 Meghan Maguire1
Meghan Maguire1 Anisa S. Khan
Anisa S. Khan Karla Georges
Karla Georges Narjol Gonzalez-Escalona
Narjol Gonzalez-Escalona
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 10 May 2022
Sec. Food Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.863104
Salmonella enterica is an important foodborne pathogen worldwide. We used long and short-read sequencing to close genomes of eight multidrug-resistant (MDR) S. enterica strains, belonging to serovars Infantis (2), Albany, Oranienburg, I 4,[5],12:i:-, Javiana, Schwarzengrund, and Kentucky from broiler chicken farms and processing plants in Trinidad and Tobago. They also belonged to seven different sequence types (STs- 32, 292, 1510, 19, 24, 152, and 96). Among the strains, seven had demonstrated multi-drug resistance with the presence of at least three AMR genes, whereas three isolates contained the quinolone resistance gene qnrB19 in plasmids (CFSAN103840, CFSAN103854, and CFSAN103872). The extended-spectrum β-lactamase genes blaCTX−M−65 (CFSAN103796) and blaTEM−1 (CFSAN103852) were detected in this study. The genomes closed in this study will be useful for future source tracking and outbreak investigations in Trinidad and Tobago and worldwide.
Salmonella enterica contamination of poultry is a major route of transmission to humans. We have closed genomes of multiple Salmonella serovars and report multiple antimicrobial resistance (AMR) genes from isolates collected in processing plants and retail markets in Trinidad and Tobago. These closed genomes will aid in monitoring AMR transmission and future outbreak investigations.
Salmonella enterica is one of the most important foodborne pathogens in the world (Gonzalez-Escalona et al., 2009). There are more than 325,000 Salmonella genomes in NCBI encompassing isolates from many countries (https://www.ncbi.nlm.nih.gov/pathogens/). Non-typhoidal Salmonella (NTS) is responsible for ~93,757,000 cases of gastroenteritis and 155,000 deaths worldwide each year. Nearly 86% are linked to foodborne transmission (Majowicz et al., 2010). Poultry, including eggs and egg products, is one of the main reservoirs of Salmonella and has often been implicated as the source of many outbreaks worldwide (Schoeni et al., 1995; Kuehn, 2010; Jackson et al., 2013; Deng et al., 2014; Antunes et al., 2016).
Salmonella serovars Enteritidis, Newport, Typhimurium, Javiana, I 4,[5],12:i:-, and Heidelberg have been among the top five serovars associated with human illness over the last 15 years (2006–2016) in the United States (https://www.cdc.gov/nationalsurveillance/pdfs/2016-Salmonella-report-508.pdf). S. serovar Infantis has shown the highest percent increase in reported cases over the same time frame (+168.5%). While, S. serovar Kentucky has become the most commonly detected serovar in chicken (Foley et al., 2011), but is not among the top 20 serovars causing human illness.
While most Salmonella infections result in gastroenteritis without the need for additional medical intervention, severe, invasive infections can require antimicrobial treatment. Multidrug resistance (MDR) in Salmonella is closely monitored in clinical and environmental isolates around the world [National Antimicrobial Resistance Monitoring System (NARMS), European Center for Disease Control and Prevention (ECDC), and European Food Safety Authority (EFSA)]. In the United States, the FDA has issued guidance on the judicious use of antimicrobial agents in food-producing animals (https://www.fda.gov/regulatory-information/search-fda-guidance-documents/cvm-gfi-209-judicious-use-medically-important-antimicrobial-drugs-food-producing-animals) and individual antimicrobial agent restrictions have resulted in the decreased prevalence of resistance-conferring genes (https://www.fda.gov/media/108304/download).
Several antimicrobial-resistant (AMR) Salmonella strains were isolated during a Salmonella and AMR surveillance program of poultry production facilities in Trinidad and Tobago from 2016 to 2019 (Khan et al., 2022b). Their genomes were sequenced by short-read sequencing, and we selected a group of eight strains belonging to multiple serovars and showing a diverse range of MDR, to completely close their genomes using a combination of long-read and short-read sequencing technologies. The availability of these closed genomes will be useful for future outbreak investigations and for understanding AMR acquisition and transmission in Salmonella within the local Trinidad and Tobago region and worldwide.
Cloacal swabs, drag swabs, whole chicken carcasses, and neck skin samples were collected from retail outlets, processing plants, and farms in Trinidad and Tobago during an AMR surveillance program from 2016 to 2019. The sample was pre-enriched in buffered peptone water (Oxoid, Ltd., Hampshire, UK) for 18–24 h at 37°C, then selectively enriched in tetrathionate broth (Oxoid), and thereafter incubated for 18–24 h at 37°C. The sample enriched in selective broth was then sub-cultured onto xylose lysine Tergitol 4 (Oxoid) and incubated aerobically at 37°C for 18–24 h. Suspected Salmonella colonies that displayed characteristic colonies on the selective agar plate were then purified on blood agar plates (Oxoid) and incubated at 37°C for 18–24 h. Pure cultures were subjected to a panel of biochemical tests that included triple sugar iron agar, lysine iron agar, urea, citrate, methyl red, sulfide-indole-motility medium, and o-nitrophenyl-b-D-galactopyranoside (Oxoid) (Andrews, 1992). Biochemically confirmed isolates were then subjected to serological typing by using Salmonella polyvalent antiserum (A-I and Vi, Difco, Detroit, MI). Complete confirmation and serotyping of Salmonella isolates were performed by using the phase reversal technique, and the results were interpreted according to the Kauffman-White scheme (https://www.pasteur.fr/sites/default/files/veng_0.pdf) at the Public Health Laboratory, Ministry of Health, St. Michael, Barbados. The single isolate strains were grown overnight in tryptic soy broth (TSB) medium at 35°C. The strains to have their genomes entirely closed were selected among 146 strains isolated by Khan et al. (2022b). The criteria employed for their selection was: (1) a representative of each predominant serovar from that study [Kentucky 20.5% (30/146), Javiana 19.2% (28/146), Infantis 13.7% (20/146), and Albany 8.9% (13/146)], (2) Infantis with a different AMR profile, and 3) uncommon serotypes isolated from poultry [Oranienburg, I 4,[5],12:i:- (a monophasic variant of Salmonella Typhimurium), and Schwarzengrund].
Conventional serotyping methods using the phase reversal technique described previously (Khan et al., 2021, 2022a) were performed at the Public Health Laboratory, Ministry of Health, St. Michael, Barbados. The antimicrobial resistance was determined by the disk diffusion method as described previously (Khan et al., 2021, 2022a). Data generated from these two methods were compared to the genomic data.
