 Heqiang Sun1†
Heqiang Sun1† Chao Wu
Chao Wu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 27 January 2022
Sec. Microbial Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.813774
Background: Helicobacter pylori can cause many kinds of gastric disorders, ranging from gastritis to gastric cancer. Cytotoxin-associated gene A (CagA)+H. pylori is more likely to cause gastric histopathologic damage than CagA–H. pylori. However, the underlying mechanism needs to be further investigated.
Materials and methods: Mice were intragastrically administered equal amounts of CagA+ or CagA–H. pylori. Four weeks later, 24 chemokines in stomachs were measured using a mouse chemokine array, and the phenotypes of the recruited gastric CD4+ T cells were analyzed. The migration pathway was evaluated. Finally, the correlation between each pair among the recruited CD4+ T cell sub-population, H. pylori colonization level, and histopathologic damage score were determined by Pearson correlation analysis.
Results: The concentration of chemokines, CCL3 and CX3CL1, were significantly elevated in CagA–H. pylori-infected gastric mucosa than in CagA+H. pylori-infected gastric mucosa. Among them, CX3CL1 secreted by gastric epithelial cells, which was elicited more effectively by CagA–H. pylori than by the CagA+ strain, dramatically promoted mucosal CD4+ T cell migration. The expression of CX3CR1, the only known receptor of CX3CL1, was upregulated on the surface of gastric CD4+ T cells in CagA–H. pylori-infected stomach. In addition, most of the CX3CR1-positive gastric CD4+ T cells were CD44+CD69–CCR7– effector memory T cells (Tem). Pearson correlation analysis showed that the recruited CX3CR1+CD4+ Tem cell population was negatively correlated with H. pylori colonization level and histopathologic damage score.
Conclusion: CagA–H. pylori promotes gastric mucosal CX3CR1+CD4+ Tem recruitment in mice.
Helicobacter pylori is a spiral-shaped, Gram-negative bacterium that resides in the gastric mucosa. It can cause many types of gastric disorders, such as gastritis, gastric ulcers, gastric mucosa-associated lymphoid tissue lymphoma, and gastric cancer. H. pylori eradication is effective in the prevention of metachronous gastric cancer (Ernst and Gold, 2000). However, the immune responses elicited by H. pylori infection are insufficient for its eradication; therefore, H. pylori can persist in the stomach for decades or even life (Hooi et al., 2017; Hu et al., 2017; Crowe, 2019). Cytotoxin-associated gene A (CagA) is an essential virulence factor in H. pylori that can be injected into host cells through the type IV secretion system (T4SS) (Odenbreit et al., 2000; Rieder et al., 2005; Frick-Cheng et al., 2016). Compared with CagA–H. pylori, CagA+H. pylori is more likely to cause gastric histopathologic damage and gastric disorders (van Doorn et al., 1998; Sayehmiri et al., 2015; Yong et al., 2015). CagA–H. pylori is less strongly linked to malignancy and may only be associated with diffuse types of the disease (Parsonnet et al., 1997). These histopathologic damages may be associated with factors, such as the number and invasiveness of H. pylori, corresponding immune responses, and HLA variety in the patients, among others (Kusters et al., 2006). These factors also interact with each other. Here, we focused our attention on the immune responses stimulated by H. pylori.
After H. pylori infection, CD4+ T cell responses are elicited, including T helper 1 cells (Th1, CD3+CD4+IFN-γ+) (Robinson et al., 2007), Th2 cells (CD3+CD4+IL4+) (Larussa et al., 2015), Th17 cells (CD3+CD4+IL17A+) (Dixon et al., 2019), and regulatory T cells (Treg; CD3+CD4+CD25+Foxp3+) (Rad et al., 2006), among others. Th1, Th2, and Th17 cells act as effector lymphocytes. Treg cells can inhibit effector lymphocyte responses. Although many studies on vaccination have indicated that effector CD4+ T cells play essential roles in protection against H. pylori infection, the roles of CD4+ T cells, particularly memory T cells, during chronic infection remain unclear (Ermak et al., 1998; Amedei et al., 2006; Chen et al., 2013). Memory CD4+ T cells are a type of cell with long-term function, which is of great significance in repeated responses to pathogen infection. Based on their phenotypes, memory T cells are primarily divided into three populations: central memory T cells (Tcm; CD44+CD62L+CCR7+CD127+CD69–CD103–), which patrol secondary lymphoid organs similar to naïve T cells and undergo rapid and robust proliferation, differentiation, and migration to the site of infection; effector memory T cells (Tem; CD44+CD62L–CCR7–CD127+CD69–CD103–), which recirculate between blood and non-lymphoid tissues and execute their effector functions rapidly similar to freshly stimulated effector T cells; and tissue-resident memory T cells (Trm; CD44+CD62L–CCR7–CD11a+CD69+CD103+), which are derived from precursors that enter the tissue during the immune effector phase and remain therein (Mueller et al., 2013; Spitaels et al., 2016). Thus, weak responses of memory T cells may account for the long-term persistence of H. pylori in the gastric mucosa.
Chemokines play an essential role in T cells recruitment (Yamaoka et al., 1998; Winter et al., 2010). They can be secreted by many types of cells, such as gastric epithelial cells, macrophages, dendritic cells, neutrophils, B cells, and NK cells, among others (Shimada and Terano, 1998; Bhattacharyya et al., 2002). CD4+ T cells expressing corresponding receptors can migrate toward chemokines. In H. pylori infection, the migration of CD4+ T cells to the gastric mucosa and the corresponding influence of CagA have not been well researched. To answer this question, we investigated the chemotaxis pathway through which CD4+ T cells migrate to H. pylori-infected gastric mucosa and the influence of CagA in a mouse model of H. pylori infection. The data demonstrated that CagA weakened the recruitment of protective gastric mucosal CX3CR+CD4+ Tem cells in H. pylori-infected mice. Our research provides new insights into the mechanism underlying the long-term colonization of H. pylori in the gastric mucosa and potential subsequent injury.
Helicobacter pylori strain NCTC11637 (GenBank accession number: AF202973, CagA+) and its isogenic mutant ΔcagA NCTC11637 (CagA–) were used in this study (Asahi et al., 2000; Suzuki et al., 2009; Li et al., 2017). They were cultured on brain–heart infusion agar containing 10% rabbit blood at 37°C under microaerobic conditions. Two days later, H. pylori strains were transferred from the plates to the Brucella broth containing 10% fetal bovine serum (FBS) for active culture. The actively growing H. pylori strains were harvested for infection and in vitro experiments. To develop a mouse model of H. pylori infection, 6–8-week-old female BALB/c mice were infected with H. pylori strain NCTC11637 (CagA+) or its isogenic mutant ΔcagA NCTC11637 (CagA–) through intragastric administration of 1 × 109 CFUs per mouse, once a day for 4 days. In the control group, PBS was used instead of H. pylori. Four weeks later, the mice were subjected to analyses. All mice were purchased from the Experimental Animal Center, Third Military Medical University and housed under specific pathogen-free (SPF) conditions. Animal maintenance and experimental procedures were performed in accordance with the National Institutes of Health Guidelines for the Use of Experimental Animals and approved by the Medicine Animal Care Committee of the Third Military Medical University.
Gastric epithelial cell line, AGS (Zhu et al., 2017; Yang et al., 2021), was obtained from the American Type Culture Collection (Manassas, VA, United States) and BGC-823 (Tang et al., 2013; Guo et al., 2020) was obtained from the Shanghai Institute CellBank, Chinese Academy of Sciences. The two cell lines were thawed and cultured in 3 ml of the complete Ham’s F12 medium (1 × 105 cells/well) in a 12-well plate. The complete Ham’s F12 medium consisted of the Ham’s F12 medium, 25 mmol/l hydroxyethyl piperazine ethanesulfonic acid (HEPES) (pH 7.2), 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% FBS. After culturing for 1 day, gastric epithelial cells were transferred to new wells and were ready for use.
Gastric lymphocytes were isolated and immunophenotyped as previously described (Ruiz et al., 2012; Sun et al., 2018). In brief, stomachs were washed twice gently with PBS and cut into pieces with sharp scissors. The stomach pieces were then placed in 10 ml of Hank’s balanced salt solution (without Ca2+, My2+) with 1 mM dithiothreitol, 1 mM ethylenediaminetetraacetate, and 2% fetal calf serum and incubated for 45 min at 37°C with gentle agitation. The mixture was passed through a steel mesh to remove the undigested tissue. The digested single-cell suspensions were harvested and washed twice with PBS. Single cells were stained with BV510-labeled anti-mouse CD45, APC-labeled anti-mouse CD3, APC/Cy7-labeled anti-mouse CD4, PerCP/Cy5.5-labeled anti-mouse CX3CR1, PE/Cy7-labeled anti-mouse CD44, BV421-labeled anti-mouse CD69, and PE-labeled anti-mouse CCR7 antibodies and measured by flow cytometry.
