 Komwit Surachat1,2*
Komwit Surachat1,2* Duangporn Kantachote3
Duangporn Kantachote3 Monwadee Wonglapsuwan3
Monwadee Wonglapsuwan3 Arnon Chukamnerd4Panchalika Deachamag3
Arnon Chukamnerd4Panchalika Deachamag3 Pimonsri Mittraparp-arthorn2,3Kongpop Jeenkeawpiam2
Pimonsri Mittraparp-arthorn2,3Kongpop Jeenkeawpiam2- 1Division of Computational Science, Faculty of Science, Prince of Songkla University, Songkhla, Thailand
- 2Molecular Evolution and Computational Biology Research Unit, Faculty of Science, Prince of Songkla University, Songkhla, Thailand
- 3Division of Biological Science, Faculty of Science, Prince of Songkla University, Songkhla, Thailand
- 4Department of Biomedical Sciences and Biomedical Engineering, Faculty of Medicine, Prince of Songkla University, Songkhla, Thailand
Lactic acid bacteria (LAB) in the genus Weissella spp. contain traits in their genome that confer versatility. In particular, Weissella cibaria encodes several beneficial genes that are useful in biotechnological applications. The complete genome of W. cibaria NH9449 was sequenced and an in silico comparative analysis was performed to gain insight into the genomic diversity among members of the genus Weissella. A total of 219 Weissella genomes were used in a bioinformatics analysis of pan-genomes, phylogenetics, self-defense mechanisms, virulence factors, antimicrobial resistance, and carbohydrate-active enzymes. These investigations showed that the strain NH9449 encodes several restriction-modification-related genes and a CRISPR-Cas region in its genome. The identification of carbohydrate-active enzyme-encoding genes indicated that this strain could be beneficial in biotechnological applications. The comparative genomic analysis reveals the very high genomic diversity in this genus, and some marked differences in genetic variation and genes among Weissella species. The calculated average amino acid identity (AAI) and phylogenetic analysis of core and accessory genes shows the possible existence of three new species in this genus. These new genomic insights into Weissella species and their biological functions could be useful in the food industry and other applications.
Introduction
Weissella, a genus of Gram-positive lactic acid bacteria (LAB), is classified as a member of the Leuconostocaceae family (Fusco et al., 2015). LAB are catalase-negative, non-endospore-forming coccoid or rod-shaped bacteria. They prosper in a wide range of environments from fermented food to soil, feces, saliva, breast milk, urine, and banana leaves (Bjorkroth et al., 2002; Fairfax et al., 2014; Fusco et al., 2015), but most Weissella spp. that have been studied were isolated from sources associated with fermented food, e.g., W. sagaensis (Li Y. Q. et al., 2020), W. hellenica (Panthee et al., 2019), W. confusa (Falasconi et al., 2020), and W. cibaria (Shin et al., 2020). In other studies, Weissella spp. have shown various functions that could be suitable for biotechnological and medical applications. They could inhibit the growth of other bacteria (Yeu et al., 2021), act as starter cultures (Fusco et al., 2015; Yeong et al., 2020) and as probiotics (Huang et al., 2020; Kang et al., 2020) and prevent carcinogenic processes (Kwak et al., 2014).
With the rapid development of next-generation sequencing technology, more genomes from different Weissella species have been sequenced to identify genetic elements, study their potential functions, and compare phylogenetic relationships with other organisms. Several research works have focused on the potential of phenotypic and genotypic traits of Weissella spp. (Panthee et al., 2019; Shin et al., 2020; Yeong et al., 2020; Yuan et al., 2021). In particular W. cibaria, this species is a key member of the genus and exhibits a variety of traits and abilities. For instance, W. cibaria can produce bacteriocins such as weissellicin 110, which has strong activity against Gram-positive bacteria (Srionnual et al., 2007; Wu et al., 2015). Some W. cibaria strains were reported to encode and harbor several genes with useful biotechnological properties (Carlosama Adriana et al., 2021; Tenea and Hurtado, 2021). They also play an important role in heterofermentative metabolism and CO2 production from carbohydrate metabolism, which makes them suitable for use in fermentation processes (Ricciardi et al., 2009; Kang et al., 2016). Even though many genomic studies of several members of the genus have been carried out, the understanding of versatility traits, genomic diversity, and the complex phylogenetic relationships among species of this genus remains incomplete. Therefore, continued comparative genomic study of the genus is needed to fill these knowledge gaps.
In this work, we sequenced the complete genome of W. cibaria NH9449 isolated from Thai fermented pork sausage and performed a comprehensive comparative genomic analysis with other Weissella species. Functional annotation was performed to predict the presence of carbohydrate-active enzymes and bacteriocin-encoding genes and highlight traits in the fermentation activity of these bacterial strains. In addition to the in silico analysis, we produced an overview of the biological evolution of this genus to provide a more precise picture of phylogenetic relationships among Weissella spp. To our knowledge, this is the first meta-analysis to use all the whole-genome sequences of the genus Weissella in a comparative genomic analysis. Our results provide important information about the genomic diversity among species of this genus and a versatility trait that could be useful in the food fermentation industry.
Materials and Methods
Bacterial Strain and DNA Extraction
Weissella cibaria NH9449 was isolated from a traditional Thai-style fermented pork sausage at the Faculty of Science, Prince of Songkla University, Thailand. A single colony of W. cibaria NH9449 was cultivated in MRS broth at 37°C for 24 h under static conditions. Genomic DNA was purified and extracted using a DNeasy extraction kit (QIAGEN, Hilden, Germany) following the manufacturer’s instructions.
Library Preparation and Whole-Genome Sequencing
We constructed two genome sequencing libraries to sequence both short and long reads using the Illumina and PacBio platforms, respectively. The purified genomic DNA was sent to be sequenced with a PacBio RSII sequencer (Pacific Biosciences, Menlo Park, CA, United States) by Macrogen (Seoul, South Korea). The sequencing library was prepared by 10 kb-protocol and P4-C2 chemistry to run in one single-molecule real-time (SMRT) cell. A total of 141,639 raw reads was obtained from the PacBio sequencer, with a total yield of 780 Mbp. The subread N50 and average subread lengths were 8,641 and 5,512, respectively. In addition, we sequenced the short-read library at Prince of Songkla University using a NextSeq 550 sequencer (Illumina, Inc., San Diego, CA, United States). Short-read sequencing was constructed with an insert size of 350 bp, using a Nextera XT DNA Library Prep Kit to generate 150-bp paired-end reads. The sequencer generated a total of 1 Gbp of paired-end reads. The Phred quality scores of the short reads reached Q30 for approximately 96% of all reads.
Genome Assembly and Plasmid Identification
We first cleaned the raw data. Low-quality long-read sequences of less than 1,000 bp were filtered out using Canu v2.2 (Koren et al., 2017). Using fastp v0.23.0 (Chen et al., 2018), the short-read sequences were trimmed to improve the overall quality of all reads. The assembly process was then performed by Unicycler v0.4.7 (Wick et al., 2017) using both the short-read and long-read sequences. We then scanned for a small plasmid that could be absent from the assembly as described previously (Surachat et al., 2021a,b). Briefly, we mapped short-read sequences to draft assembly and extracted all unmapped reads to perform de novo assembly with spades v3.12 (Bankevich et al., 2012). Finally, we selected high coverage contigs only to check the similarity with data in the NR database of The National Center for Biotechnology Information (NCBI) server. Assembly polishing was performed by the Unicycler pipeline since it integrates the Pilon pipeline (Walker et al., 2014) to correct the draft assembly using short-read sequences. Finally, the quality and genome completeness of the assembly was assessed using Quast v4.0 (Gurevich et al., 2013) and Busco v5.2.2 (Seppey et al., 2019), respectively.
To confirm the presence of small plasmids, plasmid isolation was performed according to a published protocol (Yao et al., 2019) with some modifications. Briefly, the bacterial strain was grown under anaerobic conditions in MRS broth for 20 h at 37°C. A culture sample of 1 ml was placed in a sterile tube and centrifuged at 11,000 × g for 1 min. The pellets were resuspended in 250 μl of solution P1 (supplemented with 100 mg/ml lysozyme) of the QIAprep Spin Miniprep Kit (QIAGEN, Germany), and incubated for 10 min at 37°C. To completely remove lysozyme, the suspension was centrifuged at 11,000 × g for 1 min at room temperature. The pellets were washed twice by resuspension in 500 μl of 5% glucose solution and centrifuged at 11,000 × g for 1 min. The pellets were subjected to QIAprep Spin Miniprep Kit. Plasmid DNA was evaluated by agarose gel electrophoresis.
Genome Annotation and Visualization
Genome annotations were predicted using an offline version of the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) (Tatusova et al., 2016). Functional annotation to assign Clusters of Orthologous Groups (COG) was performed using the WebMGA web service (Wu et al., 2011) on the COG database (Tatusov et al., 2003). In addition, the clustered regularly interspaced short palindromic repeats (CRISPRs) were identified by CRISPRFinder (Grissa et al., 2007). Finally, the visualization of the genomic content was performed by the CGView Server BETA (Stothard et al., 2019).
Comparative Genomic and Pan-Genome Analysis
The genomic sequences of all Weissella strains used in this study were downloaded from the NCBI server. We used FastANI v1.32 (Jain et al., 2018) to compute pairwise average nucleotide identity (ANI) among all genomes and the EzAAI pipeline (Kim et al., 2021) to calculate average amino acid identity (AAI) using all protein sequences of each genome. Core, accessory, and unique genes were identified by a pan-genome analysis performed with Roary v3.11.2 (Page et al., 2015), using a threshold of 70% BLASTp and default parameters. Core and accessory genes were identified as single nucleotide polymorphisms (SNPs) using SNP-sites (Page et al., 2016). Phylogenetic trees were constructed with the neighbor-joining method using RaxMLHPC (Stamatakis, 2006) and phylogenetic tree reliability was evaluated by the bootstrap method using 1000 replications. Tree visualization was illustrated in MEGA-X v10.1.7 (Kumar et al., 2018).
Detection of Bacteriocin-Encoding Genes, Antimicrobial Resistance Genes, Plasmids, and Virulence-Associated Genes
To obtain all the bacteriocin-encoding genes of each bacterial strain, the BAGEL database (Van Heel et al., 2018) was searched using BLASTx with a cut-off E-value of 1e-10. The AMR genes and predicted phenotype/drug resistance profile of each genome were scanned with staramr v0.7.21, which integrates several AMR gene-specific tools including ResFinder (Bortolaia et al., 2020) and PlasmidFinder (Carattoli and Hasman, 2020). Virulence factors were identified by searching the bacterial strains against the full dataset of the virulence factor database (VFDB) (Liu et al., 2019) with a cut-off E-value of 1e-30 and minimum identity of >70%.