DNA was extracted using the Maxwell RSC cultured cells DNA kit with a Maxwell RSC instrument (Promega, Madison, WI, USA) following the manufacturer's protocols for Gram-negative bacteria with additional RNase treatment. DNA concentration was determined by the Qubit 4 Fluorometer (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions.
The short reads whole-genome sequence for this strain was generated by Illumina MiSeq sequencing with the MiSeq V3 kit using 2 × 250 bp paired-end chemistry (Illumina, San Diego, CA, USA) according to the manufacturer's instructions. The libraries were constructed using 100 ng of genomic DNA using the Illumina® DNA Prep (M) Tagmentation (Illumina), according to the manufacturer's instructions. Reads were trimmed with Trimmomatic version.36 (Bolger et al., 2014).
The long reads for each strain were generated through GridION sequencing (Nanopore, Oxford, UK). The sequencing library was prepared using the rapid barcoding sequencing kit (SQK-RBK004). Each library was run in an FLO-MIN106 (R9.4.1) flow cell, according to the manufacturer's instructions, for 48 h. Default parameters were used for all software unless otherwise specified. The run was base called live with default settings (MinKNOW Core version 3.6.5, Bream version 4.3.16, and Guppy version 3.2.10). Reads <4000 bp and quality scores of <7 were discarded for downstream analysis, for an estimated genome average coverage of 59 – 250X.
The final complete genome [chromosome and plasmid(s), when present] for each strain was obtained using a previously described pipeline (Gonzalez-Escalona et al., 2018) with modifications. The final genomes were achieved by a hybrid assembly generated using both nanopore and MiSeq data with Unicycler version 4.8 (Wick et al., 2017). A second de novo genome assembly was obtained with Flye version 2.8 (Kolmogorov et al., 2019) using nanopore data. Both assemblies (Flye and Unicycler) for each strain were aligned with Mauve version 2.4 (Darling et al., 2004). If the two aligned assemblies agreed in synteny and size, then the Unicycler hybrid assembly was used as the final assembly (i.e., complete genome). Unicycler assembled the chromosomes and plasmids as circular closed and oriented the chromosomes to start at the dnaA gene. The genomes were annotated using the NCBI Prokaryotic Genomes Automatic Annotation Pipeline (PGAAP, http://www.ncbi.nlm.nih.gov/genome/annotation_prok) (Tatusova et al., 2016). In silico analysis was used to determine the multi-locus sequence type (MLST) with the MLST website for Salmonella (http://enterobase.warwick.ac.uk/species/index/senterica), serotype using SeqSero (Zhang et al., 2015) (http://www.denglab.info/Seqero) which infers the serovar from antigenic structure, and antimicrobial genes were found using ResFinder-4.1 (https://cge.cbs.dtu.dk/services/ResFinder/) (Bortolaia et al., 2020). In silico virulence profile was determined using the software Ridom Seqsphere+ version 6.0.2 (Ridom GmbH, Germany) and their task tool that uses the virulence factor database (VFDB) as a reference (Liu et al., 2019) to assign virulence factors.
To analyze the relationships among the eight NTS closed genomes we performed a core genome MLST (cgMLST) using Ridom Seqsphere+ version 6.0.2 (Ridom) as previously described (Toro et al., 2016). The genome of S. Enteritidis P125109 (NC_011294) was used as a reference genome (4,160 genes) (Thomson et al., 2008). The S. Enteritidis genome from strain EC20121175 (CP007269.2) was used for comparison with the reference genome to establish a list of core and accessory genome genes. Genes that are repeated in more than one copy in any of the two genomes were removed from the analysis as failed genes. A task template was created that contains both core and accessory genes for this reference SE strain for any future testing. Each individual locus (core or accessory genes) from strain P125109 was assigned allele number 1. The assemblies for each individual NTS closed genome in this study were queried against the task template and if the locus was found and was different from the reference genome or any other queried genome already in the database, a new number was assigned to that locus, and so on. After eliminating any loci that were missing from the genome of any strain used in our analyses, we performed the cgMLST analysis. These remaining loci were considered the core genome shared by the analyzed strains. We used Nei's DNA distance method (Nei et al., 1983) for calculating the matrix of genetic distance, taking only the number of same/different alleles in the core genes into consideration. A neighbor-joining (NJ) tree using the appropriate genetic distances was built after the cgMLST analysis.
A comparison of the CFSAN103796 blaCTX−M−65-positive plasmid with the ones reported in Tate et al. (2017) was performed and visualized with CGView (https://proksee.ca/) (Stothard and Wishart, 2005).
The complete genome sequences of the eight S. enterica strains have been deposited at GenBank under the accession listed in Table 1.
