 Changyou Song1,2
Changyou Song1,2 Haibo Wen1,2Guangxiang Liu2Xueyan Ma1Guohua Lv1Ningyuan Wu2Jianxiang Chen2Miaomiao Xue2Hongxia Li1,2*Pao Xu1,2*
Haibo Wen1,2Guangxiang Liu2Xueyan Ma1Guohua Lv1Ningyuan Wu2Jianxiang Chen2Miaomiao Xue2Hongxia Li1,2*Pao Xu1,2*- 1Key Laboratory of Freshwater Fisheries and Germplasm Resources Utilization, Ministry of Agriculture and Rural Affairs, Freshwater Fisheries Research Center, Chinese Academy of Fishery Sciences, Wuxi, China
- 2Wuxi Fisheries College, Nanjing Agricultural University, Wuxi, China
With strong demand for aquatic products, as well as a rapid decrease in global fishery resources and capture fisheries, domesticating animals to provide more high-quality proteins is meaningful for humans. Freshwater drum (Aplodinotus grunniens) is widely distributed in the wild habitats of North America. However, the research on A. grunniens and the feed domestication with diets composed of artificial compounds remains unclear. In this study, a 4-month feeding domestication experiment was conducted with A. grunniens larvae to evaluate the underlying mechanism and molecular targets responsible for alternations in the ingestion performance. The results indicated that a significant increase in the final body weight was exhibited by the feed domesticated group (DOM, 114.8 g) when compared to the group that did not ingest the feed (WT, 5.3 g) as the latest version we raised From the result, the final body weight exhibited significant increase between unfavorable with the feed (WT, 5.3 g) and feed domesticated group (DOM, 114.8 g). In addition, the enzyme activity of digestive enzymes like amylase, lipase, and trypsin was increased in DOM. Genes related to appetite and perception, such as NPY4R, PYY, and LEPR, were activated in DOM. 16s rRNA gene sequencing analysis revealed that Pseudomonas sp. increased from 58.74% to 89.77% in DOM, which accounts for the dominant upregulated microbial community at the genus level, followed by Plesiomonas. Analogously, Mycobacterium, Methylocystis, and Romboutsia also accounted for the down-regulated microbes in the diversity. Transcriptome and RT-PCR analysis revealed that feed domestication significantly improved protein digestion and absorption, inhibited apoptosis by AGE-RAGE signaling, and activated extracellular matrix remodeling by relaxin signaling. Integrated analysis of the microbiome and host transcriptome revealed that Pseudomonas-mediated ingestion capacity, protein utilization, and cellular homeostasis might be the underlying mechanism under feed domestication. These results indicate Pseudomonas and its key genes relating to food ingestion and digestion could serve as the molecular targets for feed domestication and sustainable development in A. grunniens.
HIGHLIGHTS
- DOM improves appetite and perception.
- Protein metabolism and cellular homeostasis were active to DOM.
- Pseudomonas was increased under DOM.
- Pseudomonas medicates ingestion and growth under DOM.
Introduction
With the fast-growing population and rising prosperity all over the world, producing sufficient amounts of animal-based food products has become a critical challenge for humankind (FAO, 2020). In accordance with livestock animal products, aquatic animals contribute significantly to meet the demand of protein requirements. Therein, aquaculture is, globally, the fastest growing farmed food production sector, showing a 5.3% increase annually from 2001 to 2018 (FAO, 2020). However, there are still some bottlenecks in the development of aquaculture in recent years. One of the most important limitations is that the majority of aquaculture production is from a small number of culture species (Du et al., 2021), as over 90% of the production involves just 27 species/species groups (Hallerman et al., 2021). Therefore, domestication of some species with excellent biological, nutritional, social, and economic traits will promisingly promote sustainable aquaculture development.
During the domestication of new aquatic species, feed utilization or ingestion performance is the first and vital step (Liao and Huang, 2000). Domestication with artificial feed is a process that involves adaptation to the ingestion habits, which is closely related to feeding motivation, metabolic and physiological change, neuroendocrine system regulation, and behavioral adaptations (Volkoff, 2016). The gut system is closely related to nutrient absorption and has been confirmed to impact the ingestion behavior and host appetite adaptation (Dabrowski and Portella, 2005). Therefore, to improve the heritable adaption to ingestion habits from the aspects of the gut system is the key question to answer for feed domestication.
In recent years, microorganisms have been increasingly recognized for their role in the health and overall performance of aquatic organisms (Parata et al., 2021). Gut microbiota refers to the entire population of microorganisms that colonize the intestine, and investigations on microbiota associated with a fish gut have deepened our knowledge about the complex interactions that occur between microbes and host fish (Talwar et al., 2018). Functionally, gut microbiota exerts a profound influence on immunity, nutrient-processing capacity, disease susceptibility, growth, and reproduction of the host animals (Nayak, 2010; Ghanbari et al., 2015). Inversely, the gut microbial community is also affected by the nutritional status (Xia et al., 2014), diet situations (Peredo et al., 2015), and feeding behaviors (Ye et al., 2014). Therefore, alternation of gut microbiota is vital to uncover the ingestion behavior under domestication.
Discovering the core microbiome is the primary goal for many researchers interested in understanding gut microbial communities (Turnbaugh et al., 2007), which is critical for their application to promote the host health and wellbeing by manipulating the microbes inhabiting the aquatic animals. However, defining a core microbiome has proven to be an elusive task in many species, including humans, and terrestrial and aquatic animals. Therefore, the diversity and abundance of dominated microbes are of great importance for the evaluation of the functions of gut microbiome. However, the studies on the role of gut microbiota in ingestion performance and appetite adaption are currently limited.
Freshwater drum (Aplodinotus grunniens) is a kind of fish that is endemic to North and Central America. It is the only species in the genus of Aplodinotus that exclusively inhabits freshwater for its entire life (Hernández-Gómez et al., 2021). With respect to edibility, the freshwater drum is featured to possess a higher edible proportion, with delicious and nutritious flesh rich in proteins, amino acids, and fatty acids. Moreover, the freshwater drum has no intermuscular bones, which improves the fish quality and processing of the aquatic product. Therefore, these distinct characteristics reveal freshwater drum has the potential for domestication and cultivation to provide high-quality proteins for human beings. However, the domestication, management practices, aquaculture, and even scientific research for this particular species is limited. With these prospects, we imported the larvae of freshwater drum from the United States in 2016, and have achieved a milestone in the artificial breeding technique and cultivation in 2019, which provided a break through and prospects for aquaculture. In behavior, freshwater drum is similar to carnivorous fish, and the diet for freshwater drums in the wild is generally benthic animals and composed of macroinvertebrates (mainly larvae of aquatic insects and bivalve mussels), as well as small fish, mollusks, and crayfish in certain ecosystems (Rypel, 2007; Jacquemin et al., 2014). In our cultivation, we found freshwater drum prefers live baits, and the ingestion ability and performance of artificial diets that contain high levels of plant-source proteins are limited. However, economically, the feeding behavior and food preference determine the production costs and profitability of freshwater drum aquaculture. Therefore, the domestication of a freshwater drum by an artificial diet enriched with plant-source proteins is critical for the sustainable development of the fish, particularly to expand the scale of breeding, aquaculture, and food processing.
In the present study, we evaluated the relationship between the gut microbiome and ingestion performance under feed domestication in the freshwater drum. Meanwhile, the regulation between gut microbes and encoding genes was also investigated. These results could reveal the feed ingestion regulation and could provide potential targets to improve the feed domestication and sustainable development of freshwater drums.
Materials and Methods
Ethics Statement
This study was approved by the Animal Care and Use Committee of Nanjing Agricultural University (Nanjing, China). All animal procedures were performed according to the Guidelines for the Care and Use of Laboratory Animals in China.
Experimental Animals and Feed Domestication
The embryos of A. grunniens were obtained from the Freshwater Fisheries Research Center, Chinese Academy of Fishery Sciences. Larvae were raised in tanks at 25 ± 2°C and fed with chironomid larvae (Chironomus tentans). After 21 days of hatching, about 60,000 healthy larvae were randomly transferred into three outdoor fish ponds (pond size was 667 m2, 20,000 individuals per pond, representing three biological replicates) for feed domestication experiment. During the experiment, fish in the WT and DOM groups were fed with commercial diets (45% crude protein and 8% crude lipid; Seahorse feed Ltd., China) four times a day. The domestication experiment was conducted for 4 months (16th May to 15th September), and the water source was drawn from underground. Aeration was also provided to maintain enough dissolved oxygen (DO). The water quality was maintained as follows: pH 7.6–7.8, DO > 6 mg/L, NH3 < 0.01 mg/L, and H2S < 0.01 mg/L.
Sample Collection
After 4 months of feed domestication experiment, fish were starved for 24 h to evacuate the residual feed in the alimentary tract prior to sampling. The fish that ingested the compounds of the feed exhibited increased size, while the non-ingested fish were obviously small. Therefore, 27 non-feed ingested (WT) and feed ingested (DOM) fish in each pond were randomly taken to evaluate the growth performance indices, respectively. Meanwhile, nine WT and DOM fish were randomly taken and anesthetized with tricaine mesylate (MS-222, 100 mg/L) for sample collection, respectively. Blood samples were obtained from the caudal vein and stored in tubes coated with heparin, and then centrifuged at 4,500 rpm at 4°C for 10 min to obtain the plasma. Meanwhile, the sampled fish were dissected on ice to collect the intestinal tissue. The whole intestine and intestinal contents were collected and immediately frozen in liquid nitrogen and stored at −80°C for mRNA sequencing, 16s sequencing, and RT-PCR analysis.
Growth Performance and Evaluation of Digestive Enzyme Activity
Growth performance was evaluated by measuring final body weight (FBW), condition factor (CF), viscerosomatic index (VSI), and hepatosomatic index (HSI). Each parameter was calculated as follows: CF = Final body weight/Final body length3; VSI (%) = 100 × Viscera weight/Final body weight; HSI (%) = 100 × Liver weight/Final body weight.
The glucose content in the plasma and the activity of digestive enzymes, such as amylase, lipase, and trypsin, in the intestine were determined by using assay kits according to the manufacturer’s protocol (provided by Nanjing Jiancheng Bioengineering Institute, China). Briefly, glucose was detected by hexokinase method (Category No: F006-1-1), amylase was determined by starch-iodine colorimetry method (Category No: C016-1-1), lipase was determined by colorimetric method (Category No: A054-1-1), and trypsin was determined by ultraviolet colorimetry method (Category No: A080-2-2).
RNA Extraction and Transcriptome Sequencing
Nine samples of whole-intestine tissues from WT and DOM groups were selected to conduct the high-throughput sequencing analysis. Then, three fish intestine samples from each group were randomly mixed, and three biological replicates were finally applied to extract the total RNA according to the established methods described by Song et al. (2017).
cDNA Library Construction and de novo Sequencing
The library construction and sequencing were conducted according to our previously established methods (Song et al., 2021). Briefly, total RNA was subjected to quality control with Ailent 2100 and Nanodrop (ThermoFisher Ltd.), and samples with 1.8 < OD260/280 < 2.0, 28S/18S > 1.0, and RNA integrity number (RIN) > 1.8 were treated with oligo (dT) to enrich mRNA. Next, random primers were used to splice the mRNA into short fragments (200–700 nt). Short fragments were used as templates to synthesize cDNA, and PCR amplification was performed after adding “A” tail and sequencing connector. Finally, paired-end sequencing strategies were applied for de novo high-throughput sequencing with Illumina Hiseq™ 2000 platform.
