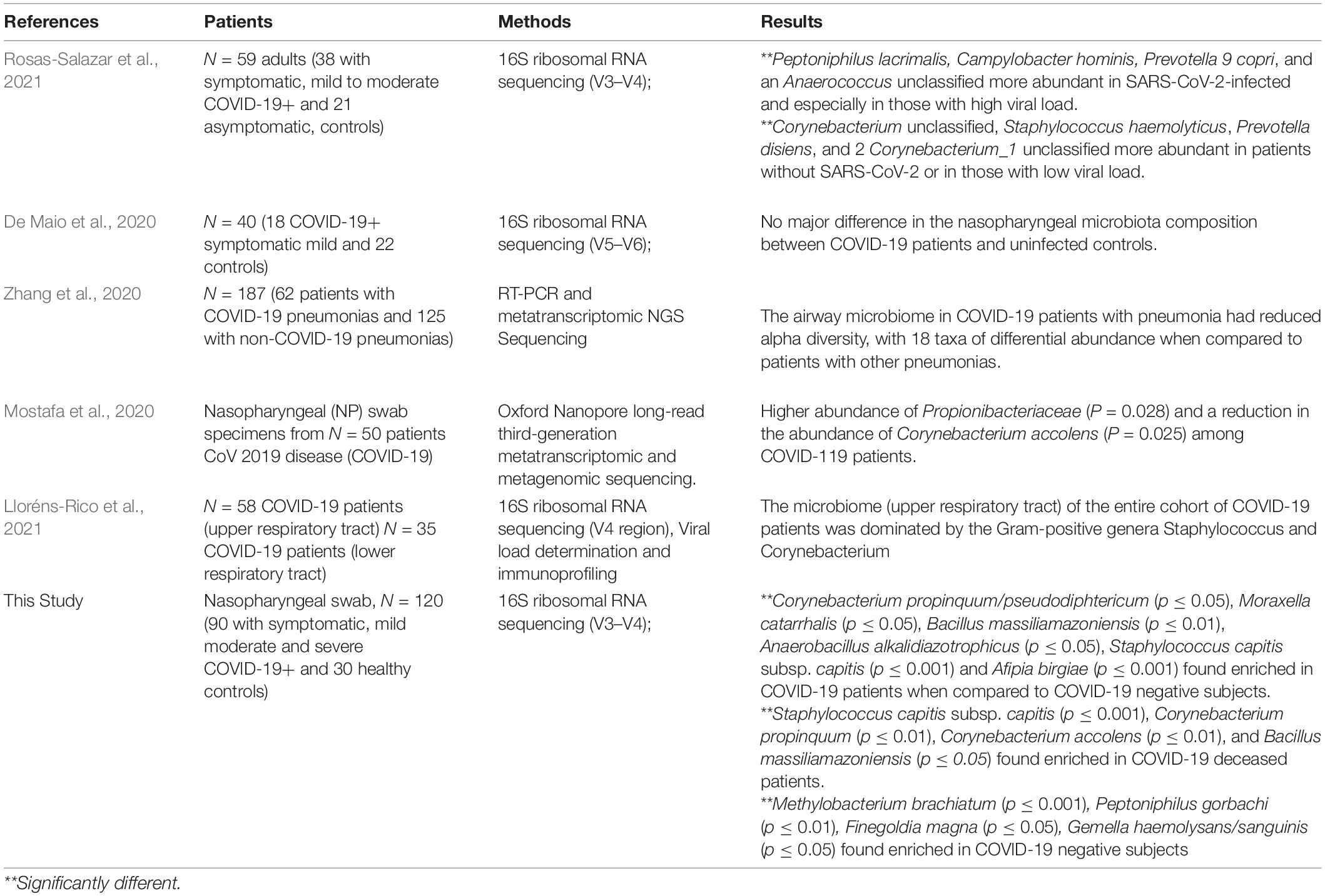
Table 2. The predominant nasopharyngeal microbiota in COVID-19 patients.
