 Katharina A. Werner1
Katharina A. Werner1 Dominik Schneider2
Dominik Schneider2 Anja Poehlein2
Anja Poehlein2 Nina Diederich1Lara Feyen1
Nina Diederich1Lara Feyen1 Katharina Axtmann3Tobias Hübner4
Katharina Axtmann3Tobias Hübner4 Nicolas Brüggemann5
Nicolas Brüggemann5 Katharina Prost5
Katharina Prost5 Rolf Daniel2
Rolf Daniel2 Elisabeth Grohmann1*
Elisabeth Grohmann1*
- 1Department of Microbiology, Faculty of Life Sciences and Technology, Berliner Hochschule für Technik, Berlin, Germany
- 2Department of Genomic and Applied Microbiology and Göttingen Genomics Laboratory, Institute of Microbiology and Genetics, Georg-August-University Göttingen, Göttingen, Germany
- 3Institute for Medical Microbiology, Immunology and Parasitology, University Hospital Bonn, Bonn, Germany
- 4Department of Environmental Microbiology, Helmholtz Centre for Environmental Research GmbH—Umweltforschungszentrum Leipzig (UFZ), Leipzig, Germany
- 5Institute of Bio- and Geosciences—Agrosphere (IBG-3), Forschungszentrum Jülich, Jülich, Germany
In times of climate change, practicing a form of sustainable, climate-resilient and productive agriculture is of primordial importance. Compost could be one form of sustainable fertilizer, which is increasing humus, water holding capacity, and nutrient contents of soils. It could thereby strengthen agriculture toward the adverse effects of climate change, especially when additionally combined with biochar. To get access to sufficient amounts of suitable materials for composting, resources, which are currently treated as waste, such as human excreta, could be a promising option. However, the safety of the produced compost regarding human pathogens, pharmaceuticals (like antibiotics) and related resistance genes must be considered. In this context, we have investigated the effect of 140- and 154-days of thermophilic composting on the hygienization of human excreta and saw dust from dry toilets together with straw and green cuttings with and without addition of biochar. Compost samples were taken at the beginning and end of the composting process and metagenomic analysis was conducted to assess the fate of antibiotic resistance genes (ARGs) and pathogenicity factors of the microbial community over composting. Potential ARGs conferring resistance to major classes of antibiotics, such as beta-lactam antibiotics, vancomycin, the MLSB group, aminoglycosides, tetracyclines and quinolones were detected in all samples. However, relative abundance of ARGs decreased from the beginning to the end of composting. This trend was also found for genes encoding type III, type IV, and type VI secretion systems, that are involved in pathogenicity, protein effector transport into eukaryotic cells and horizontal gene transfer between bacteria, respectively. The results suggest that the occurrence of potentially pathogenic microorganisms harboring ARGs declines during thermophilic composting. Nevertheless, ARG levels did not decline below the detection limit of quantitative PCR (qPCR). Thresholds for the usage of compost regarding acceptable resistance gene levels are yet to be evaluated and defined.
Introduction
Human excreta contain large amounts of key nutrients for agriculture, like nitrogen, phosphorus, and potassium. However, due to sanitation systems that treat human excreta as waste, they usually remain unused in many countries that use water-based sanitation systems. Ecological sanitation using thermophilic composting can serve as a treatment to convert human excreta into a humus-rich material with low pathogen contents that nurtures plants and increases soil fertility of agricultural fields (Dalzell et al., 1987; Amlinger et al., 2007; Rose et al., 2015; Reid, 2020; Ryals et al., 2021). The possible spread of human pathogens or ARGs is one concern toward the usage of composted human excrements (Schönning and Stenström, 2004). The positive effect of compost on soil fertility can be even additionally increased by the use of biochar-compost, i.e., compost containing co-composted charcoal pieces (Fischer and Glaser, 2012; Agegnehu et al., 2017).
Thermophilic composting is the aerobic, exothermic degradation of organic material through biological activity. The process comprises a first mesophilic, a thermophilic, a second mesophilic and a cooling or maturation phase, which are dominated by different microbial species (Bernal et al., 2009). Heat-tolerant bacteria are predominant in the thermophilic phase; fungi and yeasts proliferate when temperatures decrease again in the second mesophilic and maturation phases. Members of the bacterial phyla Firmicutes and Proteobacteria, followed by Actinobacteria, Bacteroidetes, or Planctomycetes are usually dominant throughout composting, although the composition differs with the composting conditions (Ryckeboer et al., 2003; Esperón et al., 2020). Biochar addition to composting was found to alter the microbial community through altered C:N ratios or reduced amounts of dissolved organic carbon (Jindo et al., 2012; Qiu et al., 2019). Biochar can additionally affect the removal of ARGs, but the effect seems to be dependent on the type of pyrolyzed material used for co-composting (Cui et al., 2016).
Antibiotic resistant bacteria pose a growing risk in the modern world due to limited treatment options. Increased usage of antibiotics enhances the selective pressure leading to the accumulation of multiple resistances in one strain, which has been estimated to cause at least 700,000 deaths per year (Laxminarayan et al., 2013; O’Neill, 2014). Methicillin-resistant Staphylococcus aureus (MRSA) or vancomycin-resistant enterococci (VRE) are declared high priority pathogens for research and development of new antibiotics by the WHO (Tacconelli et al., 2018). The level of ARGs in soils has increased with increasing use of antibiotics over the last decades, and transfer of ARGs from environmental to clinical strains has been reported (Lartigue et al., 2006; Walsh, 2006; Knapp et al., 2010; Graham et al., 2016). Hence, it is of great importance to interrupt possible cycles of ARG transfer. The application of untreated manure was shown to increase ARG levels in the field (Knapp et al., 2010); however, thermophilic composting as a pretreatment can decrease the load of ARGs. Lower levels of beta-lactam, macrolide, quinolone, sulfonamide and tetracycline resistance genes through thermophilic composting were reported for manure (Wang et al., 2016; Guo et al., 2017; Chen Z. et al., 2018; Esperón et al., 2020). However, variations in the reduction efficiency, inconclusive results for sewage sludge composting, and limited studies on the fate of ARGs during human waste composting, especially when co-composted together with biochar, call for more research to get better and reliable insights into the ARGs dispersal dynamics (Zhang et al., 2016; Ezzariai et al., 2018; Jang et al., 2018; Liao et al., 2018; Cui et al., 2020; Wei et al., 2020).
Virulence factors have different functions that enable bacteria to infect host cells and cause disease; their presence differentiates pathogenic from non-pathogenic strains. These functions include adherence factors, siderophores, endo- and exotoxins, invasins, as well as type III and IV secretion systems. The corresponding genes are usually located on mobile genetic elements (MGEs) and often organized in pathogenicity islands that have evolved via different mechanisms (Dobrindt et al., 2004; Gal-Mor and Finlay, 2006). Genetic material located on MGEs can be transferred between bacteria of the same or different species through horizontal gene transfer. MGEs comprise for example plasmids, transposons, integrons, and bacteriophages which can be transferred via conjugation, transformation or transduction (Dobrindt et al., 2004; Gillings, 2014).
To elucidate the effect of thermophilic composting of human excreta (feces and urine) from dry toilets together with green cuttings and straw, with and without biochar addition, we assessed the effect on ARGs and bacterial virulence genes, as well as the composition of the microbial community. Here, we present metagenomic sequencing and analysis of samples before and after 5 months of composting.
Materials and Methods
Composting Trial, Sampling, and Sample Pretreatment
The composting trial (including feedstock material properties, composition and temperature measurements), sampling and sample pretreatment were described in detail in Werner et al. (2022). In brief, thermophilic windrow composting of dry toilet contents, green cuttings, straw, and urine, with and without biochar, was conducted in Eckernförde, Germany, between September 2018 and January 2019. The trial comprised four piles and two treatments. Each of the two treatments (with and without biochar) was set up twice at an interval of 2 weeks, with E1 and E1-B referring to the replicates, which were set up first, and E2 and E2-B referring to the replicates set up 2 weeks later. “-B” indicates the biochar treatment.
Sampling was conducted at the day of the experimental set-up (“start” samples), after 2 weeks (only repetition 1) and at the end of the experimental trials after 5.5 (repetition 1) and 5 months (repetition 2), respectively.
DNA Extraction and Pooling for Metagenomic Sequencing
Extraction of total DNA was conducted from 250 mg compost using the NucleoSpin® Soil gDNA extraction kit (Macherey-Nagel) according to the manufacturer’s instructions. Lysis buffer SL2 was used without enhancer. Mechanical disruption was performed in a FastPrep-24™ 5G homogenizer (MP Biomedicals) at 5 m/s for 30 s. For DNA elution, 50 μL of nuclease-free water were applied.
For metagenomic analysis, triplicate end samples were pooled equimolarly. A total of eight samples (start and end samples of four compost piles) were sequenced at Göttingen Genomics Laboratory, Germany.
For qPCR analysis, the DNA extracts were further purified using a magnetic separation device NucleoMag® SEP (Macherey-Nagel) and CleanNGS kit (CleanNA) according to manufacturer’s instructions. DNA was stored at −20°C.
Metagenomic Sequencing
Illumina paired-end sequencing libraries were prepared using the Illumina® DNA Prep, (M) Tagmentation Kit and the Nextera™ DNA CD Indexes kit as recommended by the manufacturer (Illumina, San Diego, CA, United States). To assess quality and size of the libraries, samples were run on an Agilent Bioanalyzer 2100 using an Agilent High Sensitivity DNA Kit as recommended by the manufacturer (Agilent Technologies, Waldbronn, Germany). Concentration of the libraries were determined using the Qubit® dsDNA HS Assay Kit as recommended by the manufacturer (Life Technologies GmbH, Darmstadt, Germany). Sequencing was performed by using the HiSeq2500 instrument (Illumina Inc., San Diego, CA, United States) using the HiSeq Rapid PE Cluster Kit v2 for cluster generation and the HiSeq Raid SBS Kit (500 cycles) for sequencing in the paired-end mode and running 2 × 250 cycles.
Data Availability
The raw reads of the 8 sequenced metagenomic samples have been deposited in the NCBI Sequence Read Archive, study SRP347939, at BioProject PRJNA782422 under accession numbers SRR17041395 (E1 start), SRR17041394 (E1-B start), SRR17041392 (E2 start), SRR17041391 (E2-B start), SRR17041390 (E1 end), SRR17041389 (E1-B end), SRR17041388 (E2 end) and SRR17041387 (E2-B end). Detailed information is stated in Supplementary Table 1.
Data Analysis
Metagenomic paired-end sequences of 8 samples were quality-filtered with fastp v0.20.0 (Chen S. et al., 2018) using default settings with the addition of an increased per base phred score of 20, base pair corrections by overlap, as well as 5′- and 3′-end read trimming with a sliding window of 4, a mean quality of 20 and a minimum sequence length of 50 bp. After quality filtering, the metagenome sequences consisted of between 44,872,702 and 94,600,400 paired-end reads with an average read length of 236–239 bp (Supplementary Table 2). Taxonomic assignment was performed by a combination of Kraken2 v2.0.8-beta (Wood et al., 2019) and Kaiju v1.7.0 (Menzel et al., 2016) against the nucleotide (nt) or non-redundant protein (nr) database (retrieved on 22 July, 2020), respectively, to improve sequence classification. The Kraken2 classification was favored over Kaiju classification due to higher accuracy of nucleotide-based classification and taxonomy was added by “addTaxonNames” script of Kaiju. Functional classification of reads was performed with KaijuX (Menzel et al., 2016) with default settings against the KEGG database (Kanehisa et al., 2009) (retrieved on 2018-10-01). P-values for functional groups as assigned by the KEGG database were calculated using two-tailed, paired Student’s test. Due to the low number of replicates, start and end samples of all four compost piles were compared. P-values below 0.05 were considered significant (*), p < 0.01 (**) highly significant.
Metagenomic assembly was performed with SPAdes v3.14.0 (Bankevich et al., 2012) using defined kmers (-k 21,33,55,77,99,127) which resulted in 8 assembled datasets. Contigs with length < 1,000 bp were discarded for further processing. Open reading frames (ORFs) from all contigs were determined with PRODIGAL v2.6.3 (Hyatt et al., 2010). Subsequently, all protein sequences were combined and dereplicated with USEARCH v9.2.64 (Edgar, 2010). The protein sequences of predicted ORFs were aligned against the Resfams v1.2.2 (Gibson et al., 2015) core genes database using HMMER v3.31 with the recommended parameters to identify ARG-like ORFs which are referred to as ARGs. Relative abundance of ARGs of each sample was calculated by mapping reads with Bowtie2 v2.3.5.1 (Langmead and Salzberg, 2012) against the ARG nucleotide sequences. SAMtools v1.9 (Li et al., 2009) was used to extract mapped read counts to ARG sequences. Contigs carrying ARGs were taxonomically classified with CAT v5.1.2 (von Meijenfeldt et al., 2019) and used to determine ARG taxonomic origin.
Data basis for MaAsLin2 analysis were genes assigned by Prodigal (Hyatt et al., 2010) from assembled metagenomes. ARG annotation was conducted with HMMer against the Resfams database (Gibson et al., 2015) and taxonomic origin was assigned using CAT (von Meijenfeldt et al., 2019) on the contig carrying the ARG.
Data manipulation and visualization was performed with ampvis2 v2.6.5 (Andersen et al., 2018). Bar charts of superkingdoms were generated with ggplot2 v3.3.2 (Wickham, 2016) and read counts were converted to relative abundances. PCoA based on taxonomic composition was performed at same sequencing effort for (44,090,688 reads per sample). Diversity indices (Richness, Shannon) were calculated with ampvis2 and visualized with ggplot2. Normalization was conducted through conversion of read counts to relative abundances. 77–83% could be assigned taxonomically at kingdom level, 53–69% functionally.
Quantitative Real Time PCR
qPCR analyses were conducted on selected genes conferring resistance to different classes of antibiotics to quantify the gene concentrations in samples before, after 14-days (only repetition 1) and after composting. Genes were chosen based on previous PCR results of the samples (aph(2″)-Ia, aph(3′)-IIIa, ermA, ermB, sul1, sul2, tetL and tetS) (Werner et al., 2022). qPCR was performed using a LightCycler® 480 II instrument (Roche Diagnostics Ltd.) and the LightCycler® 480 Probes Master (Roche Diagnostics Deutschland GmbH). Preliminary tests of the DNA extracts revealed an optimal dilution of 1:100 of the template DNA to reduce inhibition of the qPCR. Primer and probe sequences, programs and bacterial strains used for the generation of positive controls are shown in Supplementary Table 3 and composition of the qPCR reactions in Supplementary Table 4. A standard curve of 101–108 gene copies was used for quantification in every run. Each sample was measured in triplicates in each run, and the average of three individual runs is presented. qPCR results were normalized to the 16S rRNA gene copy number, as well as to 1 g compost (dry weight). P-values were calculated on the normalized target copy numbers using two-tailed, paired Student‘s test. Due to the low number of replicates, start and end samples of all four compost piles were compared. For this reason, the average of the triplicates from the end of composting was used for analysis. P-values below 0.05 were considered significant (*), p < 0.01 (**) highly significant.
Results
Thermophilic Composting Changes the Microbial Community Composition
Composition of the compost community including prokaryotes, eukaryotes and viruses changed from start to end of composting, as suggested by principal coordinates analysis (PCoA). The functional profile of the metagenomic data also exhibits distinct differences, drawing a similar picture as the taxonomic PCoA (Figure 1). Bacteria represented the largest group with around 70% relative abundance in the start and 65–67% in the end samples, followed by Eukaryota that increase from 10 to 11% relative abundance. Archaea and viruses accounted for a maximum of 1% each. At the phylum level, Proteobacteria (start: 31–38%; end: 28–29%) and Actinobacteria (start: 15–22%; end: 17–19%) showed highest relative abundance in samples from both start and end of the composting. The phyla following in abundance accounted for less than 10% of the metagenomic reads (Figure 2). The composition of the bacterial community was elucidated previously based on 16S rRNA gene amplicon sequencing (Werner et al., 2022). The order of phyla (regarding their relative abundances) differed between amplicon and metagenomic data, but phyla with highest abundance were the same and general trends mostly corresponded. Proteobacteria exhibited highest relative abundances in both amplicon and metagenomic sequencing with decreases from the beginning to the end of composting. Actinobacteria increased in abundance during composting in the amplicons, whereas averaged metagenomics relative abundance revealed no differences. Firmicutes showed the most pronounced decrease in both data sets, but metagenomics detected lower overall abundances. Amplicon and metagenomics both revealed decreases in relative abundance of Bacteroidetes during composting. Data on Chloroflexi was very similar with strong, almost sixfold increases of the initial relative abundances. In both cases, Chloroflexi rose from the fifth to the fourth most abundant phylum during composting (Supplementary Figure 1).