Prediction of CRISPR-Cas, R-M Systems, and Carbohydrate-Active Enzyme
CRISPR-Cas system was identified by CRISPRFinder (Grissa et al., 2007). In addition, Restriction-Modification (R-M) systems were explored using BLASTp analysis against the Rebase database (Roberts et al., 2015), with a setting of 70% identity threshold and a cut-off E-value of 1e-30. In addition, we identified the carbohydrate-active enzyme (CAZymes) (Lombard et al., 2014) families of all bacterial strains using run_dbcan2, a standalone tool of dbCAN2 (Zhang et al., 2018), with standard parameters.
Results and Discussion
Weissella cibaria NH9449 Genomic Features
The genome of W. cibaria NH9449 consists of a circular chromosome and six plasmids (Figure 1A). The genome size is 2.6 Mb. We identified a total of 2,390 protein-coding sequences, 28 rRNA genes, and 90 tRNA genes in this genome. The general genomic features are given in Table 1. The functional annotation predicted by the COG database is given in Figure 1B. In the NH9449 genome, more COGs (1490) were assigned to the metabolism functional category than any other category. Information storage and processing, and cellular processes and signaling were represented by 989 and 930 COGs, respectively. We found six plasmids in this genome; the size of the biggest plasmid was 34,161 bp and the smallest was only 1,526 bp. The presence of this small plasmid was confirmed using PCR amplification as shown in Supplementary Figure 1. The sequence similarities of the plasmids were close to various other strains of LAB species, e.g., Lactiplantibacillus plantarum (formerly Lactobacillus plantarum), Leuconostoc mesenteroides, W. confusa, and W. soli. Besides the NH9449 genome, we also identified seven and six plasmids in the genomes of W. cibaria CH2, and W. paramesenteroides WpK4, respectively. These findings could indicate that the NH9449 strain may have evolved to survive in new environments by acquiring new plasmids from closely related bacteria.
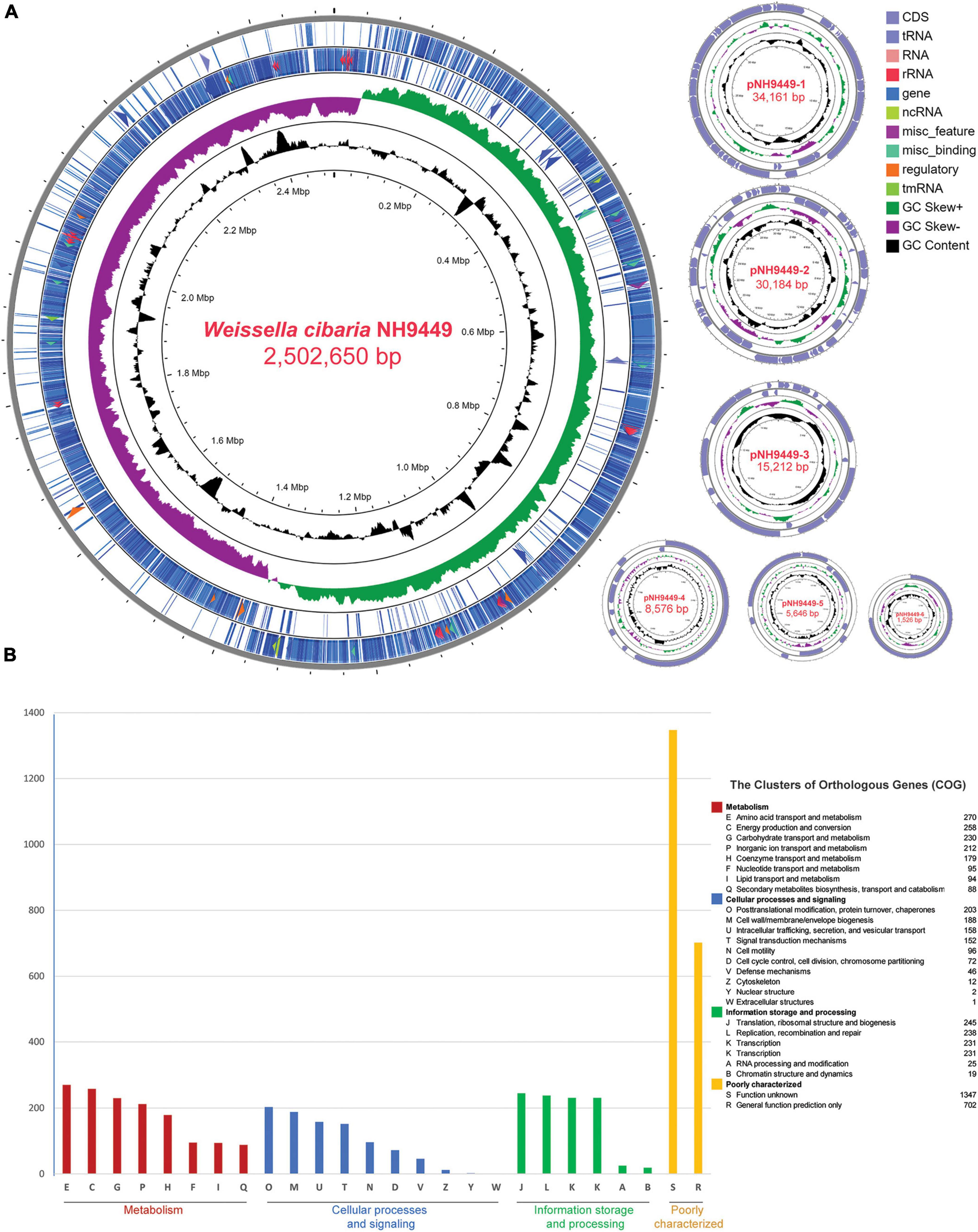
Figure 1. Genome visualization and annotation of W. cibaria NH9449. (A) Circular genome representation of chromosome and six plasmids. The element color of each circle is indicated in the legend. The following rings provide this information: forward strand CDS, reverse strand CDS, GC Skew, CG content, respectively. tRNA, RNA, rRNA, ncRNA, regulatory, and tmRNA are also located at the same ring of CDS based on their strand. (B) Clusters of Orthologous Groups (COG) identification of W. cibaria NH9449 genome.
Table 1. Genome features of W. cibaria NH9449.
Comparative Genomic Analysis of Weissella Species
To better understand the genomic diversity among all Weissella species, we performed a comparative genome analysis against all 236 genomes in the NCBI database (Supplementary Table 1). After we had removed duplicated assembly names in different versions and genomes derived from metagenome assembly, the total number of genome sequences was 218. This total included 35 complete genomes, 43 scaffold levels, and 140 contig levels (Accessed:18 Oct 2021). The numbers of sequenced genomes of different Weissella spp. are shown in Supplementary Figure 2. The genome size distribution among the genus is quite wide, varying from 1.04 to 3.02 Mbp, while the GC content is between 35.3 and 45.4%. The average genome size of all strains was 2.14 Mbp and the number of identified CDSs was 1947. The scatterplot between genome size and the number of CDSs of Weissella species is shown in Figure 2. The size distribution tends to be consistent among the same species; the biggest genome was that of Weissella cryptocerci 26KH-42T (Heo et al., 2019), the smallest that of Weissella viridescens MFPC16A2805 (Poirier et al., 2018). Naturally, genome size tends to increase as bacteria acquire mobile genetic components, transposons, and plasmids from other species during evolution (Li and Du, 2014). The size variation among the species within Weissella could also imply how these bacteria have adapted to survive in different environments by losing genes or receiving new ones (Bobay and Ochman, 2017). However, the genome size might also reflect the completeness of the assembly. Contamination, removal of contigs, and sequencing technology can affect the quality and size of the assembly.
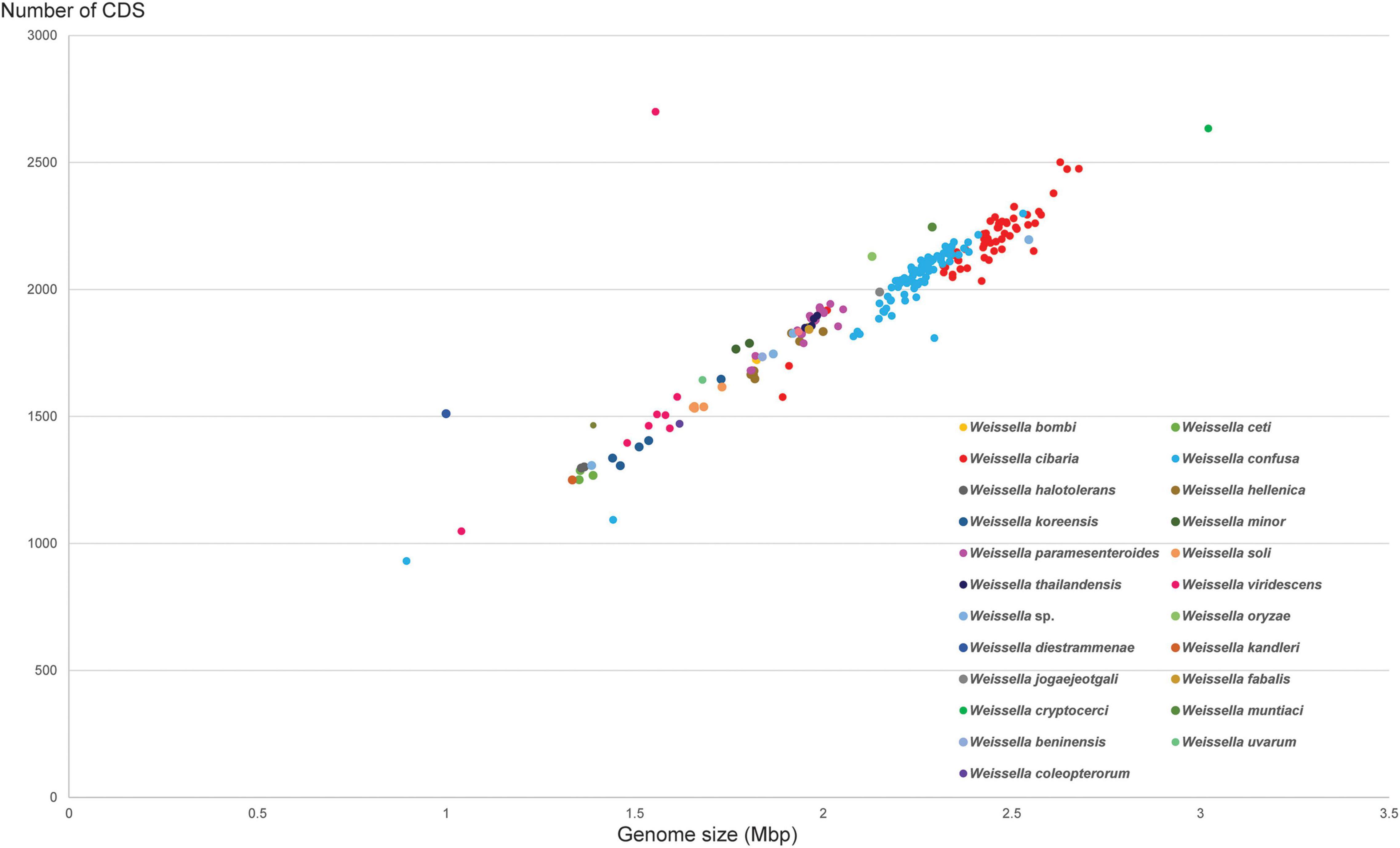
Figure 2. The genome size distribution was plotted against the number of CDSs identified in each bacterial strain among Weissella spp.
