 Li-Gang Xiang
Li-Gang Xiang Han-Cheng Wang
Han-Cheng Wang Feng Wang2*
Feng Wang2* Tom Hsiang
Tom Hsiang- 1College of Agriculture, Yangtze University, Jingzhou, China
- 2Guizhou Provincial Academician Workstation of Microbiology and Health, Guizhou Academy of Tobacco Science, Guiyang, China
- 3Guizhou Institute of Plant Protection, Guizhou Academy of Agricultural Sciences, Guiyang, China
- 4School of Environmental Sciences, University of Guelph, Guelph, ON, Canada
- 5College of Life Sciences, Yangtze University, Jingzhou, China
In the tobacco phyllosphere, some of the microbes may have detrimental effects on plant health, while many may be neutral or even beneficial. Some cannot be cultivated, so culture-independent methods are needed to explore microbial diversity. In this study, both metagenetic analysis and traditional culture-dependent methods were used on asymptomatic healthy leaves and symptomatic diseased leaves of tobacco plants. In the culture-independent analysis, asymptomatic leaves had higher microbial diversity and richness than symptomatic leaves. Both asymptomatic and symptomatic leaves contained several potentially pathogenic bacterial and fungal genera. The putative bacterial pathogens, such as species of Pseudomonas, Pantoea, or Ralstonia, and putative fungal pathogens, such as species of Phoma, Cladosporium, Alternaria, Fusarium, Corynespora, and Epicoccum, had a higher relative abundance in symptomatic leaves than asymptomatic leaves. FUNGuild analysis indicated that the foliar fungal community also included endophytes, saprotrophs, epiphytes, parasites, and endosymbionts. PICRUSt analysis showed that the dominant functions of the bacterial community in a symptomatic leaf were cellular processes and environmental information processing. In the other five foliar samples, the dominant functions of the bacterial community were genetic information processing, metabolism, and organismal systems. In the traditional culture-dependent method, 47 fungal strains were isolated from 60 symptomatic tobacco leaf fragments bearing leaf spots. Among them, 21 strains of Colletotrichum (29%), Xylariaceae (14%), Corynespora (14%), Pestalotiopsis (10%), Alternaria (10%), Epicoccum (10%), Byssosphaeria (5%), Phoma (5%), and Diaporthe (5%) all fulfilled Koch’s postulates and were found to cause disease on detached tobacco leaves in artificial inoculation tests. Symptoms on detached leaves caused by three strains of Corynespora cassiicola in artificial inoculation tests were similar to the original disease symptoms in the tobacco field. This study showed that the combined application of culture-dependent and independent methods could give comprehensive insights into microbial composition that each method alone did not reveal.
Introduction
Tobacco (Nicotiana tabacum L.) is an annual, leafy, solanaceous plant, and one of the most widely cultivated non-food crops in the world (Chen et al., 2020). In China, the annual planting area of tobacco is more than 1 million hectares, and the annual output is more than 2 million tons (Yan et al., 2021). Tobacco leaf diseases are one of the main challenges in tobacco cultivation, and these can be divided into physiological, bacterial, fungal, and viral diseases. A typical physiological leaf disease of tobacco is weather fleck caused by high levels of ozone or air pollution (Macdowall et al., 1964). The fungal leaf diseases of tobacco include brown spot (Stavely and Slana, 1975), and powdery mildew (Cai et al., 2017), among others. Important bacterial leaf diseases of tobacco include angular leaf spot disease (Fromme and Murray, 1919) and wildfire disease (Cai et al., 2019). Important viral leaf diseases of tobacco are caused by tobacco mosaic virus (Scholthof, 2004), etch virus (Hari, 1981), and cucumber mosaic virus (Wagih et al., 2021).
The phyllosphere is colonized by specific microorganisms as an essential plant-associated habitat (Vorholt, 2012). However, the environmental conditions of the phyllosphere have great impact on foliar microbial populations. Phyllosphere microorganisms face harsh environmental conditions including nutrient and free water limitations and high ultraviolet radiation (Vorholt, 2012). Even with such poor growth conditions, there are numerous microorganisms that can colonize and exploit the phyllosphere, including bacteria, fungi, yeast, protists, algae, and bacteriophages. According to Vorholt (2012), about 106–107 cells reside on 1 cm2 of leaf surface. Within these communities, some microorganisms may be beneficial or neutral to the host plant, while others may be pathogenic and antagonistic to the host plant. Pathogens can invade plant leaves through wounds, stomates, hydathodes or other openings (Hugouvieux et al., 1998).
In the past few decades, plant pathogens have traditionally been discovered and identified based on culture-dependent methods. Because these are selective methods, the results can be influenced by many factors, including surface sterilization approaches, culture medium, and incubation conditions (Dissanayake et al., 2018). Therefore, the results obtained by traditional culture-dependent method cannot comprehensively reflect the numbers and functions of foliar microorganisms or pathogens. Metagenetic analysis involving high-throughput sequencing as a culture-independent method has also been used to investigate phyllosphere microbial community and diversity (Luo et al., 2019). However, species resolution of this technique is strongly influenced by databases available since it will not identify species that are not represented in the database. Furthermore, because of variability within species and similarity between species especially of the same genus, the taxonomic assignments at the commonly used threshold of 97% similarity are not entirely accurate for species level identification (Nilsson et al., 2008). In other words, the taxonomic results derived from metagenetic analysis may be reasonable only to resolve to the genus level (Dissanayake et al., 2018). Species-level classification may be obtained using species-specific primers or by analyzing specific loci, but these methods are labor-intensive and complex.
The aims of this study included (i) characterizing fungi and bacteria associated with asymptomatic healthy leaves and symptomatic diseased leaves of tobacco plants using the internal transcribed spacer (ITS) region and the partial 16S rRNA gene sequence, (ii) isolating potential fungal pathogens from symptomatic leaves of an unknown leaf spot disease of tobacco, and (iii) comparing the results for fungi from metagenetic analysis vs. culturing and ITS sequencing from the same samples.
Materials and Methods
Sampling Strategy and Sites
Samples were collected from Tongren City (27.94N, 108.26E), Guizhou province, China in July 2020. In a field where plants of tobacco cultivar Yunyan K326 were mature, six plants at least 3 m apart were randomly chosen for sampling. Three of these plants had leaves with large yellow-brown irregular spots and lesions covering more than 25% of each leaf (Figure 1), and a leaf from each was randomly selected as representative samples for symptomatic leaves (GB1–GB3). The other three tobacco plants were healthy and used to collect asymptomatic leaves (JK1–JK3). Each leaf was divided into two sets of samples, one for fungal isolation (stored at 4°C) and another for microbial community structure and diversity analysis (stored at −80°C).

Figure 1. Field symptoms of diseased tobacco plants (A) and leaves (B).
Culture-Independent Method
DNA Extraction, Polymerase Chain Reaction Amplification and Metagenetic Analysis
Leaf samples were not disinfected so as to obtain both endophytes and epiphytes. To ensure homogeneous sampling, the leaf tissue was cut into pieces and then 0.5 g of tissue was randomly taken for DNA extraction (only necrotic leaf tissues were collected as symptomatic samples). Total genomic DNA was extracted using a FastDNA® Spin Kit (MP Biomedicals, Santa Ana, CA, United States) based on the manufacturer’s protocol. DNA quantity and quality were assessed using agarose gel electrophoresis, and the concentrations estimated using the NanoDrop-2000 (Thermo Fisher Scientific, Waltham, MA, United States). DNA was diluted to 30 ng/μL using sterile water. After DNA extraction, the bacterial V4 region of the 16S rRNA gene was amplified using primers 515F (5′-GTGYCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) (Caporaso et al., 2011), and the fungal ITS1 region was amplified using primers ITS1 (5′-TCCGTAGGTGAACCTGCGG-3′) and ITS2 (5′-GCTGCGTTCTTCATCGATGC-3′) (Zhang et al., 2010). The polymerase chain reaction (PCR) programs and total reaction volumes for ITS and 16S rRNA regions followed Wei et al. (2018). The PCR products were detected with 2% agarose gel electrophoresis using the DL2000 marker (TaKaRa, Dalian, China) for quantity examination, and purified with the Qiagen Gel Extraction Kit (Qiagen, Düsseldorf, Germany). Sequencing libraries were generated using the TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina, CA, United States) following manufacturer’s recommendations, and index codes were added. The library quality was assessed on the Qubit@ 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, United States) and Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, United States). Finally, the library was sequenced on an Ion S5 XL platform (Thermo Fisher Scientific, Waltham, MA, United States) at Novogene Bioinformatics Technology Co., Tianjin, China, targeting 250 bp paired-end reads.
Data Processing and Statistical Analyses
Paired-end reads were merged using FLASH V1.2.7 (overlap ≥10 bp, maximum mismatch density of 0.25) (Magoč and Salzberg, 2011). Quality filtering of raw reads was performed with specific criteria to obtain clean reads (Bokulich et al., 2013) based on the QIIME V1.9.1 quality control process (q ≤ 19) (Caporaso et al., 2010). The UCHIME algorithm (Edgar et al., 2011) was used to detect chimeric sequences, which were removed (Haas et al., 2011). After these operations, the sequences were considered clean reads. ITS reads were compared against the UNITE database (ver. 12.01.2017), and 16S rRNA reads were compared against the Silva database (ver. 132).
