
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 25 February 2022
Sec. Systems Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.813783
 Saisai Zhang1
Saisai Zhang1 Hein M. Tun2,3
Hein M. Tun2,3 Dengwei Zhang2
Dengwei Zhang2 Hau-Tak Chau1
Hau-Tak Chau1 Fung-Yu Huang1Hin Kwok4Danny Ka-Ho Wong1,5Lung-Yi Mak1,5Man-Fung Yuen1,5*
Fung-Yu Huang1Hin Kwok4Danny Ka-Ho Wong1,5Lung-Yi Mak1,5Man-Fung Yuen1,5* Wai-Kay Seto1,5,6*
Wai-Kay Seto1,5,6*
Non-alcoholic fatty liver disease (NAFLD), the world’s most common chronic liver disease, is increasingly linked to gut dysbiosis. Paneth cells secrete antimicrobial peptides that regulate the gut microbiome, but their role in the pathogenesis of NAFLD remains unclear. Here, we determine the changes in NAFLD development and gut microbial composition and function via the injection of dithizone that can pharmacologically deplete the granules of Paneth cells. Eight-week-old C57BL/6J male mice (n = 31) were given a high-fat diet (HFD) or standard control diet for 12 weeks. Dithizone (10 mg/kg) was intravenously injected every 3 weeks during the period of diet feeding. Metagenomic DNA was extracted from fecal samples for PacBio Single-Molecule Real-Time sequencing to identify changes in microbial composition and predicted function. We observed dithizone-treated HFD mice, when compared to non-treated HFD mice, to have significant reductions in hepatic triglyceride content (28.98 vs. 53.52 mg/g, p = 0.0419); plasma insulin level (2.18 vs. 6.63 ng/ml, p = 0.0079); and relative mRNA levels of fatty acid synthase (0.52 vs. 1.57, p = 0.0428) and stearoyl-CoA desaturase-1 (0.43 vs. 1.20, p = 0.0121). Bacterial taxonomic profiling found dithizone-treated HFD mice, when compared to non-treated HFD mice, had a lower Firmicutes/Bacteroidetes ratio (2.53 vs. 5.26, p = 0.0541); a higher relative abundance of Bacteroides ASV21 and ASV42 (1.04 vs. 0.22%, p = 0.0277 and 0.96 vs. 0.09%, p = 0.0213); and a reduction in microbes belonging to Firmicutes (all p < 0.05). Bacteroides species correlated positively with predicted microbial functions such as L-methionine (r = 0.54, p = 0.0019) and tetrahydrofolate (r = 0.52, p = 0.0029) biosynthesis. Collectively, dithizone treatment was associated with alleviation in the severity of liver steatosis in HFD mice, possibly through gut microbiome modulation involving the increase in Bacteroides, suggesting microbiome-targeted therapies may have a role in the treatment of NAFLD.
The general population with non-alcoholic fatty liver disease (NAFLD) is increasing owing to the prevalence of obesity and metabolic risk factors (Bedogni et al., 2005; Cohen et al., 2011). There is emerging evidence demonstrating the gut microbiome as a critical environmental factor that affects the onset and progression of NAFLD. Germ-free mice with a high-fat diet (HFD) were found to gain less weight than conventional mice, with weight gain replenished by microbiota transplantation (Backhed et al., 2004, 2007). The involvement of the gut microbiome may contribute to the progression from simple hepatic steatosis to liver fibrosis and cirrhosis (Henao-Mejia et al., 2012; Le Roy et al., 2013; Mouzaki et al., 2013; Boursier et al., 2016). Based on 16S rRNA gene sequencing, obese NAFLD patients showed an increase in the levels of Firmicutes Lactobacillus and a reduction in the levels of Firmicutes Oscillibacter when compared to healthy controls (Michail et al., 2015).
Many efforts were put on to explain how gut microbiome dysbiosis leads to NAFLD development. The increased intestinal permeability caused by gut microbiome dysbiosis promotes translocation of metabolites into portal blood circulation. Responses to this dysbiosis promote liver damage (Miele et al., 2009; Bashiardes et al., 2016; Poeta et al., 2017). However, the exact role of the microbiome and its interaction with the pathogenesis of NAFLD remains undetermined. Exploring underlying regulatory mechanisms may contribute a deeper understanding of NAFLD and on the potential of microbiome-based interventions.
The specialized epithelial cells in the small intestine, named Paneth cells, play a homeostatic role in establishing and maintaining the intestinal microbiota by secreting antimicrobial peptides and proteins (Wilson et al., 1999; Bevins and Salzman, 2011). A previous study indicated that the percentage of Firmicutes was significantly lower and the percentage of Bacteroidetes was significantly higher in defensin-overexpressing mice, with the opposite observed in defensin-deficient mice (Raman et al., 2013; Jiang et al., 2015). These findings directly demonstrated that alterations in Paneth cell defensin expression have a significant impact on the bacterial composition of the microbiota. Given the regulatory role of Paneth cells in the microbial microenvironment, they are largely investigated in irritable bowel syndrome and Crohn’s disease (Stappenbeck and McGovern, 2017; Zhou et al., 2020). Paneth cells may be associated with other metabolic disorders, but there is a paucity of data about their role in the pathogenesis of NAFLD. We hence aimed to determine whether Paneth cells are involved in the development of NAFLD by injection of dithizone, which can disrupt cell granulates (Sawada et al., 1991; Lueschow et al., 2018; Berger et al., 2019; Guo et al., 2019; Liu et al., 2019), and identify Paneth-cell-oriented microbial alterations.
Eight-week-old male C57BL/6J mice (n = 31, Center of Comparative Medicine Research, The University of Hong Kong) were given an HFD (containing 60% fat from lard, 20% carbohydrates, and 20% proteins, D12492, Research Diets, New Brunswick, NJ) or a standard control diet (STD, containing 10% fat from lard, 70% carbohydrates, and 20% proteins, D12450J, Research Diets, New Brunswick, NJ) for 12 weeks to establish a NAFLD mouse model. The granulates in Paneth cells were depleted pharmacologically by intravenous injection of dithizone (Sawada et al., 1991; Lueschow et al., 2018; Berger et al., 2019; Guo et al., 2019; Liu et al., 2019) (10 mg/kg body weight, dissolved in 10 mg/ml lithium carbonate solution, Sigma-Aldrich, Darmstadt, Germany) every 3 weeks during the period of diet feeding. Mice were randomly divided into four groups (n = 7–8): (i) mice fed with STD and injected with the vehicle (SC group); (ii) mice fed with STD and injected with dithizone (SD group); (iii) mice fed with HFD and injected with the vehicle (HC group); and (iv) mice fed with HFD and injected with dithizone (HD group). All mice were maintained under controlled environmental conditions (23 ± 1°C, 50–60% humidity, 12-h light/dark cycles) with food and water ad libitum. Body weight was monitored weekly. All animal experimental procedures were approved by the Committee on the Use of Live Animals in Teaching and Research at the University of Hong Kong (reference number: CULATR 4507-17) and received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals.”
After a 12-week intervention, glucose tolerance tests were performed in all mice injected with D-glucose (2 g/kg body weight) intraperitoneally after being fasted for 16 h (overnight, between 6 p.m. and 10 a.m.). For insulin tolerance tests, all mice were intraperitoneally injected with insulin (0.75 IU/kg body weight) after being fasted for 6 h (between 10 a.m. and 4 p.m.). Blood glucose levels were monitored at various time points (0, 15, 30, 60, and 120 min) with an ACCU-CHEK Glucose Meter (Roche Diabetes Care, Inc., Basel, Switzerland).
Mice were fasted for 6 h and orally administrated with fluorescein isothiocyanate (FITC)-dextran (4 kDa, 44 mg/kg bodyweight, Sigma-Aldrich, Darmstadt, Germany). After a 1-h treatment, whole blood was collected from the tail vein and centrifuged at 3,000 rpm for 15 min. FITC-dextran was diluted with 1× phosphate buffer saline into 0, 27.5, 55, 110, 220, 440, 880, and 1,760 ng/ml to make a standard curve, and plasma samples were also diluted to a 1:10 ratio. Plasma FITC-dextran concentrations were measured at an excitation wavelength of 485 nm and an emission wavelength of 535 nm as previously reported (Fernandez-Carrera et al., 2017).
After a 12-week intervention and all in vivo tests, all mice were sacrificed under anesthesia. Whole-blood samples were collected by cardiac puncture and centrifuged at 3,000 rpm for 15 min. The circulating Paneth cell-specific α-defensin 5 (DEFA5) levels (Abbexa Ltd., Cambridge, United Kingdom) and insulin levels (Antibody and Immunoassay Services, HKU, Hong Kong) were detected by way of enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions.
Mouse liver tissues were embedded with both optimal cutting temperature compound and paraffin to make frozen and paraffin sections. The paraffin sections were prepared in 5 mm sizes, dehydrated in graded ethanol and xylene, and then processed for hematoxylin and eosin (H&E) and Masson’s trichrome staining following standard protocols, with the frozen sections stained using Oil Red O (Sigma-Aldrich, Darmstadt, Germany). Mouse ileal sections were stained with H&E and immunostained with an anti-lysozyme antibody (Abbexa Ltd., Cambridge, United Kingdom).
The lipids were extracted from liver tissues using the chloroform–methanol method (Folch et al., 1957) and were measured with the responding triglyceride and total cholesterol kits according to the manufacturer’s instructions. The mRNA levels of fatty acid synthesis-related genes fatty acid synthase (FASN) and SCD1 and fatty acid β-oxidation-related gene CTP1a were measured by quantitative PCR (qPCR).