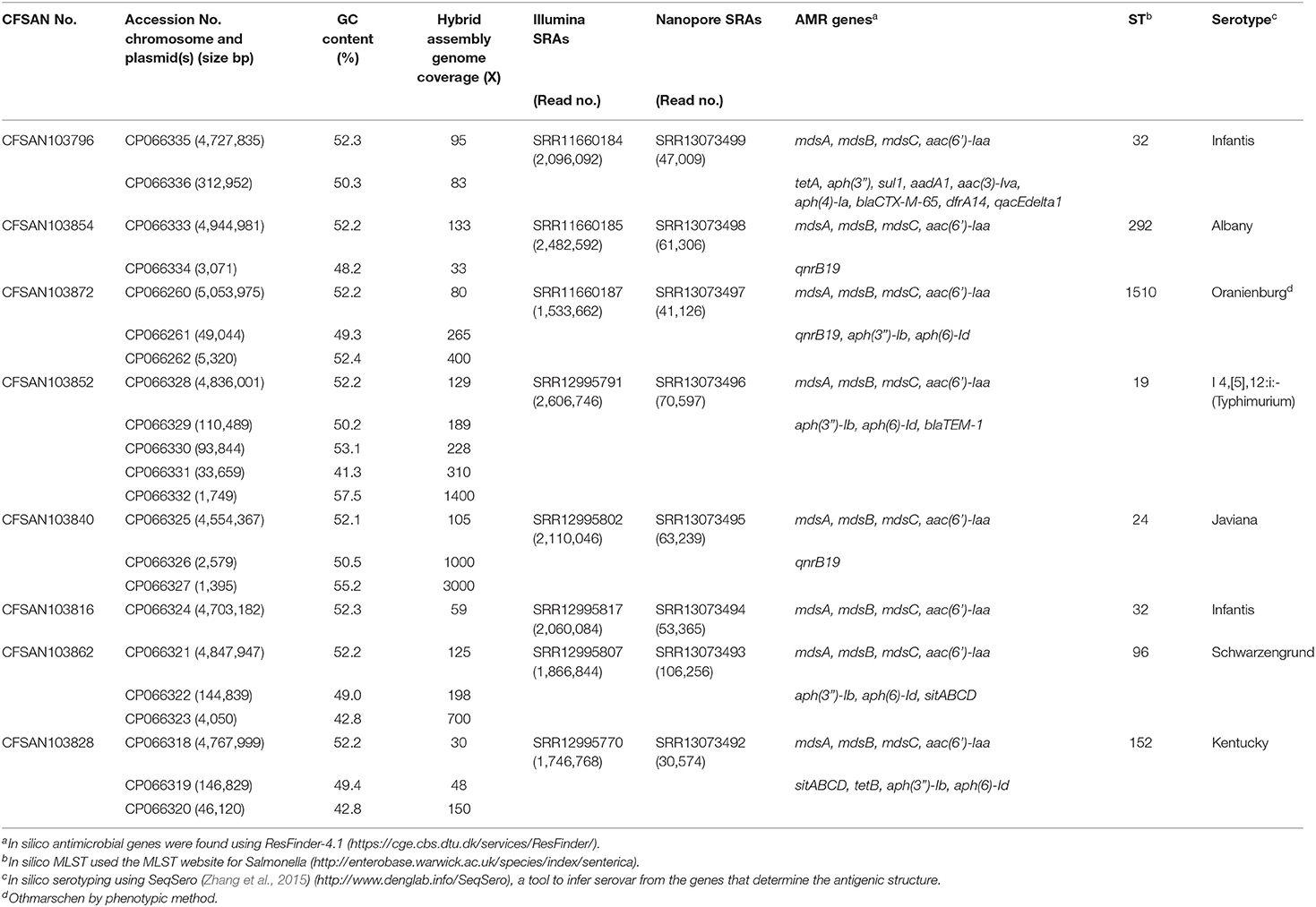
Table 1. Metadata for the eight Salmonella enterica strains isolated from chicken on poultry farms and processing plants were reported in this study.
An AMR surveillance program of poultry farms and retail outlets in Trinidad and Tobago from 2016 to 2019 produced several Salmonella isolates that were confirmed to bsse Salmonella by selective microbiological plating. Initial sequencing efforts identified the sequence type (ST) and serovar for each sample by in silico analysis. We selected eight samples from multiple serovars to completely close genomes using a hybrid assembly with Illumina MiSeq short-read and Oxford Nanopore long-read sequencing. The short-read sequencing for these eight samples was performed over three sequencing runs with an average output of 12.6 Gb and estimated average genome coverage of 50–150 ×. The multiplexed nanopore sequencing for eight samples was carried out over two sequencing runs. The output for run 1 was 7.69 Gb (2.33 million reads with a quality score of 9.48 and N50−11,835 bp) and for running 2 was 3.71 Gb (694 thousand reads with a quality score of 9.93 and N50−12,930 bp). The estimated average genome coverage was 59–250 × (Table 1).
The final closed genomes were generated by a hybrid assembly (Unicycler), which matched in synteny and size to a de novo assembly generated using only nanopore reads by the Flye assembler. In silico analysis confirmed the ST and serovar for each closed genome observed with the preliminary analysis run using the short-read sequencing (Khan et al., 2022b) (Table 1). The strains represented seven different STs and serotypes (32—Infantis, 292—Albany, 1,510—Oranienburg, 19−1,4,[5],12:i:- (Typhimurium), 24—Javiana, 96—Schwarzengrund and 152—Kentucky). There was an 88% agreement (7/8) in the serotyping results by phenotypic and in silico WGS (SeqSero) methods (Table 1). Strain CFSAN103872 was identified as Othmarschen by the phenotypic method but as Oranienburg by SeqSero. This discrepancy can be explained by the fact that both serotypes have the same antigenic formula (6,7:m,t-) and other authors showed that they were undisguisable genetically (Robertson et al., 2018). The genome size of these NTS strains varied from 4.6 Mb (Javiana, CFSAN103840) to 5.1 Mb (Oranienburg, CFSAN103872), highlighting the plasticity of NTS genomes (Table 1). A cgMLST analysis showed that these strains were highly divergent but shared at least 3,432 genes out of a total of 4,160 genes of the reference S. Enteritidis P125109 (NC_011294) strain used for this cgMLST analysis (Figure 1A; Supplementary Table 1). A minimum spanning tree of the cgMLST analysis showed that they differed in most loci, >78% (Figure 1B). The two Infantis (ST32) differed in at least 136 loci, which was expected since Infantis strain CFSAN103816 lacked the MDR plasmid, indicating that these two strains belonged to very different clones circulating in the same country at the same time.
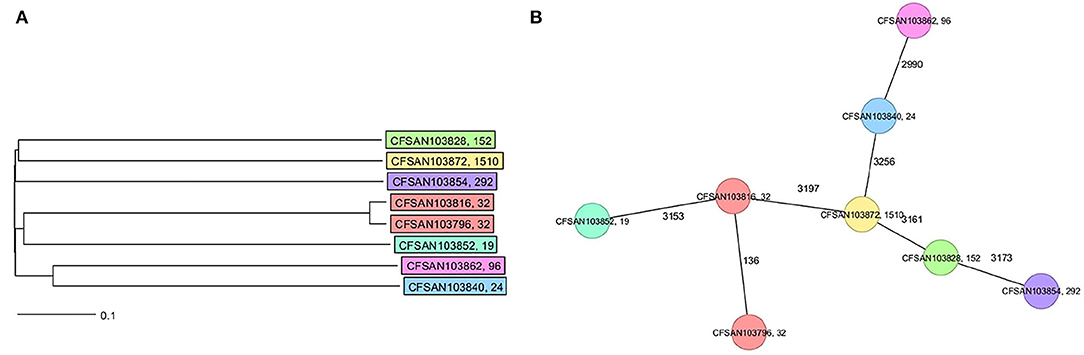
Figure 1. Core genome multi-locus sequence type (cgMLST) analysis of the eight Salmonella closed genomes based on 4,028 shared genes. (A) NJ tree showing the genetic differences among these genomes based on the shared loci. (B) Minimum spanning tree allowing to visualize the number of loci differences between these genomes. Strains are color-coded according to their serotype (ST). The ST is displayed after the name of the strain, separated by a comma. Numbers by lines represent the number of loci differing between strains. The lines are not drawn to scale.