Sequences with ≥97% similarity were assigned to the same operational taxonomic unit (OTU) using Uparse V7.0.1001 software (Edgar, 2013). Subsequently, for each representative sequence of each OTU, the UNITE and Silva databases were used based on the Mothur algorithm to annotate taxonomic information. Duncan’s multiple range tests were performed in SPSS V23.0 (IBM, Armonk, NY, United States) at p < 0.05 to compare relative abundance between microbial genera of symptomatic versus asymptomatic leaves. The OTU tables of fungal and bacterial communities were normalized by rarefying to 8000 sequences per sample for alpha and beta diversity analyses. Finally, the estimates of alpha and beta diversity of microbial communities were calculated using QIIME V1.7.0 and V1.9.1, respectively, and displayed with R software V2.15.3. The alpha index comparison between symptomatic versus asymptomatic leaves used the Wilcoxon signed rank test at p < 0.1. Principal co-ordinates analysis (PCoA) of unweighted UniFrac distance were calculated based on Bray-Curtis dissimilarity matrix. The FUNGuild database1 was used to analyze fungal trophic mode and guild (Nguyen et al., 2016). PICRUSt2 was used to predict bacterial functionality based on the Greengene database (Langille et al., 2013). The relationship of the different genera was demonstrated using network analysis based on Spearman’s rank analysis. The co-occurrence patterns of tobacco phyllosphere microbial genera were assessed based on strong (r > 0.6) and significant correlations (p < 0.05) (Li and Wu, 2018).
Culture-Dependent Method
Fungal Isolation
Each symptomatic leaf was cut into 20 small pieces (5 mm × 5 mm). All tissue pieces were surface disinfected for 30 s in 75% ethanol, and then sterilized with 1% sodium hypochlorite (NaOCl) for 1 min and washed three times in sterile distilled water (Li et al., 2017). Finally, the tissues were placed onto potato dextrose agar (PDA) and cultured at 25°C. Plates were checked daily for visible hyphal growth up to 1 week, and once the hyphal tips were visible around the leaf tissue, some tips were transferred to fresh PDA for purification via serial hyphal tip transfer. All purified isolates were stored on PDA slants at 4°C.
Molecular Identification of Isolated Strains
Mycelia cultured on PDA for a week were collected, and their genomic DNA was extracted using the DNeasy® PowerLyzer® Microbial Kit (Qiagen, Düsseldorf, Germany). The DNA concentration was assessed using the NanoDrop-2000, and diluted to 30 ng/μL using sterile water. The ITS was amplified with primers ITS1 (5′-TCCGTAGGTGAACCTGCGG-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) (White et al., 1990). The total reaction volume was 25.0 μL, including 10.5 μL of Taq PCR Master Mix, 1.0 μL each of primer (10 μM), 1.0 μL of template DNA, and 11.5 μL of double distilled water. The PCR program was used as followed: Denaturation at 94°C for 3 min, denaturation at 94°C for 30 s, annealing at 56°C for 30 s, extension at 72°C for 45 s for 30 cycles, and final extension at 72°C for 5 min. The amplified fragments (500–750 bp) were sequenced in both directions by Sango Biotech Co., Ltd., Shanghai, China and consensus sequences were derived and edited with DNAMAN (version 5.2.2). The sequences were compared to the NCBI nr database using BLASTn and the top matching hits showing >98% identity were chosen to represent the species.
Pathogenicity Testing
Fresh young detached tobacco leaves without visible disease symptoms were obtained. They were surface disinfected with 75% ethanol for 30 s, 1% NaOCl for 1 min, and finally washed three times using distilled water. A sterile piece of cotton was wrapped around the petiole of each detached leaf and moistened to prevent wilt. These leaves were each gently wounded with a sterile needle in a 4 mm2 area by lightly scratching, and a 5-mm-diameter mycelial plug was placed on the wound. Each treatment (isolated strain) was tested on three detached leaves, and fresh PDA plugs without hyphae were used as non-inoculated control. After inoculation, detached leaves were placed in plastic boxes and incubated at 28°C for 10 days in the dark, and then assessed for disease in terms of lesion area and color. Pathogenicity testing was repeated twice.
Results
Sequence Data
A total of 263,626 and 243,453 clean bacterial 250-bp reads were obtained from the asymptomatic and symptomatic samples, respectively. The paired-end reads were merged and then clustered into 304 and 272 OTUs using a cut off of 97% similarity. The OTUs were compared against the Silva database for identity. For fungi, there were 238,329 clean reads from asymptomatic samples, and paired-end reads were merged and clustered into 77 OTUs. The 269,671 clean reads from symptomatic samples were processed and clustered into 149 OTUs (Table 1). The fungal OTUs were compared against the UNITE database. The fungal and bacterial raw sequences of each sample have been deposited in the SRA database under project number PRJNA722239.
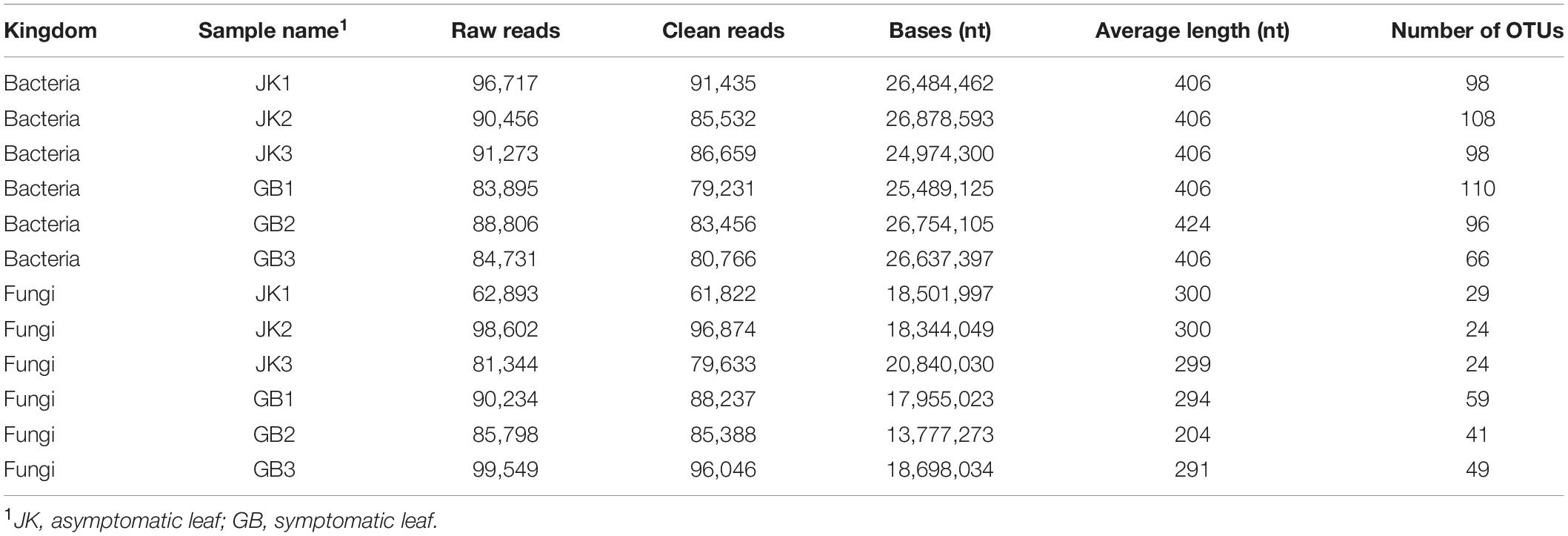
Table 1. Metagenetic sequence data from different samples of tobacco leaves.
Microbial Community Composition
The influence of disease on microbial communities of tobacco leaves was assessed in this study. A rich microbial community was observed in both symptomatic and asymptomatic leaves. The “Other” group in Figure 2 contains all microorganisms that were not identified or whose relative abundance was less than 0.1%. Aside from “others,” the dominant identified bacterial phyla in both types of leaves were Proteobacteria, Firmicutes, Fusobacteria, Bacteroidetes, and Actinobacteria, with relative abundances ranging from 0.4 to 36% (Figure 2A). The major fungal phylum found was Ascomycota which for some of the samples exceeded “Others.” Basidiomycota were also present at a much lower levels in each sample. The relative abundance of Ascomycota in asymptomatic or symptomatic leaves was 26 ± 11% or 72 ± 22%, respectively; and for Basidiomycota, it was 2.3 ± 0.7% or 1.2 ± 1.0%, respectively (Figure 2C).