Proteins and RNAs were extracted from mouse livers and the ileum using a radioimmunoprecipitation assay buffer (Sigma-Aldrich, Darmstadt, Germany) and TRIzol reagent (Invitrogen). The protein concentration was determined by using the BCA protein assay (Thermo Fisher Scientific, Waltham, MA, United States) according to the manufacturer’s instructions. Anti-lysozyme rabbit antibody (Abbexa Ltd., Cambridge, United Kingdom), anti-ZO-1 rabbit antibody (Thermo Fisher Scientific, Waltham, MA, United States), anti-occludin rabbit antibody (Thermo Fisher Scientific, Waltham, MA, United States), and anti-β-actin mouse antibody (Thermo Fisher Scientific, Waltham, MA, United States) were used. The protein bands were visualized and imaged with the ChemiDoc™ MP Imaging System (Bio-Rad, Hercules, CA, United States), with the densities analyzed by ImageJ software. qPCR was performed using the DNA Engine Opticon® 2 System for Real-Time PCR Detection (Bio-Rad, Hercules, CA, United States) with TB Green Premix Ex Taq (Tli RNase H Plus) (Takara, Kusatsu, Shiga, Japan) and specific primers. The relative levels of gene expressions were calculated by the 2–Δ Δ Ct method, after normalization with the abundance of β-actin. The sequences of primers used in this study were listed in Supplementary Table 1.
The protein level of lysozyme, a specific marker of Paneth cell (Sato et al., 2011), was determined by immunofluorescence staining and western blot analysis. Angiogenin 4 (ANG4) and regenerating islet-derived protein 3 gamma (Reg3γ) mRNA levels, antimicrobial peptides produced by Paneth cells as part of the innate immune response, were determined by qPCR.
Stool DNAs were extracted by using a QIAamp PowerFecal DNA kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Standard agarose gel electrophoresis was applied to evaluate the quality of DNA samples, and DNAs were quantified using a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States).
DNA library preparation was performed following the Full-Length 16S Amplification SMRTbell Template Preparation workflow. For generating tagged amplicons, first-round PCR (16S amplification) was performed using the 16S primers tailed with universal sequences (27F: 5AmMC6-gcagtcgaacatgtagctga ctcaggtcac-AGRGTTYGATYMTGGCTCAG and 1492R: 5Am MC6-tggatcacttgtgcaagcatcacatcgtag-RGYTACCTTGTTACGAC TT) and 2.5 ng input of gDNA with 20 PCR cycles. For generating barcoded amplicons, second-round PCR (barcoding amplification) for multiplexing reactions was performed using PacBio barcoded universal primers and 2 ng of tagged amplicons as input with nine PCR cycles. The KAPA HiFi Hot Start DNA Polymerase (KAPA Biosystems) was used to perform with denaturing at 95°C for 30 s, annealing at 57°C for 30 s, and extension at 72°C for 60 s. Post-amplification quality control was performed on a Bioanalyzer (Agilent Technologies, Santa Clara, CA, United States). SMRTbell library construction was performed by pooling the barcoded amplicons in equimolar concentrations, followed by DNA damage repair, SMRTbell ligation, and purification (SMRTbell library). Sequencing template preparation of the SMRTbell library was then performed using the binding calculator as instructed by PacBio followed by sequencing on the PacBio Sequel System.
Prior to the analysis, subreads were converted from raw PacBio sequencing data to highly accurate Q30 circular consensus sequencing (CCS) reads using Single-Molecule, Real-Time (SMRT) Link 3.1.1 software. Sequences were filtered with four full passes and a minimum predicted accuracy of 0.999. The DADA2 method (Callahan et al., 2016) was used in these processing steps: filtering, dereplication, denoising, amplicon sequence variant (ASV) inference, chimera, and merging. Taxonomy assignment was based on the naive Bayesian classifier with the SILVA v132 database (Quast et al., 2013) and BLAST searches against the NCBI nucleotide database (nt), at the genus level and species level, respectively. A minimum subsampling depth of 4,556 reads was selected for microbiota composition analysis.
Chao 1 (a measure of community richness) and Shannon’s index (a measure of richness and evenness) were used to describe the alpha diversity of microbial community. For beta diversity, unweighted UniFrac distance (a qualitative measure of community dissimilarity) was adopted. Principal coordinate analysis (PCoA) was performed using the vegan package1 of R (Version 3.4.4). The comparison of taxa between groups was tested by the Wilcoxon rank sum test when the taxa were present in more than 50% of samples; otherwise, Fisher’s exact test was adopted based on the presence/absence of taxa. The p-value was corrected using the Benjamini–Hochberg method (Benjamini and Hochberg, 1995), which controlled the false discovery rate (FDR). The log2-fold change in the relative abundance of individual ASVs was calculated manually with log2(abundance in case group + 1)/(abundance in control group + 1). ASVs were separated horizontally by genus and colored by phylum. Spearman’s rank correlation coefficient was adopted to understand the correlation, and the co-occurrence network was visualized with the Cytoscape software (Shannon et al., 2003) if |Spearman’s rank correlation coefficient| > 0.3 and adjusted p-value < 0.05. For pathway prediction, PICRUSt2 (Douglas et al., 2020) was employed for functional annotation to the MetaCyc database (SRI International, Menlo Park, CA, United States). Significantly differential pathways were calculated and visualized by STAMP (Parks et al., 2014) with the extended error bar, indicating statistically significant features along with the p-values, effect sizes, and confidence intervals. The generated sequencing data were deposited in the European Nucleotide Archive database with the accession number PRJEB46885.
All data were analyzed using GraphPad Prism 7.0 (GraphPad Software, San Diego, CA, United States) or R (Version 3.4.4) and presented as the mean ± standard error of the mean. Homeostatic Model Assessment for Insulin Resistance (HOMA-IR) was calculated according to the following formula (Matthews et al., 1985): [fasting insulin (μU/ml) × fasting glucose (mmol/L)]/22.5. To examine the significant differences between every two groups, the data were compared using unpaired two-tailed Student’s t-test as appropriate. A p-value < 0.05 was considered statistically significant.
In STD mice, the ileal protein expression of lysozyme (mean: 0.38 ± 0.05 vs. 1.00 ± 0.12, p = 0.0015, Figures 1A,B), the relative mRNA levels of antimicrobial peptides ANG4 (mean: 0.39 ± 0.07 vs. 1.00 ± 0.15, p = 0.0022, Figure 1C), Reg3γ (mean: 0.63 ± 0.04 vs. 1.00 ± 0.10, p = 0.0034, Figure 1D), and the plasma alpha-defensin 5 level (mean: 716.2 ± 60.71 vs. 1,211 ± 148.8 ng/ml, p = 0.0096, Figure 1E) were significantly reduced in dithizone-treated mice when compared to non-treated mice. Similar results were also observed between non-treated STD and non-treated HFD mice (all p < 0.05, Figures 1A–E). No significant differences were noted between non-treated HFD and dithizone-treated HFD mice (all p > 0.05) except alpha-defensin 5, one of the major antimicrobial peptides. Dithizone-treated HFD mice had a significantly lower plasma alpha-defensin 5 level than non-treated HFD mice (mean: 290.4 ± 62.28 vs. 660.4 ± 75.88 ng/ml vs. p = 0.0027, Figure 1E).
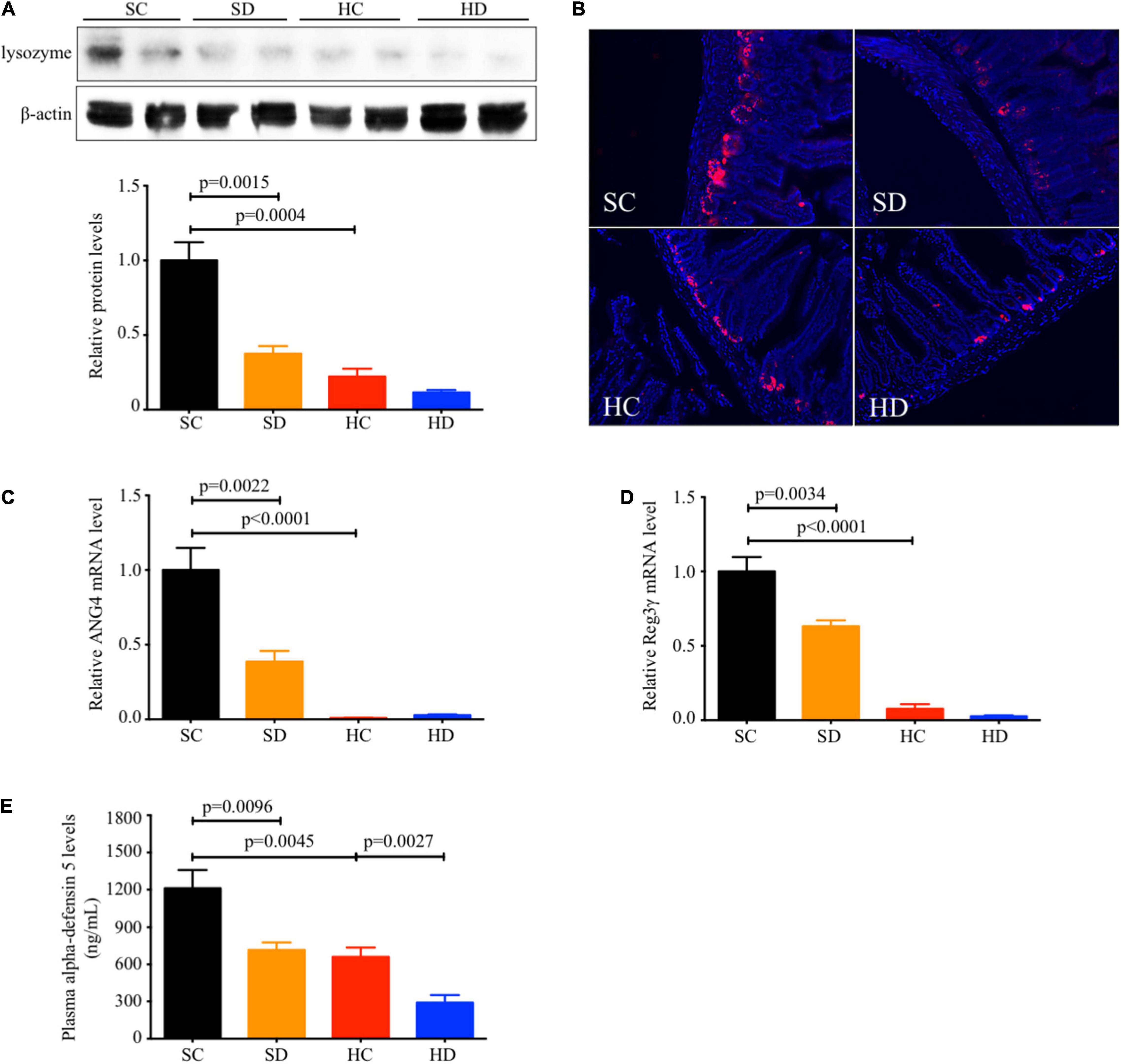
Figure 1. Both HFD and dithizone resulted in Paneth cell dysfunction. Eight-week-old male C57BL/6J mice were fed with STD or HFD for 12 weeks. The mice were intravenously injected with either vehicle or dithizone (10 mg/kg) every 3 weeks for a 12-week diet in total (n = 7–8). (A) The protein expression of lysozyme (a marker of Paneth cells) in the ileum was determined by western blot analysis and densitometric quantification of lysozyme. (B) Representative images of immunofluorescent staining of lysozyme with ×400 magnification. (C,D) The relative mRNA levels of ANG4 and Reg3γ. (E) Plasma alpha-defensin five levels were assayed by ELISA. Data are expressed as mean ± SEM for each group with unpaired Student’s t-test. SC, standard control diet/control; SD, standard control diet/dithizone; HC, high-fat diet/control; HD, high-fat diet/dithizone; ANG4, angiogenin 4; Reg3γ, regenerating islet-derived protein 3 gamma; ELISA, enzyme-linked immunosorbent assay; SEM, standard error of the mean.