Helicobacter pylori colonization in the stomach was detected by quantitative real-time PCR. Bacterial genome was extracted from the mouse stomach using a TIANamp Bacteria DNA Kit (TIANGEN, DP302). Then, H. pylori 16S rDNA copies were measured (sense primer, 5′-TTTGTTAGAGAAGATAATGACGGTATCTAAC-3′; anti-sense primer, 5′-CATAGGATTTCACACCTGACTGACTATC-3′; and H. pylori 16S probe, FAM-CGTGCCAGCAGCCGCGGT-TAMRA). For quantitative real-time PCR, the target gene with a concentration gradient was set as the standard in addition to positive and negative controls. Two replicates were used for each sample. The number of 16S rDNA copies of H. pylori were considered to depict H. pylori colonization levels in the mouse stomach, and data were presented as log10 values. To measure IFN-γ, IL-17A, and IL-4 responses of CX3CR1+CD4+ T cells in the mouse stomach, CX3CR1+CD4+ T cells were sorted by FACS; then total RNA was extracted using TRIzol and reverse-transcribed into cDNA using a PrimeScript TM 1st Strand cDNA Synthesis Kit (Takara). The expression levels of IFN-γ, IL-17A, and IL-4 were determined using quantitative real-time PCR with SYBR green staining. β-Actin was used as the reference gene (IFN-γ sense primer, 5′- GATCCTTTGGACCCTCTGACTT-3′; IFN-γ anti-sense primer, 5′-TGACTGTGCCGTGGCAGTAA-3′; IL-17A sense primer, 5′-CTCCAGAAGGCCCTCAGACTAC-3′; IL-17A anti-sense primer, 5′-GGGTCTTCATTGCGGTGG-3′; IL-4 sense primer, 5′ -GAGCTGCAGAGACTCTTTCG-3′; IL-4 anti-sense primer, 5′-ACTCATTCATGGTGCAGCTTA-3′; β-actin sense primer, 5′-CCTGCAGAGTTAAGCATGCCAG-3′; and β-actin anti-sense primer, 5′-TGCTTGATCACATGTCTCGATCC-3′).
The longitudinal strips of the stomach from H. pylori-infected or wild-type (WT) uninfected mice were formalin-fixed, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin. Histological evaluation was performed by two experienced pathologists in a blinded fashion at the Department of Pathology, Southwest Hospital, Third Military Medical University. The gastric histopathologic damage score was graded on a scale of 0–5 as previously reported (Garhart et al., 2002). The infiltrates of inflammatory cells, epithelial hyperplasia, and mucous cell metaplasia were included in the histological evaluation.
Mice were infected with H. pylori strain NCTC11637 (CagA+) or its isogenic mutant ΔcagA NCTC11637 (CagA–) by intragastric administration. Four weeks later, the stomachs were harvested, washed twice gently with PBS, cut into pieces, homogenized, and digested in a cell lysis buffer containing a protease inhibitor cocktail for 30 min. After centrifugation (12,000 rpm, 15 min, 4°C), the supernatant was collected for chemokine array detection using Quantibody® Mouse Chemokine Array 1 (Cat# QAM-CHE-1, RayBiotech, Inc., Guangzhou, China) following the manufacturer’s instructions. In brief, completely air-dried glass chips were blocked with a sample diluent. After washing, 100 μl of samples were added to each well and incubated at room temperature for 2 h. The array was washed again and incubated with detection antibody cocktail for 1.5 h. The chips were washed, followed by incubation with Cy3 equivalent dye–streptavidin for 1 h at room temperature. An InnoScan 300 Microarray Scanner was used to collect fluorescence intensities.
In gastric epithelial cells and H. pylori co-culture chemokine secretion experiments, gastric epithelial cell lines AGS and BGC823 were cultured in the complete Ham’s F12 medium for 1 day in a 37°C incubator with 5% CO2. Then, gastric epithelial cells were transferred to new wells, 1 × 105 cells/well, and 1 × 107 bacteria/well of CagA+H. pylori, CagA–H. pylori, or equal volumes of PBS were added (gastric epithelial cells: H. pylori = 1:100). After 24 h, the supernatant was collected for chemokine array measurement following the manufacturer’s instructions (Human Chemokine Array 1, Cat# AAH-CHE-G1, RayBiotech, Inc., Guangzhou, China). An InnoScan 300 Microarray Scanner was used to measure fluorescence intensities.
CD4+ T cells were sorted using flow cytometry from the mouse spleen or stomach. Then, sorted 1.0 × 105 CD4+ T cells/well were added to the upper chambers of a 24-well transwell plate. The lower chambers were filled with the complete RPMI-1640 medium containing 500 ng/ml of mouse chemokines, CCL-3 and CX3CL1, or PBS as the control. After incubation for 5 h at 37°C in a 5% CO2 incubator, the cell number in the lower chambers was counted by flow cytometry. In some cases, the blocking antibody for CX3CL1 (20 μg/ml) or the corresponding control isotype (20 μg/ml) were added into the lower chambers, and anti-CX3CR1 antibody (20 μg/ml) or corresponding control isotype (20 μg/ml) were added to the cell suspensions for 2 h and then added to the upper chambers of the plate for the transwell migration assay. Triplicate wells were established for each condition. Data represent the chemotactic index (number of cells that migrated to the chemokines divided by number of cells that migrated to the complete RPMI-1640 medium plus PBS). The complete RPMI-1640 medium consisted RPMI-1640 medium, 25 mmol/l HEPES (pH 7.2), 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% FBS.
Differences between the two groups were analyzed using independent-sample t-tests. Comparisons of three or more groups and the expression levels of each chemokine in the array data were analyzed by one-way ANOVA with the Bonferroni post-test. Pearson correlation analysis was used to analyze the correlation between the groups. Statistical analysis was performed using SPSS16 software. Differences were considered significant at p < 0.05.
Mice were infected with the H. pylori strain NCTC11637 (CagA+) or its isogenic mutant ΔcagA NCTC11637 (CagA–) via intragastric administration. In the control group, PBS was used instead of H. pylori. Four weeks later, H. pylori colonization in the stomach was detected by quantitative real-time PCR. Histopathologic damages in the stomachs were scored, and the percentage and number of CD4+ T cells in the gastric mucosa were measured by flow cytometry. The data showed that the colonization of H. pylori was significantly higher, accompanied with more severe histopathological damage in the stomach mucosa of CagA+H. pylori-infected mice than in the stomach mucosa of CagA–H. pylori-infected mice (Figures 1A,B), although an equal number of CFUs were used for infection via intragastric administration. Although the CagA+ group showed a larger CD4+ T cell population in the gastric mucosa than the PBS group, gastric mucosal CD4+ T cell population in the CagA– group was dramatically higher than that in the CagA+ group (Figure 1C). Based on these data, we found that CagA affects mucosal CD4+ T cell numbers after H. pylori infection.
