 Qiang Liu
Qiang Liu Daiyu Li1
Daiyu Li1 Gang Guo
Gang Guo Yun Shi
Yun Shi Quanming Zou
Quanming Zou- 1West China Biopharmaceutical Research Institute, West China Hospital, Sichuan University, Chengdu, China
- 2National Engineering Research Center of Immunological Products, Department of Microbiology and Biochemical Pharmacy, College of Pharmacy, Third Military Medical University, Chongqing, China
Staphylococcus aureus is a leading pathogen that is currently the most common cause of infection in hospitalized patients. An in-depth genetic analysis of S. aureus virulence genes contributing to pathogenesis is needed to develop novel antimicrobial therapies. However, tools for genetic manipulation in S. aureus are limited, particularly those for gene expression. Here, 38 highly expressed genes were identified in S. aureus USA300_FPR3757 via RNA-seq. Promoter regions from 30 of these genes were successfully cloned, of which 20 promoters exhibited a wide range of activity. By utilizing these active promoters, 20 S. aureus-Escherichia coli shuttle vectors were constructed and evaluated by expressing an egfp reporter gene. Expression of the egfp gene under the control of different promoters was confirmed and quantified by Western blotting and qPCR, which suggested that the activity of these promoters varied from 18 to 650% of the activity of PsarA, a widely used promoter for gene expression. In addition, our constructed vectors were verified to be highly compatible with gene expression in different S. aureus strains. Furthermore, these vectors were evaluated and used to overexpress two endogenous proteins in S. aureus, namely, catalase and the transcriptional repressor of purine biosynthesis (PurR). Meanwhile, the physiological functions and phenotypes of overexpressed PurR and catalase in S. aureus were validated. Altogether, this evidence indicates that our constructed vectors provide a wide range of promoter activity on gene expression in S. aureus. This set of vectors carrying different constitutive promoters developed here will provide a powerful tool for the direct analysis of target gene function in staphylococcal cells.
Introduction
Staphylococcus aureus is a major Gram-positive opportunistic pathogen causing both community-acquired and hospital-acquired infections in humans. The success of S. aureus as a leading pathogen is undoubtedly attributed to its severe antibiotic resistance as well as the extensive repertoire of virulence factors that enable it to evade the host immune system (Turner et al., 2019). An increased understanding of the pathogenicity and antibiotic resistance mechanisms of S. aureus is necessary to identify potential targets for the development of novel antimicrobial therapies. Therefore, an in-depth genetic analysis of S. aureus virulence genes contributing to pathogenesis is highly desirable.
Characterization of genes of unknown function in S. aureus has traditionally been conducted via gene knockout followed by complementation, as well as overexpression of the gene product using an inducible or constitutive promoter. Several shuttle vectors, including pBT2, pKOR1, pMAD, and a range of pRLY2, are currently available for gene disruption in S. aureus via double homologous recombination (Bruckner, 1997; Arnaud et al., 2004; Bae and Schneewind, 2006; Redder and Linder, 2012). However, the system for gene expression is relatively limited, largely due to the lack of well-characterized functional promoters in S. aureus.
Several inducible promoters have been applied in S. aureus. These include the Bacillus megaterium-derived xylose inducible promoter (Wieland et al., 1995; Kim et al., 1996; Yepes et al., 2014; Shang et al., 2019), the IPTG (isopropyl-beta-D-thiogalactopyranoside)-inducible promoter from the pSpac-lacI system (Gardete et al., 2004; Pereira et al., 2007; Liew et al., 2011), and the tetracycline-regulated hybrid promoter Pxyl–tetO (Bateman et al., 2001; Corrigan and Foster, 2009). Although these inducible promoters have been used for gene expression in S. aureus, some limitations have yet to be resolved. For example, the pSpac/lacI system is not readily adaptable to S. aureus due to its high basal promoter activity (Bateman et al., 2001; Liew et al., 2011). In addition, induction with IPTG renders this system less attractive in an animal model system. The xylose-inducible promoter system is repressible by glucose (Wieland et al., 1995), which also prohibits its use in in vivo studies, as glucose is a common constituent inside mammalian cells. The tetracycline-inducible promoter system is currently the most functional inducible system for gene expression in S. aureus (Bateman et al., 2001; Harrison et al., 2019; Goncheva et al., 2020; Kuiack et al., 2020). However, the inducers such as tetracycline or anhydrotetracycline required in this system, in our experience, affect the normal metabolism of or are even toxic to S. aureus, which results in undesirable phenotypes.
Given the deficiencies of inducible systems, constitutive promoter systems, with no need for inducer substances, possess the advantage of making them ideally suitable for functional investigation of specific genes in S. aureus, particularly in an in vivo infection model. However, only a few constitutive promoters are currently available for gene expression in S. aureus. These include the global regulator sarA gene promoter and the type 1 capsule gene promoter Pcap. The sarA gene consists of three promoters upstream of the sarA gene, the proximal P1 and distal P3 and P2 promoters (Bayer et al., 1996; Cheung and Manna, 2005). sarA-P1 has the strongest promoter activity and is thus widely employed for expressing various fluorescent reporter genes constitutively in S. aureus (Cheung et al., 1998; Malone et al., 2009; de Jong et al., 2017), which makes S. aureus able to be tracked and monitored both in vitro and in vivo (Boero et al., 2021; Yu et al., 2021). Other than the promoter sarA-P1, the serotype 1 capsule gene promoter Pcap has been characterized to have constitutively strong activity in S. aureus (Ouyang and Lee, 1997; Schwendener and Perreten, 2015). Using the Pcap promoter, an Escherichia coli–S. aureus shuttle vector pBUS1-Pcap-HC has been constructed for gene expression in both E. coli and S. aureus (Schwendener and Perreten, 2015).
Although few constitutive promoters are available for gene expression in S. aureus, it is still limited for expressing a gene constitutively at a desired level. For example, when it is needed for the expression of genes with potential toxic effects, such as some transcriptional repressors, the transcriptional level of the target gene should be controlled at an appropriate level. Protein overexpression due to a strong constitutive promoter may be lethal to cells. However, insufficient protein expression and the control of a weak promoter might lead to insignificant phenotypes, which hinder the real function of the gene to be uncovered. Therefore, constitutive promoters with a wide range of activity provide the possibility of gene expression at a desirable level and may be utilized in investigating various virulence genes in S. aureus.
In this study, we identified a set of highly expressed genes during different growth phases of S. aureus. The promoters of these highly expressed genes were cloned and evaluated in terms of their activity. Based on the active promoters, a set of vectors was constructed for gene overexpression. The promoter strength of each vector was quantified by expressing an egfp reporter gene. Additionally, the universality of these vectors was evaluated in different S. aureus strains. Furthermore, two endogenous proteins, namely, PurR and catalase, were successfully expressed using our constructed vectors. Moreover, phenotypes of S. aureus overexpressing PurR or catalase were checked and confirmed. We believe that the set of vectors we developed may be utilized in investigating the contribution of virulence genes responsible for the pathogenesis of S. aureus.
Results
Screening of Highly Expressed Genes in S. aureus USA300_FPR3757 via RNA-Seq
Thirty-eight genes highly expressed in three different growth phases were screened via RNA-seq (Figure 1). To identify the highly expressed genes in different growth phases, the growth curve of S. aureus USA300_FPR3757 in TSB medium was first drawn (Figure 1A). Bacterial cells from different time points (2, 6, or 10 h) after inoculation, corresponding to the lag phase (LP), exponential phase (EP), and stationary phase (SP), were collected for RNA extraction and subsequently submitted for RNA-seq. The transcriptome dataset, including read counts and fragments per kilobase million (FPKM) of each gene from LP, EP, and SP, is listed in Supplementary Table 1. The expression levels of all 6,030 genes during LP, EP, and SP were ranked from the most highly expressed to the least expressed according to their FPKM values (Figure 1B). Genes with FPKM values ≥ 1,500 were defined as highly expressed, and there were 152, 81, and 88 highly expressed genes in the LP, EP, and SP growth phases, respectively (Figures 1B,C). Among these genes, 38 genes were all highly expressed in all three growth phases (Figure 1C).