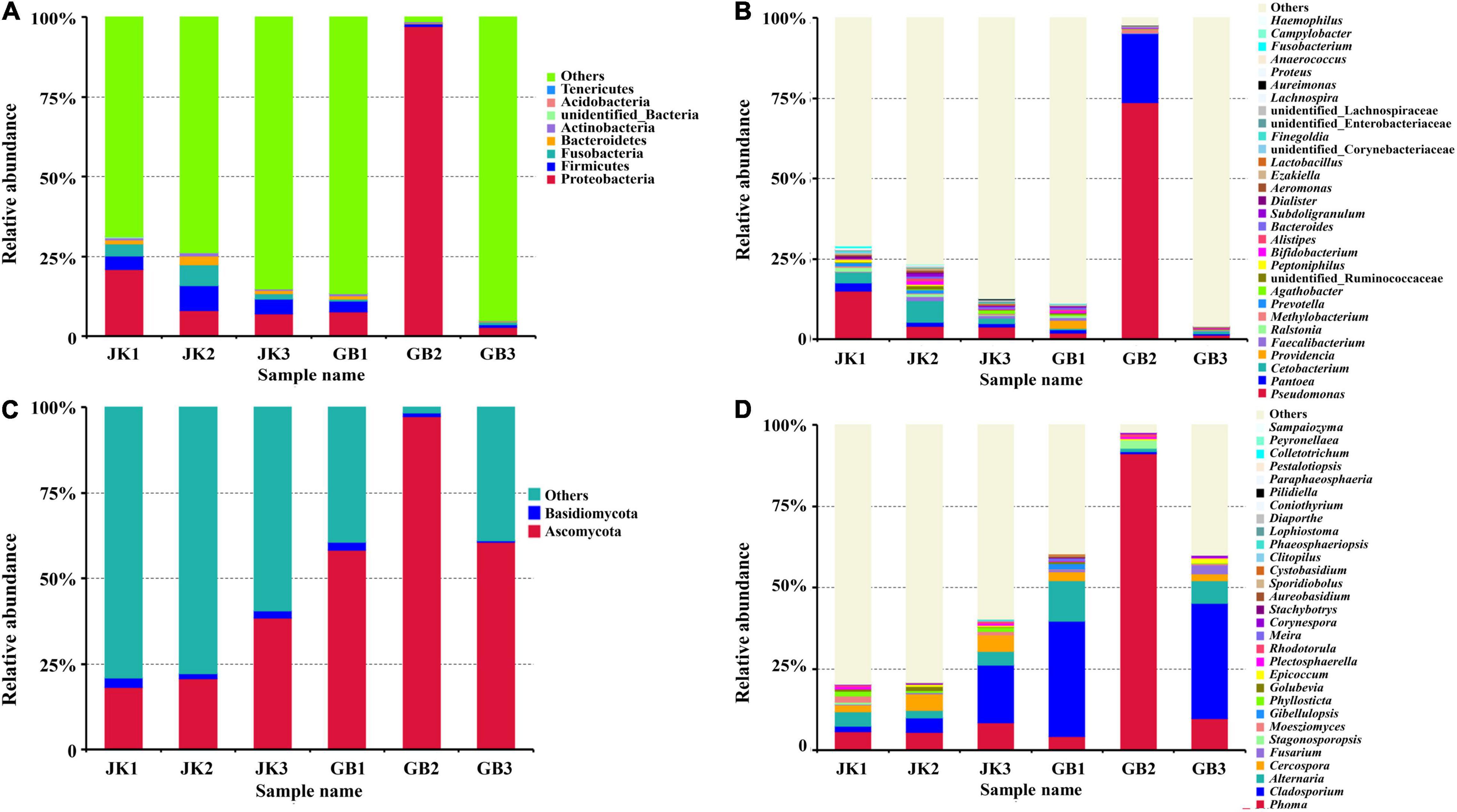
Figure 2. Microbial community composition of different samples (asymptomatic leaves JK1, JK2, JK3; symptomatic leaves GB1, GB2, GB3) at the phylum [(A) for bacteria, (C) for fungi] and genus [(B) for bacteria, (D) for fungi] levels. The relative abundance of phyla or genera making up less than 0.01% were classified as “Others.”
The relative abundance of the top 30 identified bacterial or fungal genera are shown in Figures 2B,D, respectively. Among the top 30 bacterial genera, most were non-pathogenic, and only some species from three of these genera have been classified as plant pathogens, namely Pseudomonas, Pantoea, and Ralstonia. The relative abundances of these three genera in symptomatic leaves were 25.67, 7.65, and 0.24%, respectively, and in asymptomatic leaves, they were 7.61, 1.60, and 0.73%, respectively. In addition to these genera, Prevotella, Peptoniphilus, Ezakiella, Corynebacteriaceae (family), Proteus, Anaerococcus, and Campylobacter were exclusively found in asymptomatic leaves, although in lower abundance (<0.7%).
Compared with bacterial genera, the top 30 identified fungal genera had more potential plant pathogens (Table 2). A total of 15 genera containing plant pathogens were detected in asymptomatic or symptomatic leaves. Among them, only the relative abundance of Cercospora, Moesziomyces, Phyllosticta, and Plectosphaerella in asymptomatic leaves was higher than their counterparts in symptomatic leaves. When counts were combined from asymptomatic or symptomatic leaves, the pathogenic genera with relative abundance over 1% included Phoma (6.6%/35.1%), Cladosporium (8.0%/23.9%), Alternaria (3.7%/6.9%), Cercospora (4.1%/1.5%), and Fusarium (0.22%/1.18%). Fungal genera which were found only on asymptomatic leaves included Clitopilus and Phaeosphaeriopsis, and those found only on symptomatic leaves included Gibellulopsis, Meira, Stachybotrys, Aureobasidium, and Cystobasidium.
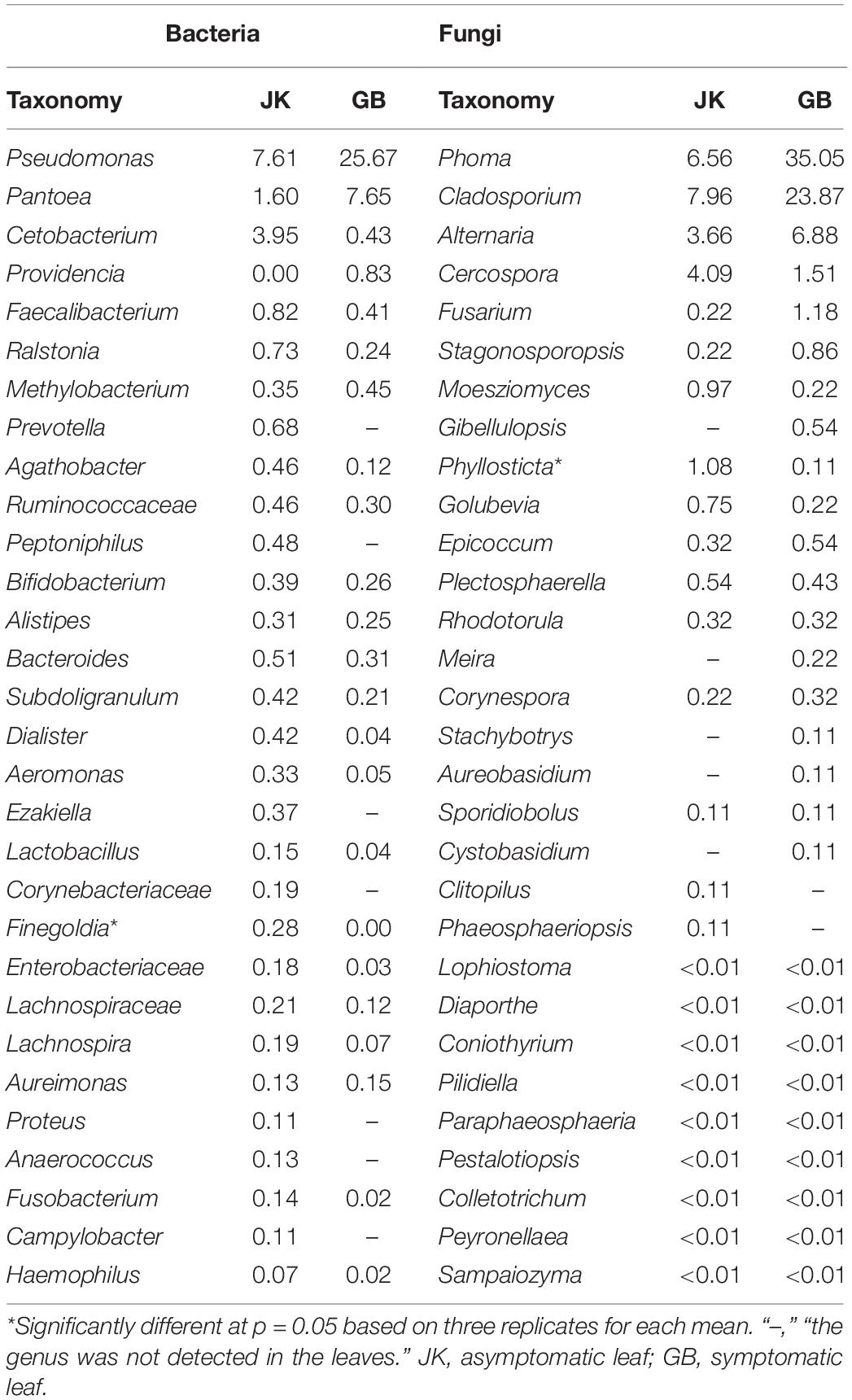
Table 2. The top 30 dominant taxa and their relative abundance (%) of fungal and bacterial community OTUs based on metagenetic analysis.
Microbial Community Diversity
Estimates of indices of richness and diversity for fungi and bacteria in asymptomatic and symptomatic leaves are shown in Table 3. For bacterial community analysis, the asymptomatic leaves show trends of higher Shannon–Weaver index, Simpson index, Chao1 index, and ACE index compared to the symptomatic leaves, but there were no statistically significant differences. For fungal community analysis, the asymptomatic leaves showed trends of higher Shannon–Weaver index and Simpson index, and a lower Chao1 index and ACE index compared to the symptomatic leaves, but there were no statistically significant differences.
