 Wenzi Ren
Wenzi Ren Reijo Penttilä2
Reijo Penttilä2 Risto Kasanen
Risto Kasanen Fred O. Asiegbu
Fred O. Asiegbu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 03 May 2022
Sec. Microbe and Virus Interactions with Plants
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.864619
This article is part of the Research Topic Insights in Microbe and Virus Interactions with Plants: 2021 View all 14 articles
The microbiome of Heterobasidion-induced wood decay of living trees has been previously studied; however, less is known about the bacteria biota of its perennial fruiting body and the adhering wood tissue. In this study, we investigated the bacteria biota of the Heterobasidion fruiting body and its adhering deadwood. Out of 7,462 operational taxonomic units (OTUs), about 5,918 OTUs were obtained from the fruiting body and 5,469 OTUs were obtained from the associated dead wood. Interestingly, an average of 52.6% of bacteria biota in the fruiting body was shared with the associated dead wood. The overall and unique OTUs had trends of decreasing from decay classes 1 to 3 but increasing in decay class 4. The fruiting body had the highest overall and unique OTUs number in the fourth decay class, whereas wood had the highest OTU in decay class 1. Sphingomonas spp. was significantly higher in the fruiting body, and phylum Firmicutes was more dominant in wood tissue. The FAPROTAX functional structure analysis revealed nutrition, energy, degradation, and plant-pathogen-related functions of the communities. Our results also showed that bacteria communities in both substrates experienced a process of a new community reconstruction through the various decay stages. The process was not synchronic in the two substrates, but the community structures and functions were well-differentiated in the final decay class. The bacteria community was highly dynamic; the microbiota activeness, community stability, and functions changed with the decay process. The third decay class was an important turning point for community restructuring. Host properties, environmental factors, and microbial interactions jointly influenced the final community structure. Bacteria community in the fruiting body attached to the living standing tree was suppressed compared with those associated with dead wood. Bacteria appear to spread from wood tissue of the standing living tree to the fruiting body, but after the tree is killed, bacteria moved from fruiting body to wood. It is most likely that some of the resident endophytic bacteria within the fruiting body are either parasitic, depending on it for their nutrition, or are mutualistic symbionts.
The genus Heterobasidion within the phylum Basidiomycota contains 13 species with saprotrophic and necrotrophic lifestyles. The species complex, H. annosum s.l., has a necrotrophic lifestyle and contains five species, namely, H. abietinum, H. annosum sensu stricto (s.s.), H. irregulare, H. parviporum, and H. occidentale (Ioos et al., 2019). Among these, H. annosum and H. parviporum are the most severe root rot disease causal agents, posing a major threat to conifer forests of the northern hemisphere (Asiegbu et al., 2005). The primary infection of Heterobasidion is mediated by basidiospores dispersal and germination on wounds or exposed stump surfaces. The secondary infection continuously spreads the disease to the adjacent healthy tree by root-to-root contact (Korhonen and Stenlid, 1998; Zaluma et al., 2019). The current control methods include silvicultural, chemical, and biological control approaches (Mesanza et al., 2016). Chemical and biological control methods are the most efficient (Pellicciaro et al., 2021). However, the application of chemical treatment such as urea is restricted in Europe due to its potential negative impacts on the environment (EU Reg. 2020/1160). In contrast, the persistent use of the biocontrol product (e.g., Rotstop), which is based on a single natural isolate of Phlebiopsis gigantea, might increase the ecological risks of pathogen tolerance (GŽibovska, 2016). Exploring other alternative efficient control approaches is therefore required.
Fruiting body is a highly nutritious multicellular structure containing spores of fungi and other microbes (Maurice et al., 2021). Its morphology, sporulation period, and longevity vary in different species (Moore et al., 2008). Heterobasidion species tend to be active when the temperature is above 5°C, and the spore deposition rate for each square meter can be over 1,000 basidiospores per hour (Gonthier et al., 2005; Müller et al., 2014). The spreading distance could reach 1,000 m (Möykkynen et al., 1997). It has been observed that spruce logs left on peat soil form the most abundant fruiting body (Gaitnieks et al., 2021), cull lumbar piece was also found to be significant to the fruiting body formation (Müller et al., 2007). The fruiting body provides habitat or food resources for diverse organisms, including bacteria and fungi (Elliott et al., 2019; Legzdina et al., 2021; Maurice et al., 2021). The interactions between Heterobasidion and associated organisms influence the development of each other and will produce a dynamic counterbalance (Deveau et al., 2018). The fruiting body associated with wood often has biochemical mechanism to defend itself against external injuries, however, with the progression of the decay process, biochemical compounds fade away, and the physical structure of fruiting body changes (Venkatesh and Keller, 2019; Legzdina et al., 2021; Maurice et al., 2021). These physical-chemical property variations influence the microbial interactions inside the fruiting body (Gohar et al., 2020). Thus, the microbiome composition of fruiting body might be of relevance for Heterobasidion primary infection. Despite the potential importance of fruiting body microbiome in the ecological interactions (Gohar et al., 2020; Maurice et al., 2021), almost no information is available on Heterobasidion fruiting body.
In contrast, deadwood is a major substrate for most wood-inhabiting fungi, and the associated fungal fruiting bodies play an important ecological role in the ecosystem. Deadwood is important for nutrient cycling, carbon sequestration, soil property, and biodiversity (Brunner and Kimmins, 2003; Litton et al., 2007; Rondeux and Sanchez, 2010; Błońska et al., 2017). Both quality and quantity of deadwood have impact on its ecological roles (Müller and Bütler, 2010). Deadwood might be also an incubator for forest diseases, since pathogens with dual lifestyles are known to survive over decades on deadwood by aid of their saprotrophic feeding (Cleary et al., 2013). Heterobasidion has been reported to remain in deadwood and retain infectious activity for over 62 years after clear cutting. Also, the Heterobasidion fruiting body can develop on 25 to 45-year-old rotted spruce wood (Piri, 1996; Gaitnieks et al., 2021). Deadwood volume, property, and mechanisms for its decay are all vital parameters for forest management (Müller and Bütler, 2010). A number of recent studies have reported on Heterobasidion's decay impact on trees, the tree resistance against Heterobasidion, the microbiome associated with the pathosystem but not much on the fruiting body microbiome (Puentes Rodriguez et al., 2013; Keilhofer et al., 2018; Probst et al., 2018; Ren et al., 2019; Pellicciaro et al., 2021).
The bacteria-fungi or fruiting body interactions (BFIs) could be beneficial, neutral, or detrimental to each other (Wargo and Hogan, 2006). The symbionts could be obligate or facultative. The obligate symbiotic bacteria are usually beneficial to its fungal host, as they are highly dependent, while the facultative bacteria have more diverse influences. Endosymbiont usually has a positive effect, while the ectosymbiont's effects on hosts are diverse (Bastías et al., 2020).
Bacillus subtilis, a soil-dwelling bacterium, can form a biofilm to attach on the fungus surface (Kjeldgaard et al., 2019). Bacterium Pseudomonas fluorescens was recorded to stimulate both colonization and growth of the fungus Laccaria bicolor (Deveau et al., 2007). In a mutual interaction, bacteria consume metabolites from the fungus and contribute amino acids in return (Lackner et al., 2011) or benefit of carbon and nitrogen exchange (Sharmin et al., 2018). The symbiont could also help the fungus (Guo et al., 2017) or bacteria (Ruiz-Lozano and Bonfante, 2000) to establish or facilitate invasion to other fungi (Spraker et al., 2016) or plant tissues (Ruiz-Lozano and Bonfante, 2000). At the other extreme, the BFIs could also suppress each other. Strain GB 4-2 from the genus Streptomyces was reported to suppress Heterobasidion infection through promoting plant resistance (Lehr et al., 2008). However, some bacteria genera have an opportunistic influence on fungal pathogenicity (e.g., Pseudomonas spp.) (GŽibovska, 2016; Lipps and Samac, 2021; Pellicciaro et al., 2021).
Bacteria-fungi or fruiting body interactions can also increase host's tolerance to stress and promote nutrient uptake as well as plant growth (Steffan et al., 2020; Asiegbu and Kovalchuk, 2021).
The structure of microbial community in this pathosystem might result as a consequence of tripartite (bacteria-fruiting body-plant) overall interactions. Understanding the structure of the community is therefore the crucial foundation to unravel the functional mechanism of the interacting partners. Other than their direct and indirect effect on each other, many other factors also contribute to the microbial structure formation. Soil properties (Pent et al., 2017), host genetics (Maurice et al., 2021), host physical and chemical properties (Maurice et al., 2021), and microclimate (Fravolini et al., 2016) have been reported to have profound influence on the microbial structure. Besides, being different from dead wood, the appearance of fruiting body may already indicate a specific environment background. For example, moisture and temperature were recorded to have a significant impact on the fungus sporulation (Moore et al., 2008). The microbes inhabiting the fruiting body could have a smaller range compared with those in dead wood. The aims of this study were to unravel (1) the bacteria biota of fruiting body and associated dead wood of different decay classes, (2) their possible nature and functional characteristics, and (3) using the network model to unravel their potential interactions among each other, as well as possible impact on Heterobasidion pathogenesis.
In each sampling spot, Heterobasidion fruiting body and the associated wood were collected; the dead wood was classified to decay classes 1 to 4 based on how deep the knife was able to penetrate the wood (Mäkinen et al., 2006). Samples were collected from a total of 38 spots. Notably, 12 of the fruiting bodies were from wood classified as decay class 1, i.e., recently dead wood; eight from decay class 2, weakly decayed; five from decay class 3, medium decayed; and 13 from decay class 4, highly decayed (decay classes represented as D1F–D4F for fruiting body and D1W–D4W for wood). All samples were collected from six Norway spruce dominated boreal forest sites in Uusimaa region, located in Viikki (Helsinki) and Myrskylä, two sites from Lapinjärvi, and two sites from Sipoo. Forest types under different management purposes were also considered in the sampling. Viikki forest site is mainly used for recreational purpose; sites from Sipoo and one site from Lapinjärvi are nature conservation areas, with or without minor human interference; and the other sites from Lapinjärvi and Myrskylä forest are managed sites.