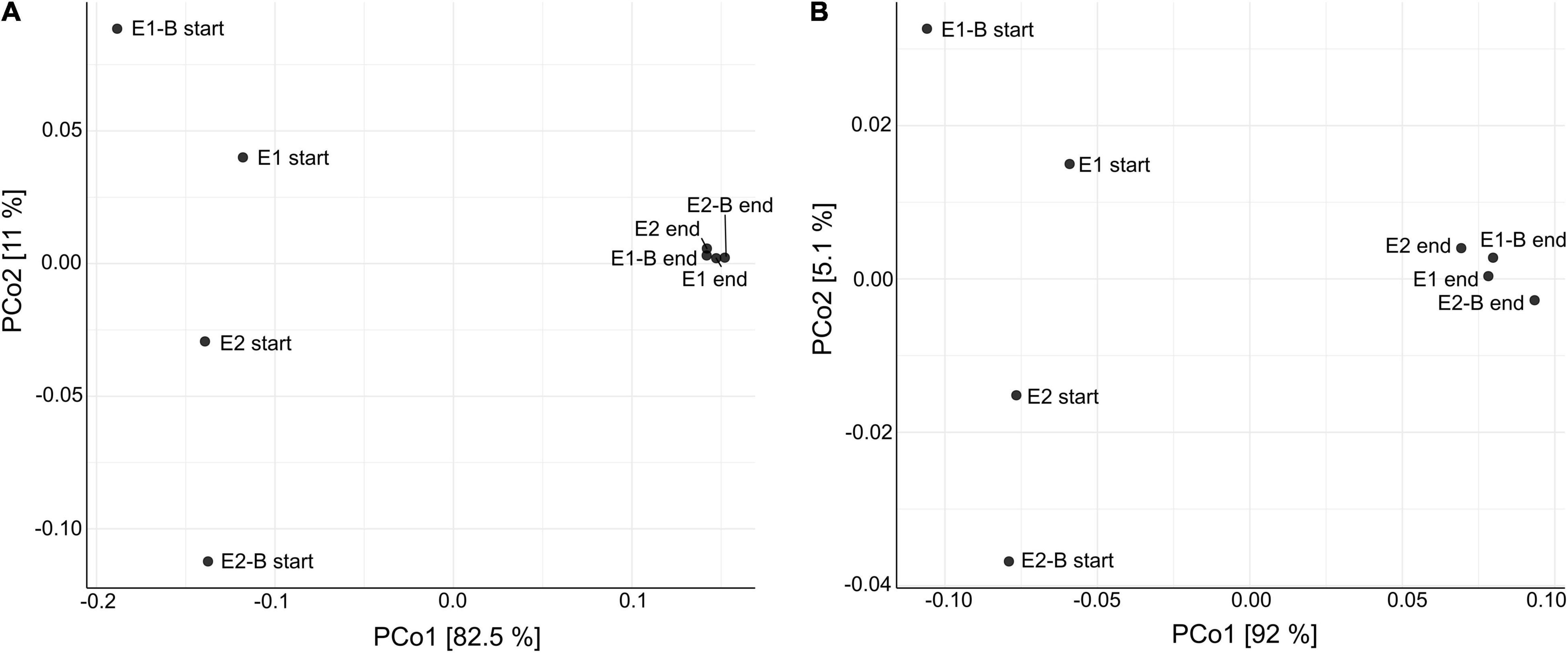
Figure 1. Principal coordinates analysis (PCoA) of the metagenomic data depicting the dissimilarity of each individual sample toward all others. (A) Dissimilarity of taxonomic data is depicted in the left diagram with PCo1 (x-axis) explaining 82.5% of differences and PCo2 (y-axis) explaining 11%, respectively. (B) Functional data in the right diagram with PCo1 explaining 92% of differences and PCo2 explaining 5.1%, respectively. Each data point represents one of the 8 compost samples of the trial {start and end samples of two treatments [without and with biochar (B)] and two repetitions (E1/E2)}.
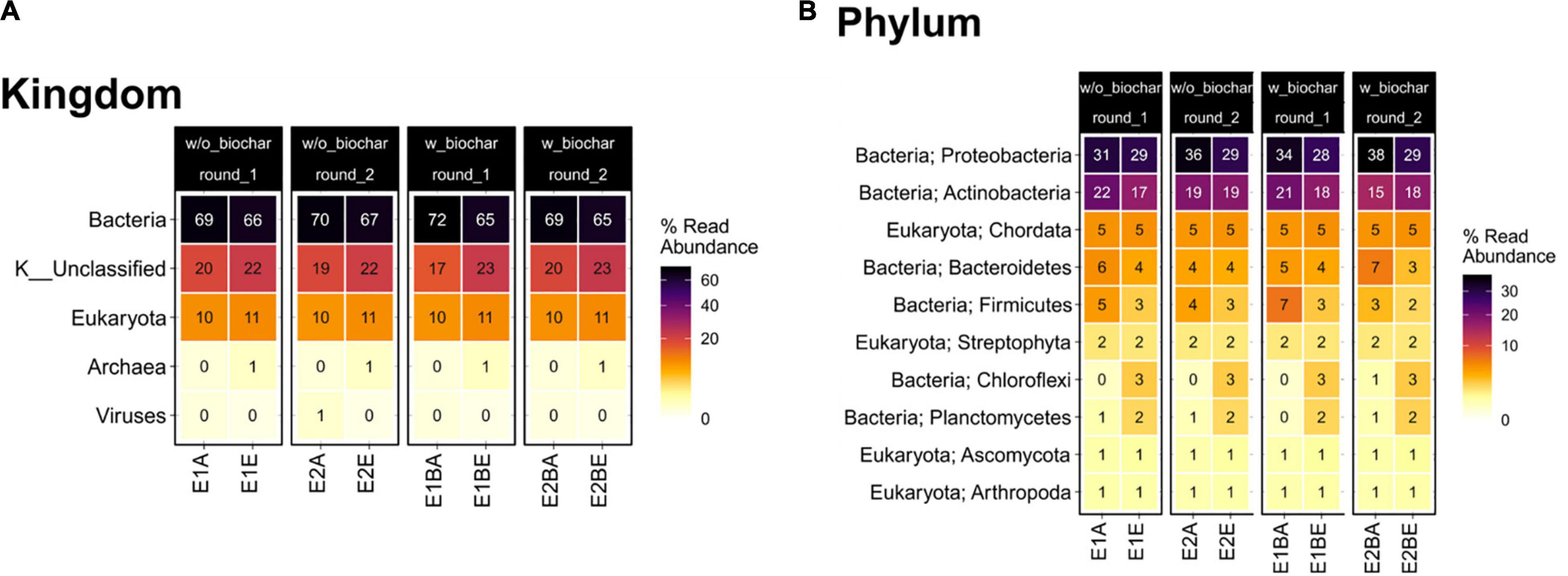
Figure 2. Heatmap depicting relative read abundance on taxonomic kingdom (A) and phylum levels (B) for the metagenomic data. Samples from start (A) and end (E) of two separate repetitions (1 and 2) of compost treatments without and with biochar (B) are depicted individually.
Since bacterial community composition was assessed before (Werner et al., 2022), here we focus on potential human pathogens. In Figure 3 the 40 most abundant genera and species with pathogenic potential are shown. Abundances of spore-forming pathogens (see Bacillus; B. cereus and Clostridium; Clostridium botulinum in Figure 3) were very low. Strong decreases were observed for Pseudomonas stutzeri, Stenotrophomonas maltophilia, Pseudomonas aeruginosa, Pseudomonas oryzihabitans, Pseudomonas alcaligenes, Alcaligenes faecalis, Pseudomonas syringae, Listeria monocytogenes, Staphylococcus spp., Enterococcus spp., as well as for Enterococcus faecium and Staphylococcus sciuri. Proteus mirabilis was detected only in the start sample of one compost pile (E1B) with a relative abundance of 0.64%. The relative abundance of Ralstonia, Burkholderia and Leishmania species increased, as well as that of Legionella pneumophila and Roseomonas gilardii. Amplicon sequences were resolved up to the genus level and revealed the same trends for Pseudomonas, Staphylococcus, Enterococcus, Proteus, and Legionella spp., respectively. The other mentioned bacterial genera were not detected in the amplicon sequencing data.
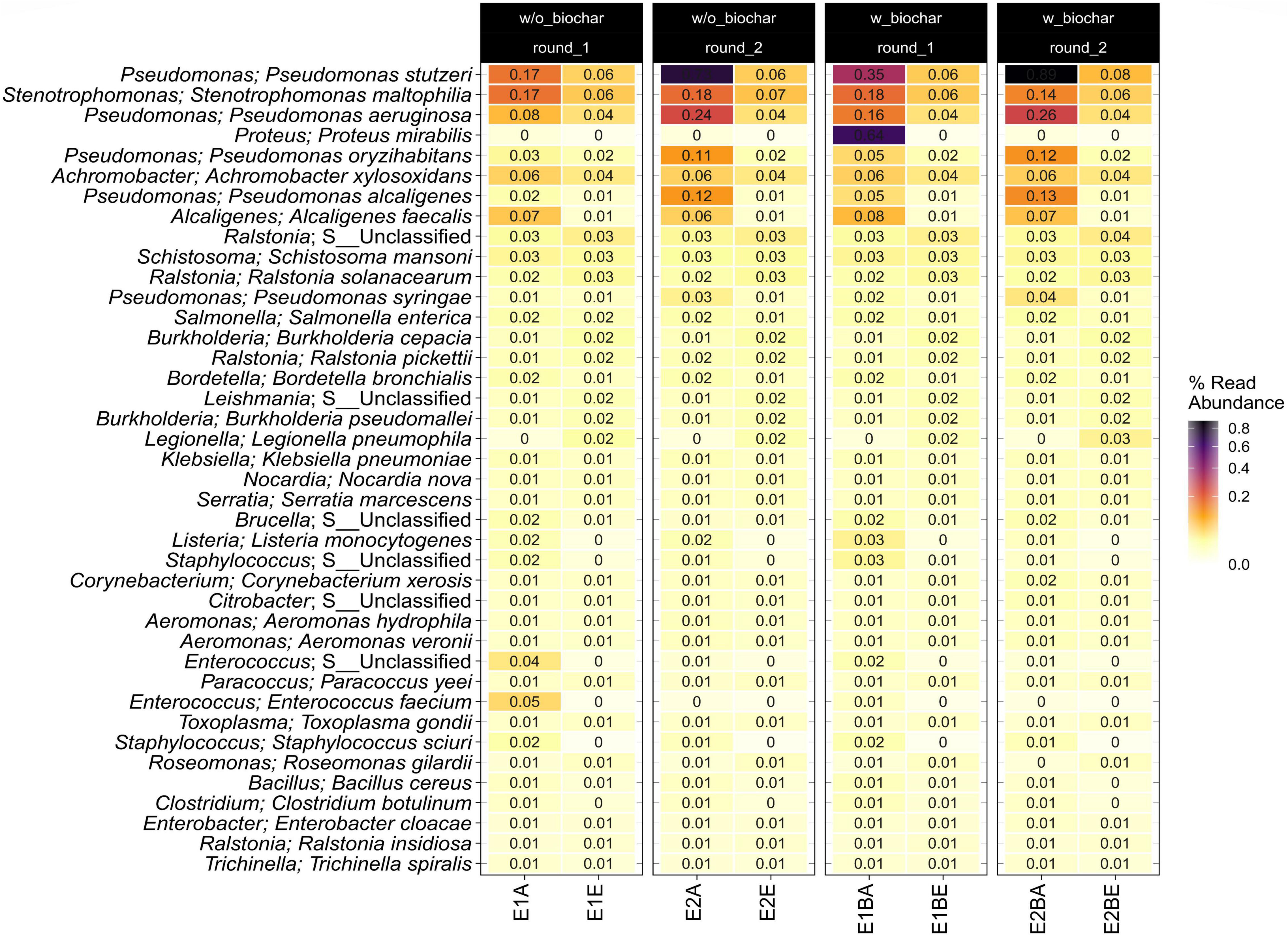
Figure 3. Heatmap showing the 40 most abundant potential pathogenic species and genera in the metagenomic data set as relative read abundance. Samples from start (A) and end (E) of two separate repetitions (1 and 2) of compost treatments without and with biochar (B) are depicted individually.
Decrease of Antibiotic Resistance Gene Abundances During Thermophilic Composting
The presence of ARGs was assessed by means of the KEGG database. When all genes conferring resistance to the same class of antibiotic were combined, relative abundance of ARGs decreased during composting. The least abundant group of fosfomycin resistance genes was an exception, showing an increase in abundance during composting (Figure 4). Trimethoprim and sulfamethoxazole resistance genes exhibited strongest decreases of 99 and 95%, respectively, followed by the resistance genes to rifamycin with 70%, chloramphenicol with 63%, quinolones with 60%, kanamycin with 56%, tetracycline with 54%, aminoglycosides with 39%, MLSB with 37%, beta-lactam antibiotics with 25% and vancomycin with 21% decreases. In addition, the relative abundance of ABC transporters and multidrug efflux pumps decreased, as well (Supplementary Figure 2).
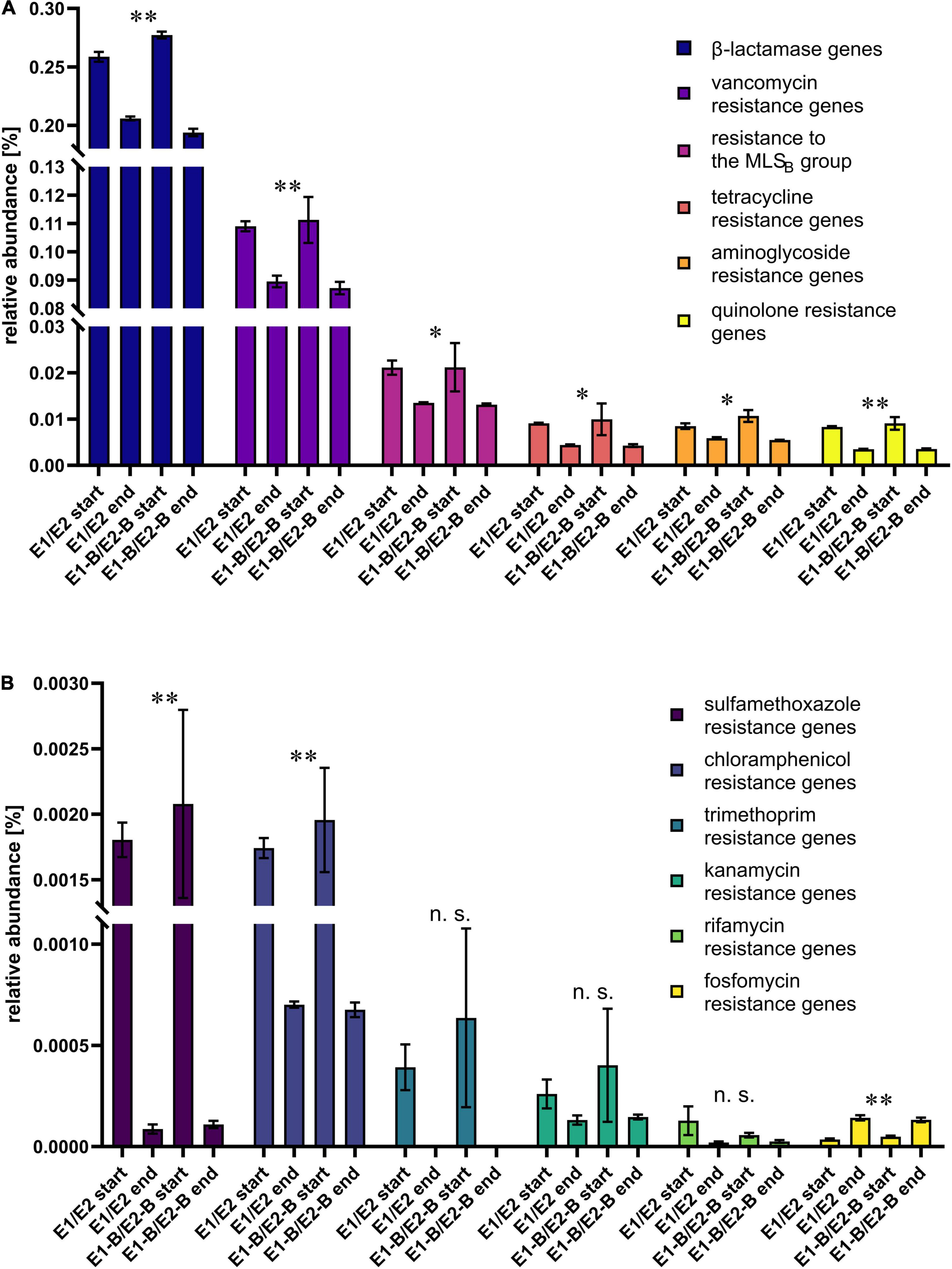
Figure 4. Relative abundance of resistance genes toward different antibiotic classes as assigned by KEGG database. The bars depict the mean and standard deviation of the replicate compost piles without (E1/E2) and with biochar (E1B/E2B) for start and end of composting. Higher abundant genes are depicted in (A), low abundant genes in (B). The numbers of assigned genes differ: 94 β-lactamase, 21 vancomycin, 19 MLSB, 11 tetracycline, 32 aminoglycoside, 2 quinolone, 3 sulfamethoxazole, 6 chloramphenicol, 3 trimethoprim, 2 kanamycin, 3 rifamycin and 3 fosfomycin resistance genes. Panel A shows the six highest abundant ARG groups, B the six lower abundant groups. Asterisks indicate the p-values obtained from Student’s t-test (**p < 0.01, *p < 0.05) representing the statistical significance of the data. Non-significant values are indicated by “n.s.” P-values were calculated to compare start with end samples of composting for all four compost piles combined (no differentiation of the treatments).