Raw Data Dominate, de novo Assembly, and Annotation
After sequencing, adaptors and low-quality reads were removed by using FastQC software (version: 0.10.8) to get the clean reads. Clean reads were assembled with Trinity, and the transcripts were mapped to NR, Swiss Port, Pfam, KOG, and GO databases for annotation.
Enrichment of Differentially Expressed Genes
Read Count and FPKM (fragments per kilobase million) for each annotated transcript were calculated using RSEM (version: 1.3.1), and transcripts with | Fold Change| ≥ 2 and corrected P-value < 0.05 were recognized as the differentially expressed genes (DEGs). Meanwhile, DEGs were also subjected to gene ontology (GO) and KEGG enrichment analysis using Blast2GO.
Gut Microbe Identification
Microbial DNA Extraction and 16S Sequencing
The intestinal tissues and contents from three fishes were randomly pooled as a replicate, and five replicates in each group were selected for gut microbiome analysis. Microbial DNA from each replicate was extracted by using E.Z.N.A.® Soil DNA Kit (Omega Bio-Tek, Norcross, GA, United States) according to the manufacturer’s protocol (Sun et al., 2020). The integrity of isolated DNA was measured by Nanodrop ND2000 spectrophotometer (Thermo Scientific, United States) and 1% agarose gel electrophoresis. Meanwhile, the concentration of isolated DNA was measured with Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen, United States) and fluorometer, and diluted to 20 ng/μl for sequencing.
The full length of 16S rRNA was amplified using the primers 338 F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806 R (5′-GGACTACHVGGGTWTCTAAT-3′) by PCR and sequenced in PacBio Sequel System using SMRT cell protocol. Sequences were denoised using DADA2 (version 1.8) and assembled into amplicon sequence variants (ASVs). A representative sequence of each ASV was assigned to a taxonomic level in the Ribosomal Database Project (RDP) database using the RDP classifier. Principal component analysis and heatmap analysis were performed by using the R package (version 3.1.0).
16s Sequencing Data Analysis
The alpha diversity was calculated using QIIME software (version 1.9.1), which included Ace, Chao, Shannon, and Simpson. One-way analysis of variance (ANOVA) was performed for all diversity indices, followed by a Tukey’s post hoc test when statistically significant differences were observed (P < 0.05) using SPSS 25.0. The enriched chord diagram and PCoA diagram were drawn using the R package (version 3.1.0). A genera abundance table was loaded into Primer v5, and a similarity percentage (SIMPER) analysis was performed to determine the genera responsible for differences between the groups. The cut-off for low contributions was set to a default of 90%. We then used SourceTracker2 (Knights et al., 2011), a Bayesian community-level microbial source-tracking tool, to estimate the proportion of sequences in different groups (Liu et al., 2021).
Integrated Analysis Between Differentially Expressed Genes and Microbes
To explore the relationship between differential DEGs and microbes, the functional prediction of differential OTUs and DEGs was integrated. With Pearson analysis, the same or similar pathways that were shared between the two sets of data were selected, and the obtained pathways were regarded as DEG-related functions that were affected by the intestinal microbiota.
RT-PCR Analysis
Total RNA was extracted from nine whole-intestine or brain tissues in each group using RNAiso Plus reagent (Dalian Takara Co., Ltd., China), and was incubated with RNase-free DNase (Dalian Takara Co., Ltd., China) to remove the contaminating genomic DNA. Quantity and quality of the RNA were assessed by OD260/280 method and electrophoresis in 1.5% agarose gel. Primers (listed in Table 1) for each gene were designed using Primer Premier 5.0 based on the mRNA sequences obtained from A. grunniens intestine genome database in our lab. All primers were synthesized by Shanghai Generay Biotechnology, Co., Ltd., China. Real-time quantitative PCR (RT-PCR) was performed with the SYBR® Primix Ex TaqTM II (TliRNase Plus) Kit using ABI 7500 Real-time PCR System according to the manufacturer’s protocol. The relative expression levels of the target genes were normalized to the housekeeping A. grunniens gene β-actin and further calculated using the 2–ΔΔCT method.
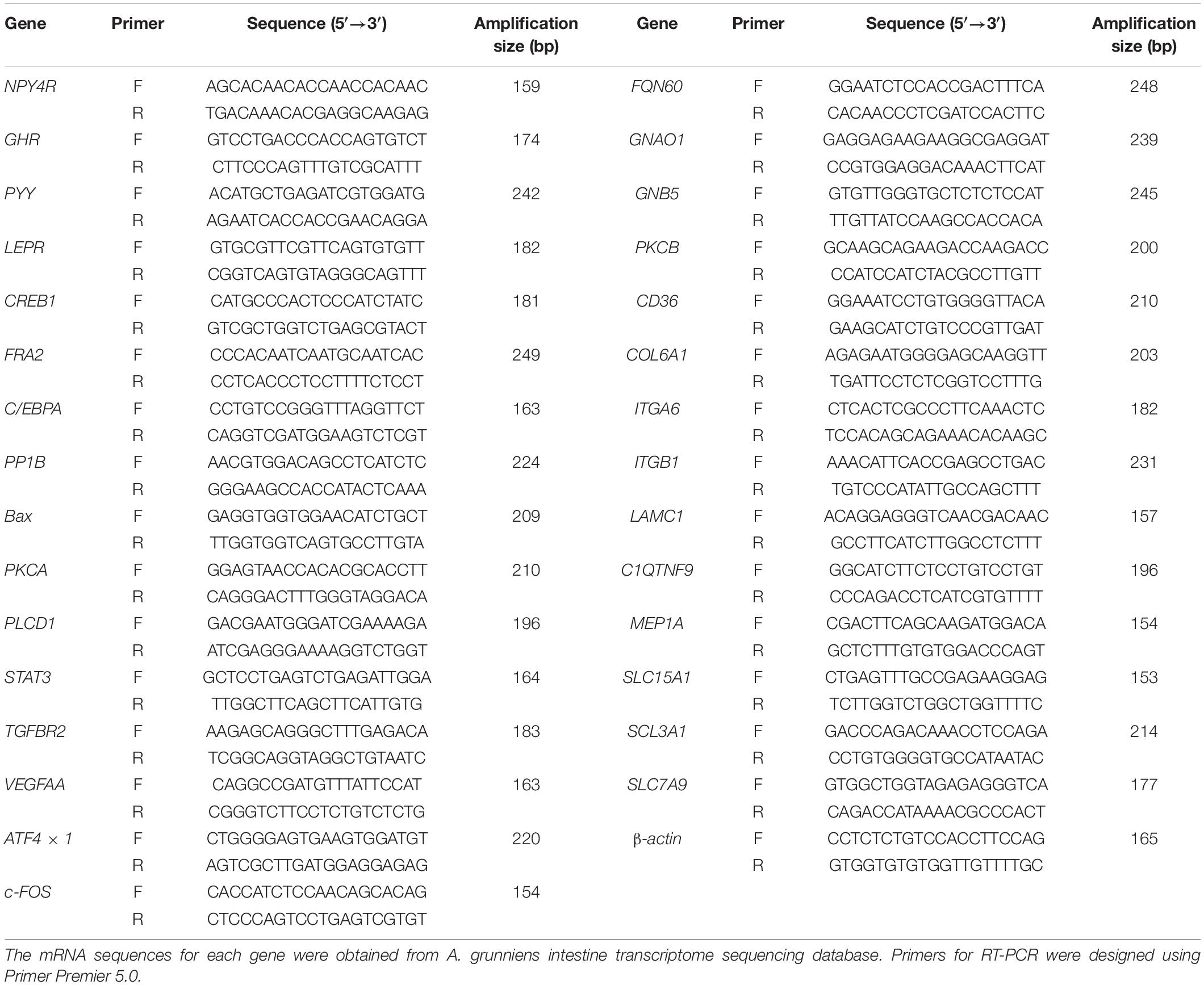
Table 1. Primers and sequences referred in the experiments.
Statistical Analysis
Independent samples t-test was conducted to analyze the transcriptional expression of related genes in SPSS 25.0. Meanwhile, all the data were validated for normality and homogeneity for variances. All results were expressed as the mean ± standard error of the mean (mean ± SEM).
Results
Feed Domestication Improves Growth Performance and Digestive Enzyme Activity in A. grunniens
The growth performance was first evaluated. We found the fish domesticated with compound feed (DOM) showed a significant improvement in the final body weight (FBW, Figure 1A, P = 4.395 × 10–10), increased by about 21.66-fold when compared to the fish that were unfavorable with the feed (WT). Meanwhile, the condition factor (CF, Figure 1B, P = 0.0020), viscerosomatic index (VSI, Figure 1C, P = 1.028 × 10–7), and hepatosomatic index (HSI, Figure 1D, P = 0.0001) were all significantly increased in the DOM group when compared to the WT group.
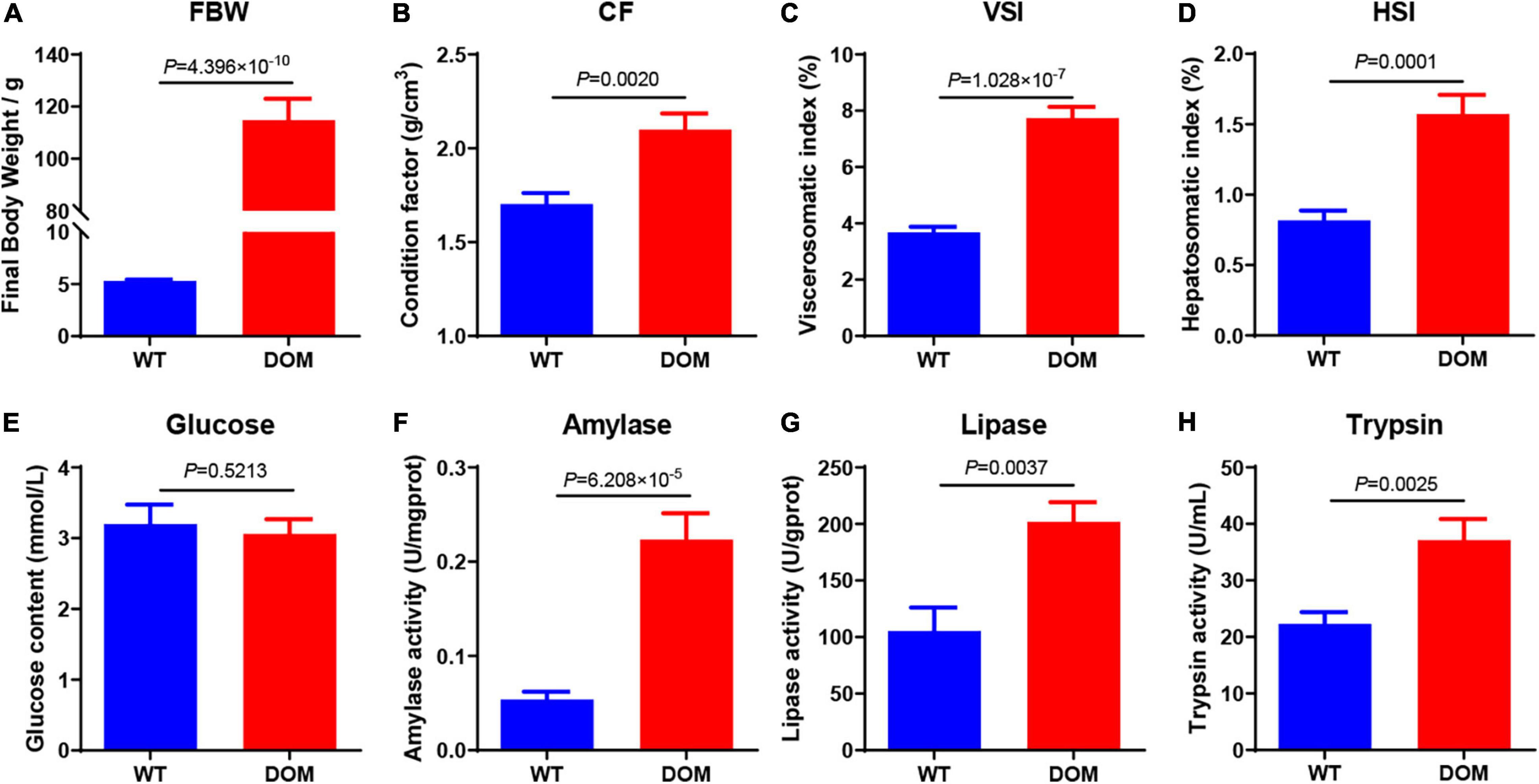
Figure 1. Feed domestication improves growth performance and digestive enzyme activity in A. grunniens. Panels (A–D) represent the growth performance, panel (E) represents the glucose content in the plasma, and panels (F–H) represent the digestive enzyme activity in the intestine. (A) Final body weight, FBW; (B) Condition factor, CF; (C) Viscerosomatic index, VSI; (D) Hepatosomatic index, HSI; (E) Glucose; (F) Amylase; (G) Lipase; and (H) Trypsin. Data were analyzed by Student’s t-test, and results were indicated as mean ± SEM; (A–D) n = 27; (E–H) n = 9.