The fruiting body and associated dead wood samples were collected from stumps, logs, and standing tree wood. Wood tissue immediately below and attached to the basidiocarp was collected, with the aid of a sharp knife and an ax if necessary. The stand age of sampling sites ranged from 0 to 109 years, the canopy cover ranged from 0% to over 71%. The elevation of sites ranged from 7.42 to 102.67 m above the sea level. The diameter of selected wood ranged from 9 to 40 cm for managed forest and from 15 to 40 cm for unmanaged forest. Stand age and canopy cover of each spot were obtained from National Land Survey of Finland (Supplementary Table 1; https://kartta.paikkatietoikkuna.fi/). Samples were stored in a cooling box before storage in −20°C. Sample collections were conducted in June 2020.
A combination of visual morphological identification and ITS-based analysis was used to confirm that the collected fruiting bodies were Heterobasidion spp., and that the associated deadwood was infected by it.
All samples were prewashed over running water followed by immersion in 70% ethanol (EtOH) for 1 min and 1% commercial bleach for 5 min and rinsed several times with sterile distilled water in order to remove surface adhering microbes. Samples were dried with paper towel and air-dried before homogenization (Moreno et al., 2014). To avoid bias, subsamples from the fruiting body were collected from four directions as well as the middle part, whereas the entire wood sample was fully homogenized. Samples were extracted following a standard cetyltrimethylammonium bromide (CTAB) method (Chang et al., 1993) with modifications (Terhonen et al., 2011). The concentration and purity of DNA were measured using the NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, USA). PCR amplification of the bacterial 16S rDNA V3–V4 region and sequencing were performed at Novogene (Cambridge Science Park, UK). The PCR products were purified and sequenced with the Illumina NovaSeq platform. The amplification primers were 341F 5′-CCTAYGGGRBGCASCAG-3′ and 806R 5′-GGACTACNNGGGTATCTAAT-3′. Approximately 250 nucleotides per sequencing reads were produced. Raw sequences obtained in this study are available from the Sequence Read Archive (SRA) of National Center for Biotechnology Information (NCBI) under project number PRJNA800074.
The raw 16S rRNA sequences were preprocessed at Novogene, UK. The paired-end reads were merged using FLASH Version 1.2.7 (Magoč and Salzberg, 2011); the pair-end reads were spliced when reads overlapped with the same fragment. The sequence data were filtered by Qiime pipeline (Caporaso et al., 2010) to get high-quality sequences. Chimera was detected by comparing to Gold database with the UCHIME algorithm (Haas et al., 2011) and removed using the UPARSE software (Edgar, 2013). The preprocessed data were analyzed and grouped into operational taxonomic units (OTUs) using UPARSE. Sequences with ≥97% similarity were assigned to the same OTUs (Wang et al., 2007). Representative sequence for each OTU was screened for further annotation. OTUs were assigned to taxonomic groups using the Mothur software by performing sequence against the SSUrRNA database of SILVA Database (Quast et al., 2013) (taxonomic rank threshold: 0.8–1). Normalization was conducted using a standard of sequence number corresponding to the sample with the least sequences. The subsequent analysis was performed based on this normalized data. Chao1 (Chao, 1984), Shannon diversity index (H) (Shannon and Weaver, 1949), and Shannon evenness index (SEI) (Heip, 1974) were chosen to represent community species richness, diversity, and evenness. One-way ANOVA tests were conducted to identify the differences in community richness, diversity, evenness, and abundance of taxonomic groups. Principal coordinates analysis (PCoA) was used to visualize the bacterial community structure with weighted unifrac distance matrix using relative abundance of OTUs calculated by the QIIME software (Version 1.7.0). PCoA was performed by vegan package and ggplot2 package in the R software (Version 2.15.3). Functional annotation of prokaryotic taxa (FAPROTAX) database was used to identify bacterial function via the annotation of 16S sequence classification (Louca et al., 2016; Sansupa et al., 2021). Linear discriminant analysis (LDA) effect size (LEfSe) and one-way ANOVA tests were used to identify bacterial taxonomic and functional groups difference between different decay class samples (Segata et al., 2011). We implemented microbiome ecological network (de Vries et al., 2018) by calculating the spearman correlation of OTUs abundance among different groups through psych package in R and visualized the result by Gephi (Version 0.9.2).
There were 6,198,431 high-quality sequences in total after denoising and quality filtering. After filtering out unclassified sequences, singletons, and sequences assigned to plant and animal specific sequences, a core set of 5,921,960 sequences was obtained. Due to the technical problem of PCR amplification and sequencing, eight samples with low read numbers were excluded; the remaining 68 samples were used for further analysis. Details on the deleted samples are stated in Supplementary Table 1. The remaining samples had sequence numbers ranging from 49,011 to 114,758 with the average number as 87,088 ± 12,577 (mean ± standard deviation). A random normalization was conducted based on the smallest sample size of 49,011 sequences among all samples and was used in the further analysis. All samples had over 99% of average Good's coverage. The rarefaction curves in Supplementary Figure 1 showed the sequencing depth.
The bacterial species richness (Chao1), diversity (Shannon diversity), and evenness (Shannon even) did not differ neither among the fruiting body and dead wood in the four decay classes nor between fruiting body and wood under the same decay class (P > 0.05; Figure 1).
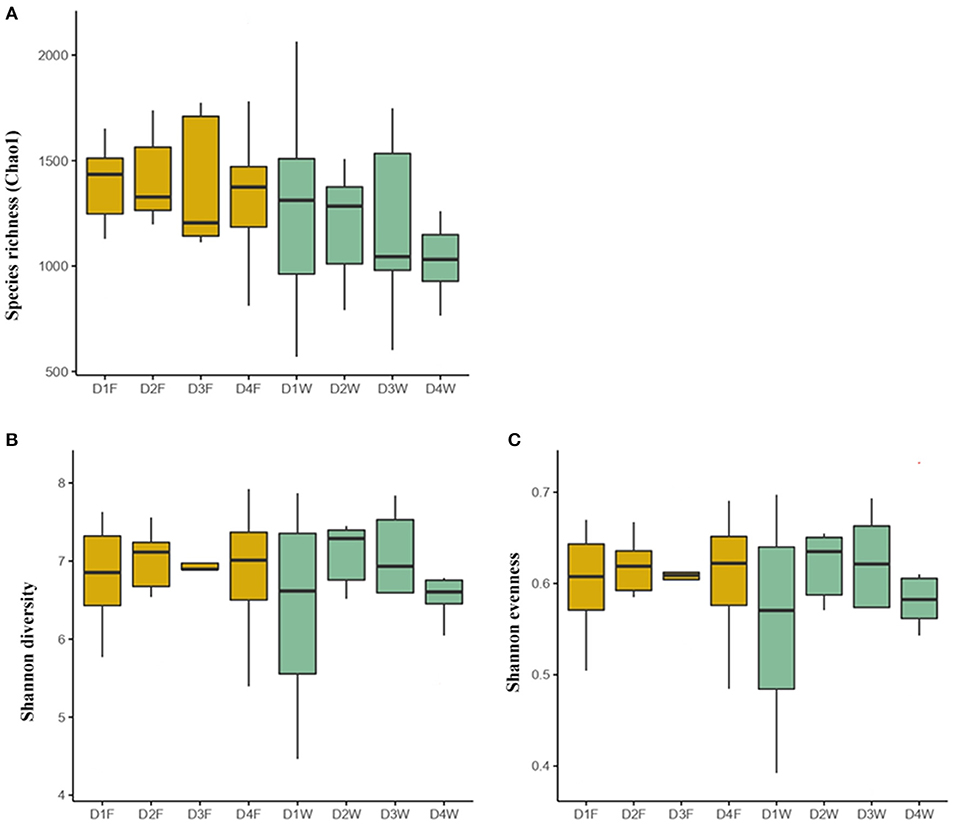
Figure 1. The bacterial community indices in the fruiting body and wood of different decay classes. (A) Species richness, (B) Shannon diversity, and (C) Shannon evenness. The outliers were removed. D1F and D1W refer to the fruiting body and wood from the first decay class; D2F and D2W refer to the fruiting body and wood from the second decay class; D3F and D3W refer to the fruiting body and wood from the third decay class; D4F and D4W refer to the fruiting body and wood from the fourth decay class.
Across all samples, a total of 7,462 OTUs were obtained, where 5,918 OTUs were obtained from fruiting body and 5,469 OTUs were obtained from wood, 52.6% of all OTUs was shared between the two substrates. The Venn diagram revealed that the overall, common, and unique OTUs from both substrates had a varying trend of high abundance in decay class 1 with decreases in decay classes 2 and 3, and high again in decay class 4 (Figures 2A,B).
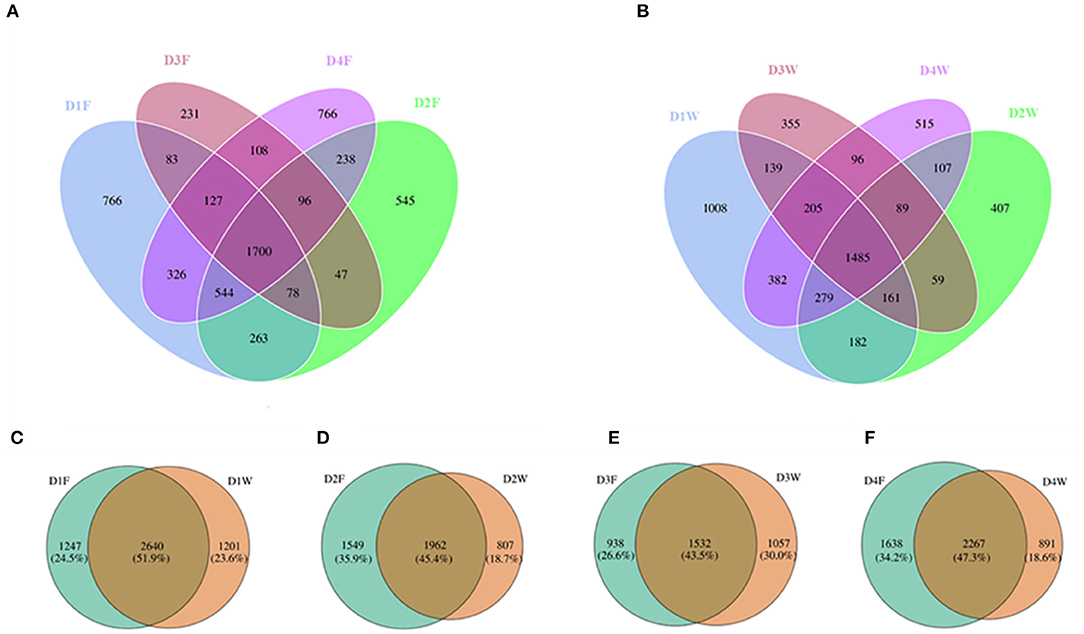
Figure 2. Venn diagram showing the unique and shared operational taxonomic units (OTUs) among different decay classes from (A) fruiting body, (B) dead wood, between fruiting body and dead wood from decay classes 1 (C), 2 (D), 3 (E), and 4 (F).