Trends of individual genes, however, differed: “Serine Protease Do” (serine endoprotease, degP, htrA [EC 3.4.21.107], involved in the two-component system in cell envelope protein folding and protein degradation, and cationic antimicrobial peptide (CAMP) resistance of periplasmic protein folding and degrading factors) and several genes involved in oligopeptide transport systems of beta-lactam resistance, ABC transporters and quorum sensing (oppA/mppA, oppB, oppC, oppD, oppF) showed increases from the beginning to the end of composting (Figure 5).
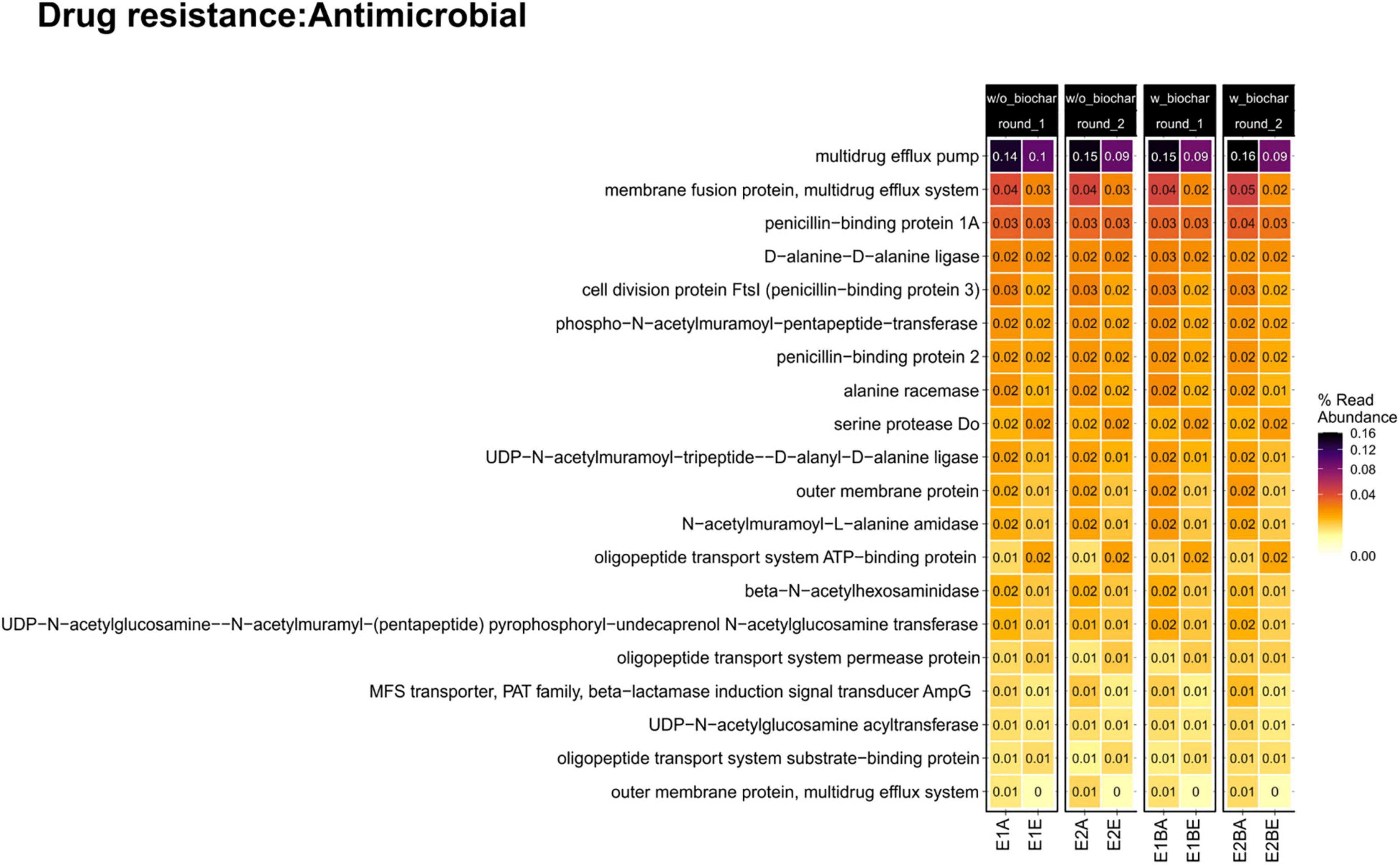
Figure 5. Heatmap of abundant genes conferring resistance to antibiotics in the metagenomic dataset. The compost trial comprises two repetitions (E1, E2) of two treatments without and with biochar (B) as compost supplement. Samples from start (A) and end (E) of composting are compared.
MaAsLin2 analysis (Mallick et al., 2021) revealed strong differences in abundance of antibiotic resistance mechanisms, assigned to a bacterial phylum, over composting and as a function of the treatment (with or without biochar) (Figure 6). Effects of the biochar treatment were observed only for beta-lactamase genes in Gemmatimonadetes, with lower abundance in the biochar treatment. All genes revealed differences in terms of the composting time. Beta-lactamases, ABC transporters, RND (resistance nodulation division) efflux pumps, acetyltransferases and antibiotic inactivation genes mostly increased toward the end of composting, and all entries of “gene modulating resistance” rose as well. Only phosphotransferases that modify, e.g., macrolide and aminoglycoside targets for inactivation of the antibiotic (Ramirez and Tolmasky, 2010; Golkar et al., 2018), declined in two cases. However, more resistance-related functions decreased: nucleotidyltransferases [transfer of a nucleotide monophosphate to a hydroxyl group of aminoglycoside or lincosamide antibiotics (Wright, 2005)], MFS (major facilitator superfamily) transporters (efflux pumps), rRNA methyltransferases [methylation of the antibiotic target site in the 16S ribosomal RNA (Osterman et al., 2020)] and target protection [association of a resistance protein and the antibiotic target site (Wilson et al., 2020)]. Besides unclassified phyla, ARGs associated with Chloroflexi, followed by Gemmatimonadetes and Bacteroidetes were the most represented taxonomic groups. Proteobacteria were assigned to three resistance mechanisms (RND antibiotic efflux pumps, phosphotransferases, nucleotidyltransferases), all decreased in abundance over composting. Actinobacteria were assigned to two resistance mechanisms that decreased (rRNA methyltransferases, beta-lactamases). Three out of four resistance functions associated with Bacteroidetes decreased (ABC transporters, antibiotic inactivation, acetyltransferases vs. genes modulating resistance). Apart from unclassified phyla, all other phyla were associated with increased resistance functions.

Figure 6. Top 50 strongly increased or decreased genes related to antibiotic resistance associated with bacterial phyla as assessed by MaAsLin2 analysis. Samples were tested regarding composting timepoint (“time” = start vs. end of composting) and regarding treatment (“biochar” = with vs. without biochar). Red color indicates increase, blue decrease of the respective gene abundance.
Decrease of Abundance of Genes Involved in Type III, Type IV, and Type VI Secretion Systems
Concerning the potential of horizontal gene transfer and infection of eukaryotic cells, we assessed the fate of genes that are involved in type III (T3SS), IV (T4SS), and VI (T6SS) secretion systems. Relative abundance decreased for all three systems (Figure 7). Strongest decreases to around 25% of the initial level were observed for T3SS, followed by T6SS and T4SS with decreases to an average of 43 and 55%, respectively. Most individual genes of the three secretion systems followed this trend.
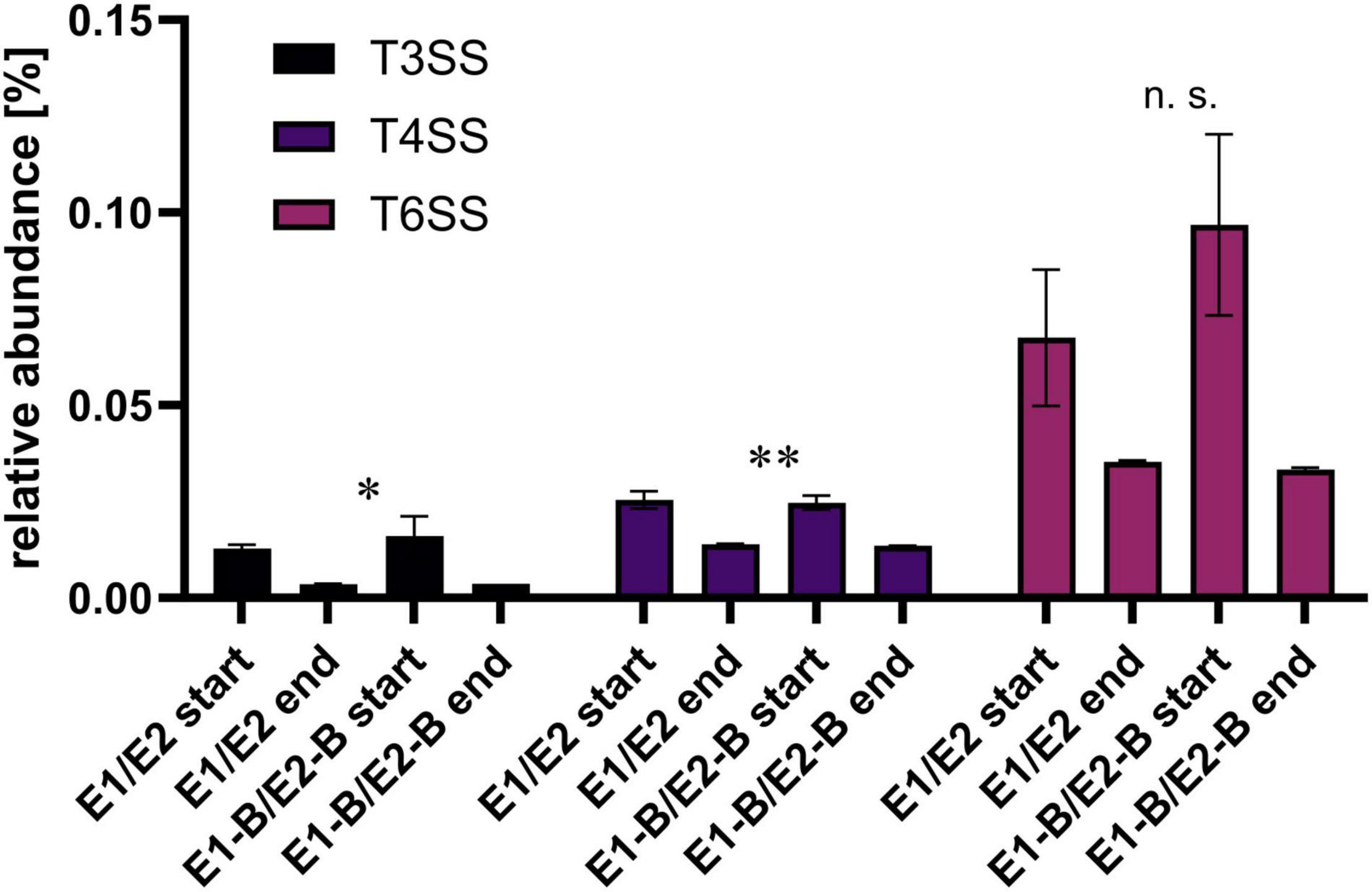
Figure 7. Relative abundance of type III, IV and VI secretion systems genes as assigned by KEGG database. The bars depict the mean and standard deviation of the replicate compost piles without (E1/E2) and with biochar (E1-B/E2-B) for start and end of composting. 33 genes were assigned to T3SS, 26 to T4SS and 25 toT6SS. Asterisks indicate the p-values obtained from Student’s t-test (**p < 0.01, *p < 0.05) representing the statistical significance of the data. Non-significant values are indicated by “n.s.” P-values were calculated to compare start with end samples of composting for all four compost piles combined (no differentiation of the treatments).
Decrease in Virulence Factor Abundances During Thermophilic Composting
To evaluate the potential of bacterial pathogenicity before and after composting, relative abundance of genes involved in quorum sensing, bacterial motility, flagellar assembly, chemotaxis, bacterial toxins, and invasion of epithelial cells was assessed. The relative abundance of quorum sensing genes remained almost stable, whereas the other groups exhibited a decline over composting. Chemotaxis genes decreased by around 37% in abundance, bacterial motility genes showed a statistically significant decrease by 34%, flagellar assembly genes decreased by 29%, genes for invasion of epithelial cells by 22% and bacterial toxins genes exhibited significant decreases by 20% (Figure 8). Moreover, the relative abundance of biofilm-related genes for P. aeruginosa, Vibrio cholerae and Escherichia coli decreased from start to end of the composting process (Figure 8). The decrease in relative abundance of these factors for P. aeruginosa and V. cholerae was significant and more pronounced than for E. coli, but relative abundance levels of the samples taken at the end of composting were similar for all three species (0.11–0.13%).