Both body weight (Figure 2A) and liver-to-body weight ratio (Figure 2B) were significantly increased by HFD feeding (mean: 31.30 ± 0.51 vs. 48.95 ± 0.70 g, p < 0.0001; 43.86 ± 0.30 vs. 54.48 ± 4.14 mg/g, p = 0.0336) and partially reversed with borderline significance by dithizone treatment in HFD mice (mean: 48.95 ± 0.70 vs. 42.21 ± 2.90 g, p = 0.0864; 54.48 ± 4.14 vs. 37.98 ± 6.78 mg/g, p = 0.0801). Based on H&E staining and Oil Red O staining, HFD-induced hepatic lipid accumulation was significantly decreased in dithizone-treated HFD mice compared with non-treated HFD mice (19.38 ± 0.59% vs. 42.99 ± 4.72%, p = 0.0290, Figures 2C,D). Similarly, dithizone-treated HFD mice had a significant reduction in mean intrahepatic triglyceride (28.98 ± 7.40 vs. 53.52 ± 5.93 mg/g, p = 0.0419) and total cholesterol content (4.62 ± 0.68 vs. 8.96 ± 1.16 mg/g, p = 0.018) when compared to non-treated HFD mice (Figure 2E). No aforementioned significant differences were noted between non-treated and dithizone-treated STD mice.
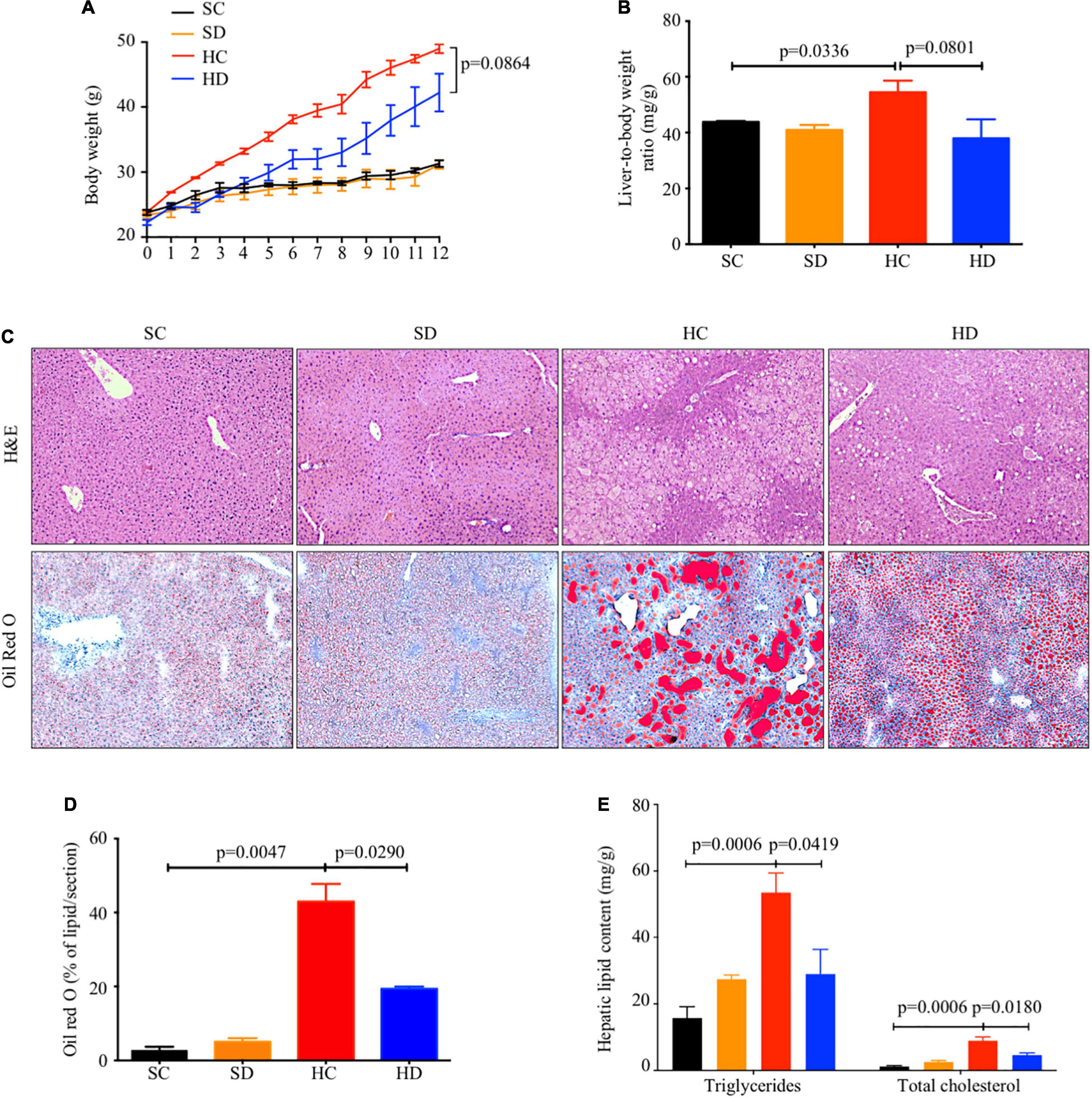
Figure 2. The inhibitory effects of dithizone on HFD-induced lipid accumulation. Eight-week-old male C57BL/6J mice were fed with STD or HFD for 12 weeks. The mice were intravenously injected with either vehicle or dithizone (10 mg/kg) every 3 weeks for a 12-week diet in total (n = 7–8). (A) Dynamic changes in body weight during the 12-week intervention. (B) Liver-to-body weight ratio after the 12-week intervention. (C) Representative images of hematoxylin and eosin (upper) and Oil Red O staining (lower) in the liver sections with ×100 magnification. (D) Quantification of Oil Red O-stained area using ImageJ software. (E) Intrahepatic triglyceride and total cholesterol content. Data are expressed as mean ± SEM for each group with unpaired Student’s t-test. SC, standard control diet/control; SD, standard control diet/dithizone; HC, high-fat diet/control; HD, high-fat diet/dithizone; H&E, hematoxylin and eosin; ORO, Oil Red O; SEM, standard error of the mean.
The relative mRNA levels of fatty acid synthesis-related genes FASN and stearoyl-CoA desaturase-1 (SCD1) were significantly downregulated in dithizone-treated HFD mice when compared to non-treated HFD mice (0.52 ± 0.18 vs. 1.57 ± 0.43, p = 0.0428; 0.43 ± 0.12 vs. 1.20 ± 0.23, p = 0.0121), whereas fatty acid β-oxidation-related gene carnitine palmitoyltransferase 1a (CTP1a) was upregulated (1.67 ± 0.27 vs. 1.05 ± 0.14, p = 0.0653, Supplementary Figure 1).
Pancreatic weight was reduced in dithizone-treated HFD mice when compared to non-treated HFD mice (mean: 0.17 ± 0.02 vs. 0.26 ± 0.04 g, p = 0.0609, Figure 3A), while no significant differences in fasting blood glucose level was observed (Figure 3B). In addition, non-treated HFD mice exhibited progressive development of glucose intolerance, hyperinsulinemia, and insulin resistance when compared to STD mice (Figures 3C–H). This was not seen in dithizone-treated HFD mice, with significantly lower insulin levels (mean: 2.18 ± 0.36 vs. 6.63 ± 1.29 ng/ml, p = 0.0079, Figure 3C), HOMA-IR (mean: 18.31 ± 1.87 vs. 64.90 ± 8.71, p = 0.0002, Figure 3D), and glucose intolerance (mean area under curve: 2,220 ± 230.1 vs. 3,163 ± 181.0, p = 0.0323, Figures 3E,F) when compared to non-treated HFD mice. Insulin resistance also showed a similar decrease (mean area under curve: 809.3 ± 52.53 vs. 1,119 ± 134.4, p = 0.0981, Figures 3G,H). No aforementioned significant differences were observed between non-treated and dithizone-treated STD mice.