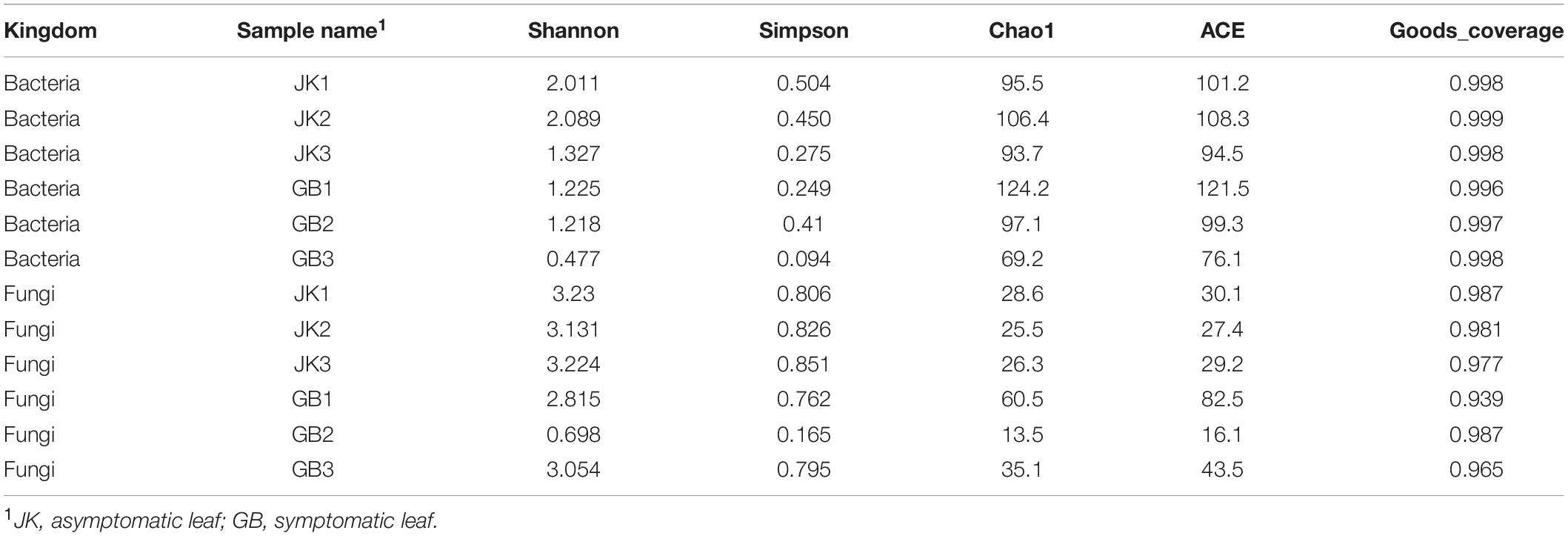
Table 3. Alpha diversity indexes of phyllosphere microbial community.
Principal co-ordinates analysis for bacterial communities did not show separate clustering for symptomatic vs. asymptomatic samples (Figure 3A), which meant that the bacterial community composition was similar between symptomatic and asymptomatic groups. However, for fungal communities, the asymptomatic samples did cluster together in a PCoA quadrant, while the symptomatic samples were more scattered (Figure 3B), which implied that there were fewer differences in fungal community composition of asymptomatic leaves compared to symptomatic leaves.
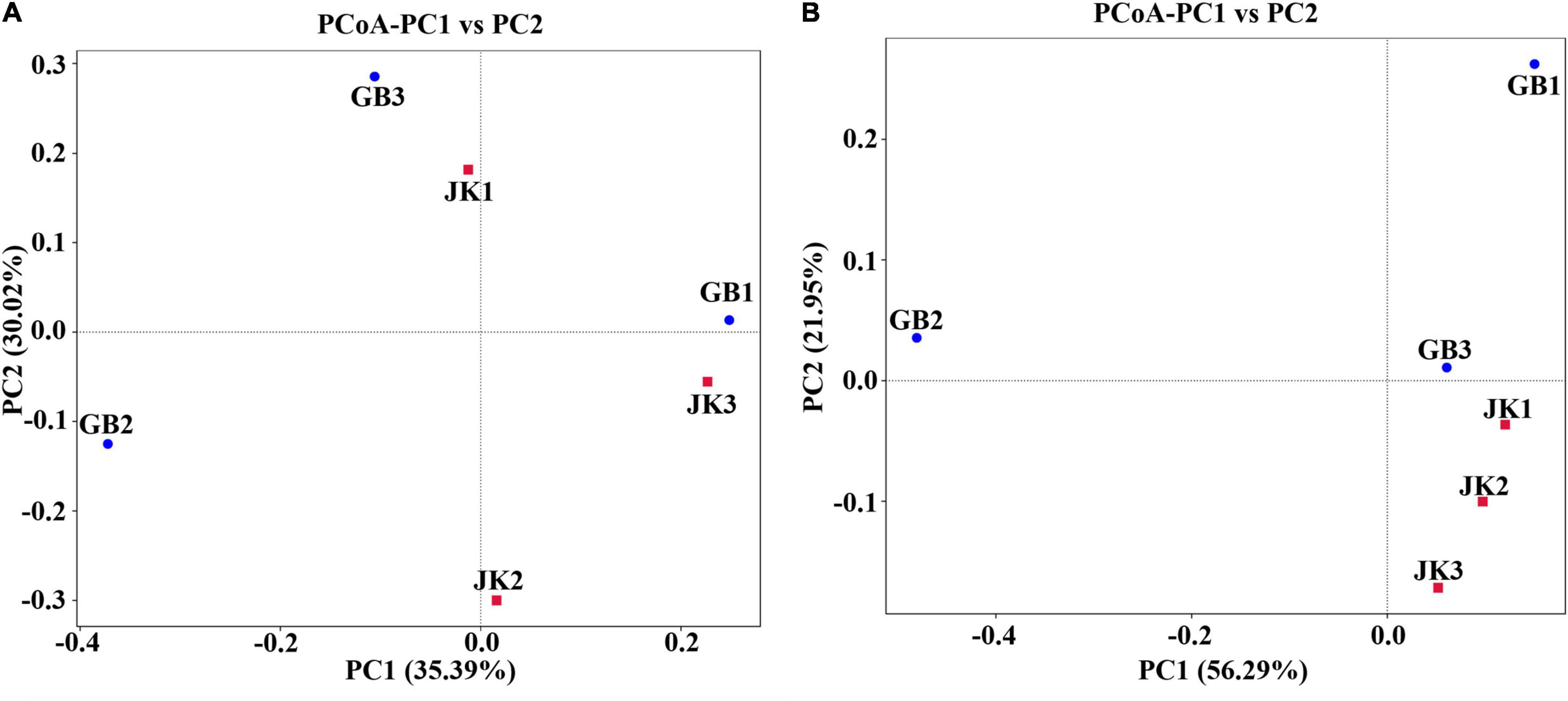
Figure 3. Principal Co-ordinate Analysis (PCoA) of the bacterial (A) and fungal (B) communities of different samples (asymptomatic leaves JK1, JK2, JK3; symptomatic leaves GB1, GB2, GB3).
Microbial Community Functional Characteristics
FUNGuild was used to predict the ecological functions of fungi in the samples based on their composition (Figure 4). The dominant ecological functions of fungal communities in asymptomatic leaves were classified as endophyte, plant pathogen, and wood saprotroph (JK1); fungal parasite and plant pathogen (JK2); and plant pathogen, endophyte, wood saprotroph, and animal endosymbiont (JK3). The dominant ecological functions of fungal communities in symptomatic leaves were fungal parasite, animal pathogen, endophyte, epiphyte, plant pathogen, and saprotroph (GB1); animal endosymbiont, saprotroph, and plant pathogen (GB2); and fungal parasite, plant pathogen, soil saprotroph, wood saprotroph, and endophyte (GB3).

Figure 4. Dendrogram of fungal functional categories in symptomatic (GB1, GB2, GB3) and asymptomatic samples (JK1, JK2, JK3) analyzed using FUNGuild.
PICRUSt was designed to estimate the gene families in metagenetic analysis from bacteria or archaea identified using 16S rRNA sequencing (Figure 5). In this study, genetic information processing, metabolism, and organismal systems were the common bacterial community functions in JK1, JK2, JK3, GB1, and GB3. In addition to the common functions, these were specific bacterial community in symptomatic leaves: unclassified, environment information processing, cellular processes, and human disease.