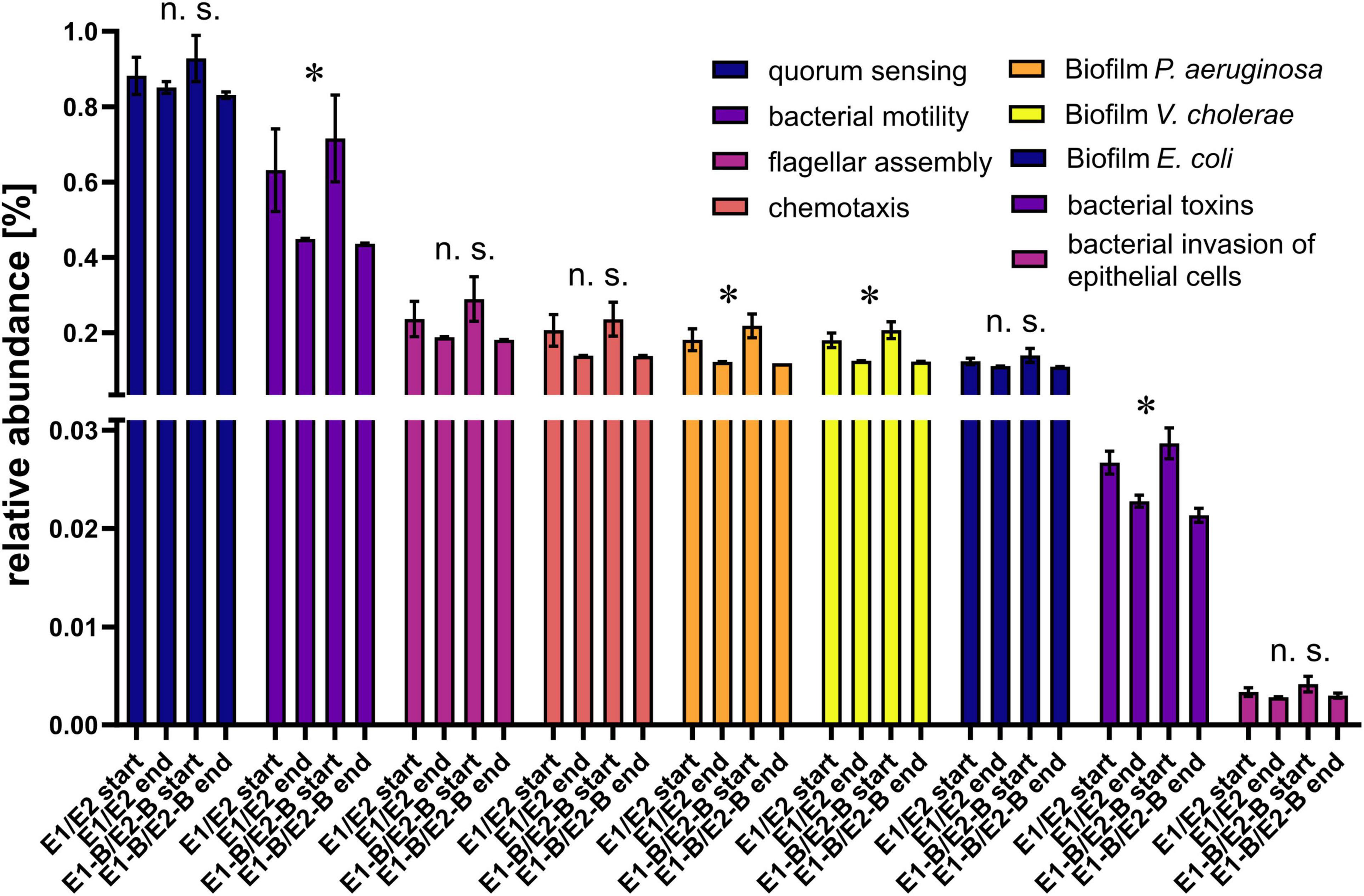
Figure 8. Relative abundance of genes related to quorum sensing, bacterial motility, flagellar assembly, chemotaxis, biofilm-formation for P. aeruginosa, V. cholerae and E. coli, bacterial toxins, and invasion of epithelial cells as assigned by KEGG database. The bars depict the mean and standard deviation of the replicate compost piles without (E1/E2) and with biochar (E1-B/E2-B) for start and end of composting. 145 genes were assigned to quorum sensing, 132 to bacterial motility, 39 to flagellar assembly, 26 to chemotaxis, 87 to biofilm-formation in P. aeruginosa, 78 to biofilm-formation in V. cholerae, 47 to biofilm-formation in E. coli, 40 to bacterial toxins and 65 to invasion of epithelial cells. Asterisks indicate the p-values obtained from Student’s t-test (*p < 0.05) representing the statistical significance of the data. Non-significant values are indicated by “n.s.” P-values were calculated to compare start with end samples of composting for all four compost piles combined (no differentiation of the treatments).
Decline of Antibiotic Resistance Gene Copy Numbers During Thermophilic Composting
The occurrence of individual ARGs was quantified using qPCR. Copy numbers normalized to the 16S rRNA gene and to 1 g compost showed decreases for all genes tested. The samples after 14 days of composting (available for repetition 1) exhibited decreases relative to the start samples for all ARGs tested (Figures 9–12).
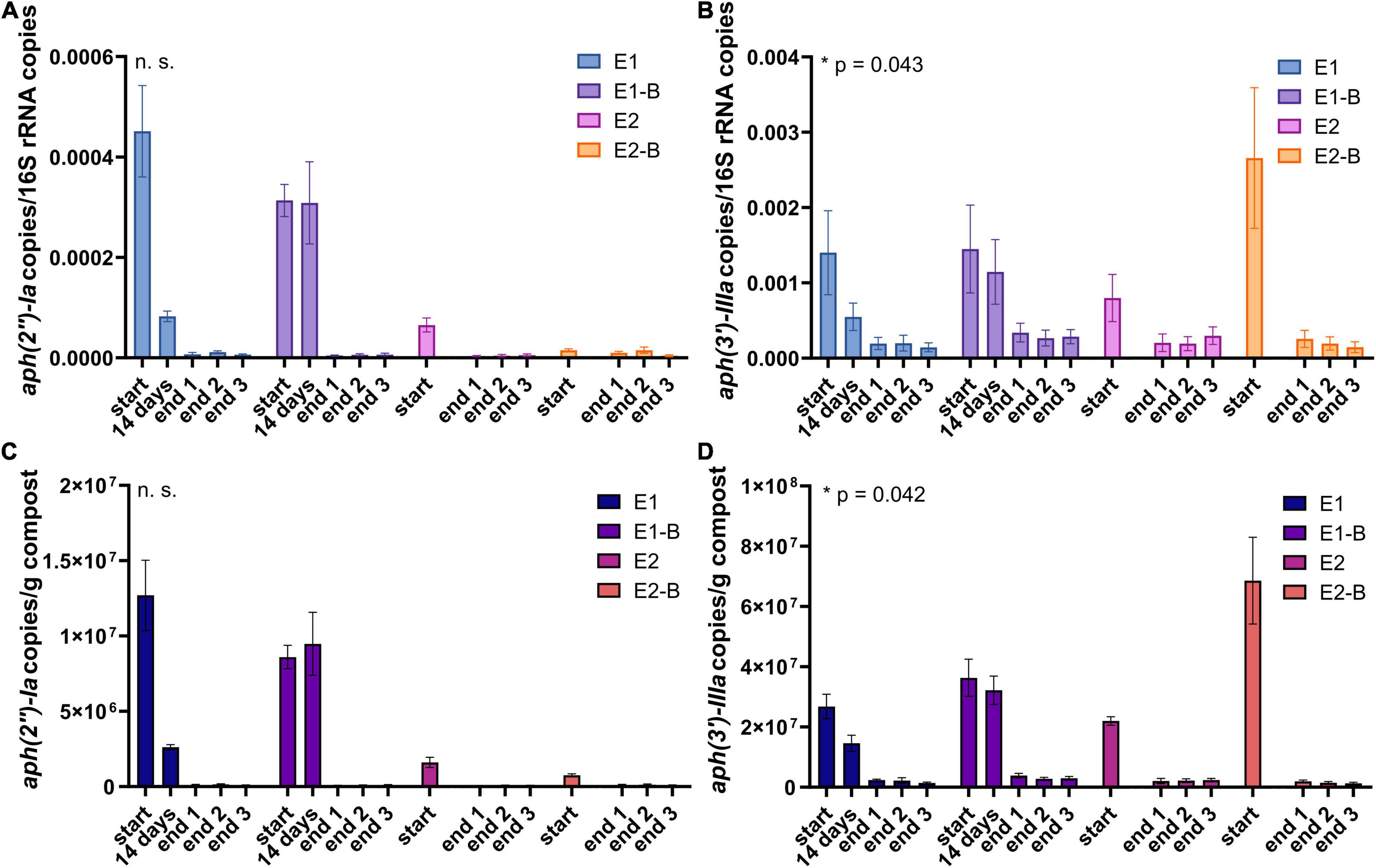
Figure 9. Copy numbers of aminoglycoside ARGs aph(2″)-Ia (A,C) and aph(3′)-IIIa (B,D) as assessed by qPCR. Copy numbers were normalized to 16S rRNA gene copy numbers (upper charts) and to 1 g compost (lower charts). Bars represent the average of three individual qPCR measurements with the standard deviation. Start, 14 days (only repetition 1) and end samples are depicted individually for the first (E1, E1-B) and second repetition of composting (E2, E2-B). End samples comprise replicates from front (end 1), middle (end 2) and back (end 3) of each compost pile. Asterisks indicate the p-values obtained from Student’s t-test (*p < 0.05) representing the statistical significance of the data. Non-significant values are indicated by “n.s.” P-values were calculated to compare start with end samples of composting for all four compost piles combined (average of the triplicate end samples, no differentiation of the treatments).
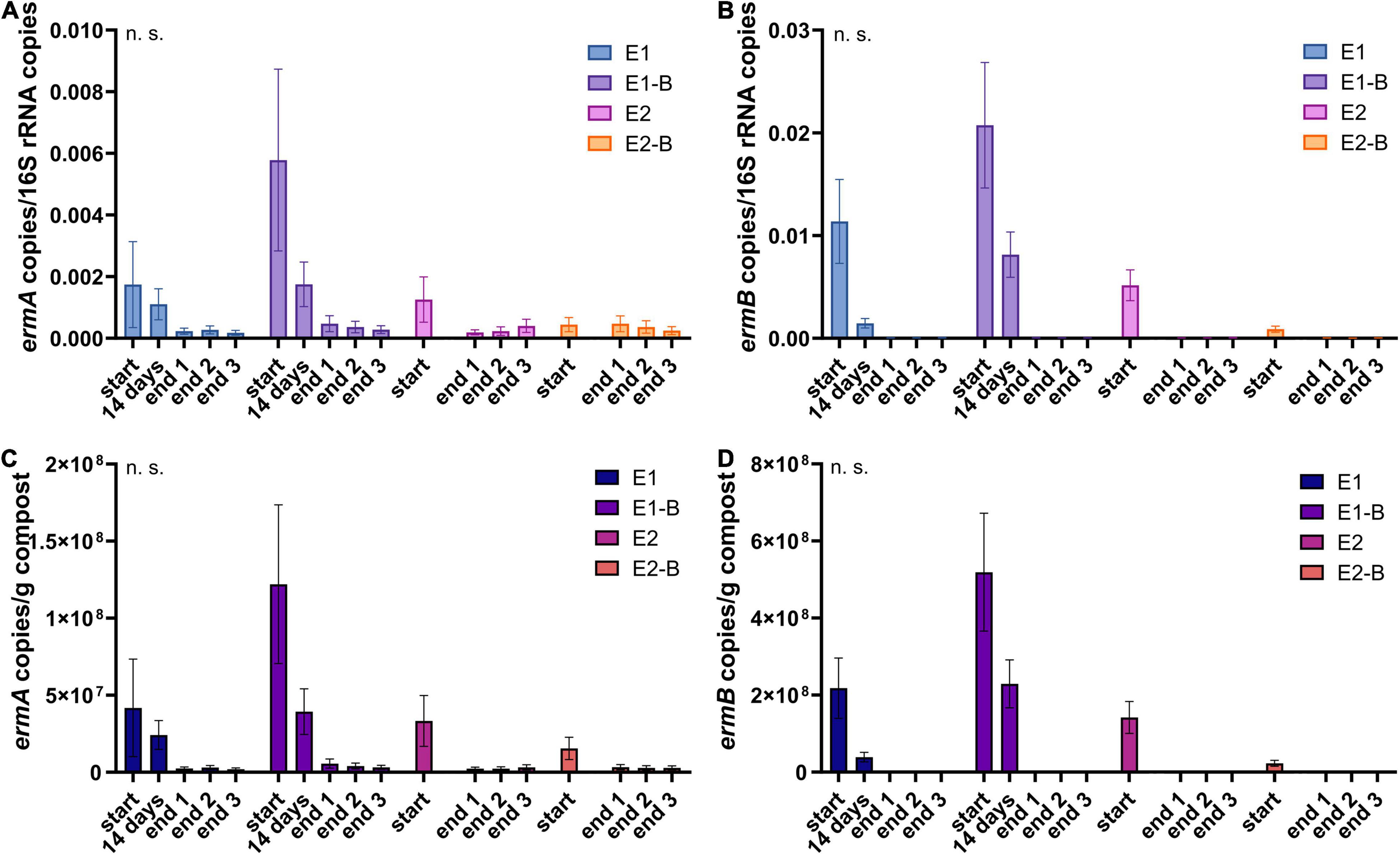
Figure 10. Copy numbers of erythromycin ARGs ermA (A,C) and ermB (B,D) as assessed by qPCR. Copy numbers were normalized to 16S rRNA gene copy numbers (upper charts) and to 1 g compost (lower charts). Bars represent the average of three individual qPCR measurements with the standard deviation. Start, 14 days (only repetition 1) and end samples are depicted individually for the first (E1, E1-B) and second repetition of composting (E2, E2-B). End samples comprise replicates from front (end 1), middle (end 2) and back (end 3) of each compost pile. Student’s t-test was conducted to test for statistical significance. Non-significant values are indicated by “n.s.” P-values were calculated to compare start with end samples of composting for all four compost piles combined (average of the triplicate end samples, no differentiation of the treatments).
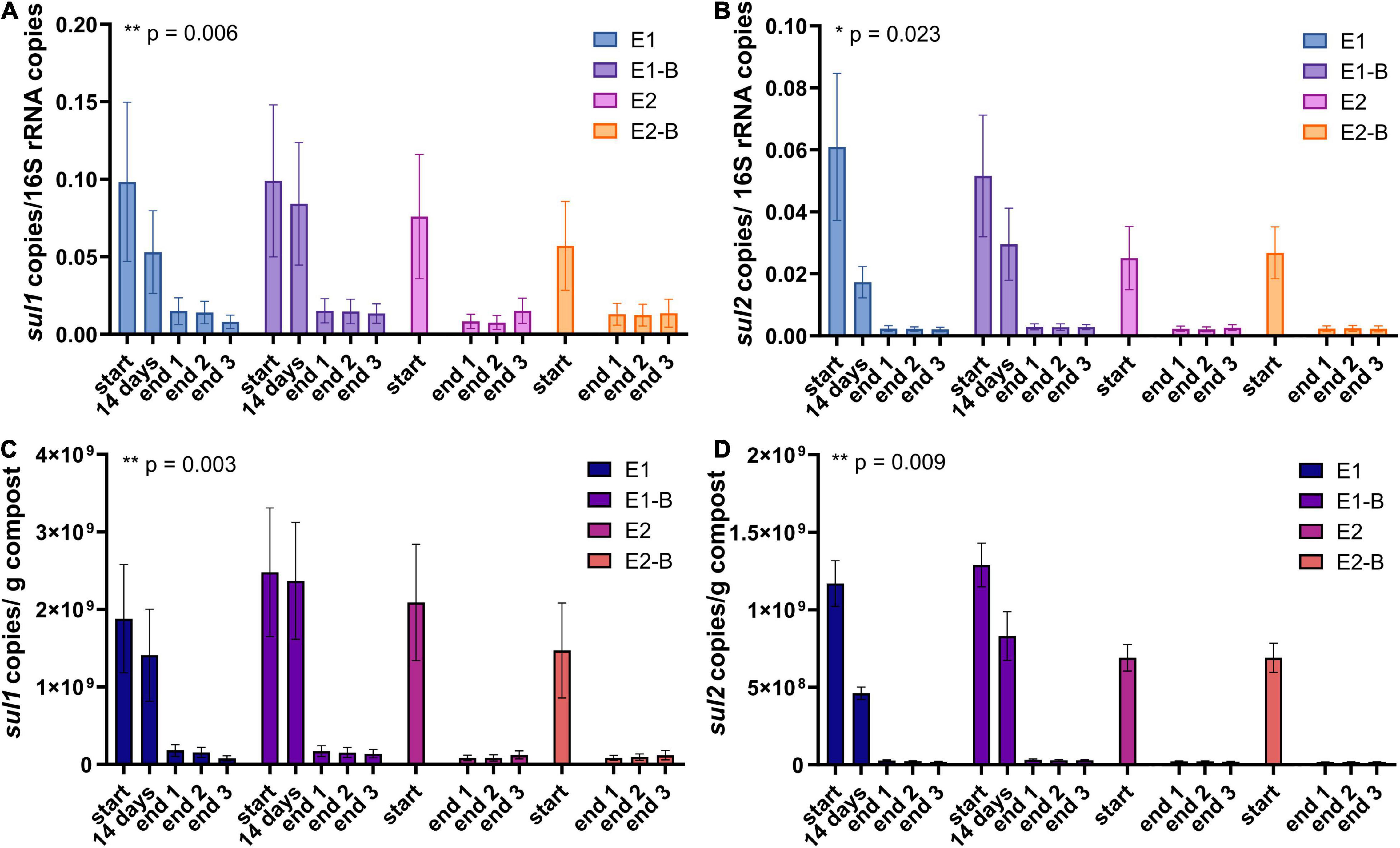
Figure 11. Copy numbers of sulfonamide ARGs sul1 (A,C) and sul2 (B,D) as assessed by qPCR. Copy numbers were normalized to 16S rRNA gene copy numbers (upper charts) and to 1 g compost (lower charts). Bars represent the average of three individual qPCR measurements with the standard deviation. Start, 14 days (only repetition 1) and end samples are depicted individually for the first (E1, E1-B) and second repetition of composting (E2, E2-B). End samples comprise replicates from front (end 1), middle (end 2) and back (end 3) of each compost pile. Asterisks indicate the p-values obtained from Student’s t-test (**p < 0.01, *p < 0.05) representing the statistical significance of the data. P-values were calculated to compare start with end samples of composting for all four compost piles combined (average of the triplicate end samples, no differentiation of the treatments).