In accordance with the growth performance, digestibility and absorption property were also detected. Plasma glucose content exhibited no significant difference between the groups (Figure 1E, P = 0.5213). However, the activities of intestinal amylase (Figure 1F, P = 6.208 × 10–5), lipase (Figure 1G, P = 0.0037), and trypsin (Figure 1H, P = 0.0025) were significantly increased in the DOM group.
Feed Domestication Improves Appetite and Perception in A. grunniens
To obtain an overview regarding the growth performance, digestive capacity, and food performance, we evaluated the transcriptional expression of appetite- and perception-related genes. Key genes related to appetite, including NYP4R (Figure 2A, P = 0.0017), GHR (Figure 2B, P = 0.0109), PYY (Figure 2C, P = 0.0175), and LEPR (Figure 2D, P = 0.0156), were all significantly upregulated in the intestine of DOM. The expression of learning- and memory-related genes that function to feed perception were also detected; the expression of CREB1 (Figure 2E, P = 0.0163), C/EBPA (Figure 2F, P = 0.0330), and PP1B (Figure 2G, P = 0.0189) were upregulated, while that of FRA2 (Figure 2H, P = 0.0119) was downregulated in the brain tissue of DOM.
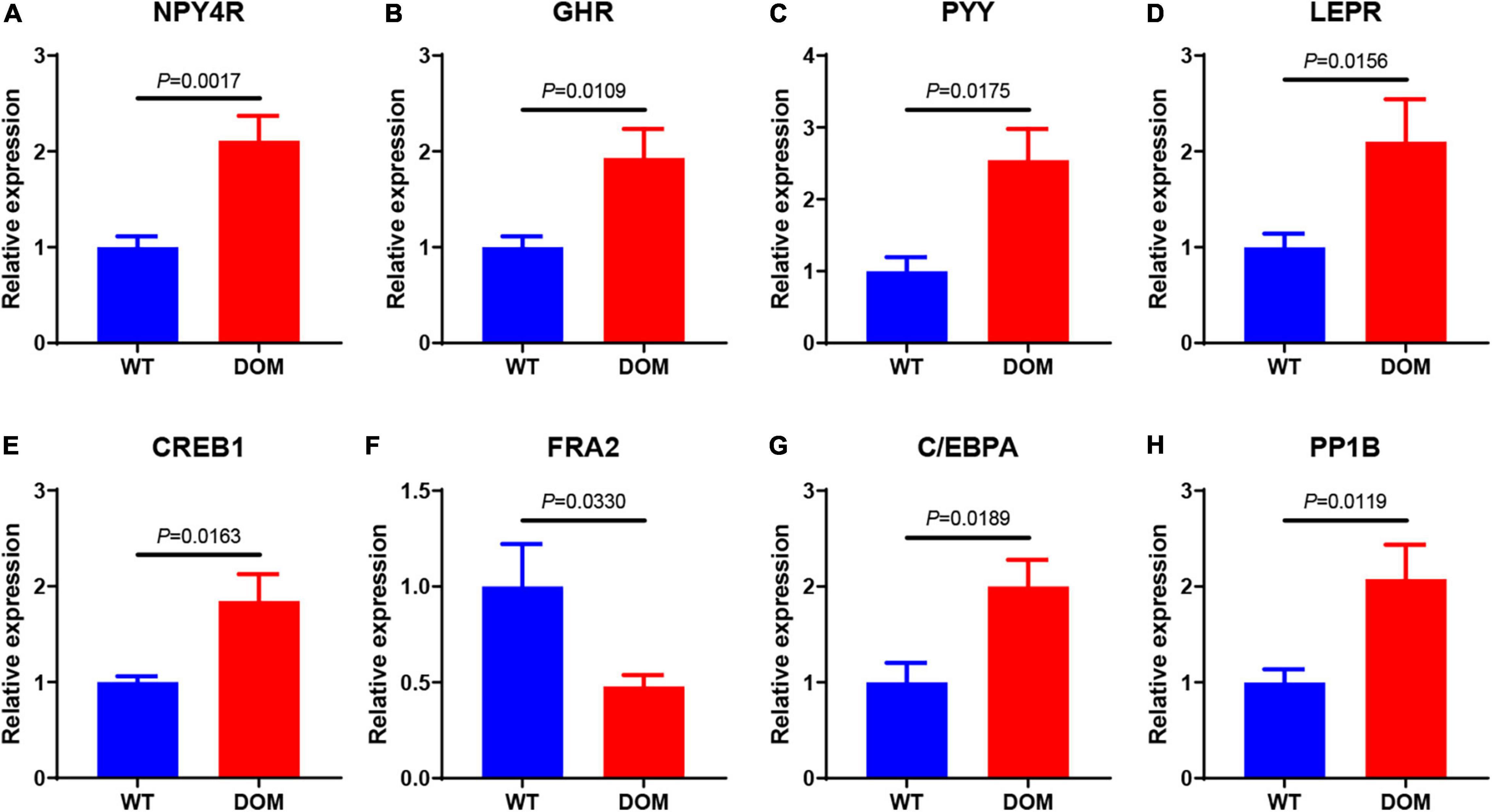
Figure 2. Feed domestication improves appetite and perception in the brain of A. grunniens. Transcriptional expression of appetite-related (A–D) and perception-related (E–H) genes. (A) NPY4R, (B) GHR, (C) PYY, (D) LEPR, (E) CREB1, (F) FRA2, (G) C/EBPA, and (H) PP1B. Data were analyzed by Student’s t-test, and results were indicated as mean ± SEM, n = 9.
Transcriptomic Analysis of A. grunniens Intestine Domesticated With Compound Feed
To reveal the underlying mechanism of food performance, we performed de novo transcriptomic analysis with high-throughput sequencing. When compared to WT, a total of 1,035 differentially expressed genes (DEGs) were identified and annotated in DOM (| Fold Change| ≥ 2 and corrected P-value < 0.05), including 451 upregulated and 584 downregulated genes (Figures 3A–C and Supplementary Tables 1A,B). To uncover the molecular functions of these DEGs, GO and KEGG enrichment analyses were conducted. With the threshold of corrected P-value < 0.05, a total of 9 GO items (Figure 3D and Supplementary Table 2A) and 26 KEGG pathways (Figure 3E and Supplementary Table 2B) were enriched. Specifically, collagen trimer, structural constituent of extracellular matrix, and lipid transporter activity were the most significantly enriched GO items (corrected P-value < 0.01), indicating DOM altered the cytoskeleton and cellular lipid transport. In accordance with GO enrichment, ECM–receptor interaction, relaxin signaling pathway, protein digestion and absorption, and AGE-RAGE signaling pathway were the most significantly enriched KEGG pathways (corrected P-value < 1 × 10–10), revealing DOM affected protein utilization, extracellular structure, and cell fate determination.
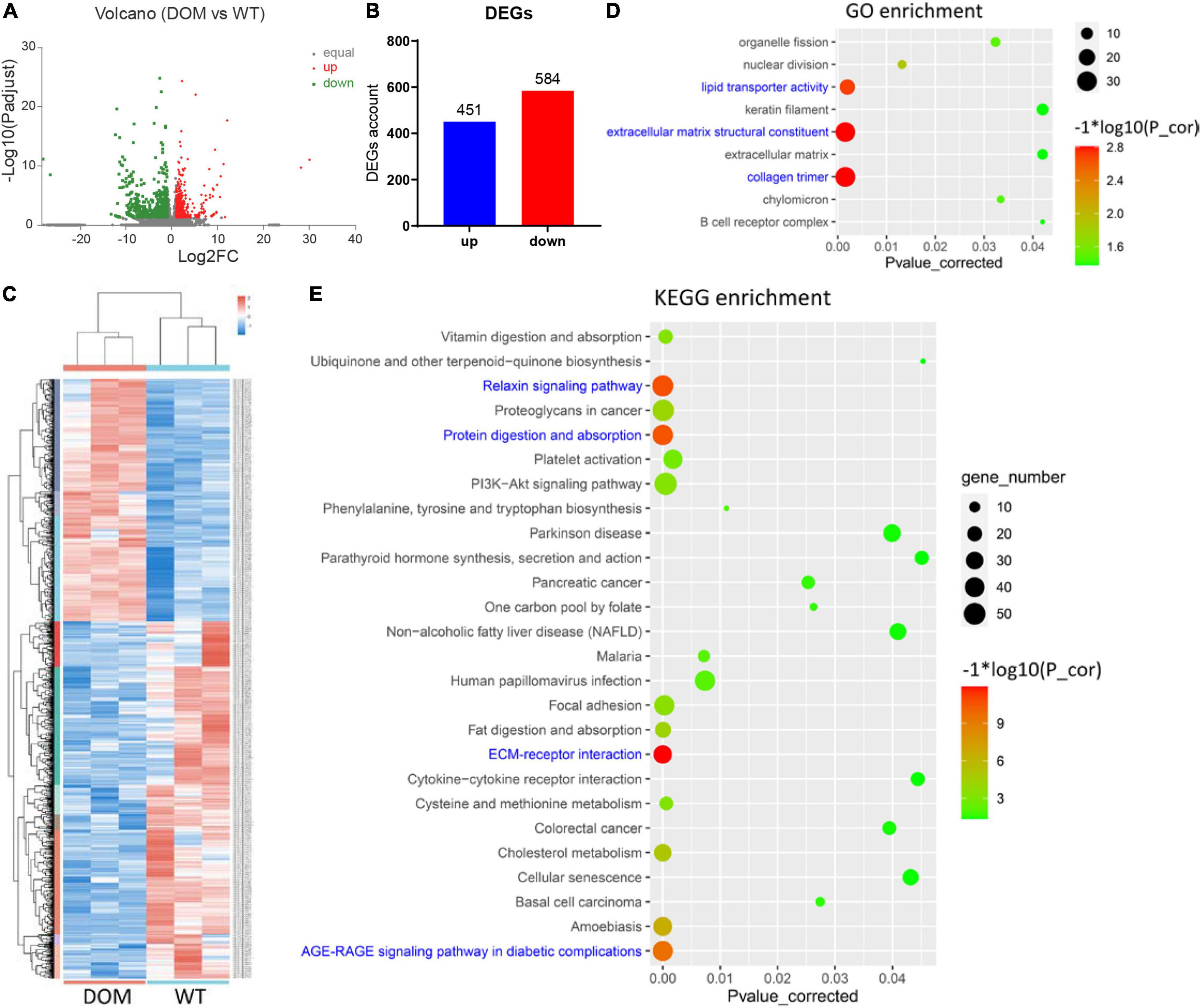
Figure 3. Transcriptomic analysis of A. grunniens intestine domesticated with compound feed. (A) Volcano plot of unigenes; (B) Statistics of differentially expressed genes (DEGs); (C) Heatmap cluster of DEGs; (D) GO annotation of DEGs; and (E) KEGG enrichment of DEGs.