When comparing OTU numbers between wood and fruiting body in the same decay class, decay class 1 showed the highest proportion of OTUs that were common to both fruiting body and wood, and decay class 3 showed the lowest (Figures 2C,E). Wood had the maximum overall and unique OTUs in D1W and minimum in D3W and D2W (Figures 2C–E). The fruiting body had the maximum overall and unique OTUs in D4F and minimum in D3F (Figures 2E,F). In the last decay class, the fruiting body had 391 more OTUs compared with those in the initial decay stages, while wood had 310 less.
Principal coordinates analysis based on OTUs data explained 39.4% of the variation, the clusters were distinct between fruiting body and dead wood in all decay class except decay class 1 (Figure 3). The subsequent PERMANOVA confirmed the significant differences of all classes (P < 0.05). However, the differences were not significant among decay classes in the same substrate (Supplementary Figures 2, 3; P > 0.05).
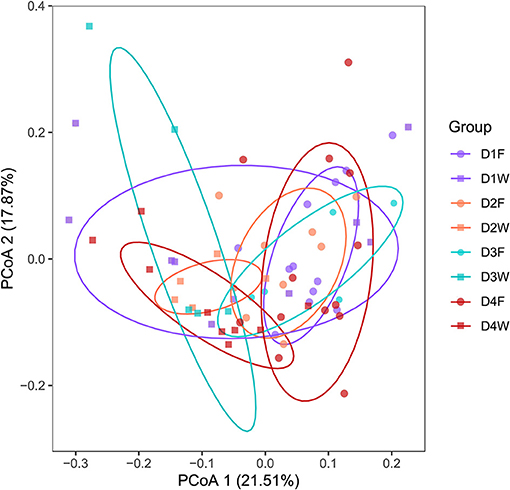
Figure 3. Principal-coordinate analysis (PCoA) showing the bacterial community structures of fruiting body and wood from four decay classes.
In the top 50 most abundant OTUs, several OTUs contributed to the significant differences between fruiting body and dead wood in decay classes 1, 3, and 4. In classes 1 and 3, the significant patterns were higher bacteria abundance in fruiting body than dead wood. In decay class 4, both substrates had two significant abundant OTUs (Table 1A).
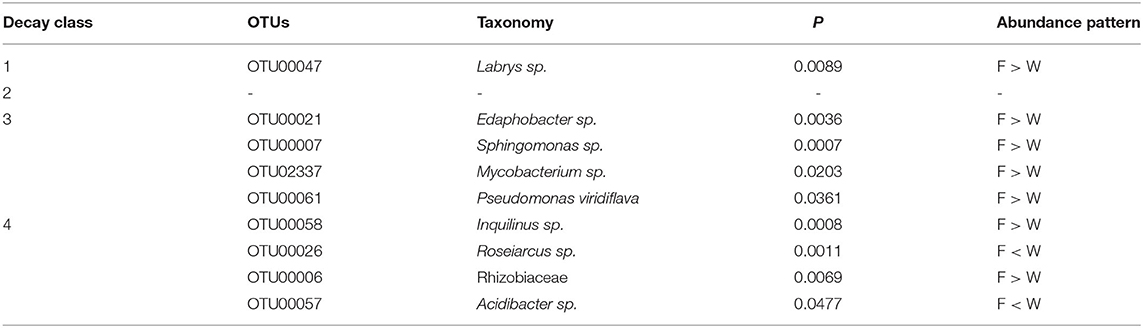
Table 1A. The most abundant operational taxonomic units (OTUs) in top 50 showing significant different patterns between fruiting body (F) and wood (W) in each decay class.
Considering the same substrate, only the third decay class had significantly higher abundant OTUs than the other decay classes in the fruiting body, while in dead wood, all decay classes except class 1 had significant abundant OTUs (Table 1B).
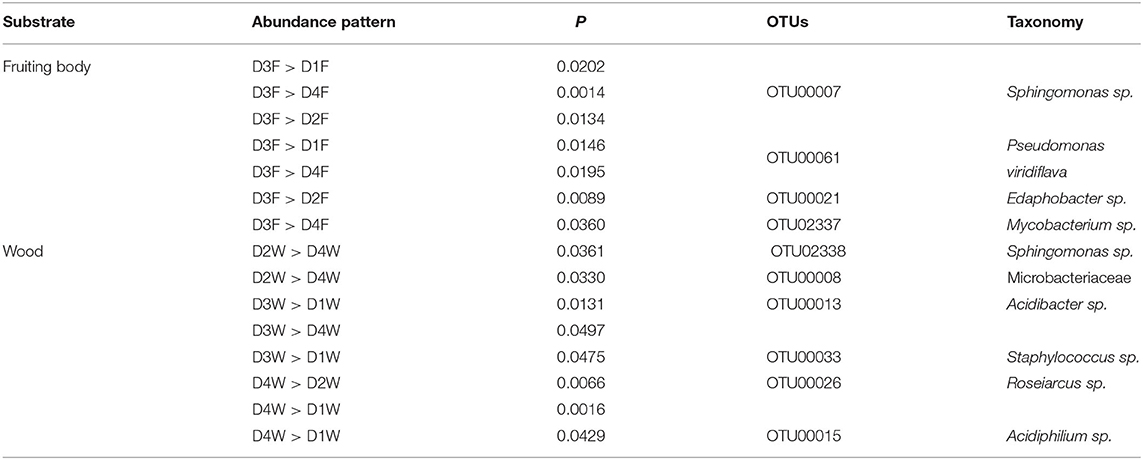
Table 1B. The most abundant OTUs in top 50 showing significant different patterns among four decay classes (D) from fruiting body (F) and wood (W).
Sequences were classified to 34 phyla, Proteobacteria was the most dominant of all groups, followed by Actinobacteria, Acidobacteria, Bacteroidetes, and Firmicutes, where they accounted for over 93.7% of the total sequences. The changing patterns of relative abundance in both substrates were the same in phyla Actinobacteria, Acidobacteria, and Cyanobacteria, while phyla Firmicutes and Bacteroidetes had the opposite trend (Figure 4A).
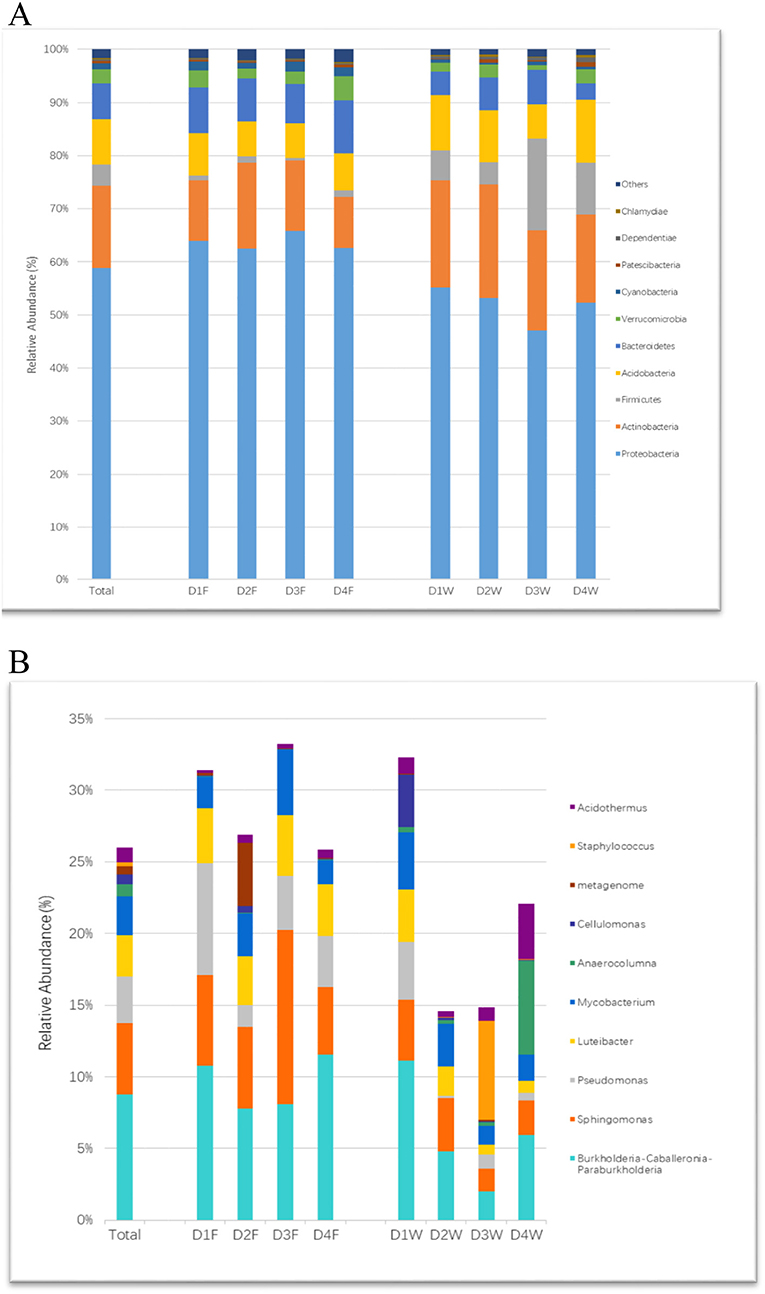
Figure 4. The relative abundance of bacterial community at different taxonomic levels in study groups. (A) Phylum and (B) genus.