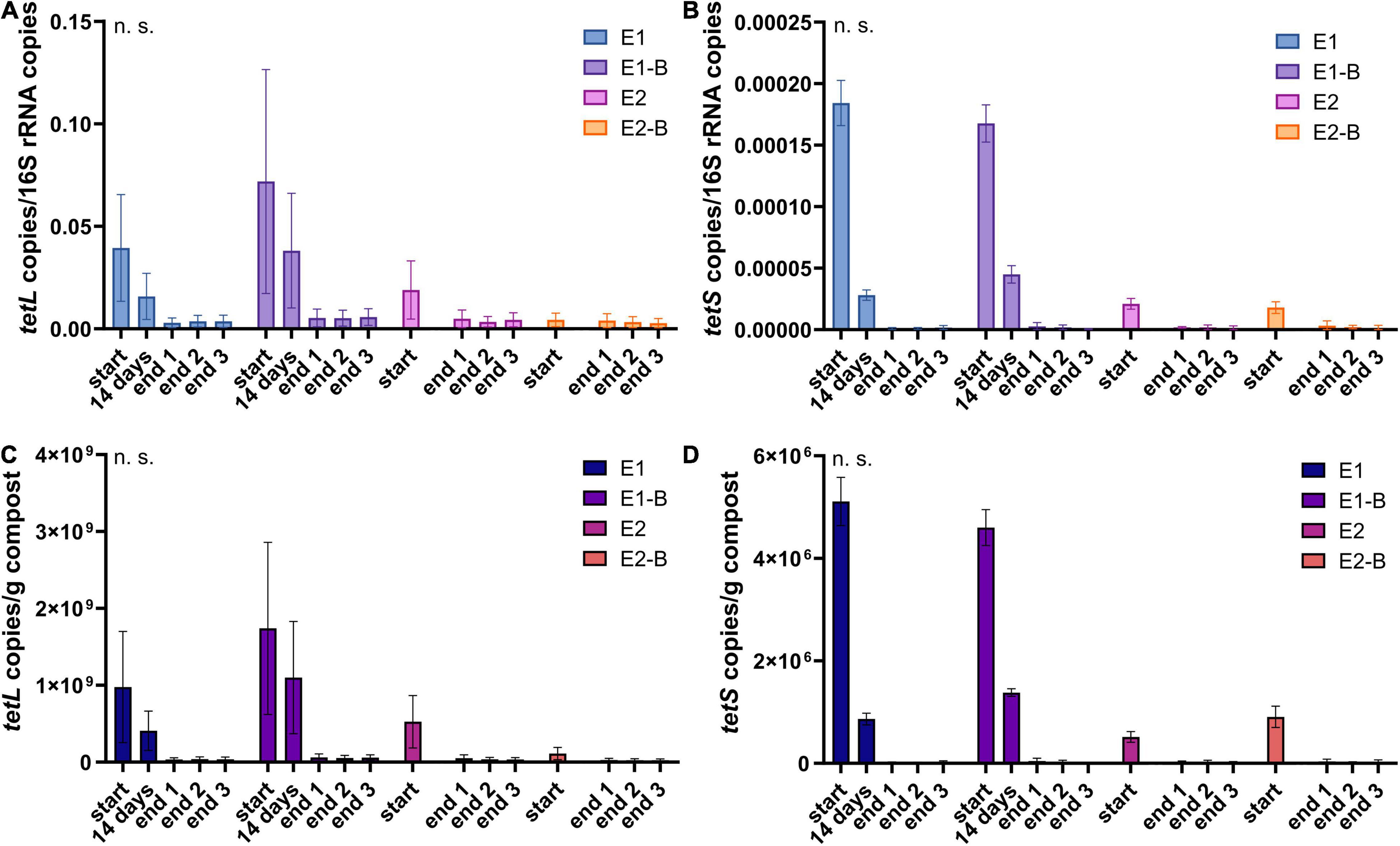
Figure 12. Copy numbers of tetracycline ARGs tetL (A,C) and tetS (B,D) as assessed by qPCR. Copy numbers were normalized to 16S rRNA gene copy numbers (upper charts) and to 1 g compost (lower charts). Bars represent the average of three individual qPCR measurements with the standard deviation. Start, 14 days (only repetition 1) and end samples are depicted individually for the first (E1, E1-B) and second repetition of composting (E2, E2-B). End samples comprise replicates from front (end 1), middle (end 2) and back (end 3) of each compost pile. Student’s t-test was conducted to test for statistical significance. Non-significant values are indicated by “n.s.” P-values were calculated to compare start with end samples of composting for all four compost piles combined (average of the triplicate end samples, no differentiation of the treatments).
The abundance of the sulfonamide resistance gene sul1 decreased from a mean of 2.0 × 109 to 1.2 × 108 copies per gram compost, sul2 from 9.6 × 108 to 2.4 × 107, the tetracycline resistance gene tetL from 8.4 × 108 to 4.1 × 107, tetS from 2.8 × 106 to 2.4 × 104, the aminoglycoside resistance genes aph(3′)-IIIa from 3.8 × 107 to 2.3 × 106, aph(2″)-Ia from 5.9 × 106 to 9.4 × 104, the erythromycin resistance gene ermA from 5.3 107 to 3.1 × 106, and ermB from 2.3 × 108 to 4.1 × 105 gene copies per gram compost (Supplementary Tables 5–8). Decreases of sul1, sul2 and aph(3′)-IIIa genes were statistically significant. Strongest decreases in relative abundance from start to end of composting were observed for ermB, followed by tetS, sul2, sul1, aph(3′)-IIIa, aph(2″)-Ia, tetL and ermA (Table 1). In general, piles exhibiting peaking gene copies in the starting material were more often found in the first repetition (E1, E1-B), whereas low starting concentrations were mostly found in the second repetition (E2, E2-B).
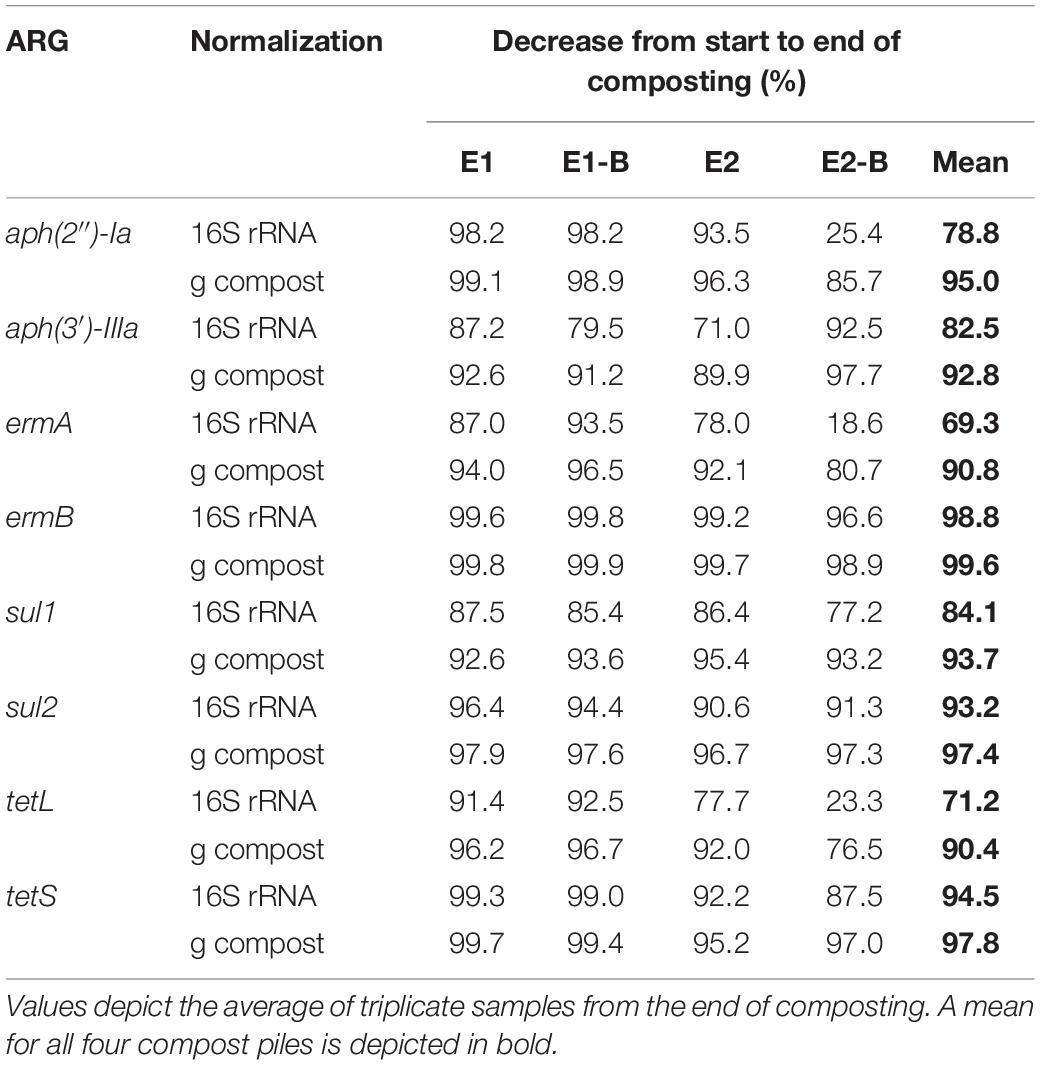
Table 1. Decreases of gene copy numbers normalized to the 16S rRNA gene and to 1 g compost from start to end of composting.
Discussion
Metagenomic sequencing was conducted on samples before and after thermophilic composting of the composting substrate (human excreta, green cuttings, and straw with and without addition of biochar) to assess possible changes in the bacterial community. We were especially interested in the fate of antibiotic resistance and bacterial pathogenicity genes.
The compost community including prokaryotes, eukaryotes, and viruses and thus its functional profile was reshaped through the composting process as suggested by PCo analyses. PCoA drew a similar picture of the dissimilarity of samples in terms of taxonomy, as well as functional genes. Differences between the treatments with and without biochar are not apparent; mature compost samples cluster close to each other indicating that the treatment had little influence on the compost community.
Decrease of Relative Abundance of Many Human Pathogens During Thermophilic Composting
Bacteria account for most organisms in the compost. The group declined in relative abundance toward the end of composting in favor of unclassified organisms, Eukaryota and Archaea. The most abundant bacterial phyla and the changes in abundance over composting mostly correspond to the 16S rRNA gene amplicons from the same samples (Supplementary Figure 1). Differences in relative abundances of bacterial phyla might be due to possible primer and amplification biases in 16S rRNA gene amplicon sequencing (Liu et al., 2008; Větrovský and Baldrian, 2013; Yang et al., 2016; Gołębiewski and Tretyn, 2020). Besides, different databases for taxonomic classification were used that might have caused differences (Silva, NCBI).
Cultivation-based assessment of indicator organisms for human pathogens (MPN technique for Salmonella and E. coli) was described in Werner et al. (2022). E. coli MPNs were found to be below the German threshold values for fertilizers, whereas Salmonella was detected in one of the four compost piles not matching German regulations for fertilizer. Moreover, an isolation study focusing on human pathogens was performed on the mature compost. In the metagenomic study, the relative abundance of human pathogenic microorganisms mostly declined during composting. Several Pseudomonas species showed a strong decrease in relative abundance, as well as A. faecalis, S. maltophilia, L. monocytogenes, E. faecium, Enterococcus spp., and Staphylococcus spp. Pseudomonas species and S. maltophilia are known pathogens. They are mostly opportunistic and have been isolated from diverse habitats, like soil, the rhizosphere and water (Aujoulat et al., 2012; Brooke, 2012; De-la-Peña and Loyola-Vargas, 2014). A. faecalis is a rather uncommon nosocomial pathogen that has also been isolated from soil, water, or the human gut. It has also been found in compost, where it is involved in nitrification and denitrification processes (Tena et al., 2015; Chen et al., 2021). L. monocytogenes, a common foodborne pathogen, present in soil and aquatic environments, is especially harmful to elderly or immunocompromised patients (Schlech, 2019). Enterococci and staphylococci pose a high risk to public health due to strains harboring resistances to many different antibiotic classes. Vancomycin-resistant E. faecium and methicillin-resistant and vancomycin- intermediate and -resistant S. aureus were listed as high priority pathogens for research and development of new antibiotics by the WHO (Tacconelli et al., 2018).
Species increasing in relative abundance during composting belong to the genera Ralstonia, Burkholderia, and Leishmania. The first are rather known as environmental bacteria, although few species can cause disease in patients with preconditions that are often difficult to treat due to multiple resistances (Ryan and Adley, 2014). Burkholderia is likewise considered as an environmental clade of mostly saprophytic species with high potential for bioremediation due to the degradation of aromatic compounds. B. mallei is the only obligate pathogen, several others are known as opportunistic and are often resistant to several antimicrobials (Rhodes and Schweizer, 2016; Morya et al., 2020). Ralstonia and especially Burkholderia spp. have been found dominant during poultry manure composting as assessed by 16S rRNA gene DGGE (Omar et al., 2015). Leishmania is a genus of obligate protozoan parasites with a lifecycle in sand flies and mammals. Approximately 20 species are pathogenic to humans. The increase in Leishmania DNA during composting is interesting, since the organism needs a host to proliferate. One explanation could be infected rodents that cause entry of Leishmania or their genes into the compost heaps (Akhoundi et al., 2016). Another reason could be breeding sandflies. Breeding and larvae habitats of sandflies are not well understood. Eggs have been found in diverse non-aquatic environments, usually moist habitats rich in organic matter (Killick-Kendrick, 1999; Feliciangeli, 2004; Vivero et al., 2015).
L. pneumophila is the etiological agent of the Legionnaires’ disease and Pontiac fever and is usually found in water systems and aquatic environments. Biofilms or a parasitic lifestyle in amoeba, protozoa or nematode hosts allow the species to survive in natural habitats. Moreover, L. pneumophila can change between a transmissive and a replicative phase. The latter takes place in nutrient-rich settings, which might explain the rise of abundance of this microorganism in our study (Abu Khweek and Amer, 2018). Besides, temperatures of 45–50°C were found optimal for L. pneumophila in water distribution systems and only temperatures of 55°C prevent outbreaks (Lin et al., 1998; Darelid et al., 2002). Legionella spp., amongst others L. pneumophila, have been isolated from different green waste composts particularly in low-temperature composts (Casati et al., 2010; Conza et al., 2013). It can therefore be assumed that the poor temperature development in the compost trial with peak temperatures between 51 and 57°C was insufficient to reduce the abundance of L. pneumophila. Roseomonas gilardii is a rare opportunistic pathogen causing bacteremia with low morbidity and mortality in humans. It was originally isolated from drinking water and has since then mostly been described in clinical samples. The natural habitat is not known, the bacterium might, however, be associated with humans as a commensal. Due to its ability to utilize urea, the compost environment could have favored its growth (Rihs et al., 1993; Shokar et al., 2002; Schlappi et al., 2020).