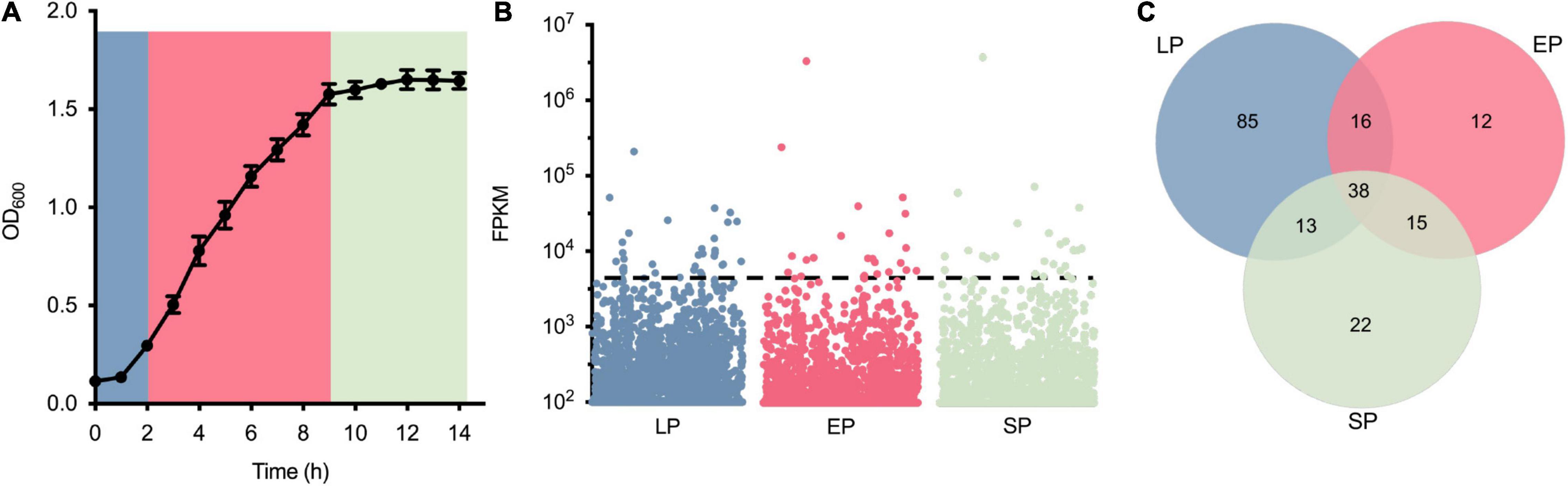
Figure 1. Identification of highly expressed genes from Staphylococcus aureus USA300_FPR3757. (A) Growth curve of S. aureus USA300_FPR3757 growing at 37°C in TSB medium. (B) Normalized expression level (FPKM) of genes in the LP, EP, and SP growth phases. Each point represents a gene, and the dashed line indicates the cutoff value of FPKM 1,500. (C) Venn diagram representing the number of shared highly expressed genes (FPK ≥ 1,500) in the LP, EP, and SP growth phases.
Cloning of the Promoter Regions of Identified Highly Expressed Genes
Since the exact -35 and -10 sequences of these promoters were unknown, the gap non-coding sequence between the highly expressed gene and its upstream gene was defined as the potential promoter sequence. Among these 38 highly expressed genes, there were eight genes with very short promoter regions or no promoter region, making them difficult or impossible to clone. Therefore, the promoter regions of the other 30 genes were selected for cloning and evaluation (Supplementary Table 3). If the promoter sequence was longer than 450 bp, then only 450 bp was chosen as a promoter sequence. In addition, the commonly used constitutive promoter of the transcriptional regulator gene sarA PsarA (P31) was also cloned as a positive control (Bayer et al., 1996; Cheung and Manna, 2005). The length of the selected promoters ranged from 120 to 434 bp, and each promoter was named the gene locus of its corresponding gene. Information on the cloned promoters, including their length, coding products of their corresponding genes, and the transcriptional level (FPKM value) of the selected gene, is shown in Table 1. In short, among these 38 highly expressed genes, the promoter regions of 30 genes were successfully cloned (Table 1).
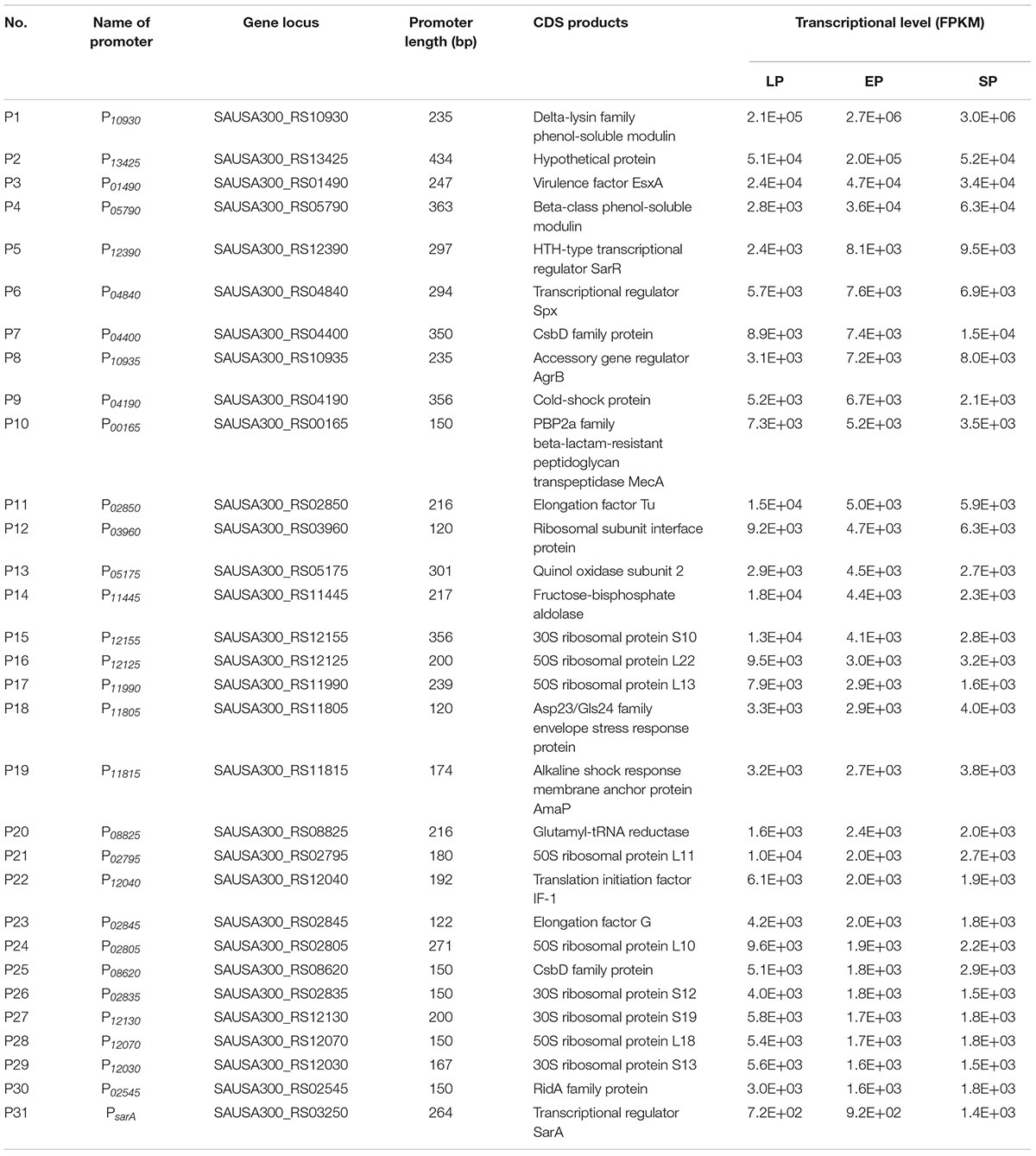
Table 1. Cloned promoter regions of highly expressed genes of S. aureus USA300_FPR3757.
Characterization of the Cloned Promoters by Beta-Galactosidase Assay
To check the activity of the selected promoters, the promoter-probe vector pQLV1003 carrying the lacZ reporter gene was constructed as described in the methods and materials (Table 2). All the selected promoters were amplified and cloned into pQLV1003 upstream of a lacZ reporter gene (Figure 2A). The generated plasmids were transformed into S. aureus USA300, and the activity of each cloned promoter was evaluated by measuring the beta-galactosidase activity of each transformed strain.
Table 2. Bacterial strains and plasmids used in this study.