Microbial Composition of A. grunniens Intestine Domesticated With Compound Feed
To further explore the potential regulatory mechanism, 16s rRNA gene amplicon sequencing was used to determine the composition of the microbial community, and alpha and beta diversity indices present in the fish gut contents. A total of 943 operational taxonomic units (OTUs) were identified (Supplementary Table 3), corresponding to 26 phyla, 78 classes, 189 orders, 286 families, 458 genera, and 657 species. Ace, Chao, Shannon, and Simpson’s analysis were applied to evaluate the alpha diversity of OTUs, and the results indicate that the OTUs exhibited a significant difference between the WT and DOM groups (Figure 4A, P < 0.05). Meanwhile, hierarchical clustering, PCA, and PLS-DA analyses were also conducted to evaluate the impact of DOM on the intestinal microbiome composition. Results indicate the microbes in the WT and DOM groups were clustered into different subsets at the phylum level due to the microbial diversity (Figures 4B–D). Additionally, microbes of Proteobacteria, Firmicutes, and Actinobacteriota show the most abundant diversity at the phylum level in both the WT and DOM groups. Notably, the proportion of the members of the phylum Proteobacteria was significantly increased from 74.48% (WT) to 97.22% (DOM) (P = 0.0007, Figure 4B).
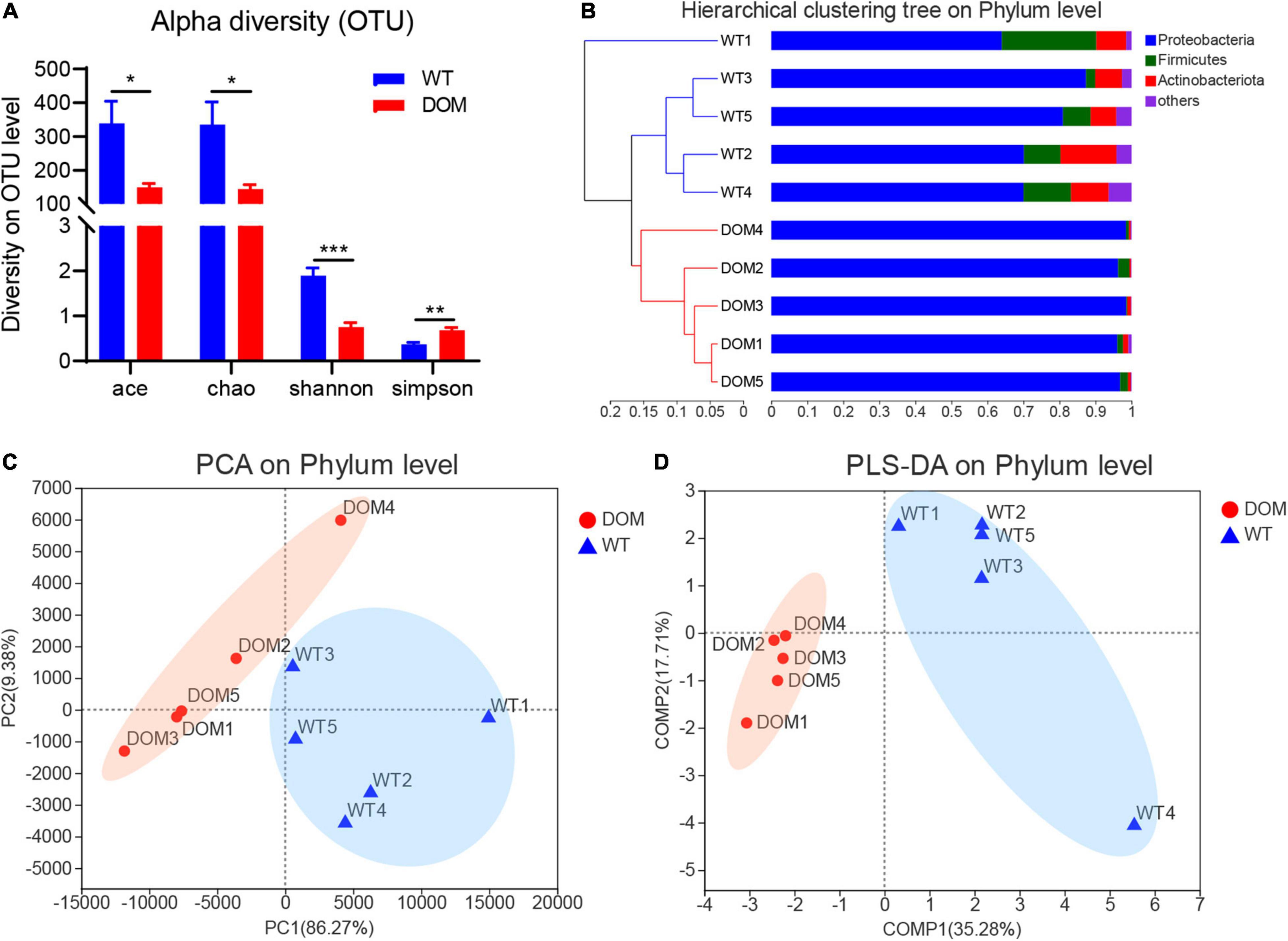
Figure 4. Microbial composition of A. grunniens intestine domesticated with compound feed. (A) Alpha density of the operational taxonomic units (OTUs); (B) Hierarchical cluster at phylum level; (C), Principal coordinates analysis (PCA) at the phylum level; and (D) Partial least-squares discriminant analysis (PLS-DA) at the phylum level. * represent P < 0.05; ** represent P < 0.01; and *** represent P < 0.001.
Microbial Comparison and Interaction Analysis at Phylum Level in A. grunniens Intestine Domesticated With Compound Feed
Next, the microbial composition and interaction analysis was conducted to reveal the dominant microbiota in both WT and DOM groups. A total of nine phyla were identified as the dominant microbes in the intestine of the DOM group, including upregulated Proteobacteria and Spirochaetota, and downregulated Firmicutes, Actinobacteriota, Cyanobacteria, Chloroflexi, Verrucomicrobiota, Myxococcota, and SAR324_cladeMarine_group_B (P < 0.05, Figure 5A). The interactions between microbes and samples are shown in Figure 5B, which indicates the members of phyla Proteobacteria, Firmicutes, and Actinobacteriota were dominant in both the WT and DOM groups, while Cyanobacteria, Chloroflexi, and Verrucomicrobiota were mainly dominant in the WT groups. Additionally, the phylogenetic analysis of these dominant microbes was also conducted, which revealed Proteobacteria exhibited a far distance from other microbes at the phylum level in evolution (Figure 5C).
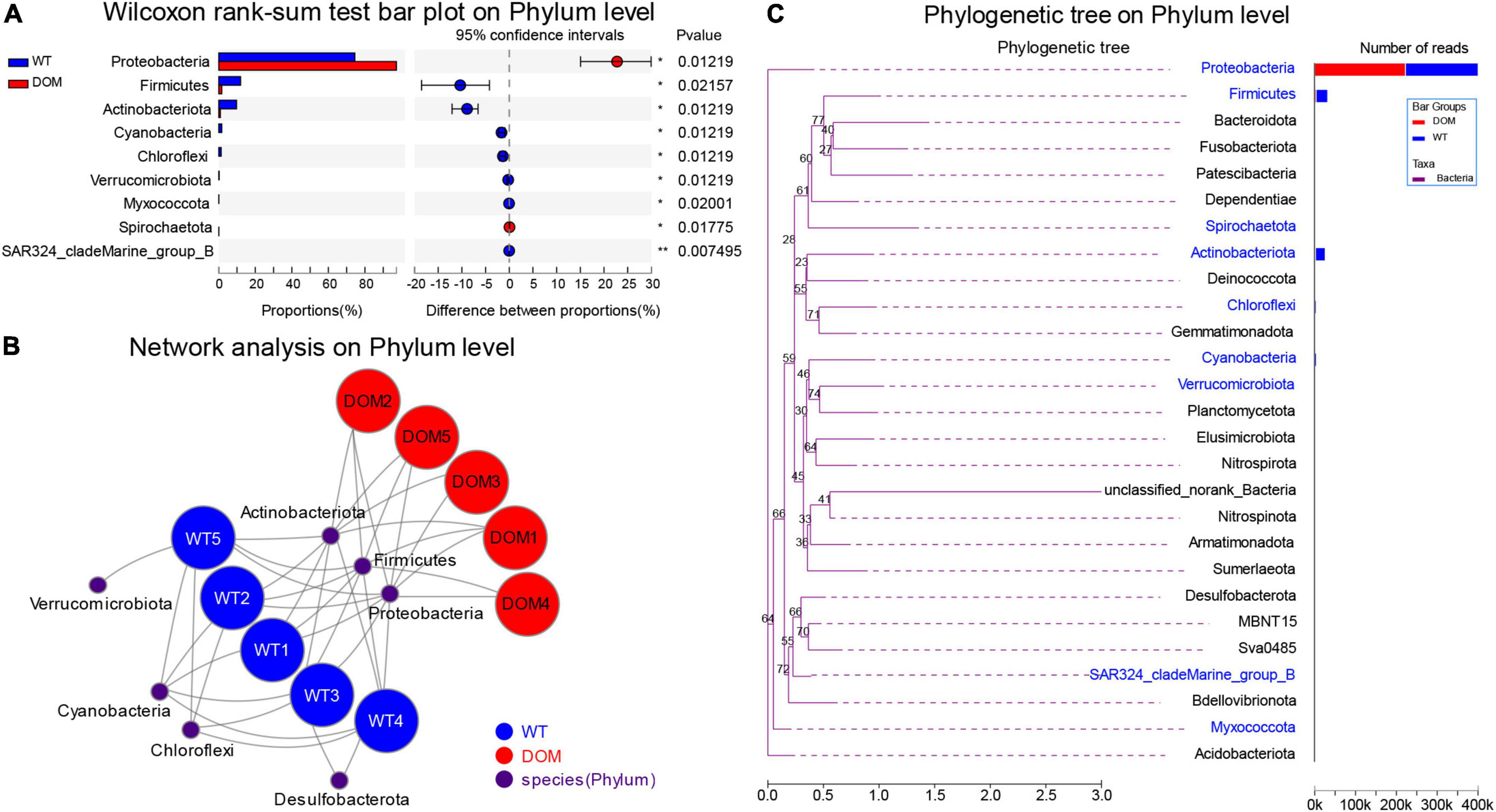
Figure 5. Microbial comparison and interaction analysis at phylum level in A. grunniens intestine domesticated with compound feed. (A) Differentially colonized microbial communities at phylum level; (B) interaction network between microbial communities and A. grunniens individuals; and (C) Phylogenetic analysis at the phylum level.