The metagenomic data suggested the putative presence of several human pathogens. Severe pathogens showed a decline during composting. Mostly environmental genera with few opportunistic representatives or species less common as pathogens (besides L. pneumophila) increased in relative abundance over composting. L. pneumophila growth was presumably linked to poor temperature development during the composting trial. The treatments with and without biochar did not exhibit differences regarding the microbial community or pathogenic strains.
Thermophilic Composting as an Effective Treatment to Decrease Bacterial Pathogenicity and the Dissemination of Antibiotic Resistance Genes
ARGs of major classes of antibiotics decreased in relative abundance from start to end of composting. In a recently published, comprehensive study Keenum et al. (2021) compared start and mature compost from dairy and cattle manure under antibiotic administration and without administration. They analyzed relative abundance (16S rRNA gene normalized) of ARGs using metagenomic sequencing. Strongest decreases from start to end of composting were observed for trimethoprim, tetracycline and MLSB resistance genes. In our study, trimethoprim resistance genes also exhibited strongest decreases of 99% from start to end, followed by sulfamethoxazole and rifamycin resistance genes. Tetracycline (54%) and MLSB resistance genes (37%) showed less pronounced declines in relative abundance through composting.
In a metagenomic study on agricultural soils fertilized with untreated and composted cattle manure, total relative ARG abundance did not reveal differences. However, application of composted manure reduced the ARG diversity in the soil by 21%, whereas increases in diversity were detected for raw manure application (Wind et al., 2021). Guron et al. (2019) compared ARG abundance and diversity on lettuce and radish fertilized with raw and composted cattle manure, and fertilizer as a control. Vegetable type had the strongest influence on the ARG abundance and diversity, followed by soil type. Composting as a pre-treatment resulted in a different ARG profile, but not in different relative abundances when compared with raw manure. Another metagenomic study of ARGs on vegetables grown in soil treated with raw or composted chicken litter revealed decreased relative abundance of total ARGs for compost-application, despite increases of specific ARGs (Subirats et al., 2021). Although fertilization with compost differs from the composting process itself and therefore cannot be compared directly, the field trials with raw vs. composted manure show interesting similarities with our study (starting material vs. finished compost). In our study, the reduction of ARGs differed for the different classes of ARGs, which likewise changed the resistance profile from start to end of the composting process, during which the total relative ARG abundance decreased. Single genes exhibited a contrary tendency, which agrees with the findings on compost-fertilized vegetables by Subirats et al. (2021). Metagenomic analyses on animal manure compost and its influence on agricultural soils and vegetables show advantages of composting over raw manure application in terms of ARG abundance and diversity.
qPCR results on the fate of individual ARGs during animal manure composting differ in the literature. Decreases were reported for beta-lactam, macrolide, sulfonamide, tetracycline and quinolone resistance genes during swine and poultry manure composting (Wang et al., 2016; Guo et al., 2017; Chen Z. et al., 2018; Esperón et al., 2020), while increases in aminoglycoside, tetracycline and sulfonamide resistance genes (especially sul1) were detected during cattle, swine and chicken manure composting (Li et al., 2017; Cheng et al., 2019; Duan et al., 2019; Esperón et al., 2020). Studies on sewage sludge composting detected a variety of beta-lactam, macrolide, sulfonamide, tetracycline and quinolone resistance genes with different tendencies of increase or decrease during the composting process (Jang et al., 2018; Liao et al., 2018; Cui et al., 2020; Wei et al., 2020). Mechanisms of ARG reduction in animal raw and treated waste are mostly unknown and should be focused on in future research (Pereira et al., 2021).
The marker genes analyzed in the present study showed strong decreases of copy numbers during composting (> 90% for all genes normalized to 1 g compost, > 70% when normalized to 16S rRNA gene copy numbers). Copy numbers of the same gene usually ranged within the same order of magnitude in the end samples despite different levels in the start samples. Highest decreases of around 99% were observed for erythromycin resistance gene ermB, which also exhibited low copy numbers in the end samples (104–105/g compost). This is in accordance with conventional PCR results from the same samples, which showed ermB in the start samples and after 14 days of composting but could not detect the gene in the end samples (Werner et al., 2022). ermB is a transferable erythromycin resistance gene present in many clinically relevant Enterobacteriaceae, in which it is often found on plasmids carrying different erm genes (Roberts, 2004; Gomes et al., 2017). ermA on the other hand was detected in all samples using conventional PCR (Werner et al., 2022), and still exhibited copy numbers of 106 in the end samples. The gene is associated with transposons, and frequently found in S. aureus, including MRSA, where it is often integrated in its chromosome (Kadlec et al., 2012; Sarrou et al., 2019). Besides Staphylococci, Actinobacillus, Peptostreptococcus, and Streptococcus host the ermA gene (Roberts, 2004). All these genera are represented in the metagenomic data in both start and end samples (data not shown), which might explain the comparatively high copy numbers in the mature compost.
The relative abundance of the tetracycline resistance gene tetL decreased least over composting and showed rather high copy numbers of 107/g compost in the end samples, compared with other genes analyzed. Correspondingly, tetL detection via the less sensitive conventional PCR revealed positive results for both start and end samples with few exceptions (Werner et al., 2022). The TetL protein functions as an antiporter not only for tetracycline, but also for sodium and potassium ions and is therefore beneficial for K+ acquisition and during Na+ and alkali stresses (Krulwich et al., 2001). This multifunctional role might explain the high level of tetL after composting. In addition, the gene is not only present on plasmids, but was also detected in the genome of Bacillus subtilis (Krulwich et al., 2001). 16S rRNA gene amplicon sequencing of the same sample set revealed Bacilli as the class highest in relative abundance, and B. subtilis as the most frequently isolated species from mature compost (Werner et al., 2022). TetS follows a different mode of action as the antiporter TetL. It functions as a protection protein at the ribosomal binding site of tetracycline antibiotics. It was found in both Gram-positive and Gram-negative genera, all of which have been detected in the metagenome (data not shown): Citrobacter, Enterococcus, Klebsiella, Lactobacillus, Lactococcus, Listeria, Staphylococcus, Streptococcus, and Veillonella. However, the gene was not originally associated with mobile genetic elements and is not among the frequently detected tet-genes (Roberts, 2005; Roberts and Schwarz, 2016). This corresponds with lowest abundances among all analyzed genes in both the start material and the mature compost. The same trends were found using conventional PCR: tetS was only detected in E1 and E1-B in the start and 14-days samples (Werner et al., 2022).
qPCR revealed decreases of aph(2″)-Ia to comparatively low abundances of 104 copies/g compost in the mature compost. The gene is located downstream of aac(6′)-Ie on Tn5281 which is often localized on conjugative plasmids resulting in a bifunctional enzyme aac(6′)-Ie-aph(2″)-Ia conferring resistance to most aminoglycoside antibiotics (Vakulenko and Mobashery, 2003; Hegstad et al., 2010; Rosvoll et al., 2012). It is associated with Enterococcus, Streptococcus, and Staphylococcus spp. (Vakulenko and Mobashery, 2003), which were found to decrease in the metagenome. Kanamycin/amikacin resistance gene aph(3′)-IIIa revealed comparatively low levels in both start and end samples with intermediate decreases, which matches the PCR results showing aph(3′)-IIIa in samples taken at the start and after 14 days of composting, but not in samples taken at the end (Werner et al., 2022). Aph(3′) is the largest group of clinically relevant aminoglycoside phosphotransferases. aph(3′)-III has been frequently found in pathogens like enterococci and staphylococci, including MRSA, and is often located in the vicinity of genes conferring resistance to other antibiotics (Vakulenko and Mobashery, 2003).
Conventional PCR analysis of the sulfonamide resistance genes sul1 and sul2 was positive for all samples (Werner et al., 2022). Using qPCR, both genes exhibited significant decreases during composting. Before composting, gene copy numbers were high for both genes (108–109/g compost). sul1 was still present with an average of 108 copies/g compost in the mature compost, whereas sul2 was reduced to 107 copies/g compost. sul1 and sul2 are located on transposons and plasmids of Gram-negative bacteria and are often associated with other resistances. Their dissemination is therefore very effective, which might be a reason that the genes have often been found after composting, as well (Enne et al., 2001; Sköld, 2001). Besides, the sulfonamide sulfamethazine is harder to degrade through composting than other antibiotics. Thus, selective pressure maintains and promotes the persistence of sul genes (Youngquist et al., 2016).
The qPCR results complement our conventional PCR study (Werner et al., 2022) by demonstrating pronounced decreases of clinically relevant resistance genes for gentamicin, kanamycin/amikacin, sulfonamide, and tetracycline during thermophilic composting of human excreta. Differences between the two treatments with and without biochar were not observed, possibly due to rather low biochar contents. Studies comparing different biochar contents found stronger decreases of ARG abundances in composts containing biochar than in composts without biochar. However, increments of the biochar content did not significantly improve ARG decreases. This was shown for swine manure compost with 6, 12, and 24% biochar, where 6% biochar improved ARG removal and higher percentages revealed the opposite effect (Wang et al., 2018). Li et al. (2017) compared chicken manure compost supplemented with 0, 5, 10, and 20% bamboo charcoal. 10% biochar was found to achieve the best results regarding ARG removal (Li et al., 2017). In a recent study on municipal sludge three treatments apart from the control without additives were tested: addition of hyperthermophiles, hyperthermophiles with 2% biochar, hyperthermophiles with 5% biochar. Biochar significantly increased ARG removal rates, but no significant differences between the different biochar contents were found (Fu et al., 2021). Further research would be needed to understand the mechanisms behind different biochar or compost types and biochar contents on ARG removal.
T4SSs enable horizontal gene transfer through conjugation in Gram-negative and Gram-positive bacteria, as well as in some archaea. They have been encountered in most bacterial species and are therefore crucial, when studying horizontal gene transfer in clinically relevant bacteria like Enterococcus, Clostridium, Staphylococcus, or Streptococcus spp. Beside their role in the dissemination of ARGs, T4SSs are involved in the transfer of virulence factors, in biofilm formation, competition with other microbes through killing toxins, and injection of effector proteins into host cells (Grohmann et al., 2003, 2018; Goessweiner-Mohr et al., 2014). The decrease in relative abundance of T4SS genes over composting in the present study indicates that the potential for horizontal gene transfer, as well as bacterial pathogenicity is reduced. This has been shown for manure composts before by quantifying the integrase gene intI1 and transposase genes as markers for horizontal gene transfer (Wang et al., 2021; Zhou et al., 2021). Biochar addition to the compost increased intI1 removal efficiency during composting (Zhou et al., 2021). This might be due to higher temperatures and a longer thermophilic phase through biochar (Godlewska et al., 2017). The importance of high temperatures for removal efficiencies of integrase genes during composting was demonstrated by Qian et al. (2016). In the present study biochar did not increase temperatures during the thermophilic phase, and temperature development was poor in general, which might explain that relative T4SS gene abundances did not differ between the treatments with and without the addition of biochar.
The decrease in pathogenicity was further demonstrated by the decrease in genes associated with T3SSs and T6SSs in our study. T3SSs are common in many Gram-negative bacteria and have often been acquired through horizontal gene transfer. Pathogens like Pseudomonas, Yersinia, Shigella or E. coli use T3SSs to inject effector proteins into eukaryotic cells (Büttner, 2012; Green and Mecsas, 2016). Functional diversity of T6SSs could explain their comparatively high relative abundance in start and end samples, respectively. The primary function of T6SSs is effector protein translocation to competing bacteria of the same or different species, and fungi. They are also involved in transformation of naturally competent bacteria and aid in metal scavenging providing survival benefits. In addition, effector proteins can be translocated from bacteria to eukaryotic cells and thereby they contribute to bacterial virulence (Borgeaud et al., 2015; Green and Mecsas, 2016; Coulthurst, 2019).
Virulence factors comprise a broad group of functions that are used by pathogens to cause infection and enhance their pathogenicity. They include toxin production, motility, adhesins, capsules, quorum sensing and biofilm formation, as well as secretion systems. Strategies are diverse, complex and virulence factor categories are not clearly defined, e.g., quorum sensing being essential for bacteria to form biofilms (Casadevall and Pirofski, 2009; Hill, 2012).
We assessed the abundance of the functional groups, quorum sensing, bacterial motility, flagellar assembly, chemotaxis, toxins, invasion of epithelial cells, and biofilm formation, and found decreases in relative abundance for all groups of genes. Studies on compost and virulence mainly focus on plant pathogenicity rather than on the impact on human health. They usually assess plant-waste compost, which is not well comparable to our study (Lim et al., 2014; Antoniou et al., 2017; Andreazza et al., 2019). Indicator organisms for plant pathogenicity according to German and European regulations for fertilizer comprise the genus Tobamovirus, especially Tobacco mosaic viruses, the clubroot pathogen Plasmodiophora brassicae, as well as the fungal species Synchytrium endobioticum, Rhizoctonia solani, and Sclerotinia spp. (BioAbfV, 1998; Carrington, 2001; DüMV, 2012). All these indicators were detected in the start and mature compost samples (data not shown).
Sidhu et al. (2017) compared raw and treated sludge from a wastewater treatment plant and found decreases of microbial genes involved in signal transduction, cell motility and chemotaxis. Although the treatment was different from our study, the results indicate that fecal material is usually rich in potential virulence factors that decline through treatment changing the conditions for the microbial community. As virulence factors are not solely used by pathogenic microorganisms, it is not likely that the respective genes disappear completely (Hill, 2012). In fact, bacterial secretion systems or some classes of ARGs exhibited stronger decreases than other groups of virulence genes. Especially the relative abundance of quorum sensing genes exhibited only minor declines. This is most likely due to its involvement in numerous bacterial processes that are not exclusively used for infection, but also for competition and cooperation with other microbes (Zhao et al., 2020). N-Acyl homoserine lactone (AHL)-mediated quorum sensing has also been described in soils supplemented with compost, which might be a hint for the natural occurrence of quorum sensing systems in compost (Burmølle et al., 2005).
In summary, we have conducted a metagenomic analysis of compost samples before and after thermophilic composting of human excreta from dry toilets together with green cuttings and straw, with and without biochar, to assess the influence of the process on human pathogens, antibiotic resistance, and bacterial virulence. In terms of pathogenicity and antibiotic resistance, the addition of biochar had no positive effect. Severe pathogenic species decreased in relative abundance, the reverse tendency was generally found in less pathogenic or opportunistic species. The overall level of ARGs was lower after composting. Resistance gene analyses using qPCR showed strong decreases of genes conferring resistance to erythromycin, aminoglycosides, sulfonamide, and tetracyclines over composting. Declines in the relative abundance of T4SSs indicate that the potential for horizontal gene transfer was also reduced. Pathogenicity strategies like T3SSs, biofilm formation capacity, chemotaxis or bacterial motility also exhibited decreases in relative abundance, thereby suggesting that the infection potential of the bacterial community was reduced through composting. All these strategies are also used by non-pathogenic bacteria to compete with the surrounding microbes. Thus, it would be interesting to assess a natural “background” level in a non-anthropogenic environment by evaluating the impact of compost fertilization in comparison to other organic fertilizers concerning the potential spread of antibiotic resistances. Since the study used DNA-based analysis, the results reflect the general potential of the compost community, but not the actually expressed functions. Metatranscriptomic analysis would be interesting to assess how the composting process changes functional gene expression.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in Supplementary Table S1: NCBI—PRJNA782422.
Author Contributions
EG and KW designed and supervised the laboratory experiments. KW, DS, AP, ND, LF, and KA performed the laboratory experiments and analyzed the data. KP, NB, and TH designed the composting trial. TH conducted process control. KW, DS, AP, TH, and EG wrote the manuscript and designed all figures and tables. RD contributed with insightful discussions on analysis and interpretation of the data. All authors interpreted the results, read, revised the manuscript, contributed to the article, and approved the submitted version.
