 Xiu Pei Koh1
Xiu Pei Koh1 Zhiyong Shen2
Zhiyong Shen2 Chun Fai Woo1
Chun Fai Woo1 Yanping Yu2
Yanping Yu2 Hau In Lun2
Hau In Lun2 Sze Wan Cheung3
Sze Wan Cheung3 Joseph Kai Cho Kwan1
Joseph Kai Cho Kwan1 Stanley Chun Kwan Lau2*
Stanley Chun Kwan Lau2*- 1Division of Environment and Sustainability, The Hong Kong University of Science and Technology, Kowloon, Hong Kong SAR, China
- 2Department of Ocean Science, The Hong Kong University of Science and Technology, Kowloon, Hong Kong SAR, China
- 3Division of Life Science, The Hong Kong University of Science and Technology, Hong Kong, Hong Kong SAR, China
Escherichia coli not only inhabit the large intestines of human and warm-blooded animals but could also persist in the external environment. However, current knowledge was largely based on host-associated strains. Moreover, cryptic Escherichia clades that were often misidentified as E. coli by conventional diagnostic methods were discovered. Failure to distinguish them from E. coli sensu stricto could lead to inaccurate conclusions about the population genetics of E. coli. Based on seven housekeeping genes, we determine the genetic and ecological diversity of E. coli and cryptic clades as they occupy aquatic habitats with different characteristics and human impact levels in subtropical Hong Kong. Contrary to previous reports, clade II was the most abundant cryptic lineage co-isolated with E. coli, being especially abundant in relatively pristine subtropical aquatic environments. The phylogenetically distinct cryptic clades and E. coli showed limited recombination and significant genetic divergence. Analyses indicated that these clade II strains were ecologically differentiated from typical E. coli; some may even represent novel environmental Escherichia clades that were closely related to the original clade II strains of fecal origins. E. coli of diverse origins exhibited clonality amidst divergent genotypes STs, echoing other studies in that recombination in housekeeping genes was insufficient to disrupt phylogenetic signals of the largely clonal E. coli. Notably, environmental E. coli were less diverse than fecal isolates despite contributing many new alleles and STs. Finally, we demonstrated that human activities influenced the distribution of E. coli and clade II in a small aquatic continuum. Moving from relatively pristine sites toward areas with higher human disturbance, the abundance of clade II isolates and new E. coli genotypes reduces, while E. coli bearing class I integrons and belonging to CCs of public health concern accumulates. Altogether, this work revealed the new genetic diversity of E. coli and cryptic clades embedded in selected subtropical aquatic habitats, especially relatively pristine sites, which will aid a more thorough understanding of the extent of their genetic and functional variations in relation to diverse habitats with varied conditions.
Introduction
Variations in DNA sequences underlie the evolution and divergence of the physiological, metabolic, and functional traits of organisms as they adapt to different habitats (Maiden et al., 1998; Spratt, 1999; Reid et al., 2000; Schloter et al., 2000; Yang et al., 2004; Tenaillon et al., 2010). Thus, probing the correspondence between genetic diversification and ecological differentiation is fundamental to construe the evolutions that caused the formation of permanently separated genetic clusters of closely related bacteria with respective ecological roles (Fraser et al., 2007; Hunt et al., 2008). Ecological differentiation occurs not only between species but also within species (Cohan and Perry, 2007; Hunt et al., 2008; Coleman and Chisholm, 2010). Many bacterial species contained multiple ecologically distinct populations, as observed in Escherichia coli (Gordon and Cowling, 2003; Touchon et al., 2020), Legionella pneumophila (Cianciotto, 2006), Vibrio splendidus (Hunt et al., 2008), Bacillus simplex (Koeppel et al., 2008), etc.
Current knowledge about the genus Escherichia largely stems from its type species, E. coli, which commonly inhabits the gastrointestinal tracts of human and warm-blooded animals (primary habitats). While many strains are considered transient in external environments (secondary habitats) after being egested from their hosts, some could persist or even become naturalized in diverse environmental matrices such as waters, soils, sediments, and periphyton (Ishii and Sadowsky, 2008; Touchon et al., 2020). In line with its diverse habitats, E. coli displays considerable phenotypic and genetic variations (Yang et al., 2004; Chaudhuri and Henderson, 2012). Its population is divided into four major phylogroups A, B1, B2, and D, and four rarer phylogroups C, E, F, and G that are differentially distributed among habitats (Clermont et al., 2015, 2019; Waters et al., 2020). Phylogroup A contained many of the human commensal strains. While B2 strains have characteristics that enabled better adaptation to the mammalian gut, they were found at higher proportion in wild animals than domesticated animals such as farm and zoo animals. On the other hand, strains that prevail in aquatic environments predominantly belong to phylogroup B1 (Ratajczak et al., 2010; Tenaillon et al., 2010; Berthe et al., 2013; Petit et al., 2017; Touchon et al., 2020).
Habitat-specific conditions such as the degree of anthropogenic influences have significantly shaped E. coli’s diversity and population structure by selecting for particular phenotypes and genotypes (Walk et al., 2007; Tenaillon et al., 2010; Van Elsas et al., 2011; Perchec-Merien and Lewis, 2013). For example, the widespread utilization of antibiotics in clinical and agricultural settings has enriched E. coli strains carrying class 1 integrons in human and animal hosts and also in external environments that are impacted by human activities (wastewater discharge, agriculture runoff, etc.) (Gillings et al., 2015). Owing to the fact that E. coli is used as a fecal pollution indicator bacteria, the vast majority of the studies on the genetic characteristics of the species have focused on environments that are impacted by human activities. Sites that are relatively undisturbed have received much less attention (Dusek et al., 2018). However, previous studies have shown that less disturbed sites were more likely to harbor novel genetically distinct strains of E. coli (Bermúdez and Hazen, 1988; Dusek et al., 2018). Extensive genome-based analyses suggested that the full genetic diversity and population structure of E. coli are indeed yet to be resolved entirely (Gonzalez-Alba et al., 2019; Touchon et al., 2020).
Population studies of E. coli became more perplexed with the discovery of the cryptic clades I to VI in Escherichia (Walk et al., 2009; Clermont et al., 2011; Gangiredla et al., 2018). These cryptic lineages are often misidentified as E. coli due to their resemblance to E. coli in many phenotypic and genetic characters (Cohan and Kopac, 2011; Deng et al., 2014; Walk, 2015). However, the cryptic lineages are in fact genetically divergent from E. coli sensu stricto and many are ecologically distinct from host-associated E. coli due to historical habitat differences. The members of clades III to V display characteristics of ex vivo adaptation, including higher prevalence in aquatic environments, low virulence and antibiotic resistance, poor GI-colonizing ability (except clade V), but better biofilm-forming ability than host-associated Escherichia. Contrarily, clade I, which is regarded as a subspecies of E. coli, appeared to be primarily host-associated, potentially pathogenic, and more resistant to antibiotics (Ingle et al., 2011; Luo et al., 2011; Perchec-Merien and Lewis, 2013; Deng et al., 2014; Quero et al., 2015; Vignaroli et al., 2015; Walk, 2015; Petit et al., 2017). These studies unraveled notable variations in the physiology, ecology, and clinical implications of these lineages as they establish different lifestyles. Such characteristics remained unknown for clade II because they remain rare despite thousands of host and non-host E. coli-like isolates had been screened in previous studies (Walk et al., 2009; Clermont et al., 2011; Lescat et al., 2013; Walk, 2015). Clade VI has been recently discovered, represented by an isolate that originated from a dog (Gangiredla et al., 2018).
The discovery of the cryptic clades and their primary association with non-host habitats (members of clades III to V specifically) reflected fragmentary comprehension of the evolution histories and ecology of Escherichia as a result of the under-sampling of external environments (Walk, 2015). More efforts to genetically characterize populations in understudied non-host habitats are required to obtain a holistic understanding of the biodiversity of E. coli and the cryptic clades. Examples are the tropical and subtropical aquatic environments where resources, stresses, and biotic factors are different from those in the temperate regions, in which the majority of relevant studies were conducted (Quero et al., 2015; Rochelle-Newall et al., 2015). Therefore, it is plausible that the distribution and genetic diversity of E. coli and cryptic clades and their ecological differentiation in such environments are different from those observed in temperate regions. From a practical perspective, the amplitude to which the presence of cryptic clades will confound E. coli-based water quality monitoring of these aquatic environments also needs further investigations.