These results are in line with other studies (Rosas-Salazar et al., 2021) that have observed a modification of the nasopharyngeal microbiota between symptomatic COVID-19 patients and negative subjects (Table 2).
Regarding the clinical course of the disease, we found a significant difference in the microbial profile of each sample grouped by severity index in the COVID-19 case groups, when compared to COVID-19 negative group (p ≤ 0.006), using principal component analysis. This diversity of the microbial community in the COVID-19 patient groups according to the severity index was observed by Mostafa et al. (2020) in a study conducted on nasopharyngeal swabs from 2 distinct groups of patients (a COVID-19 positive group and a COVID-19 negative group) (Table 2). Similar findings have been reported regarding infections involving other respiratory viruses (Edouard et al., 2018).
At the species level, we found 8 taxa enriched in the nasopharyngeal microbiota of COVID-19 positive patients when compared to COVID-19 negative subjects including C. propinquum/pseudodiphtericum, M. catarrhalis, S. pneumoniae, K. pneumoniae, and E. faecalis. Interestingly, these pathogens were previously found to be enriched in patients with respiratory infections involving other viruses (DeMuri et al., 2018; Edouard et al., 2018), suggesting that their overrepresentation may not be specific to COVID-19. However, a recent study conducted on a cohort of COVID-19 patients showed that the upper respiratory tract microbiome of COVID-19 patients is dominated by the Corynebacterium and Staphylococcus genera (Lloréns-Rico et al., 2021)(Table 2). In addition, C. propinquum and C. accolens belong to the 4 taxa enriched in deceased patients when compared to COVID-19 negative individuals. Interestingly, the latter was found to be more abundant in non-COVID-19 subjects when compared to COVID-19 patients in another study (Mostafa et al., 2020) (Table 2). C. propinquum was, however, much less detected among asymptomatic subjects than in patients who died or had a favorable outcome, raising the question of its involvement in the onset of symptoms. The genus Corynebacterium is predominant in the nasopharynx (Maxime et al., 2018) and the species C. propinquum primarily colonizes the nasopharyngeal region and is frequently isolated from respiratory specimens in patients with infection with pulmonary involvement (Riegel et al., 1993; Motomura et al., 2004). S. pneumoniae, which was increased among COVID-19 subjects, is a common respiratory pathogen and an agent of superinfections during viral episodes. Nasopharyngeal colonization with S. pneumoniae was associated with an impaired antiviral immune response in COVID-19 patients (Mitsi et al., 2022), while we did not observe this in our subgroups. E. faecalis, more frequent among deceased patients than in COVID-19 negative subjects, is surprisingly a main agent of bacteremia among COVID-19 patients, particularly in ICU settings (Giacobbe et al., 2021; Porto et al., 2022). Among the potentially protective species, we highlighted a decrease of A. muciniphila in deceased subjects when compared to those with a favorable outcome. To our knowledge, this species has been very rarely detected in respiratory samples but is well known for its anti-inflammatory properties, among others (Cani and de Vos, 2017). Finally, we have no explanation for the increase in Proteobacteria in asymptomatic subjects, as the presence of this phylum in the respiratory tract is frequently associated with inflammation or disease (Dickson et al., 2016).
Our study is comprehensive because it includes a large population size according to the clinical course of the disease, a total of 120 patients. We also acknowledge some limitations, such as the absence of a test for other respiratory viruses in our samples, which could have influenced our results. In addition, we cannot exclude that the changes in the composition of the nasopharyngeal microbiota observed in the present study are linked with alterations in the flora of other respiratory sites, such as the oral cavity or the lung. Indeed, the respiratory tract harbors a homogeneous microbiota whose biomass decreases from the upper to the lower tract (Charlson et al., 2011). However, the study demonstrates in the setting of COVID-19 a difference between upper respiratory tract bacteria obtained from nasopharyngeal swab and lower respiratory tract bacteria obtained from bronchoalveolar lavage (BAL) samples (Lloréns-Rico et al., 2021). Finally, we acknowledge that the analysis of the nasopharyngeal microbiota in our patients was performed after the diagnosis of COVID-19 disease. Therefore, we do not know if these changes were pre-existing before the SARS-CoV-2 infection. This is a topic for investigation, as the microbiota predisposing to respiratory viral infections is known from the literature for other pathogens (Allen et al., 2014; Maxime et al., 2018; Dubourg et al., 2019).