Furthermore, we computed and generated heatmaps of ANI values (Supplementary Figure 3) and AAI values (Supplementary Figure 4 and Supplementary Table 2) of 219 Weissella strains (including NH9449). Generally, taxonomic classification in the NCBI server is determined by ANI values (Ciufo et al., 2018). If the ANI value of a species compared to a reference strain is more than 95%, the species can be assigned as the same species as the reference in the taxonomic classification. As a result, some bacterial strains contained ANI values lower than 70%, we then analyzed the data based on AAI instead. The W. cibaria NH9449 strain shared sequence similarities from 96.25 to 99.23% with other W. cibaria strains. The average intraspecies similarity of all species is given in Table 2. Notably, we found several bacterial strains in W. ceti (1), W. hellenica (3), W. paramesenteroides (2), Weissella sp. (3), and W. viridescens (1), that presented AAI values lower than 95% among their groups, suggesting that species reclassification might be needed to reflect the updated data more accurately. For example, the shared AAI between Weissella sp. DD23 and W. cibaria ranged from 95.77 to 99.26%, suggesting this strain could be classified as W. cibaria. One of the findings of this analysis was that the AAI values between W. ceti CECT 7719 and all other W. ceti strains ranges from 60.59 to 85.0%. This has a certain relevance to the classification of these species since the taxonomic status of all W. ceti strains in the NCBI server, except CECT 7719, is “Inconclusive,” which means the ANI/AAI values of these strains are lower than 95%. Since W. ceti CECT 7719 was announced as a new species in 2011 (Vela et al., 2011), we suggest based on this analysis, that the other W. ceti strains, including NC36 (Ladner et al., 2013), WS08 (Figueiredo et al., 2014b), WS74, and WS105 (Figueiredo et al., 2014a), might also be new species and should be further investigated. In addition, analysis of the average AAI showed that interspecies similarities are between 59.81 and 97.05% (Figure 3). We found that W. jogaejeotgali and W. thailandensis, in agreement with a previous report (Panthee et al., 2019), present very high average similarity values over 97%. However, W. jogaejeotgali was reclassified as a heterotypic synonym of W. thailandensis in 2019 (Kwak et al., 2019). This analysis revealed that at least 11 of the studied strains presented AAI values lower than the criterion for taxonomic classification (95%). This finding could lead to the discovery of at least three new species/subspecies in this genus.
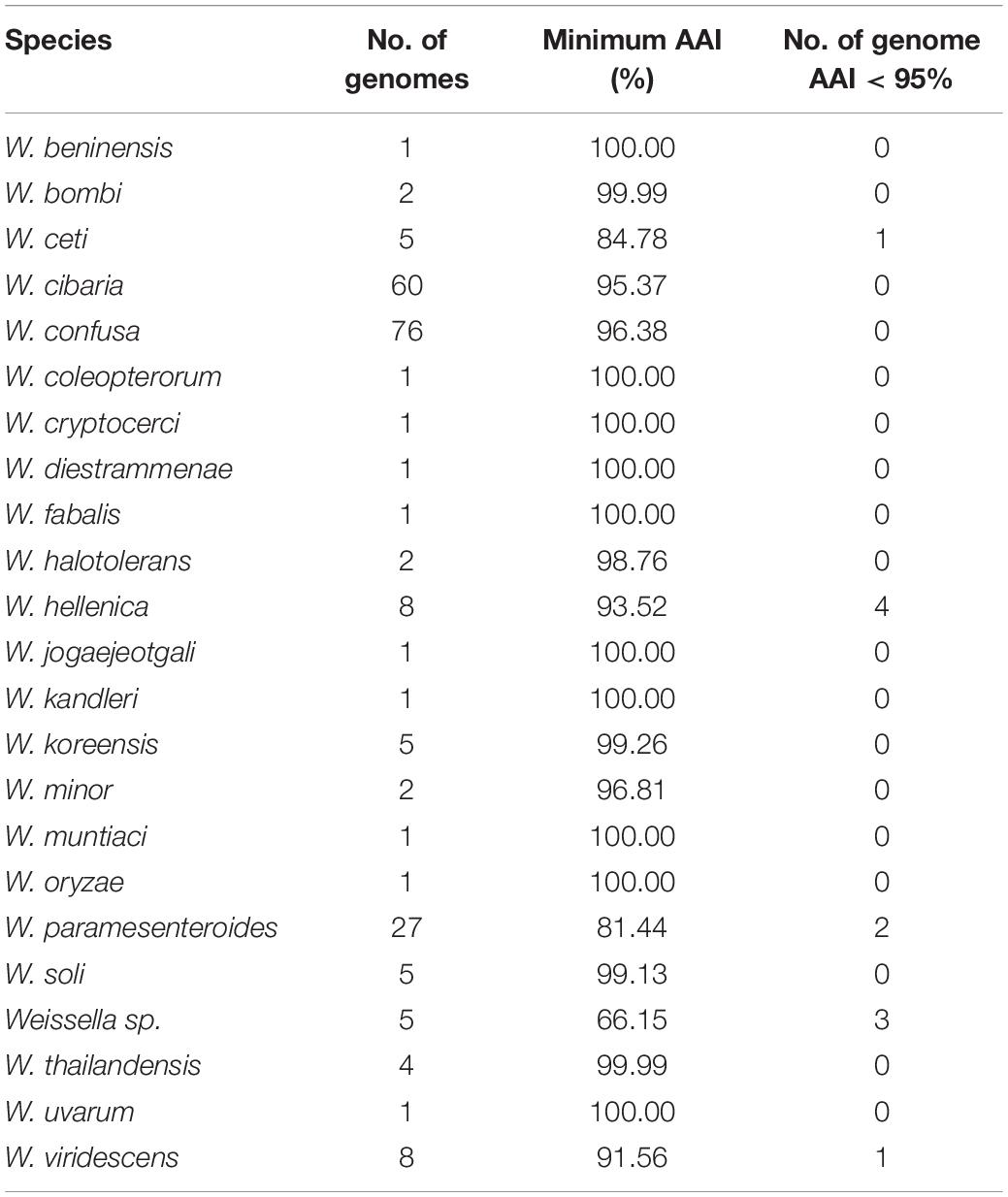
Table 2. Average amino acid identity information among Weissella spp.
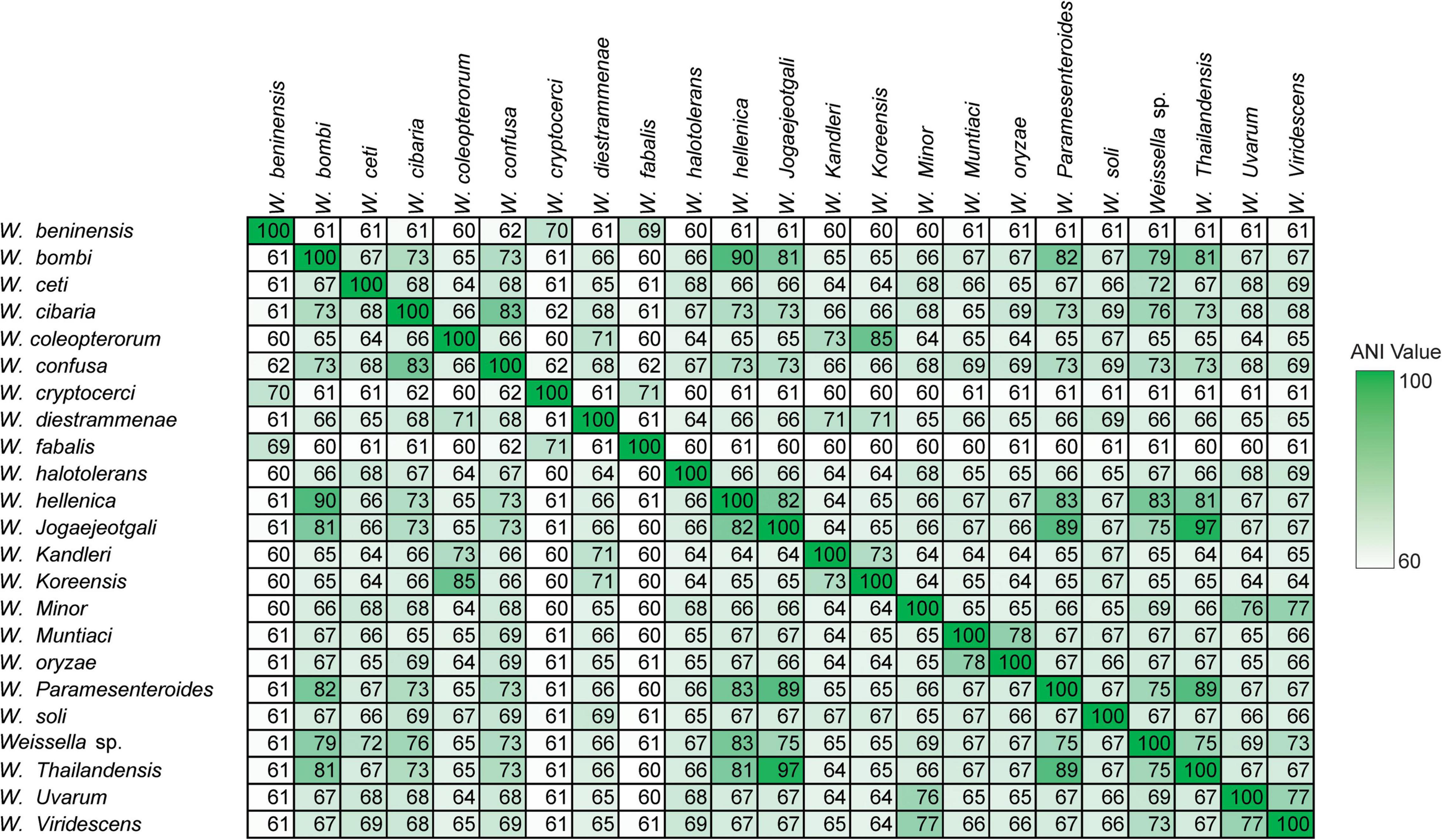
Figure 3. Interspecies similarities among the genus Weissella.