To fill the knowledge gap, we analyzed the DNA sequences of seven housekeeping genes in 708 isolates of E. coli and cryptic clades obtained from multiple locations with different habitats and levels of human impacts in subtropical Hong Kong, China. We aimed at determining the genetic diversity and population structures of E. coli and cryptic clades present in different habitats as well as the roles of recombination and the nature of selection in generating genetic variations, and the extent of linkage disequilibrium and genetic differentiation among populations. Furthermore, using a larger dataset containing strains from other studies for a more comprehensive representation of habitat diversity, we inferred the ecological differentiation within E. coli and between members of Escherichia using an ecological partitioning tool. Focusing on a small watershed with contrasting level of human impact, we then elucidated how human activities shaped the genetic diversity and differential distribution of populations along this aquatic continuum. Ultimately, this study would reveal the diversity of E. coli and cryptic clades embedded in selected subtropical aquatic habitats, which will help achieve a more thorough understanding of the extent of their genetic and functional variations in relation to diverse habitats with varied conditions.
Methodology
Bacterial Isolate and Source of Isolation
This study analyzed 708 E. coli-like isolates recovered from samples collected from 18 locations around Hong Kong within the years 2008–2017. The isolates resembled typical E. coli (blue colonies) on CHROMagar™ ECC (CHROMagar Microbiology, Paris, France). Most isolates (n = 625) originated from water, periphyton, or sediment of freshwater and marine habitats. The remaining 83 isolates were of fecal origin (Table 1, Supplementary Figure 1, and Supplementary File 1). Reference strains (n = 105; 72 E. coli from ECOR collection, 30 clade I–VI, one each for E. albertii, E. fergusonii, and Salmonella enterica) were included for comparison whenever appropriate (Supplementary File 2).
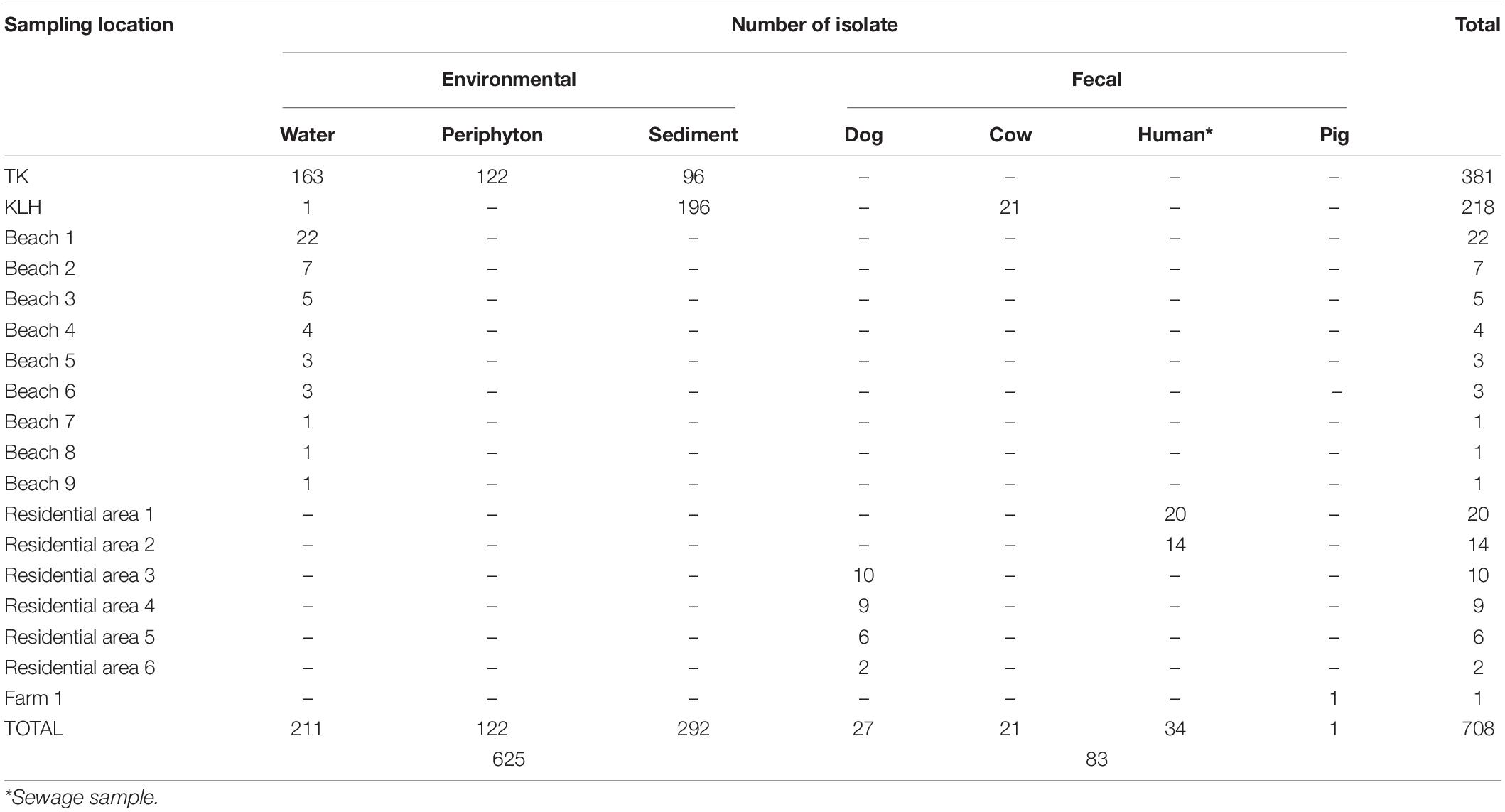
Table 1. Source and sample type of the 708 isolates used in this study.
Most environmental isolates were from two locations: TK, a small freshwater watershed connected to a beach, and KLH, an intertidal mudflat (Supplementary File 1: detailed site description and sampling campaign). The remaining environmental isolates were from water samples randomly collected from nine beaches in 2014. For the TK watershed (381 isolates), 258 isolates (from water, periphyton, and sediment) were collected during the dry season of 2016 from 13 sites that spanned a gradient of human impact, from relatively undisturbed upstream catchwater and streams to downstream more impacted village and beach (low impact, 103 isolates; high impact, 155 isolates). This subset was further investigated for the influence of human activities on the distribution of clade II and E. coli. Another 123 isolates (39 from a storm outfall and 84 from the beach) were obtained from water samples of a follow-up investigation during the wet season of 2017.
The distinction of human impact level at the TK watershed was made based on on-site observations, such as distance from housing, beach activities, and human and vehicle traffic (Supplementary Figure 2 and Supplementary File 1). Six low human impact sites were situated upstream along the edge of a country park. Five of these sites were in three streams; the remaining site is a roadside drain receiving water from the country park located uphill. Seven high human impact sites were located downhill. One is a catchwater of stormwater that was channeled from uphill, situated in the vicinity of human settlement, including village houses and squatters, north of the TK village located downhill. Another three were two stormwater outfalls and a surface channel in the TK village, which is located next to the TK beach. The stormwater outfalls discharge directly into the beach water (three sampling sites). The beach area was perceived to receive the highest level of human disturbance due to beach activities, effluent from the Stonecutters Island Sewage Treatment Work, the largest sewage treatment plant in Hong Kong, and possible leakage from failing sewage infrastructure or cross-connected sewage and stormwater networks.
For KLH, a triangle-shaped, semi-enclosed intertidal mudflat, isolates were recovered from sediment at a single site, repeatedly sampled over 8 months spanning May 2009 to October 2010. The seaward margin of the mudflat faces north while a hill bounds its southeast margin and its southwestern margin bordered shrubland that is adjacent to a hill. Twenty-two houses were scattered around the shrubland, with the house closest to the mudflat being at least 100 m away. The mudflat is a sink for terrestrial runoff in the wet season due to the steep surrounding terrains. Observed fecal sources at the sampling site included a herd of approximately 15–20 feral cows and a few pet dogs. The maximum water depth of the mudflat is less than 2 m during high tides, whereas the sediment is fully exposed during low tides. The KLH isolates were screened with repetitive element palindromic genotyping to maximize genetic diversity while reducing sample size (see Supplementary Figure 3 and Supplementary File 1). Therefore, analyses that included the number of isolates (for example, frequency of genotype) shall be treated cautiously when the KLH dataset is included.