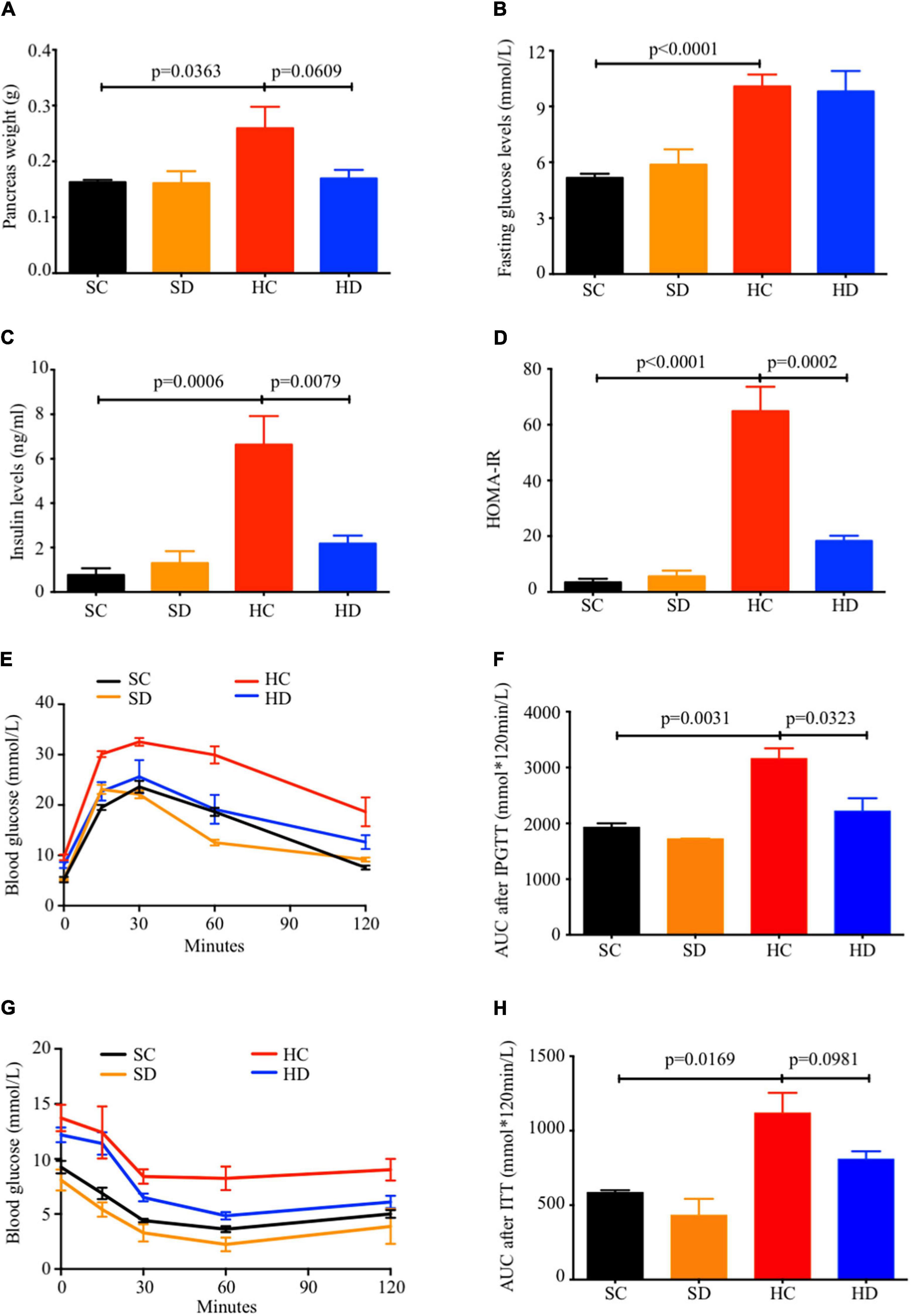
Figure 3. Dithizone ameliorates HFD-induced glucose intolerance and insulin resistance. Eight-week-old male C57BL/6J mice were fed with STD or HFD for 12 weeks. The mice were intravenously injected with either vehicle or dithizone (10 mg/kg) every 3 weeks for a 12-week diet in total (n = 7–8). (A) Pancreas weight after the 12-week intervention. (B) Fasting blood glucose level after the 12-week intervention. (C) Fasting insulin levels after the 12-week intervention. (D) HOMA-IR was calculated according to the following formula: [fasting insulin (μU/L) × fasting glucose (nmol/L)]/22.5. (E,F) Glucose tolerance was determined by an intraperitoneal glucose tolerance test (IPGTT), and the total area under the curve (AUC) for the 0–120 min period was calculated. (G,H) Insulin resistance was determined by an insulin tolerance test (ITT) and quantification of AUC. Data are expressed as mean ± SEM for each group with unpaired Student’s t-test. SC, standard control diet/control; SD, standard control diet/dithizone; HC, high-fat diet/control; HD, high-fat diet/dithizone; AUC, area under the curve; SEM, standard error of the mean.
An in vivo gut permeability test showed that the FITC-dextran concentration was significantly increased by HFD feeding in non-treated mice (715.2 ± 13.86 vs. 1,091 ± 147.6 ng/ml, p = 0.0239). Dithizone-treated HFD mice had a numeric reduction in FITC-dextran concentration (855.6 ± 36.46 vs. 1,091 ± 147.6 ng/ml, p = 0.1708) when compared to non-treated HFD mice (Supplementary Figure 2A). The expression of tight junction proteins zonula occludens (ZO)-1, a marker of intestinal permeability, was downregulated by HFD feeding (mean: 1.00 ± 0.01 vs. 0.49 ± 0.09, p = 0.0326), and partially restored with borderline significance by dithizone treatment (mean: 1.95 ± 0.37, p = 0.0604, Supplementary Figure 2B).
Altogether, 247,277 reads were obtained from PacBio SMRT sequencing. In terms of bacterial taxonomic profiling at the phylum level (Figure 4A), no considerable differences were observed in the alpha diversity indices (Chao 1 and Shannon’s index) within these four groups (Supplementary Figure 3A). PCoA based on unweighted UniFrac distance demonstrated that the gut microbiota was significantly altered in dithizone-treated STD mice when compared to those in non-treated STD mice (p = 0.0015, Supplementary Figure 3B). The difference was also observed between non-treated and dithizone-treated HFD mice (p = 0.0958, Supplementary Figure 3B). The compositions of gut microbiota in non-treated HFD mice and treated STD mice differ greatly from those in non-treated STD mice (Supplementary Figure 4). The Firmicutes/Bacteroidetes ratio was significantly increased by HFD feeding (mean: 5.26 ± 0.92 vs. 0.41 ± 0.02, p = 0.0002) and partially reversed with borderline significance by dithizone treatment (mean: 2.53 ± 0.34, p = 0.0541, Figure 4B).
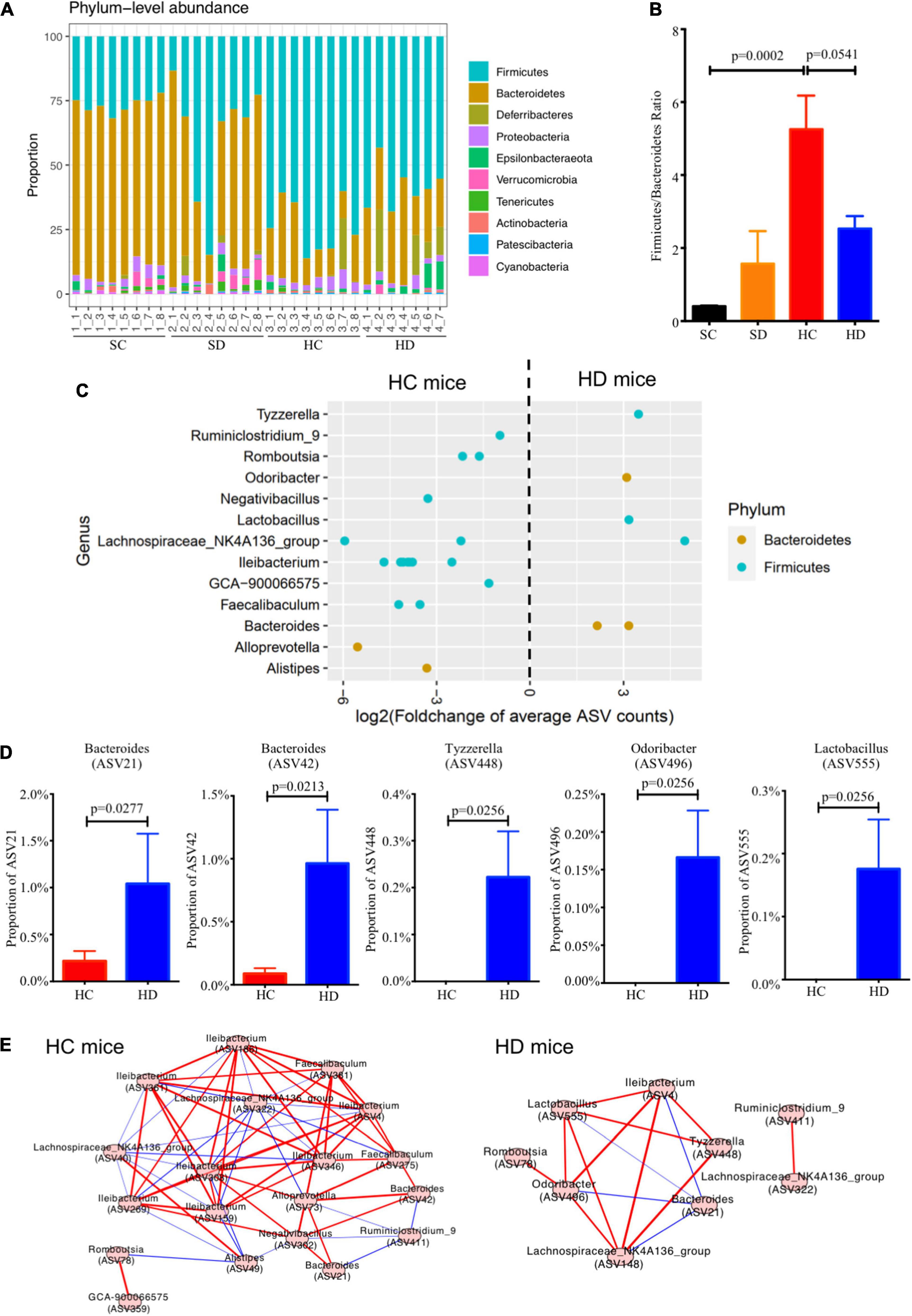
Figure 4. Dithizone modulates gut microbiota in HFD-fed mice. Mouse fecal microbiome from four groups were analyzed by using PacBio SMRT sequencing (n = 7–8 for each group). (A) The microbial profiles at the genus level. (B) The ratio of Firmicutes to Bacteroidetes. (C) Log2-fold change in the relative abundance of ASVs was used to identify the altered bacterial genera. ASVs were separated horizontally by genus and colored by phylum. (D) The proportion of altered ASVs. (E) The co-occurrence networks were used to understand the bacterial interaction at the genus level under different groups. Spearman’s rank correlation coefficient was adopted to understand the correlation, and the co-occurrence network was visualized if | Spearman’s rank correlation coefficient| was > 0.3 and p-value was < 0.05. The comparison was tested by the Wilcoxon rank sum test. The p-value was corrected using the Benjamini–Hochberg method. A red line indicates a positive correlation, and a blue line indicates a negative correlation. SC, standard control diet/control; SD, standard control diet/dithizone; HC, high-fat diet/control; HD, high-fat diet/dithizone; ASVs, individual amplicon sequence variants.