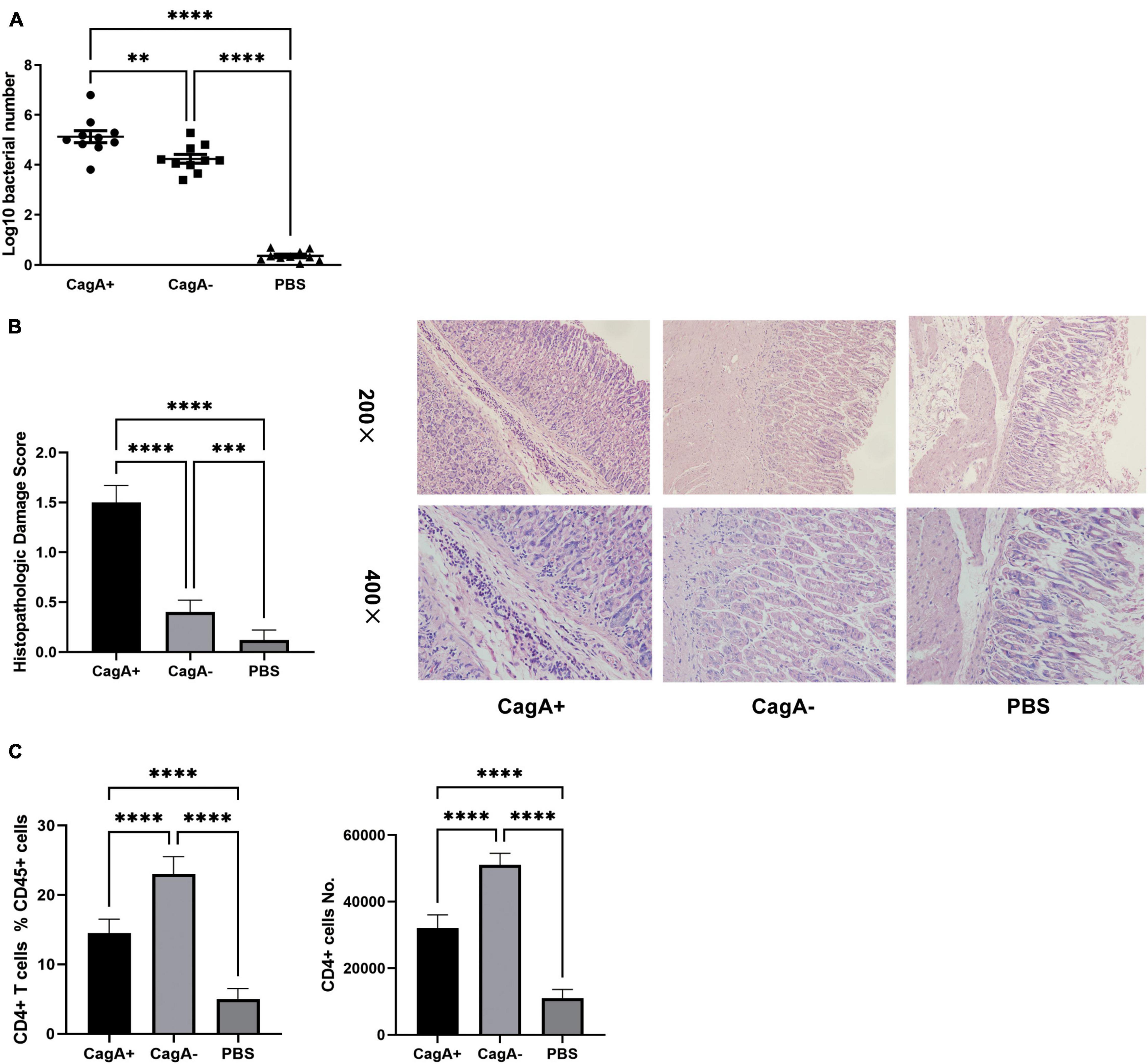
Figure 1. CagA+H. pylori induced stronger inflammation but less mucosal CD4+ T cell formation than CagA– strain. Six- to eight-weeks old mice were infected with H. pylori strain NCTC11637 (CagA+) or its isogenic mutant ΔcagA NCTC11637 (CagA–) via intragastric administration with 109 colony-forming units (CFUs) per mouse once daily for 4 days. In the control group, PBS was used instead of H. pylori. (A) Four weeks later, H. pylori colonization in the stomachs were detected by quantitative real-time PCR. (B) Mice stomach histopathologic damage score and representative histological images are shown. (C) The percentage and number of mucosal CD4+ T cells were measured by flow cytometry. Data are shown as mean ± SD. N = 10. **p < 0.01, ***p < 0.001, and ****p < 0.0001. Results are representative of three individual experiments.
To investigate whether chemokines were involved in the difference in the gastric mucosal CD4+ T cell population between CagA+H. pylori and CagA–H. pylori-infected mice, stomach tissue was harvested from the CagA+ and CagA– groups 4 weeks after H. pylori infection. A series of mouse chemokines were measured in the stomach using Quantibody® Mouse Chemokine Array 1. Several chemokines increased significantly after H. pylori infection compared to that in the PBS control (Figure 2A). CCL3 and CX3CL1 showed significant differences between the CagA+ and CagA– groups (Figure 2A). Moreover, the expression of CX3CL1 was much higher than that of CCL3. Next, CX3CR1, the only known CX3CL1 receptor on CD4+ T cell surface, was measured by flow cytometry. In the stomach mucosa of mice infected with CagA–H. pylori, the CX3CR1+CD4+ T cell population percentage and number were significantly elevated compared with the stomach mucosa of mice infected with CagA+H. pylori (Figure 2B, left and middle). Spleen CD4+ T cells showed no significant difference in CX3CR1 expression between the CagA+ and CagA– groups (Figure 2B, right). In addition to CX3CR1, the expression of another 10 chemokine receptors on mucosal CD4+ T cells was detected (Supplementary Figure 1). Two of the 11 measured chemokine receptors, CX3CR1 and CXCR4 showed increased expression levels in CagA–H. pylori-infected mice compared with those in CagA+ H. pylori-infected mice. But the percentage of CXCR4+CD4+ T cells (around 0.2–0.5%) was very low compared with CX3CR1+CD4+ T cells (around 12–14%). Thus, we focused our attention on CX3CR1+CD4+ T cells and concluded that CagA primarily affected the percentage of mucosal CX3CR1+CD4+ T cells.
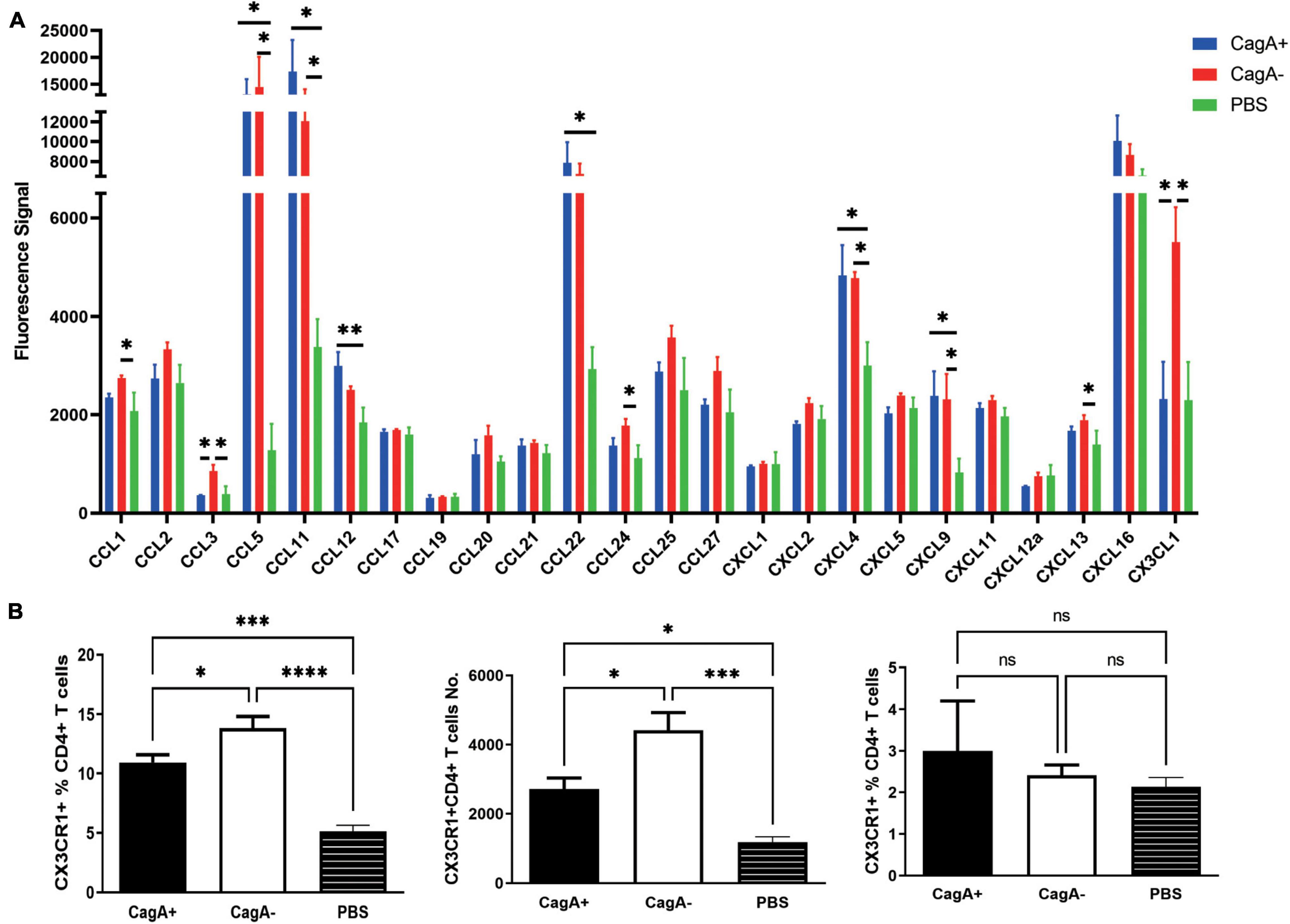
Figure 2. Gastric chemokine CX3CL1 expression and the number of CX3CR1+CD4+ T cells were significantly elevated in CagA–H. pylori-infected mice. Mice were infected with H. pylori strain NCTC11637 (CagA+) or its isogenic mutant ΔcagA NCTC11637 (CagA–) via intragastric administration. Four weeks later, the stomachs and spleens were harvested. (A) A series of mouse mucosal chemokines were measured. (B) The percentage of CX3CR1+ cells in gastric mucosal CD4+ T cells and the number of CX3CR1+CD4+ T cells were detected (left and middle). The percentage of splenic CX3CR1+CD4+ T cells in CD4+ T cells is also shown (right). Data are shown as mean ± SD. N = 5, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Data in (A) are representative of two similar experiments. Data in (B) are representative of three individual experiments.