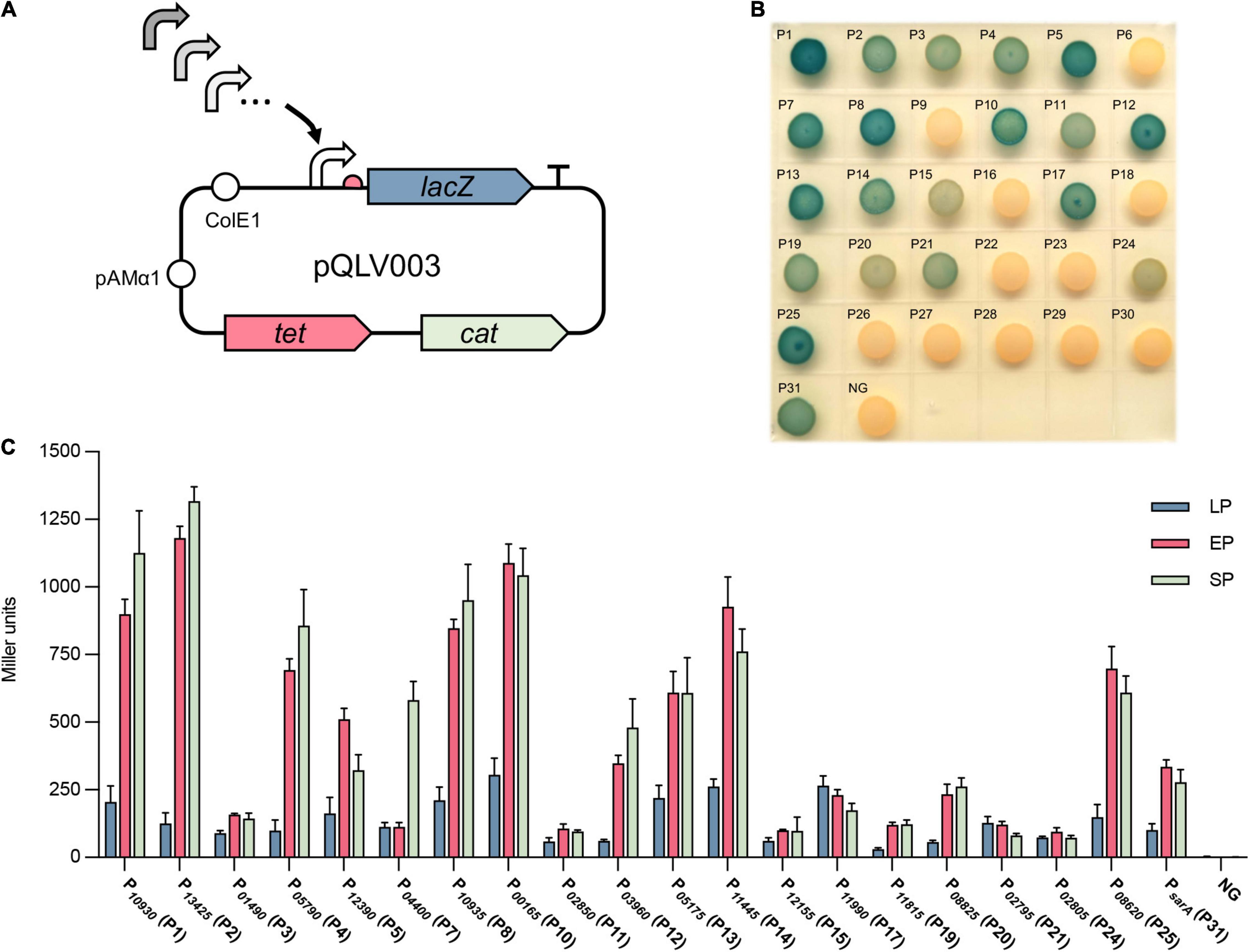
Figure 2. Evaluation of the activity of selected promoters by beta-galactosidase assay. (A) Selected active promoters were cloned into the promoter-probe vector pQLV003 carrying a lacZ reporter gene. The pQLV003 vector contains the Escherichia coli origin ColE1, pAMα1 replicon for propagation in Gram-positive bacteria, the selectable marker tetracycline (tet) in E. coli and chloramphenicol (cat) in S. aureus. (B) The vector pQLV003 carrying each selected promoter in front of the lacZ gene was transformed into S. aureus USA300. The resulting strains were grown on TSB X-gal plates at 37°C for 24 h. (C) The beta-galactosidase activity of the cell lysate of S. aureus USA300 transformed with each promoter_lacZ plasmid at lag phase (LP), exponential phase (EP), and stationary phase (SP). NG represents the negative control in which the egfp gene was driven by no promoter.
First, the beta-galactosidase activity of the transformed strains was tested on a TSB X-gal plate. The results showed that 20 strains exhibited blue colonies on the plate (Figure 2B), with the corresponding 20 promoters (P1, P2, P3, P4, P5, P7, P8, P10, P11, P12, P13, P14, P15, P17, P19, P20, P21, P24, and P25) and the positive control PsarA (P31). Subsequently, the strength of these 20 verified active promoters was quantified by measuring the beta-galactosidase activity of their corresponding bacterial cell lysates (Figure 2C). Overall, the activity of most promoters in EP (6 h) and SP (10 h) was significantly stronger than that in early growing phase LP (2 h), except P3, P11, P15, P17, P19, P21, and P24, which exhibited a similar activity in all growing phases. The activity of most promoters exhibited no significant difference (p-value ≥ 0.05) in EP (6 h) and SP (10 h) with the exception of P1, P2, P4, P5, P7, and P12, among which P1, P2, P4, P7, and P12 had stronger activity in SP and P5 had stronger activity in EP. Compared with the commonly used promoter PsarA (P31), 11 promoters displayed stronger activity. Overall, the activity of the cloned 20 promoters varied from 25 to 500% of the activity of PsarA. Vector pQLV1003 without a promoter served as a negative control (NG), which exhibited no beta-galactosidase activity. In summary, the activity of these 30 selected promoters was evaluated in the native host, and 20 promoters showed a wide range of activity.
Construction of Overexpression Vectors Based on the Selected Promoters
Using the identified 20 active promoters, 20 vectors derived from pBUS1_Pcap_HC were constructed for constitutive protein expression in S. aureus. pBUS1_Pcap_HC is a high-copy S. aureus-E. coli shuttle vector used for protein overexpression in S. aureus using the promoter of type 1 capsule gene 1A (Pcap) (Schwendener and Perreten, 2015) (Figure 3A). It contains the tetracycline marker for plasmid selection in both E. coli and S. aureus. Because some clinical S. aureus strains, such as S. aureus USA300 and S. aureus MW2, are resistant to tetracycline, an additional selection marker, the chloramphenicol cassette (cat), was amplified from pBT2 (Bruckner, 1997) and introduced into pBUS1_Pcap_HC at the BglII site, generating the vector pBUS1_Pcap_HC_cat (pQLV1002) (Figure 3B).
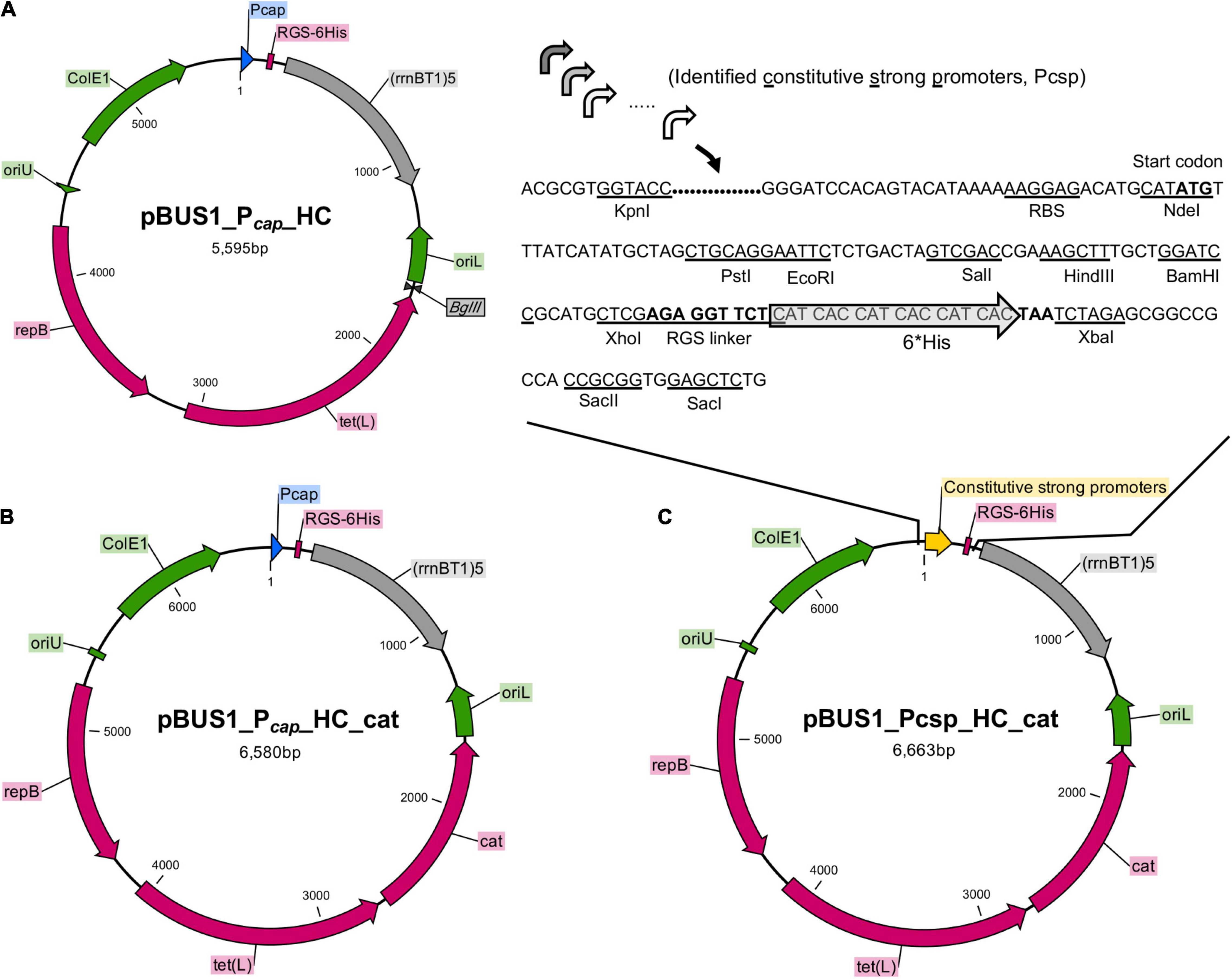
Figure 3. Construction of overexpression vectors by using the active promoters. (A) The backbone vector pBUS1_Pcap_HC was used for the development of the set of overexpression vectors. The elements in the backbone vector pBUS1_Pcap_HC include the Pcap promoter, E. coli origin ColE1, the terminator sequence (rrnB T1)5 and the selectable marker tetracycline tet (L). The elements required for rolling-circle replication are indicated: the replication initiator protein gene (repB), the double-strand origin (oriU), and the single-strand origin (oriL). (B) The chloramphenicol cassette (cat) was inserted into pBUS1_Pcap_HC at the BglII site to generate the plasmid pBUS1_Pcap_HC_cat. (C) The DNA fragment containing each individual selected promoter sequence and a ribosomal-binding site (RBS) was cloned into pBUS1_Pcap_HC_cat at the KpnI and NdeI sites, which replaced the original Pcap promoter sequence, to generate a set of expression vectors carrying different constitutive and active promoters.