At the genus level, Bacteroides, Odoribacter, Lactobacillus, and Tyzzerella were significantly enriched whereas Ruminiclostridiumin_9, Romboutsia, Ileibacterium, Faecalibaculum, and GCA-900066575, which all belong to Firmicutes were decreased in dithizone-treated HFD mice when compared to non-treated HFD mice (all p < 0.05, Figure 4C). Among them, dithizone-treated HFD mice had a significantly higher mean abundance of Bacteroides ASV21 and ASV42 (1.04 ± 0.54% vs. 0.22 ± 0.11%, p = 0.0277, and 0.96 ± 0.43% vs. 0.09 ± 0.04%, p = 0.0213), Tyzzerella ASV448, Odoribacter ASV496, and Lactobacillus ASV555 (0.22 ± 0.10%, 0.17 ± 0.06%, and 0.18 ± 0.08% vs. 0.00 ± 0.005, all p = 0.0256) than non-treated HFD mice (Figure 4D).
The co-occurrence network in dithizone-treated HFD mice differed from that in non-treated HFD mice, displaying that Bacteroides ASV21 correlated negatively with Odoribacter (r = -0.85, p = 0.0148) and Lactobacillus (r = -0.93, p = 0.0027) (Figure 4E). Bacteroides ASV42 was also correlated negatively with the expression of antimicrobial peptides, including ANG4, Reg3γ, and DEFA5 (r = -0.45, -0.62, and -0.59, p = 0.0116, 0.0002, and 0.0007, Figure 5). The decreases in genera belonging to Firmicutes (Ruminiclostridiumin_9, Romboutsia ASV78 and ASV840, Faecalibaculum ASV275, and Lachnospiraceae_NK4A136_group ASV40) correlated positively with the corresponding reductions in intrahepatic triglycerides and total cholesterol levels (all r > 0.3 and p < 0.05, Figure 5).
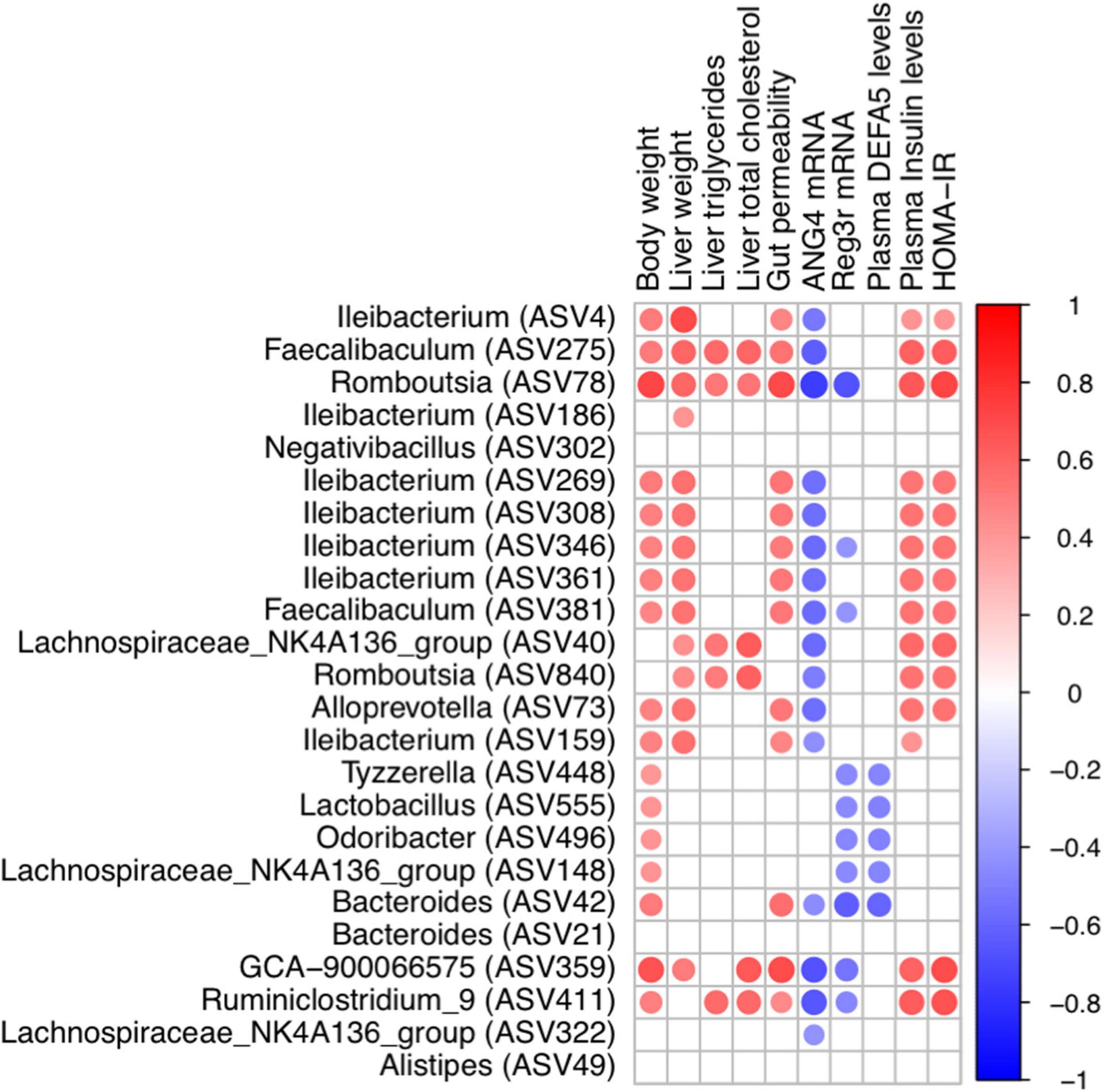
Figure 5. Correlation between metabolic indices and bacterial genera. Heatmap of Spearman’s rank correlation coefficient between metabolic indices and bacterial genera in HC and HD groups was filtered by rho > 0.3 and p-value < 0.05. A red line indicates a positive correlation, and a blue line indicates a negative correlation. SC, standard control diet/control; SD, standard control diet/dithizone; HC, high-fat diet/control; HD, high-fat diet/dithizone.
Multiple predicted pathways were altered by dithizone treatment in HFD mice. Among them, the superpathways of L-methionine and tetrahydrofolate biosynthesis were significantly enriched (p = 0.024 and 0.05, Figure 6A) in dithizone-treated HFD mice when compared to non-treated HFD mice. Correlation analysis demonstrated that Bacteroides ASV42 had the strongest positive correlation with the superpathway of L-methionine biosynthesis (by sulfhydrylation) (r = 0.54, p = 0.0019) and superpathway of tetrahydrofolate biosynthesis (r = 0.52, p = 0.0029) (Figure 6B). In addition, microbiome functional analysis also predicted that the purine/pyrimidine nucleotide biosynthesis-related pathways were significantly decreased in dithizone-treated HFD mice when compared to non-treated HFD mice (all p < 0.05, Figure 6A). Purine/pyrimidine nucleotide biosynthesis-related pathways correlated positively with genera belonging to Firmicutes (Romboutsia ASV78, Ruminiclostridiumin_9, and GCA-900066575; all r > 0.3 and p < 0.05, Figure 6B) and also correlated positively with intrahepatic triglycerides and total cholesterol levels (all r > 0.3 and p < 0.05, Supplementary Figure 5).