In summary, we have shown that the microbiota of COVID-19 patients may have a signature at different stages of the disease. Although we did not consider other respiratory infections during our evaluation, further studies considering co-infections with other viruses should be conducted to confirm whether several species, especially C. propinquum, could influence the course of the disease in order to include it as a predictor of COVID-19 severity. Nevertheless, we cannot state with certainty whether these bacteria will predict the development of secondary infections.
Conclusion and Perspectives
In this study, we were able to observe a specific microbial signature of the nasopharyngeal microbiota of COVID-19 patients (Figure 5), in particular a significant decrease in anaerobes. Among the species of interest, the role of C. propinquum/diphteriticum, M. catarrhalis, and C. accolens remains to be elucidated. As all COVID-19 patients were included during the first pandemic and were thereby infected with the same variant, it would be interesting to further investigate a possible correlation between the modification of the nasopharyngeal microbiota according to the different COVID-19 variants.
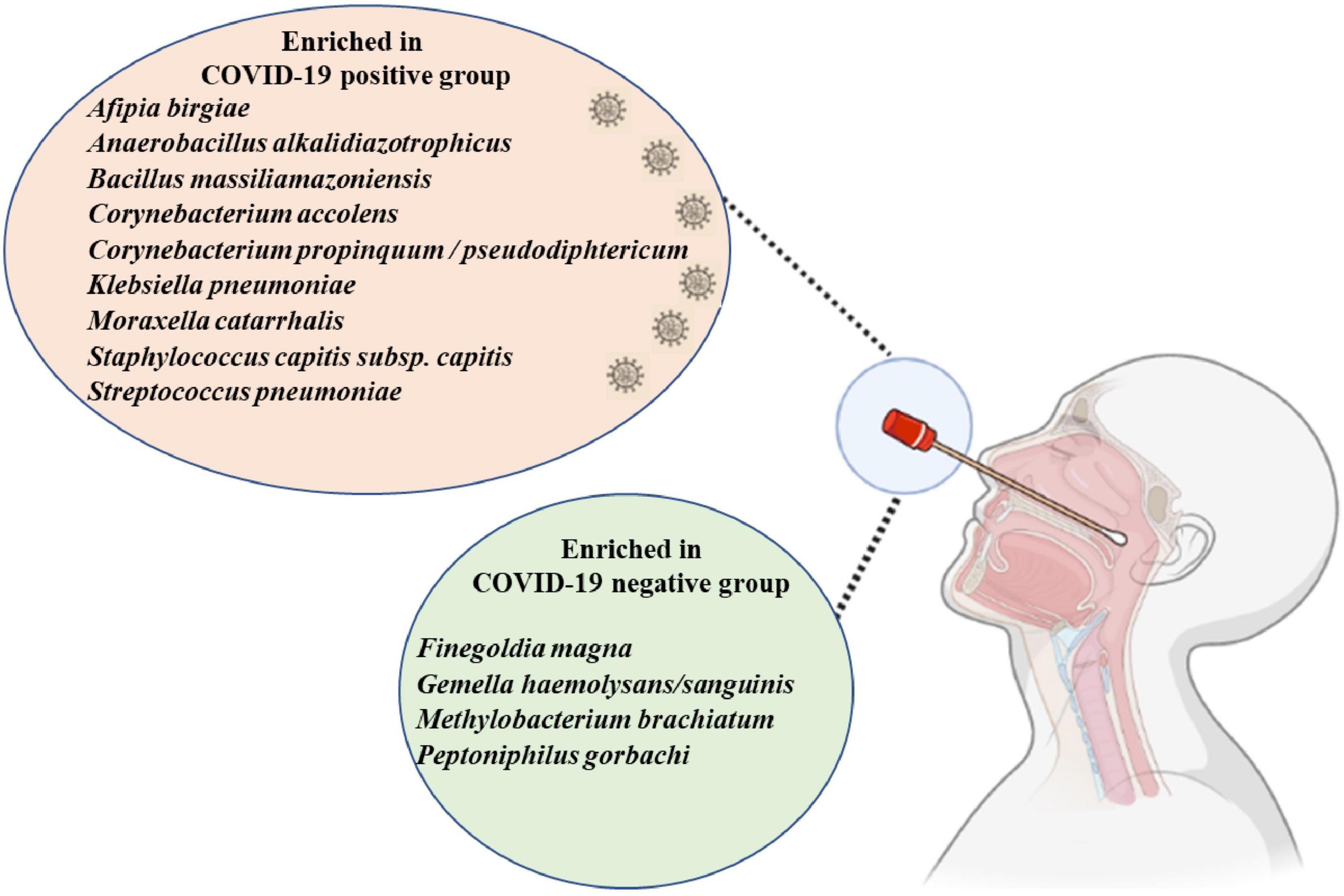
Figure 5. The enriched nasopharyngeal microbiota bacteria in the COVID-19 positive group and COVID-19 negative group. Generated using Biorender (https://biorender.com).
Data Availability Statement
The datasets presented in this study can be found in the NCBI Sequence Read Archive under BioProject ID: PRJNA801106.
Ethics Statement
This study consisted in studying pseudonymized samples not specifically obtained for this context but rather clinical samples remaining from diagnostic screenings. The patients were informed of the possible use of their samples for research purposes and retained their right to refuse approval at any point. According to the French Jardé Law (Law No. 2012–300 of March 05, 2012 and Decree No. 2016–1537 of November 16, 2016 published in the “Journal Officiel de la République Française”), as this study did not involve a specific collection