Microbial Comparison and Interaction Analysis at Genus Level in A. grunniens Intestine Domesticated With Compound Feed
To further explore the underlying microbial regulation on feed domestication, the alternation at the genus level was analyzed. The microbes of the phylum Proteobacteria, Pseudomonas, Plesiomonas, and Methylocystis were the most abundant and dominant microbes in the DOM group (Figure 6A). Meanwhile, unclassified_c_Bacilli and Romboutsia were the most abundant and dominant microbes in the DOM group for the phylum Firmicutes (Figure 6B), and Mycobacterium was the most abundant and dominant microbe in the DOM group for the phylum Actinobacteriota (Figure 6C). Additionally, analysis of the community diversity revealed that the genus Pseudomonas was the most dominant and diverse microbe under feed domestication, which increased from 58.47% in the WT group to 89.77% in the DOM group (Figure 6D).
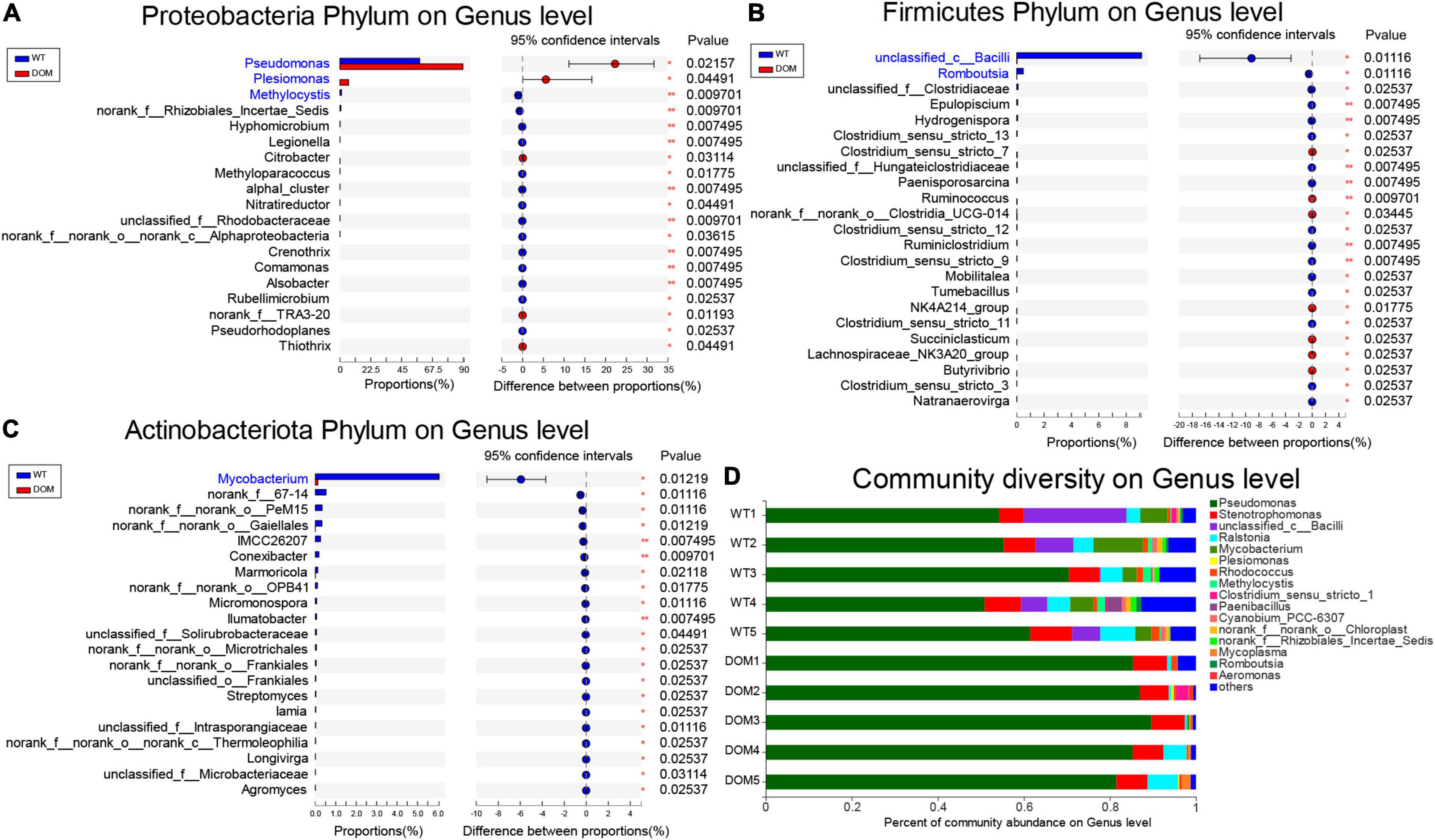
Figure 6. Microbial comparison and interaction analysis at genus level in A. grunniens intestine domesticated with compound feed. (A–C) Differentially colonized microbial communities at genus level of (A) phylum Proteobacteria; (B) phylum Firmicutes; and (C) phylum Actinobacteriota. (D) Community diversity at the genus level.
Integrated Analysis Between Differentially Expressed Genes and Microbes Under Feed Domestication in A. grunniens
The integrated network analysis between DEGs and microbes was performed to uncover the underlying mechanism of feed domestication. With Pearson analysis, a total of 67 DEGs co-related with six dominant microbes were applied to the heatmap cluster. Results revealed that the microbes were clustered into three sub-clusters based on their correlations with DEGs: Pseudomonas and Plesiomonas that belong to the phylum Proteobacteria were clustered into the same sub-cluster, Romboutsia and unclassified c Bacilli that belong to the phylum Firmicutes were clustered into the same sub-cluster, while Methylocystis in the phylum Proteobacteria and Mycobacterium in the phylum Actinbacteriota were clustered into another sub-cluster (Figure 7A). Meanwhile, DEGs (P-value < 0.05 in correlation with key microbes) from the heatmap cluster were enriched in the integrated network analysis, shown in Figure 7B. From the results, 44 DEGs were found to functionally interact with the microbes, including 25 DEGs that were closely related to Pseudomonas. Unexpectedly, the microbe Plesiomonas, which belongs to the phylum Proteobacteria along with Pseudomonas, did not directly interact with Pseudomonas in the network, revealing their different regulatory mechanisms under domestication. Additionally, the expression of Pseudomonas-related DEGs retrieved form RNA-seq was also analyzed, which indicated the regulatory role of Pseudomonas in appetite, perception, protein digestion and absorption, AGE-RAGE, relaxin, and ECM–receptor signaling pathways (Figure 7C).
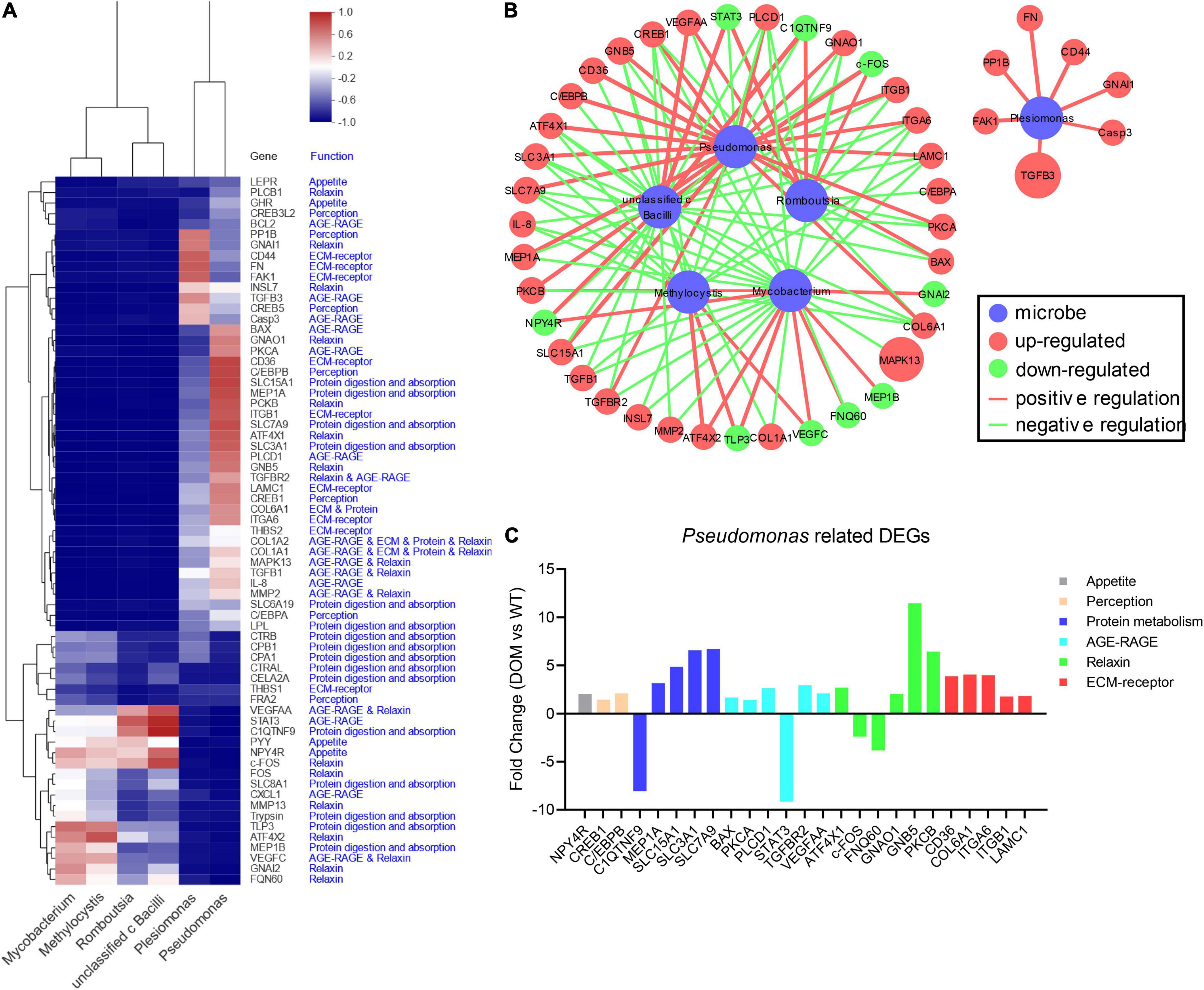
Figure 7. Integrated analysis between DEGs and microbes under feed domestication in A. grunniens. (A) Heatmap cluster between key microbes at genus level and enriched genes. (B) Interacting regulatory network between key microbes and key enriched genes. In the network, nodes in red color represent the upregulated DEGs, in green color represent the downregulated DEGs, in blue color represent the microbes. The size of a node represents the mRNA abundance retrieved from RNA-seq, and the width of the connecting line represents the correlation intensity between DEGs and microbes. (C) Transcriptome profiles of Pseudomonas-mediated DEGs relate to appetite, perception, protein digestion and absorption (protein metabolism), AGE-RAGE, relaxin, and ECM–receptor signaling.