The closed genomes were further analyzed for the presence of AMR genes using the ResFinder database (Table 1) (Bortolaia et al., 2020). All strains were found to contain mdsA, mdsB, and mdsC components of the MdsABC complex in the chromosome. MdsABC confers resistance to multiple toxic compounds, including novobiocin, acriflavine, crystal violet, ethidium bromide, methylene blue, rhodamine 6G, tetraphenylphosphonium bromide, benzalkonium bromide, and SDS and oxidative stress-inducing compounds (diamide, H2O2, Paraquat), and is necessary for colonization of host cells (Nishino et al., 2006; Song et al., 2015). Seven of the strains demonstrated multi-drug resistance with the presence of at least three AMR genes. Aminoglycoside, tetracycline, and sulfonamide resistance genes were detected in seven strains: CFSAN103796 [aac (6′), tetA, aph(3″), sul1, aadA1, aac(3)-lva, aph(4)-la], CFSAN103854 [aac(6′)-Iaa], CFSAN103872 [aac(6′)-Iaa, aph(3″)-lb, aph(6)-ld], CFSAN103852 [aac(6′)-Iaa, aph(3″)-lb, aph(6)-ld], CFSAN103816 [aac(6′)-Iaa], CFSAN103862 [aac(6′)-Iaa, aph(3″)-lb, aph(6)-ld], and CFSAN103828 [aac(6′)-Iaa, aph(3″)-lb, aph(6)-ld, tetB] (Table 1).
In clinical Salmonella isolates, combined resistance to ampicillin, chloramphenicol, streptomycin, sulfonamides, and tetracycline (ACSSuT) decreased from 8.2% in 1996 to 3.6% in 2009 (Medalla et al., 2013), but other medically relevant antibiotics have become a recent focus of study. Synthetically derived fluoroquinolones were introduced to combat ACSSuT resistance. Three isolates contained the quinolone resistance gene qnrB19 in plasmids (CFSAN103840, CFSAN103854, and CFSAN103872) of different sizes. Plasmid-mediated quinolone resistance was not identified in retail meat products until 2017 (Sjölund-Karlsson et al., 2010; Tyson et al., 2017). The presence of these small plasmids carrying the qnrB19 gene is worrisome since they have been identified in other countries in Latin America, such as Peru, Bolivia, Colombia, and Argentina, and appear to be highly mobile among different microorganisms (Tran et al., 2012).
The β-lactams, including ampicillin and cephalosporin, are another treatment for invasive salmonellosis, particularly in children. Ceftriaxone resistance increased in the US from 10% in 2002 to 38% in 2010 in chickens, prompting the prohibition of cephalosporin by the FDA in 2012. The extended-spectrum β-lactamase genes blaCTX−M−65 (CFSAN103796) and blaTEM−1 (CFSAN103852) were detected in this study on plasmid pCFSA103796 (312,952 bp) and on the chromosome, respectively. An alignment of pCFSA103796 against several blaCTX−M−65−containing plasmids reported by Tate et al. (2017), showed pCFSAN103796 was highly similar but differed, besides size and SNPs, in two of the main AMR sites from the other reported plasmids (Table 2; Figure 2). There were at least two genes missing in Site 1 that were present in the other plasmids (floR, and fosA3) while pCFSAN103796 has acquired a qacEdelta1 gene on Site 2 (Table 2).
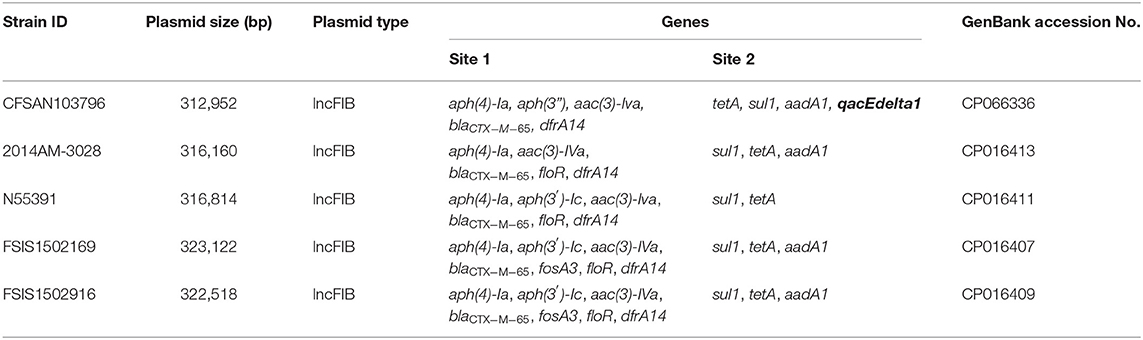