Also, AAI similarity was identified among only 60 W. cibaria strains (Figure 4). The similarities among these W. cibaria strains ranged from 96.08 to 100%. Every strain showed consistent similarities which were all over 95%. The bacterial strains isolated from similar sources seemed to share closer AAI values than strains isolated from different sources. For instance, the species with the AAI values farthest from and closest to NH9449 were W. cibaria SP7 isolated from cow feces, and W. cibaria AV1 isolated from fermented food, respectively. Habitat differences have a direct influence on which genetic elements are acquired or lost through horizontal gene transfer (HGT) to and from the environment. This process affects genetic variation and diversity in the same species (Wiedenbeck and Cohan, 2011; Lee et al., 2022).

Figure 4. Average amino acid identity analysis of 60 W. cibaria strains. The numbers display the percentage similarity between W. cibaria strains, where the colors vary from white (low similarity) to green (high similarity).
Pan-Genome and Phylogenetic Analysis
The diversity of the genus Weissella was clear from the pan-genome analysis. The analysis clustered genes from all genomes of interest into subgroups, namely core genes (found in >99%), soft core genes (found in 95–99%), shell genes (found in 15–95%), and cloud genes (found in <15%). The number of core genes, soft core genes, shell genes and cloud genes identified at the genus level were 165, 151, 3,055, and 25,224, respectively. It is clear that W. cibaria, W. confusa, W. hellenica, W. paramesenteroides, W. jogaejeotgali and W. thailandensis tend to share more core genes if other species are removed from the analysis (Figure 5). In contrast, other Weissella species, e.g., W. oryzae, W. cryptocerci, and W. fabalis, carried several unique genes that spread over the top of matrix, producing a large portion of cloud genes. Those genes were mostly predicted as hypothetical proteins while the annotated genes were diverse and could be found in several organisms. For example, many genes commonly found in Bacillus subtilis, Escherichia coli, Lactococcus lactis and other bacteria, were also identified among the cloud genes, e.g., adhR, mhqA, rhaS, and lcnD. These gene families might be acquired from microorganisms encountered by Weissella spp. during their evolution in a diverse range of ecological niches.
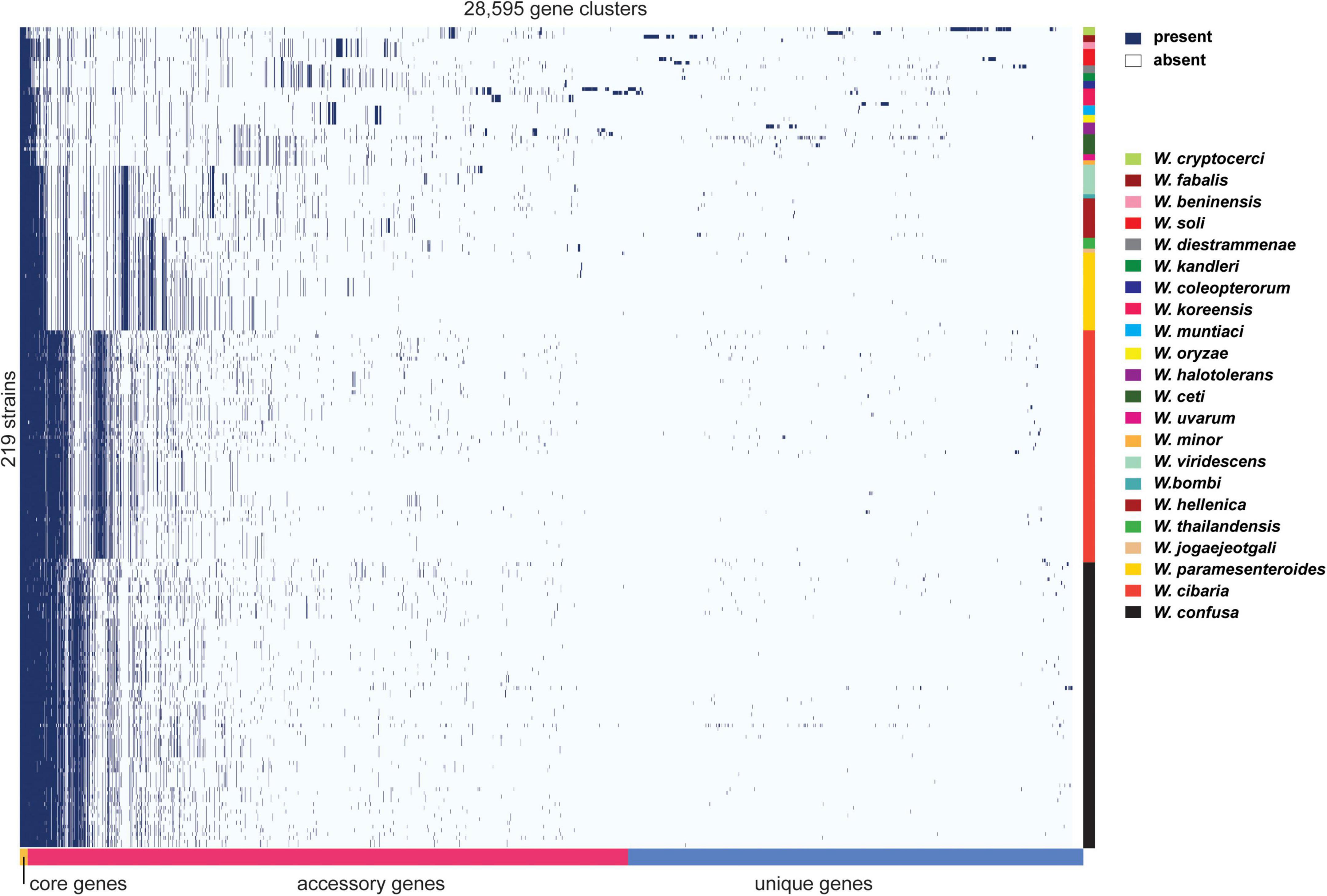
Figure 5. Pan-genome matrix of 219 Weissella genomes from 22 species. The right-hand color bars indicate the species boundary in the matrix. Lime, cherry, rose, red, gray, emerald, dark blue, pink, blue, yellow, violet, basil, magenta, cream, mint, teal, apple, green, wood, gold, orange, and black colors (from top to bottom) represent W. cryptocerci, W. fabalis, W. beninensis, W. soli, W. diestrammenae, W. kandleri, W. coleopterorum, W. koreensis, W. muntiaci, W. oryzae, W. halotolerans, W. ceti, W. uvarum, W. minor, W. viridescens, W. bombi, W. hellenica, W. thailandensis, W. jogaejeotgali, W. paramesenteroides, W. cibaria, and W. confusa, respectively.
At the species level, W. cibaria had 1,187 core genes, 344 soft core genes, 1,403 shell genes and 3,880 cloud genes. In addition, the presence/absence matrix showed a big portion of homologous genes that were present in the cluster containing W. cibaria and W. confusa. These two species, which were the closest among all the species, shared 747 core genes and 146 soft core genes. Ongoing HGT and gene loss may have helped one of the two evolve from the other (Polz et al., 2013). Unsurprisingly, we found that 40, 30, and 10% of core genes of W. cibaria and W. confusa were, respectively, assigned into the “Metabolism,” “Information storage and processing,” “Cellular processes and signaling” categories of COG classification. Most core genes were classified in several subcategories, e.g., “Translation, ribosomal structure and biogenesis (J)”, “Amino acid transport and metabolism (E),” and “Carbohydrate transport and metabolism (G).” The three COG categories accessory genes were most commonly assigned to were “Cell wall/membrane/envelope biogenesis (M),” “Replication, recombination and repair (L),” and “Carbohydrate transport and metabolism (G).” We found that core genes and accessory genes both contained a high abundance of genes involved in carbohydrate transport and metabolism. We then further identified those genes using the CAZymes database to explore more related functions in Weissella spp. As a result, we found that W. cibaria NH9449 encodes a total of 105 CAZyme domain sequences in five families, including Glycoside Hydrolase (GH), Glycosyl Transferase (GT), Carbohydrate Esterase (CE), Auxiliary Activity (AA), and Carbohydrate-Binding Module (CBM) (Figure 6A). The highest frequency of CAZyme predicted in the NH9449 genome was found in the GH family (43) followed by the GT family (33). This investigation could indicate the ability of this bacterial strain to metabolize carbohydrate, which could be beneficial in several biotechnological applications. We investigated this characteristic among all members of the genus Weissella, by searching all strains against the CAZyme database. W. cibaria 85 and W. viridescens NCTC13645 presented the highest and the lowest number of CAZyme domain sequences of 112, and 21, respectively. The average numbers of CAZyme genes identified in each Weissella species are presented in Figure 6B. W. cibaria (101), W. confusa (88), and W. jogaejeotgali (83) are the three species that encode most CAZyme genes in their genomes. Our identification results are consistent with those of a previous study (Yuan et al., 2021), which found that W. confusa and W. cibaria metabolize carbohydrates and could be beneficial in several applications. These findings could also imply that most Weissella species, and especially W. cibaria and W. confusa, specialize in carbohydrate metabolism, which helps control mass degradation in the fermentation process.