of samples, neither institutional ethics approval nor individual patient consent was required for this non-invasive study. However, this general approach of retrospective study was validated by the Ethics Committee of the Méditerranée Infection Institute, under agreement no. 2022-011. The analysis of collected data followed the MR-004 reference methodology registered under no. 2020-152 and 2020-151 in the AP-HM register.
Author Contributions
DR, J-CL, and GD conceived and designed the experiments. OT and VB contributed materials and analysis tools. OT, GD, and AY analyzed the data. OT and GD wrote the manuscript. J-CL and DR reviewed the manuscript. All authors approved the final version of the manuscript.
Funding
This work was supported by the French Government under the “Investments for the Future” program managed by the National Agency for Research (ANR), Méditerranée-Infection 10-IAHU-03, and was also supported by Région Provence Alpes Côte d’Azur and European funding FEDER PRIMMI (Fonds Européen de Développement Régional-Plateformes de Recherche et d’Innovation Mutualisées Méditerranée Infection), FEDER PA 0000320 PRIMMI.
Conflict of Interest
DR is a scientific board member of Eurofins company and a founder of a microbial culture company (Culture Top) and was a consultant for Hitachi High-Technologies Corporation, Tokyo, Japan, from 2018 to 2020.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are very grateful to the genomic platform for training and support in the development of the protocols and the analysis of the sequences, in particular, Pierre Edouard Fournier and Ludivine Brechard. We also thank the Bioinformatics engineers, namely, Aurelia Caputo, Marielle Bedotto, and Jeremy Delerce. Finally, for their participation in technical help, we thank Maryam Tidjani Alou, Souad Belkacemi, Camille Valles, Fatou Samba Diouf, Salimata Konate, and Luis Tsakou Gouaffo.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.871627/full#supplementary-material
Supplementary Figure 1 | Venn diagram illustrating the different taxa specific to each group of COVID-19 patients.