To generate the overexpression vectors by applying our identified constitutive promoters, the Pcap promoter sequence from the pBUS1_Pcap_HC_cat plasmid was replaced with a new DNA fragment, which contained an individual selected promoter sequence and a typical ribosomal-binding site (RBS) (Figures 3B,C). This generated a set of vectors harboring the identified strong constitutive promoter individually (Figure 3C). Downstream of the promoter, the multiple cloning site (MCS) and the RGS-6 × His coding sequence were the same as those in the original vector pBUS1_Pcap_HC_cat, which enables the expression of the target protein with a C-terminal 6 × His tag (Figure 3C). Briefly, 20 vectors carrying different constitutive promoters were developed for gene expression and the production of tagged fusion proteins.
Evaluation and Quantification of the Constructed Vectors by Expressing an egfp Reporter Gene
To evaluate the effect and efficiency of the constructed expression vectors on gene expression, an egfp reporter gene was cloned into the MCS of each vector at the NdeI and XhoI sites, which allowed the egfp gene to be expressed with an RGS_6 × His tag under the control of different promoters. The verified plasmid was transformed into S. aureus USA300, and the fluorescence and OD600 value of each resulting strain were measured in a bioreader. Promoter activity was analyzed by calculating the ratio of RLU/OD600 (Supplementary Table 4), and a heatmap based on the RLU/OD600 value in different growth phases was generated (Figure 4A). All tested promoters were active in LP, EP, or SP compared to the negative control, in which the egfp gene was driven by no promoter (Figure 4A).
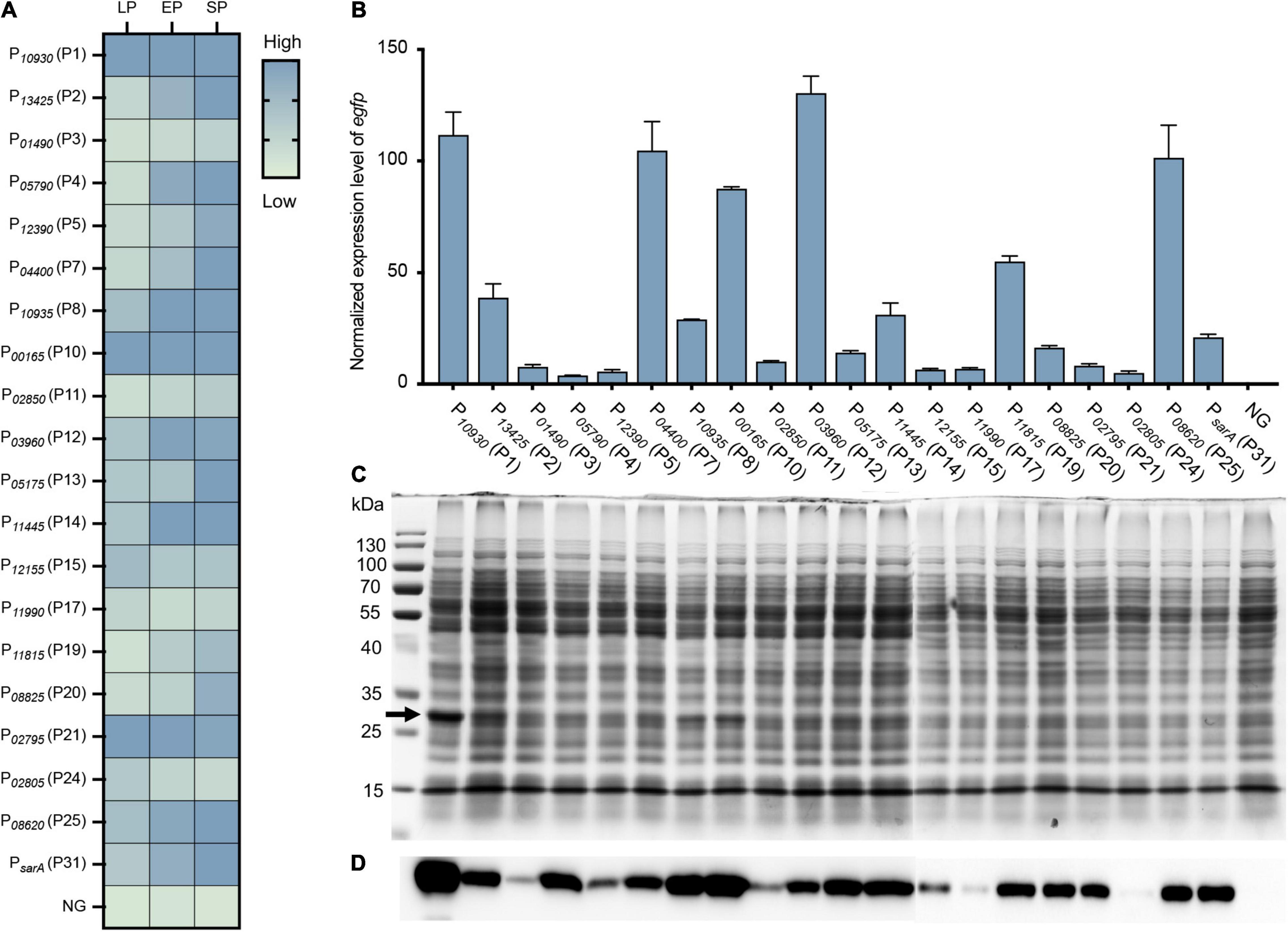
Figure 4. Evaluation and quantification of the strength of constructed vectors by expressing an egfp reporter gene. (A) Fluorescence-based promoter activity assay. The egfp reporter gene was cloned into each vector under the control of different promoters. The resulting strain was transformed into S. aureus USA300, and the fluorescence intensity and OD600 value of each strain were measured by a Bioreader at the LP, EP, and SP growth phases. The value of the ratio fluorescence intensity/OD600 ratio was calculated. (B) Additionally, the transcriptional level of the egfp gene under different promoters was assessed by RT-qPCR. The expression level was normalized to the internal control sigA gene. Data shown are the mean ± SD of three experiments. (C) The production of recombinant eGFPs was checked by SDS-PAGE and (D) Western blotting using an anti-6 × His tag antibody. NG represents the negative control wherein the egfp gene was driven by no promoter.
To further quantify the strength of these vectors on gene expression, the transcriptional level of the egfp gene in each vector was quantified by RT-qPCR. The egfp gene was overexpressed in all vectors compared to the negative control (NG) (Figure 4B), which further confirmed the effect of our constructed vectors on gene expression. When compared to the commonly used promoter PsarA (P31), nine promoters (P1, P2, P7, P8, P10, P12, P14, P19, and P25) were stronger than PsarA (P31), while 10 promoters were weaker than PsarA (P31) (Figure 4B). Overall, the activity of our selected promoters in the overexpression vectors varied from 18 to 650% of the activity of PsarA (P31) (Figure 4B).
Additionally, the expression of the eGFP was confirmed by SDS-PAGE and western blotting. Overproduction of the recombinant eGFPs (28 kDa) from some strong promoters, such as P10930 (P1), P10935 (P8), and P00165 (P10), was clearly visible on the SDS-PAGE gel (Figure 4C). Owing to the eGFP being expressed with a 6 × His tag at its C-terminal, the expression of the eGFP was further confirmed by western blotting using an anti-6 × His tag antibody. The recombinant eGFP was detected in all strains at various levels (Figure 4D), most of which corresponded to their transcriptional level (Figures 4C,D). Collectively, all the overexpression vectors constructed here efficiently expressed their target genes. More importantly, this set of vectors displayed a wide range of activity in their effects on gene expression.
Evaluation of the Gene Overexpression Vectors in Different S. aureus Strains
To evaluate the universality of the constructed expression vectors, an egfp gene was cloned into each vector and transformed into different S. aureus strains, which included S. aureus USA300, S. aureus MW2, S. aureus Newman, S. aureus RN4220, S. aureus NCTC_8325, and S. aureus ATCC 10832. As shown, the eGFP was successfully overexpressed in all vectors compared to the negative control, in which the egfp gene was regulated without a promoter (Figure 5). However, the fluorescence of eGFP expressed from the few vectors exhibited variance in different strains, such as P10935 (P8), which displayed strong promoter activity in all tested strains but not in S. aureus RN4220 (Figure 5). Importantly, most of the vectors expressed eGFPs with the same intensity in all treated S. aureus strains, which suggested that the promoters screened from S. aureus USA300 were functional in the same manner as those in other strains. These results indicated that the promoters we selected from S. aureus USA300 are conserved in S. aureus species, and the overexpression vectors constructed based on the identified active promoters were functional in different S. aureus strains with slight variance in their activity.