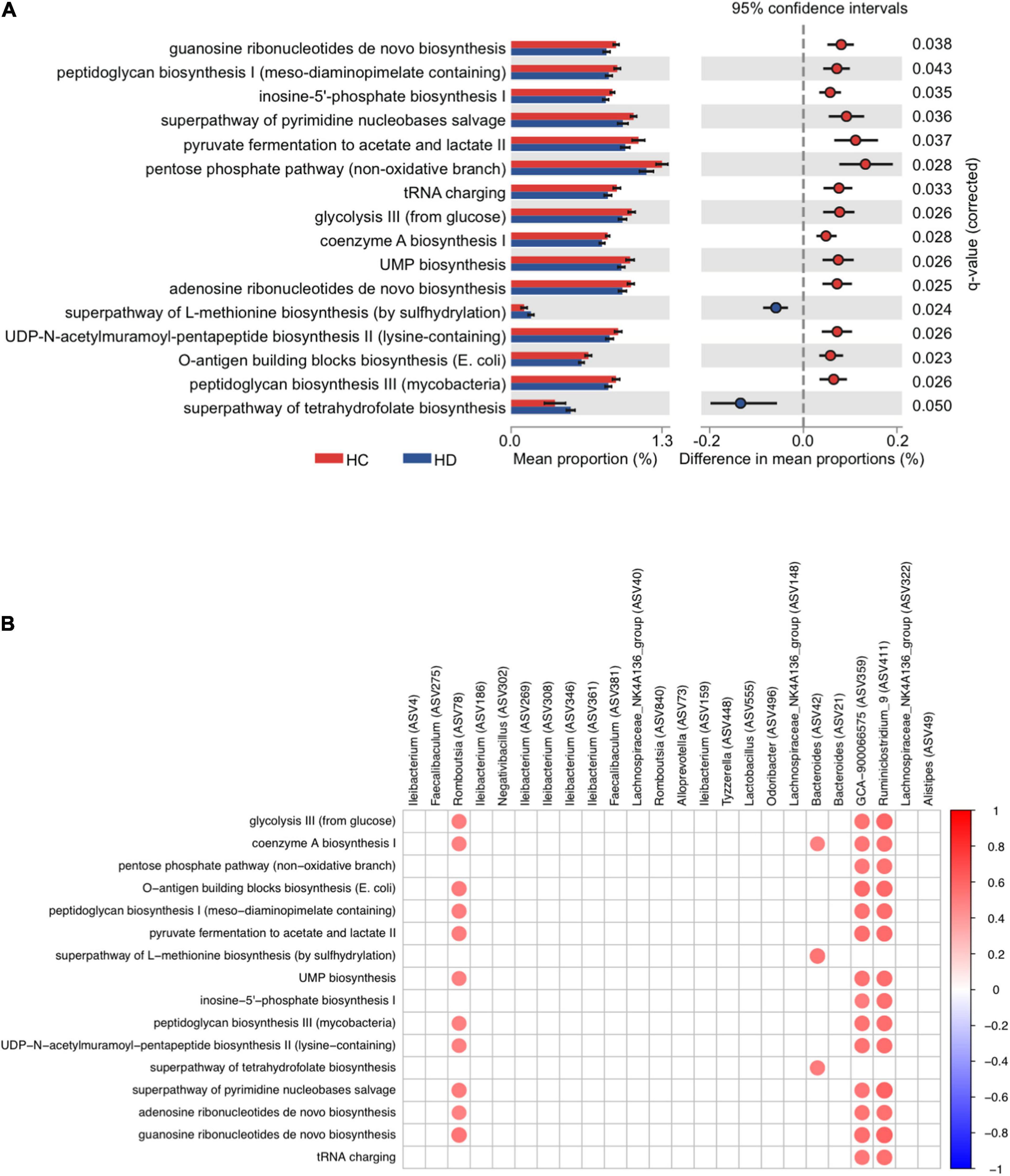
Figure 6. The microbial function of altered microbiota in HFD-fed mice. Mouse fecal microbiome from four groups was analyzed by using PacBio SMRT sequencing (n = 7–8 for each group). (A) The significantly differential pathways were calculated and visualized with the extended error bar, indicating statistically significant features along with the p-values, effect sizes, and confidence intervals. (B) Heatmap of Spearman’s rank correlation coefficient between genera and differential pathways was filtered by | Spearman’s rank correlation coefficient| > 0.3 and p-value < 0.05. A red dot indicates a positive correlation, and a blue dot indicates a negative correlation. SC, standard control diet/control; SD, standard control diet/dithizone; HC, high-fat diet/control; HD, high-fat diet/dithizone.
While a great deal of success had been achieved in explaining how gut microbiome dysbiosis leads to NAFLD development, the exact role remains undetermined. The present study had several important findings. First, administration of dithizone significantly disrupted Paneth cells and altered the microbial composition in STD mice; however, no significant disturbance to liver steatosis and glucose metabolism was caused. Second, dithizone significantly reduced HFD-induced hepatic triglycerides and total cholesterol, with improvements in glucose intolerance and insulin resistance and modulatory effects on fatty acid processes. Third, microbial dysbiosis caused by HFD was markedly altered by dithizone, associated with a decrease in the Firmicutes/Bacteroidetes ratio and an increase in genus Bacteroides. Last, the presence of Bacteroides in dithizone-treated HFD mice may result in the upregulated production of L-methionine and folate via the superpathway of L-methionine and tetrahydrofolate biosynthesis. Our study findings suggest that dithizone-associated microbial alterations in HFD mice may play a beneficial role in the disease process of NAFLD (Figure 7).
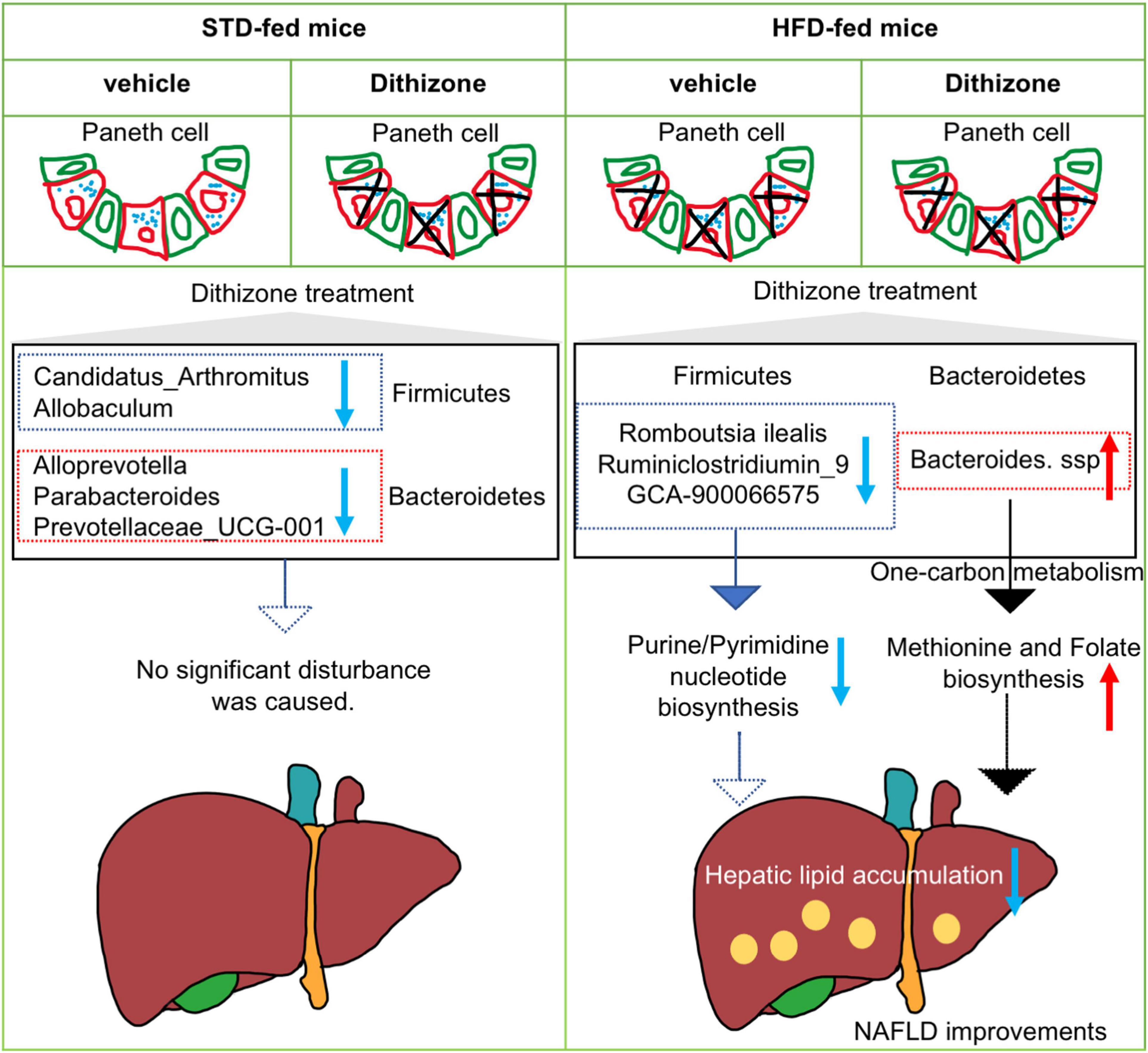
Figure 7. Schematic diagram of the involvement of dithizone-modulated microbial alterations in NAFLD. In STD mice, administration of dithizone significantly disrupted the Paneth cell and altered the microbial composition with no significant disturbance to liver steatosis and glucose metabolism. In HFD mice, dithizone significantly reduced HFD-induced hepatic lipid accumulation. The protective effect of dithizone on NAFLD was mediated by gut microbiome modulation, associated with a notable increase in certain Bacteroides species and a decrease in certain Firmicutes genera. The presence of Bacteroides may result in the upregulated production of L-methionine and folate via the superpathway of L-methionine and tetrahydrofolate biosynthesis.
Hepatic steatosis, the major histopathological change with HFD feeding, was alleviated by dithizone treatment. Compared to non-treated HFD mice, dithizone-treated HFD mice had a significantly reduced hepatic triglyceride and total cholesterol content (Figure 2). These results, supported by the modulatory effect of dithizone on lipogenesis and β-oxidation in HFD mice (Supplementary Figure 1), were consistent with previous reports (Postic and Girard, 2008; Xiong et al., 2021).