In the fruiting body, Burkholderia, Sphingomonas, and Pseudommonas were the most abundant identified genera. The genus Luteibacter had stable abundance associated with all decay classes of the fruiting body. In wood tissue, each genus had quite balanced proportions in the first decay stage, but the proportion of most genera reduced with prolonged decay (Figure 4B).
The Genus Staphylococcus had the highest relative abundance in D3W and was significantly higher than D1F, D4F, and D1W among all the group comparisons (Figure 4B; P < 0.05). The genus Sphingomonas was significantly higher in D3F than D1W–D4W (Figure 4B; P < 0.05).
Linear discriminant analysis effect size revealed that decay classes 2, 3, and 4 had significant higher taxonomic groups in the fruiting body and wood. In the fruiting body, decay class 4 had more abundant taxonomies, but wood had more in decay class 2 (LDA > 2.0, P < 0.05; Figures 5A,B). Similar differences also existed between both substrates in all classes (LDA > 4.0, P < 0.05; Figures 5C–F).
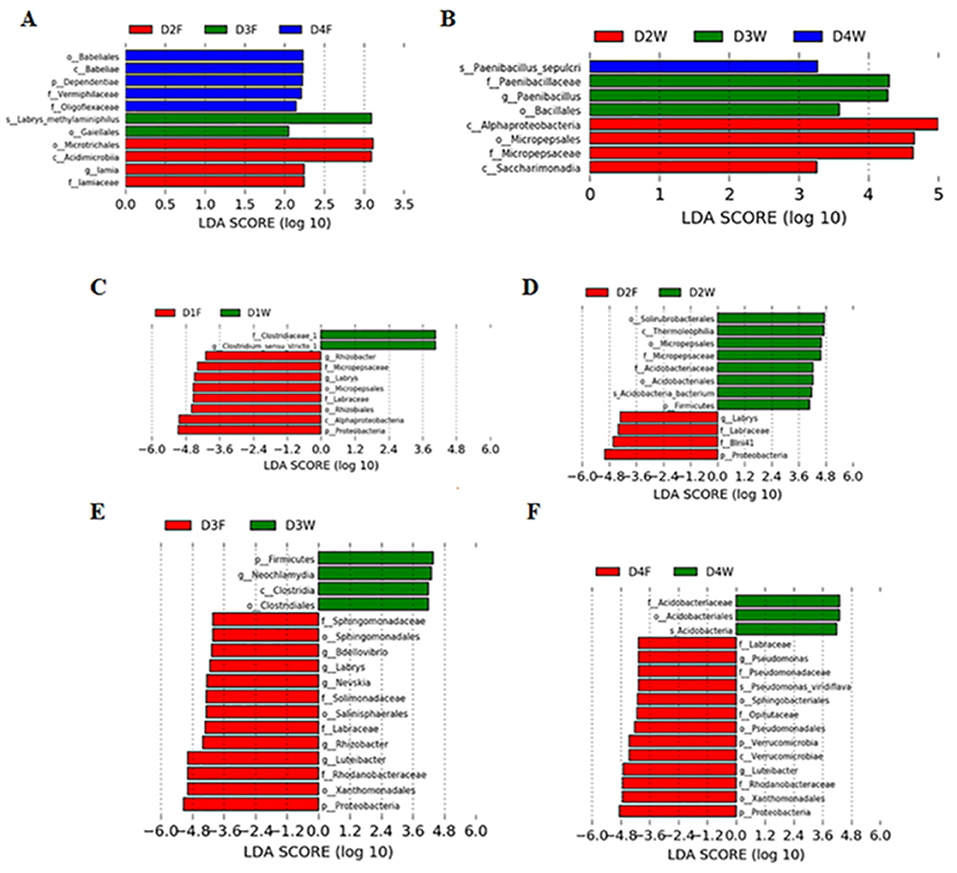
Figure 5. Linear discriminant analysis (LDA) effect size (LEfSe) showing bacterial phyla, class, order, family, genus, and species, which were significantly different in (A) fruiting body, (B) wood under different decay classes, and between fruiting body and wood in the same decay class, (C) class 1, (D) class 2, (E) class 3, and (F) class 4. p, phylum; c, class; o, order; f, family; g, genus; s, species.
The top 252 OTUs, which cover 80.0% of all sequences, were assigned to 16 functional groups in FAPROTAX analysis. In total, 251 records (37.9%) were assigned to at least one group (Li et al., 2021). Chemoheterotrophy (42.2%) and aerobic chemoheterotrophy (41.4%) were the most abundant functions and were involved in carbon cycle, followed by nitrogen fixation (7.3%) and ureolysis (2.2%), which were involved in N cycle, together with fermentation (1.4%), cellulolysis (1.1%), non-photosynthetic Cyanobacteria (1.0%), plant pathogen related (0.8%), and hydrocarbon degradation (0.2%). The detailed information is found in Supplementary Table 2.
There was no functional group differences among bacteria biota observed among the decay classes in wood samples, however, in the fruiting body, phototrophy showed a pattern of D4F < D2F (P < 0.05); plant pathogen-related function was higher in D3F compared with that in D1F and D4F (P < 0.05).
Comparing between wood and fruiting body in the same decay class, classes 1, 3, and 4 had significant abundant functional groups (LDA > 3.0, P < 0.05; Figure 6), and they changed with progress of decay. The fruiting body had more diverse abundant functional groups, while dead wood only had fermentation group in the third and fourth classes (Figure 6).
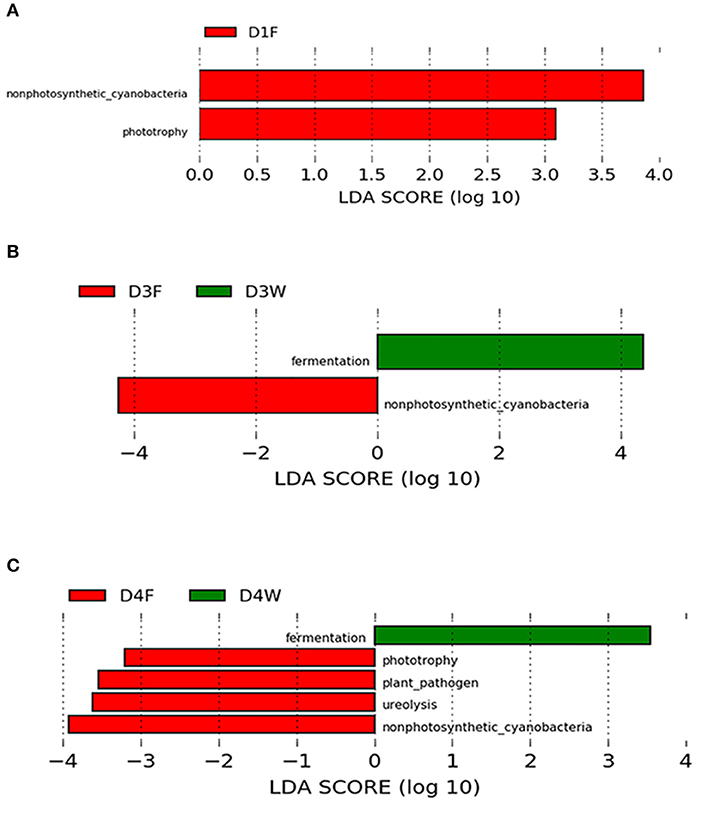
Figure 6. LEfSe (LDA > 3.0, P < 0.05) showing the predicted functional groups significantly abundant in decay classes 1 (A), 3 (B), and 4 (C).
The complex microbial network was unraveled by the nature of its topology characteristics (Blondel et al., 2008; Paranyushkin, 2012a). The most important topology parameters of connectivity, modularity, and Po/Ne ratio of the communities' network were selected and used to explain its activeness (Hartman et al., 2018), function-related modularization (Newman, 2006), and stability (de Vries et al., 2018). The top 252 OTUs with ρ > 0.7, P < 0.001 of Spearman correlation values were used for the network analysis (Supplementary Tables 3, 4). Overall, OTU numbers that satisfied the threshold ranged from 164 to 207 in the fruiting body and 155 to 200 in the dead wood (Supplementary Table 3).
The connectivity in both substrates had the changing pattern of increasing before the third decay stage and decreasing at the fourth stage. The third decay class had the peak connectivity in both substrates, the lowest point was observed in D1F and D4W (Table 2; Figure 7). Connectivity in wood was higher than in the fruiting body in all the decay classes, which means that bacteria hosted by the decayed wood were more actively interacting with each other (Hartman et al., 2018). The modularity had a pattern of decrease-increase-decrease in the fruiting body, where D3F reached the peak and D4F was the lowest. In wood, the modularity had the opposite pattern, where D2W had the highest value, and D4W was the lowest (Table 2). The value of modularity indicated the modularization of community, which could be related to their function (Newman, 2006) and stability (Paranyushkin, 2012b).