Multilocus Sequence Typing
DNA was extracted using crude cell lysate method, followed by polymerase chain reaction (PCR) of the Achtman MLST scheme (Wirth et al., 2006) targeting seven loci: adk (adenylate kinase), fumC (fumarate hydratase), gyrB (DNA gyrase), icd (isocitrate/isopropylmalate dehydrogenase), mdh (malate dehydrogenase), purA (adenylosuccinate dehydrogenase), and recA (ATP/GTP binding motif).
The nucleotide sequences of amplicons were obtained through Sanger sequencing. Sequences were checked for quality, assembled, and trimmed using the Multilocus Sequence Typing (MLST) script in BioNumerics v7.1 (Applied Maths, Kortrijk, Belgium). Sequence-based and allele-based analyses were performed using the resulting sequences. For the allele-based approach, each distinct sequence at a locus represents a unique allele and was assigned an arbitrary allelic number. Each distinct combination of seven allelic numbers gives rise to a unique allelic profile (MLST genotype) denoted as sequence type (ST). Allelic and ST number was assigned using EnteroBase1 (Alikhan et al., 2018). Novel sequences were assigned new allelic numbers and STs locally.
Phylogenetic Analysis
Phylogeny of the isolates was inferred by a sequence-based approach. Concatenated sequences of 3,423 bp each (adk-fumC-gyrB-icd-mdh-purA-recA) were generated for all isolates and reference strains. Resultant sequences were loaded into MEGA7 software v7 (Kumar et al., 2016) to build a multiple sequence alignment, followed by construction of a maximum likelihood (ML) tree (general time reversible G + I model, 500 bootstrap replications) and calculation of average evolutionary divergence (p-distance). S. enterica Typhimurium LT2 was used as outgroup to root the tree. The tree was used in the subsequent AdaptML analysis. Another simplified tree was built with the same parameters, but with each sequence type represented only once for the isolates.
AdaptML Analysis
The AdaptML algorithms were used to investigate ecological differentiation within Escherichia in relation to their habitats (Hunt et al., 2008). An ML tree (generated as in the phylogenetic analysis section, from the concatenated MLST sequences of 708 isolates and 105 references) and sample sources were used as inputs for analysis. Isolates were categorized into 11 sample sources from human, animal (wild, domesticated, livestock, captive, avian, and unknown), and environments (freshwater, marine, freshwater marine mix, and unknown). While no further demarcations were made for human hosts, animal hosts were further differentiated. The animal hosts were mammalians and avian. The mammals were divided into five categories based on different lifestyles that affect the general likelihood of exposure to anthropogenic influences: livestock, domesticated, captive (zoo), wild, and unknown (lifestyles unknown or stated only as mammals). Environmental sources were further divided into four categories mainly based on salinity, i.e., freshwater, marine, freshwater marine mix (freshwater with occasional seawater intrusion), and unknown (for other environments, i.e., soil and where sources were only stated as environment or water). Default AdaptML parameters were used. ML tree overlaid with inferred ecological differentiation and metadata (phylogeny and sample sources) was visualized using iTOL (Letunic and Bork, 2019).
Population Structure
Population structure was determined by allele-based approach. Single locus variants (SLVs), i.e., ST pairs sharing 6 alleles, were identified and assigned into clonal complexes (CCs) using geoBURST algorithm (Francisco et al., 2009) as implemented in PHYLOViZ v2.0 (Nascimento et al., 2016). A CC contained at least three STs, and its evolutionary descent patterns were inferred by identifying a founding genotype and its SLVs. Minimum spanning tree (MST) based on allelic profiles was generated using BioNumerics v7.1 (Applied Maths, Kortrijk, Belgium) and PHYLOViZ v2.0 (Nascimento et al., 2016).
Population Genetics
The extent of recombination was visualized with a splits network using the NeighborNet algorithm (uncorrected p distance) in Splits Tree4 v4.14.6 (Huson, 1998; Huson and Bryant, 2005). The network was overlaid with E. coli phylogroup information. The assignment of phylogroup was done based on the correspondence between ST and phylogroup information in literature and MLST database. An E. coli ST was assigned into one of eight phylogroups according to information in Clermont et al. (2015, 2019) and EnteroBase (Alikhan et al., 2018). When phylogroup information is not readily available for a particular ST, its phylogroup was inferred by its genetic relatedness with STs of known phylogroup. Two STs were assumed to belong to the same phylogroup only if they are SLVs or double locus variants (DLVs). No assignment was made if criteria were not met. Subsequently the phi test (Bruen et al., 2006) implemented in the same program was used to test statistically for the role of recombination in generating (a) allelic variation at each locus (within gene recombination), and (b) different STs (within gene recombination + assortive recombination).
To determine if significant linkage disequilibrium (non-random association of alleles at loci) is present, the standardized index of association (ISA) was calculated (Haubold and Hudson, 2000), followed by the null hypothesis testing of linkage equilibrium in START2 (Jolley et al., 2001). Expected value for ISA is zero when alleles are in linkage equilibrium. A reduced dataset (each ST represented only once) was analyzed to minimize linkage disequilibrium introduced by sampling bias and/or possible existence of adaptive clones (Maynard Smith et al., 1993).
The nature of selection in generating allelic variation in each locus was assessed using the dN/dS ratio by comparing the percentage of non-synonymous substitutions per non-synonymous site (dN) relative to synonymous substitutions per synonymous site (dS) (Nei and Kumar, 2000). Analysis was performed using START2 (Jolley et al., 2001). A dN/dS ratio of 1 indicated neutral selection, < 1 suggested purifying selection, whereas > 1 implied adaptive or diversifying selection.
The fixation index, FST, was calculated to assess pairwise genetic differentiation between different populations using Arlequin v3.0 (Excoffier et al., 2005). The lower the FST value, the higher the genetic similarity of two populations, indicating more gene flow.
Detection of Class 1 Integron-Integrase (intI1) Gene
Clinical variant of the intI1 gene was detected using PCR with the primer pair IntI1F165/IntI1R476 as described by Gillings et al. (2015). The assay was performed on the 258 isolates collected from the TK watershed during the dry season of 2016.
Statistical Analysis
Statistical analyses were performed using the Minitab v18.1 software (Minitab Inc., Pennsylvania, United States).
Results
Phylogenetic Analysis
The phylogeny of the isolates was established using an ML tree generated from the concatenated sequences of seven MLST loci. The resulting tree constituted several highly supported clusters (> 75% bootstrap support), which corresponded to known groups of Escherichia (Figure 1). Among the 708 isolates, 660 isolates (93%) constituted a major cluster with all reference E. coli from the ECOR collection (Ochman and Selander, 1984), clades I and VI with high bootstrap support (96%). The remaining 48 isolates (7%) formed monophyletic clusters with the reference strains of cryptic clades II, IV, and V, respectively. The majority of the cryptic clade isolates (n = 44) were associated with clade II, forming a highly supported (100%) cluster with three references of the clade, namely, ROAR019, B1147, and EC5350. The remaining four isolates belonged to clade IV (n = 3) and clade V (n = 1).