Funding
Financial support of BMBF (Federal Ministry of Education and Research) is highly acknowledged (ClimEtSan-Pathogene, grant 01DG18005 to EG, being part of the ClimEtSan-Project, funded by BMBF grant 01DG17010 and the German Academic Exchange Service, DAAD, grant 57354010).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We want to express our gratitude to Finizio GmbH and Goldeimer gGmbH, especially Florian Augustin and Enno Schröder, for the opportunity to join the composting trial, as well as their helpfulness and dedication throughout the project. We highly acknowledge the possibility to conduct the composting trials at the “Recyclinghof Eckernförde.” We thank Melanie Heinemann for technical support. KW thanks the “Agriculture and Environment Foundation” for the Ph.D. fellowship.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.826071/full#supplementary-material
Footnote
- ^ hmmsearch, http://hmmer.org/
References
Abu Khweek, A., and Amer, A. O. (2018). Factors mediating environmental biofilm formation by Legionella pneumophila. Front. Cell. Infect. Microbiol. 8:38. doi: 10.3389/fcimb.2018.00038
Agegnehu, G., Srivastava, A. K., and Bird, M. I. (2017). The role of biochar and biochar-compost in improving soil quality and crop performance: a review. Appl. Soil Ecol. 119, 156–170. doi: 10.1016/j.apsoil.2017.06.008
Akhoundi, M., Kuhls, K., Cannet, A., Votýpka, J., Marty, P., Delaunay, P., et al. (2016). A historical overview of the classification, evolution, and dispersion of Leishmania parasites and sandflies. PLoS Negl. Trop. Dis. 10:e0004349. doi: 10.1371/journal.pntd.0004349
Amlinger, F., Peyr, S., Geszti, J., Dreher, P., Karlheinz, W., and Nortcliff, S. (2007). Beneficial effects of compost application on fertility and productivity of soils. Austria: Federal Ministry for Agriculture and Forestry, Environment and Water Management, 32–75.
Andersen, K. S., Kirkegaard, R. H., Karst, S. M., and Albertsen, M. (2018). ampvis2: an R package to analyse and visualise 16S rRNA amplicon data. BioRxiv [Preprint]. doi: 10.1101/299537
Andreazza, A. P., Cardoso, R. L. A., Cocco, J., Guizelini, D., Faoro, H., Tadra-Sfeir, M. Z., et al. (2019). Genome analysis of entomopathogenic Bacillus sp. ABP14 isolated from a lignocellulosic compost. Genome Biol. Evol. 11, 1658–1662. doi: 10.1093/gbe/evz114
Antoniou, A., Tsolakidou, M. D., Stringlis, I. A., and Pantelides, I. S. (2017). Rhizosphere microbiome recruited from a suppressive compost improves plant fitness and increases protection against vascular wilt pathogens of tomato. Front. Plant Sci. 8:2022. doi: 10.3389/fpls.2017.02022
Aujoulat, F., Roger, F., Bourdier, A., Lotthé, A., Lamy, B., Marchandin, H., et al. (2012). From environment to man: genome evolution and adaptation of human opportunistic bacterial pathogens. Genes 3, 191–232. doi: 10.3390/genes3020191
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bernal, M. P., Alburquerque, J. A., and Moral, R. (2009). Composting of animal manures and chemical criteria for compost maturity assessment. A review. Bioresour. Technol. 100, 5444–5453. doi: 10.1016/j.biortech.2008.11.027
BioAbfV (1998). Bioabfallverordnung in der Fassung der Bekanntmachung vom 4. April 2013 (BGBl. I S. 658), die Zuletzt durch Artikel 3 Absatz 2 der Verordnung vom 27. September 2017 (BGBl. I S. 3465) Geändert Worden ist. Bonn: Federal Ministry for the Environment, Nature Conservation, Nuclear Safety and Consumer Protection.
Borgeaud, S., Metzger, L. C., Scrignari, T., and Blokesch, M. (2015). The type VI secretion system of Vibrio cholerae fosters horizontal gene transfer. Science 347, 63–67. doi: 10.1126/science.1260064
Brooke, J. S. (2012). Stenotrophomonas maltophilia: an emerging global opportunistic pathogen. Clin. Microbiol. Rev. 25, 2–41. doi: 10.1128/CMR.00019-11
Burmølle, M., Hansen, L. H., and Sørensen, S. J. (2005). Use of a whole-cell biosensor and flow cytometry to detect AHL production by an indigenous soil community during decomposition of litter. Microb. Ecol. 50, 221–229. doi: 10.1007/s00248-004-0113-8
Büttner, D. (2012). Protein export according to schedule: architecture, assembly, and regulation of type III secretion systems from plant- and animal-pathogenic bacteria. Microbiol. Mol. Biol. Rev. 76, 262–310. doi: 10.1128/mmbr.05017-11
Carrington, E. G. (2001). Evaluation of sludge treatments for pathogen reduction - final report. European Commission. Report No. CO 5026/1. Available Online at: https://ec.europa.eu/environment/archives/waste/sludge/pdf/sludge_eval.pdf (accessed December 22, 2020).
Casadevall, A., and Pirofski, L. A. (2009). Virulence factors and their mechanisms of action: the view from a damage-response framework. J. Water Health 7, 2–18. doi: 10.2166/wh.2009.036
Casati, S., Conza, L., Bruin, J., and Gaia, V. (2010). Compost facilities as a reservoir of Legionella pneumophila and other Legionella species. Clin. Microbiol. Infect. 16, 945–947. doi: 10.1111/j.1469-0691.2009.03009.x
Chen, L., Chen, L., Pan, D., Lin, H., Ren, Y., Zhang, J., et al. (2021). Heterotrophic nitrification and related functional gene expression characteristics of Alcaligenes faecalis SDU20 with the potential use in swine wastewater treatment. Bioproc. Biosyst. Eng. 44, 2035–2050. doi: 10.1007/s00449-021-02581-z
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Chen, Z., Wang, Y., and Wen, Q. (2018). Effects of chlortetracycline on the fate of multi-antibiotic resistance genes and the microbial community during swine manure composting. Environ. Pollut. 237, 977–987. doi: 10.1016/j.envpol.2017.11.009
Cheng, D., Feng, Y., Liu, Y., Xue, J., and Li, Z. (2019). Dynamics of oxytetracycline, sulfamerazine, and ciprofloxacin and related antibiotic resistance genes during swine manure composting. J. Environ. Manag. 230, 102–109. doi: 10.1016/j.jenvman.2018.09.074
Conza, L., Pagani, S. C., and Gaia, V. (2013). Presence of Legionella and free-living amoebae in composts and bioaerosols from composting facilities. PLoS One 8:e68244. doi: 10.1371/journal.pone.0068244
Coulthurst, S. (2019). The type VI secretion system: a versatile bacterial weapon. Microbiology 165, 503–515. doi: 10.1099/mic.0.000789
Cui, E., Wu, Y., Zuo, Y., and Chen, H. (2016). Effect of different biochars on antibiotic resistance genes and bacterial community during chicken manure composting. Bioresour. Technol. 203, 11–17. doi: 10.1016/j.biortech.2015.12.030
Cui, P., Bai, Y., Li, X., Peng, Z., Chen, D., Wu, Z., et al. (2020). Enhanced removal of antibiotic resistance genes and mobile genetic elements during sewage sludge composting covered with a semi-permeable membrane. J. Hazard. Mater. 396:122738. doi: 10.1016/j.jhazmat.2020.122738
Dalzell, H. W., Dalzell, H. E., Biddlestone, A. J., Gray, K. R., and Thurairajan, K. (1987). Soil management: compost production and use in tropical and subtropical environments. In FAO Soils Bulletin, 56th Edn. Rome: Food and Agriculture Organization of the United Nations.
Darelid, J., Löfgren, S., and Malmvall, B. E. (2002). Control of nosocomial Legionnaires’ disease by keeping the circulating hot water temperature above 55°C: experience from a 10-year surveillance programme in a district general hospital. J. Hosp. Infect. 50, 213–219. doi: 10.1053/jhin.2002.1185
De-la-Peña, C., and Loyola-Vargas, V. M. (2014). Biotic interactions in the rhizosphere: a diverse cooperative enterprise for plant productivity. Plant Physiol. 166, 701–719. doi: 10.1104/pp.114.241810
Dobrindt, U., Hochhut, B., Hentschel, U., and Hacker, J. (2004). Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2, 414–424. doi: 10.1038/nrmicro884
Duan, M., Zhang, Y., Zhou, B., Wang, Q., Gu, J., Liu, G., et al. (2019). Changes in antibiotic resistance genes and mobile genetic elements during cattle manure composting after inoculation with Bacillus subtilis. Bioresour. Technol. 292:122011. doi: 10.1016/j.biortech.2019.122011
DüMV (2012). Düngemittelverordnung vom 5. Dezember 2012 (BGBl. I S. 2482), die zuletzt durch Artikel 3 der Verordnung vom 26. Mai 2017 (BGBl. I S. 1305) geändert worden ist. Available Online at: https://www.gesetze-im-internet.de/d_mv_2012/DüMV.pdf (accessed December 22, 2020).
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Enne, V. I., Livermore, D. M., Stephens, P., and Hall, L. M. C. (2001). Persistence of sulphonamide resistance in Escherichia coli in the UK despite national prescribing restriction. Lancet 357, 1325–1328. doi: 10.1016/S0140-6736(00)04519-0
Esperón, F., Albero, B., Ugarte-Ruíz, M., Domínguez, L., Carballo, M., Tadeo, J. L., et al. (2020). Assessing the benefits of composting poultry manure in reducing antimicrobial residues, pathogenic bacteria, and antimicrobial resistance genes: a field-scale study. Environ. Sci. Pollut. Res. 27, 27738–27749. doi: 10.1007/s11356-020-09097-1
Ezzariai, A., Hafidi, M., Khadra, A., Aemig, Q., El Fels, L., Barret, M., et al. (2018). Human and veterinary antibiotics during composting of sludge or manure: global perspectives on persistence, degradation, and resistance genes. J. Hazard. Mater. 359, 465–481. doi: 10.1016/j.jhazmat.2018.07.092
Feliciangeli, M. D. (2004). Natural breeding places of phlebotomine sandflies. Med. Vet. Entomol. 18, 71–80. doi: 10.1111/j.0269-283X.2004.0487.x
Fischer, D., and Glaser, B. (2012). “Synergisms between compost and biochar for sustainable soil amelioration,” in Management of Organic Waste, ed. S. Kumar (London, UK: In Tech), doi: 10.5772/31200
Fu, Y., Zhang, A., Guo, T., Zhu, Y., and Shao, Y. (2021). Biochar and hyperthermophiles as additives accelerate the removal of antibiotic resistance genes and mobile genetic elements during composting. Materials 14:5428. doi: 10.3390/ma14185428
Gal-Mor, O., and Finlay, B. B. (2006). Pathogenicity islands: a molecular toolbox for bacterial virulence. Cell. Microbiol. 8, 1707–1719. doi: 10.1111/j.1462-5822.2006.00794.x
Gibson, M. K., Forsberg, K. J., and Dantas, G. (2015). Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J. 9, 207–216. doi: 10.1038/ismej.2014.106
Gillings, M. R. (2014). Integrons: past, present, and future. Microbiol. Mol. Biol. Rev. 78, 257–277. doi: 10.1128/mmbr.00056-13
Godlewska, P., Schmidt, H. P., Ok, Y. S., and Oleszczuk, P. (2017). Biochar for composting improvement and contaminants reduction. A review. Bioresour. Technol. 246, 193–202. doi: 10.1016/j.biortech.2017.07.095
Goessweiner-Mohr, N., Arends, K., Keller, W., and Grohmann, E. (2014). Conjugation in gram-positive bacteria. Microbiol. Spectr. 2:PLAS-0004-2013. doi: 10.1128/microbiolspec.PLAS-0004-2013
Gołębiewski, M., and Tretyn, A. (2020). Generating amplicon reads for microbial community assessment with next-generation sequencing. J. Appl. Microbiol. 128, 330–354. doi: 10.1111/jam.14380
Golkar, T., Zielinski, M., and Berghuis, A. M. (2018). Look and outlook on enzyme-mediated macrolide resistance. Front. Microbiol. 9:1942. doi: 10.3389/fmicb.2018.01942
Gomes, C., Martínez-Puchol, S., Palma, N., Horna, G., Ruiz-Roldán, L., Pons, M. J., et al. (2017). Macrolide resistance mechanisms in Enterobacteriaceae: focus on azithromycin. Crit. Rev. Microbiol. 43, 1–30. doi: 10.3109/1040841X.2015.1136261
Graham, D. W., Knapp, C. W., Christensen, B. T., McCluskey, S., and Dolfing, J. (2016). Appearance of β-lactam resistance genes in agricultural soils and clinical isolates over the 20th century. Sci. Rep. 6:21550. doi: 10.1038/srep21550
Green, E. R., and Mecsas, J. (2016). Bacterial secretion systems: an overview. Microbiol. Spectr. 4:10.1128/microbiolspec.VMBF-0012-2015. doi: 10.1128/microbiolspec.VMBF-0012-2015
Grohmann, E., Christie, P. J., Waksman, G., and Backert, S. (2018). Type IV secretion in gram-negative and gram-positive bacteria. Mol. Microbiol. 107, 455–471. doi: 10.1111/mmi.13896
Grohmann, E., Muth, G., and Espinosa, M. (2003). Conjugative plasmid transfer in gram-positive bacteria. Microbiol. Mol. Biol. Rev. 67, 277–301. doi: 10.1128/mmbr.67.2.277-301.2003
Guo, A., Gu, J., Wang, X., Zhang, R., Yin, Y., Sun, W., et al. (2017). Effects of superabsorbent polymers on the abundances of antibiotic resistance genes, mobile genetic elements, and the bacterial community during swine manure composting. Bioresour. Technol. 244, 658–663. doi: 10.1016/j.biortech.2017.08.016
Guron, G. K. P., Arango-Argoty, G., Zhang, L., Pruden, A., and Ponder, M. A. (2019). Effects of dairy manure-based amendments and soil texture on lettuce- and radish-associated microbiota and resistomes. mSphere 4, e00239–19. doi: 10.1128/mSphere.00239-19
Hegstad, K., Mikalsen, T., Coque, T. M., Werner, G., and Sundsfjord, A. (2010). Mobile genetic elements and their contribution to the emergence of antimicrobial resistant Enterococcus faecalis and Enterococcus faecium. Clin. Microbiol. Infect. 16, 541–554. doi: 10.1111/j.1469-0691.2010.03226.x
Hill, C. (2012). Virulence or niche factors: what’s in a name? J. Bacteriol. 194, 5725–5727. doi: 10.1128/JB.00980-12
Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Jang, H. M., Lee, J., Kim, Y. B., Jeon, J. H., Shin, J., Park, M.-R., et al. (2018). Fate of antibiotic resistance genes and metal resistance genes during thermophilic aerobic digestion of sewage sludge. Bioresour. Technol. 249, 635–643. doi: 10.1016/j.biortech.2017.10.073
Jindo, K., Sánchez-Monedero, M. A., Hernández, T., García, C., Furukawa, T., Matsumoto, K., et al. (2012). Biochar influences the microbial community structure during manure composting with agricultural wastes. Sci. Total Environ. 416, 476–481. doi: 10.1016/j.scitotenv.2011.12.009
Kadlec, K., Feßler, A. T., Hauschild, T., and Schwarz, S. (2012). Novel and uncommon antimicrobial resistance genes in livestock-associated methicillin-resistant Staphylococcus aureus. Clin. Microbiol. Infect. 18, 745–755. doi: 10.1111/j.1469-0691.2012.03842.x
Kanehisa, M., Goto, S., Furumichi, M., Tanabe, M., and Hirakawa, M. (2009). KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 38, 355–360. doi: 10.1093/nar/gkp896
Keenum, I., Williams, R. K., Ray, P., Garner, E. D., Knowlton, K. F., and Pruden, A. (2021). Combined effects of composting and antibiotic administration on cattle manure–borne antibiotic resistance genes. Microbiome 9:81. doi: 10.1186/s40168-021-01006-z
Killick-Kendrick, R. (1999). The biology and control of Phlebotomine sand flies. Clin. Dermatol. 17, 279–289. doi: 10.1016/S0738-081X(99)00046-2
Knapp, C. W., Dolfing, J., Ehlert, P. A. I., and Graham, D. W. (2010). Evidence of increasing antibiotic resistance gene abundances in archived soils since 1940. Environ. Sci. Technol. 44, 580–587. doi: 10.1021/es901221x
Krulwich, T. A., Jin, J., Guffanti, A. A., and Bechhofer, D. H. (2001). Functions of tetracycline efflux proteins that do not involve tetracycline. J. Mol. Microbiol. Biotechnol. 3, 237–246.