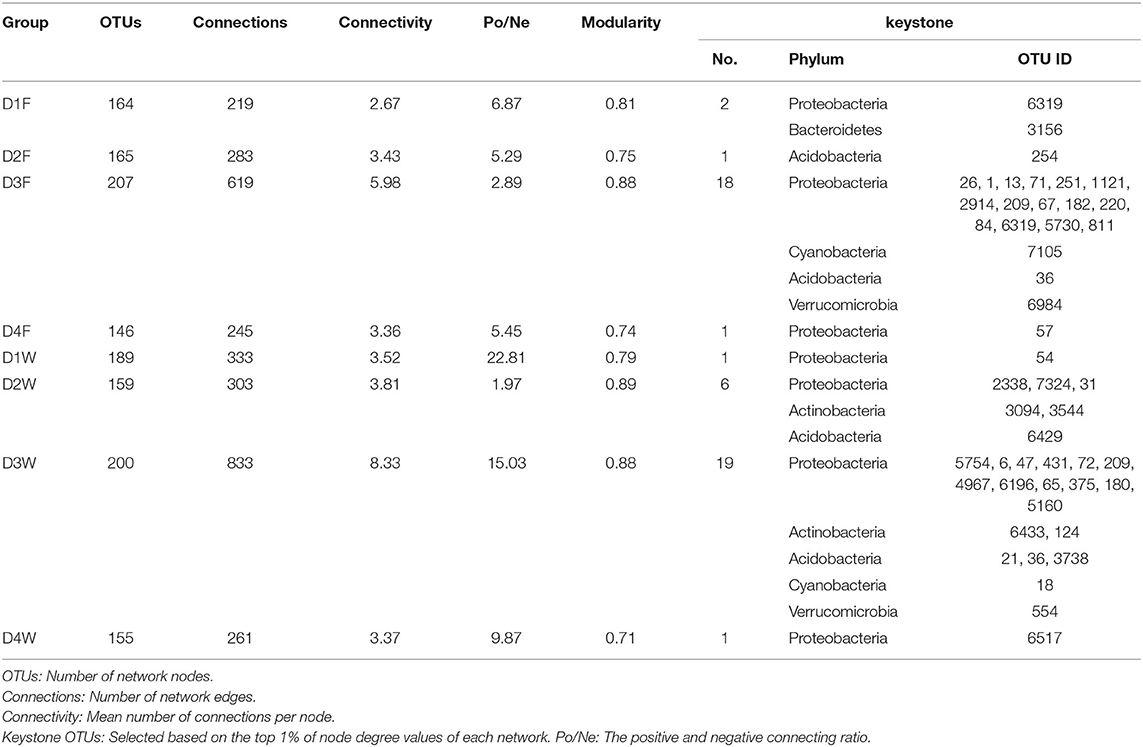
Table 2. Detailed network properties of study groups.
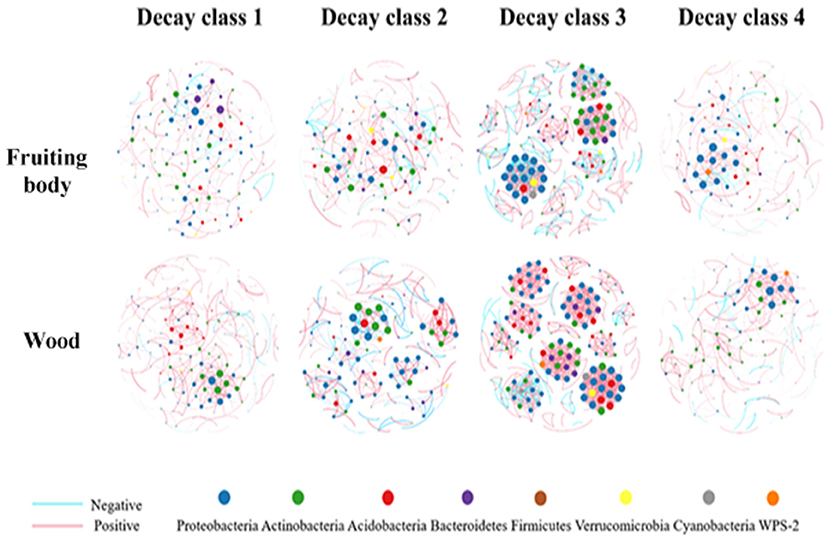
Figure 7. Co-occurrence networks of top 252 OTUs showing the significant correlations in fruiting body and wood over decay. Nodes represent individual OTUs; edges represent significant Spearman correlations (ρ > 0.7, P < 0.001). The size of node represents its link degree, the larger node has more connections with other nodes. The thickness of edge represents the correlation level. The red and blue edges show the positive and negative connections between nodes separately.
The positive and negative connecting ratio (Po/Ne) used to represent connection stability (de Vries et al., 2018) had the opposite changing pattern to their modularity in both substrates. D3F and D2W had the minimum value, and D1F and D1W reached the peak (Table 2; Figure 7). Our result indicated that the communities in the fruiting body were more stable compared with wood as their Po/Ne values had smaller changing range (Table 2). Moreover, the thicker linking edges in the middle decay classes in both substrates also showed stronger connections (Figure 7). We selected nodes with top 1% connectivity in each group as the keystone OTUs (Hartman et al., 2018). We found that phylum Proteobacteria existed in all networks except D2F and was the only phylum that existed at the end of the decay process (Table 2). Among all selected OTUs, 11 of them were in top 50 abundant group; only OTU6319 reoccurred in D1F and D3F; OTU209 appeared in D3F and D3W; and all the rest were unique (Table 2).
The alpha diversity and beta diversity of bacterial community did not show any significant differences neither in wood tissue nor in fruiting body. However, community structures, activeness, stabilities, and functions kept changing. At the end of the fourth decay process, new bacterial communities replaced the old ones previously constructed in living standing tree. Changes from host chemical-physical properties (Pent et al., 2020), interactions between hosts (Steffan et al., 2020) and microbiome (Bastías et al., 2020) might be the key parameters contributing to the bacterial community restructuring. This could be partly because the environmental factors such as soil property, moisture, and temperature were quite similar in the fruiting body as well as its adhering wood tissue.
At the first decay stage, bacterial biota in the fruiting body were more abundant, unique, active, and functional, however, the opposite trend was observed in the wood tissue. We assumed that in standing living tree, bacterial community of the fruiting body associated to it may have been suppressed, compared with dead wood tissue. However, the effect and trend weakened with progress of decay. It is very probable that the active defense system and associated chemical compounds in living trees (Asiegbu et al., 2005; Arnerup et al., 2010) may have contributed to the migration of the bacteria to the fruiting body. It is also likely that in decay class 1, bacteria were able to metabolize easily degradable sugars and carbohydrates, as well as utilizing the opportunity to source for nutrients directly from woody tissues (Clausen, 1996). Ray cell, which provides this kind of nutrients, was considered to be partly decomposed during this stage. Additionally, the increasing permeability of cellulosic substrates provides more chance for lateral bacterial groups to invade (Greaves, 1971). At the second decay class, a considerable number of bacteria groups dead from the first decay class serves as an important nitrogen resource, which lack in wood tissue. With further progress of the decay, more nitrogen and carbon resources become available (Greaves, 1971; King and White, 1976). Based on these, increasing number of nitrogen fixing bacteria start to be active with a better niche and able to have access to more resources (Johnston et al., 2016). In contrast to the living wood, plant endophytes and mycorrhiza helper bacteria (Lehr et al., 2007; Terhonen et al., 2016; Lipps and Samac, 2021) have been reported to support diverse microbial communities in that niche.
The third decay class was an important turning point for the whole decay process where diversity indicators, OTUs structures, the network topological parameters reached the extreme points, and functional groups started to be differentiated between the two study materials. According to previous studies (Mäkinen et al., 2006), the wood density and stem mass start to have a sharp decrease for conifer tissues at this stage. The physical status of deadwood directly changes the accessible oxygen for wood inhabiting microbiome. The aerobic bacteria have better chance to thrive, while the anaerobic bacteria are much more at a disadvantage in the community. It is therefore possible that at this decay class, the community might reach the limitation range due to constraints of nutrition (Hacquard et al., 2015), host property (Swedjemark et al., 1998; Gohar et al., 2020), and environmental factors (Probst et al., 2018).
We observed a dramatic change in stability indicator (Po/Ne) between D2W and D3W. Besides the accumulated nutritional factors, external perturbations rather than the gradual microenvironmental changes could also be the possible reason. The previous study showed that though a higher modularity indicated a higher network stability when facing external perturbations, the network would be susceptible when targeted attack happens on community's key stone OTUs (Paranyushkin, 2012b) or by uncertain perturbations (Kitano, 2004). The obvious keystone OTUs change between D2W and D3W, and the activated Cyanobacteria and Verrucomicrobia in D3W support this concept.
In the last decay stage, as the nutrition from the old resources diminishes, a new successional group of bacteria community was observed. The new bacterial structures and functional groups were well shaped in both substrates. However, the increased instability (Po/Ne) may indicate a new community restructuring phase, as the decomposed wood turns into the soil organic layer, and soil properties take the dominant role to influence the microbial community (Fierer et al., 2007; Johnston et al., 2016; Uroz et al., 2016; Dong et al., 2021).
We also found several soil- or plant-dwelling bacteria on Heterobasidion fruiting body, Bacillus spp. (Kjeldgaard et al., 2019), for example. As discussed in previous studies, the fruiting body microbiome was also mobile as mobility is a significant microbial feature (Mitchell and Kogure, 2006; Son et al., 2015). It is most probable that bacteria might be moving from living wood tissue to the fruiting body attached or associated with it. This is reflected in the increased number of bacteria OTUs in the fruiting body. However, when the tree is dead, the active bacteria in the fruiting body start to invade dead wood with extended duration or progress of decay process. It is also possible that the low numbers of OTUs in living wood are due to the less nutritious properties of wood (Maurice et al., 2021). The higher community activity and decreasing OTUs abundance in wood might also imply that the community members were more stable in wood, making it difficult for new species to join the community. The predominant bacteria in wood might be highly selected because of a tougher niche due to various plant secondary metabolites (Garzoli et al., 2021).
In this study, the environmental factors had no significant impact on the decay classes (Pent et al., 2017).
The FAPROTEX analysis confirmed nutrition, energy, degradation, and plant pathogen-related functions of the bacterial community. Several dominant taxonomic groups or those that showed significant changes were also of interest, as their functions are closely related to the community structure (Fierer et al., 2007).
Burkholderia spp. was the most abundant phylum in both substrates and was active in all decay classes (Figure 4; Supplementary Table 3). The abundance decreased with decay process in wood, though it was more consistent in the fruiting body. This result is similar to a previous observation on bacteria biota of the decayed wood (Probst et al., 2018). The genus' typical characteristic of being a fungal symbiont with associated fungal host protecting capability might explain its consistence in the fruiting body (Partida-Martinez and Hertweck, 2005; Marupakula et al., 2016; Sharmin et al., 2018). Furthermore, its general functions as nitrogen fixing (Sun et al., 2014), cellulose, and aromatic degrading (Kielak et al., 2016) made it lose competitive advantage in wood.