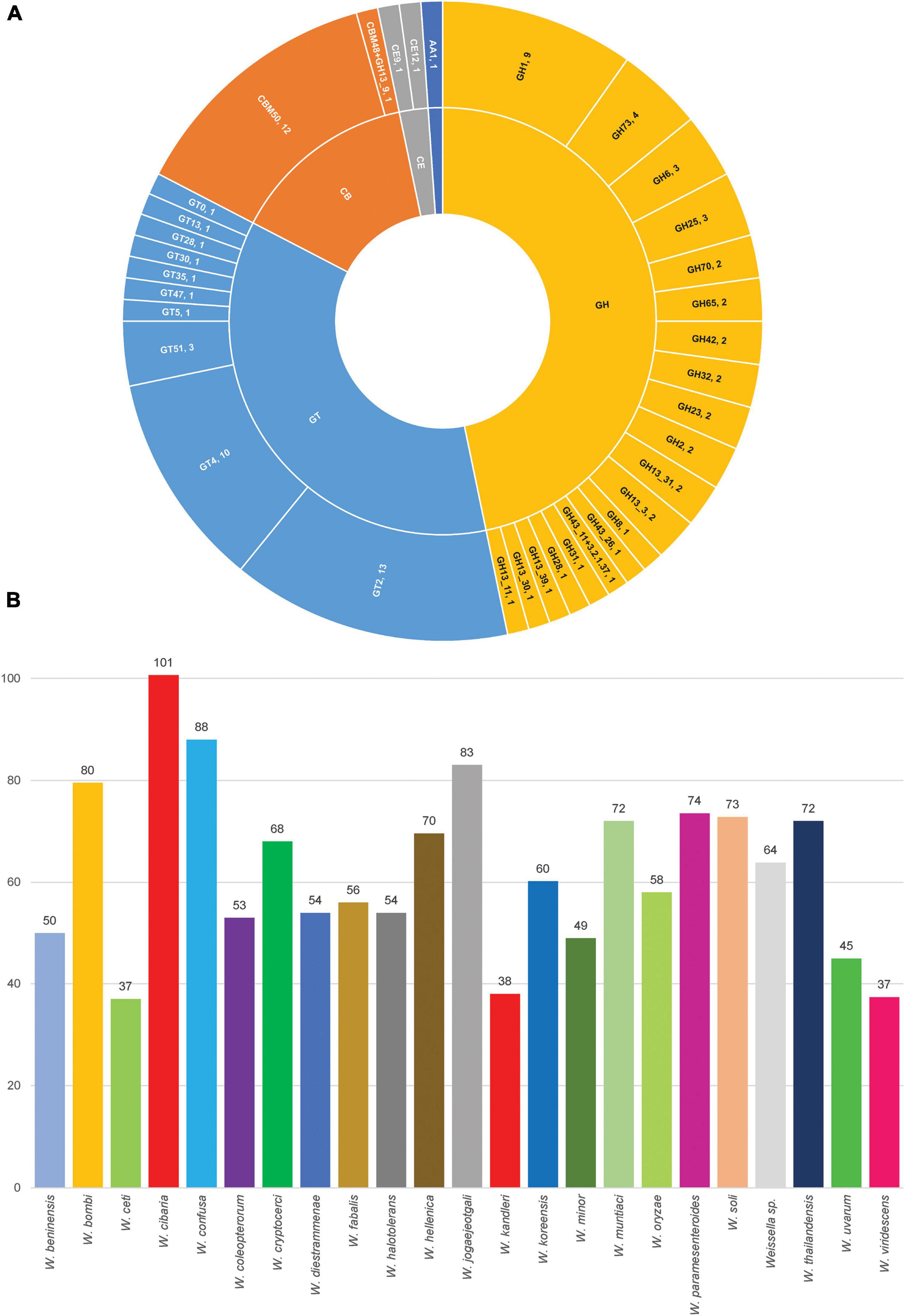
Figure 6. Carbohydrate-active enzyme analysis. (A) CAZymes distribution in W. cibaria NH9449. (B) Average count of CAZymes distribution in Weissella spp.
The openness of the pan-genome of the genus Weissella was apparent from the pan-genome analysis (Supplementary Figure 5A). When more genomes were included in the analysis, the pan-genome plots of W. cibaria strains tended to be closed (Supplementary Figure 5B). Therefore, sequencing more W. cibaria strains might not be useful since the chance of discovering new genetic elements is probably slim. However, W. cibaria was not the only species whose pan-genome showed a tendency to be closed. In a previous report (Yuan et al., 2021), the pan-genome of 39 W. confusa strains was also open but in this study, when we analyzed 77 strains of W. confusa, the pan-genome status tended to be closed. In addition, we observed that the core genome sizes of W. cibaria strains seemed to be stable after sequencing around 50 genomes including ≈ 1,200 genes (Supplementary Figure 5B). At the genus level, the core genome plot of Weissella reached the stable stage after sampling around 150 genomes at ≈ 165 genes (Supplementary Figure 5A). However, when more strains were added, the core genome size of the genus remains very small while the pan-genome size increases. We suggest that sequencing other Weissella species would be more likely to reveal new gene families and there are many genomes of other species that have not yet been fully investigated.
The phylogenetic analyses were constructed based on core genes (Figure 7) and accessory genes (Supplementary Figure 6) of all Weissella strains. The NH9449 was grouped in the same clade as W. cibaria strains 7.8.34, BM2, 142, 85, 92, and AV1, respectively, implying that these bacterial strains shared close genetic components since they were all isolated from the same ecological niche. Furthermore, both phylogenetic trees formed a distinct clade for each Weissella species, enabling their taxonomic classification in this genus. We found that some bacterial strains were misplaced in the trees. This finding was consistent with our AAI analysis. Notably, W. hellenica, W. viridescens, and W. ceti branches were divided into two distinct clades and showed AAI similarities below the species criterion (95%) (Table 3). W. hellenica formed two distinct groups in both phylogenetic trees, based on core genes and accessory genes. W. hellenica CCUG 33494 (representative strain) grouped with strains NBRC 15553 and R-53116. In contrast, in the other clade, W. hellenica strains Wikim14, CBA3632, 0916-4-2, and 1.2.50 grouped in the same branch together with W. paramesenteroides strains DmW_118 and DmW_118119, and Weissella sp. X0278, X0401, and X0750. Since the similarity of 16S rRNA sequences of Weissella spp. are very close, previous taxonomic classifications could have been directly affected. So, we suggest that these two W. paramesenteroides strains and three Weissella sp. could be reclassified as W. hellenica. Based on our AAI calculations and proposed phylogenetic tree analysis, we suggest that the findings of this analysis might lead to the discovery of new species/subspecies in the genus Weissella. Also, using only the 16S rRNA marker might not be enough to classify species in this genus, and more markers could be considered for a better resolution in the taxonomic classification.

Figure 7. The phylogenetic tree is based on the core genes of 219 Weissella genomes.
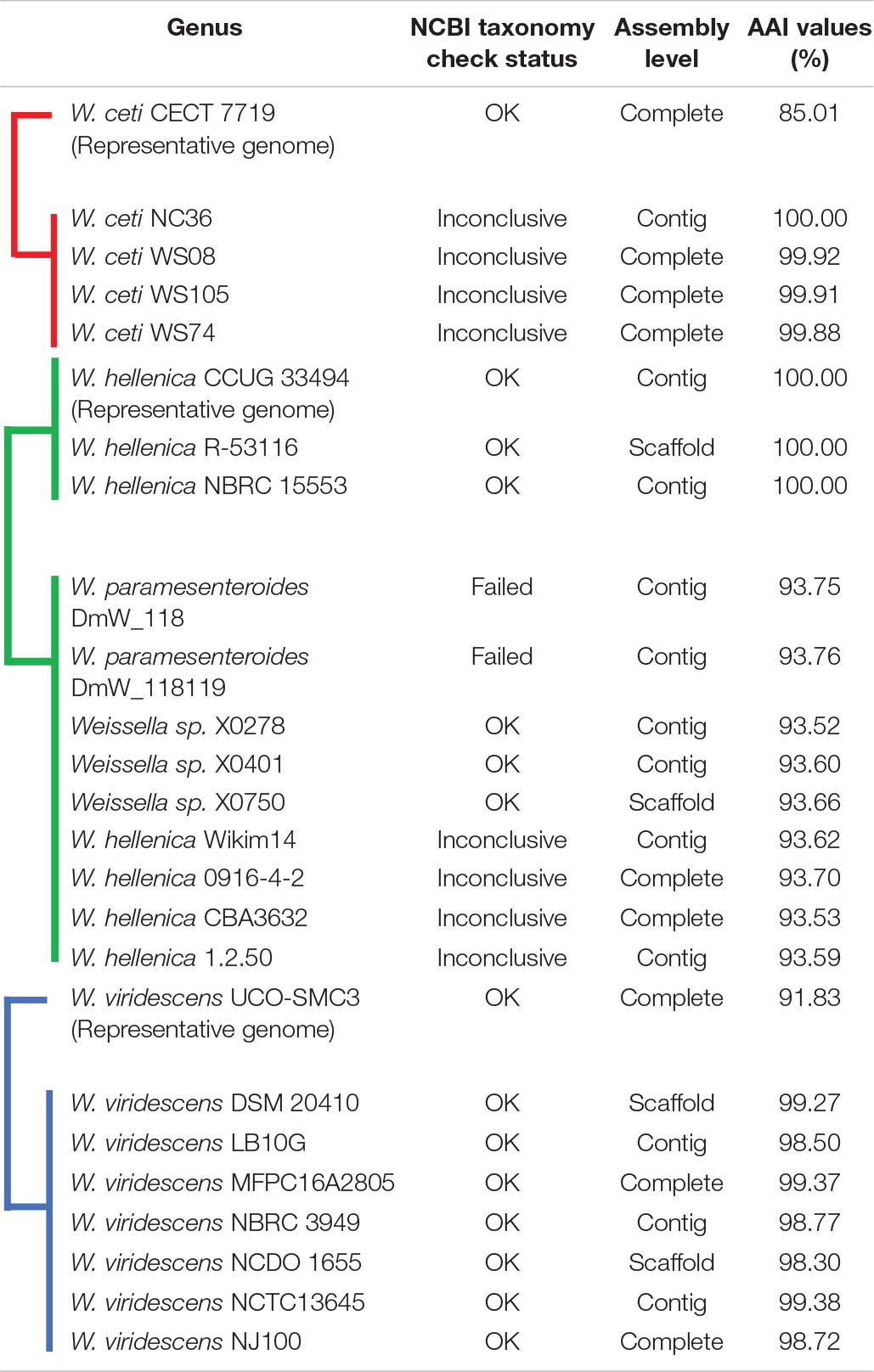
Table 3. NCBI taxonomy status and AAI values.
Bacteriocin Diversity in Weissella spp.
Bacteriocins are ribosomally synthesized antibacterial peptides/proteins produced by bacteria to inhibit the growth of other bacterial strains (De Vuyst and Leroy, 2007). Since LAB contain several bacteriocin-encoding genes in many bacterial strains (Riley and Wertz, 2002), they have been deployed to perform important functions in microbiology applications. Therefore, we looked to identify the bacteriocin-encoding genes in all Weissella strains based on a BLASTx analysis against the bacteriocin database and the BAGEL server. We found no bacteriocin-encoding gene in the NH9449 genome and that other Weissella spp. encoded bacteriocin-encoding genes only in classes I and III. In class I, we found only two strains that contained bacteriocin-encoding genes. These strains were W. cryptocerci 26KH-42 (nukacinISK-1 and lactococcinDR) and W. cibaria 1001713B170221_170320_B3 (cytolysin_ClyLs). We did not identify any class II bacteriocin in any strain. Surprisingly, weisselin A (Papagianni and Papamichael, 2012), weissellicin M and Y (Masuda et al., 2012), which were originally W. paramesenteroides DX and W. hellenica QU13, were not found in any Weissella strain. In class III, eight strains from three species, including W. thailandensis (4), W. soli (3), and W. minor (1), were found to encode large protein bacteriocins (Table 4). In addition, weissellicin 110 (Srionnual et al., 2007), which was not included in the BAGEL database, was identified in only four strains of W. cibaria. This analysis clarified the frequency of bacteriocin-encoding genes in Weissella spp. We found that only a few bacterial strains in the genus encode ribosomally synthesized antibacterial peptides, while LAB in other genera encode a larger diversity of bacteriocin-encoding genes: for example, Pediococcus (Jiang et al., 2020; Surachat et al., 2021b), Lactobacillus (Kaskoniene et al., 2017; Surachat et al., 2021a), and Enterococcus (Nes et al., 2007; Lopez-Cuellar et al., 2016). Even though bacteriocins are a crucial biological tool for inhibiting pathogens and other microorganisms, most Weissella spp. use genomic traits such as CRISPR and R-M systems to maintain their stability.