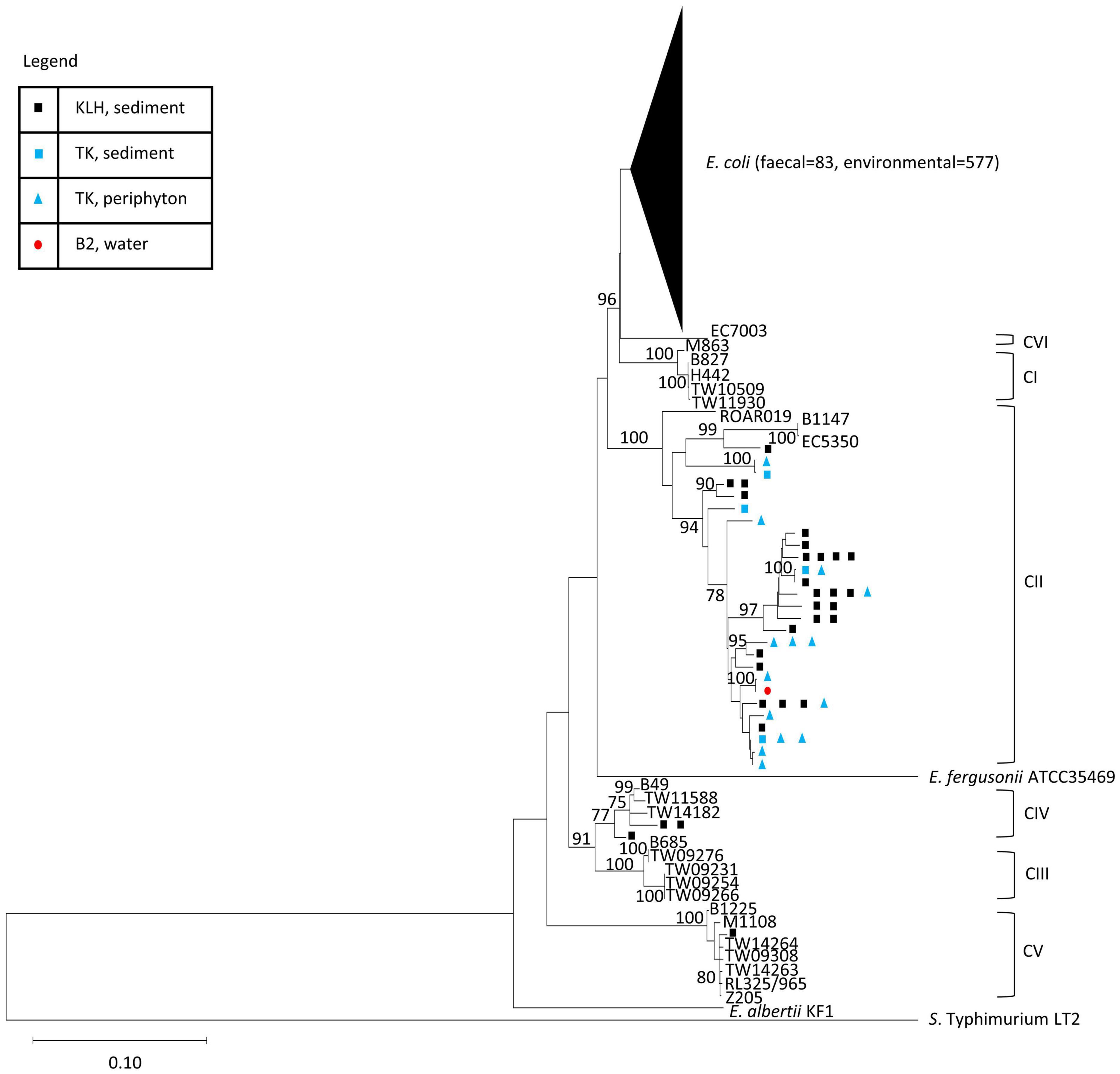
Figure 1. Phylogenetic position of the isolates in study in reference to strains of Escherichia spp. reported in previous studies. ML tree was built using concatenated sequences of seven MLST loci from the isolates and reference strains (general time reversible G + I model, 500 bootstrap replications). Bootstrap supports > 75% are indicated at the nodes. The genetic cluster containing all E. coli was collapsed (size of triangle not proportionate to number of entries) for ease of visualization of the whole tree. Each sequence type of the isolates was represented once. S. enterica Typhimurium LT2 was used as an outgroup and the reference strains for the cryptic clades were indicated. Complete tree containing all entries (708 isolates and 105 references) were presented in Figure 2. The isolation sources of the cryptic clades in this study were indicated at their respective terminal nodes.
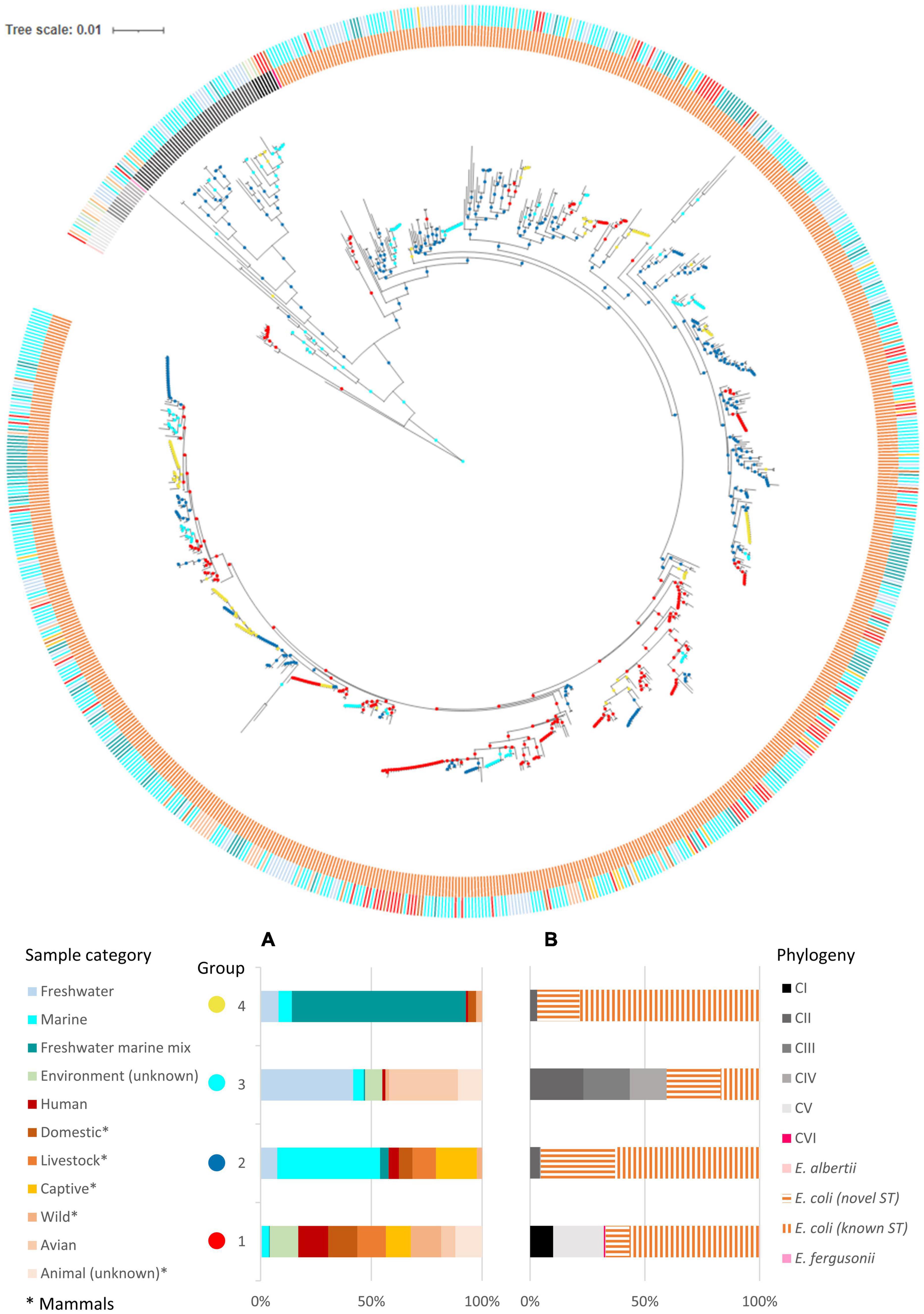
Figure 2. Inferred habitat associations of ecologically distinct groups of Escherichia. Metadata were mapped onto the ML tree built using concatenated sequences of seven MLST loci (general time reversible G + I model, 500 bootstrap replications). The outer ring indicated the sample category of the isolates while the inner ring indicated their phylogeny. Inferred ecologically distinct groups are shown as colored circles at the nodes. (A) Inferred ecologically distinct groups, defined by distinct distributions of isolates over the sample sources. (B) Distribution of phylogeny for each inferred group. All distributions are normalized by the total counts in each sample source to account for uneven sampling across sample categories.
All fecal isolates were E. coli sensu stricto, whereas environmental isolates consisted of both E. coli and cryptic clades. The cryptic clades (n = 48, 8% of the 625 environmental isolates) were mainly recovered from KLH and TK, except for a clade II isolate (from seawater of Beach 2). Clade II (n = 25) was the most abundant cryptic clades recovered from KLH, followed by clade IV (n = 3) and clade V (n = 1). Only clade II (100%, n = 18) was isolated from the TK watershed, during the dry season in 2016.
The average nucleotide divergence within group (p-distance) was 1.40% for E. coli, 0.59% for clade I, 2.22% for clade II, 0.73% for clade III, 0.94% for clade IV, and 0.27% for clade V. Notably, not only was the divergence within clade II higher than the other groups, the divergence between the clade II references alone (ROAR019 vs. B1147 and EC5350) was 3.7%. On the other hand, the nucleotide divergence between each pair of known sister clades was 2.6% (E. coli vs. clade I) and 2.3% (clade III vs. clade IV). These observations suggested finer classification of clade II into subclades similar to the above sister clades. However, confirmation of distinct subclades requires taxonomic analyses with higher resolution such as DNA–DNA hybridization and average nucleotide identity (Chun et al., 2018), and some of the branches within the clade II cluster lacked bootstrap support (< 75%). Therefore, we investigated the clade II-associated group in its entirety while recognizing the possible existence of subgroups.