Supplementary Figure 2 | Relative abundance of phyla observed in all COVID-19 patients and in each group: an increase of Proteobacteria was observed in COVID-19 asymptomatic patients, compared to COVID-19 negative subjects (p ≤ 0.006). A decrease of Firmicutes was observed in COVID-19 asymptomatic patients compared to deceased patients (p ≤ 0.04) and patients with a favorable outcome (p ≤ 0.004).
Supplementary Figure 3 | Association of specific taxa in the airway microbiota of COVID-19 patients. Enrichments of specific taxa in the respiratory microbiota of COVID-19 patients in (A) negative and COVID-19 patients (asymptomatic patients, deceased patients, and patients with a favorable outcome groups) and in (B) asymptomatic and deceased patients by presence-absence.
Supplementary Figure 4 | The number of reads per sample representing each group of COVID-19 patients.
Supplementary Figure 5 | All respiratory pathogens detected in our COVID-19 patients. Red: Specific real-time PCR. Green: Routine microbiological analysis.
Supplementary Table 1 | Primers design: Corynebacterium propinquum.
Abbreviations
ACE2, angiotensin converting enzyme 2; COVID, corona virus disease; DNA, desoxyribonucleic acid; IHU, Institut Hospitalo-Universitaire; LDA, linear discriminant analysis; LEfSe, linear discriminant analysis effect size; OTUs, operational taxonomic units; PCoA, principal coordinates analysis; PCR, polymerase chain reaction; rRNA, ribosomal ribonucleic acid; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
References
Allameh, S. F., Nemati, S., Ghalehtaki, R., Mohammadnejad, E., Aghili, S. M., Khajavirad, N., et al. (2020). Clinical characteristics and outcomes of 905 COVID-19 patients admitted to Imam Khomeini hospital complex in the capital city of Tehran. Iran. Arch. Iran. Med. 23, 766–775. doi: 10.34172/aim.2020.102
Allen, E. K., Koeppel, A. F., Hendley, J. O., Turner, S. D., Winther, B., and Sale, M. M. (2014). Characterization of the nasopharyngeal microbiota in health and during rhinovirus challenge. Microbiome 2:22. doi: 10.1186/2049-2618-2-22
Amrane, S., Hocquart, M., Afouda, P., Kuete, E., Pham, T. P. T., Dione, N., et al. (2019). Metagenomic and culturomic analysis of gut microbiota dysbiosis during Clostridium difficile infection. Sci. Rep. 9:12807. doi: 10.1038/s41598-019-49189-8
Angelakis, E., Bachar, D., Henrissat, B., Armougom, F., Audoly, G., Lagier, J. C., et al. (2016). Glycans affect DNA extraction and induce substantial differences in gut metagenomic studies. Sci. Rep. 6:26276. doi: 10.1038/srep26276
Bogaert, D., Keijser, B., Huse, S., Rossen, J., Veenhoven, R., van Gils, E., et al. (2011). Variability and diversity of nasopharyngeal microbiota in children: a metagenomic analysis. PLoS One 6:e17035. doi: 10.1371/journal.pone.0017035
Brugger, S. D., Frei, L., Frey, P. M., Aebi, S., Muhlemann, K., and Hilty, M. (2012). 16S rRNA terminal restriction fragment length polymorphism for the characterization of the nasopharyngeal microbiota. PLoS One 7:e52241. doi: 10.1371/journal.pone.0052241
Cani, P. D., and de Vos, W. M. (2017). Next-generation beneficial microbes: the case of Akkermansia muciniphila. Front. Microbiol. 8:1765. doi: 10.3389/fmicb.2017.01765
Charlson, E. S., Bittinger, K., Haas, A. R., Fitzgerald, A. S., Frank, I., Yadav, A., et al. (2011). Topographical continuity of bacterial populations in the healthy human respiratory tract. Am. J. Respir. Crit. Care Med. 184, 957–963. doi: 10.1164/rccm.201104-0655OC
Chong, J., Liu, P., Zhou, G. Y., and Xia, J. G. (2020). Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 15, 799–821. doi: 10.1038/s41596-019-0264-1
Colson, P., Lagier, J. C., Baudoin, J. P., Khalil, J. B., La Scola, B., and Raoult, D. (2020). Ultrarapid diagnosis, microscope imaging, genome sequencing, and culture isolation of SARS-CoV-2. Eur. J. Clin. Microbiol. Infect. Dis. 39, 1601–1603. doi: 10.1007/s10096-020-03869-w
Dahyot, A., Baruch, J., Aubert, R., Ferrer, M., Sanchez, L., Dagorn, G., et al. (2021). Covid-19: The Dashboard of the Epidemic. Available online at: https://www.lemonde.fr/les-decodeurs/article/2020/05/05/coronavirus-age-mortalite-departements-pays-suivez-l-evolution-de-l-epidemie-en-cartes-etgraphiques_6038751_4355770.html (accessed May 18, 2021).