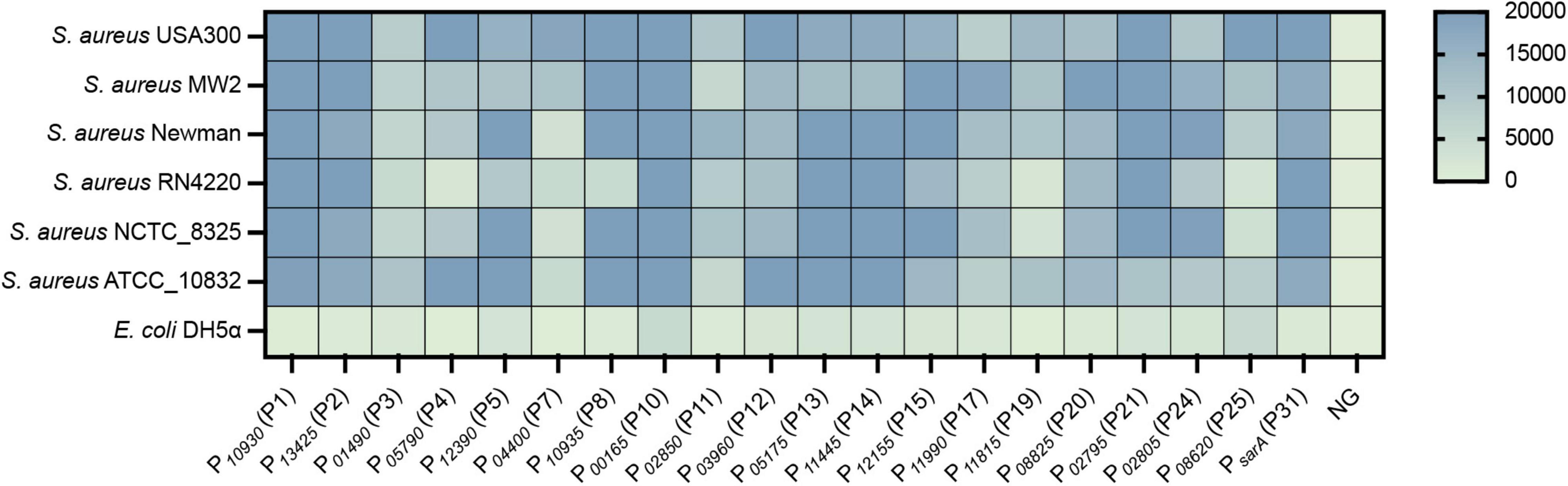
Figure 5. The constructed vectors were evaluated in different S. aureus strains. The egfp reporter gene was cloned into each vector under the control of different promoters. The resulting plasmid was transformed into different S. aureus strains, and the fluorescence intensity and OD600 value of each strain were measured by a Bioreader at the exponential phase. The fluorescence intensity/OD600 ratio was calculated. NG represents the negative control in which the egfp gene was driven by no promoter.
Evaluation and Application of Developed Expression Vectors to Endogenous Gene Expression in S. aureus
To further evaluate and apply our constructed overexpression system, the endogenous gene purR, which encodes a transcriptional repressor, was expressed and validated in terms of its function in S. aureus. The PurR protein has been proven to be a transcriptional repressor of purine biosynthesis that inhibits the transcription of fibronectin-binding protein-coding genes fnbA and fnbB (Goncheva et al., 2019). Three overexpression vectors carrying promoters P00165 (P10), P08620 (P25), and P04400 (P7) were randomly selected for overexpression of the purR gene. RT-qPCR analysis revealed that the purR gene was overexpressed in the three vectors compared to that in S. aureus WT (Figure 6A). The production of PurR protein (31.7 kDa) from strains overexpressing the purR gene with the P00165 (P10), P08620 (P25), or P04400 (P7) promoter was further confirmed by SDS-PAGE and western blotting (Figure 6B). The signal detected from western blotting coincided with the transcriptional level of the purR gene as quantified by RT-qPCR, with the vector harboring P04400 (P7) having the highest output, followed by P08620 (P25) and P00165 (P10). In addition, the physiological function of the overexpressed PurR protein was verified by checking the transcriptional level of its target genes fnbA and fnbB (Figure 6C). As shown, transcription of the fnbA and fnbB genes was significantly repressed in PurR overexpression strains compared to the WT (Figure 6C).
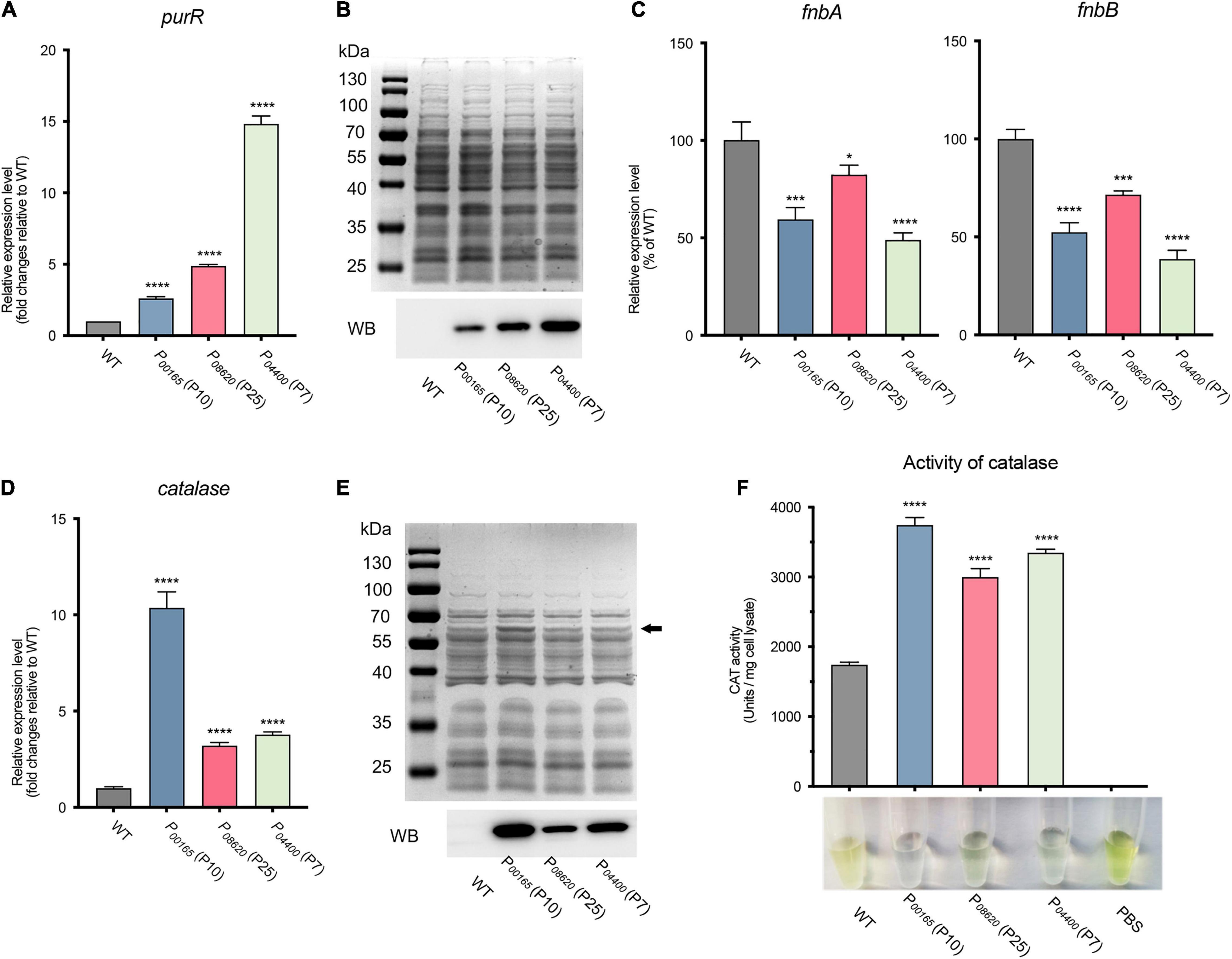
Figure 6. Evaluation and application of the developed expression vectors on endogenous gene expression in S. aureus. (A) The expression level of the purR gene in three overexpression vectors was validated by RT-qPCR. (B) The production of the PurR protein was evaluated by SDS-PAGE and western blotting. (C) The expression levels of the fnbA and fnbB genes, the target genes repressed by the transcriptional repressor PurR, were assessed by RT-qPCR in three purR overexpression and WT strains. (D) The expression level of the catalase gene in three overexpression vectors was assessed by RT-qPCR. (E) The production of PurR protein was evaluated by SDS-PAGE and western blotting. (F) The catalase activity of cell lysates from each overexpression strain and WT was measured by H2O2 and ammonium molybdate-based assays. The undecomposed hydrogen peroxide reacts with ammonium molybdate to produce a yellowish color. More transparency in the reacted mixture represents less hydrogen peroxide left, indicating higher catalase activity. Data shown are the mean ± SD of three experiments. (****P ≤ 0.0001, ***P ≤ 0.005; *P ≤ 0.05 relative to the WT).
Furthermore, the endogenous catalase of S. aureus was overexpressed to further evaluate the constructed vectors. Similarly, overexpression of catalase using three vectors under the control of promoters P00165 (P10), P08620 (P25), and P04400 (P7) was confirmed by RT-qPCR and western blotting (Figures 6D,E). The results showed that the transcription of the catalase gene in the overexpression strains was 3- to 10-fold higher than that in the WT, in which P00165 (P10) possessed the highest transcription level, followed by P04400 (P7) and P08620 (P25) (Figure 6D). The same expression patterns of catalase were observed and confirmed by SDS-PAGE and western blotting (Figure 6E). In addition, the activity of catalase overproduced from different vectors was measured by H2O2 and ammonium molybdate-based assays. The undecomposed H2O2 reacts with ammonium molybdate to produce a yellowish color, i.e., more transparency of the reacted mixture represents less hydrogen peroxide left, which indicates higher catalase activity. The cell lysates from the overexpression strains exhibited significantly stronger catalase activity than those from the WT strain (Figure 6F). In line with the expression level, the vector carrying P00165 (P10) that possessed the highest expression exhibited the highest catalase activity (Figure 6F).