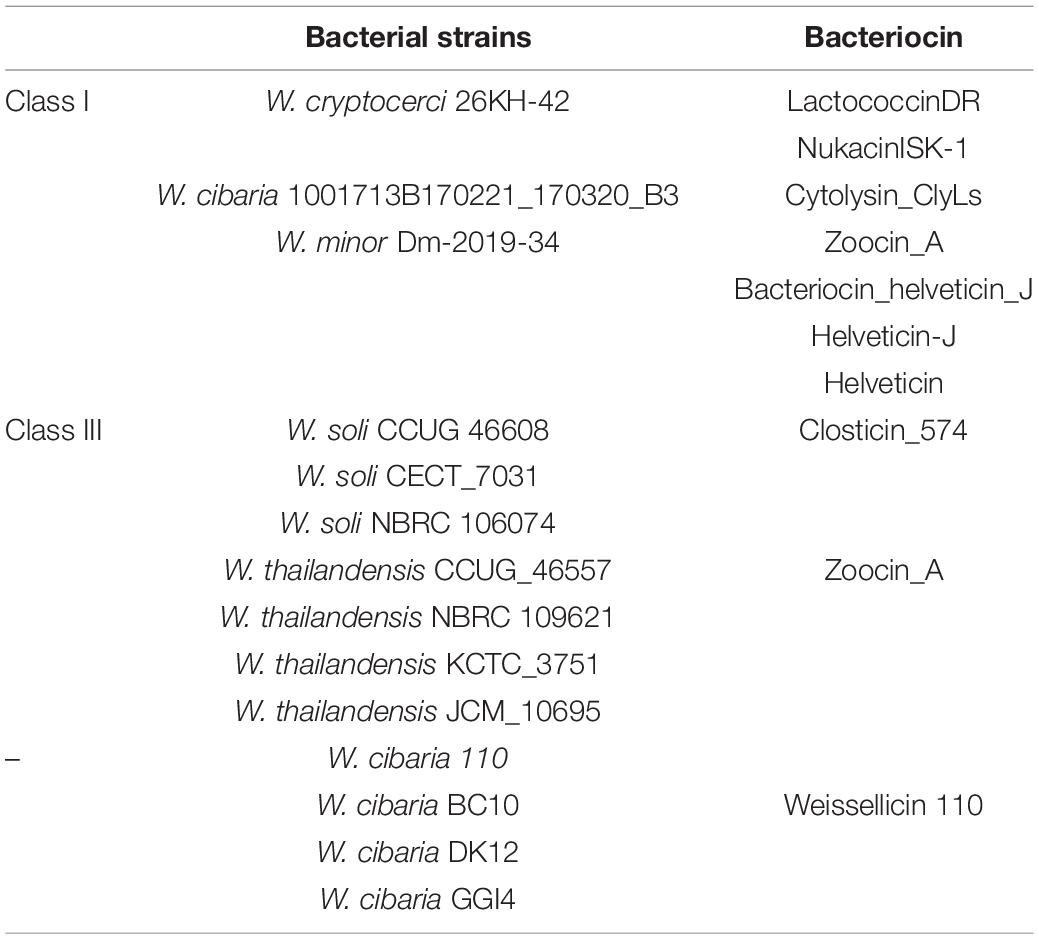
Table 4. Bacteriocin-encoding genes identified in Weissella spp.
CRISPR-Cas and R-M Systems
We explored CRISPR-Cas regions and R-M systems in all bacterial strains used in this study (Supplementary Figure 7 and Supplementary Table 3). From the CRISPR-Cas prediction, we found that 94.5 and 68.5% of all strains, respectively, contained the CRISPR array and the cas locus in their genome. Most of the cas loci were detected with CRISPR arrays, while some of them were detected without CRISPR arrays. Overall, type I-E, II-A, III-A, and III-C CRISPR-Cas systems were identified in 2, 36, 1, and 4 Weissella genomes, respectively, whereas we could not identify the subtype of type I CRISPR-Cas system in most of the Weissella genomes (n = 160). The combinations of type I and II-A (9.6%), type I and III-C (1.8%), and type I-E, II-A, and III-A (0.5%) CRISPR-Cas systems were also identified in these Weissella genomes. A CRISPR region at 2,231,486 to 2,231,580 (94 bp) with a DR length of 23 bp was found in the NH9449 genome. For R-M prediction, we found that 76.3, 66.0, 30.6, and 26.3% of all strains contained type I, II, III, and IV R-M systems, respectively. Three types of R-M systems, I, II, and III, were predicted in W. cibaria NH9449. We did not find any type V R-M system in any strain in this analysis. This finding might indicate that these Weissella genomes have been infected with bacteriophages. Furthermore, it could imply that the presence of CRISPR-Cas and R-M regions in most Weissella genomes probably provides adaptive immunity against foreign genetic elements from bacteriophages or extracellular plasmids (Abriouel et al., 2017; Xing et al., 2017).
Antimicrobial Resistance and Virulence Factors in the Genus Weissella
To assess antimicrobial resistance (AMR), we investigated the presence of AMR genes in all 219 Weissella strains. An AMR gene was found only in W. cibaria 142 and W. cibaria AV1, which, respectively, encoded the blaTEM–1A and blaTEM–162 genes. These 2 β-lactamase genes may provide resistance to ampicillin. Thus, we propose that contamination with blaTEM-carrying strains should be a concern in food production. Additionally, we still need to clarify how these AMR genes are acquired as well as the location of the genes. If AMR genes are located on mobile genetic elements (such as plasmids), they can be transferred to other Weissella spp., especially pathogenic species. This study identified the ColRNAI plasmid in W. cibaria 92. The ColRNAI plasmid is predominantly found in Klebsiella pneumoniae, especially multidrug- and carbapenem-resistant strains (Dong et al., 2018; Agyepong et al., 2019; Yu et al., 2019; Li R. C. et al., 2020) and has also been detected in Escherichia coli and Salmonella spp. (Litrup et al., 2017; Hadziabdic et al., 2018). Therefore, the discovery of the ColRNAI plasmid in W. cibaria 92 implies acquisition from K. pneumoniae, Escherichia coli, or Salmonella spp. Previous studies have reported that the ColRNAI plasmid carries many AMR genes, including the carbapenem resistance gene (blaKPC–2) and colistin resistance gene (mcr-3) as well as other AMR genes (blaCTX–M–55, aac(3)-IId, tet(A), qnrS1, sul2, catA2, floR, strA) (Litrup et al., 2017; Hadziabdic et al., 2018; Agyepong et al., 2019; Yu et al., 2019; Shelenkov et al., 2020). Although no AMR gene was identified in this ColRNAI plasmid, we still need to be concerned about this ColRNAI-harboring strain. Since neither AMR genes nor plasmids were detected in W. cibaria NH9449 in our study, the NH9449 may not be harmful to humans and can perhaps be used in biotechnological applications.
Virulence-related factors in the Weissella genomes were identified based on a BLASTn search against the full dataset of virulence factors in the VFDB database. We found 27 matches of putative virulence factor genes in the NH9449 genome, including tufA (elongation factor Tu), lisR (two-component response regulator), hasC (DP-glucose pyrophosphorylase) and SMU_322c (glucose-1-phosphate uridylyltransferase). These genes were previously reported in W. hellenica 0916-4-2 (Panthee et al., 2019), and W. cibaria UTNGt21O (Tenea and Hurtado, 2021). W. confusa FS54 encodes 133 virulence-associated genes, which is the highest number in this genus. The average number of occurrences of virulence factors of all members was found to be only 23, which were almost the same putative virulence factor genes that we found in NH9449. These results show that most Weissella genomes encode common virulence-associated genes which have been widely identified in Lactobacillus, Pediococcus, Enterococcus and other LAB species (Muñoz-Atienza et al., 2013; Chajęcka-Wierzchowska et al., 2017; Colautti et al., 2022). However, further study should be conducted to explore the functions and characteristics of these virulence genes.
Conclusion
The complete genome of W. cibaria NH9449 was sequenced and a comparative genomic analysis was performed against all members in the genus Weissella. We found several self-defense mechanisms in the NH9449 genome, including CRISPR-Cas and R-M systems. Also, the significantly higher abundance of CAZyme encoding genes in this bacterial strain compared to other Weissella strains indicates its greater ability to metabolize carbohydrate. The absence of virulence factors and AMR genes in the NH9449 genome imply that the strain could be safe for use in food processing. Moreover, the comparative genomic analysis revealed an extremely high genomic diversity among this genus and a high variation in the genome content. Besides W. cibaria NH9449, several other Weissella spp. did not exhibit AMR genes or harmful virulence factors and could possibly be considered as safe. Some strains produce antimicrobial substances that can inhibit a broad spectrum of pathogens, and we found that Weissella species, especially W. cibaria and W. confusa, carried several CAZyme genes. These properties could be useful for biotechnological applications, especially in food fermentation and probiotics. However, these bacterial strains have to be carefully selected and tested before use since Weissella spp. are still not Generally Recognized As Safe (GRAS). Furthermore, the results of AAI and phylogenetic analyses suggest that several Weissella spp. should be reclassified and that reclassification might uncover new species of W. ceti, W. hellenica and W. viridescens. However, further investigations are still needed to confirm this assumption. This study provides new information about W. cibaria NH9449 and the genomic diversity among Weissella spp. and could be a guideline for future studies.
Data Availability Statement
The complete chromosome and six plasmids of W. cibaria NH9449 have been deposited at GenBank with accession numbers CP061503–CP061509. The BioProject and BioSample accession numbers are PRJNA663405 and SAMN16131644, respectively. The sequence raw reads of short reads and long reads were deposited in SRA with the respective accession numbers SRR18492148 and SRR18492149.
Author Contributions
KS, DK, and MW designed the study. KS and PD performed the genome sequencing and annotation. KS and KJ performed the comparative genomic analysis. KS, DK, PM-A, AC, and MW wrote the manuscript. DK and KS integrated the research and critically revised the manuscript for important intellectual content. KS, DK, PM-A, and MW approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the National Science, Research and Innovation Fund (NSRF) and Prince of Songkla University (Grant No. SCI6505013S) and the Faculty of Science, Prince of Songkla University, Thailand (Grant No. SCI64040135).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the Medical Science Research and Innovation Institute (MSRI), Prince of Songkla University, for providing W. cibaria NH9449 short-read sequences by the NextSeq 550 Sequencing System.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.826683/full#supplementary-material
Supplementary Figure 1 | PCR amplification of plasmid size 1,526 bp.