To further study whether CD4+ T cells could migrate toward CX3CL1, a transwell migration assay was performed. Purified splenic CD4+ T cells from WT mice were added to the upper chambers of a transwell plate. The lower chambers were filled with the complete RPMI-1640 medium supplemented with chemokines CCL-3 or CX3CL1, which were significantly increased in CagA–H. pylori-infected mouse stomachs compared with in the stomach of CagA+H. pylori-infected mice. PBS was used as the control. Even though the data in Figure 2 drew our attention on CX3CR1, which was the only known chemokine receptor of CX3CL1, to be rigorous, we selected another increased chemokine as control and CCL-3 was the most distinguished one with nearly twofold increase in CagA– group than CagA+ group. As a result, CX3CL1 was found to be a more powerful chemokine for mouse CD4+ T cell migration (Figure 3A). Subsequently, gastric mucosal CD4+ T cells from CagA–H. pylori-infected mice were harvested for the transwell assay. We found that CX3CR1 and CX3CL1 antibodies inhibited the migration of mucosal CD4+ T cells (Figure 3B). Thus, our data confirmed that the CX3CL1–CX3CR1 axis contributes to the accumulation of mucosal CD4+ T cells after H. pylori infection.
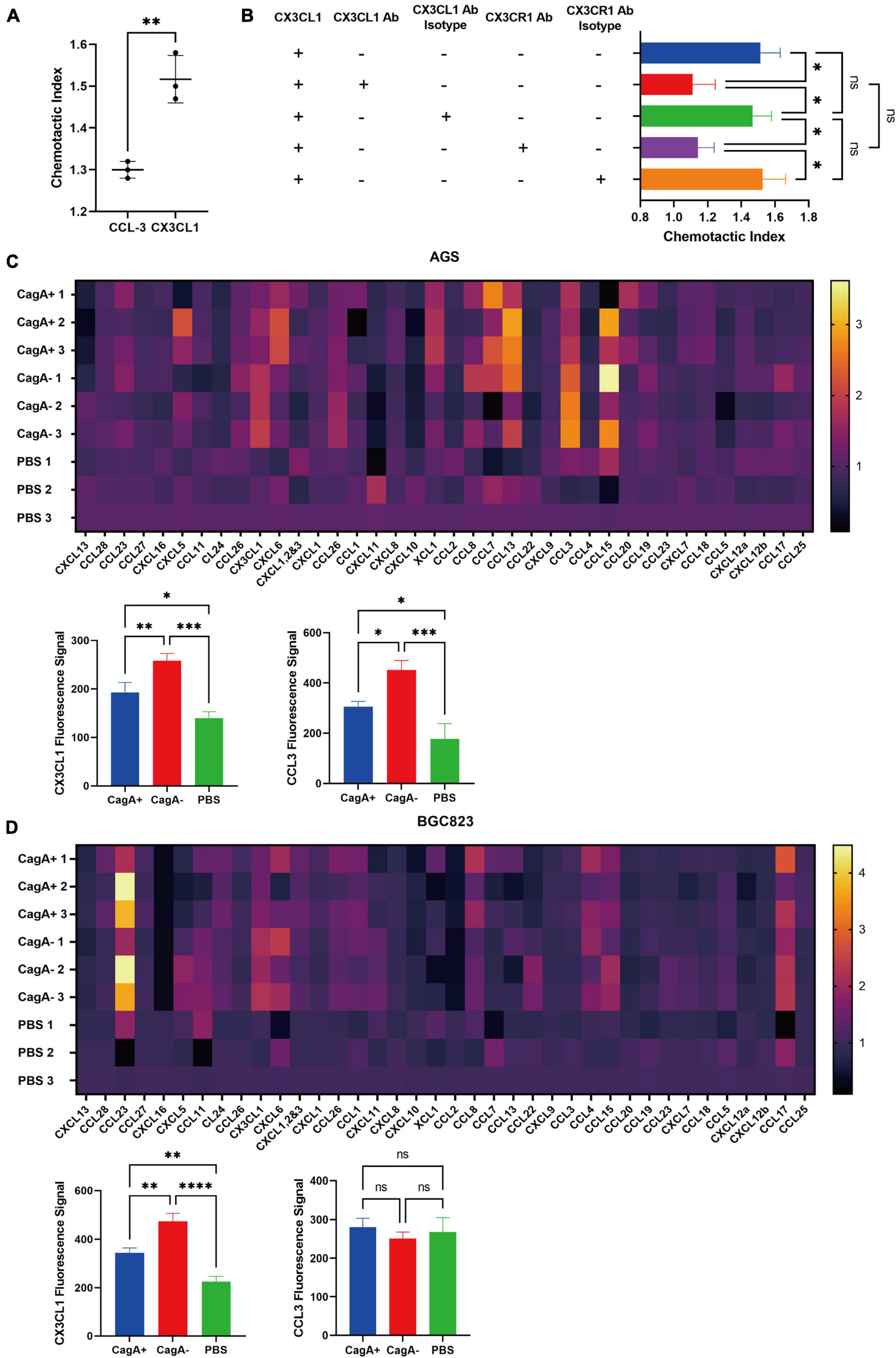
Figure 3. Chemotaxis assay and chemokines secreted by gastric epithelial cell lines. (A) Transwell migration assay. Purified splenic CD4+ T cells from WT mice were added to the upper chambers of a transwell plate. The lower chambers were filled with the complete RPMI-1640 medium containing 500 ng/ml of mouse chemokine CCL-3, CX3CL1, or PBS as the control. Data are shown as the chemotactic index (number of cells that migrated to the medium containing chemokine divided by the number of cells that migrated to the medium containing PBS). (B) Gastric CD4+ T cells were isolated from stomachs of CagA–H. pylori-infected mice, and their migration was assessed via transwell assays. Chemotactic indices are shown. (C,D) Gastric epithelial cell lines AGS and BGC823were co-cultured with active H. pylori strain NCTC11637(CagA+) or its isogenic mutant ΔcagA NCTC11637 (CagA–) at a ratio of 1:100 (gastric epithelial cells: H. pylori) for 24 h. PBS was used as the control. Then, chemokines were detected in the supernatant. Heat maps showed the increased percentage of each chemokine in the H. pylori stimulated medium compared with that in the medium containing PBS. The fluorescence signals of CX3CL1 and CCL3 are shown separately. Data are shown as mean ± SD. N = 3, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. The results in (A) are representative of four similar experiments. The results of (B–D) are representative of two similar experiments.
In H. pylori-infected mice, gastric epithelial cells are the primary cells that bacteria interact with. In addition, gastric epithelial cells can produce many types of chemokines (Sieveking et al., 2004). To investigate whether gastric epithelial cells could produce CX3CL1 and the influence of CagA+ and CagA–H. pylori, we cocultured the gastric epithelial cell lines, AGS and BGC823, with H. pylori strain NCTC11637 (CagA+) or its isogenic mutant ΔcagA NCTC11637 (CagA–). PBS was used as the control. A series of chemokines were measured in the supernatant. As a result, the concentration of CX3CL1 in the CagA– group was higher than that in the CagA+ group for both epithelial cell lines, AGS and BGC823 (Figures 3C,D). CCL3 levels increased in the AGS cell line but not in BCG823 after CagA–H. pylori stimulation (Figures 3C,D), which indicated different gastric epithelial cell lines would affect CCL3 expression stimulated by H. pylori. But as proved above, high CCL3 expression was not related to more recruitment of CX3CR1+CD4+ T cells in gastric mucosa. Therefore, we concluded that CagA can affect the expression of CX3CL1 by gastric epithelial cells, which plays important role in CX3CR1+CD4+ T cells recruitment.
To investigate the phenotype of CD4+ T cells migrating to the stomach through the CX3CL1/CX3CR1 pathway, gastric lymphocytes were isolated at 4 weeks post CagA+ or CagA–H. pylori infection, and cell surface markers were detected. We found that the mucosal CD3+CD4+CX3CR1+ cell population from H. pylori-infected mice or PBS controls primarily showed a phenotype of CD44+, CD69–, and CCR7–, which were regarded as Tem (Figure 4A). The percentage of CD3+CD4+CX3CR1+CD44+CD69–CCR7– population (CX3CR1+CD4+ Tem) in mouse gastric CD4+ T cells was higher in the CagA– group than in the CagA+ group (Figure 4A).