Table 2. Comparison of blaCTX−M−65-positive plasmids from our study and four other US strains.
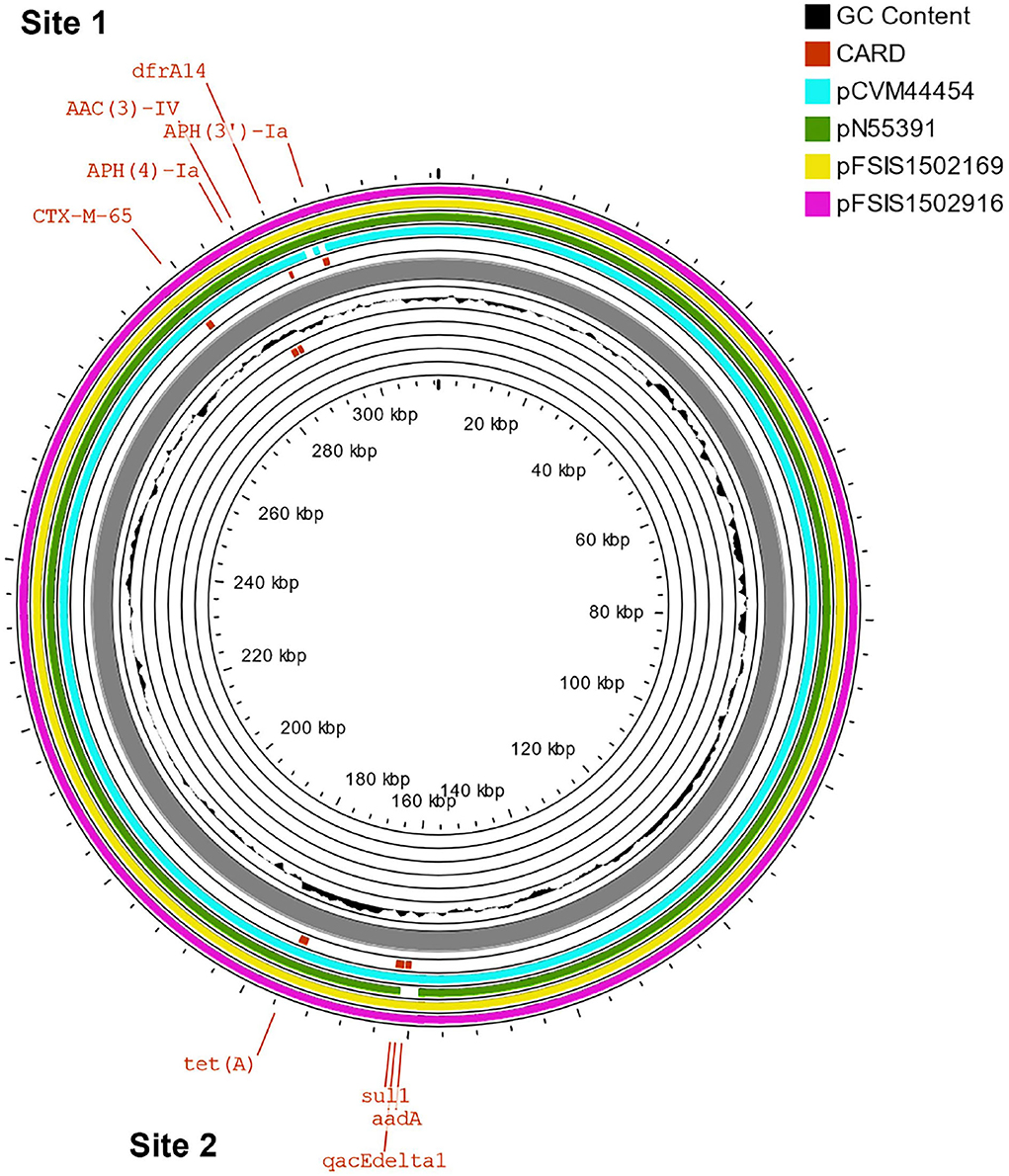
Figure 2. Comparison of blaCTX−M−65-positive plasmids from our study and four other US strains from Tate et al. (2017). Genetic map of the CFSAN103782 plasmid generated with CGView (25). The gray ring is the pCFSAN103796 plasmid. Blue block arrows in the outer circle denote coding regions in the plasmid indicating ORF transcription direction. The G+C content is shown in the middle circle and the deviation from the average G+C content in the innermost circle. BLAST comparisons with four other blaCTX-M-65-positive plasmids are shown in cyan (CP016413—strain 2014AM-3028), green (CP016411—strain N55391), yellow (CP016407—strain FSIS1502169), and pink (CP016409—strain FSIS1502916). The two sites containing the AMR genes mentioned in Table 2 are indicated.
Core genome multi-locus sequence type analysis of these 5 Infantis strains (Table 2) and CFSAN103816 (as an outgroup) showed that the 4 strains differed from CFSAN103796 by a minimum of 19 loci differences, further confirming the divergent evolution of this strain from the other ones isolated in the US (Figure 3). Furthermore, Brown et al. (2018) described the presence of blaCTX−M−65−containing Salmonella enterica serotype Infantis isolates in the US that were grouped in the same phylogenic clade as MDR Peruvian isolates (Brown et al., 2018), illustrating the intercontinental spread of this AMR gene. This latter fact highlights the importance of closing the genomes of strains carrying this important AMR gene to track the spreading and evolution of this plasmid through gene acquisitions and SNP accumulations during all these years and in different regions of the world. In turn, this type of analysis could allow establishing the route of dissemination around the world for this strain/plasmid combination as has been done for S. Enteritidis (Li et al., 2021). We have additionally detected the presence of the mcr-9 gene in another strain isolated from this overall study (Maguire et al., 2021). The mcr-9 gene variant was described in 2019 (Carroll et al., 2019) and appears not to confer colistin resistance in Salmonella and Escherichia coli (Tyson et al., 2020).
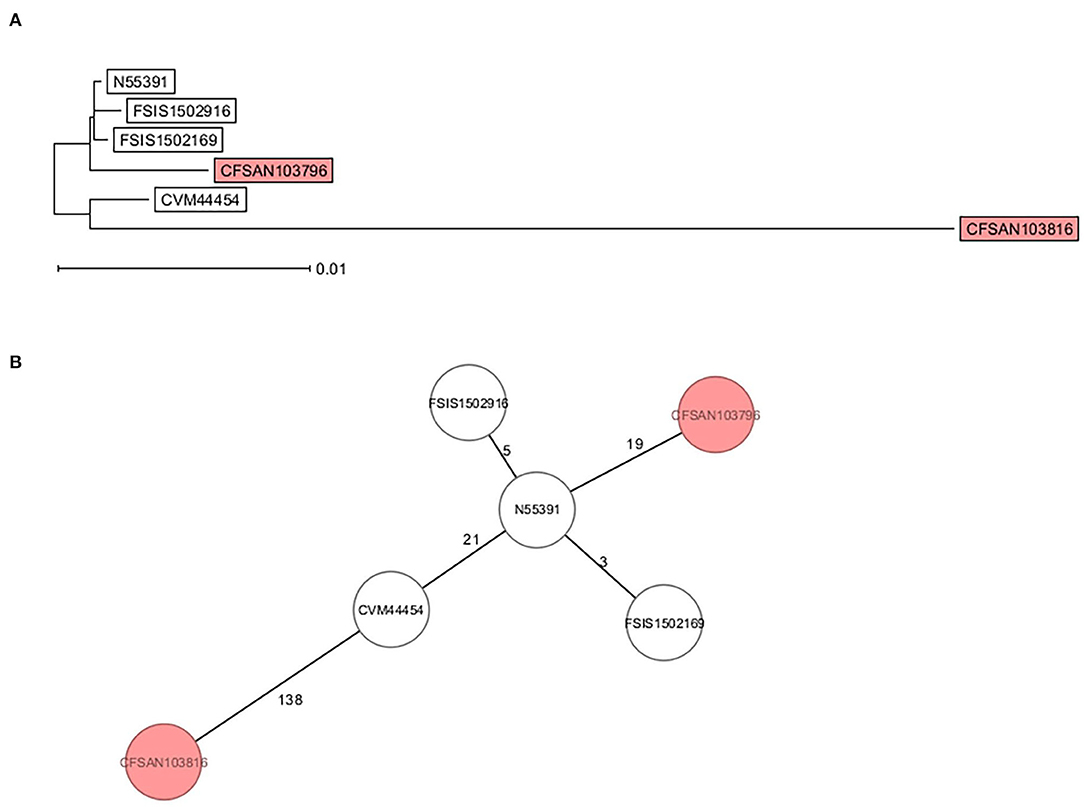
Figure 3. cgMLST analysis of six S. Infantis strains showing the divergent evolution of CFSAN103796 from the US strains (2014AM-3028, N55391, FSIS1502169, and FSIS1502916) and the other S. Infantis closed genome from Trinidad and Tobago (CFSAN103816). CFSAN103816 genome was used as an outgroup based on 3768 shared genes. (A) NJ tree showing the genetic differences among these genomes based on the shared loci rooted in CFSAN103816. (B) Minimum spanning tree allowing to visualize the number of loci differences between these genomes. Numbers by lines represent the number of loci differing between strains. The two S. Infantis from Trinidad and Tobago are shown in the colored boxes. The lines are not drawn to scale.