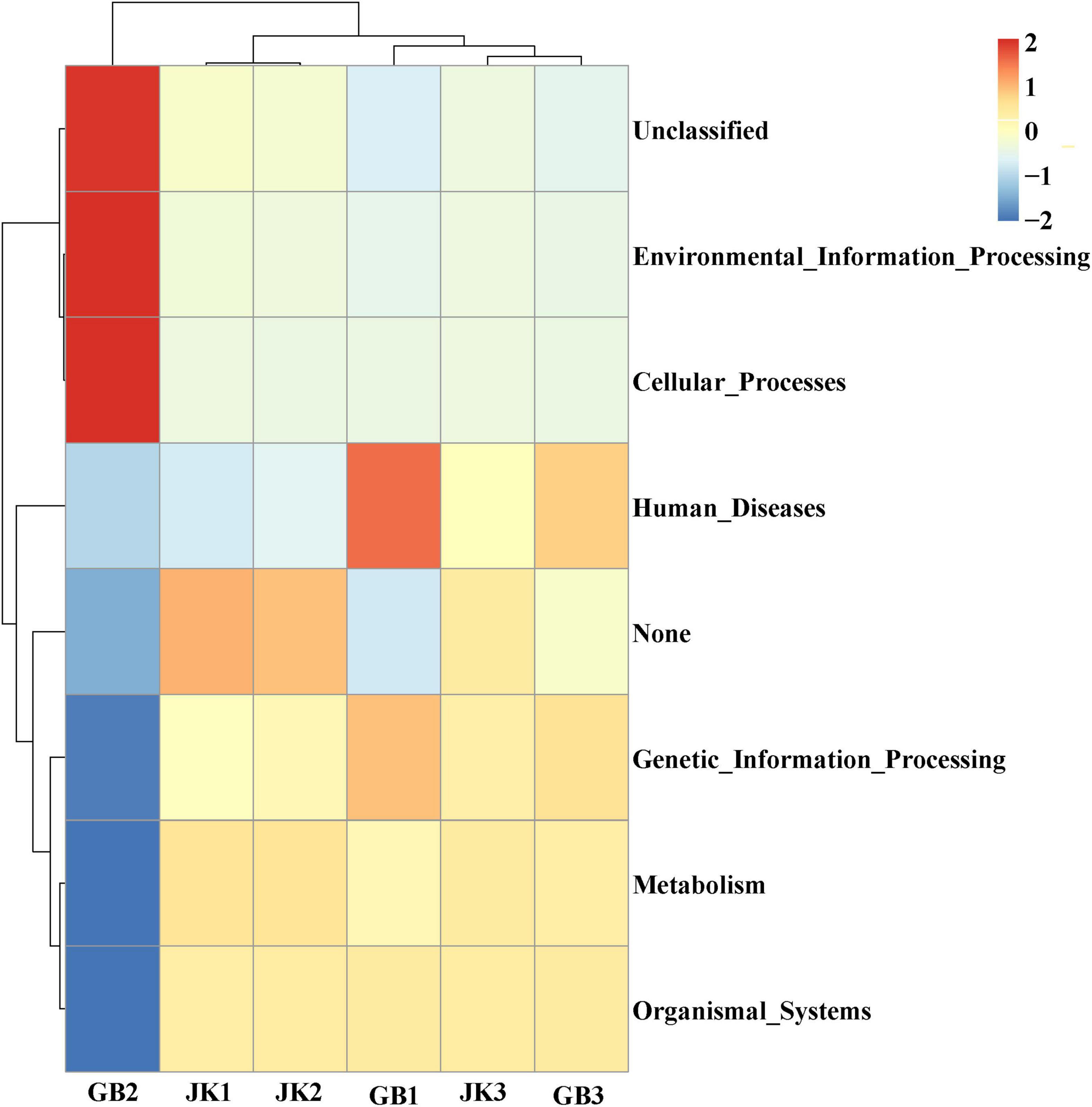
Figure 5. Dendrogram of bacterial functional categories in symptomatic (GB1, GB2, GB3) and asymptomatic samples (JK1, JK2, JK3) analyzed using PICRUSt.
Co-occurrence Network Analysis
Co-occurrence network analysis is widely used to study the interaction of organisms in complex microbial communities. A total of five strong negative correlations and 124 strong positive correlations were identified from 46 bacterial genera (Figure 6). The co-occurring genera were distributed in Firmicutes (50%, targeted nodes/total nodes), Proteobacteria (30.4%), Actinobacteria (6.5%), Bacteroidetes (6.5%), Fusobacteria (4.4%), and unidentified_Bacteria (2.2%). Dialister, Peptoniphilus, and Prevotella were identified as the top three genera based on high centrality score. The only five strong negative correlations in the bacterial communities were Peptoniphilus-Lactobacillus, Aeromonas-Aureimonas, Lactobacillus-Campylobacter, Lactobacillus-Facklamia, and Aureimonas-Vibrio.
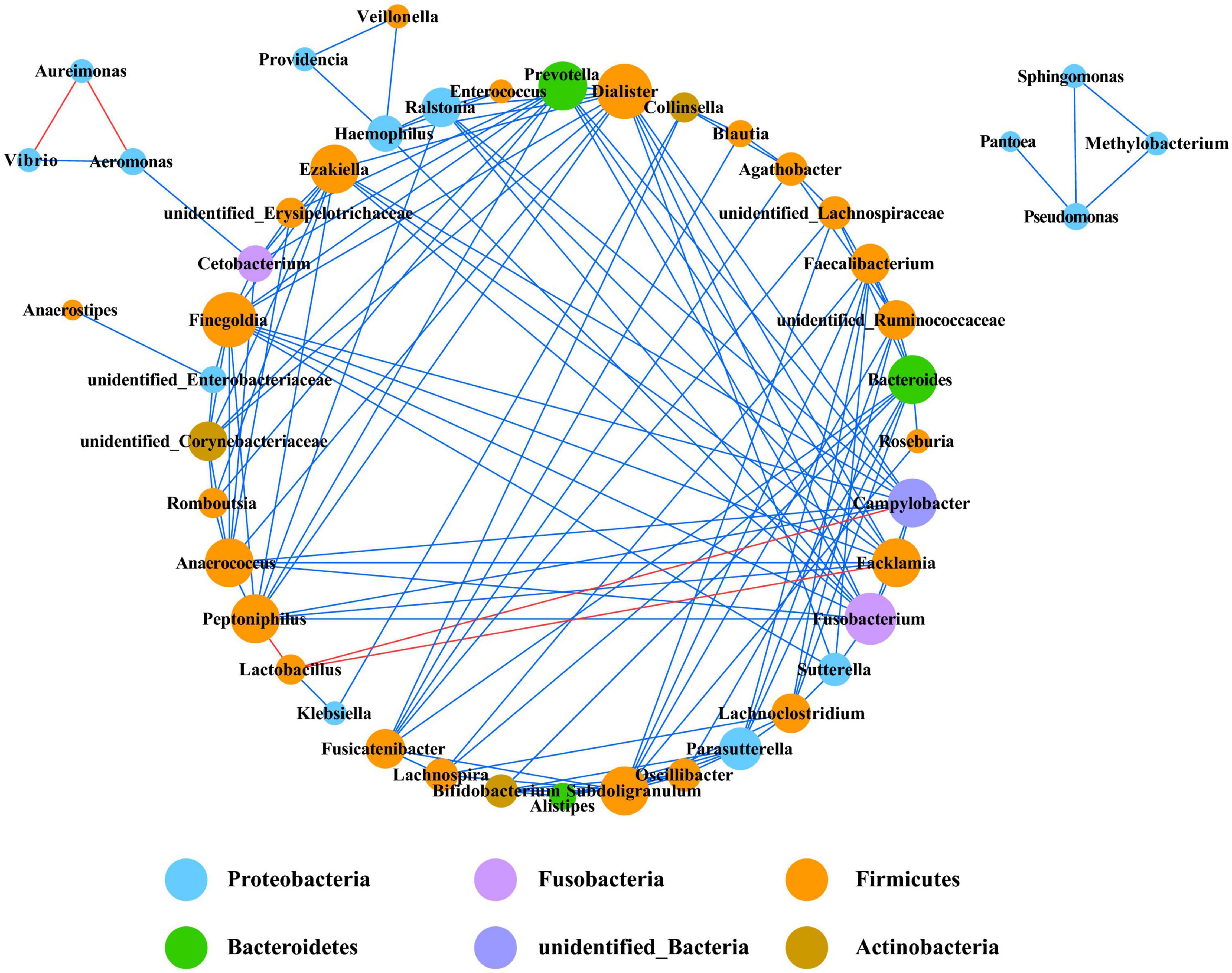
Figure 6. Co-occurrence network of bacteria at genera level. Only p values less than 0.05 and corrections coefficients higher than 0.6 were constructed in the network. The size of each node is proportional to the number of connections, the color of each node is correlates with taxonomy. Blue edges indicate positive correlations, and red edges indicate negative correlations.
In the fungal community, only eight genera had strong and significant correlations (Figure 7) (p < 0.05, r > 0.6). There were significant negative correlations between Cladosporium and Stagonosporopsis, Phoma and Golubevia, Phoma and Sporidiobolus, and Rhodotorula and Fusarium. Only Cladosporium and Alternaria had a strong and significant positive correlation in co-occurrence network analysis.