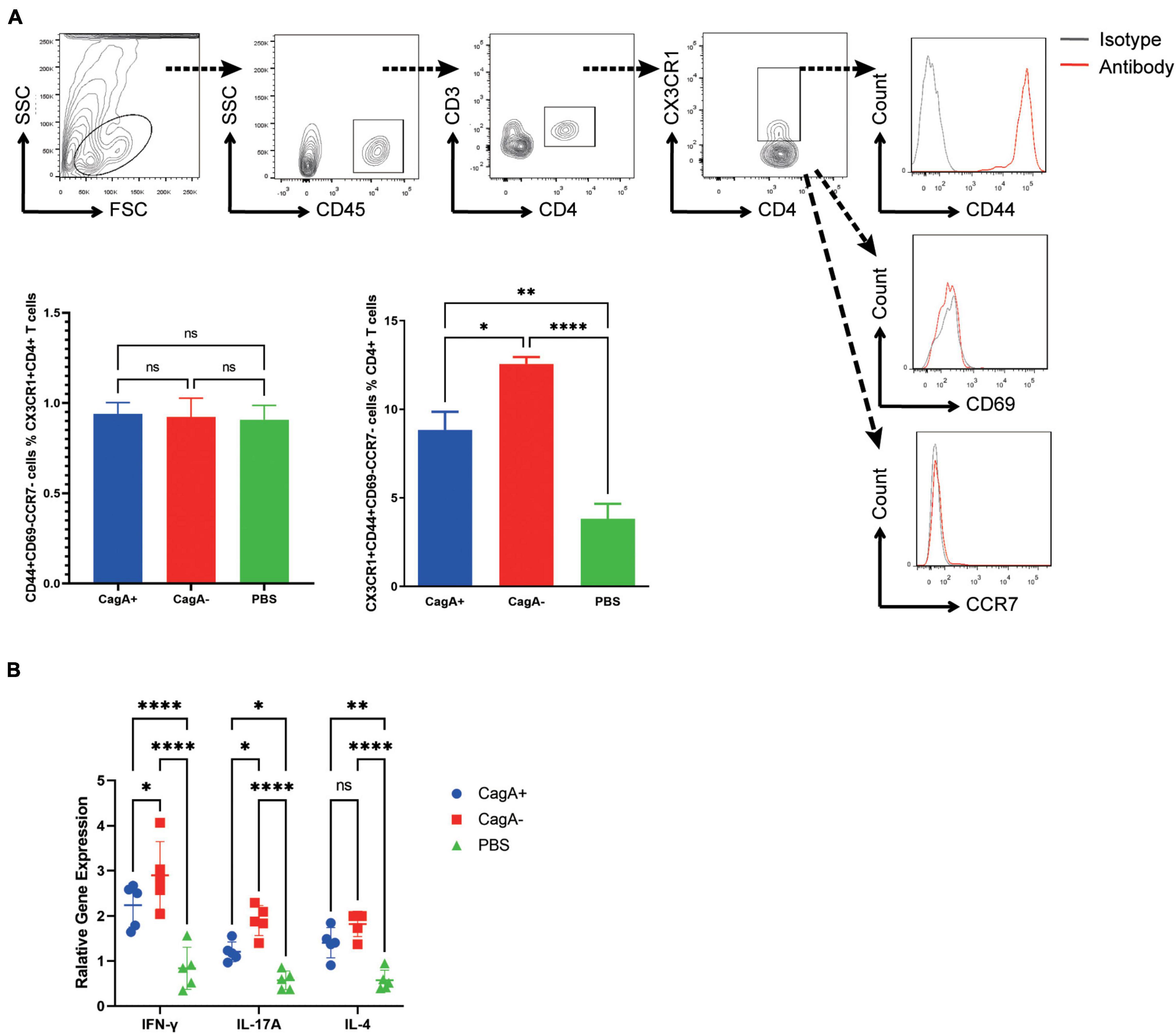
Figure 4. Phenotype of mucosal CX3CR1+CD4+ T cells in H. pylori-infected mice. Mice were infected with H. pylori strain NCTC11637 (CagA+) or its isogenic mutant ΔcagA NCTC11637 (CagA–) by intragastric administration. PBS was used as the control. Four weeks later, stomachs were harvested. (A) The expression of CD45, CD3, CD4, CX3CR1, CD44, CD69, and CCR7 in gastric cells was assessed. The percentages of CD44+CD69–CCR7– population in gastric CX3CR1+CD4+ T cells and percentages of CD3+CD4+CX3CR1+CD44+CD69–CCR7– population in gastric CD4+ T cells are shown. (B) The expression levels of IFN-γ, IL-17A, and IL-4 in sorted CX3CR1+CD4+ T cells from H. pylori-infected mice stomachs were determined by quantitative real-time PCR. β-actin was used as the reference gene. Data are shown as mean ± SD. N = 5, *p < 0.05, **p < 0.01, and ****p < 0.0001. Results are representative of four individual experiments.
To further determine the function, gastric CX3CR1+CD4+ T cells were sorted and RNA was extracted for cytokine detection. We found that IFN-γ, IL-17A, and IL-4 levels increased significantly after H. pylori infection. The higher levels of IFN-γ and IL-17A in mucosal CX3CR1+CD4+ T cells were detected in the CagA– group than in the CagA+ group (Figure 4B). These data indicated that CagA–H. pylori induced more functional CX3CR1+CD4+ Tem cell response than CagA+H. pylori.
Effector memory T cells are usually regarded as protective T cell responses (Hansen et al., 2009; Roestenberg et al., 2011). Our data also showed that more CX3CR1+CD4+ Tem cells were recruited to the stomach in the CagA– group than in the CagA+ group. Thus, to determine the function of CX3CR1+CD4+ Tem cells, we performed a Pearson correlation analysis between each pair among the CX3CR1+CD4+ Tem cell number, H. pylori colonization level, and histopathologic damage score. We found that the CX3CR1+CD4+ Tem cell population was negatively correlated with H. pylori colonization level and histopathologic damage score (Figure 5). The H. pylori colonization level and the histopathologic damage score were positively correlated with each other (Figure 5). These data indicated that CX3CR1+CD4+ Tem cells in the mouse stomach may play a protective role in clearing H. pylori.
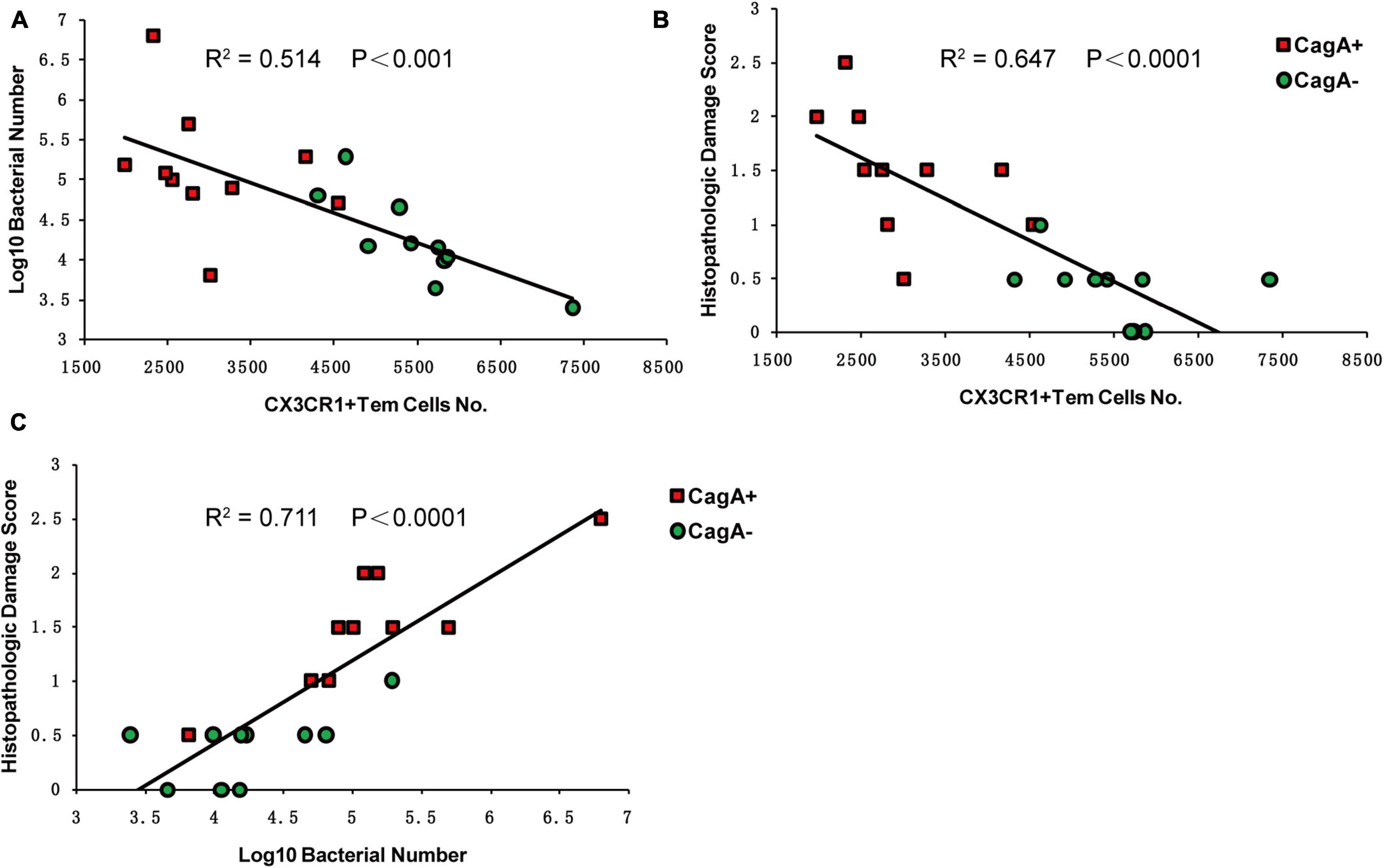
Figure 5. Pearson correlation analysis of CX3CR1+CD4+ Tem cell number, histopathologic damage score, and H. pylori colonization level in mice stomachs. Four weeks post H. pylori infection, CX3CR1+CD4+ Tem (CD3+CD4+CX3CR1+CD44+CD69–CCR7–) cell population, histopathologic damage score, and H. pylori colonization in the mice stomachs were measured. (A) Pearson correlation analysis of H. pylori colonization and CX3CR1+CD4+ Tem population. (B) Pearson correlation analysis of the histopathologic damage score and CX3CR1+CD4+ Tem population. (C) Pearson correlation analysis of the histopathologic damage score and H. pylori colonization. N = 20, the values of R2 and P are shown. Results are representative of three separate experiments.