In conclusion, the endogenous genes purR and catalase were successfully overexpressed by using our constructed overexpression vectors, and their related functions and phenotypes were confirmed in S. aureus USA300.
Discussion
Currently, there are only a few genetic tools available for constitutive gene expression in S. aureus, which is largely due to the lack of characterized strong and reliable constitutive promoters. Here, we screened and evaluated 20 strong constitutive promoters for gene expression at a wide range of transcriptional levels in S. aureus.
A decent promoter used for constitutive gene overexpression principally possesses the characteristics of strong promoter activity in host cells. Thus, the top 1.5% highly expressed genes (RPKM ≥ 1,500) identified via RNA-seq in S. aureus were selected as the source of candidate promoter selection. Considering that the transcriptional level of a gene probably significantly varies during different growth phases (Mader et al., 2016), highly expressed genes in the lag phase, exponential phase, and stationary phases were filtered for promoter screening.
Since the exact -35 and -10 regions of the promoter from these genes are unknown, the gap non-coding sequence between the candidate gene and its upstream gene was defined as the potential promoter sequence. After the selected promoter sequences were cloned into a promoterless vector upstream of the beta-galactosidase reporter gene, the activity of these promoters was evaluated by measuring the beta-galactosidase activity of a strain transformed with the individual plasmid. As expected, not all selected promoters exhibited activity in the beta-galactosidase assay. This observation is presumably due to the incorrect selection of the promoter region, which is probably located within the coding region of the upstream gene, or the upstream gene utilizing the same promoter. Therefore, further work could be performed to identify the transcriptional start site of each highly expressed gene so that we can determine the exact -35 and -10 positions of these promoters. However, two-thirds of our cloned promoter sequences are active in S. aureus, which provides great potential for the development of a constitutive gene overexpression system.
Using the selected active promoters, a set of vectors was constructed for gene expression in S. aureus. All these generated vectors here are derived from the plasmid pBUS1_Pcap_HC a S. aureus-E. coli shuttle vector constructed for gene expression under the control of the constitutive promoter Pcap (Schwendener and Perreten, 2015). This vector is a promising backbone source for developing gene expression systems due to its characteristics of high copy number and high segregational stability in S. aureus (Schwendener and Perreten, 2015). Additionally, an RGS-6 × His coding sequence downstream of MCS enables target gene expression with a C-terminal polyhistidine-tag, allowing it to be directly detected or purified directly from S. aureus. Therefore, our developed overexpression vectors were constructed by replacing the Pcap promoter sequence on pBUS1_Pcap_HC with our identified promoters individually while keeping the other elements the same as the original.
After the expression vectors were constructed, the promoter strength or the ability of these vectors to express protein expression was evaluated and quantified by expressing an egfp reporter gene (Figure 4). Unexpectedly, the promoter activity evaluated by eGFP fluorescence did not all correspond to that from RNA-seq. For example, P04400 (P7) had lower activity than P01490 (P3) in RNA-seq (Table 1), while P01490 (P7) had higher promoter activity than P01490 (P3) in the eGFP fluorescence assay (Figure 4). This observation was probably because some regulatory regions of the promoter were not included in our selected promoter sequences, which resulted in increasing or decreasing the strength of the promoter. In addition, eGFP expression does not always correlate with its transcript level, which could probably be caused by posttranscriptional regulation. The mRNAs of the egfp gene transcribed from different promoters have a different 5′ untranslated region (5′UTR). Some of the 5′ UTR regions contain regulatory elements that negatively or positively affect translation efficiency. Therefore, mRNAs with the same abundances might be translated to different amounts of proteins due to their different 5′ UTRs. In addition, it is worth noting that all these vectors were tested in S. aureus under lab growth conditions in TSB medium. If these are used in any other conditions, such as growing S. aureus in the presence of antibiotics or in a different medium, it would be better to evaluate these vectors first because the strength of a promoter may vary in different conditions (Mader et al., 2016). Nevertheless, our developed vectors with a wide range of promoter activity make it possible to constitutively express a certain protein at a desired level.
Because the promoters were screened from the strain S. aureus USA300, the universality of the vectors was evaluated by expressing the egfp gene in other clinical or laboratory strains. The expression levels of the egfp gene from a few vectors varied among different strains (Figure 5), which was probably caused by the slight differences in the genome sequence among each strain. For example, S. aureus RN4220 harbors many mutations in the genes encoding numerous regulators compared to S. aureus NCTC_8325 (Nair et al., 2011), which largely affects the transcriptional efficiency of their target promoters. However, most of the promoters exhibit the same strength as that of the native host S. aureus USA300, suggesting that our developed system is suitable for gene expression in different S. aureus strains. In addition, two endogenous genes, purR and catalase, were successfully expressed with our constructed vectors. The biological functions of the overexpressed proteins PurR and catalase were verified in S. aureus (Goncheva et al., 2019), which provides two practical examples for characterizing the function of unknown genes in S. aureus by using our developed overexpression system.
In the present study, we identified 20 strong constitutive promoters in S. aureus that exhibit a wide range of promoter strengths. Based on these promoters, a set of vectors for gene expression were constructed and evaluated. This system provides many choices for constitutive gene expression at different levels, thereby resolving previously limited genetic tools for gene expression. We believe that the expression system developed here will promote more in-depth studies on virulence genes or the function of unknown genes in S. aureus.
Materials and Methods
Bacterial Strains and Growth Conditions
The bacterial strains and plasmids used in this study are described and listed in Table 2. E. coli strains were grown in Luria-Bertani (LB) broth with constant shaking at 220 rpm or on LB agar plates at 37°C. S. aureus strains were cultured in tryptic soy broth (TSB) with shaking at 220 rpm or on TSB agar plates (TSA) at 37°C. Plasmids used for S. aureus transformation were modified by S. aureus RN4220. All S. aureus transformants were obtained through electroporation as described previously (Schenk and Laddaga, 1992). E. coli and S. aureus transformants were selected on agar plates containing 10 μg/mL tetracycline or 25 μg/mL chloramphenicol, and antibiotics were also used to maintain the plasmids in the cells.
RNA Isolation and RNA-Seq
The day culture of S. aureus USA300 was prepared by inoculating 100 μL of an overnight culture into 10 mL of TSB medium in a 100-mL flask. Two milliliters of bacterial cells were collected after 2, 6, and 10 h of cultivation by centrifugation at 5,000 g for 10 min at 4°C. Bacterial cells were resuspended in 100 μL RNase-free cell lysis buffer (20 mM Tris-Cl, pH 8.0; 2 mM sodium EDTA; 1.2% Triton X-100; 50 μg/mL lysostaphin) and incubated at 37°C for 15 min. Then, RNA extraction was performed using an RNApure Bacteria Kit (CwBIO, Jiangsu, China) following the manufacturer’s instructions. The rRNA of the RNA samples was removed with a Ribo-Zero rRNA Removal Kit (Gram-positive Bacteria, Illumina) according to the manufacturer’s instructions. cDNA library preparation and sequencing were performed by Personalbio Co. (Shanghai, China).
DNA Preparation and PCR Analysis
The isolation of genomic DNA and plasmid preparation using E. coli or S. aureus were performed with a bacterial genomic DNA kit and PurePlasmid Mini Kit, respectively (CwBIO, Jiangsu, China). Oligonucleotide primers were synthesized by Sangon Biotech (Shanghai, China). All primers used in this study are listed in Supplementary Table 2. For analytical purposes, PCRs were performed using OneTaq 2 × Master Mix (NEB, Ipswich, England). PCRs for plasmid construction were performed using Q5 High-Fidelity 2 × Master Mix from NEB according to the manufacturers’ instructions. The PCR products were purified using the DNA Clean-up Kit (CwBIO, Jiangsu, China). For cloning or plasmid construction, the plasmid was linearized by the related restriction enzymes from NEB. Cloning was performed using the ClonExpress II One Step Cloning Kit (Vazyme, Nanjing, China), which is based on homologous recombination.
Construction of Vectors pQLV1001, pQLV1002, and pQLV1003
An entire chloramphenicol resistance cassette was amplified from plasmid pBT2 (Bruckner, 1997) using primers QL0230/0231 (Supplementary Table 2) and cloned into S. aureus–E. coli shuttle vectors pBUS1-HC and pBUS1-Pcap-HC (Schwendener and Perreten, 2015) at the BglII site, which generated vectors pQLV1001 and pQLV1002, respectively. To generate a promoterless beta-galactosidase reporter vector, a DNA fragment containing the red fluorescence protein gene (rfp) and beta-galactosidase gene (lacZ) was amplified from the pBS1C_lacZ plasmid (Popp et al., 2017) with primers QL409/0410 (Supplementary Table 2). The rfp_lacZ fragment was inserted into pQLV1001 linearized with HindIII and BamHI to generate the lacZ promoter-probe vector pQLV1003.
Promoter Cloning
The promoter regions of highly expressed genes (Supplementary Table 3) were amplified from the genomic DNA of S. aureus USA300 by PCR using the primers listed in Supplementary Table 2. Then, the promoter DNA fragment was individually cloned into the lacZ promoter-probe vector pQLV1003, which was linearized with EcoRI and PstI. After confirmation by PCR and sequencing, the generated plasmid was modified by RN4220 and subsequently transformed into S. aureus USA300 for the beta-galactosidase-based promoter activity assay.