Firmicutes was much more abundant in wood tissue than in fruiting body across all decay classes and was significantly abundant in D3W. This result was similar to previous studies, where Firmicutes takes less proportion in mycorrhizosphere but is abundant in soil and wood tissue (Vik et al., 2013; Timonen et al., 2017). This might be because bacteria from this phylum have strong resilient capability in anaerobic condition (Hartmann et al., 2014). Studies from Shen et al. (2015) showed that enrichment of Firmicutes (Bacillus) suppresses Fusarium wilt disease. Studies by Trivedi et al. (2017) and Xiong et al. (2017) also indicate that Firmicutes is more abundant in disease-suppressed soil. Interestingly, this phylum acted non-remarkably in all decay stages and were mostly interacting within the phylum (Supplementary Figure 4). It is very probable that its powerful pathogen-suppressing capability contributed to its high stability and abundance.
Cyanobacteria and Verrucomicrobia only appeared in the third decay class in both substrates and only co-existed within the same module (Figure 7). Environmental factors were shown to be the primary driver for cyanobacterial community structuring (Rigonato et al., 2012). The decayed wood tissue might be the trigger driving the community restructuring as well as the secondary metabolites produced by Cyanobateria could suppress the pathogenic activity (Suurnäkki, 2015). Verrucomicrobia was proved to be an important phylum for plant health (Buée et al., 2009; Da Rocha et al., 2013), nutrient availability is the key factor for verrucomicrobial assemblage (Da Rocha et al., 2013; Uroz et al., 2013). Cyanobacteria provided nutrition by fixing nitrogen and carbon (Rigonato et al., 2012), which was also crucial for the activeness of both groups.
Pseudomonas spp. was abundant in both substrates in the first decay class; however, it decreased dramatically with progress of wood decay (Figure 4; Supplementary Tables 3, 4). Its ecological function might be different in the two substrates and in the different decay classes. In woody tissue, it could be endobacteria in a living tree (GŽibovska, 2016). The tree might also have supported its existence, but the beneficial interaction stopped when the tree was killed. Consequently, the genus had difficulty to survive on dead wood tissue (Pellicciaro et al., 2021). In the fruiting body, Pseudomonas decreased in relative abundance at the start of decay, which later increased considerably. Some species in this genus are also known to be opportunistic pathogens (Lipps and Samac, 2021). It is likely that Pseudomonas viridiflava, identified in this study, could be a pathogen, endophyte, or saprotroph (Lipps and Samac, 2021). The species might live as an endophyte when tree was alive but shifted to pathogenic behavior triggered by bacterial competition (Lipps and Samac, 2021). With no suppression from tree resistance, resulting in its increased abundance (Supplementary Table 3).
Microbiome analysis of Heterobasidion associated organisms has been widely conducted on the group level (e.g., fungi, bacteria, or virus) and specific species level; however, not much has been explored at the network level. Network metrics in this study indicated more detailed ecological (Bascompte, 2010; Thébault and Fontaine, 2010) and evolutionary (Olesen et al., 2007) information. The changing characteristics of the community were evident in this study. To our knowledge, this is the first study on the network analysis of bacteria from Heterobasidion fruiting body and its associated decaying wood. Our study also revealed a more complete changing trend of important bacteria throughout the various decay stages. However, the participation of fungi in both substrates merits to be investigated as it might provide further useful insight on the community structure, functions, and interaction with bacteria as well as the complete microbiome picture.
The bacterial communities inhabiting the fruiting body and wood were found to be highly dynamic through the entire wood decay process. Physical-chemical properties of fruiting body and wood, properties of bacteria species, and the interactions among fruiting body-wood-bacteria might probably have had influence in the community assembly. The third decay class was an important shifting point for both study materials as reflected by network metrics and functional groups' differentiation. The bacterial community assembled in the course of the decay process in both fruiting body and wood tend to start a new round of successional changes reflecting the transition from living tree to dead wood and vice versa.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
FA and WR designed the study. RP, RK, and WR collected samples in the field. WR performed the experiments, analyzed the data, and wrote the manuscript. FA conceived the study and contributed in drafting the manuscript. All authors edited and approved the manuscript.
This study was supported by Ministry of Agriculture and Forestry (Grant number 4400T-2002). This study was also supported by the Chinese Scholarship Council Doctoral Stipendium.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to acknowledge National Land Survey of Finland for kindly providing us with the stand details.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.864619/full#supplementary-material
Supplementary Figure 1. Rarefaction curve of study groups showing the sequence depth.
Supplementary Figure 2. PCoA analysis of bacterial communities in four decay stages in fruiting body.
Supplementary Figure 3. PCoA analysis of bacterial communities in four decay stages in wood tissue.
Supplementary Figure 4. Co-occurrence networks of Firmicutes from top 252 OTUs in fruiting body and wood over decay.
Supplementary Table 1. The basic information of each sampling point.
Supplementary Table 2. The relative abundance of functional groups.
Supplementary Table 3. The basic information of OTUs Node in the networks.
Supplementary Table 4. The basic information of co-occurrence edge in networks.
Arnerup, J., Swedjemark, G., Elfstrand, M., Karlsson, B., and Stenlid, J. (2010). Variation in growth of Heterobasidion parviporum in a full-sib family of Picea abies. Scand. J. Forest Res. 25, 106–110. doi: 10.1080/02827581003730799
Asiegbu, F. O., Adomas, A., and Stenlid, J. (2005). Conifer root and butt rot caused by Heterobasidion annosum (Fr.) Bref. s.l. Mol. Plant Pathol. 6, 395–409. doi: 10.1111/j.1364-3703.2005.00295.x
Asiegbu, F. O., and Kovalchuk, A. (2021). Forest Microbiology: Volume 1: Tree Microbiome: Phyllosphere, Endosphere, and Rhizosphere. London: Elsevier Publishers.
Bascompte, J. (2010). Structure and dynamics of ecological networks. Science. 329, 765–766. doi: 10.1126/science.1194255
Bastías, D. A., Johnson, L. J., and Card, S. D. (2020). Symbiotic bacteria of plant-associated fungi: friends or foes? Curr. Opin. Plant Biol. 56, 1–8. doi: 10.1016/j.pbi.2019.10.010
Blondel, V. D., Guillaume, J. L., Lambiotte, R., and Lefebvre, E. (2008). Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 1–12. doi: 10.1088/1742-5468/2008/10/P10008
Błońska, E., Kacprzyk, M., and Spólnik, A. (2017). Effect of deadwood of different tree species in various stages of decomposition on biochemical soil properties and carbon storage. Ecol. Res. 32, 193–203. doi: 10.1007/s11284-016-1430-3
Brunner, A., and Kimmins, J. P. (2003). Nitrogen fixation in coarse woody debris of Thuja plicata and Tsuga heterophylla forests on northern Vancouver Island. Can. J. For. Res. 33, 1670–1682. doi: 10.1139/x03-085
Buée, M., de Boer, W., Martin, F., van Overbeek, L., and Jurkevitch, E. (2009). The rhizosphere zoo: an overview of plant-associated communities of microorganisms, including phages, bacteria, archaea, and fungi, and of some of their structuring factors. Plant Soil 321, 189–212. doi: 10.1007/s11104-009-9991-3
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chang, S., Puryear, J., and Cairney, J. (1993). A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Rep. 11, 113–116. doi: 10.1007/BF02670468
Chao, A. (1984). Nonparametric estimation of the number of classes in a population. Scand. J. Statis. 11, 265–270.
Clausen, C. A. (1996). Bacterial associations with decaying wood: a review. Int. Biodeterior. Biodegrad. 37, 101–107. doi: 10.1016/0964-8305(95)00109-3
Cleary, M. R., Arhipova, N., Morrison, D. J., Thomsen, I. M., Sturrock, R. N., Vasaitis, R., et al. (2013). Stump removal to control root disease in Canada and Scandinavia: a synthesis of results from long-term trials. For. Ecol. Manage. 290, 5–14. doi: 10.1016/j.foreco.2012.05.040
Da Rocha, U. N., Plugge, C. M., George, I., Van Elsas, J. D., and Van Overbeek, L. S. (2013). The rhizosphere selects for particular groups of Acidobacteria and Verrucomicrobia. PLoS ONE 8, e82443. doi: 10.1371/journal.pone.0082443
de Vries, F. T., Griffiths, R. I., Bailey, M., Craig, H., Girlanda, M., Gweon, H. S., et al. (2018). Soil bacterial networks are less stable under drought than fungal networks. Nat. Commun. 9, 3033. doi: 10.1038/s41467-018-05516-7
Deveau, A., Bonito, G., Uehling, J., Paoletti, M., Becker, M., Bindschedler, S., et al. (2018). Bacterial-fungal interactions: ecology, mechanisms and challenges. FEMS Microbiol. Rev. 42, 31–48. doi: 10.1093/femsre/fuy008
Deveau, A., Palin, B., Delaruelle, C., Peter, M., Kohler, A., Pierrat, J. C., et al. (2007). The mycorrhiza helper Pseudomonas fluorescens BBc6R8 has a specific priming effect on the growth, morphology and gene expression of the ectomycorrhizal fungus Laccaria bicolor S238N. New Phytol. 175, 743–755. doi: 10.1111/j.1469-8137.2007.02148.x
Dong, M., Kowalchuk, G. A., Liu, H., Xiong, W., Deng, X., Zhang, N., et al. (2021). Microbial community assembly in soil aggregates: a dynamic interplay of stochastic and deterministic processes. Appl. Soil Ecol. 163, 103911. doi: 10.1016/j.apsoil.2021.103911
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods. 10, 996–998. doi: 10.1038/nmeth.2604
Elliott, T. F., Jusino, M. A., Trappe, J. M., Lepp, H., Ballard, G. A., Bruhl, J. J., et al. (2019). A Global Review of the Ecological Significance of Symbiotic Associations Between Birds and Fungi. Heidelberg: Springer Netherlands. doi: 10.1007/s13225-019-00436-3
Fierer, N., Bradford, M. A., and Jackson, R. B. (2007). Toward an ecological classification of soil bacteria. Ecology 88, 1354–1364. doi: 10.1890/05-1839
Fravolini, G., Egli, M., Derungs, C., Cherubini, P., Ascher-Jenull, J., Gómez-Brandón, M., et al. (2016). Soil attributes and microclimate are important drivers of initial deadwood decay in sub-alpine Norway spruce forests. Sci. Total Environ. 569–570, 1064–1076. doi: 10.1016/j.scitotenv.2016.06.167
Gaitnieks, T., Bruna, L., Zaluma, A., Burnevica, N., Klavina, D., Legzdina, L., et al. (2021). Development of Heterobasidion spp. fruit bodies on decayed Piecea abies. For. Ecol. Manage. 12, 1–12. doi: 10.1016/j.foreco.2020.118835
Garzoli, S., Masci, V. L., Caradonna, V., Tiezzi, A., Giacomello, P., and Ovidi, E. (2021). Liquid and vapor phase of four conifer-derived essential oils: comparison of chemical compositions and antimicrobial and antioxidant properties. Pharmaceuticals 14, 1–15. doi: 10.3390/ph14020134
Gohar, D., Pent, M., Poldmaa, K., Bahram, M., Aittaleb, M., Gao, G., et al. (2020). Bacterial community dynamics across developmental stages of fungal fruiting bodies. Cell. Signal. 96, 1–8. doi: 10.1093/femsec/fiaa175
Gonthier, P., Garbelotto, M. M., and Nicolotti, G. (2005). Seasonal patterns of spore deposition of Heterobasidion species in four forests of the western Alps. Phytopathology 95, 759–767. doi: 10.1094/PHYTO-95-0759
Greaves, H. (1971). The bacterial factor in wood decay. Wood Sci. Technol. 5, 6–16. doi: 10.1007/BF00363116
Guo, H., Glaeser, S. P., Alabid, I., Imani, J., Haghighi, H., Kämpfer, P., et al. (2017). The abundance of endofungal bacterium Rhizobium radiobacter (syn. Agrobacterium tumefaciens) increases in its fungal host Piriformospora indica during the tripartite sebacinalean symbiosis with higher plants. Front. Microbiol. 8, 629. doi: 10.3389/fmicb.2017.00629
GŽibovska, Z. (2016). Evaluation of Phlebiopsis gigantea and Pseudomonas spp. for biocontrol of Heterobasidion spp. in Norway spruce (Master thesis). Swedish University of Agricultural Sciences, Uppsala, Sweden.