De Maio, F., Posteraro, B., Ponziani, F. R., Cattani, P., Gasbarrini, A., and Sanguinetti, M. (2020). Nasopharyngeal microbiota profiling of SARS-CoV-2 infected patients. Biol. Proced. Online 22:18. doi: 10.1186/s12575-020-00131-7
DeMuri, G. P., Gern, J. E., Eickhoff, J. C., Lynch, S. V., and Wald, E. R. (2018). Dynamics of bacterial colonization with Streptococcus pneumoniae, Haemophilus influenzae, and Moraxella catarrhalis during symptomatic and asymptomatic viral upper respiratory tract infection. Clin. Infect. Dis. 66, 1045–1053. doi: 10.1093/cid/cix941
Dhar, D., and Mohanty, A. (2020). Gut microbiota and Covid-19-possible link and implications. Virus Res. 285:198018. doi: 10.1016/j.virusres.2020.198018
Dickson, R. P., Singer, B. H., Newstead, M. W., Falkowski, N. R., Erb-Downward, J. R., Standiford, T. J., et al. (2016). Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat. Microbiol. 1:16113. doi: 10.1038/nmicrobiol.2016.113
Ding, Q., Lu, P. P., Fan, Y. H., Xia, Y. J., and Liu, M. (2020). The clinical characteristics of pneumonia patients coinfected with 2019 novel coronavirus and influenza virus in Wuhan, China. J. Med. Virol. 92, 1549–1555. doi: 10.1002/jmv.25781
Dubourg, G., Abat, C., Rolain, J. M., and Raoult, D. (2015). Correlation between sputum and bronchoalveolar lavage fluid cultures. J. Clin. Microbiol. 53, 994–996. doi: 10.1128/jcm.02918-14
Dubourg, G., Baron, S., Cadoret, F., Couderc, C., Fournier, P. E., Lagier, J. C., et al. (2018). From culturomics to clinical microbiology and forward. Emerg. Infect. Dis. 24, 1683–1690. doi: 10.3201/eid2409.170995
Dubourg, G., Edouard, S., and Raoult, D. (2019). Relationship between nasopharyngeal microbiota and patient’s susceptibility to viral infection. Expert Rev. Anti Infect. Ther. 17, 437–447. doi: 10.1080/14787210.2019.1621168
Edelstein, P. H., Jorgensen, C. S., and Wolf, L. A. (2020). Performance of the ImmuView and BinaxNOW assays for the detection of urine and cerebrospinal fluid Streptococcus pneumoniae and Legionella pneumophila serogroup 1 antigen in patients with Legionnaires’ disease or pneumococcal pneumonia and meningitis. PLoS One 15:e0238479. doi: 10.1371/journal.pone.0238479
Edouard, S., Million, M., Bachar, D., Dubourg, G., Michelle, C., Ninove, L., et al. (2018). The nasopharyngeal microbiota in patients with viral respiratory tract infections is enriched in bacterial pathogens. Eur. J. Clin. Microbiol. Infect. Dis. 37, 1725–1733. doi: 10.1007/s10096-018-3305-8
Gautret, P., Million, M., Jarrot, P.-A., Camoin-Jau, L., Colson, P., Fenollar, F., et al. (2020). Natural history of COVID-19 and therapeutic options. Expert Rev. Clin. Immunol. 16, 1159–1184. doi: 10.1080/1744666X.2021.1847640
Giacobbe, D. R., Labate, L., Tutino, S., Baldi, F., Russo, C., Robba, C., et al. (2021). Enterococcal bloodstream infections in critically ill patients with COVID-19: a case series. Ann. Med. 53, 1779–1786. doi: 10.1080/07853890.2021.1988695