Supplementary Figure 2 | The chart shows the number of genomes of each Weissella sp. used in this study.
Supplementary Figure 3 | In the results of the ANI analysis of 219 Weissella strains, similarity levels are represented by red, orange, yellow, lime, and green. Below and including 60% similarity is represented by red, 61 to 70% by orange, 71 to 80% by yellow, 81 to 90% by lime, and 91 to 100% by green.
Supplementary Figure 4 | In the AAI analysis of 219 Weissella strains, similarity levels from low to high are represented by red, orange, yellow, lime, and green.
Supplementary Figure 5 | The plots were produced from the pan-genome analysis at the genus (A) and species (B) level. The plots show the number of genes found when adding more genomes in the dataset of the genus and the dataset of W. cibaria strains only.
Supplementary Figure 6 | The phylogenetic tree is based on accessory genes of 219 Weissella genomes.
Supplementary Figure 7 | The gene presence/absence matrix of R-M system related genes, CRISPR-cas systems, and bacteriocins was produced from analysis of all the strains studied in this work.
Supplementary Table 1 | List of bacterial strains used in this study.
Supplementary Table 2 | AAI Analysis of 219 Weissella strains.
Supplementary Table 3 | CRISPR identification in Weissella strains.
Footnotes
References
Abriouel, H., Perez Montoro, B., Casado Munoz, M. D. C., Knapp, C. W., Galvez, A., and Benomar, N. (2017). In silico genomic insights into aspects of food safety and defense mechanisms of a potentially probiotic lactobacillus pentosus MP-10 isolated from brines of naturally fermented alorena green table olives. PLoS One 12:e0176801. doi: 10.1371/journal.pone.0176801
Agyepong, N., Govinden, U., Owusu-Ofori, A., Amoako, D. G., Allam, M., Janice, J., et al. (2019). Genomic characterization of multidrug-resistant ESBL-producing Klebsiella pneumoniae isolated from a ghanaian teaching hospital. Int. J. Infect. Dis. 85, 117–123. doi: 10.1016/j.ijid.2019.05.025
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bjorkroth, K. J., Schillinger, U., Geisen, R., Weiss, N., Hoste, B., Holzapfel, W. H., et al. (2002). Taxonomic study of Weissella confusa and description of Weissella cibaria sp. nov., detected in food and clinical samples. Int. J. Syst. Evol. Microbiol. 52, 141–148. doi: 10.1099/00207713-52-1-141
Bobay, L. M., and Ochman, H. (2017). The evolution of bacterial genome architecture. Front Genet 8:72. doi: 10.3389/fgene.2017.00072
Bortolaia, V., Kaas, R. S., Ruppe, E., Roberts, M. C., Schwarz, S., Cattoir, V., et al. (2020). ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob Chemother 75, 3491–3500. doi: 10.1093/jac/dkaa345
Carattoli, A., and Hasman, H. (2020). PlasmidFinder and In Silico pMLST: Identification and typing of plasmid replicons in whole-genome sequencing (WGS). Methods Mol. Biol. 2075, 285–294. doi: 10.1007/978-1-4939-9877-7_20
Carlosama Adriana, M., Rodriguez Misael, C., Londono Guillermo, C., Sanchez Fernando, O., and Cock Liliana, S. (2021). Optimization of the reproduction of Weissella cibaria in a fermentation substrate formulated with agroindustrial waste. Biotechnol. Rep. (Amst) 32, e00671. doi: 10.1016/j.btre.2021.e00671
Chajęcka-Wierzchowska, W., Zadernowska, A., and Łaniewska-Trokenheim, Ł (2017). Virulence factors of Enterococcus spp. presented in food. LWT 75. Poland: University of Warmia and Mazury, 670–676.
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Ciufo, S., Kannan, S., Sharma, S., Badretdin, A., Clark, K., Turner, S., et al. (2018). Using average nucleotide identity to improve taxonomic assignments in prokaryotic genomes at the NCBI. Int. J. Syst. Evol. Microbiol. 68, 2386–2392. doi: 10.1099/ijsem.0.002809
Colautti, A., Arnoldi, M., Comi, G., and Iacumin, L. (2022). Antibiotic resistance and virulence factors in lactobacilli: something to carefully consider. Food Microbiol 103, 103934. doi: 10.1016/j.fm.2021.103934
De Vuyst, L., and Leroy, F. (2007). Bacteriocins from lactic acid bacteria: production, purification, and food applications. J Mol Microbiol Biotechnol 13, 194–199. doi: 10.1159/000104752
Dong, N., Yang, X. M., Zhang, R., Chan, E. W. C., and Chen, S. (2018). Tracking microevolution events among ST11 carbapenemase-producing hypervirulent Klebsiella pneumoniae outbreak strains. Emerg. Microbes Infect. 7, 146. doi: 10.1038/s41426-018-0146-6
Fairfax, M. R., Lephart, P. R., and Salimnia, H. (2014). Weissella confusa: problems with identification of an opportunistic pathogen that has been found in fermented foods and proposed as a probiotic. Front. Microbiol. 5:254. doi: 10.3389/fmicb.2014.00254
Falasconi, I., Fontana, A., Patrone, V., Rebecchi, A., Duserm Garrido, G., Principato, L., et al. (2020). Genome-assisted characterization of lactobacillus fermentum, Weissella cibaria, and Weissella confusa strains isolated from sorghum as starters for sourdough fermentation. Microorganisms 8:1388. doi: 10.3390/microorganisms8091388
Figueiredo, H. C., Leal, G., Pereira, F. L., Soares, S. C., Dorella, F. A., Carvalho, A. F., et al. (2014b). Whole-genome sequence of Weissella ceti strain WS08, isolated from diseased rainbow trout in Brazil. Genome Announc 2, e851–e814. doi: 10.1128/genomeA.00851-14
Figueiredo, H. C., Leal, C. A., Dorella, F. A., Carvalho, A. F., Soares, S. C., Pereira, F. L., et al. (2014a). Complete genome sequences of fish pathogenic Weissella ceti strains WS74 and WS105. Genome Announc 2, e1014–e1014. doi: 10.1128/genomeA.01014-14
Fusco, V., Quero, G. M., Cho, G. S., Kabisch, J., Meske, D., Neve, H., et al. (2015). The genus Weissella: taxonomy, ecology and biotechnological potential. Front. Microbiol. 6:155. doi: 10.3389/fmicb.2015.00155
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hadziabdic, S., Fischer, J., Malorny, B., Borowiak, M., Guerra, B., Kaesbohrer, A., et al. (2018). In vivo transfer and microevolution of avian native IncA/C2blaNDM-1-carrying plasmid pRH-1238 during a broiler chicken infection study. Antimicrob Agents Chemother 62, e2128–e2117. doi: 10.1128/AAC.02128-17
Heo, J., Hamada, M., Cho, H., Weon, H. Y., Kim, J. S., Hong, S. B., et al. (2019). Weissella cryptocerci sp. nov., isolated from gut of the insect cryptocercus kyebangensis. Int. J. Syst. Evol. Microbiol. 69, 2801–2806. doi: 10.1099/ijsem.0.003564
Huang, L., Cui, K., Mao, W., Du, Y., Yao, N., Li, Z., et al. (2020). Weissella cibaria attenuated lps-induced dysfunction of intestinal epithelial barrier in a caco-2 cell monolayer model. Front. Microbiol. 11:2039. doi: 10.3389/fmicb.2020.02039
Jain, C., Rodriguez, R. L., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114.
Jiang, J., Yang, B., Ross, R. P., Stanton, C., Zhao, J. X., Zhang, H., et al. (2020). Comparative genomics of pediococcus pentosaceus isolated from different niches reveals genetic diversity in carbohydrate metabolism and immune system. Front. Microbiol. 11:253. doi: 10.3389/fmicb.2020.00253
Kang, B. K., Cho, M. S., and Park, D. S. (2016). Red pepper powder is a crucial factor that influences the ontogeny of Weissella cibaria during kimchi fermentation. Sci. Rep. 6:28232. doi: 10.1038/srep28232
Kang, M. S., Lee, D. S., Lee, S. A., Kim, M. S., and Nam, S. H. (2020). Effects of probiotic bacterium Weissella cibaria CMU on periodontal health and microbiota: a randomised, double-blind, placebo-controlled trial. BMC Oral Health 20:243. doi: 10.1186/s12903-020-01231-2
Kaskoniene, V., Stankevicius, M., Bimbiraite-Surviliene, K., Naujokaityte, G., Serniene, L., Mulkyte, K., et al. (2017). Current state of purification, isolation and analysis of bacteriocins produced by lactic acid bacteria. Appl Microbiol Biotechnol 101, 1323–1335. doi: 10.1007/s00253-017-8088-9
Kim, D., Park, S., and Chun, J. (2021). Introducing EzAAI: a pipeline for high throughput calculations of prokaryotic average amino acid identity. J. Microbiol. 59, 476–480. doi: 10.1007/s12275-021-1154-0
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol.Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kwak, M. J., Choi, S. B., Kim, B. Y., and Chun, J. (2019). Genome-based reclassification of weissella jogaejeotgali as a later heterotypic synonym of Weissella thailandensis. Int. J. Syst. Evol. Microbiol. 69, 3672–3675. doi: 10.1099/ijsem.0.003315
Kwak, S. H., Cho, Y. M., Noh, G. M., and Om, A. S. (2014). Cancer preventive potential of kimchi lactic acid bacteria (Weissella cibaria). (Lactobacillus plantarum). J. Cancer. Prev. 19, 253–258. doi: 10.15430/JCP.2014.19.4.253
Ladner, J. T., Welch, T. J., Whitehouse, C. A., and Palacios, G. F. (2013). Genome sequence of Weissella ceti NC36, an emerging pathogen of farmed rainbow trout in the United States. Genome Announc 1, e187–e112. doi: 10.1128/genomeA.00187-12
Lee, I. P. A., Eldakar, O. T., Gogarten, J. P., and Andam, C. P. (2022). Bacterial cooperation through horizontal gene transfer. Trends Ecol. Evol. 37, 223–232. doi: 10.1016/j.tree.2021.11.006
Li, Y. Q., Tian, W. L., and Gu, C. T. (2020). Weissella sagaensis sp. nov., isolated from traditional chinese yogurt. Int. J. Syst. Evol. Microbiol. 70, 2485–2492. doi: 10.1099/ijsem.0.004062
Li, R. C., Cheng, J., Dong, H. Y., Li, L. Y., Liu, W. T., Zhang, C. L., et al. (2020). Emergence of a novel conjugative hybrid virulence multidrug-resistant plasmid in extensively drug-resistant Klebsiella pneumoniae ST15. Int J Antimicrob Agents. 55, 105952. doi: 10.1016/j.ijantimicag.2020.105952