Haas, B. J., Gevers, D., Earl, A. M., Feldgarden, M., Ward, D. V., Giannoukos, G., et al. (2011). Genome Research. Available online at: genome.cshlp.org (accessed April 12, 2022).
Hacquard, S., Garrido-Oter, R., González, A., Spaepen, S., Ackermann, G., Lebeis, S., et al. (2015). Microbiota and host nutrition across plant and animal kingdoms. Cell Host Microbe 17, 603–616. doi: 10.1016/j.chom.2015.04.009
Hartman, K., van der Heijden, M. G. A., Wittwer, R. A., Banerjee, S., Walser, J. C., and Schlaeppi, K. (2018). Cropping practices manipulate abundance patterns of root and soil microbiome members paving the way to smart farming. Microbiome 6, 1–15. doi: 10.1186/s40168-017-0389-9
Hartmann, M., Niklaus, P. A., Zimmermann, S., Schmutz, S., Kremer, J., Abarenkov, K., et al. (2014). Resistance and resilience of the forest soil microbiome to logging-associated compaction. ISME J. 8, 226–244. doi: 10.1038/ismej.2013.141
Ioos, R., Chrétien, P., Perrault, J., Jeandel, C., Dutech, C., Gonthier, P., et al. (2019). Multiplex real-time PCR assays for the detection and identification of Heterobasidion species attacking conifers in Europe. Plant Pathol. 68, 1493–1507. doi: 10.1111/ppa.13071
Johnston, S. R., Boddy, L., and Weightman, A. J. (2016). Bacteria in decomposing wood and their interactions with wood-decay fungi. FEMS Microbiol. Ecol. 92, fiw179. doi: 10.1093/femsec/fiw179
Keilhofer, N., Nachtigall, J., Kulik, A., Ecke, M., Hampp, R., Süssmuth, R. D., et al. (2018). Streptomyces AcH 505 triggers production of a salicylic acid analogue in the fungal pathogen Heterobasidion abietinum that enhances infection of Norway spruce seedlings. Antonie van Leeuwenhoek, Int. J. Gen. Mol. Microbiol. 111, 691–704. doi: 10.1007/s10482-018-1017-9
Kielak, A. M., Scheublin, T. R., Mendes, L. W., van Veen, J. A., and Kuramae, E. E. (2016). Bacterial community succession in pine-wood decomposition. Front. Microbiol. 7, 231. doi: 10.3389/fmicb.2016.00231
King, B., and White, J. (1976). Translocation of nitrogen to wood by fungi. Znt. Biodet. Bull. 15, 29–35.
Kjeldgaard, B., Listian, S. A., Ramaswamhi, V., Richter, A., Kiesewalter, H. T., and Kovács, Á. T. (2019). Fungal hyphae colonization by Bacillus subtilis relies on biofilm matrix components. Biofilm 1, 100007. doi: 10.1016/j.bioflm.2019.100007
Korhonen, K., and Stenlid, J. (1998). Biology of Heterobasidion annosum. Wallingford: CAB Int. 43–70.
Lackner, G., Moebius, N., Partida-Martinez, L. P., Boland, S., and Hertweck, C. (2011). Evolution of an endofungal Lifestyle: deductions from the Burkholderia rhizoxinica genome. BMC Genomics 12, 210. doi: 10.1186/1471-2164-12-210
Legzdina, L., Spungis, V., Burnevica, N., Gaitnieks, T., and Menkis, A. (2021). Invertebrates in fruitbodies of Heterobasidion spp., infected picea abies logs and adjacent soil. Forests 12, 1–12. doi: 10.3390/f12081100
Lehr, N. A., Schrey, S. D., Bauer, R., Hampp, R., and Tarkka, M. T. (2007). Suppression of plant defence response by a mycorrhiza helper bacterium. New Phytol. 174, 892–903. doi: 10.1111/j.1469-8137.2007.02021.x
Lehr, N. A., Schrey, S. D., Hampp, R., and Tarkka, M. T. (2008). Root inoculation with a forest soil streptomycete leads to locally and systemically increased resistance against phytopathogens in Norway spruce. New Phytol. 177, 965–976. doi: 10.1111/j.1469-8137.2007.02322.x
Li, H., La, S., Zhang, X., Gao, L., and Tian, Y. (2021). Salt-induced recruitment of specific root-associated bacterial consortium capable of enhancing plant adaptability to salt stress. ISME J. 15, 2865–2882. doi: 10.1038/s41396-021-00974-2
Lipps, S. M., and Samac, D. A. (2021). Pseudomonas viridiflava: an internal outsider of the Pseudomonas syringae species complex. Mol. Plant Pathol. 23, 3–15. doi: 10.1111/mpp.13133
Litton, C. M., Raich, J. W., and Ryan, M. G. (2007). Carbon allocation in forest ecosystems. Glob. Chang. Biol. 13, 2089–2109. doi: 10.1111/j.1365-2486.2007.01420.x
Louca, S., Parfrey, L. W., and Doebeli, M. (2016). Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277. doi: 10.1126/science.aaf4507
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Mäkinen, H., Hynynen, J., Siitonen, J., and Sievänen, R. (2006). Predicting the decomposition of scots pine, norway spruce, and birch stems in Finland. Ecol. Appl. 16, 1865–1879. doi: 10.1890/1051-0761(2006)0161865:PTDOSP2.0.CO;2
Marupakula, S., Mahmood, S., and Finlay, R. D. (2016). Analysis of single root tip microbiomes suggests that distinctive bacterial communities are selected by Pinus sylvestris roots colonized by different ectomycorrhizal fungi. Environ. Microbiol. 18, 1470–1483. doi: 10.1111/1462-2920.13102
Maurice, S., Arnault, G., Nordén, J., Botnen, S. S., Miettinen, O., and Kauserud, H. (2021). Fungal sporocarps house diverse and host-specific communities of fungicolous fungi. ISME J. 15, 1445–1457. doi: 10.1038/s41396-020-00862-1
Mesanza, N., Iturritxa, E., and Patten, C. L. (2016). Native rhizobacteria as biocontrol agents of Heterobasidion annosum s.s. and Armillaria mellea infection of Pinus radiata. Biol. Control 101, 8–16. doi: 10.1016/j.biocontrol.2016.06.003
Mitchell, J. G., and Kogure, K. (2006). Bacterial motility: links to the environment and a driving force for microbial physics. FEMS Microbiol. Ecol. 55, 3–16. doi: 10.1111/j.1574-6941.2005.00003.x
Moore, D., Gange, A. C., Gange, E. G., and Boddy, L. (2008). Chapter 5 Fruit bodies: their production and development in relation to environment. Br. Mycol. Soc. Symp. Ser. 28, 79–103. doi: 10.1016/S0275-0287(08)80007-0
Moreno, D., Gil, M., and Piccoli, P. (2014). Bacteria isolated from roots and rhizosphere of Vitis vinifera retard water losses, induce abscisic acid accumulation and synthesis of defense-related terpenes in in vitro. Physiol. Plant 151, 359–374. doi: 10.1111/ppl.12117
Möykkynen, T., Von Weissenberg, K., and Pappinen, A. (1997). Estimation of dispersal gradients of S- and P-type basidiospores of Heterobasidion annosum. Eur. J. For. Pathol. 27, 291–300. doi: 10.1111/j.1439-0329.1997.tb01083.x
Müller, J., and Bütler, R. (2010). A review of habitat thresholds for dead wood: a baseline for management recommendations in European forests. Eur. J. For. Res. 129, 981–992. doi: 10.1007/s10342-010-0400-5
Müller, M. M., Heinonen, J., and Korhonen, K. (2007). Occurrence of Heterobasidion basidiocarps on cull pieces of Norway spruce left on cutting areas and in mature spruce stands. For. Pathol. 37, 374–386. doi: 10.1111/j.1439-0329.2007.00516.x
Müller, M. M., Sievänen, R., Beuker, E., Meesenburg, H., Kuuskeri, J., Hamberg, L., et al. (2014). Predicting the activity of Heterobasidion parviporum on Norway spruce in warming climate from its respiration rate at different temperatures. For. Pathol. 44, 325–336. doi: 10.1111/efp.12104
Newman, M. E. J. (2006). Modularity and community structure in networks. Proc. Natl. Acad. Sci. U. S. A. 103, 8577–8582. doi: 10.1073/pnas.0601602103
Olesen, J. M., Bascompte, J., Dupont, Y. L., and Jordano, P. (2007). The modularity of pollination networks. Proc. Natl. Acad. Sci. U. S. A. 104, 19891–19896. doi: 10.1073/pnas.0706375104
Paranyushkin, D. (2012a). Informational Epidemics and Synchronized Viral Contagion in Social Networks. Nodus Labs. Available online at: https://noduslabs.com/
Paranyushkin, D. (2012b). Metastability of Cognition in the Body-Mind-Environment Network ! Nodus Labs. 1–35.