Beta-Galactosidase-Based Promoter Activity Assay
The activity of the cloned promoters was first evaluated on the plate. Briefly, day cultures of S. aureus USA300 strains harboring the plasmid constructed above were prepared by inoculating 100 μL of the overnight culture into 10 mL of TSB medium. After the OD600 value reached 0.6, 5 μL of the bacterial culture was pipetted out and dropped onto a TSA agar plate containing 25 μg/mL chloramphenicol and 200 μg/mL X-Gal (5-bromo-4-chloro-3-indolyl-β-D-galactoside). The plate was incubated at 30°C, and the color of the colony was monitored and photographed after 48 h.
To further quantify the promoter activity, analysis of beta-galactosidase activity using O-nitrophenyl-β-D-galactopyranoside (ONPG) as substrate was performed and modified as previously described (Zhu et al., 2020). A single blue colony on the TSA X-Gal plate was inoculated into chloramphenicol-containing TSB medium overnight culture preparation. The next day, the day culture was prepared as described above. After 2, 6, or 10 h of cultivation, 100 μL of the bacterial culture was collected for OD600 value measurement in a 96-well plate by using a Synergy H1 microplate reader (Vermont, United States).
Meanwhile, bacterial cells were harvested from the 200 μL culture at each time point by centrifugation at 5,000 g for 10 min at 4°C. The cells were resuspended in 100 μL of lysis buffer (60 mM K2HPO4, 40 mM KH2PO4, 100 mM NaCl, 0.1% Triton X-100, 50 μg/mL lysostaphin) for total protein extraction. The suspension was incubated at 37°C until the bacterial cells had completely lysed. Then, 50 μL of the lysate was pipetted onto a 96-well plate for OD420 measurement. After that, 100 μL of a reaction buffer (60 mM K2HPO4, 40 mM KH2PO4, 100 mM NaCl, 0.1% Triton X-100, 5 mg/mL ONPG) was added to each well and mixed with the cell lysate. The 96-well plate was incubated in the Bioreader at 37°C with constant shaking, and the OD420 was measured every 5 min. The slope of the linear part of the spectrophotometric output was used to calculate the specific activity as follows: Miller units = (1,000 × slope)/(V × OD600), where V is the volume (in milliliters) of the culture used in the assay. This assay was repeated three times for each strain.
Construction of Overexpression Vectors and Their Derived Plasmids
The promoter sequences of the validated active sequences (Supplementary Table 3) were amplified by PCR from the pQLV1003-derived plasmids carrying different promoters using the primers listed in Supplementary Table 2. A fragment “AGGAGGTTTATCATATG” that contained a typical RBS (underlined), a spacer, and an NdeI site (italic) was introduced downstream of each promoter from the primer. This will lead the target gene to be cloned at the NdeI site and other restriction enzyme sites downstream of NdeI. The amplified promoter was cloned into vectors pQLV1002 linearized with KpnI and NdeI to generate a series of overexpression vectors.
To evaluate the constructed expression vectors, the egfp reporter gene was PCR amplified from plasmid pTH100 (de Jong et al., 2017) with primer pairs QL0558/0559 and inserted into each constructed expression vector that was linearized with NdeI and XhoI. The endogenous transcriptional repressor purR or catalase gene of S. aureus USA300 was PCR amplified with the primer pairs QL0609/0610 and QL0613/0614, respectively. These were cloned into the overexpression vectors at the NdeI and XhoI sites for PurR and catalase expression with an RGS_6 × His tag at their C-termini.
Fluorescence-Based Promoter Activity Assay
A day-culture was prepared as described above. Bacterial cells from the 200 μL day-culture after 2, 6, and 10 h of inoculation were harvested by centrifugation. Cells were washed twice with 500 μL of PBS buffer and suspended in PBS to an OD600 value of approximately 0.3. The OD600 value and relative light unit (RLU) of GFP fluorescence (excitation, 485 nm; emission, 512 nm) were measured using a Synergy H1 plate reader. Promoter activity was analyzed by calculating the ratio of RLU/OD600. The assay for each strain was performed in three independent experiments, and the mean value of RLU/OD600 was used to generate the heatmap.
Real-Time Quantitative PCR
The qPCR was performed in a CFX-96 Touch Real-Time PCR system (Bio-Rad, Hercules, CA, United States) using SYBR green master mix TB Green Premix Ex Ta II (Takara, Beijing, China). Total RNA from S. aureus was isolated as described above. Approximately 1 μg of total RNA was used for reverse transcription using a PrimeScript™ RT reagent kit with gDNA Eraser (Takara, Beijing, China) according to the manufacturer’s instructions. After the transcribed cDNAs were diluted fivefold, 2 μL of the cDNA was used as DNA template in 15-μL amplification volumes with 400 nM of each primer and 7.5 μL of SYBR green master mix using the following cycling parameters: 95°C 30 s; followed by 40 cycles of 5 s at 95°C, 30 s at 55°C, and 30 s at 72°C. The primer pairs QL0615/QL0616, QL0619/QL0620, QL0633/QL0634, QL0635/QL0636, QL0637/QL0638, and QL0645/QL0646 were used to amplify the egfp, purR, catalase, fnbA, fnbB, and sigA genes, respectively, by qPCR. The endogenous gene sigA, which encodes the housekeeping RNA polymerase sigma factor, was used as an internal control for promoter characterization. The expression levels of the egfp gene under different promoters were normalized to the expression of the internal control.
Western Blot Analysis
Total protein extracts of S. aureus were prepared as described in the beta-galactosidase assay. The protein concentration was measured using a Bradford assay kit (Thermo Scientific, Waltham, MA, United States). Twenty micrograms of the total protein extract was separated by 12% SDS-PAGE and transferred onto a nitrocellulose membrane (Bio-Rad). The membrane was blocked with TBS-T buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% [vol/vol] Tween-20) containing 5% non-fat dry milk at room temperature (RT) for 1 h. Detection of the target proteins eGFP_6 × His, PurR_6 × His, and catalase_6 × His overexpressed in S. aureus was performed by incubating the membrane with the horseradish peroxidase (HRP)-labeled His-Tag (27E8) mouse mAb (CST, Danvers, MA, United States) (diluted 1:2,000 in TBS-T buffer) at RT for 1 h. After four washing steps (10 min per step) with TBS-T buffer, the signals were detected with a ChemiDoc MP (Bio-Rad) imaging system using Pierce ECL (Thermo Scientific, Waltham, MA, United States) as a chemiluminescence substrate.
Catalase Activity Assay
Bacterial cells from the 200 μL day-culture were collected in the middle of the lag growth phase (6 h after inoculation) by centrifugation. The precipitated cells were resuspended and lysed in 800 μL of phosphate-buffered saline (PBS, pH 7.4) by sonication on a SCIENTZ-IID system (SCIENTZ, Ningbo, China) according to the user guide. The protein concentration of the cell lysate was measured as described above. To measure the activity of catalase in the lysate, 20 μL of the cell lysate was mixed with 100 μL of 200 mM H2O2 (diluted with PBS) and incubated at 25°C for 10 min. Then, the reaction was stopped by adding 180 μL of ammonium molybdate (50 mM in H2O) into the mixture. After incubation at room temperature for 10 min, the absorbance of the yellow complex of molybdate and undecomposed hydrogen peroxide was measured at a wavelength of 405 nm in a 96-well plate (ASample) using the plate reader Synergy H1 (BioTek, Vermont, VT, United States). One unit of catalase activity was defined as 1 μM of H2O2 catalyzed and hydrolyzed by 1 mg lysate in 1 min at 25°C. Thereby, the catalase activity of the lysate (CAT) was calculated as follows: CAT (U/mg) = [(ΔAStandard − ΔASample)/ΔAStandard] × N/m/T × F, where ΔAStandard = AStandard − ABlank and ΔASample = ASample − AControl. AControl is the absorbance of the control group, in which H2O2 was replaced by PBS. AStandard is the absorbance of the standard group, in which the cell lysate was replaced by PBS, while ABlank is the absorbance of the blank group, in which cell lysate and H2O2 were both replaced by PBS. N represents the molar mass of H2O2 (micromole) used in the assay, and m is the mass of the cell lysate (mg) used in the assay. T is the reaction time in minutes, and F is the dilution factor of the cell lysate. The assay for each strain was performed in three independent experiments.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
QL: funding acquisition, project administration, investigation, methodology, writing, review, and editing. DL, NW, GG, YS, and QZ: data curation, formal analysis, investigation, methodology, visualization, writing original draft, and editing. XZ: project investigation, administration, writing, review, and editing. All authors contributed to the article and approved the submitted version.