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lartigue, M. F., Poirel, L., Aubert, D., and Nordmann, P. (2006). In vitro analysis of ISEcp1B-mediated mobilization of naturally occurring β-lactamase gene blaCTX–M of Kluyvem ascorbata. Antimicrob. Agents Chemother. 50, 1282–1286. doi: 10.1128/AAC.50.4.1282-1286.2006
Laxminarayan, R., Duse, A., Wattal, C., Zaidi, A. K. M., Wertheim, H. F. L., Sumpradit, N., et al. (2013). Antibiotic resistance-the need for global solutions. Lancet Infect. Dis. 13, 1057–1098. doi: 10.1016/S1473-3099(13)70318-9
Li, H., Duan, M., Gu, J., Zhang, Y., Qian, X., Ma, J., et al. (2017). Effects of bamboo charcoal on antibiotic resistance genes during chicken manure composting. Ecotoxicol. Environ. Saf. 140, 1–6. doi: 10.1016/j.ecoenv.2017.01.007
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Liao, H., Lu, X., Rensing, C., Friman, V. P., Geisen, S., Chen, Z., et al. (2018). Hyperthermophilic composting accelerates the removal of antibiotic resistance genes and mobile genetic elements in sewage sludge. Environ. Sci. Technol. 52, 266–276. doi: 10.1021/acs.est.7b04483
Lim, Y. L., Ee, R., Yin, W. F., and Chan, K. G. (2014). Quorum sensing activity of Aeromonas caviae strain YL12, a bacterium isolated from compost. Sensors 14, 7026–7040. doi: 10.3390/s140407026
Lin, Y. E., Stout, J. E., Yu, V. L., and Vidic, R. D. (1998). Disinfection of water distribution systems for Legionella. Semin. Respir. Infect. 13, 147–159.
Liu, Z., Desantis, T. Z., Andersen, G. L., and Knight, R. (2008). Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res. 36:e120. doi: 10.1093/nar/gkn491
Mallick, H., Rahnavard, A., McIver, L. J., Ma, S., Zhang, Y., Tickle, T. L., et al. (2021). Multivariable association discovery in population-scale meta-omics studies. BioRxiv [Preprint]. doi: 10.1101/2021.01.20.427420
Menzel, P., Ng, K. L., and Krogh, A. (2016). Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 7:11257. doi: 10.1038/ncomms11257
Morya, R., Salvachúa, D., and Thakur, I. S. (2020). Burkholderia: an untapped but promising bacterial genus for the conversion of aromatic compounds. Trends Biotechnol. 38, 963–975. doi: 10.1016/j.tibtech.2020.02.008
Omar, F. N., Xiang, L. Y., Lie, L. Y., Dzulkornain, C. A., Mohammed, M. A. P., and Baharuddin, A. S. (2015). Investigation of physico-chemical properties and microbial community during poultry manure co-composting process. J. Environ. Sci. 28, 81–94. doi: 10.1016/j.jes.2014.07.023
O’Neill, J. (2014). Review on Antimicrobial Resistance. Antimicrobial Resistance: tackling a Crisis for the Health and Wealth of Nations. Available Online at: http://www.jpiamr.eu/wp-content/uploads/2014/12/AMR-Review-Paper-Tackling-a-crisis-for-the-health-and-wealth-of-nations_1-2.pdf
Osterman, I. A., Dontsova, O. A., and Sergiev, P. V. (2020). rRNA methylation and antibiotic resistance. Biochemistry 85, 1335–1349. doi: 10.1134/S000629792011005X
Pereira, A. R., Paranhos, A. G., de, O., de Aquino, S. F., Silva, S., and de, Q. (2021). Distribution of genetic elements associated with antibiotic resistance in treated and untreated animal husbandry waste and wastewater. Environ. Sci. Pollut. Res. 28, 26380–26403. doi: 10.1007/s11356-021-13784-y
Qian, X., Sun, W., Gu, J., Wang, X. J., Zhang, Y. J., Duan, M. L., et al. (2016). Reducing antibiotic resistance genes, integrons, and pathogens in dairy manure by continuous thermophilic composting. Bioresour. Technol. 220, 425–432. doi: 10.1016/j.biortech.2016.08.101
Qiu, X., Zhou, G., Zhang, J., and Wang, W. (2019). Microbial community responses to biochar addition when a green waste and manure mix are composted: a molecular ecological network analysis. Bioresour. Technol. 273, 666–671. doi: 10.1016/j.biortech.2018.12.001
Ramirez, M. S., and Tolmasky, M. E. (2010). Aminoglycoside modifying enzymes. Drug Resist. Updates 13, 151–171. doi: 10.1016/j.drup.2010.08.003
Reid, M. (2020). Sanitation and climate. Nat. Clim. Chang. 10, 496–497. doi: 10.1038/s41558-020-0787-z
Rhodes, K. A., and Schweizer, H. P. (2016). Antibiotic resistance in Burkholderia species. Drug Resist. Updates 28, 82–90. doi: 10.1016/j.drup.2016.07.003
Rihs, J. D., Brenner, D. J., Weaver, R. E., Steigerwalt, A. G., Hollis, D. G., and Yu, V. L. (1993). Roseomonas, a new genus associated with bacteremia and other human infections. J. Clin. Microbiol. 31, 3275–3283. doi: 10.1128/jcm.31.12.3275-3283.1993
Roberts, M. C. (2004). Resistance to macrolide, lincosamide, streptogramin, ketolide, and oxazolidinone antibiotics. Mol. Biotechnol. 28, 47–62. doi: 10.1385/MB:28:1:47
Roberts, M. C. (2005). Update on acquired tetracycline resistance genes. FEMS Microbiol. Lett. 245, 195–203. doi: 10.1016/j.femsle.2005.02.034
Roberts, M. C., and Schwarz, S. (2016). Tetracycline and phenicol resistance genes and mechanisms: importance for agriculture, the environment, and humans. J. Environ. Qual. 45, 576–592. doi: 10.2134/jeq2015.04.0207
Rose, C., Parker, A., Jefferson, B., and Cartmell, E. (2015). The characterization of feces and urine: a review of the literature to inform advanced treatment technology. Crit. Rev. Environ. Sci. Technol. 45, 1827–1879. doi: 10.1080/10643389.2014.1000761
Rosvoll, T. C. S., Lindstad, B. L., Lunde, T. M., Hegstad, K., Aasnæs, B., Hammerum, A. M., et al. (2012). Increased high-level gentamicin resistance in invasive Enterococcus faecium is associated with aac(6′)Ie-aph(2″)Ia-encoding transferable megaplasmids hosted by major hospital-adapted lineages. FEMS Immunol. Med. Microbiol. 66, 166–176. doi: 10.1111/j.1574-695X.2012.00997.x
Ryals, R., Bischak, E., Porterfield, K. K., Heisey, S., Jeliazovski, J., Kramer, S., et al. (2021). Toward zero hunger through coupled ecological sanitation-agriculture systems. Front. Sustain. Food Syst. 5:716140. doi: 10.3389/fsufs.2021.716140
Ryan, M. P., and Adley, C. C. (2014). Ralstonia spp.: emerging global opportunistic pathogens. Eur. J. Clin. Microbiol. Infect. Dis. 33, 291–304. doi: 10.1007/s10096-013-1975-9
Ryckeboer, J., Mergaert, J., Vaes, K., Klammer, S., De Clercq, D., Coosemans, J., et al. (2003). A survey of bacteria and fungi occurring during composting and self-heating processes. Ann. Microbiol. 53, 349–410.
Sarrou, S., Malli, E., Tsilipounidaki, K., Florou, Z., Medvecky, M., Skoulakis, A., et al. (2019). MLSB-Resistant Staphylococcus aureus in central Greece: rate of resistance and molecular characterization. Microb. Drug Resist. 25, 543–550. doi: 10.1089/mdr.2018.0259
Schlappi, C., Bernstock, J. D., Ricketts, W., Nix, G. A., Poole, C., Lebensburger, J., et al. (2020). Roseomonas gilardii bacteremia in a patient with HbSβ0-thalassemia: clinical implications and literature review. J. Pediatr. Hematol. Oncol. 42, e385–e387. doi: 10.1097/MPH.0000000000001476
Schlech, W. F. (2019). Epidemiology and clinical manifestations of Listeria monocytogenes infection. Microbiol. Spectr. 7, 1–13.
Schönning, C., and Stenström, T. A. (2004). Guidelines for the safe use of urine and faeces in ecological sanitation systems. In EcoSanRes Publication Series. Stockholm: Stockholm Environment Institute.
Shokar, N. K., Shokar, G. S., Islam, J., and Cass, A. R. (2002). Roseomonas gilardii infection: case report and review. J. Clin. Microbiol. 40, 4789–4791. doi: 10.1128/JCM.40.12.4789-4791.2002
Sidhu, C., Vikram, S., and Pinnaka, A. K. (2017). Unraveling the microbial interactions and metabolic potentials in pre- and post-treated sludge from a wastewater treatment plant using metagenomic studies. Front. Microbiol. 8:1382. doi: 10.3389/fmicb.2017.01382
Sköld, O. (2001). Resistance to trimethoprim and sulfonamides. Vet. Res. 32, 261–273. doi: 10.1051/vetres:2001123
Subirats, J., Murray, R., Yin, X., Zhang, T., and Topp, E. (2021). Impact of chicken litter pre-application treatment on the abundance, field persistence, and transfer of antibiotic resistant bacteria and antibiotic resistance genes to vegetables. Sci. Total Environ. 801:149718. doi: 10.1016/j.scitotenv.2021.149718
Tacconelli, E., Carrara, E., Savoldi, A., Harbarth, S., Mendelson, M., and Monnet, D. L. (2018). Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 18, 318–327. doi: 10.1016/S1473-3099(17)30753-3
Tena, D., Fernáandez, C., and Lago, M. R. (2015). Alcaligenes faecalis: an unusual cause of skin and soft tissue infection. Jpn. J. Infect. Dis. 68, 128–130. doi: 10.7883/yoken.JJID.2014.164
Vakulenko, S. B., and Mobashery, S. (2003). Versatility of aminoglycosides and prospects for their future. Clin. Microbiol. Rev. 16, 430–450. doi: 10.1128/CMR.16.3.430-450.2003
Větrovský, T., and Baldrian, P. (2013). The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One 8:e57923. doi: 10.1371/journal.pone.0057923
Vivero, R. J., Torres-Gutierrez, C., Bejarano, E. E., Peña, H. C., Estrada, L. G., Florez, F., et al. (2015). Study on natural breeding sites of sand flies (Diptera: phlebotominae) in areas of Leishmania transmission in Colombia. Parasit. Vectors 8:116. doi: 10.1186/s13071-015-0711-y
von Meijenfeldt, F. A. B., Arkhipova, K., Cambuy, D. D., Coutinho, F. H., and Dutilh, B. E. (2019). Robust taxonomic classification of uncharted microbial sequences and bins with CAT and BAT. Genome Biol. 20:217. doi: 10.1186/s13059-019-1817-x
Walsh, T. R. (2006). Combinatorial genetic evolution of multiresistance. Curr. Opin. Microbiol. 9, 476–482. doi: 10.1016/j.mib.2006.08.009
Wang, J., Sui, B., Shen, Y., Meng, H., Zhao, L., Zhou, H., et al. (2018). Effects of different biochars on antibiotic resistance genes during swine manure thermophilic composting. Int. J. Agric. Biol. Eng. 11, 166–171. doi: 10.25165/j.ijabe.20181106.4667
Wang, R., Zhang, J., Sui, Q., Wan, H., Tong, J., Chen, M., et al. (2016). Effect of red mud addition on tetracycline and copper resistance genes and microbial community during the full scale swine manure composting. Bioresour. Technol. 216, 1049–1057. doi: 10.1016/j.biortech.2016.06.012
Wang, Y., Chen, Z., Wen, Q., and Ji, Y. (2021). Variation of heavy metal speciation, antibiotic degradation, and potential horizontal gene transfer during pig manure composting under different chlortetracycline concentration. Environ. Sci. Pollut. Res. 28, 1224–1234. doi: 10.1007/s11356-020-10557-x
Wei, H., Ma, J., Su, Y., and Xie, B. (2020). Effect of nutritional energy regulation on the fate of antibiotic resistance genes during composting of sewage sludge. Bioresour. Technol. 297:122513. doi: 10.1016/j.biortech.2019.122513
Werner, K. A., Poehlein, A., Schneider, D., El-Said, K., Wöhrmann, M., Linkert, I., et al. (2022). Thermophilic composting of human feces: development of bacterial community composition and antimicrobial resistance gene pool. Front. Microbiol. 13:824834. doi: 10.3389/fmicb.2022.824834
Wickham, H. (2016). ggplot2: elegant graphics for data analysis. Berlin/Heidelberg, Germany: Springer-Verlag.
Wilson, D. N., Hauryliuk, V., Atkinson, G. C., and O’Neill, A. J. (2020). Target protection as a key antibiotic resistance mechanism. Nat. Rev. Microbiol. 18, 637–648. doi: 10.1038/s41579-020-0386-z
Wind, L., Krometis, L. A., Hession, W. C., and Pruden, A. (2021). Cross-comparison of methods for quantifying antibiotic resistance in agricultural soils amended with dairy manure and compost. Sci. Total Environ. 766:144321. doi: 10.1016/j.scitotenv.2020.144321
Wood, D. E., Lu, J., and Langmead, B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20:257. doi: 10.1186/s13059-019-1891-0
Wright, G. D. (2005). Bacterial resistance to antibiotics: enzymatic degradation and modification. Adv. Drug Deliv. Rev. 57, 1451–1470. doi: 10.1016/j.addr.2005.04.002
Yang, B., Wang, Y., and Qian, P. Y. (2016). Sensitivity and correlation of hypervariable regions in 16S rRNA genes in phylogenetic analysis. BMC Bioinformatics 17:135. doi: 10.1186/s12859-016-0992-y
Youngquist, C. P., Mitchell, S. M., and Cogger, C. G. (2016). Fate of antibiotics and antibiotic resistance during digestion and composting: a review. J. Environ. Qual. 45, 537–545. doi: 10.2134/jeq2015.05.0256
Zhang, J., Chen, M., Sui, Q., Tong, J., Jiang, C., Lu, X., et al. (2016). Impacts of addition of natural zeolite or a nitrification inhibitor on antibiotic resistance genes during sludge composting. Water Res. 91, 339–349. doi: 10.1016/j.watres.2016.01.010
Zhao, X., Yu, Z., and Ding, T. (2020). Quorum-sensing regulation of antimicrobial resistance in bacteria. Microorganisms 8:425. doi: 10.3390/microorganisms8030425
Keywords: compost, ecological sanitation, human excreta, bacterial community, antibiotic resistance, pathogenicity, metagenomics, qPCR (quantitative PCR)
Citation: Werner KA, Schneider D, Poehlein A, Diederich N, Feyen L, Axtmann K, Hübner T, Brüggemann N, Prost K, Daniel R and Grohmann E (2022) Metagenomic Insights Into the Changes of Antibiotic Resistance and Pathogenicity Factor Pools Upon Thermophilic Composting of Human Excreta. Front. Microbiol. 13:826071. doi: 10.3389/fmicb.2022.826071
Received: 30 November 2021; Accepted: 17 February 2022;
Published: 31 March 2022.
Edited by:
Ed Topp, Agriculture and Agri-Food Canada (AAFC), CanadaReviewed by:
Jagat Rathod, National Cheng Kung University, TaiwanJincai Ma, College of New Energy and Environment/Jilin Unversity, China
Copyright © 2022 Werner, Schneider, Poehlein, Diederich, Feyen, Axtmann, Hübner, Brüggemann, Prost, Daniel and Grohmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elisabeth Grohmann, ZWdyb2htYW5uQGJodC1iZXJsaW4uZGU=