Partida-Martinez, L. P., and Hertweck, C. (2005). Pathogenic fungus harbours endosymbiotic bacteria for toxin production. Nature 437, 884–888. doi: 10.1038/nature03997
Pellicciaro, M., Lione, G., Giordano, L., and Gonthier, P. (2021). Biocontrol potential of Pseudomonas protegens against Heterobasidion species attacking conifers in Europe. Biol. Control 157, 104583. doi: 10.1016/j.biocontrol.2021.104583
Pent, M., Bahram, M., and Põldmaa, K. (2020). Fruitbody chemistry underlies the structure of endofungal bacterial communities across fungal guilds and phylogenetic groups. ISME J. 14, 2131–2141. doi: 10.1038/s41396-020-0674-7
Pent, M., Põldmaa, K., and Bahram, M. (2017). Bacterial communities in boreal forest mushrooms are shaped both by soil parameters and host identity. Front. Microbiol. 8, 836. doi: 10.3389/fmicb.2017.00836
Piri, T. (1996). The spreading of the S type of Heterobasidion annosum from Norway spruce stumps to the subsequent tree stand. Eur. J. For. Pathol. 26, 193–204. doi: 10.1111/j.1439-0329.1996.tb00839.x
Probst, M., Gómez-Brandón, M., Bardelli, T., Egli, M., Insam, H., and Ascher-Jenull, J. (2018). Bacterial communities of decaying Norway spruce follow distinct slope exposure and time-dependent trajectories. Environ. Microbiol. 20, 3657–3670. doi: 10.1111/1462-2920.14359
Puentes Rodriguez, Y., Morales, L., Willför, S., Pulkkinen, P., Peltola, H., and Pappinen, A. (2013). Wood decay caused by Heterobasidion parviporum in juvenile wood specimens from normal- and narrow-crowned Norway spruce. Scand. J. For. Res. 28, 331–339. doi: 10.1080/02827581.2012.746387
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
Ren, F., Kovalchuk, A., Mukrimin, M., Liu, M., Zeng, Z., Ghimire, R. P., et al. (2019). Tissue microbiome of Norway spruce affected by Heterobasidion-induced wood decay. Microb. Ecol. 77, 640–650. doi: 10.1007/s00248-018-1240-y
Rigonato, J., Alvarenga, D. O., Andreote, F. D., Dias, A. C. F., Melo, I. S., Kent, A., et al. (2012). Cyanobacterial diversity in the phyllosphere of a mangrove forest. FEMS Microbiol. Ecol. 80, 312–322. doi: 10.1111/j.1574-6941.2012.01299.x
Rondeux, J., and Sanchez, C. (2010). Review of indicators and field methods for monitoring biodiversity within national forest inventories. Core variable: Deadwood. Environ. Monit. Assess. 164, 617–630. doi: 10.1007/s10661-009-0917-6
Ruiz-Lozano, J. M., and Bonfante, P. (2000). A Burkholderia strain living inside the arbuscular mycorrhizal fungus Gigaspora margarita possesses the vacB gene, which is involved in host cell colonization by bacteria. Microb. Ecol. 39, 137–144. doi: 10.1007/s002480000008
Sansupa, C., Wahdan, S. F. M., Hossen, S., Disayathanoowat, T., Wubet, T., and Purahong, W. (2021). Can we use functional annotation of prokaryotic taxa (Faprotax) to assign the ecological functions of soil bacteria? Appl. Sci. 11, 1–17. doi: 10.3390/app11020688
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, 1–18. doi: 10.1186/gb-2011-12-6-r60
Shannon, C., and Weaver, W. (1949). The Mathematical Theory of Communication. Urbana, IL: University of Illinois Press. doi: 10.1145/584091.584093
Sharmin, D., Guo, Y., Nishizawa, T., Ohshima, S., Sato, Y., Takashima, Y., et al. (2018). Comparative genomic insights into endofungal lifestyles of two bacterial endosymbionts, mycoavidus cysteinexigens and burkholderia rhizoxinica. Microbes Environ. 33, 66–76. doi: 10.1264/jsme2.ME17138
Shen, Z., Ruan, Y., Wang, B., Zhong, S., Su, L., Li, R., et al. (2015). Effect of biofertilizer for suppressing Fusarium wilt disease of banana as well as enhancing microbial and chemical properties of soil under greenhouse trial. Appl. Soil Ecol. 93, 111–119. doi: 10.1016/j.apsoil.2015.04.013
Son, K., Brumley, D. R., and Stocker, R. (2015). Live from under the lens: exploring microbial motility with dynamic imaging and microfluidics. Nat. Rev. Microbiol. 13, 761–775. doi: 10.1038/nrmicro3567
Spraker, J. E., Sanchez, L. M., Lowe, T. M., Dorrestein, P. C., and Keller, N. P. (2016). Ralstonia solanacearum lipopeptide induces chlamydospore development in fungi and facilitates bacterial entry into fungal tissues. ISME J. 10, 2317–2330. doi: 10.1038/ismej.2016.32
Steffan, B. N., Venkatesh, N., and Keller, N. P. (2020). Let's get physical: bacterial-fungal interactions and their consequences in agriculture and health. J. Fungi 6, 1–18. doi: 10.3390/jof6040243
Sun, H., Terhonen, E., Kasanen, R., and Asiegbu, F. O. (2014). Diversity and community structure of primary wood-inhabiting bacteria in Boreal forest. Geomicrobiol. J. 31, 315–324. doi: 10.1080/01490451.2013.827763
Suurnäkki, S. (2015). Anatoxin-a and Odorous Metabolites in Cyanobacteria: Molecular Detection of the Producers (Doctoral thesis). University of Helsinki, Finland. Available online at: https://helda.helsinki.fi/handle/10138/156512
Swedjemark, G., Stenlid, J., and Karlsson, B. (1998). Genetic variation among clones of Picea abies in resistance to growth of Heterobasidion annosum. Silvae Genet. 46, 369–374.
Terhonen, E., Marco, T., Sun, H., Jalkanen, R., Kasanen, R., Vuorinen, M., et al. (2011). The effect of latitude, season and needle-age on the mycota of scots pine. Silva Fenn. 45, 301–317. doi: 10.14214/sf.104
Terhonen, E., Sipari, N., and Asiegbu, F. O. (2016). Inhibition of phytopathogens by fungal root endophytes of Norway spruce. Biol. Control 99, 53–63. doi: 10.1016/j.biocontrol.2016.04.006
Thébault, E., and Fontaine, C. (2010). Stability of ecological communities and the architecture of mutualistic and trophic networks. Science. 329, 853–856. doi: 10.1126/science.1188321
Timonen, S., Sinkko, H., Sun, H., Sietiö, O. M., Rinta-Kanto, J. M., Kiheri, H., et al. (2017). Ericoid roots and mycospheres govern plant-specific bacterial communities in Boreal forest humus. Microb. Ecol. 73, 939–953. doi: 10.1007/s00248-016-0922-6
Trivedi, P., Delgado-Baquerizo, M., Trivedi, C., Hamonts, K., Anderson, I. C., and Singh, B. K. (2017). Keystone microbial taxa regulate the invasion of a fungal pathogen in agro-ecosystems. Soil Biol. Biochem. 111, 10–14. doi: 10.1016/j.soilbio.2017.03.013
Uroz, S., Ioannidis, P., Lengelle, J., Cébron, A., Morin, E., Buée, M., et al. (2013). Functional assays and metagenomic analyses reveals differences between the microbial communities inhabiting the soil horizons of a norway spruce plantation. PLoS ONE 8, e55929. doi: 10.1371/journal.pone.0055929
Uroz, S., Oger, P., Tisserand, E., CéBron, A., Turpault, M. P., Bueé, M., et al. (2016). Specific impacts of beech and Norway spruce on the structure and diversity of the rhizosphere and soil microbial communities. Sci. Rep. 6, 1–11. doi: 10.1038/srep27756
Venkatesh, N., and Keller, N. P. (2019). Mycotoxins in conversation with bacteria and fungi. Front. Microbiol. 10, 1–10. doi: 10.3389/fmicb.2019.00403
Vik, U., Logares, R., Blaalid, R., Halvorsen, R., Carlsen, T., Bakke, I., et al. (2013). Different bacterial communities in ectomycorrhizae and surrounding soil. Sci. Rep. 3, 3471. doi: 10.1038/srep03471
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Wargo, M. J., and Hogan, D. A. (2006). Fungal-bacterial interactions: a mixed bag of mingling microbes. Curr. Opin. Microbiol. 9, 359–364. doi: 10.1016/j.mib.2006.06.001
Xiong, W., Li, R., Ren, Y., Liu, C., Zhao, Q., Wu, H., et al. (2017). Distinct roles for soil fungal and bacterial communities associated with the suppression of vanilla Fusarium wilt disease. Soil Biol. Biochem. 107, 198–207. doi: 10.1016/j.soilbio.2017.01.010
Keywords: wood decay process, bacteria community, Heterobasidion, fruiting body, network analysis
Citation: Ren W, Penttilä R, Kasanen R and Asiegbu FO (2022) Bacteria Community Inhabiting Heterobasidion Fruiting Body and Associated Wood of Different Decay Classes. Front. Microbiol. 13:864619. doi: 10.3389/fmicb.2022.864619
Received: 28 January 2022; Accepted: 04 April 2022;
Published: 03 May 2022.
Edited by:
Marco Scortichini, Council for Agricultural and Economics Research (CREA), ItalyReviewed by:
Ari Mikko Hietala, Norwegian Institute of Bioeconomy Research (NIBIO), NorwayCopyright © 2022 Ren, Penttilä, Kasanen and Asiegbu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fred O. Asiegbu, ZnJlZC5hc2llZ2J1QGhlbHNpbmtpLmZp
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.