Habitat Prediction by AdaptML
We analyzed our data with the AdaptML algorithms (Hunt et al., 2008) to probe the ecological differentiation within Escherichia. The primary ecology questions under consideration were whether the members of Escherichia were ecologically differentiated in relation to their habitats (represented by different sample sources), in particular whether clade II was ecologically distinct from E. coli. From a dataset that contained STs from multiple sources spanning different hosts and environments, AdaptML inferred four ecologically distinct groups with different spectra of habitat associations (Figure 2). Each of these groups (namely, 1, 2, 3, and 4) is composed of sequence types (STs) with common characteristics in relation to their habitats.
The habitat spectra of groups 1 and 4 suggested clear ecological specialization. Group 1 reflected a primarily host-associated lifestyle, composed almost exclusively of E. coli, E. albertii, E. fergusonii, CI, and CVI STs isolated from diverse hosts and geographical locations. Clade V STs (originated from both hosts and environment), which were suggested to be capable of dual lifestyle in both host and environment (Vignaroli et al., 2015), were also included in this group. Group 4, on the other hand, is conspicuously enriched with isolates that could survive in a freshwater environment that was under occasional seawater intrusion. Majority of the group 4 STs were from the TK watershed, especially from sites encountering seawater intrusion (Supplementary Figure 4 and Supplementary File 3). Notably, group 4 contained only isolates from this study. Majority of the STs were E. coli while few clade II were present.
The habitat spectra of groups 2 and 3 suggested a more cosmopolitan characteristic, as both groups contained roughly an equal mixture of STs from both environment and hosts. Nonetheless, their constituent STs and habitats were markedly different. Group 2 contained mostly isolates from marine environments and animal hosts that are domesticated, captive (zoo animals), or grown as livestock. Similar to group 4, the isolates were almost exclusively E. coli, with few clade II present. On the other hand, unlike the other groups that contained predominantly E. coli, group 3 contained roughly an equal portion of E. coli, CII, CIII, and CIV. These isolates were mainly of freshwater and avian origins. Notably, STs of animal origins belonged to CII, CIII, and CIV. Majority of CII STs were contained in group 3. Also, while novel E. coli STs can be found in all groups (constituted 10, 32, 23, and 18% of their respective group, from group 1 to 4), slightly more than half (58%) of the E. coli STs in group 3 were novel. Many of these novel E. coli STs originated from the freshwater sites of the TK watershed, which remained largely undisturbed by human activities. Contrarily, most E. coli STs in the other three groups were known STs previously reported in the EnteroBase (Alikhan et al., 2018), which constituted 85% (group 1), 66% (group 2), and 81% (group 4) of the E. coli STs in their respective group. Our results thus demonstrated ecological differentiation within E. coli that were isolated from diverse habitats, and that in general clade II STs shared ecological similarities with only a small fraction of E. coli, many of which were newly described STs.
Population Structure
In total, 380 STs were detected out of the 708 isolates (Table 2). E. coli (n = 660) contributed to 350 STs, 318 STs by E. coli from environmental source (n = 577) and 53 STs by fecal isolates (n = 83), with 21 common STs shared by both sources. Another 30 STs (44 clade II isolates contributed 27 STs; 3 clade IV, 2 STs; and 1 clade V, 1 ST) were contributed by the 48 cryptic clade isolates.
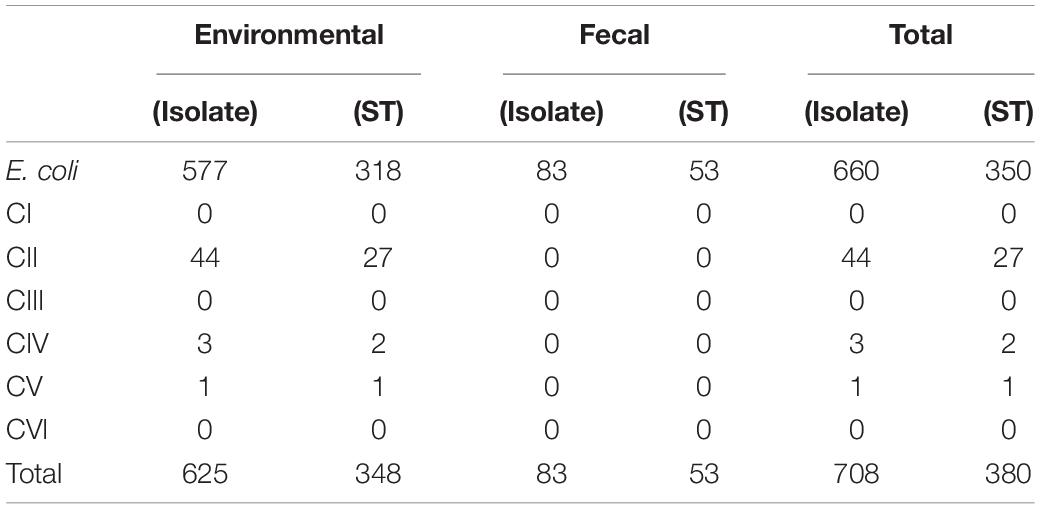
Table 2. Number of STs contributed by E. coli and cryptic clades.
The population structure of all 708 isolates was visualized using allele-based MST (Figure 3). MST revealed a largely clonal E. coli population structure containing a mixture of closely related STs (CCs, SLVs, and DLVs) and distant STs. E. coli of same phylogroup (mainly A, B1, B2, or D) generally clustered together, forming four main branches on the MST (see Supplementary Figure 6 and Supplementary File 4 for the same MST overlaid with phylogroup information). No clear demarcation of isolates according to geographical locations, sampling period, or sample types was observed, as many animal, human, and aquatic environment isolates of different geographical and temporal origins shared identical STs or were SLVs and DLVs.
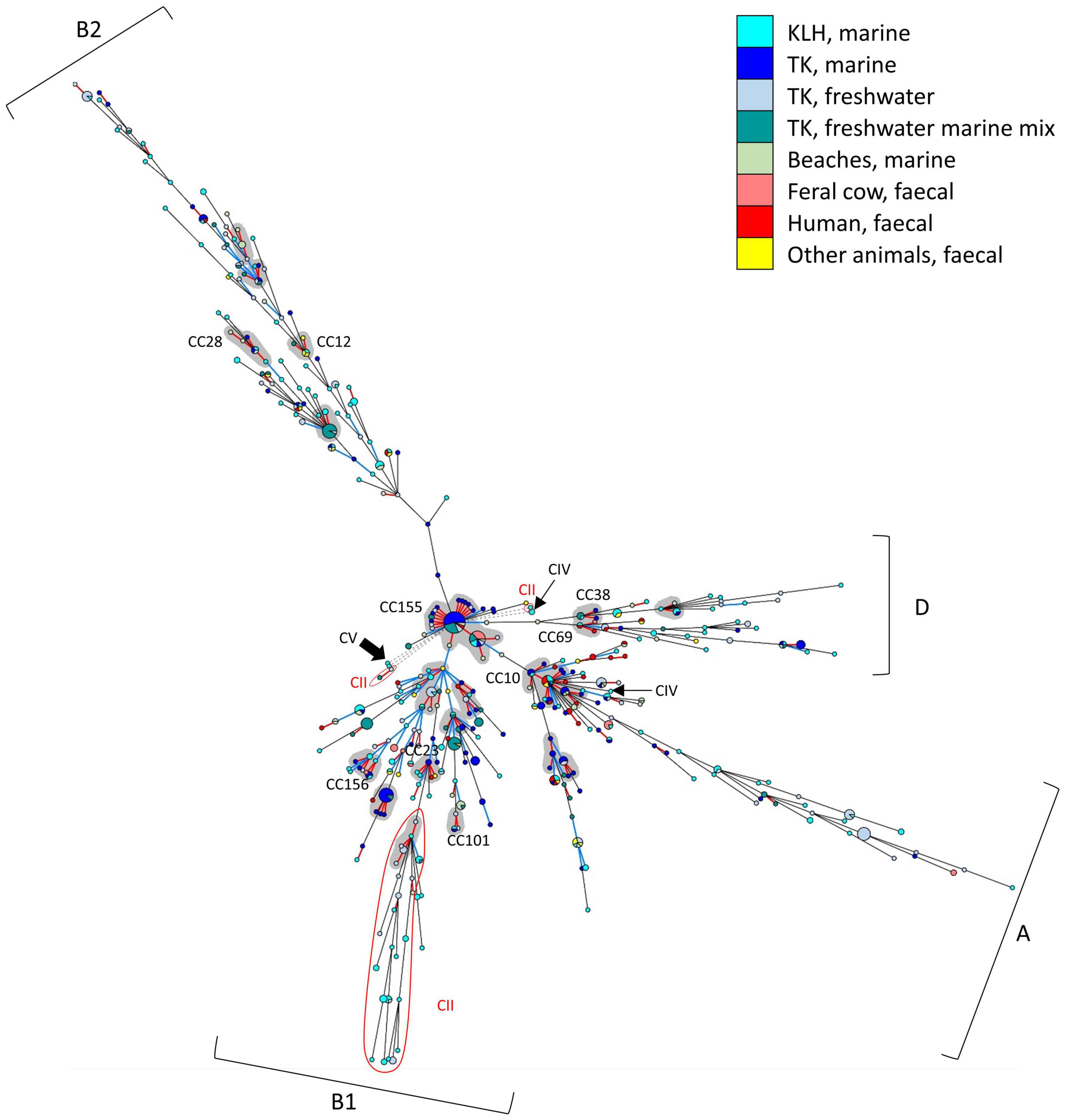
Figure 3. Minimum spanning tree (MST) displaying the genetic relatedness of the 708 isolates. Each node represented one ST. Node size is proportionate to the number of isolates contained in the node. Branch length is proportionate to distance between nodes, with red branch denoted 1 locus difference; blue: 2 loci; black: 3–6 loci; and gray dashed branch: no loci in common. CCs were shaded with gray background and nine well-established E. coli CCs that could also be identified in this study were labeled. E. coli phylogroups (A,B1,B2,D) for majority of the STs in each of the four main branches were indicated. The cryptic clade II STs were highlighted and indicated with red fonts; clade IV STs were indicated with thin black arrows; and a thick arrow indicated the sole clade V in this study.