Paneth cells release their microbicidal granule contents into the gut lumen to maintain gut homeostasis. The function of Paneth cells was disturbed by HFD feeding, evidenced by the decreased ileal lysozyme level and antimicrobial peptides ANG4, Reg3γ, and DEFA5 (Figure 1). Similar results were also noted in STD mice after dithizone administration. Both HFD feeding and dithizone disrupted Paneth cells and downregulated the level of antimicrobial peptides. Recent studies have elucidated that Paneth cells secrete lysozyme via secretory autophagy (Bel et al., 2017; Bel and Hooper, 2018; Delorme-Axford and Klionsky, 2018); the disruption of Paneth cells by dithizone administration may also be mediated through an autophagy-related mechanism (Lueschow et al., 2018). Although dithizone treatment resulted in Paneth cell dysfunction in STD mice, no significant disturbance to markers of liver steatosis was caused (Figure 2), and the altered genus induced by dithizone administration (Supplementary Figure 4A) significantly differed from that induced by HFD feeding (Supplementary Figure 4B), suggesting that Paneth cell dysfunction per se is not the causal factor contributing to NAFLD development.
The beneficial effects of dithizone only become relevant in an HFD environment. In the normal condition, the effects of dithizone on Paneth cells, which downregulate the levels of antimicrobial peptides (Figure 1), are considered as a kind of side effects; no other additional effects on liver function were observed. However, the side effect of dithizone is not prominent in an HFD environment, since HFD feeding also disrupted the function of Paneth cells. In this case, besides dithizone functioning as an inhibitor of antimicrobial peptides, our results showed that dithizone treatment alleviates the severity of liver steatosis by modulating the gut microbiome in HFD mice.
An increase in the ratio of two domain phyla Firmicutes and Bacteroidetes has been widely considered a signature of gut dysbiosis and associated with NAFLD (Ley et al., 2006; Porras et al., 2017). In the current study, dithizone treatment led to a decreased Firmicutes/Bacteroidetes ratio in HFD mice. Moreover, Bacteroides presented in dithizone-treated HFD mice with higher abundance than in non-treated HFD mice. However, in STD mice, no significant difference was observed in the abundance of genus Bacteroides between non-treated and dithizone-treated STD mice. Whether dithizone had a direct effect on promoting the growth of Bacteroides species needs to be further determined by an in vitro growth curve study. Prior studies have shown that several Bacteroides species alleviate lipid metabolic disorders; Bacteroides thetaiotaomicron, vulgatus, and acidifaciens were associated with reductions in diet-induced body weight gain, attenuations in atherosclerosis, and improvements in insulin sensitivity, respectively (Liu et al., 2017; Yang et al., 2017; Yoshida et al., 2018). Meanwhile, Ruminiclostridiumin_9, Romboutsia, and GCA-900066575, all microbes belonging to Firmicutes and with associations to obesity (Gao et al., 2020; Horne et al., 2020), were decreased in dithizone-treated HFD mice when compared to non-treated HFD mice. Romboutsia ASV78 was further annotated as Romboutsia ilealis (matched with 100% identity by NCBI BLAST); a recently published study has demonstrated that supplementation with Romboutsia ilealis in mice resulted in glucose intolerance (Rodrigues et al., 2021).
One-carbon metabolism comprises interrelated methionine and folate pathways (Clare et al., 2019). Folate has the beneficial effects of alleviating hepatic steatosis (Qiao et al., 2020), while its depletion may cause oxidative stress in the liver and lead to the development of NAFLD (Huang et al., 2001). Methionine is an essential amino acid involved in DNA methylation, with its depletion leading to liver steatosis in mice (Pacana et al., 2015). Indeed, a methionine- and choline-deficient diet is usually used to establish an animal NAFLD model covering the whole spectrum of liver pathologies from steatosis to hepatic fibrosis (Stephenson et al., 2018). In our study, we demonstrated that one species from the genus Bacteroides, ASV42 (matched to Bacteroides vulgatus with 99.931% identity by NCBI BLAST), had a positive relationship with producing folate and L-methionine. However, the intervention study and the mechanistic insights of Bacteroides species and its metabolites folate and L-methionine in the prevention of NAFLD development will need further confirmation.
Our study findings would lead to an intriguing clinical question: can therapies aimed at these microbial alterations be effective in treating NAFLD? This question is especially relevant given the paucity of pharmaceutical options currently available for the disease. Microbiome-based therapies targeting different Bacteroides species may be a possible approach. That being said, our study is limited by the inadequate microbial classification of Bacteroides. The two Bacteroides species found in this study would require further classification via full-length sequencing, as well as microbial isolation and identification. Moreover, further animal experiments are required to investigate whether Paneth cell dysfunction-related microbial changes are the causal factor contributing to NAFLD development by colonizing mice with fecal microbiota from dithizone-treated mice. Other limitations include the lack of analysis for actual microbiome functional pathways which may provide better mechanistic insight on Paneth cell dysfunction and NAFLD and the lack of tissue metabolomic data to further clarify the role of bacteria-fermented metabolites in the development of NAFLD.
In summary, dithizone alleviated the severity of NAFLD, manifesting as a decrease in intrahepatic lipid accumulation, accompanying improvements in glucose intolerance and insulin resistance. The protective effect of dithizone on NAFLD was mediated by gut microbiome modulation, potentially associated with the notable increase in Bacteroides species and decrease in some Firmicutes genera. Our study strengthens the understanding of gut-related mechanisms in NAFLD development. Future research will be needed to study if Bacteroides species can emerge as a novel therapeutic option for NAFLD.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The animal study was reviewed and approved by the Committee on the Use of Live Animals in Teaching and Research at the University of Hong Kong (reference number: CULATR 4507-17).
SZ was involved in study concept and design, laboratory measurements, acquisition of data, analysis, interpretation of data, and drafting of the manuscript. HT, DZ, H-TC, F-YH, HK, DW, and L-YM were involved in the acquisition of data, analysis, and interpretation of data. M-FY was involved in the study concept and design and critical revision of the manuscript. W-KS was involved in study concept and design, securing research funding, analysis and interpretation of data, critical revision of the manuscript, and overall study supervision. All authors declare that they have participated in the preparation of the manuscript and have seen and approved the final version.
This study was supported by the Guangdong Natural Science Fund, Guangdong Province Science and Technology Department (ref no: 2019A1515012003).
M-FY is an advisory board member and/or received research funding from AbbVie, Arbutus Biopharma, Assembly Biosciences, Bristol Myers Squibb, Dicerna Pharmaceuticals, GlaxoSmithKline, Gilead Sciences, Janssen, Merck Sharp and Dohme, Clear B Therapeutics, Springbank Pharmaceuticals; and received research funding from Arrowhead Pharmaceuticals, Fujirebio Incorporation, and Sysmex Corporation. W-KS received speaker’s fees from AstraZeneca and Mylan, is an advisory board member of CSL Behring, is an advisory board member and received speaker’s fees from AbbVie, and is an advisory board member, received speaker’s fees and researching funding from Gilead Sciences.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the Centre of PanorOmic Sciences, The University of Hong Kong for the PacBio sequencing of fecal samples.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.813783/full#supplementary-material
Backhed, F., Ding, H., Wang, T., Hooper, L. V., Koh, G. Y., Nagy, A., et al. (2004). The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci U.S.A. 101, 15718–15723. doi: 10.1073/pnas.0407076101
Backhed, F., Manchester, J. K., Semenkovich, C. F., and Gordon, J. I. (2007). Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. U.S.A. 104, 979–984. doi: 10.1073/pnas.0605374104
Bashiardes, S., Shapiro, H., Rozin, S., Shibolet, O., and Elinav, E. (2016). Non-alcoholic fatty liver and the gut microbiota. Mol. Metab. 5, 782–794. doi: 10.1016/j.molmet.2016.06.003
Bedogni, G., Miglioli, L., Masutti, F., Tiribelli, C., Marchesini, G., and Bellentani, S. (2005). Prevalence of and risk factors for nonalcoholic fatty liver disease: the Dionysos nutrition and liver study. Hepatology 42, 44–52. doi: 10.1002/hep.20734
Bel, S., and Hooper, L. V. (2018). Secretory autophagy of lysozyme in Paneth cells. Autophagy 14, 719–721. doi: 10.1080/15548627.2018.1430462
Bel, S., Pendse, M., Wang, Y., Li, Y., Ruhn, K. A., Hassell, B., et al. (2017). Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science 357, 1047–1052. doi: 10.1126/science.aal4677