Further analysis of the presence or absence of reported virulence genes for S. enterica was determined by an in silico approach using the data available in the virulence factor database (VFDB) for that bacterium (Liu et al., 2019) (Table 3). As reported in that table (the complete virulence gene repertoire is available in Supplementary Table 2), all closed Salmonella genomes from this study carried both Salmonella pathogenicity islands SPI-1 and SPI-2, as reported previously by others (Hoffmann et al., 2016; Lou et al., 2019). SPI-1 contains genes encoding various components of a type III secretion system (T3SS), its regulators, and its secreted effectors (Marcus et al., 2000). SPI-1 enables S. typhimurium to efficiently penetrate the intestinal epithelium and its role may be limited to the colonization of the gut (Marcus et al., 2000). While SPI-2 contains a second T3SS that is essential for survival within the macrophage and for the establishment of systemic infection (Kombade, 2021). The type III secretion system of SPI-2 is structurally and functionally distinct from that of SPI-1. These two SPIs are necessary for Salmonella intestinal epithelial invasion and virulence. An interesting finding was that strain CFSAN103862 serotype Schwarzengrund lacked prgH, -I, -J, -K and orgA genes in the SPI-1. These genes are required for the secretion of invasion effector proteins and mutant strains lacking these genes were attenuated for invasion in a standard invasion assay of HEp-2 cell monolayers (Fahlen et al., 2000). Some did not carry the lpf operon (CFSAN103840—Javiana, and CFSAN103862—Schwarzengrund) which has been shown to be crucial for the attachment of Salmonella enterica serovar Typhimurium to murine Peyer's patches (Bäumler et al., 1996). The lack of these many genes in this particular Schwarzengrund strain suggests that it might be less pathogenic to humans since invasion and attachment might be impaired. However proper experiments must be conducted in the future to prove this theory.
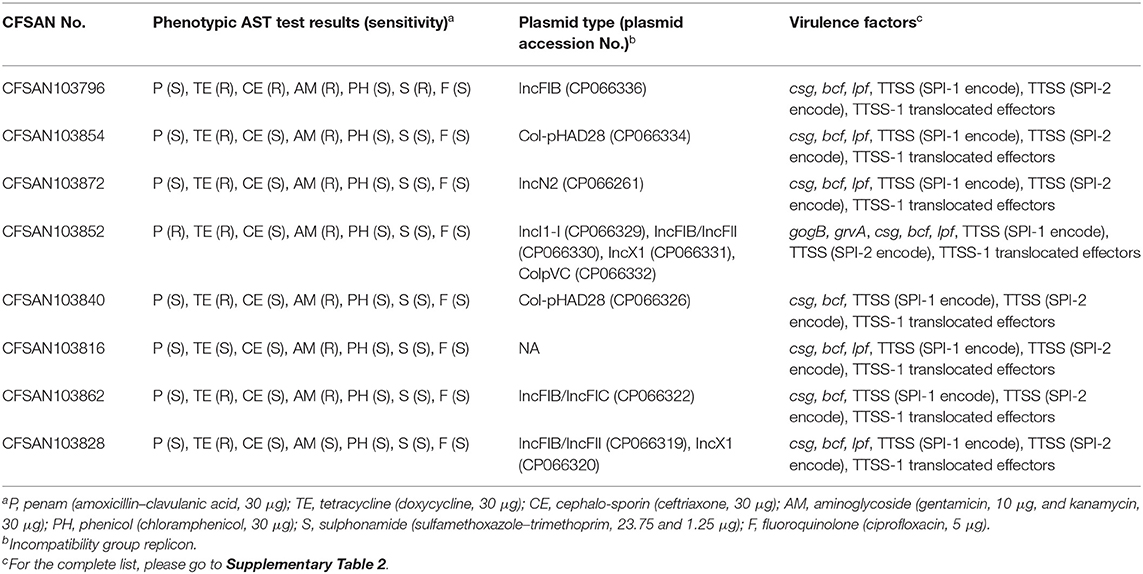
Table 3. Phenotypic and plasmid classification per strain from this study.
Strain CFSAN103852, a monophasic variant of S. Typhimurium was the only one carrying both gogB and grvA genes. gogB, an anti-inflammatory effector, helps Salmonella regulate inflammation-enhanced colonization by limiting tissue damage during infection (Pilar et al., 2012). While grvA, an anti-virulence gene located on the Gifsy-2 phage of S. Typhimurium, participates actively in modulating its pathogenicity (Ho and Slauch, 2001). The presence of these genes only in this monophasic S. Typhimurium confirms that the pathogenicity of different Salmonella serotypes is a complex system and might include redundant systems. Most of these eight strains appear to be pathogenic to humans, although with some differences in virulence profiles.
We further reported the plasmid type and the phenotypic AST test results for the eight strains sequenced in this study. The plasmids carried by these strains contained at least six different incompatibility group replicons, with four of the strains carrying plasmids with an IncFIB incompatibility group replicon (Table 3), which has been reported in many MDR plasmids (Tate et al., 2017).
The presence of MDR NTS in poultry environments remains a significant global challenge for public health. With the increasing reports of newer variants of AMR genes that confer resistance to diverse types of antibiotics, the need to enact active AMR gene surveillance studies is paramount. With these types of studies, we can not only detect the presence and spread of important AMR genes in poultry, but also identify the strains carrying these AMR genes and their location (whether in the chromosome or plasmids). Here we closed eight genomes of several MDR NTS, determine their ST, serotype, and associated AMR gene profiles, which will be useful for future source tracking, and outbreak investigations in Trinidad and Tobago and worldwide.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
MM and NG-E analyzed the data and wrote the manuscript. MM performed laboratory experiments. MM, AK, AA, KG, and NG-E provided materials and critically reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by funding from the Chief Scientist Challenge Grants Program Proposal Number 2021-200F07A and the FDA Foods Program Intramural Funds. The University of the West Indies, St. Augustine Campus Research and Publication Fund Committee (Research Grant Number 2660-457522) supported the collection of Salmonella from poultry sources in Trinidad and Tobago.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.863104/full#supplementary-material
Andrews, W (1992). Manual of food quality control. 4. Rev. 1. Microbiological analysis. Food And Drug Administration. FAO Food Nutr. Pap. 14, 1–338.