Han, M. S., Byun, J. H., Cho, Y., and Rim, J. H. (2021). RT-PCR for SARS-CoV-2: quantitative versus qualitative. Lancet Infect. Dis. 21:165. doi: 10.1016/s1473-3099(20)30424-2
Hasegawa, K., Mansbach, J. M., Ajami, N. J., Espinola, J. A., Henke, D. M., Petrosino, J. F., et al. (2016). Association of nasopharyngeal microbiota profiles with bronchiolitis severity in infants hospitalised for bronchiolitis. Eur. Respir. J. 48, 1329–1339. doi: 10.1183/13993003.00152-2016
Hoang, V. T., Goumballa, N., Dao, T. L., Ly, T. D. A., Ninove, L., Ranque, S., et al. (2019). Respiratory and gastrointestinal infections at the 2017 Grand Magal de Touba, Senegal: a prospective cohort survey. Travel Med. Infect. Dis. 32:101410. doi: 10.1016/j.tmaid.2019.04.010
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., Erichsen, S., et al. (2020). SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8. doi: 10.1016/j.cell.2020.02.052
Lloréns-Rico, V., Gregory, A. C., Van Weyenbergh, J., Jansen, S., Van Buyten, T., Qian, J., et al. (2021). Clinical practices underlie COVID-19 patient respiratory microbiome composition and its interactions with the host. Nat. Commun. 12:6243. doi: 10.1038/s41467-021-26500-8
Lundstrom, A., Ziegler, L., Havervall, S., Rudberg, A. S., von Meijenfeldt, F., Lisman, T., et al. (2021). Soluble angiotensin-converting enzyme 2 is transiently elevated in COVID-19 and correlates with specific inflammatory and endothelial markers. J. Med. Virol. 93, 5908–5916. doi: 10.1002/jmv.27144
Man, W. H., Clerc, M., Piters, W., van Houten, M. A., Chu, M., Kool, J., et al. (2019). Loss of microbial topography between oral and nasopharyngeal microbiota and development of respiratory infections early in life. Am. J. Respir. Crit. Care Med. 200, 760–770. doi: 10.1164/rccm.201810-1993OC
Maxime, P., Lina, B., and Josset, L. (2018). Caractérisation et impact du microbiote bactérien respiratoire sur les maladies virales. Virologie 22, 161–172. doi: 10.1684/vir.2018.0739
Mitsi, E., Reiné, J., Urban, B. C., Solorzano, C., Nikolaou, E., Hyder-Wright, A. D., et al. (2022). Streptococcus pneumoniae colonization associates with impaired adaptive immune responses against SARS-CoV-2. J. Clin. Invest. 132:e157124. doi: 10.1172/jci157124
Mostafa, H. H., Fissel, J. A., Fanelli, B., Bergman, Y., Gniazdowski, V., Dadlani, M., et al. (2020). Metagenomic next-generation sequencing of nasopharyngeal specimens collected from confirmed and suspect COVID-19 patients. Mbio 11:e01969-20. doi: 10.1128/mBio.01969-20
Motomura, K., Masaki, H., Terada, M., Onizuka, T., Shimogama, S., Furumoto, A., et al. (2004). [Three adult cases with Corynebacterium propinquum respiratory infections in a community hospital]. Kansenshogaku Zasshi 78, 277–282. doi: 10.11150/kansenshogakuzasshi1970.78.277