Funding
This work was financed by the National Science Fund for Distinguished Young Scholars (32000094), the China Postdoctoral Science Foundation (2021M692311), the Postdoctoral Research Project, West China Hospital, Sichuan University (20HXBH017), and the 1⋅3⋅5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYXY21004).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Vincent Perreten for providing the pBUS1-Pcap-HC vector.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.818307/full#supplementary-material
References
Arnaud, M., Chastanet, A., and Debarbouille, M. (2004). New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl. Environ. Microbiol. 70, 6887–6891. doi: 10.1128/AEM.70.11.6887-6891.2004
Baba, T., Bae, T., Schneewind, O., Takeuchi, F., and Hiramatsu, K. (2008). Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J. Bacteriol. 190, 300–310. doi: 10.1128/JB.01000-07
Bae, T., and Schneewind, O. (2006). Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55, 58–63. doi: 10.1016/j.plasmid.2005.05.005
Bateman, B. T., Donegan, N. P., Jarry, T. M., Palma, M., and Cheung, A. L. (2001). Evaluation of a tetracycline-inducible promoter in Staphylococcus aureus in vitro and in vivo and its application in demonstrating the role of sigB in microcolony formation. Infect. Immun. 69, 7851–7857. doi: 10.1128/IAI.69.12.7851-7857.2001
Bayer, M. G., Heinrichs, J. H., and Cheung, A. L. (1996). The molecular architecture of the sar locus in Staphylococcus aureus. J. Bacteriol. 178, 4563–4570. doi: 10.1128/jb.178.15.4563-4570.1996
Boero, E., Brinkman, I., Juliet, T., van Yperen, E., van Strijp, J. A. G., Rooijakkers, S. H. M., et al. (2021). Use of Flow Cytometry to Evaluate Phagocytosis of Staphylococcus aureus by Human Neutrophils. Front. Immunol. 12:635825. doi: 10.3389/fimmu.2021.635825
Bruckner, R. (1997). Gene replacement in Staphylococcus carnosus and Staphylococcus xylosus. Fems. Microbiol. Lett. 151, 1–8.
Cheung, A. L., and Manna, A. C. (2005). Role of the distal sarA promoters in SarA expression in Staphylococcus aureus. Infect. Immun. 73, 4391–4394. doi: 10.1128/IAI.73.7.4391-4394.2005
Cheung, A. L., Nast, C. C., and Bayer, A. S. (1998). Selective activation of sar promoters with the use of green fluorescent protein transcriptional fusions as the detection system in the rabbit endocarditis model. Infect. Immun. 66, 5988–5993. doi: 10.1128/IAI.66.12.5988-5993.1998
Corrigan, R. M., and Foster, T. J. (2009). An improved tetracycline-inducible expression vector for Staphylococcus aureus. Plasmid 61, 126–129. doi: 10.1016/j.plasmid.2008.10.001
de Jong, N. W., van der Horst, T., van Strijp, J. A., and Nijland, R. (2017). Fluorescent reporters for markerless genomic integration in Staphylococcus aureus. Sci. Rep. 7:43889. doi: 10.1038/srep43889
Gardete, S., Ludovice, A. M., Sobral, R. G., Filipe, S. R., de Lencastre, H., and Tomasz, A. (2004). Role of murE in the Expression of beta-lactam antibiotic resistance in Staphylococcus aureus. J. Bacteriol. 186, 1705–1713. doi: 10.1128/JB.186.6.1705-1713.2004
Goncheva, M. I., Conceicao, C., Tuffs, S. W., Lee, H. M., Quigg-Nicol, M., Bennet, I., et al. (2020). Staphylococcus aureus Lipase 1 Enhances Influenza A Virus Replication. mbio 11, e00975–20. doi: 10.1128/mBio.00975-20
Goncheva, M. I., Flannagan, R. S., Sterling, B. E., Laakso, H. A., Friedrich, N. C., Kaiser, J. C., et al. (2019). Stress-induced inactivation of the Staphylococcus aureus purine biosynthesis repressor leads to hypervirulence. Nat. Commun. 10:775. doi: 10.1038/s41467-019-08724-x
Harrison, E. M., Ba, X., Coll, F., Blane, B., Restif, O., Carvell, H., et al. (2019). Genomic identification of cryptic susceptibility to penicillins and beta-lactamase inhibitors in methicillin-resistant Staphylococcus aureus. Nat. Microbiol. 4, 1680–1691. doi: 10.1038/s41564-019-0471-0
Kim, L., Mogk, A., and Schumann, W. (1996). A xylose-inducible Bacillus subtilis integration vector and its application. Gene 181, 71–76. doi: 10.1016/s0378-1119(96)00466-0
Kuiack, R. C., Veldhuizen, R. A. W., and McGavin, M. J. (2020). Novel Functions and Signaling Specificity for the GraS Sensor Kinase of Staphylococcus aureus in Response to Acidic pH. J. Bacteriol. 202, e00219–20. doi: 10.1128/JB.00219-20
Liew, A. T. F., Theis, T., Jensen, S. O., Garcia-Lara, J., Foster, S. J., Firth, N., et al. (2011). A simple plasmid-based system that allows rapid generation of tightly controlled gene expression in Staphylococcus aureus. Microbiology 157, 666–676. doi: 10.1099/mic.0.045146-0
Mader, U., Nicolas, P., Depke, M., Pane-Farre, J., Debarbouille, M., van der Kooi-Pol, M. M., et al. (2016). Staphylococcus aureus Transcriptome Architecture: from Laboratory to Infection-Mimicking Conditions. PLoS Genet. 12:e1005962. doi: 10.1371/journal.pgen.1005962
Malone, C. L., Boles, B. R., Lauderdale, K. J., Thoendel, M., Kavanaugh, J. S., and Horswill, A. R. (2009). Fluorescent reporters for Staphylococcus aureus. J. Microbiol. Methods 77, 251–260. doi: 10.1016/j.mimet.2009.02.011
Nair, D., Memmi, G., Hernandez, D., Bard, J., Beaume, M., Gill, S., et al. (2011). Whole-Genome Sequencing of Staphylococcus aureus Strain RN4220, a Key Laboratory Strain Used in Virulence Research, Identifies Mutations That Affect Not Only Virulence Factors but Also the Fitness of the Strain. J. Bacteriol. 193, 2332–2335. doi: 10.1128/Jb.00027-11
Ouyang, S., and Lee, C. Y. (1997). Transcriptional analysis of type 1 capsule genes in Staphylococcus aureus. Mol. Microbiol. 23, 473–482. doi: 10.1046/j.1365-2958.1997.d01-1865.x
Pereira, S. F., Henriques, A. O., Pinho, M. G., de Lencastre, H., and Tomasz, A. (2007). Role of PBP1 in cell division of Staphylococcus aureus. J. Bacteriol. 189, 3525–3531. doi: 10.1128/JB.00044-07
Popp, P. F., Dotzler, M., Radeck, J., Bartels, J., and Mascher, T. (2017). The Bacillus BioBrick Box 2.0: expanding the genetic toolbox for the standardized work with Bacillus subtilis. Sci. Rep. 7:15058. doi: 10.1038/s41598-017-15107-z
Redder, P., and Linder, P. (2012). New Range of Vectors with a Stringent 5-Fluoroorotic Acid-Based Counterselection System for Generating Mutants by Allelic Replacement in Staphylococcus aureus. Appl. Environ. Microbiol. 78, 3846–3854. doi: 10.1128/Aem.00202-12
Schenk, S., and Laddaga, R. A. (1992). Improved method for electroporation of Staphylococcus aureus. FEMS Microbiol. Lett. 73, 133–138. doi: 10.1016/0378-1097(92)90596-g
Schwendener, S., and Perreten, V. (2015). New shuttle vector-based expression system to generate polyhistidine-tagged fusion proteins in Staphylococcus aureus and Escherichia coli. Appl. Environ. Microbiol. 81, 3243–3254. doi: 10.1128/AEM.03803-14
Shang, W., Rao, Y., Zheng, Y., Yang, Y., Hu, Q., Hu, Z., et al. (2019). beta-Lactam Antibiotics Enhance the Pathogenicity of Methicillin-Resistant Staphylococcus aureus via SarA-Controlled Lipoprotein-Like Cluster Expression. Mbio 10:3. doi: 10.1128/mBio.00880-19
Turner, N. A., Sharma-Kuinkel, B. K., Maskarinec, S. A., Eichenberger, E. M., Shah, P. P., Carugati, M., et al. (2019). Methicillin-resistant Staphylococcus aureus: an overview of basic and clinical research. Nat. Rev. Microbiol. 17, 203–218. doi: 10.1038/s41579-018-0147-4
Wieland, K. P., Wieland, B., and Gotz, F. (1995). A promoter-screening plasmid and xylose-inducible, glucose-repressible expression vectors for Staphylococcus carnosus. Gene 158, 91–96. doi: 10.1016/0378-1119(95)00137-u
Yepes, A., Koch, G., Waldvogel, A., Garcia-Betancur, J. C., and Lopez, D. (2014). Reconstruction of mreB expression in Staphylococcus aureus via a collection of new integrative plasmids. Appl. Environ. Microbiol. 80, 3868–3878. doi: 10.1128/AEM.00759-14
Yu, J., Jiang, F., Zhang, F., Hamushan, M., Du, J., Mao, Y., et al. (2021). Thermonucleases Contribute to Staphylococcus aureus Biofilm Formation in Implant-Associated Infections-A Redundant and Complementary Story. Front Microbiol 12:687888. doi: 10.3389/fmicb.2021.687888
Keywords: S. aureus, highly expressed gene, constitutive promoter, gene expression, PurR, catalase
Citation: Liu Q, Li D, Wang N, Guo G, Shi Y, Zou Q and Zhang X (2022) Identification and Application of a Panel of Constitutive Promoters for Gene Overexpression in Staphylococcus aureus. Front. Microbiol. 13:818307. doi: 10.3389/fmicb.2022.818307
Received: 19 November 2021; Accepted: 14 February 2022;
Published: 28 February 2022.
Edited by:
José E. Barboza-Corona, University of Guanajuato, MexicoReviewed by:
Richard A. Daniel, Newcastle University, United KingdomKarl Hassan, The University of Newcastle, Australia
Copyright © 2022 Liu, Li, Wang, Guo, Shi, Zou and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiang Liu, liuqiangdyy@163.com; Xiaokai Zhang, zhangxkk@163.com