Antunes, P., Mourao, J., Campos, J., and Peixe, L. (2016). Salmonellosis: the role of poultry meat. Clin. Microbiol. Infect. 22, 110–121. doi: 10.1016/j.cmi.2015.12.004
Bäumler, A.J., Tsolis, R. M., and Heffron, F. (1996). The Lpf fimbrial operon mediates adhesion of Salmonella typhimurium to murine peyer's patches. Proc. Natl. Acad. Sci. USA 93, 279–283. doi: 10.1073/pnas.93.1.279
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bortolaia, V., Kaas, R. S., Ruppe, E., Roberts, M. C., Schwarz, S., Cattoir, V., et al. (2020). Resfinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 75, 3491–3500. doi: 10.1093/jac/dkaa345
Brown, A. C., Chen, J. C., Watkins, L. K. F., Campbell, D., Folster, J. P., Tate, H., et al. (2018). Ctx-M-65 extended-spectrum β-lactamase-producing Salmonella enterica serotype infantis, United States. Emerg. Infect. Dis. 24, 2284–2291. doi: 10.3201/eid2412.180500
Carroll, L. M., Gaballa, A., Guldimann, C., Sullivan, G., Henderson, L. O., and Wiedmann, M. (2019). Identification of novel mobilized colistin resistance gene Mcr-9 in a multidrug-resistant, colistin-susceptible Salmonella enterica serotype typhimurium isolate. Mbio 10, e00853-19. doi: 10.1128/mBio.00853-19
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genom. Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Deng, X., Desai, P. T., Den Bakker, H. C., Mikoleit, M., Tolar, B., Trees, E., et al. (2014). Genomic epidemiology of salmonella enterica serotype enteritidis based on population structure of prevalent lineages. Emerg. Infect. Dis. 20, 1481–1489. doi: 10.3201/eid2009.131095
Fahlen, T. F., Mathur, N., and Jones, B. D. (2000). Identification and characterization of mutants with increased expression of Hila, the invasion gene transcriptional activator of Salmonella typhimurium. Fems. Immunol. Med. Microbiol. 28, 25–35. doi: 10.1111/j.1574-695X.2000.tb01453.x
Foley, S. L., Nayak, R., Hanning, I. B., Johnson, T. J., Han, J., and Ricke, S. C. (2011). Population dynamics of Salmonella enterica serotypes in commercial egg and poultry production. Appl. Environ. Microbiol. 77, 4273–4279. doi: 10.1128/AEM.00598-11
Gonzalez-Escalona, N., Haendiges, J., Miller, J. D., and Sharma, S. K. (2018). Closed genome sequences of two Clostridium botulinum strains obtained by nanopore sequencing. Microbiol. Resour. Announc. 7, e01075–18. doi: 10.1128/MRA.01075-18
Gonzalez-Escalona, N., Hammack, T. S., Russell, M., Jacobson, A. P., De Jesus, A. J., Brown, E. W., et al. (2009). Detection of live Salmonella sp. cells in produce by a taqman-based quantitative reverse transcriptase real-time Pcr targeting inva Mrna. Appl. Environ. Microbiol. 75, 3714–3720. doi: 10.1128/AEM.02686-08
Ho, T. D., and Slauch, J. M. (2001). Characterization Of Grva, an antivirulence gene on the Gifsy-2 phage in Salmonella enterica serovar typhimurium. J. Bacteriol. 183, 611–620. doi: 10.1128/JB.183.2.611-620.2001
Hoffmann, M., Luo, Y., Monday, S. R., Gonzalez-Escalona, N., Ottesen, A. R., Muruvanda, T., et al. (2016). Tracing origins of the Salmonella bareilly strain causing a food-borne outbreak in the United States. J. Infect. Dis. 213, 502–508. doi: 10.1093/infdis/jiv297
Jackson, B. R., Griffin, P. M., Cole, D., Walsh, K. A., and Chai, S. J. (2013). Outbreak-associated Salmonella enterica serotypes and food commodities, United States, 1998–2008. Emerg. Infect. Dis. 19, 1239–1244. doi: 10.3201/eid1908.121511
Khan, A. S., Georges, K., Rahaman, S., Abebe, W., and Adesiyun, A. A. (2021). Characterization Of Salmonella isolates recovered from stages of the processing lines at four broiler processing plants in trinidad and tobago. Microorganisms 9, 1048. doi: 10.3390/microorganisms9051048
Khan, A. S., Georges, K., Rahaman, S., Abebe, W., and Adesiyun, A. A. (2022a). Occurrence, risk factors, serotypes, and antimicrobial resistance of Salmonella strains isolated from imported fertile hatching eggs, hatcheries, and broiler farms in trinidad and tobago. J. Food Prot. 85, 266–277. doi: 10.4315/JFP-21-236
Khan, A. S., Pierneef, R. E., Gonzalez-Escalona, N., Maguire, M., Li, C., Tyson, G. H., et al. (2022b). Molecular characterization of Salmonella detected along the broiler production chain in Trinidad and Tobago. Microorganisms 10, 570. doi: 10.3390/microorganisms10030570
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Kuehn, B. M (2010). Salmonella cases traced to egg producers: findings trigger recall of more than 500 million eggs. JAMA 304, 1316. doi: 10.1001/jama.2010.1330
Li, S., He, Y., Mann, D. A., and Deng, X. (2021). Global spread of Salmonella enteritidis via centralized sourcing and international trade of poultry breeding stocks. Nat. Commun. 12, 5109. doi: 10.1038/s41467-021-25319-7
Liu, B., Zheng, D., Jin, Q., Chen, L., and Yang, J. (2019). Vfdb 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic. Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Lou, L., Zhang, P., Piao, R., and Wang, Y. (2019). Salmonella pathogenicity island 1 (Spi-1) and its complex regulatory network. Front Cell Infect. Microbiol. 9, 270. doi: 10.3389/fcimb.2019.00270
Maguire, M., Khan, A. S., Adesiyun, A. A., Georges, K., and Gonzalez-Escalona, N. (2021). Closed genome sequence of a Salmonella Enterica serotype senftenberg strain carrying the Mcr-9 gene isolated from broken chicken eggshells in Trinidad and Tobago. Microbiol. Resour. Announc. 10, E0146520. doi: 10.1128/MRA.01465-20
Majowicz, S. E., Musto, J., Scallan, E., Angulo, F. J., Kirk, M., O'brien, S. J., et al. (2010). The global burden of nontyphoidal Salmonella gastroenteritis. Clin. Infect. Dis. 50, 882–889. doi: 10.1086/650733