Among E. coli, 153 STs (44%) very closely related, manifested as SLVs among which 21 CCs (containing 97 STs, 28% of all E. coli STs) were identified. Nine E. coli CCs were well-established CCs in EnteroBase (Alikhan et al., 2018), namely, CC10, CC12, CC23, CC28, CC38, CC69, CC101, CC155, and CC156. The two largest CCs in our dataset were CC155 (18 STs) and CC10 (12 STs), containing both fecal and environmental E. coli of different geographical and temporal origins. E. coli belonging to CCs of public health importance such as CC10, CC28, CC69, CC73, CC95, CC131, and CC155 (Riley, 2014; Skurnik et al., 2015; Stoesser et al., 2016; Reid et al., 2018) were present in both marine and freshwater habitats.
The clade II population was also clonal; 11 STs (41%) were SLVs and one CC was present, containing isolates mainly from the freshwater sites of TK (5 STs, 19%). The main clade II cluster was manifested as STs distantly related to E. coli phylogroup B1. The connecting clade II ST shared a common allele at recA (allele 7) with E. coli ST. Some clade II isolates from KLH, TK, and Beach 2 shared identical STs or were SLVs despite being isolated from different sample types and geographical origins. Majority of remaining STs exhibited ≥ 5 loci difference with each other.
A few STs were very distantly related to the rest of the isolates, without any allele in common. These STs included one E. coli ST, a pair of clade II SLVs, a clade II ST, one clade IV ST, and the sole clade V in our collection. The remaining clade IV ST shared a common allele with E. coli ST10 at recA (allele 2).
Genetic Diversity and Differentiation of Different Populations
After looking at the expansive population structure of E. coli and clade II in their entirety, analyses were done at different population levels to yield ecologically meaningful comparisons. Populations were stratified into different levels (i.e., from the entire collection to subpopulations according to phylogeny, sampling dimensions such as host vs. environment, TK vs. KLH) to demarcate patterns of genetic variation, purifying selection strength, linkage disequilibrium, recombination, and genetic differentiation at/between different levels.
Sequence Type and Allelic Variation
Forty-five percent (n = 172) of STs were known STs available at EnteroBase (Alikhan et al., 2018) while the remaining 55% (n = 208) were novel (Table 3). New STs were significantly more abundant among environmental isolates than among fecal isolates (58 and 15%, respectively, p < 0.001). Number of alleles ranged from 74 for purA (frequency = 0.1) to 130 for fumC (frequency = 0.18) for all isolates.
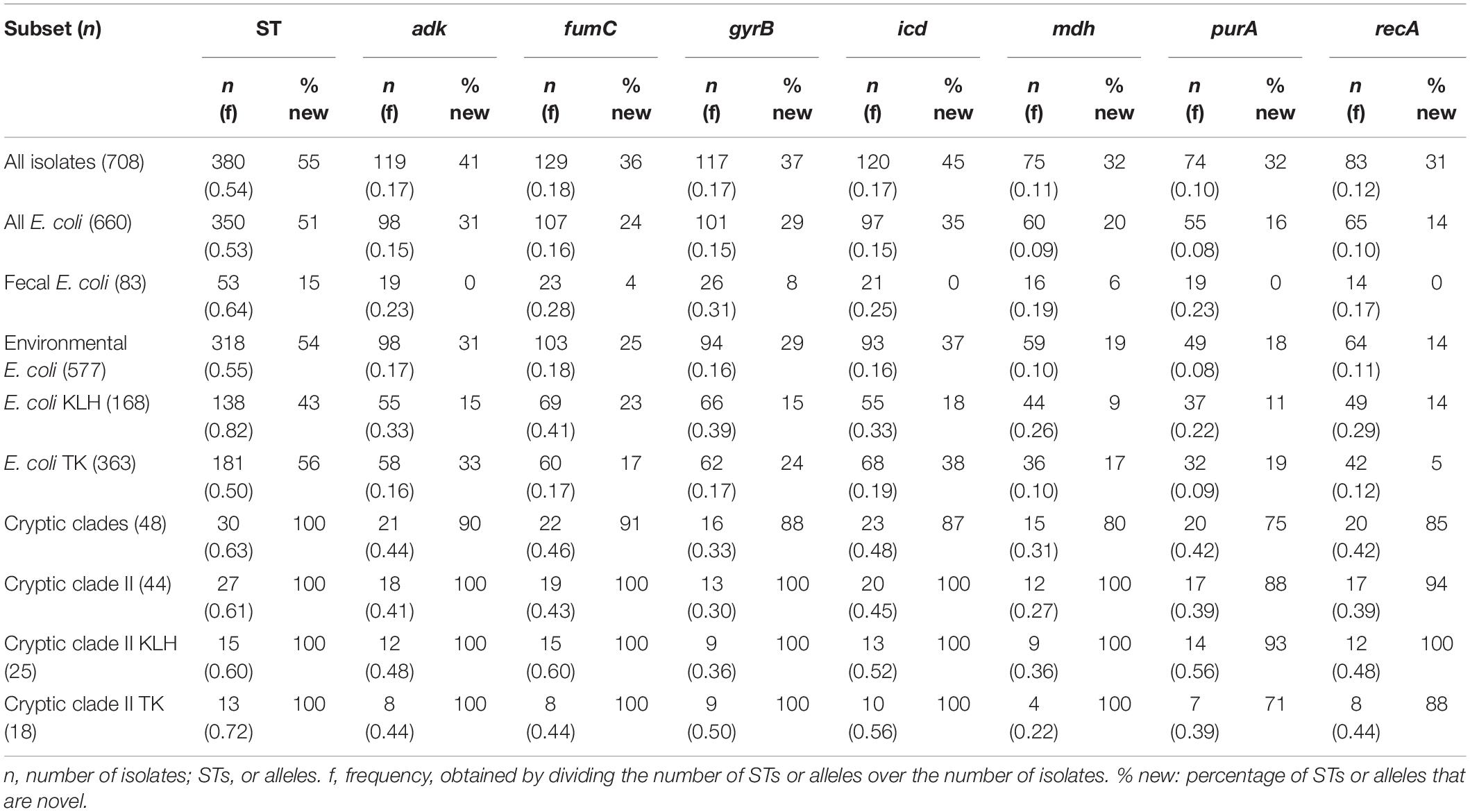
Table 3. ST and allelic variation of data subset.