Protein Digestion and Absorption, and AGE-RACE-Mediated Apoptosis Were Active to Feed Domestication in A. grunniens
The above-mentioned results indicate that cellular homeostasis (featured as AGE-RACE, relaxin, and ECM-receptor signaling) and protein digestion and absorption were involved in feed domestication. Next, we evaluated the expression of the key genes in these signaling pathways. The genes related to AGE-RAGE signaling (Figure 8A), such as Bax (P = 0.0190), PKCA (P = 0.0288), PLCD1 (P = 0.0012), TGFBR2 (P = 0.0187), and VEGFAA (P = 0.0364) were up-regulated in DOM, while STAT3 (P = 0.0497) was down-regulated in DOM. Meanwhile, genes related to relaxin signaling (Figure 8B), such as ATF4X1 (P = 0.0308), GNAO1 (P = 0.0358), GNB5 (P = 0.0156), and PKCB (P = 0.0072) were up-regulated, while c-FOS (P = 0.0448) and FQN60 (P = 0.0319) were down-regulated in DOM. Accordingly, genes related to ECM-receptor signaling (Figure 8C), such as CD36 (P = 0.0431), COL6A1 (P = 0.0201), ITGA6 (P = 0.0280), ITGB1 (P = 0.0377), and LAMC1 (P = 0.0230) were all up-regulated in DOM. Additionally, genes related to protein digestion and absorption (Figure 8D), such as C1QTNF9 (P = 0.0127), MEP1A (P = 0.0358), SCL15A1 (P = 0.0110), SLC3A1 (P = 0.0390), and SLC7A9 (P = 0.0091) were all up-regulated in DOM. These data reveal that Pseudomonas activates protein utilization and cellular homeostasis in response to feed domestication.
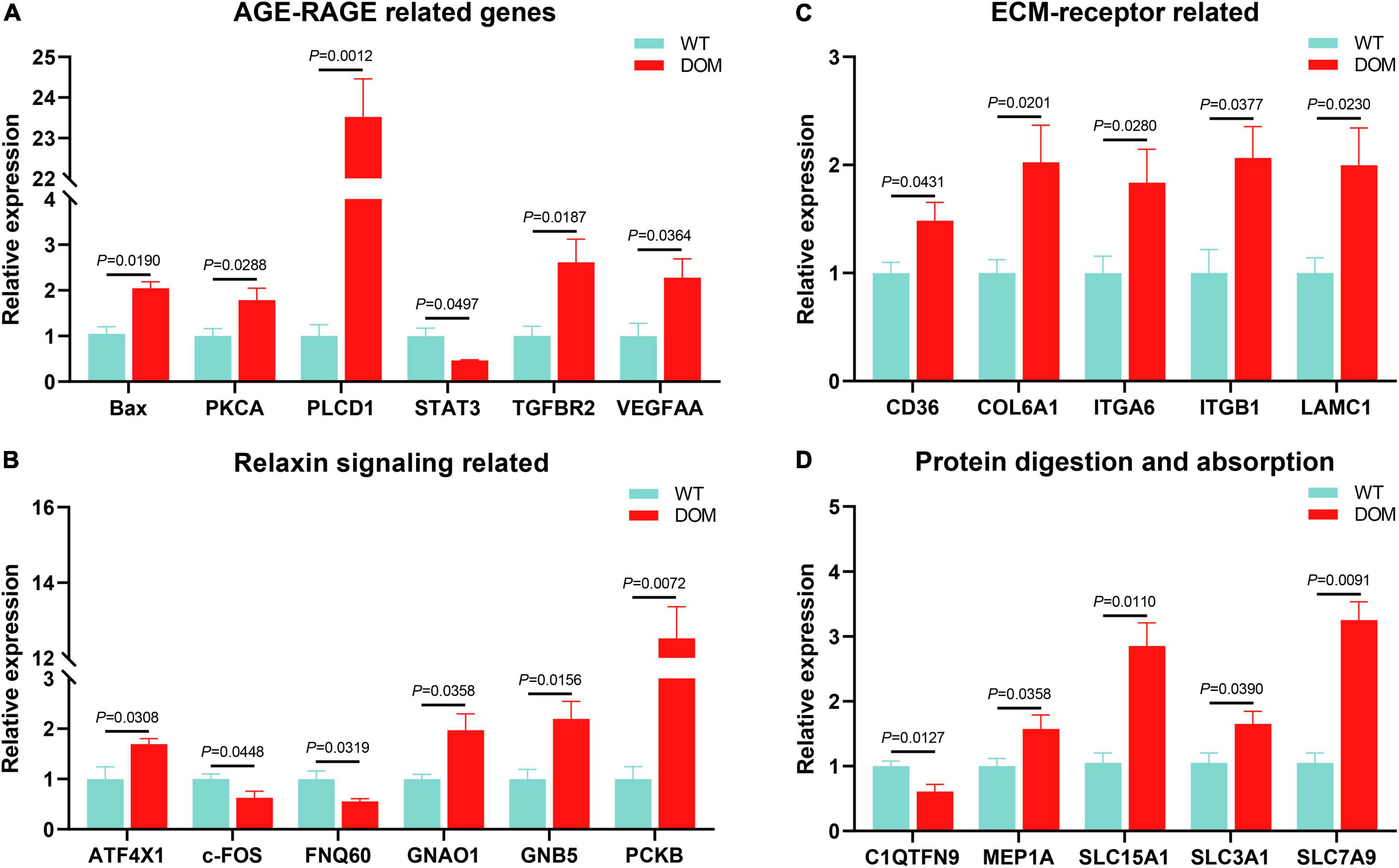
Figure 8. Pseudomonas medicated AGE-RAGE, Relaxin, ECM-receptor, and Protein digestion and absorption were active to feed domestication in A. grunniens. Transcriptional expression of AGE-RAGE related (A), Relaxin signaling related (B), ECM-receptor related (C), and protein digestion and absorption related (D) genes. Data were analyzed by Students’ t-test, results were indicated as mean ± SEM, n = 9.
”
Hypothetical Schematic of Pseudomonas-Mediated Ingestion and Growth Performance Regulated by Feed Domestication in A. grunniens
Based on the above-mentioned results, we raised the hypothetical regulation schematic of microbiota Pseudomonas (Figure 9). Feed domestication improved the food perception in the brain tissue, thereby transducing the signal to the intestine. In the intestine, DOM enhanced the abundance of the microbiota of Pseudomonas, which specifically plays a vital role in the activation of the appetite, protein digestion and absorption, and cellular homeostasis capacity. Therefore, Pseudomonas is the dominant microbiota, and its functions are important to improve the ingestion and growth performance during feed domestication.
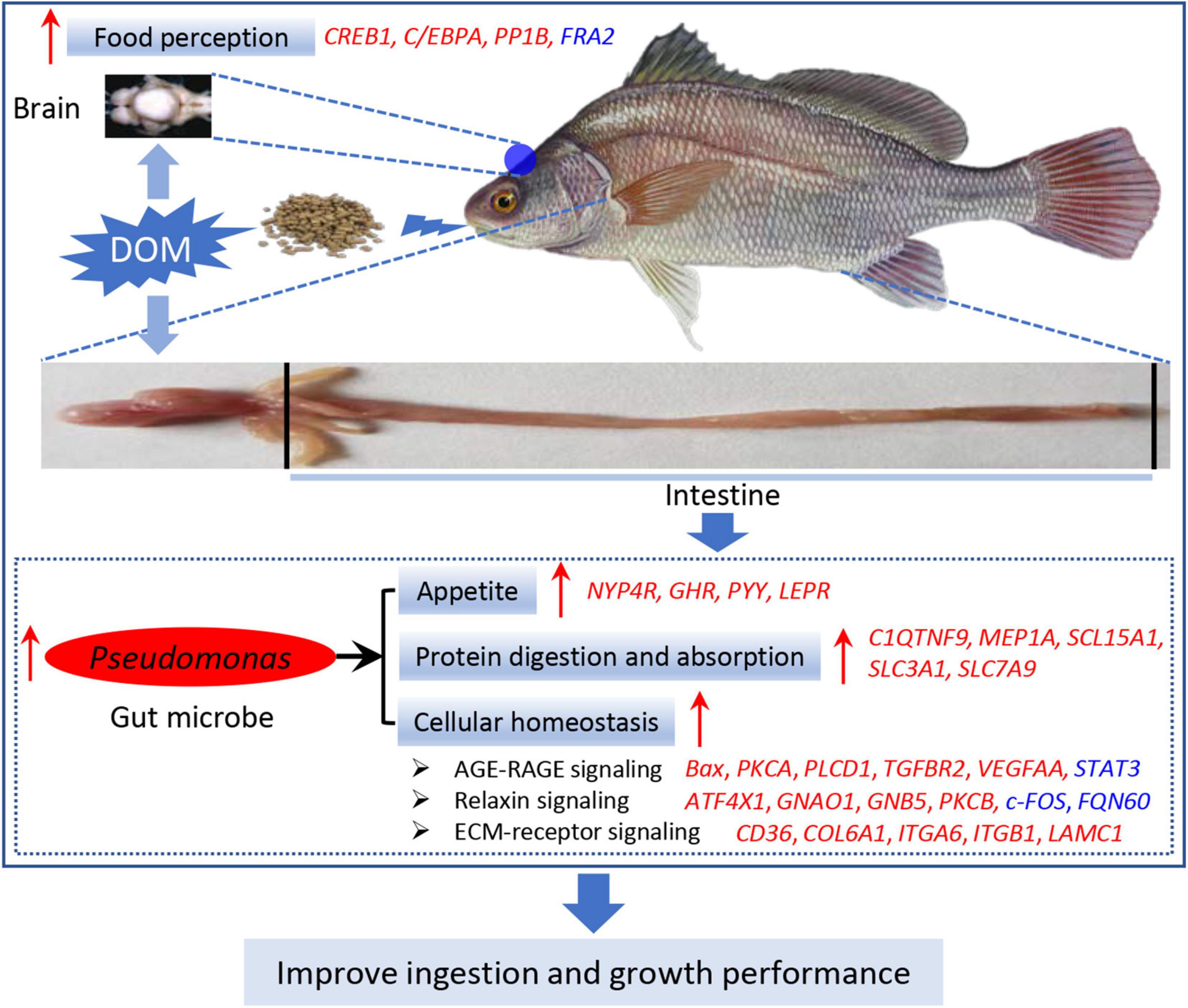
Figure 9. Hypothetical schematic of Pseudomonas-mediated ingestion and growth performance regulated by feed domestication in A. grunniens. Hypothetical regulation of Pseudomonas on ingestion and growth performance was raised based on the results in this study. Feed domestication improved feed perception in the brain tissue, thereby transducing the signal to the intestine. In the intestine, DOM improved the abundance of microbiota of Pseudomonas, eventually improving the ingestion and growth performance by enhancing appetite, protein digestion and absorption, and cellular homeostasis capacity. Arrows in red color represent the positive regulation or activation, italic letters in red color represent the upregulated genes, and italic letters in blue color represent the downregulated genes.