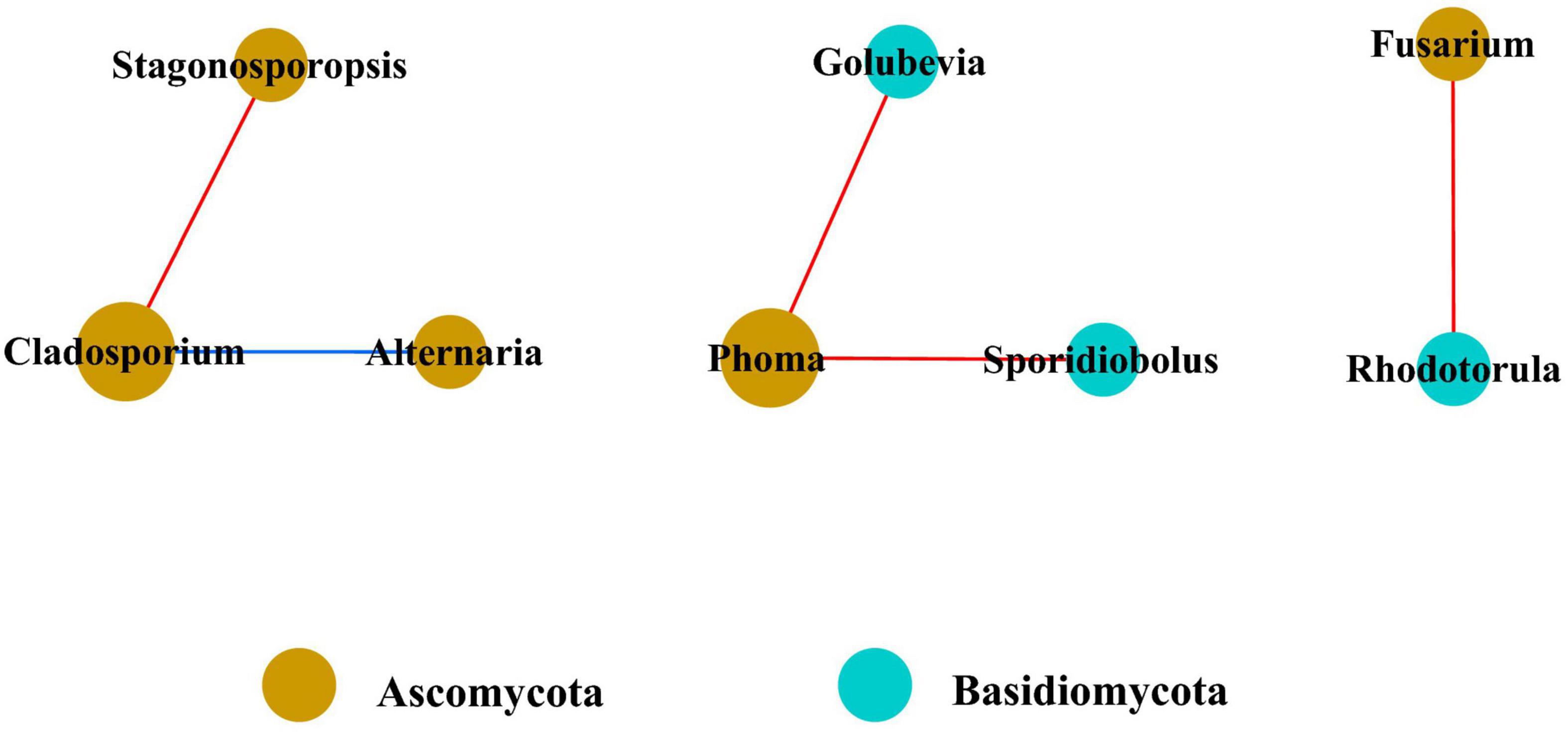
Figure 7. Co-occurrence network of fungi at genera level. Only p values less than 0.05 and corrections coefficients higher than 0.6 were constructed in the network. The size of each node is proportional to the number of connections, the color of each node is correlates with taxonomy. Blue edges indicate positive correlations, and red edges indicate negative correlations.
Culture-Based Fungal Diversity
A total of 47 fungal isolates belonging to 18 genera were obtained by tissue isolation, and their ITS sequences have been deposited in NCBI GenBank (Table 4). The pathogenicity of all 47 isolates was tested on detached tobacco leaves. Afterward, leaves were wounded, inoculated, and then incubated for 10 days at 28°C. The non-inoculated control and 26 isolates did not yield any symptoms on detached leaves, and the remaining 21 isolates caused varying degrees of symptoms on the detached leaves. Disease symptoms on detached leaves progressed from inoculation sites to surrounding areas and formed yellowish-brown spots with irregular shapes within a week (Figure 8). Among the 21 pathogenic isolates, six (wz16, wz17, wz19, wz45, wz50, and wz52) had strong pathogenicity (lesion area >3.14 cm2); seven (wz20, wz38, wz39, wz40, wz48, wz55, and wz60) had medium pathogenicity; and eight (wz21, wz32, wz35, wz43, wz49, wz51, wz56, and wz65) had weak pathogenicity (noticeable lesions <0.79 cm2). The above 21 strains were classified as Colletotrichum (wz35/49/51/52/55/60), Xylariaceae (wz21/38/40), Corynespora (wz16/17/45), Pestalotiopsis (wz19/56), Alternaria (wz20/39), Epicoccum (wz48/50), Byssosphaeria (wz43), Phoma (wz32), or Diaporthe (wz65). The symptoms caused by three of the six highly virulent isolates (wz16, wz17, and wz45) were consistent with those of originally collected diseased leaves, and these were all identified as Corynespora cassiicola.
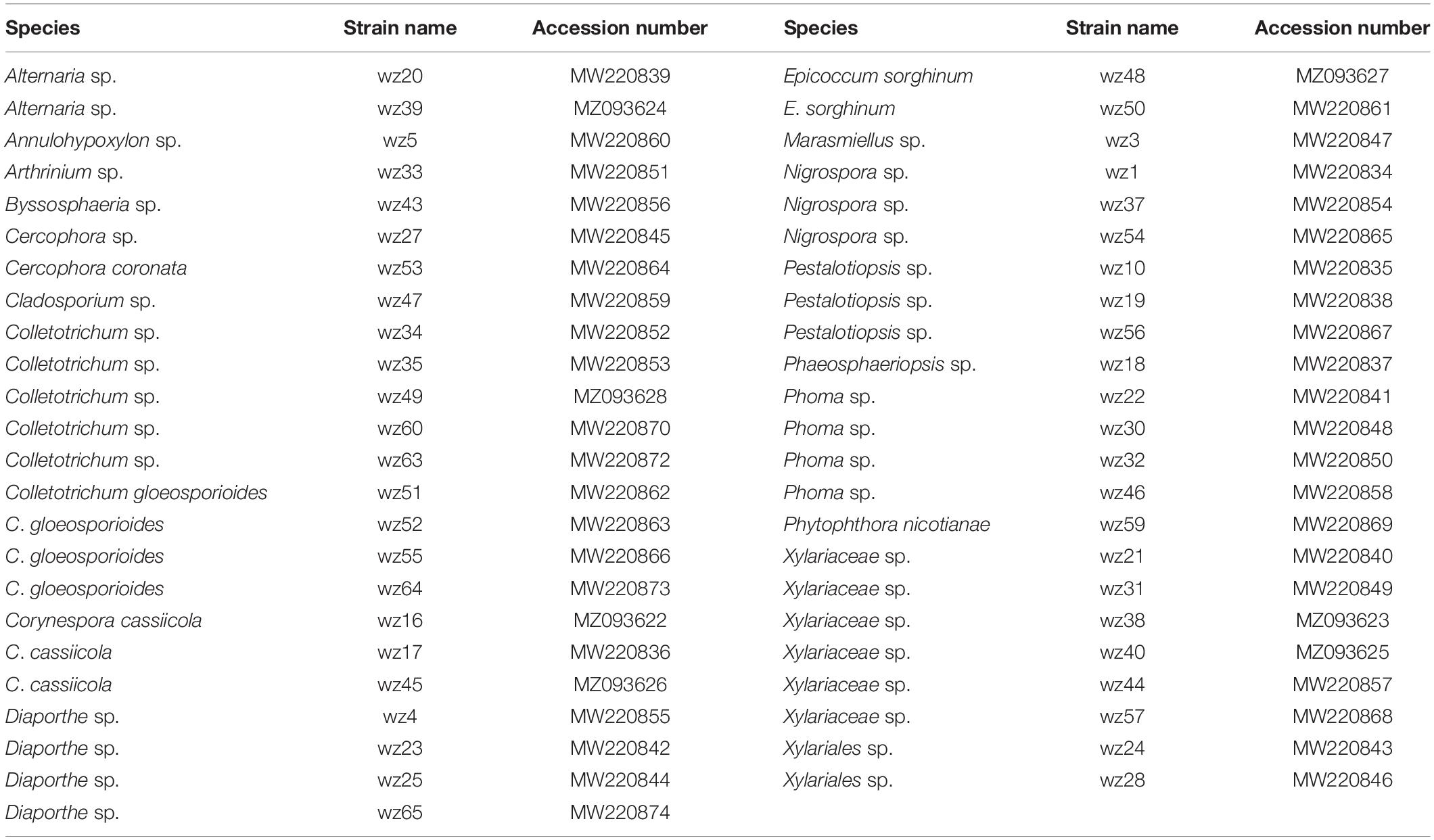
Table 4. Fungi isolated from symptomatic leaves, and their identity based on ITS sequences, and their GenBank accession numbers.

Figure 8. Symptoms on inoculated tobacco leaves at 10 days after inoculation and incubation at 28°C. These isolates came from symptomatic leaves and their identification based on ITS sequences is shown in Table 4. The white mesh board behind each leaf is the same size (25 cm × 10 cm).