Considering only E. coli, the percentage of new STs among environmental isolates remained significantly higher than that of fecal isolates (54 vs. 15%, p < 0.05) (Table 3). Moreover, environmental E. coli isolates noticeably displayed more new alleles for all loci (from 14% for recA to 37% for icd) than fecal isolates (from 0% for adk, icd, purA, and recA to 8% for gyrB). However, environmental isolates have fewer alleles across all seven loci than fecal isolates despite contributing more new alleles. E. coli generally has lower diversity for mdh, purA, and recA.
All 30 cryptic clade STs were newly discovered. Most were distantly related to that of the reference cryptic clades (≤ 1 allele in common), except for the sole clade V isolate, which is a DLV of ST2721 (ST of reference clade V strain E1118). All clade II isolates have no common allele with the reference clade II STs (Supplementary Figure 5 and Supplementary File 4). For cryptic clades, at least 75% of alleles for each locus were new alleles. Clade II exhibited lowest diversity for mdh. Compared to E. coli, generally more allele per locus was observed for cryptic clades collectively and for clade II specifically.
Nature of Selection
A low dN/dS value was observed for all loci, indicating strong purifying selection acting at these loci to limit amino acid polymorphisms (Table 4). Thus, allelic variations at the housekeeping loci were mainly contributed by silent mutation or recombination. Different strength of purifying selection was observed across the seven loci, with recA under strongest purifying selection across all subsets.
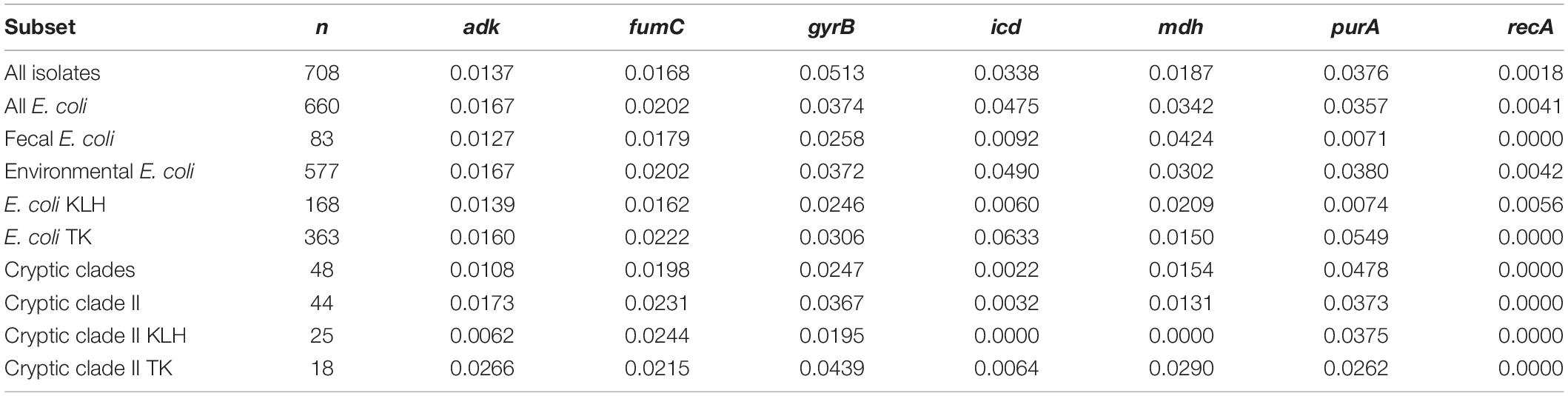
Table 4. dN/dS ratio of the seven housekeeping genes.
For E. coli, fecal isolates showed ∼5-fold lower dN/dS ratios for icd and purA than environmental isolates. Among environmental E. coli, dN/dS for icd and purA were ∼10- and 7-fold lower, respectively, for isolates from KLH than those from TK. Within clade II, isolates from KLH generally underwent stronger purifying selection at more than half of the loci. Disparate dN/dS were most evident at icd and mdh between isolates from TK and KLH. When comparing E. coli and clade II, dN/dS for icd were more dissimilar, with clade II having ∼15-fold lower ratio. Specifically, comparing E. coli and clade II from KLH, which were co-isolated from a single site, clade II experienced stronger purifying selection for most loci except for fumC and purA.
Linkage Disequilibrium
Extensive linkage disequilibrium was detected (ISA > 0 and p = 00.00 for all subsets), indicating the presence of clonality in population structure at all tested levels (Table 5).
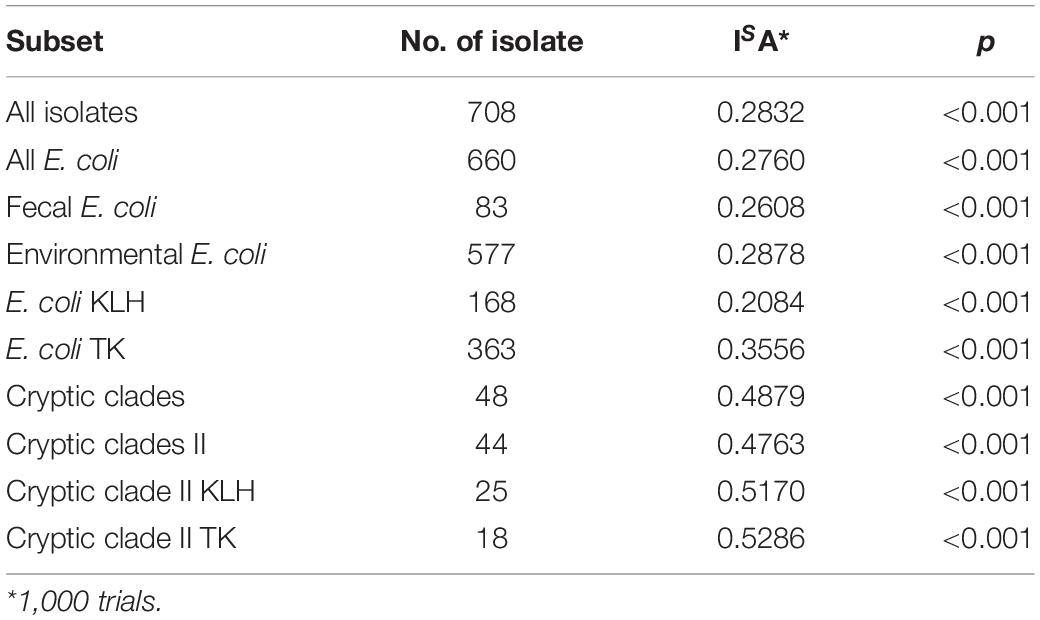
Table 5. Test of linkage equilibrium.
Recombination
Network analysis revealed that majority of E. coli STs clustered according to phylogroups, except for phylogroup A, which was split into two main clusters (Figure 4). The cryptic clades formed clusters distinct from E. coli. The network largely resembled a tree-like structure although parallelograms representing conflicting phylogenetic signals (mainly due to recombination) were present. Intra-phylogroup recombination was more evident, which contrasted limited inter-phylogroups recombination. Recombination was also evident within cryptic clades but essentially rare between cryptic clades and E. coli.