Discussion
Aquaculture is the fastest growing food-producing sector worldwide (FAO, 2020). The continued success of this sector relies on the productivity of fish culture by overcoming common challenges, such as the availability of excellent germplasm resources, advanced breeding management, and reasonable nutritional and environmental management programs (FAO, 2020). However, despite the fact that the cultivation practice has been well developed, over 90% of the production involves just 27 species (Hallerman et al., 2021). Therefore, the limited availability of well-developed species might become another bottleneck for aquaculture development in the next few decades. Herein, the domestication of aquatic species with high prolificacy, disease resistance, and rapid growth rate is of great importance (Teletchea and Fontaine, 2014; Xiong et al., 2014; Chen et al., 2017). Accumulating evidence has shown that inherited differences are closely associated with the feeding habits in the aquatic animals, as demonstrated in tilapia (Xiong et al., 2014), sticklebacks (Purnell et al., 2007), Eurasian perches (Chen et al., 2017), and mandarin fish (Shen et al., 2021). This inspired us to develop adaptive traits from the natural precious species to meet the high-quality protein requirements of humans.
One of the mechanisms influencing the feeding behavioral traits could be the changes in the host gut microbiome. Evidence suggests that microbial diversity is a marker of a healthy intestinal microbiome (Lozupone et al., 2012). Generally, microbiome diversity is relatively lower in diseased individuals compared to that of healthy individuals in humans (Cortes et al., 2022) and mice (Zhang et al., 2021). However, studies also reveal that starvation could increase the intestinal microbial diversity in desert locusts (Dillon et al., 2010) and fish (Xia et al., 2014). In addition, individuals with pure diets had less diverse microbiota than the fish feeding with mixed diets (Bolnick et al., 2014). These contrasting results indicate gut microbiota exerts different colonization resistance patterns under different enteric threats. In our present study, DOM decreased the microbial diversity in compare with WT, which might be a multiple effects of ingestion behavior, dietary intake, and intestinal microbiome rebalance.
In addition to microbial diversity, the relative abundance of microbes directly reveals the function of specific gut microbes. Generally, all species have species-specific gut microbiome composition, which influences their adaptation and diversification behaviors, including food preference, phenotypic plasticity, and innate and adaptive immunity (Glazko et al., 2021). The intestinal microbiota is either indigenous or transient. The indigenous genera are mainly obtained from the parents (Thomas and Percival, 2009), while the transient ones are largely dependent on the diet (Ringø et al., 2016). Fish with different dietary habits have distinct transient gut microbial communities: carnivorous fish are typically abundant in the phylum Proteobacteria, which functions importantly in the digestion and absorption of proteins (Ray et al., 2012). Meanwhile, herbivorous fish are enriched with Clostridiales, Bacteroidales, and Verrucomicorbiales; omnivorous fish had increased populations of Rhizobiales, Fusobacteriales, and Planctomycetales; while both omnivorous and carnivorous fish have abundant populations of Desulfovibrionales and Aeromonadales (Sullam et al., 2012; Liu et al., 2016). Additionally, dietary intake could affect the structure of the fish gut microbiome. In general, plant-derived dietary proteins have been linked to significantly reduced diversity of microbiota (Desai et al., 2012), with an increase in the relative abundance of Lactobacillales, Bacillales, and Pseudomonadales (Michl et al., 2017), while animal-derived proteins nurture Bacteroidales, Clostridiales, Vibrionales, Fusobacteriales, and Alteromonadales in the gut (Michl et al., 2017). In the present study, phylum Proteobacteria was significantly increased in the gut of freshwater drum, indicating that the protein digestion and absorption capacity were enhanced under feed domestication, which was in accordance with the results of digestive enzyme activity in the present study. Consistently, the abundance of the phylum Verrucomicrobiota, which is commonly found in herbivorous fish, was decreased. In addition, our data are in line with the previous studies that reported Proteobacteria, Firmicutes, and Actinobacteria (in this order of abundance) as the most common phyla of intestinal bacteria in fish (Butt and Volkoff, 2019; Silva-Brito et al., 2021).
The phylum Proteobacteria has the largest phylogenetic composition, comprising 116 validated bacterial families (Shin et al., 2015). Meanwhile, the population of Proteobacteria is the most unstable among the four main phyla (Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria) in the gut microbiota over time (Faith et al., 2013). The expansion of Proteobacteria is sensitive to environmental factors, such as diet (Caporaso et al., 2011). In humans, the abundance of Proteobacteria in the gut is low under healthy conditions, while an abnormal expansion of Proteobacteria is associated with inflammation or disease conditions, such as gastric bypass (Liou et al., 2013), metabolic disorders (Fei and Zhao, 2013), and inflammation and cancer (Morgan et al., 2012). However, the exact function of Proteobacteria in the fish remains unclear. Biologically, Proteobacteria are the major source to induce gene dysregulation in animals (Bradley and Pollard, 2017). To assess which taxa contributed to the variable expression of genes, we first conducted interaction and community diversity analyses at the genus level. The results indicate that the most abundant genus Pseudomonas was increased and occupies the most proportion of alternation.
Over the past decades, the crosstalk between the gut microbiota and the brain has received dramatic attention (Zheng et al., 2021). The microbiota–gut–brain axis is a bidirectional signaling pathway mediating the crosstalk between the microbiota, the intestine, and the central nervous system (CNS) (Cryan et al., 2019). CNS regulates the activities of the intestine and the microbiota, while the signals originating from the microbiota and the intestine affect the development and the function of the CNS inversely (Lynch and Pedersen, 2016). A wide array of signaling molecules that convey information about whole-body nutritional status have been identified and studied, including neuropeptides, hormones, nutrients, and metabolites produced by peripheral and CNS tissues in response to changes in the nutrition and environment (Richards and Proszkowiec-Weglarz, 2007). In the present study, the levels of metabolic enzymes like amylase, lipase, and trypsin; neuropeptides of NPY4R and PYY; hormones of GHR; functional proteins of LEPR, CREB1, FRA2, C/EBPA, and PP1B were all increased in the DOM group, indicating the appetite and perception were activated. Meanwhile, Pseudomonas was confirmed to regulate appetite-related NPY4R and perception-related CREB1 and C/EBPB in the study, indicating the crosstalk between microbiome Pseudomonas and CNS. As the appetite and perception are regulated by the central nervous system (Isganaitis and Lustig, 2005), the gut microbiota Pseudomonas might improve the ingestion performance of freshwater drums by crosstalk with the central nervous system.
Additionally, Pseudomonas was identified to have the genes involved in protein metabolism and cellular homeostasis signaling (AGE-RAGE, relaxin, and ECM-receptor signaling), which was in accordance with the results of RNA-seq analysis. However, there are limited reports demonstrating the regulation between gut Pseudomonas and the host protein metabolism and cellular homeostasis. In addition, reports indicate metabolic homeostasis is associated with appetite regulation (Sloth et al., 2007; Karl et al., 2016; Hopkins and Blundell, 2017). Therefore, we hypothesize that Pseudomonas impacts protein metabolism and cellular homeostasis by the mediator of CNS. These data reveal the ingestion behavior of freshwater drums is closely related to the central nervous system, the gut, and the gut microbiota Pseudomonas. Based on these results, future efforts in the development of feed demonstration strategies for a freshwater drum by targeting Pseudomonas and its medicated genes are warranted.
Conclusion
In this study, feed domestication improves ingestion, digestion, and growth performance, enhances protein digestion and absorption, and cellular homeostasis (Relaxin, ECM-receptor, and AGE-RAGE signaling). Pseudomonas was the most abundant microbe interacting with DEGs in response to feed domestication. These results indicate the Pseudomonas-mediated protein utilization and cellular homeostasis play a prominent role in ingestion performance during feed domestication. Consequently, Pseudomonas and related DEGs could be the target to improve the food performance and domestication in freshwater drum A. grunniens.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI BioProject, PRJNA801055.
Ethics Statement
The animal study was reviewed and approved by the Animal Care and Use Committee of Nanjing Agricultural University (Nanjing, China).
Author Contributions
CS: conceptualization, methodology, investigation, software, writing–original draft, and writing–review and editing. HW: data curation and resources. GaL: investigation and validation. XM, GoL, and MX: investigation and editing. NW: investigation and visualization. JC: investigation and software. HL: supervision and writing–original draft. PX: funding acquisition and supervision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Central Public-Interest Scientific Institution Basal Research Fund, CAFS (grant numbers: 2020JBFR03, 2020JBMR02, 2021JBFM13, and 2020TD62), the Innovation Project of Jiangsu Agricultural Science and Technology [grant number: CX(20)2025], and the Science and Technology Development Fund of Wuxi City (grant number: N20203008).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.861705/full#supplementary-material
Supplementary Table 1 | Differentially expressed genes (DEGs) under feed domestication in A. grunniens intestine. (A) Upregulated DEGs; (B) downregulated DEGs. Transcripts with | fold change| ≥ 2 and corrected P-value < 0.05 were identified as DEGs.
Supplementary Table 2 | GO and KEGG enrichment of DEGs under feed domestication in A. grunniens intestine. (A) GO items of DEGs; (B) KEGG items of DEGs.
Supplementary Table 3 | Operational taxonomic units (OTUs) of the microbiota identified from the intestine of A. grunniens under feed domestication. OTUs were identified and annotated at the level of phylum, class, order, family, genus, and species, respectively.
Supplementary Table 4 | Pearson analysis between DEGs and microbes.
References
Bolnick, D. I., Snowberg, L. K., Hirsch, P. E., Lauber, C. L., Knight, R., Caporaso, J. G., et al. (2014). Individuals’ diet diversity influences gut microbial diversity in two freshwater fish (threespine stickleback and Eurasian perch). Ecol. Lett. 17, 979–987. doi: 10.1111/ele.12301
Bradley, P. H., and Pollard, K. S. (2017). Proteobacteria explain significant functional variability in the human gut microbiome. Microbiome 5:36. doi: 10.1186/s40168-017-0244-z
Butt, R. L., and Volkoff, H. (2019). Gut microbiota and energy homeostasis in fish. Front. Endocrinol. 10:9. doi: 10.3389/fendo.2019.00009
Caporaso, J. G., Lauber, C. L., Costello, E. K., Berg-Lyons, D., Gonzalez, A., Stombaugh, J., et al. (2011). Moving pictures of the human microbiome. Genome Biol. 12:R50. doi: 10.1186/gb-2011-12-5-r50
Chen, X., Wang, J., Qian, L., Gaughan, S., Xiang, W., Ai, T., et al. (2017). Domestication drive the changes of immune and digestive system of Eurasian perch (Perca fluviatilis). PLoS One 12:e0172903. doi: 10.1371/journal.pone.0172903
Cortes, G. M., Marcialis, M. A., Bardanzellu, F., Corrias, A., Fanos, V., and Mussap, M. (2022). Inflammatory bowel disease and COVID-19: How microbiomics and metabolomics depict two sides of the same coin. Front. Microbiol. 13:856165. doi: 10.3389/fmicb.2022.856165
Cryan, J. F., O’riordan, K. J., Cowan, C. S. M., Sandhu, K. V., Bastiaanssen, T. F. S., Boehme, M., et al. (2019). The microbiota-gut-brain axis. Physiol. Rev. 99, 1877–2013.
Dabrowski, K., and Portella, M. C. (2005). Feeding plasticity and nutritional physiology in tropical fishes. Fish Physiol. 21, 155–224.
Desai, A. R., Links, M. G., Collins, S. A., Mansfield, G. S., Drew, M. D., Van Kessel, A. G., et al. (2012). Effects of plant-based diets on the distal gut microbiome of rainbow trout (Oncorhynchus mykiss). Aquaculture 35, 134–142.