Li, X. Q., and Du, D. (2014). Variation, evolution, and correlation analysis of C+G content and genome or chromosome size in different kingdoms and phyla. PLoS One 9:e88339. doi: 10.1371/journal.pone.0088339
Litrup, E., Kiil, K., Hammerum, A. M., Roer, L., Nielsen, E. M., and Torpdahl, M. (2017). Plasmid-borne colistin resistance gene mcr-3 in Salmonella isolates from human infections, denmark, 2009-17. Euro. Surveill 22:30587. doi: 10.2807/1560-7917.ES.2017.22.31.30587
Liu, B., Zheng, D. D., Jin, Q., Chen, L. H., and Yang, J. (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Lombard, V., Ramulu, H. G., Drula, E., Coutinho, P. M., and Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. doi: 10.1093/nar/gkt1178
Lopez-Cuellar, M. D., Rodriguez-Hernandez, A. I., and Chavarria-Hernandez, N. (2016). LAB bacteriocin applications in the last decade. Biotechnol. Biotechnol. Equip. 30, 1039–1050. doi: 10.1080/13102818.2016.1232605
Masuda, Y., Zendo, T., Sawa, N., Perez, R. H., Nakayama, J., and Sonomoto, K. (2012). Characterization and identification of weissellicin y and weissellicin m, novel bacteriocins produced by Weissella hellenica QU 13. J. Appl. Microbiol. 112, 99–108. doi: 10.1111/j.1365-2672.2011.05180.x
Muñoz-Atienza, E., Gómez-Sala, B., Araújo, C., Campanero, C., Del Campo, R., Hernández, P. E., et al. (2013). Antimicrobial activity, antibiotic susceptibility and virulence factors of Lactic acid bacteria of aquatic origin intended for use as probiotics in aquaculture. BMC Microbiol. 13:15. doi: 10.1186/1471-2180-13-15
Nes, I. F., Diep, D. B., and Holo, H. (2007). Bacteriocin diversity in streptococcus and enterococcus. J. Bacteriol. 189, 1189–1198. doi: 10.1128/JB.01254-06
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Page, A. J., Taylor, B., Delaney, A. J., Soares, J., Seemann, T., Keane, J. A., et al. (2016). SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microbial Genomics 2:e000056. doi: 10.1099/mgen.0.000056
Panthee, S., Paudel, A., Blom, J., Hamamoto, H., and Sekimizu, K. (2019). Complete genome sequence of Weissella hellenica 0916-4-2 and its comparative genomic analysis. Front. Microbiol. 10:1619. doi: 10.3389/fmicb.2019.01619
Papagianni, M., and Papamichael, E. M. (2012). Production of the antimicrobial protein weisselin a by Weissella paramesenteroides dx in batch fermentations: the type of carbohydrate used as the C-source in the substrate affects the association of production with growth. Appl. Biochem. Biotechnol 168, 1212–1222. doi: 10.1007/s12010-012-9851-0
Poirier, S., Coeuret, G., Champomier-Verges, M. C., and Chaillou, S. (2018). Draft genome sequences of nine strains of Brochothrix thermosphacta, Carnobacterium divergens, Lactobacillus algidus, lactobacillus fuchuensis, Lactococcus piscium, Leuconostoc gelidum subsp. gasicomitatum, Pseudomonas lundensis, and Weissella viridescens, a collection of psychrotrophic species involved in meat and seafood spoilage. Genome Announc 6:e479–e418. doi: 10.1128/genomeA.00479-18
Polz, M. F., Alm, E. J., and Hanage, W. P. (2013). Horizontal gene transfer and the evolution of bacterial and archaeal population structure. Trends Genet 29, 170–175. doi: 10.1016/j.tig.2012.12.006
Ricciardi, A., Parente, E., and Zotta, T. (2009). Modelling the growth of Weissella cibaria as a function of fermentation conditions. J. Appl. Microbiol. 107, 1528–1535. doi: 10.1111/j.1365-2672.2009.04335.x
Riley, M. A., and Wertz, J. E. (2002). Bacteriocins: evolution, ecology, and application. Annu. Rev. Microbiol. 56, 117–137. doi: 10.1146/annurev.micro.56.012302.161024
Roberts, R. J., Vincze, T., Posfai, J., and Macelis, D. (2015). REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 43, D298–D299.
Seppey, M., Manni, M., and Zdobnov, E. M. (2019). BUSCO: assessing genome assembly and annotation completeness. Methods Mol Biol 1962, 227–245. doi: 10.1007/978-1-4939-9173-0_14
Shelenkov, A., Mikhaylova, Y., Yanushevich, Y., Samoilov, A., Petrova, L., Fomina, V., et al. (2020). molecular typing, characterization of antimicrobial resistance, virulence profiling and analysis of whole-genome sequence of clinical Klebsiella pneumoniae isolates. Antibiotics (Basel) 9:261. doi: 10.3390/antibiotics9050261
Shin, J. I., Oh, S. G., Bang, M. S., Jeong, H. W., Lee, Y. J., Lee, S. C., et al. (2020). Complete genome sequence of Weissella cibaria strain BM2, isolated from korean kimchi. Microbiol. Resour Announc 9, e534–e520. doi: 10.1128/MRA.00534-20
Srionnual, S., Yanagida, F., Lin, L. H., Hsiao, K. N., and Chen, Y. S. (2007). Weissellicin 110, a newly discovered bacteriocin from Weissella cibaria 110, isolated from plaa-som, a fermented fish product from Thailand. Appl. Env. Microbiol. 73, 2247–2250. doi: 10.1128/AEM.02484-06
Stamatakis, A. (2006). RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Stothard, P., Grant, J. R., and Van Domselaar, G. (2019). Visualizing and comparing circular genomes using the CGView family of tools. Brief. Bioinformatics 20, 1576–1582. doi: 10.1093/bib/bbx081
Surachat, K., Kantachote, D., Deachamag, P., and Wonglapsuwan, M. (2021b). Genomic insight into Pediococcus acidilactici HN9, a potential probiotic strain isolated from the traditional thai-style fermented beef nhang. Microorganisms 9, 1–23. doi: 10.3390/microorganisms9010050
Surachat, K., Deachamag, P., Kantachote, D., Wonglapsuwan, M., Jeenkeawpiam, K., and Chukamnerd, A. (2021a). In silico comparative genomics analysis of Lactiplantibacillus plantarum DW12, a potential gamma-aminobutyric acid (GABA)-producing strain. Microbiol. Res. 251:126833. doi: 10.1016/j.micres.2021.126833
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. doi: 10.1186/1471-2105-4-41
Tatusova, T., Dicuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Tenea, G. N., and Hurtado, P. (2021). Next-generation sequencing for whole-genome characterization of Weissella cibaria UTNGt21O strain originated from wild Solanum quitoense Lam. fruits: an atlas of metabolites with biotechnological significance. Front. Microbiol. 12:675002. doi: 10.3389/fmicb.2021.675002
Van Heel, A. J., De Jong, A., Song, C. X., Viel, J. H., Kok, J., and Kuipers, O. P. (2018). BAGEL4: a user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 46, W278–W281. doi: 10.1093/nar/gky383
Vela, A. I., Fernandez, A., De Quiros, Y., Herraez, P., Dominguez, L., and Fernandez-Garayzabal, J. F. (2011). Weissella ceti sp. nov., isolated from beaked whales (Mesoplodon bidens). Int. J. Syst. Evol. Microbiol. 61, 2758–2762. doi: 10.1099/ijs.0.028522-0
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Wiedenbeck, J., and Cohan, F. M. (2011). Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiol. Rev. 35, 957–976. doi: 10.1111/j.1574-6976.2011.00292.x
Wu, H. C., Srionnual, S., Yanagida, F., and Chen, Y. S. (2015). Detection and characterization of Weissellicin 110, a bacteriocin produced by Weissella cibaria. Iran. J. Biotechnol. 13, 63–67. doi: 10.15171/ijb.1159
Wu, S., Zhu, Z., Fu, L., Niu, B., and Li, W. (2011). WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics 12:444. doi: 10.1186/1471-2164-12-444
Xing, Z., Geng, W., Li, C., Sun, Y., and Wang, Y. (2017). Comparative genomics of Lactobacillus kefiranofaciens ZW3 and related members of Lactobacillus. spp reveal adaptations to dairy and gut environments. Sci. Rep. 7:12827. doi: 10.1038/s41598-017-12916-0
Yao, F., Xu, X. Y., and Pan, Q. (2019). A modified method for plasmid extraction from Lactobacillus plantarum contained lysozyme removal step. Anal Biochem 566, 37–39. doi: 10.1016/j.ab.2018.11.005
Yeong, M. S., Hee, M. S., and Choon, C. H. (2020). Characterization of high-ornithine-producing Weissella koreensis db1 isolated from kimchi and its application in rice bran fermentation as a starter culture. Foods 9:1545. doi: 10.3390/foods9111545
Yeu, J. E., Lee, H. G., Park, G. Y., Lee, J., and Kang, M. S. (2021). Antimicrobial and antibiofilm activities of Weissella cibaria against pathogens of upper respiratory tract infections. Microorganisms 9:1181. doi: 10.3390/microorganisms9061181
Yu, X. L., Zhang, W., Zhao, Z. P., Ye, C. S., Zhou, S. Y., Wu, S. G., et al. (2019). Molecular characterization of carbapenem-resistant Klebsiella pneumoniae isolates with focus on antimicrobial resistance. Bmc Genomics 20:822. doi: 10.1186/s12864-019-6225-9
Yuan, S., Wang, Y., Zhao, F., and Kang, L. (2021). Complete genome sequence of Weissella confusa LM1 and comparative genomic analysis. Front Microbiol 12:749218. doi: 10.3389/fmicb.2021.74
Keywords: Weissella cibaria, pan-genome, phylogenetic analysis, lactic acid bacteria, probiotic
Citation: Surachat K, Kantachote D, Wonglapsuwan M, Chukamnerd A, Deachamag P, Mittraparp-arthorn P and Jeenkeawpiam K (2022) Complete Genome Sequence of Weissella cibaria NH9449 and Comprehensive Comparative-Genomic Analysis: Genomic Diversity and Versatility Trait Revealed. Front. Microbiol. 13:826683. doi: 10.3389/fmicb.2022.826683
Received: 01 December 2021; Accepted: 12 April 2022;
Published: 19 May 2022.
Edited by:
Apichai Tuanyok, University of Florida, United StatesReviewed by:
Zhichao Xu, Northeast Forestry University, ChinaMeichen Pan, North Carolina State University, United States
Copyright © 2022 Surachat, Kantachote, Wonglapsuwan, Chukamnerd, Deachamag, Mittraparp-arthorn and Jeenkeawpiam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Komwit Surachat, a29td2l0LnNAcHN1LmFjLnRo