Na, Z., Zhang, D., Wang, W., Li, X., Yang, B., Song, J., et al. (2020). A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733. doi: 10.1056/NEJMoa2001017
Oh, A. C., Lee, J. K., Lee, H. N., Hong, Y. J., Chang, Y. H., Hong, S. I., et al. (2013). Clinical utility of the Xpert MRSA assay for early detection of methicillin-resistant Staphylococcus aureus. Mol. Med. Rep. 7, 11–15. doi: 10.3892/mmr.2012.1121
Porto, A. P. M., Borges, I. C., Buss, L., Machado, A., Bassetti, B. R., Cocentino, B., et al. (2022). Healthcare-associated infections on the ICU in 21 Brazilian hospitals during the early months of the COVID-19 pandemic: an ecological study. Infect. Control Hosp Epidemiol 1–37. doi: 10.1017/ice.2022.65
Riegel, P., de Briel, D., Prévost, G., Jehl, F., and Monteil, H. (1993). Proposal of Corynebacterium propinquum sp. nov. for Corynebacterium group ANF-3 strains. FEMS Microbiol. Lett. 113, 229–234. doi: 10.1111/j.1574-6968.1993.tb06519.x
Rosas-Salazar, C., Kimura, K. S., Shilts, M. H., Strickland, B. A., Freeman, M. H., Wessinger, B. C., et al. (2021). SARS-CoV-2 infection and viral load are associated with the upper respiratory tract microbiome. J. Allergy Clin. Immunol. 147, 1226–1233.e2. doi: 10.1016/j.jaci.2021.02.001
Shilts, M. H., Rosas-Salazar, C., Strickland, B. A., Kimura, K. S., Asad, M., Sehanobish, E., et al. (2021). Severe COVID-19 is associated with an altered upper respiratory tract microbiome. Front. Cell. Infect. Microbiol. 11:781968. doi: 10.3389/fcimb.2021.781968
Weinstock, G. M. (2012). Genomic approaches to studying the human microbiota. Nature 489, 250–256. doi: 10.1038/nature11553
Wen, Z. X., Xie, G., Zhou, Q., Qiu, C. Z., Li, J., Hu, Q., et al. (2018). Distinct nasopharyngeal and Oropharyngeal microbiota of children with influenza a virus compared with healthy children. Biomed Res. Int. 2018:6362716. doi: 10.1155/2018/6362716
Keywords: COVID-19, SARS-CoV-2, nasopharyngeal microbiota, 16S rRNA sequencing, metagenomics
Citation: Tchoupou Saha Olf, Dubourg G, Yacouba A, Bossi V, Raoult D and Lagier J-C (2022) Profile of the Nasopharyngeal Microbiota Affecting the Clinical Course in COVID-19 Patients. Front. Microbiol. 13:871627. doi: 10.3389/fmicb.2022.871627
Received: 08 February 2022; Accepted: 11 April 2022;
Published: 17 May 2022.
Edited by:
George Grant, University of Aberdeen, United KingdomReviewed by:
Kundlik Gadhave, Johns Hopkins University, United StatesShigefumi Okamoto, Kanazawa University, Japan
Alessandra Pierangeli, Sapienza University of Rome, Italy
Gislane Lelis Vilela de Oliveira, São Paulo State University, Brazil
Yogesh Singh, University of Tübingen, Germany
Copyright © 2022 Tchoupou Saha, Dubourg, Yacouba, Bossi, Raoult and Lagier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Christophe Lagier, amVhbi1jaHJpc3RvcGhlLmxhZ2llckB1bml2LWFtdS5mcg==; Grégory Dubourg, Z3JlZy5kdWJvdXJnQGdtYWlsLmNvbQ==