Helicobacter pylori is a Gram-negative bacterium in the stomach that infects half of the world’s population (Del Giudice et al., 2001). H. pylori infection is a key factor in the etiology of various gastric disorders, ranging from chronic gastritis without clinical symptoms to peptic ulceration and even gastric carcinoma (Suerbaum and Michetti, 2002). H. pylori virulence factor CagA, the most extensively studied virulence factor, is a 120–145 kDa protein encoded by the 40-kb cag pathogenicity island (PAI). It has been reported that approximately 60% of H. pylori strains isolated in Western countries carry cag PAI and almost all East Asian isolates are cag PAI-positive (Hatakeyama and Higashi, 2005). CagA can be injected directly from the bacterium into bacterium-attached gastric epithelial cells via T4SS (Schuelein et al., 2011). After being injected into host cells, CagA is able to activate SHP-2 tyrosine phosphatase (Hayashi et al., 2017) and interact with the human tumor suppressor apoptosis-stimulating protein of p53-2 (ASPP2) (Buti et al., 2011; Nesic et al., 2014), which favors H. pylori colonization (Buti et al., 2020). In this study, higher bacterial colonization was detected in the CagA+H. pylori infection group than in the CagA–H. pylori group.
Cytotoxin-associated gene A can upregulate NF-κB transcriptional activity and promote inflammation (Yamaoka, 2010). In this study, we found higher histopathological damage scores in CagA+ strain-infected mice than in CagA– strain-infected mice (Figure 1). Inflammatory cells comprise neutrophils, eosinophils, lymphocytes, and other cells. Lymphocytes, particularly CD4+ T cells, are vital during H. pylori infection (Chen et al., 2013). In addition to CD4+ T cells, a number of CD8+ T cells can be observed in the stomachs of H. pylori-infected mice (Tan et al., 2008; Becher et al., 2010; Ruiz et al., 2012). The effects of CagA on inflammatory cells, including CD4+ and CD8+ T cells, should be different. These may be the reasons why higher histopathological damage scores accompanied by fewer CD4+ T cells were detected in CagA+ strain-infected mice than in CagA– strain-infected mice (Figure 1C). In addition, the effects of CagA on T cells remain unclear. CagA and peptides from CagA have been confirmed to induce gastric/mesenteric lymph node and CD4+ T cells effectively secrete IFN-γ, which sensitizes mice to preneoplastic immunopathology rather than protecting them from H. pylori infection (Arnold et al., 2011). However, phytohemagglutinin-induced T cell proliferation can be inhibited by CagA (Paziak-Domanska et al., 2000). Bagheri et al. (2017) counted CD4+ T cells by immunohistochemical staining of mucosal biopsies from areas of antral gastric mucosa of patients with H. pylori infection. Their data showed that more CD4+ T cells were detected in mucosal biopsies from CagA+H. pylori-infected patients than in mucosal biopsies from CagA–H. pylori-infected patients (Bagheri et al., 2017). However, CagA has been shown to promote T cell death in a Fas-dependent manner (Larussa et al., 2015). In this study, we measured CD4+ T cells in the whole stomach by digesting the stomach into a single-cell suspension and found that mucosal CD4+ T cells increased post H. pylori infection and CagA–H. pylori recruited more CD4+ T cells in the stomach mucosa than CagA+ H. pylori.
Primed lymphocytes must migrate to the infection site to execute their function. Chemokines and their corresponding receptors play essential roles in CD4+ T cell recruitment. To confirm why more CD4+ T cells migrate to the gastric mucosa in CagA–H. pylori-infected mice than to the gastric mucosa in CagA+H. pylori-infected mice, we measured 24 chemokines in CagA+ or CagA–H. pylori-infected mice and found that compared with those in the CagA+ group, higher concentrations of CCL3 and CX3CL1 were detected in the CagA– group and the concentration of CX3CL1 was much higher than that of CCL3 (Figure 2). The in vitro stimulation of gastric epithelial cells by H. pylori strains further confirmed that chemokine secretion can be affected by CagA+/CagA– strains, particularly CX3CL1 (Figures 3C,D). CX3CL1 is primarily expressed in endothelial cells, which potently chemoattracts T cells and monocytes through the receptor CX3CR1. Therefore, in this study, CX3CR1 expression in mucosal lymphocytes was determined. We found that the number of CX3CR1+CD4+ T cells increased significantly after H. pylori infection. Moreover, the CagA–H. pylori strain induced more CX3CR1+CD4+ T cells in stomachs than the CagA+H. pylori strain (Figure 2B). In the chemotaxis assay, gastric CD4+ T cell migration to CX3CL1 was blocked by the CX3CL1 and CX3CR1 antibodies (Figure 3B). These results suggest that CagA+/CagA– strains affect the recruitment of gastric CX3CR1+CD4+ T cells by changing CX3CL1 expression.
Memory T cells are a subset of “experienced” infection- or cancer-fighting T cells that have previously encountered and responded to their cognate antigen during a prior infection, encounter with cancer, or previous vaccination. They show greatly improved immune efficacy with a rapid and strong immune response. Until now, memory T cell populations have been considered to comprise Tcm, which are restricted to the secondary lymphoid tissues and blood; Tem, which broadly migrate between peripheral tissues, blood, and spleen; and Trm, which occupy tissues without recirculation (Tan et al., 2008). To phenotype the increased number of CX3CR1+CD4+ T cells in CagA–H. pylori-infected mice, we detected a series of memory cell markers and found that CX3CR1+CD4+ T cells were primarily CD44+CD69–CCR7–, a Tem phenotype, indicating that CX3CR1+CD4+ T cells were Tem cells (Figure 4A). Tem has been demonstrated to provide long-term protection against pathogenic infections. Hansen et al. (2009) reported that Tem responses were associated with the protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Roestenberg et al.’s (2011) research also showed the long-term persistence of Tem responses in protected volunteers. In this study, we found that the number of CX3CR1+CD4+ Tem cells increased significantly after H. pylori infection, and these cells were detected in the CagA–H. pylori group (Figure 4). Thus, we wondered whether the increased number of CX3CR1+CD4+ Tem cells account for the low level of H. pylori colonization in CagA–H. pylori-infected mice. Pearson correlation analysis showed that the CX3CR1+CD4+ Tem cell number was negatively correlated with the H. pylori colonization level and the histopathologic damage score (Figure 5). Thus, CX3CR1+CD4+ Tem cells reducing H. pylori colonization and decreasing pathological damage may represent a possible mechanism. However, more work is needed to reveal the underlying mechanisms of Tem function.
Helicobacter pylori-associated gastric disorders can be derived from many factors, such as number and invasiveness of H. pylori, corresponding immune responses, including protective responses to clear H. pylori and inflammatory responses to cause immunopathological injury, and a variety of patient HLA, among others (Kusters et al., 2006). These factors also interact in various ways with each other, which are very complex. Minimally, however, we provide insight into the mechanism by which CagA–H. pylori causes less pathological damage than CagA+H. pylori.
BALB/c mice were used in the present study, considering that they contain two classical MHC-II molecules (I-A and I-E), but only the I-A allele exists in C57BL/6 mice. There should be no I-E-restricted CD4+ T cell responses in C57BL/6 mice. In this study, we focused on CD4+ T cells, despite no MHC restrictions. To avoid missing the I-E-restricted Hp-specific CD4+ T cell subsets artificially, BALB/c mice were chosen instead of C57BL/6 mice. Unfortunately, our conclusion has not been validated using knockout mice or other mouse strains. This is a limitation of our study. We are pushing the research forward, including obtaining more evidence from human subjects.