Marcus, S. L., Brumell, J. H., Pfeifer, C. G., and Finlay, B. B. (2000). Salmonella pathogenicity islands: big virulence in small packages. Microbes Infect. 2, 145–156. doi: 10.1016/S1286-4579(00)00273-2
Medalla, F., Hoekstra, R. M., Whichard, J. M., Barzilay, E. J., Chiller, T. M., Joyce, K., et al. (2013). Increase in resistance to ceftriaxone and nonsusceptibility to ciprofloxacin and decrease in multidrug resistance among Salmonella strains, United States, 1996-2009. Foodborne Pathog. Dis 10, 302–309. doi: 10.1089/fpd.2012.1336
Nei, M., Tajima, F., and Tateno, Y. (1983). Accuracy of estimated phylogenetic trees from molecular data. Ii. Gene Frequency Data. J. Mol. Evol 19, 153–170. doi: 10.1007/BF02300753
Nishino, K., Latifi, T., and Groisman, E. A. (2006). Virulence and drug resistance roles of multidrug efflux systems of Salmonella enterica serovar typhimurium. Mol. Microbiol. 59, 126–141. doi: 10.1111/j.1365-2958.2005.04940.x
Pilar, A. V. C., Reid-Yu, S. A., Cooper, C. A., Mulder, D. T., and Coombes, B. K. (2012). Gogb is an anti-inflammatory effector that limits tissue damage during salmonella infection through interaction with human Fbxo22 And Skp1. PloS Pathogens 8, E1002773. doi: 10.1371/journal.ppat.1002773
Robertson, J., Yoshida, C., Kruczkiewicz, P., Nadon, C., Nichani, A., Taboada, E. N., et al. (2018). Comprehensive assessment of the quality of Salmonella whole genome sequence data available in public sequence databases using the Salmonella in silico typing resource (Sistr). Microb. Genom. 4, e000151. doi: 10.1099/mgen.0.000151
Schoeni, J. L., Glass, K. A., Mcdermott, J. L., and Wong, A. C. (1995). Growth and penetration of Salmonella enteritidis, Salmonella heidelberg and Salmonella typhimurium in eggs. Int. J. Food Microbiol. 24, 385–396. doi: 10.1016/0168-1605(94)00042-5
Sjölund-Karlsson, M., Howie, R., Rickert, R., Krueger, A., Tran, T. T., Zhao, S., et al. (2010). Plasmid-mediated quinolone resistance among non-typhi Salmonella enterica isolates, USA. Emerg. Infect Dis. 16, 1789–1791. doi: 10.3201/eid1611.100464
Song, S., Lee, B., Yeom, J. H., Hwang, S., Kang, I., Cho, J. C., et al. (2015). Mdsabc-mediated pathway for pathogenicity in Salmonella enterica serovar typhimurium. Infect. Immun. 83, 4266–4276. doi: 10.1128/IAI.00653-15
Stothard, P., and Wishart, D. S. (2005). Circular genome visualization and exploration using Cgview. Bioinformatics 21, 537–539. doi: 10.1093/bioinformatics/bti054
Tate, H., Folster, J. P., Hsu, C. H., Chen, J., Hoffmann, M., Li, C., et al. (2017). Comparative analysis of extended-spectrum-beta-lactamase Ctx-M-65-producing Salmonella enterica serovar infantis isolates from humans, food animals, and retail chickens in the United States. Antimicrob. Agents Chemother. 61, e00488-17. doi: 10.1128/AAC.00488-17
Tatusova, T., Dicuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). Ncbi prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Thomson, N. R., Clayton, D. J., Windhorst, D., Vernikos, G., Davidson, S., Churcher, C., et al. (2008). Comparative genome analysis of Salmonella Enteritidis Pt4 And Salmonella Gallinarum 287/91 provides insights into evolutionary and host adaptation pathways. Genome Res. 18, 1624–1637. doi: 10.1101/gr.077404.108
Toro, M., Retamal, P., Ayers, S., Barreto, M., Allard, M., Brown, E. W., et al. (2016). Whole-genome sequencing analysis of Salmonella enterica serovar enteritidis isolates in chile provides insights into possible transmission between gulls, poultry, and humans. Appl. Environ. Microbiol. 82, 6223–6232. doi: 10.1128/AEM.01760-16
Tran, T., Andres, P., Petroni, A., Soler-Bistue, A., Albornoz, E., Zorreguieta, A., et al. (2012). Small plasmids harboring qnrb19: a model for plasmid evolution mediated by site-specific recombination at Orit and Xer sites. Antimicrob. Agents Chemother. 56, 1821–1827. doi: 10.1128/AAC.06036-11
Tyson, G. H., Li, C., Hsu, C. H., Ayers, S., Borenstein, S., Mukherjee, S., et al. (2020). The Mcr-9 gene of Salmonella and Escherichia coli is not associated with colistin resistance in the United States. Antimicrob. Agents Chemother. 64, e00573-20. doi: 10.1128/AAC.00573-20
Tyson, G. H., Tate, H. P., Zhao, S., Li, C., Dessai, U., Simmons, M., et al. (2017). Identification of plasmid-mediated quinolone resistance in Salmonella isolated from swine ceca and retail pork chops in the United States. Antimicrob. Agents Chemother. 61, e01318-17. doi: 10.1128/AAC.01318-17
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13, E1005595. doi: 10.1371/journal.pcbi.1005595
Keywords: Salmonella, multi-drug resistance, complete genome, nanopore sequencing, poultry, Trinidad and Tobago
Citation: Maguire M, Khan AS, Adesiyun AA, Georges K and Gonzalez-Escalona N (2022) Genomic Comparison of Eight Closed Genomes of Multidrug-Resistant Salmonella enterica Strains Isolated From Broiler Farms and Processing Plants in Trinidad and Tobago. Front. Microbiol. 13:863104. doi: 10.3389/fmicb.2022.863104
Received: 26 January 2022; Accepted: 08 April 2022;
Published: 10 May 2022.
Edited by:
Marciane Magnani, Federal University of Paraíba, BrazilReviewed by:
Andrea Isabel Moreno Switt, Pontificia Universidad Católica de Chile, ChileCopyright © 2022 Maguire, Khan, Adesiyun, Georges and Gonzalez-Escalona. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Narjol Gonzalez-Escalona, bmFyam9sLmdvbnphbGV6LWVzY2Fsb25hQGZkYS5oaHMuZ292
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.