Discussion
In the past, research on plant pathogens and phyllosphere microorganisms was often based on traditional culture-dependent methods, which may have underestimated the number of microbial communities and the functions of non-pathogenic and non-culturable microorganisms (Dissanayake et al., 2018). This study provided new insights into potential phyllosphere pathogens and into the microbial community structure and diversity in asymptomatic and symptomatic tobacco leaves. In the present study, we found a correlation between the health status of tobacco leaves and phyllosphere microbial community structure and composition, but this correlation was not very strong (r < 0.6). This finding implied that foliar disease may affect the phyllosphere by promoting or inhibiting the growth of certain microorganisms.
Among the dominant taxa, we identified a large number of bacterial and fungal genera which have been reported as phytopathogens or plant growth promoters, as well as a several neutral bacteria and fungi. The neutral bacterial genera such as Cetobacterium, Providencia, Faecalibacterium, and Methylobacterium have been reported from leaves of different plants as endophytes or epiphytes. In previous studies, Sphingomonas, Bacillus, and Cyanobacteria were considered to be beneficial to plant growth and to protect plants from pathogens (Innerebner et al., 2011; Han et al., 2016). But in our study, they were not detected from tobacco leaves. This difference may result from differences in climate and geography and in host species (Finkel et al., 2011).
Pseudomonas, Pantoea, and Ralstonia are typical phytopathogenic genera associated with tobacco. Pseudomonas as a pathogenic genus, has a wide host range (Morris et al., 2008). Wildfire disease of tobacco is caused by Pseudomonas syringae pv. tabaci (Ramegowda et al., 2013). Some Pantoea species can cause human diseases, while others can cause plant diseases (Cruz et al., 2007). Species of Ralstonia are pathogens causing bacterial wilt in many plants (Genin and Denny, 2012) including R. solanacearum causing tobacco bacterial wilt. PICRUSt predictions showed that the functions of most bacteria were associated with regular physiological activities, such as metabolism, genetic information processing, cellular processes, and organismal systems. Among bacterial communities, some species of Pseudomonas, Pantoea, and Providencia can cause human diseases, but it is uncommon for human pathogenic bacteria to be found on plant leaves, and we were only able to resolve to genera so the human pathogenic species might not have been present.
In contrast to bacteria, the fungal community contained more phytopathogenic genera. Phoma is an ubiquitous genus among plants, and 110 species are plant pathogens (Deb et al., 2020). They can invade many economically important crops, including wheat, tobacco, oilseed rape, and tea, among others. There were other fungi besides Phoma, including Alternaria species which are known to cause tobacco brown spot (Hou et al., 2016), Cercospora nicotianae which causes tobacco frogeye (Fajola and Alasoadura, 1973), and Fusarium species which cause tobacco wilt (LaMondia, 2015). In addition to these genera, Cladosporium, Stagonosporopsis, Moesziomyces, Gibellulopsis, Phyllosticta, Epicoccum, Plectosphaerella, and Corynespora have been reported to invade plants as phytopathogenic genera. Based on the functional predictions of FUNGuild, most fungal communities were plant pathogens and saprotrophs, and few communities were endophytes, parasites or animal pathogens. The structure of fungal communities on tobacco leaves also supported this conclusion.
Plants with a higher microbial diversity were less prone to pathogen invasion than those with less microbial diversity (Berg et al., 2017). This is likely due to more diverse microbial communities having higher competition for nutrient sources, free water and ecological niches than potential pathogens (Shade, 2017). Therefore, leaves with higher microbial diversity may be associated with reduce chances of disease outbreaks (Yang et al., 2017). Disease severity is one of the many factors that affect phyllosphere microorganisms. Tobacco leaves of different disease index infected by Didymella segeticola have different microbial diversity, and the relative abundance of dominant genera in the phyllosphere increased with increasing disease index (Huang et al., 2021). The abundance of Ascomycota and Podosphaera increased as pumpkin powdery mildew severity increased from L1 to L4, and the diversity and richness of the fungal community increased from L1 to L2, and then declined from L2 to L4 (Zhang et al., 2017). Therefore, if we had tested tobacco leaves with different levels of symptoms or different disease stages, their phyllosphere microbial community diversity may also have been different.
The traditional culture-dependent method yielded community composition results which differed from the metagenetic analysis. Among the 47 strains isolated from culturing, Colletotrichum was the dominant genus, which accounted for 19%, followed as Xylariaceae (13%), Phoma (9%), Diaporthe (9%), and Cladosporium (2%). We only checked for fungi growing up to 1 week, but a longer monitoring period such as up to 4 weeks may have revealed other fungal species. Based on the metagenetic analysis, the dominant fungi in the symptomatic leaves were found to be species of Phoma (35.05%), Cladosporium (23.87%) and Alternaria (6.88%), and the relative abundances of Colletotrichum and Xylariaceae were less than 0.01%. Therefore, the strains with high isolation frequency obtained by culture-dependent methods were not necessarily the dominant fungi in the leaf layer, and the strains with low isolation frequency may also be dominant fungi based on metagenetic analysis. Pathogenicity testing showed that in the same genus, some strains had strong pathogenicity, some strains had weak pathogenicity, and some strains even had no pathogenicity. In this study, we used wounded detached leaves for pathogenicity testing, which may have overestimated pathogenicity for opportunistic fungi, but in previous research in this lab, we have found that there were no significant differences in lesion size on attached leaves vs. wounded detached leaves in tests with a variety of tobacco pathogens (unpublished data). Some fungi isolated by culturing were not detected in metagenetic analysis, which may be because these fungi had low relative abundance or some other property such as prolific sporulation exaggerated their abundance. Therefore, the combination of metagenetic analysis and traditional culture-dependent methods perhaps can more comprehensively reflect the composition of phyllosphere microbial communities. Based on our previous observations of tobacco foliar diseases, we suspected that this was likely a fungal pathogen especially since the lesions were not vein limited, and hence we targeted fungi for isolation and did not attempt to culture bacteria. Future research will focus on confirming the causal agent or agents of this disease whether a bacterium may be involved.
Conclusion
This study demonstrated that asymptomatic and symptomatic leaves of tobacco plants may differ in microbial community composition and potential pathogens. Based on isolation of culturable fungi from symptomatic leaves followed by pathogenicity testing, 21 potential pathogenic strains belonging to Colletotrichum, Xylariaceae, Corynespora, Pestalotiopsis, Alternaria, Epicoccum, Byssosphaeria, Phoma, and Diaporthe were found on symptomatic leaves, and the causal agent of this tobacco leaf spot may be Corynespora cassiicola. Metagenetic analysis of symptomatic leaves showed the following pathogen-containing genera on tobacco leaves: Pseudomonas, Prevotella, Peptoniphilus, Gibellulopsis, and Meira, among others. Metagenetic analysis also revealed that asymptomatic leaves harbored pathogenic genera such as Pseudomonas, Pantoea, Cercospora, and Phaeosphaeriopsis among others. Asymptomatic leaves had a higher microbial diversity and richness than symptomatic leaves. The combination of the traditional culture-dependent method and metagenetic analysis provided insights into the composition of microbial communities and potential pathogens on tobacco leaves more comprehensively than either one of these methods alone. These findings are significant for the prevention and control of tobacco diseases.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, PRJNA722239.
Author Contributions
L-GX, H-CW, and Z-HY contributed to conception, design of the study, and wrote the first draft of the manuscript. L-GX organized the database and performed the statistical analysis. TH wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This research was financed by the China National Tobacco Corporation [110202001035(LS-04) and 110202101048(LS-08)], National Natural Science Foundation of China (31960550, 32160522, and 31570022), Guizhou Science Technology Foundation [ZK(2021)Key036], Guizhou Provincial Academician Workstation of Microbiology and Health [(2020)4004], and Guizhou Tobacco Company (2020XM03 and 2020XM22).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
Berg, G., Köberl, M., Rybakova, D., Müller, H., Grosch, R., and Smalla, K. (2017). Plant microbial diversity is suggested as the key to future biocontrol and health trends. FEMS Microbiol. Ecol. 93, 1–9. doi: 10.1093/femsec/fix050
Bokulich, N. A., Subramanian, S., Faith, J. J., Gevers, D., Gordon, J. I., Knight, R., et al. (2013). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59. doi: 10.1038/nmeth.2276
Cai, L., Liu, Z., Zhou, W., Chen, F., and Ding, W. (2017). Inhibitive and control efficacy of Bacillus subtilis against powdery mildew on tobacco. Southwest China J. Agric. Sci. 30, 1795–1801.