Dillon, R. J., Webster, G., Weightman, A. J., and Keith Charnley, A. (2010). Diversity of gut microbiota increases with aging and starvation in the desert locust. Antonie Van Leeuwenhoek 97, 69–77. doi: 10.1007/s10482-009-9389-5
Du, Z. Y., Nie, P., and Liu, J. (2021). Genetic improvement for aquaculture species: a promising approach for aquaculture challenges and development. Rev. Aquac. 13, 1756–1757. doi: 10.1016/j.fsi.2021.10.034
Faith, J. J., Guruge, J. L., Charbonneau, M., Subramanian, S., Seedorf, H., Goodman, A. L., et al. (2013). The long-term stability of the human gut microbiota. Science 341:1237439.
Fei, N., and Zhao, L. (2013). An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J. 7, 880–884. doi: 10.1038/ismej.2012.153
Ghanbari, M., Kneifel, W., and Domig, K. J. (2015). A new view of the fish gut microbiome: Advances from next-generation sequencing. Aquaculture 448, 464–475.
Glazko, V. I., Zybaylov, B. L., Kosovsky, Y. G., Glazko, G. V., and Glazko, T. T. (2021). Domestication and microbiome. Holocene 31, 1635–1645. doi: 10.1146/annurev-micro-090817-062524
Hallerman, E., Hilsdorf, A. W. S., Bartley, D. M., and Mair, G. C. (2021). Towards increasing production of genetically improved farmed types in aquaculture. Rev. Aquac. 13, 2112–2116.
Hernández-Gómez, R. E., Contreras-Sánchez, W. M., Hernández-Franyutti, A., Perera-García, M. A., and Torres-Martínez, A. (2021). Testicular structure and development of the male germinal epithelium in the freshwater drum Aplodinotus grunniens (Perciformes: Sciaenidae) from the Usumacinta River, Southern Mexico. Acta Zool. 00:1–19.
Hopkins, M., and Blundell, J. E. (2017). “Energy Metabolism and Appetite Control,” in Appetite and Food Intake: Central Control, ed. R. B. S. Harris (Boca Raton, FL: CRC Press).
Isganaitis, E., and Lustig, R. H. (2005). Fast food, central nervous system insulin resistance, and obesity. Arterioscler. Thromb. Vasc. Biol. 25, 2451–2462. doi: 10.1161/01.ATV.0000186208.06964.91
Jacquemin, S. J., Pyron, M., Allen, M., and Etchison, L. (2014). Wabash river freshwater drum Aplodinotus grunniens diet: effects of body size, sex, and river gradient. J. Fish Wildl. Manage. 5, 133–140.
Karl, J. P., Smith, T. J., Wilson, M. A., Bukhari, A. S., Pasiakos, S. M., McClung, H. L., et al. (2016). Altered metabolic homeostasis is associated with appetite regulation during and following 48-h of severe energy deprivation in adults. Metabolism 65, 416–427. doi: 10.1016/j.metabol.2015.11.001
Knights, D., Kuczynski, J., Charlson, E. S., Zaneveld, J., Mozer, M. C., Collman, R. G., et al. (2011). Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 8, 761–765.
Liao, I. C., and Huang, Y. S. (2000). Methodological Approach used for the Domestication of Potential Candidates for Aquaculture. Recent Advances in Mediterranean Aquaculture Finfish Species Diversification: Cahiers Option Méditerranéennes. Zaragoza: CIHEAM.
Liou, A. P., Paziuk, M., Luevano, J. M., Machineni, S., Turnbaugh, P. J., and Kaplan, L. M. (2013). Conserved shifts in the gut microbiota due to gastric bypass reduce host weight and adiposity. Sci. Transl. Med. 5:178ra41. doi: 10.1126/scitranslmed.3005687
Liu, B., Song, C., Gao, Q., Zhou, Q., Sun, C., Zhang, H., et al. (2021). Maternal and environmental microbes dominate offspring microbial colonization in the giant freshwater prawn Macrobrachium rosenbergii. Sci. Total Environ. 790:148062. doi: 10.1016/j.scitotenv.2021.148062
Liu, H., Guo, X., Gooneratne, R., Lai, R., Zeng, C., Zhan, F., et al. (2016). The gut microbiome and degradation enzyme activity of wild freshwater fishes influenced by their trophic levels. Sci. Rep. 6:24340. doi: 10.1038/srep24340
Lozupone, C., Stombaugh, J. I., Gordon, J. I., Jansson, J. K., and Knight, R. (2012). Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230. doi: 10.1038/nature11550
Lynch, S. V., and Pedersen, O. (2016). The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 375, 2369–2379.
Michl, S. C., Ratten, J. M., Beyer, M., Hasler, M., La Roche, J., and Schulz, C. (2017). The malleable gut microbiome of juvenile rainbow trout (Oncorhynchus mykiss): Dietdependent shifts of bacterial community structures. PLoS One 12:e0177735. doi: 10.1371/journal.pone.0177735
Morgan, X. C., Tickle, T. L., Sokol, H., Gevers, D., Devaney, K. L., Ward, D. V., et al. (2012). Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13:R79. doi: 10.1186/gb-2012-13-9-r79
Parata, L., Sammut, J., and Egan, S. (2021). Opportunities for microbiome research to enhance farmed freshwater fish quality and production. Rev. Aquac. 13, 2027–2037.
Peredo, A. M., Buentello, A., Gatlin, D. M., and Hume, M. E. (2015). Evaluation of a dairy-yeast prebiotic in the diet of juvenile nile tilapia, Oreochromis niloticus. J. World Aquac. Soc. 46, 92–101.
Purnell, M. A., Bell, M. A., Baines, D. C., Hart, P. J. B., and Travis, M. P. (2007). Correlated evolution and dietary change in fossil stickleback. Science 317:1887. doi: 10.1126/science.1147337
Ray, A. K., Ghosh, K., and Ringø, E. (2012). Enzyme-producing bacteria isolated from fish gut: a review. Aquac. Nutr. 8, 465–492.
Richards, M. P., and Proszkowiec-Weglarz, M. (2007). Mechanisms regulating feed intake, energy expenditure, and body weight in poultry. Poult. Sci. 86, 1478–1490. doi: 10.1093/ps/86.7.1478
Ringø, E., Zhou, Z., Vecino, J. L. G., Wadsworth, S., Romero, J., and Krogdahl, et al. (2016). Effect of dietary components on the gut microbiota of aquatic animals, A never-ending story? Aquac. Nutr. 22, 219–282.
Shen, Y., Li, H., Zhao, J., Tang, S., Zhao, Y., Bi, Y., et al. (2021). The digestive system of mandarin fish (Siniperca chuatsi) can adapt to domestication by feeding with artificial diet. Aquaculture 538:736546.
Shin, N. R., Whon, T. W., and Bae, J. W. (2015). Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 33, 496–503. doi: 10.1016/j.tibtech.2015.06.011
Silva-Brito, F., Alexandrino, D. A. M., Jia, Z., Mo, Y., Kijjoa, A., Abreu, H., et al. (2021). Fish performance, intestinal bacterial community, digestive function and skin and fillet attributes during cold storage of gilthead seabream (Sparus aurata) fed diets supplemented with Gracilaria by-products. Aquaculture 541:736808.
Sloth, B., Holst, J. J., Flint, A., Gregersen, N. T., and Astrup, A. (2007). Effects of PYY1-36 and PYY3-36 on appetite, energy intake, energy expenditure, glucose and fat metabolism in obese and lean subjects. Am. J. Physiol. Endocrinol. Metab. 292, E1062–E1068. doi: 10.1152/ajpendo.00450.2006
Song, C., Liu, B., Xie, J., Ge, X., Zhao, Z., Zhang, Y., et al. (2017). Comparative proteomic analysis of liver antioxidant mechanisms in Megalobrama amblycephala stimulated with dietary emodin. Sci. Rep. 7:40356. doi: 10.1038/srep40356
Song, C., Liu, B., Xu, P., Ge, X., Li, H., Tang, Y., et al. (2021). miR-144 is the epigenetic target for emodin to ameliorate oxidative stress induced by dietary oxidized fish oil via Nrf2 signaling in Wuchang bream, Megalobrama amblycephala. Aquaculture 534:736357.
Sullam, K. E., Essinger, S. D., Lozupone, C. A., O’Connor, M. P., Rosen, G. L., Knight, R., et al. (2012). Environmental and ecological factors that shape the gut bacterial communities of fish: A meta-analysis. Mol. Ecol. 21, 3363–3378. doi: 10.1111/j.1365-294X.2012.05552.x
Sun, Y., Han, W., Liu, J., Huang, X., Zhou, W., Zhang, J., et al. (2020). Bacterial community compositions of crab intestine, surrounding water, and sediment in two different feeding modes of Eriocheir sinensis. Aquac. Rep. 16:100236.
Talwar, C., Nagar, S., Lal, R., and Negi, R. K. (2018). Fish Gut Microbiome: Current Approaches and Future Perspectives. Indian J. Microbiol. 58, 397–414. doi: 10.1007/s12088-018-0760-y
Teletchea, F., and Fontaine, P. (2014). Levels of domestication in fish: Implications for the sustainable future of aquaculture. Fish Fish. 15, 181–195. doi: 10.1016/j.marpolbul.2014.09.016
Thomas, J. G., and Percival, S. L. (2009). Indigenous microbiota and association with the host. Microbiology and Aging. Totowa, NJ: Humana Press, 15–37.
Turnbaugh, P. J., Ley, R. E., Hamady, M., Fraser-Liggett, C. M., Knight, R., and Gordon, J. I. (2007). The human microbiota project. Nature 449, 804–810. doi: 10.3390/md17060374
Volkoff, H. (2016). The neuroendocrine regulation of food intake in fish: A review of current knowledge. Front. Neurosci. 10:540. doi: 10.3389/fnins.2016.00540
Xia, J. H., Lin, G., Fu, G. H., Wan, Z. Y., Lee, M., Wang, L., et al. (2014). The intestinal microbiome of fish under starvation. BMC Genomics 15:266. doi: 10.1186/1471-2164-15-266
Xiong, Y., Huang, J., Li, X., Zhou, L., Dong, F., Ye, H., et al. (2014). Deep sequencing of the tilapia (Oreochromis niloticus) liver transcriptome response to dietary protein to starch ratio. Aquaculture 433, 299–306.
Ye, L., Amberg, J., Chapman, D., Gaikowski, M., and Liu, W. T. (2014). Fish gut microbiota analysis differentiates physiology and behavior of invasive Asian carp and indigenous American fish. ISME J. 8, 541–551.
Zhang, X., Coker, O. O., Chu, E. S., Fu, K., Lau, H. C., Wang, Y. X., et al. (2021). Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut 70, 761–774. doi: 10.1136/gutjnl-2019-319664
Keywords: feed domestication, microbe, Pseudomonas, protein utilization, Aplodinotus grunniens
Citation: Song C, Wen H, Liu G, Ma X, Lv G, Wu N, Chen J, Xue M, Li H and Xu P (2022) Gut Microbes Reveal Pseudomonas Medicates Ingestion Preference via Protein Utilization and Cellular Homeostasis Under Feed Domestication in Freshwater Drum, Aplodinotus grunniens. Front. Microbiol. 13:861705. doi: 10.3389/fmicb.2022.861705
Received: 26 January 2022; Accepted: 02 May 2022;
Published: 26 May 2022.
Edited by:
Weiqi He, Soochow University, ChinaReviewed by:
Min Jin, Ningbo University, ChinaYuan-Yuan Cai, China Pharmaceutical University, China
Copyright © 2022 Song, Wen, Liu, Ma, Lv, Wu, Chen, Xue, Li and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongxia Li, bGloeEBmZnJjLmNu; Pao Xu, eHVwQGZmcmMuY24=