Benjamini, Y., and Hochberg, Y. (1995). Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Stat. Methods 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Berger, J. N., Gong, H., Good, M., and Mcelroy, S. J. (2019). Dithizone-induced Paneth cell disruption significantly decreases intestinal perfusion in the murine small intestine. J. Pediatr. Surg. 54, 2402–2407. doi: 10.1016/j.jpedsurg.2019.02.021
Bevins, C. L., and Salzman, N. H. (2011). Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol. 9, 356–368. doi: 10.1038/nrmicro2546
Boursier, J., Mueller, O., Barret, M., Machado, M., Fizanne, L., Araujo-Perez, F., et al. (2016). The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 63, 764–775. doi: 10.1002/hep.28356
Callahan, B. J., Mcmurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Clare, C. E., Brassington, A. H., Kwong, W. Y., and Sinclair, K. D. (2019). One-Carbon Metabolism: linking Nutritional Biochemistry to Epigenetic Programming of Long-Term Development. Annu. Rev. Anim. Biosci. 7, 263–287. doi: 10.1146/annurev-animal-020518-115206
Cohen, J. C., Horton, J. D., and Hobbs, H. H. (2011). Human fatty liver disease: old questions and new insights. Science 332, 1519–1523. doi: 10.1126/science.1204265
Delorme-Axford, E., and Klionsky, D. J. (2018). Secretory autophagy holds the key to lysozyme secretion during bacterial infection of the intestine. Autophagy 14, 365–367. doi: 10.1080/15548627.2017.1401425
Douglas, G. M., Maffei, V. J., Zaneveld, J. R., Yurgel, S. N., Brown, J. R., Taylor, C. M., et al. (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688. doi: 10.1038/s41587-020-0548-6
Fernandez-Carrera, A., Vigo, E., Regueiro-Rodriguez, C., Gonzalez-Fernandez, A., Olivieri, D., and Aroeira, L. S. (2017). Sensitive and non-invasive method for the in vivo analysis of membrane permeability in small animals. Lab. Invest. 97, 1114–1120. doi: 10.1038/labinvest.2017.66
Folch, J., Lees, M., and Sloane Stanley, G. H. (1957). A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226, 497–509. doi: 10.1016/s0021-9258(18)64849-5
Gao, X., Chang, S., Liu, S., Peng, L., Xie, J., Dong, W., et al. (2020). Correlations between alpha-Linolenic Acid-Improved Multitissue Homeostasis and Gut Microbiota in Mice Fed a High-Fat Diet. mSystems 5:6. doi: 10.1128/mSystems.00391-20
Guo, Y., Huang, C., Liu, L., Fu, X., Lu, Y., Zheng, J., et al. (2019). Paneth Cell Ablation Aggravates Pancreatic and Intestinal Injuries in a Rat Model of Acute Necrotizing Pancreatitis after Normal and High-Fat Diet. Mediators Inflamm. 2019:8474523. doi: 10.1155/2019/8474523
Henao-Mejia, J., Elinav, E., Jin, C., Hao, L., Mehal, W. Z., Strowig, T., et al. (2012). Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185. doi: 10.1038/nature10809
Horne, R. G., Yu, Y., Zhang, R., Abdalqadir, N., Rossi, L., Surette, M., et al. (2020). High Fat-High Fructose Diet-Induced Changes in the Gut Microbiota Associated with Dyslipidemia in Syrian Hamsters. Nutrients 12:3557. doi: 10.3390/nu12113557
Huang, R. F., Hsu, Y. C., Lin, H. L., and Yang, F. L. (2001). Folate depletion and elevated plasma homocysteine promote oxidative stress in rat livers. J. Nutr. 131, 33–38. doi: 10.1093/jn/131.1.33
Jiang, W., Wu, N., Wang, X., Chi, Y., Zhang, Y., Qiu, X., et al. (2015). Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci. Rep. 5:8096. doi: 10.1038/srep08096
Le Roy, T., Llopis, M., Lepage, P., Bruneau, A., Rabot, S., Bevilacqua, C., et al. (2013). Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 62, 1787–1794. doi: 10.1136/gutjnl-2012-303816
Ley, R. E., Turnbaugh, P. J., Klein, S., and Gordon, J. I. (2006). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023. doi: 10.1038/4441022a
Liu, L., Guo, Y., Zheng, J., Lu, Y., Shen, Y., Huang, C., et al. (2019). Paneth cell ablation increases the small intestinal injury during acute necrotizing pancreatitis in rats. Mol. Med. Rep. 20, 473–484. doi: 10.3892/mmr.2019.10274
Liu, R., Hong, J., Xu, X., Feng, Q., Zhang, D., Gu, Y., et al. (2017). Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat. Med. 23, 859–868. doi: 10.1038/nm.4358
Lueschow, S. R., Stumphy, J., Gong, H., Kern, S. L., Elgin, T. G., Underwood, M. A., et al. (2018). Loss of murine Paneth cell function alters the immature intestinal microbiome and mimics changes seen in neonatal necrotizing enterocolitis. PLoS One 13:e0204967. doi: 10.1371/journal.pone.0204967
Matthews, D. R., Hosker, J. P., Rudenski, A. S., Naylor, B. A., Treacher, D. F., and Turner, R. C. (1985). Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28, 412–419. doi: 10.1007/BF00280883
Michail, S., Lin, M., Frey, M. R., Fanter, R., Paliy, O., Hilbush, B., et al. (2015). Altered gut microbial energy and metabolism in children with non-alcoholic fatty liver disease. FEMS Microbiol. Ecol. 91, 1–9. doi: 10.1093/femsec/fiu002
Miele, L., Valenza, V., La Torre, G., Montalto, M., Cammarota, G., Ricci, R., et al. (2009). Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 49, 1877–1887. doi: 10.1002/hep.22848
Mouzaki, M., Comelli, E. M., Arendt, B. M., Bonengel, J., Fung, S. K., Fischer, S. E., et al. (2013). Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 58, 120–127. doi: 10.1002/hep.26319
Pacana, T., Cazanave, S., Verdianelli, A., Patel, V., Min, H. K., Mirshahi, F., et al. (2015). Dysregulated Hepatic Methionine Metabolism Drives Homocysteine Elevation in Diet-Induced Nonalcoholic Fatty Liver Disease. PLoS One 10:e0136822. doi: 10.1371/journal.pone.0136822
Parks, D. H., Tyson, G. W., Hugenholtz, P., and Beiko, R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Poeta, M., Pierri, L., and Vajro, P. (2017). Gut-Liver Axis Derangement in Non-Alcoholic Fatty Liver Disease. Children 4:66. doi: 10.3390/children4080066
Porras, D., Nistal, E., Martinez-Florez, S., Pisonero-Vaquero, S., Olcoz, J. L., Jover, R., et al. (2017). Protective effect of quercetin on high-fat diet-induced non-alcoholic fatty liver disease in mice is mediated by modulating intestinal microbiota imbalance and related gut-liver axis activation. Free Radic. Biol. Med. 102, 188–202. doi: 10.1016/j.freeradbiomed.2016.11.037
Postic, C., and Girard, J. (2008). Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J. Clin. Invest. 118, 829–838. doi: 10.1172/JCI34275
Qiao, S., Bao, L., Wang, K., Sun, S., Liao, M., Liu, C., et al. (2020). Activation of a Specific Gut Bacteroides-Folate-Liver Axis Benefits for the Alleviation of Nonalcoholic Hepatic Steatosis. Cell Rep. 32:108005. doi: 10.1016/j.celrep.2020.108005
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Raman, M., Ahmed, I., Gillevet, P. M., Probert, C. S., Ratcliffe, N. M., Smith, S., et al. (2013). Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 11, e861–e863. doi: 10.1016/j.cgh.2013.02.015
Rodrigues, R. R., Gurung, M., Li, Z., Garcia-Jaramillo, M., Greer, R., Gaulke, C., et al. (2021). Transkingdom interactions between Lactobacilli and hepatic mitochondria attenuate western diet-induced diabetes. Nat. Commun. 12:101. doi: 10.1038/s41467-020-20313-x
Sato, T., Van Es, J. H., Snippert, H. J., Stange, D. E., Vries, R. G., Van Den Born, M., et al. (2011). Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469, 415–418. doi: 10.1038/nature09637
Sawada, M., Takahashi, K., Sawada, S., and Midorikawa, O. (1991). Selective killing of Paneth cells by intravenous administration of dithizone in rats. Int. J. Exp. Pathol. 72, 407–421.
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genom. Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Stappenbeck, T. S., and McGovern, D. P. (2017). Paneth cell alterations in the development and phenotype of Crohn’s disease. Gastroenterology 152, 322–326. doi: 10.1053/j.gastro.2016.10.003
Stephenson, K., Kennedy, L., Hargrove, L., Demieville, J., Thomson, J., Alpini, G., et al. (2018). Updates on Dietary Models of Nonalcoholic Fatty Liver Disease: current Studies and Insights. Gen. Expr. 18, 5–17. doi: 10.3727/105221617X15093707969658
Wilson, C. L., Ouellette, A. J., Satchell, D. P., Ayabe, T., Lopez-Boado, Y. S., Stratman, J. L., et al. (1999). Regulation of intestinal alpha-defensin activation by the metalloproteinase matrilysin in innate host defense. Science 286, 113–117. doi: 10.1126/science.286.5437.113
Xiong, Y., Chen, Y., Huang, X., Yang, Z., Zhang, J., Yu, X., et al. (2021). Icaritin ameliorates hepatic steatosis via promoting fatty acid beta-oxidation and insulin sensitivity. Life Sci. 268:119000. doi: 10.1016/j.lfs.2020.119000
Yang, J. Y., Lee, Y. S., Kim, Y., Lee, S. H., Ryu, S., Fukuda, S., et al. (2017). Gut commensal Bacteroides acidifaciens prevents obesity and improves insulin sensitivity in mice. Mucosal. Immunol. 10, 104–116. doi: 10.1038/mi.2016.42
Yoshida, N., Emoto, T., Yamashita, T., Watanabe, H., Hayashi, T., Tabata, T., et al. (2018). Bacteroides vulgatus and Bacteroides dorei Reduce Gut Microbial Lipopolysaccharide Production and Inhibit Atherosclerosis. Circulation 138, 2486–2498. doi: 10.1161/CIRCULATIONAHA.118.033714
Keywords: NAFLD, MAFLD, microbiota, Bacteroides, metabolic, steatosis
Citation: Zhang S, Tun HM, Zhang D, Chau H-T, Huang F-Y, Kwok H, Wong DK-H, Mak L-Y, Yuen M-F and Seto W-K (2022) Alleviation of Hepatic Steatosis: Dithizone-Related Gut Microbiome Restoration During Paneth Cell Dysfunction. Front. Microbiol. 13:813783. doi: 10.3389/fmicb.2022.813783
Received: 12 November 2021; Accepted: 24 January 2022;
Published: 25 February 2022.
Edited by:
Sunny H. Wong, Nanyang Technological University, SingaporeReviewed by:
Qiuxiang Zhang, Jiangnan University, ChinaCopyright © 2022 Zhang, Tun, Zhang, Chau, Huang, Kwok, Wong, Mak, Yuen and Seto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Man-Fung Yuen, bWZ5dWVuQGhrdWNjLmhrdS5oaw==; Wai-Kay Seto, d2tzZXRvQGhrdS5oaw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.