Overall, from our research, we found that more CD4+ Tem cells were recruited to the stomach mucosa of mice infected with CagA–H. pylori than to the stomach mucosa of mice infected with CagA+H. pylori. CD4+ Tem cell recruitment occurred primarily through the CX3CL1/CX3CR1 pathway, and the higher secretion of CX3CL1 was elicited by gastric epithelial cells infected with CagA–H. pylori than by the cells infected with CagA+ H. pylori. Although the gastric Tem cell number was negatively correlated with the bacterial colonization level in H. pylori-infected mice, this population cannot completely protect against H. pylori infection. H. pylori can reside in the gastric mucosa, causing chronic and persistent infection in mice and humans. The exact functions and mechanisms of CX3CR+CD4+ Tem cells remain unclear, and further evidence is warranted in the future.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The animal study wasprotect reviewed and approved by the Medicine Animal Care Committee of the Third Military Medical University.
CW and BL conceived theprotect study. HS, TH, and YW performed the experiments. HS, BL, and CW analyzed the data. HY, JN, and ZZ prepared the material. XD and CW provided the facility. BL and CW wrote the manuscript with the assistance of other authors. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Nos. 81671571 and 82001751).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.813774/full#supplementary-material
Supplementary Figure 1 | Chemokine receptors on gastric CD4+ T cells. Mice were infected with H. pylori strain NCTC11637 (CagA+) or its isogenic mutant ΔcagA NCTC11637 (CagA–) via intragastric administration. Four weeks later, the stomachs were harvested, and chemokine receptors expressed on gastric mucosal CD4+ T cells were determined. Data are shown as mean ± SD. N = 5, *p < 0.05, **p < 0.01, and ***p < 0.001.
Amedei, A., Cappon, A., Codolo, G., Cabrelle, A., Polenghi, A., Benagiano, M., et al. (2006). The neutrophil-activating protein of Helicobacter pylori promotes Th1 immune responses. J. Clin. Invest. 116, 1092–1101. doi: 10.1172/JCI27177
Arnold, I. C., Hitzler, I., Engler, D., Oertli, M., Agger, E. M., and Muller, A. (2011). The C-terminally encoded, MHC class II-restricted T cell antigenicity of the Helicobacter pylori virulence factor CagA promotes gastric preneoplasia. J. Immunol. 186, 6165–6172. doi: 10.4049/jimmunol.1003472
Asahi, M., Azuma, T., Ito, S., Ito, Y., Suto, H., Nagai, Y., et al. (2000). Helicobacter pylori CagA protein can be tyrosine phosphorylated in gastric epithelial cells. J. Exp. Med. 191, 593–602. doi: 10.1084/jem.191.4.593
Bagheri, N., Shirzad, H., Elahi, S., Azadegan-Dehkordi, F., Rahimian, G., Shafigh, M., et al. (2017). Downregulated regulatory T cell function is associated with increased peptic ulcer in Helicobacter pylori-infection. MicrobPathog 110, 165–175. doi: 10.1016/j.micpath.2017.06.040
Becher, D., Deutscher, M. E., Simpfendorfer, K. R., WijburgOL, P. J. S., Lew, A. M., Strugnell, R. A., et al. (2010). Local recall responses in the stomach involving reduced regulation and expanded help mediate vaccine-induced protection against Helicobacter pylori in mice. Eur. J. Immunol. 40, 2778–2790. doi: 10.1002/eji.200940219
Bhattacharyya, A., Pathak, S., Datta, S., Chattopadhyay, S., Basu, J., and Kundu, M. (2002). Mitogen-activated protein kinases and nuclear factor-kappaB regulate Helicobacter pylori-mediated interleukin-8 release from macrophages. Biochem. J. 368, 121–129. doi: 10.1042/BJ20020555
Buti, L., Ruiz-Puig, C., Sangberg, D., Leissing, T. M., Brewer, R. C., Owen, R. P., et al. (2020). CagA-ASPP2 complex mediates loss of cell polarity and favors H. pylori colonization of human gastric organoids. Proc. Natl. Acad. Sci. U. S. A. 117, 2645–2655. doi: 10.1073/pnas.1908787117
Buti, L., Spooner, E., Van der Veen, A. G., Rappuoli, R., Covacci, A., and Ploegh, H. L. (2011). Helicobacter pylori cytotoxin-associated gene A (CagA) subverts the apoptosis-stimulating protein of p53 (ASPP2) tumor suppressor pathway of the host. Proc. Natl. Acad. Sci. U. S. A 108, 9238–9243. doi: 10.1073/pnas.1106200108
Chen, L., Li, B., Yang, W. C., He, J. L., Li, N. Y., Hu, J., et al. (2013). A dominant CD4(+) T-cell response to Helicobacter pylori reduces risk for gastric disease in humans. Gastroenterology 144, 591–600. doi: 10.1053/j.gastro.2012.12.002
Del Giudice, G., Covacci, A., Telford, J. L., Montecucco, C., and Rappuoli, R. (2001). The design of vaccines against Helicobacter pylori and their development. Annu. Rev. Immunol. 19, 523–563. doi: 10.1146/annurev.immunol.19.1.523
Dixon, B., Hossain, R., Patel, R. V., and Algood, H. M. S. (2019). Th17 Cells in Helicobacter pylori Infection: a Dichotomy of Help and Harm. Infect. Immun. 87:e00363–19. doi: 10.1128/IAI.00363-19
Ermak, T. H., GiannascaPJ, Nichols R, Myers, G. A., Nedrud, J., Weltzin, R., Lee, C. K., et al. (1998). Immunization of mice with urease vaccine affords protection against Helicobacter pylori infection in the absence of antibodies and is mediated by MHC class II-restricted responses. J. Exp. Med. 188, 2277–2288. doi: 10.1084/jem.188.12.2277
Ernst, P. B., and Gold, B. D. (2000). The disease spectrum of Helicobacter pylori: the immunopathogenesis of gastroduodenal ulcer and gastric cancer. Annu. Rev. Microbiol. 54, 615–640. doi: 10.1146/annurev.micro.54.1.615
Frick-Cheng, A. E., Pyburn, T. M., Voss, B. J., McDonald, W. H., Ohi, M. D., and Cover, T. L. (2016). Molecular and Structural Analysis of the Helicobacter pylori cag Type IV Secretion System Core Complex. Mbio 7, e02001–e02015.
Garhart, C. A., Redline, R. W., Nedrud, J. G., and Czinn, S. J. (2002). Clearance of Helicobacter pylori Infection and Resolution of Postimmunization Gastritis in a Kinetic Study of Prophylactically Immunized Mice. Infect. Immun. 70, 3529–3538. doi: 10.1128/IAI.70.7.3529-3538.2002
Guo, Y., Zhang, T., Shi, Y., Zhang, J., Li, M., Lu, F., et al. (2020). Helicobacter pylori inhibits GKN1 expression via the CagA/p-ERK/AUF1 pathway. Helicobacter 25:e12665. doi: 10.1111/hel.12665
Hansen, S. G., Vieville, C., Whizin, N., Coyne-Johnson, L., Siess, D. C., Drummond, D. D., et al. (2009). Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat. Med. 15, 293–299. doi: 10.1038/nm.1935
Hatakeyama, M., and Higashi, H. (2005). Helicobacter pylori CagA: a new paradigm for bacterial carcinogenesis. Cancer Sci. 96, 835–843. doi: 10.1111/j.1349-7006.2005.00130.x
Hayashi, T., Senda, M., Suzuki, N., Nishikawa, H., Ben, C., Tang, C., et al. (2017). Differential Mechanisms for SHP2 Binding and Activation Are Exploited by Geographically Distinct Helicobacter pylori CagA Oncoproteins. Cell Rep. 20, 2876–2890. doi: 10.1016/j.celrep.2017.08.080
Hooi, J. K. Y., Lai, W. Y., Ng, W. K., Suen, M. M. Y., Underwood, F. E., Tanyingoh, D., Malfertheiner, P., et al. (2017). Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 153, 420–429.
Hu, Y., Wan, J. H., Li, X. Y., Zhu, Y., Graham, D. Y., and Lu, N. H. (2017). Systematic review with meta-analysis: the global recurrence rate of Helicobacter pylori. Aliment Pharmacol. Ther. 46, 773–779. doi: 10.1111/apt.14319
Kusters, J. G., van Vliet, A. H., and Kuipers, E. J. (2006). Pathogenesis of Helicobacter pylori infection. ClinMicrobiol. Rev. 19, 449–490.