Cai, X. H., Chen, T., Wang, R. Y., Fan, Y. J., Li, Y., Hu, S. N., et al. (2019). Forecasting wildfire disease on tobacco: toward developing a high-accuracy prediction model for disease index using local climate factors and support vector regression. Theor. Appl. Climatol. 137, 2139–2149. doi: 10.1007/s00704-018-2708-x
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108(Suppl. 1) 4516–4522. doi: 10.1073/pnas.1000080107
Chen, Q. L., Cai, L., Wang, H. C., Cai, L. T., Goodwin, P., Ma, J., et al. (2020). Fungal composition and diversity of the tobacco leaf phyllosphere during curing of leaves. Front. Microbiol. 11:2136. doi: 10.3389/fmicb.2020.554051
Cruz, A. T., Cazacu, A. C., and Allen, C. H. (2007). Pantoea agglomerans, a plant pathogen causing human disease. J. Clin. Microbiol. 45, 1989–1992. doi: 10.1128/JCM.00632-07
Deb, D., Khan, A., and Dey, N. (2020). Phoma diseases: epidemiology and control. Plant Pathol. 69, 1203–1217. doi: 10.1111/ppa.13221
Dissanayake, A. J., Purahong, W., Wubet, T., Hyde, K. D., Zhang, W., Xu, H., et al. (2018). Direct comparison of culture-dependent and culture-independent molecular approaches reveal the diversity of fungal endophytic communities in stems of grapevine (Vitis vinifera). Fungal Divers. 90, 85–107. doi: 10.1007/s13225-018-0399-3
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fajola, A. O., and Alasoadura, S. O. (1973). Chemical control of the frog-eye disease (Cercospora nicotianae) of tobacco (Nicotiana tabacum) in Nigeria. Ann. Appl. Biol. 74, 219–224. doi: 10.1111/j.1744-7348.1973.tb07741.x
Finkel, O. M., Burch, A. Y., Lindow, S. E., Post, A. F., and Belkin, S. (2011). Geographical location determines the population structure in phyllosphere microbial communities of a salt-excreting desert tree. Appl. Environ. Microb. 77, 7647–7655. doi: 10.1128/AEM.05565-11
Fromme, F. D., and Murray, T. J. (1919). Angular leaf spot of tobacco, an undescribed bacterial disease. J. Agric. Res. 16, 219–228.
Genin, S., and Denny, T. P. (2012). Pathogenomics of the Ralstonia solanacearum species complex. Annu. Rev. Phytopathol. 50, 67–89. doi: 10.1146/annurev-phyto-081211-173000
Haas, B. J., Gevers, D., Earl, A. M., Feldgarden, M., Ward, D. V., Giannoukos, G., et al. (2011). Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21, 494–504. doi: 10.1101/gr.112730.110
Han, T., You, C., Zhang, L., Feng, C., Zhang, C., and Wang, J. (2016). Biocontrol potential of antagonist Bacillus subtilis Tpb55 against tobacco black shank. BioControl 61, 195–205. doi: 10.1007/s10526-015-9705-0
Hari, V. (1981). The RNA of tobacco each virus: further characterization and detection of protein linked to RNA. Virology 112, 391–399. doi: 10.1016/0042-6822(81)90286-5
Hou, Y., Ma, X., Wan, W., Long, N., Zhang, J., Tan, Y., et al. (2016). Comparative genomics of pathogens causing brown spot disease of tobacco: Alternaria longipes and Alternaria alternata. PLoS One 11:e0155258. doi: 10.1371/journal.pone.0155258
Huang, Y., Wang, H. C., Cai, L. T., Li, W. H., Pan, D. W., Xiang, L. G., et al. (2021). Phyllospheric microbial composition and diversity of the tobacco leaves infected by Didymella segeticola. Front. Microbiol. 12:699699. doi: 10.3389/fmicb.2021.699699
Hugouvieux, V., Barber, C. E., and Daniels, M. J. (1998). Entry of Xanthomonas campestris pv. campestris into hydathodes of Arabidopsis thaliana leaves: a system for studying early infection events in bacterial pathogenesis. Mol. Plant Microbe Internact. 11, 537–543. doi: 10.1094/MPMI.1998.11.6.537
Innerebner, G., Knief, C., and Vorholt, J. A. (2011). Protection of Arabidopsis thaliana against leaf-pathogenic Pseudomonas syringae by Sphingomonas strains in a controlled model system. Appl. Environ. Microb. 77, 3202–3210. doi: 10.1128/AEM.00133-11
LaMondia, J. A. (2015). Fusarium wilt of tobacco. Crop Prot. 73, 73–77. doi: 10.1016/j.cropro.2015.03.003
Langille, M. G., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Li, F., Yuan, G., Liao, T., Li, Q., and Lin, W. (2017). Leaf spot of tobacco caused by Fusarium proliferatum. J. Gen. Plant Pathol. 83, 264–267. doi: 10.1007/s10327-017-0714-y
Li, S., and Wu, F. Z. (2018). Diversity and co-occurrence patterns of soil bacterial and fungal communities in seven intercropping systems. Front. Microbiol. 9:1521. doi: 10.3389/fmicb.2018.01521
Luo, L., Zhang, Z., Wang, P., Han, Y., Jin, D., Su, P., et al. (2019). Variations in phyllosphere microbial community along with the development of angular leaf-spot of cucumber. AMB Express 9, 1–13. doi: 10.1186/s13568-019-0800-y
Macdowall, F. D. H., Mukammal, E. I., and Cole, A. F. W. (1964). Direct correlation of air-polluting ozone and tobacco weather fleck. Can. J. Plant Sci. 44, 410–417. doi: 10.4141/cjps64-081
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Morris, C. E., Sands, D. C., Vinatzer, B. A., Glaux, C., Guilbaud, C., Buffiere, A., et al. (2008). The life history of the plant pathogen Pseudomonas syringae is linked to the water cycle. ISME J. 2, 321–334. doi: 10.1038/ismej.2007.113
Nguyen, N. H., Song, Z., Bates, S. T., Branco, S., Tedersoo, L., Menke, J., et al. (2016). FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20, 241–248. doi: 10.1016/j.funeco.2015.06.006
Nilsson, R. H., Kristiansson, E., Ryberg, M., Hallenberg, N., and Larsson, K. H. (2008). Intraspecific ITS variability in the kingdom Fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol. Bioinform. 4, 193–201. doi: 10.4137/ebo.s653
Ramegowda, V., Senthil-Kumar, M., Ishiga, Y., Kaundal, A., Udayakumar, M., and Mysore, K. S. (2013). Drought stress acclimation imparts tolerance to Sclerotinia sclerotiorum and Pseudomonas syringae in Nicotiana benthamiana. Int. J. Mol. Sci. 14, 9497–9513. doi: 10.3390/ijms14059497
Scholthof, K. B. G. (2004). Tobacco mosaic virus: a model system for plant biology. Annu. Rev. Phytopathol. 42, 13–34. doi: 10.1146/annurev.phyto.42.040803.140322
Shade, A. (2017). Diversity is the question, not the answer. ISME J. 11, 1–6. doi: 10.1038/ismej.2016.118
Stavely, J. R., and Slana, L. J. (1975). Initiation of tobacco brown spot. Phytopathology 65, 897–901. doi: 10.1094/phyto-65-897
Vorholt, J. A. (2012). Microbial life in the phyllosphere. Nat. Rev. Microbiol. 10, 828–840. doi: 10.1038/nrmicro2910
Wagih, E. E., Zalat, M. M., and Kawanna, M. A. (2021). Partial characterisation of two isolates of cucumber mosaic virus (CMV). Sci. J. Agric. Sci. 3, 144–157.
Wei, Y. J., Wu, Y., Yan, Y. Z., Zou, W., Xue, J., Ma, W. R., et al. (2018). High-throughput sequencing of microbial community diversity in soil, grapes, leaves, grape juice and wine of grapevine from China. PLoS One 13:e0193097. doi: 10.1371/journal.pone.0193097
White, T. J., Bruns, T., Lee, S. J. W. T., and Taylor, J. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR Protocols: A Guide to Methods and Applications, Vol. 18, eds M. A. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White (San Diego, CA: Academic Press), 315–322. doi: 10.1016/b978-0-12-372180-8.50042-1
Yan, X. F., Kong, J. S., Luo, A. N., Wang, X., Wang, Y. H., and Li, Y. H. (2021). Changes in planting scale of flue-cured tobacco production regions in China in recent 20 years. Chin. Tobacco Sci. 42, 92–101.
Yang, H., Li, J., Xiao, Y., Gu, Y., Liu, H., Liang, Y., et al. (2017). An integrated insight into the relationship between soil microbial community and tobacco bacterial wilt disease. Front. Microbiol. 8:2179. doi: 10.3389/fmicb.2017.02179
Zhang, Y. J., Zhang, S., Liu, X. Z., Wen, H. A., and Wang, M. (2010). A simple method of genomic DNA extraction suitable for analysis of bulk fungal strains. Lett. Appl. Microbiol. 51, 114–118. doi: 10.1111/j.1472-765X.2010.02867.x
Keywords: tobacco, microbial diversity, pathogen, metagenetic analysis, culture-dependent method
Citation: Xiang L-G, Wang H-C, Wang F, Cai L-T, Li W-H, Hsiang T and Yu Z-H (2022) Analysis of Phyllosphere Microorganisms and Potential Pathogens of Tobacco Leaves. Front. Microbiol. 13:843389. doi: 10.3389/fmicb.2022.843389
Received: 25 December 2021; Accepted: 11 April 2022;
Published: 29 April 2022.
Edited by:
Yong Wang, Guizhou University, ChinaReviewed by:
Jia Liu, Chongqing University of Arts and Sciences, ChinaChuanqing Zhang, Zhejiang A&F University, China
Shengming Liu, Henan University of Science and Technology, China
Guoqing Li, Huazhong Agricultural University, China
Copyright © 2022 Xiang, Wang, Wang, Cai, Li, Hsiang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Han-Cheng Wang, eGlhb2JhaXlhbmcxMjZAaG90bWFpbC5jb20=; Feng Wang, NTg0MjI4OTkyQHFxLmNvbQ==; Zhi-He Yu, emhpaGV5dUBob3RtYWlsLmNvbQ==