Larussa, T., Leone, I., Suraci, E., Imeneo, M., and Luzza, F. (2015). Helicobacter pylori and T Helper Cells: Mechanisms of Immune Escape and Tolerance. J. Immunol. Res. 2015:981328. doi: 10.1155/2015/981328
Li, N., Tang, B., JiaYP, Zhu P, Zhuang, Y., Fang, Y., Li, Q., et al. (2017). Helicobacter pylori CagA Protein Negatively Regulates Autophagy and Promotes Inflammatory Response via c-Met-PI3K/Akt-mTOR Signaling Pathway. Front. Cell Infect. Microbiol. 7:417. doi: 10.3389/fcimb.2017.00417
Mueller, S. N., Gebhardt, T., Carbone, F. R., and Heath, W. R. (2013). Memory T cell subsets, migration patterns, and tissue residence. Annu. Rev. Immunol. 31, 137–161. doi: 10.1146/annurev-immunol-032712-095954
Nesic, D., Buti, L., Lu, X., and Stebbins, C. E. (2014). Structure of the Helicobacter pylori CagA oncoprotein bound to the human tumor suppressor ASPP2. Proc. Natl. Acad. Sci. U. S. A 111, 1562–1567. doi: 10.1073/pnas.1320631111
Odenbreit, S., Puls, J., Sedlmaier, B., Gerland, E., Fischer, W., and Haas, R. (2000). Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287, 1497–1500. doi: 10.1126/science.287.5457.1497
Parsonnet, J., Friedman, G. D., Orentreich, N., and Vogelman, H. (1997). Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut 40, 297–301. doi: 10.1136/gut.40.3.297
Paziak-Domanska, B., Chmiela, M., Jarosinska, A., and Rudnicka, W. (2000). Potential role of CagA in the inhibition of T cell reactivity in Helicobacter pylori infections. Cell Immunol. 202, 136–139. doi: 10.1006/cimm.2000.1654
Rad, R., Brenner, L., Bauer, S., Schwendy, S., Layland, L., da Costa, C. P., et al. (2006). CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology 131, 525–537. doi: 10.1053/j.gastro.2006.05.001
Rieder, G., Merchant, J. L., and Haas, R. (2005). Helicobacter pylori cag-type IV secretion system facilitates corpus colonization to induce precancerous conditions in Mongolian gerbils. Gastroenterology 128, 1229–1242. doi: 10.1053/j.gastro.2005.02.064
Robinson, K., Argent, R. H., and Atherton, J. C. (2007). The inflammatory and immune response to Helicobacter pylori infection. Best Pract. Res. Clin. Gastroenterol. 21, 237–259. doi: 10.1016/j.bpg.2007.01.001
Roestenberg, M., Teirlinck, A. C., McCall, M. B., Teelen, K., Makamdop, K. N., Wiersma, J., et al. (2011). Long-term protection against malaria after experimental sporozoite inoculation: an open-label follow-up study. Lancet 377, 1770–1776. doi: 10.1016/S0140-6736(11)60360-7
Ruiz, V. E., Sachdev, M., Zhang, S., Wen, S., and Moss, S. F. (2012). Isolating, immunophenotyping and ex vivo stimulation of CD4+ and CD8+ gastric lymphocytes during murine Helicobacter pylori infection. J. Immunol. Methods 384, 157–163. doi: 10.1016/j.jim.2012.07.002
Sayehmiri, F., Kiani, F., Sayehmiri, K., Soroush, S., Asadollahi, K., Alikhani, M. Y., et al. (2015). Prevalence of cagA and vacA among Helicobacter pylori-infected patients in Iran: a systematic review and meta-analysis. J. Infect Dev. Ctries 9, 686–696. doi: 10.3855/jidc.5970
Schuelein, R., Everingham, P., and Kwok, T. (2011). Integrin-mediated type IV secretion by Helicobacter: what makes it tick? Trends Microbiol. 19, 211–216. doi: 10.1016/j.tim.2011.01.010
Shimada, T., and Terano, A. (1998). Chemokine expression in Helicobacter pylori-infected gastric mucosa. J. Gastroenterol. 33, 613–617. doi: 10.1007/s005350050146
Sieveking, D., Mitchell, H. M., and Day, A. S. (2004). Gastric epithelial cell CXC chemokine secretion following Helicobacter pylori infection in vitro. J. Gastroenterol. Hepatol. 19, 982–987. doi: 10.1111/j.1440-1746.2004.03413.x
Spitaels, J., Roose, K., and Saelens, X. (2016). In fluenza and Memory T Cells: how to Awake the Force. Vaccines 4:33. doi: 10.3390/vaccines4040033
Suerbaum, S., and Michetti, P. (2002). Helicobacter pylori infection. N. Engl. J. Med. 347, 1175–1186.
Sun, H., Yuan, H., Tan, R., Li, B., Guo, G., Zhang, J., et al. (2018). Immunodominant antigens that induce Th1 and Th17 responses protect mice against Helicobacter pylori infection. Oncotarget 9, 12050–12063. doi: 10.18632/oncotarget.23927
Suzuki, M., Mimuro, H., Kiga, K., Fukumatsu, M., Ishijima, N., Morikawa, H., et al. (2009). Helicobacter pylori CagA phosphorylation-independent function in epithelial proliferation and inflammation. Cell. Host Microbe. 5, 23–34. doi: 10.1016/j.chom.2008.11.010
Tan, M. P., Pedersen, J., Zhan, Y., Lew, A. M., Pearse, M. J., Wijburg, O. L., et al. (2008). CD8+ T cells are associated with severe gastritis in Helicobacter pylori-infected mice in the absence of CD4+ T cells. Infect. Immun. 76, 1289–1297. doi: 10.1128/iai.00779-07
Tang, B., Peng, Z. H., Yu, P. W., Yu, G., Qian, F., Zeng DZ, Zhao YL, et al. (2013). Aberrant expression of Cx43 is associated with the peritoneal metastasis of gastric cancer and Cx43-mediated gap junction enhances gastric cancer cell diapedesis from peritoneal mesothelium. PLoS One 8:e74527. doi: 10.1371/journal.pone.0074527
van Doorn, L. J., Figueiredo, C., Sanna, R., Plaisier, A., Schneeberger, P., de Boer, W., et al. (1998). Clinical relevance of the cagA, vacA, and iceA status of Helicobacter pylori. Gastroenterology 115, 58–66. doi: 10.1016/s0016-5085(98)70365-8
Winter, S., Loddenkemper, C., Aebischer, A., Rabel, K., Hoffmann, K., Meyer, T. F., et al. (2010). The chemokine receptor CXCR5 is pivotal for ectopic mucosa-associated lymphoid tissue neogenesis in chronic Helicobacter pylori-induced inflammation. J. Mol. Med.. 88, 1169–1180. doi: 10.1007/s00109-010-0658-6
Yamaoka, Y. (2010). Mechanisms of disease: helicobacter pylori virulence factors. Nat. Rev. Gastroenterol. Hepatol. 7, 629–641. doi: 10.1038/nrgastro.2010.154
Yamaoka, Y., Kita, M., Kodama, T., Sawai, N., Tanahashi, T., Kashima, K., et al. (1998). Chemokines in the gastric mucosa in Helicobacter pylori infection. Gut 42, 609–617. doi: 10.1136/gut.42.5.609
Yang, J. Y., Kim, J. B., Lee, P., and Kim, S. H. (2021). Evodiamine Inhibits Helicobacter pylori Growth and Helicobacter pylori-Induced Inflammation. Int. J. Mol. Sci. 22:3385. doi: 10.3390/ijms22073385
Yong, X., Tang, B., Li, B. S., Xie, R., Hu, C. J., Luo, G., et al. (2015). Helicobacter pylori virulence factor CagA promotes tumorigenesis of gastric cancer via multiple signaling pathways. Cell. Commun. Signal. 13:30. doi: 10.1186/s12964-015-0111-0
Keywords: Helicobacter pylori, CagA, effector memory T cell, CX3CL1, CX3CR1
Citation: Sun H, He T, Wu Y, Yuan H, Ning J, Zhang Z, Deng X, Li B and Wu C (2022) Cytotoxin-Associated Gene A-Negative Helicobacter pylori Promotes Gastric Mucosal CX3CR1+CD4+ Effector Memory T Cell Recruitment in Mice. Front. Microbiol. 13:813774. doi: 10.3389/fmicb.2022.813774
Received: 12 November 2021; Accepted: 07 January 2022;
Published: 27 January 2022.
Edited by:
Juarez Antonio Simões Quaresma, Universidade do Estado do Pará, BrazilReviewed by:
Kathryn Haley, Grand Valley State University, United StatesCopyright © 2022 Sun, He, Wu, Yuan, Ning, Zhang, Deng, Li and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chao Wu, d3VjaGFvOTkyNjFAMTYzLmNvbQ==; Bin Li, bGliaW4yMDA1MDE3QDE2My5jb20=
†These authors share first authorship
‡These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.