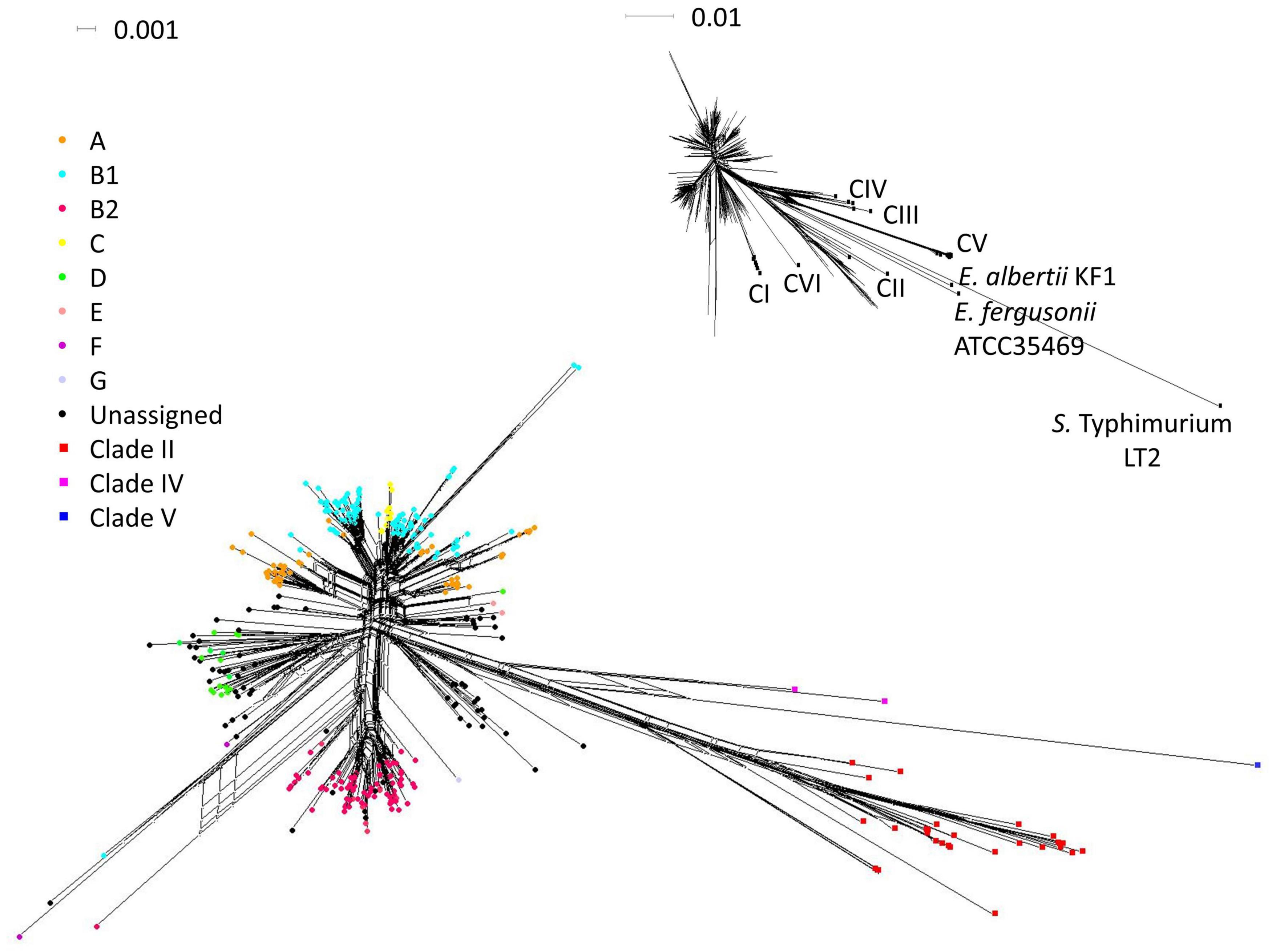
Figure 4. Phylogenetic network of the 380 STs based on NeighborNet algorithm using concatenated sequence. Parallelogram denotes incompatible partition within sequence data (conflicting phylogenetic signals due to recombination or recurrent mutation). Insert is the network generated using the same dataset plus reference sequences as used in Figure 2. References of cryptic clades I–VI, E. albertii KF1, E. fergusonii ATCC 35469, and Salmonella enterica Typhimurium LT2 were indicated.
The phi test (Bruen et al., 2006) was performed to detect statistically significant recombination: within different E. coli populations, between E. coli and cryptic clades, within cryptic clades, and within clade II (Table 6). Recombination was significant in generating different STs for all subsets (p < 0.05). Generally, with cryptic clades included, different extent of recombination existed across all seven loci, with adk having the least recombination and fumC having the most extensive recombination. Recombination within E. coli and within cryptic clades was more significant compared to recombination between the two groups. Similarly, recombination was more significant within fecal E. coli and within environmental E. coli than between these two populations. Among E. coli, recombination was significant both within and between population subsets for fumC and icd, but was only significant within subpopulations for mdh and purA.
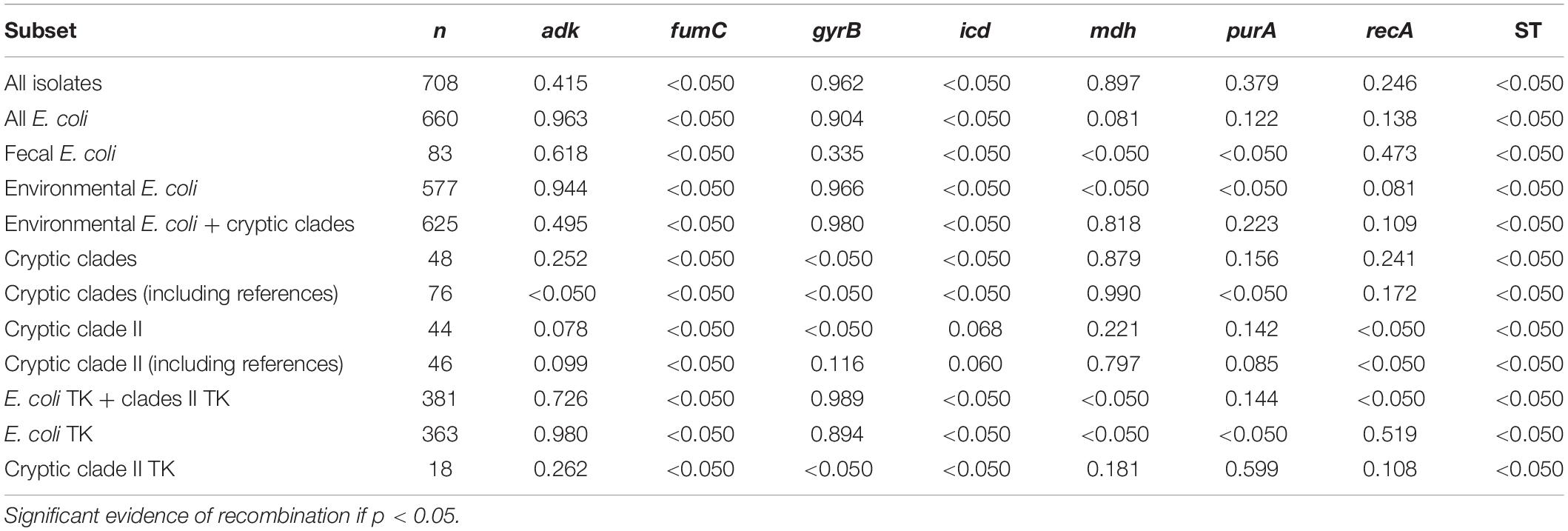
Table 6. Recombination analysis by phi test (Bruen et al., 2006) with p-values indicated.
Genetic Differentiation
All pairwise FST values between different E. coli populations (same subsets as in Table 3) were less than 0.05000 (p < 0.05), indicating a lack of genetic differentiation between tested populations (Supplementary File 5). The environmental and fecal E. coli subsets only showed little genetic difference (FST = 0.01539, p < 0.01). On the other hand, gene flow between the cryptic clades (collectively and clade II specifically) and E. coli were infrequent (FST > 0.5, p < 0.01). Moderate genetic difference existed between cryptic clade II from KLH and TK (FST = 0.09912, p < 0.01).
Distribution of Escherichia coli and Cryptic Clade II in a Watershed With Distinct Human Impact
To investigate the distribution of clade II and E. coli in relation to human impact, we further examined isolates that were collected from two differentially impacted areas in the TK watershed during the dry season of 2016. We examined the genetic differentiation of isolates from the two areas by first including both E. coli and clade II. Moderate genetic differentiation existed between the two groups of isolates under differential human impact (FST = 0.07031, p < 0.01). However, when clade II isolates were removed, little genetic differences were observed between the two groups of E. coli sensu stricto (FST = 0.04846, p < 0.01).
In fact, majority of the clade II isolates (89%, n = 16) were recovered from the low human impact sites. Prevalence of clade II isolates was significantly higher (p < 0.01) at the low human impact sites (16% of 103 isolates) than at the high human impact sites (1% of 155 isolates). Clade II isolates were present in 83% of periphyton and 40% of sediment samples collected from the low human impact sites, constituting 10–60% of analyzed isolates per sample when present. The two clade II isolates from high human impact sites were from periphyton and sediment of storm outfall 1 in TK Village, each constituting 3% of analyzed isolates of respective sample (Supplementary Table 1 and Supplementary File 1). Besides clade II, E. coli phylogroups were also differentially distributed in the two areas (Figure 5). Notably, the low impact sites contained more E. coli without phylogroup assignment.
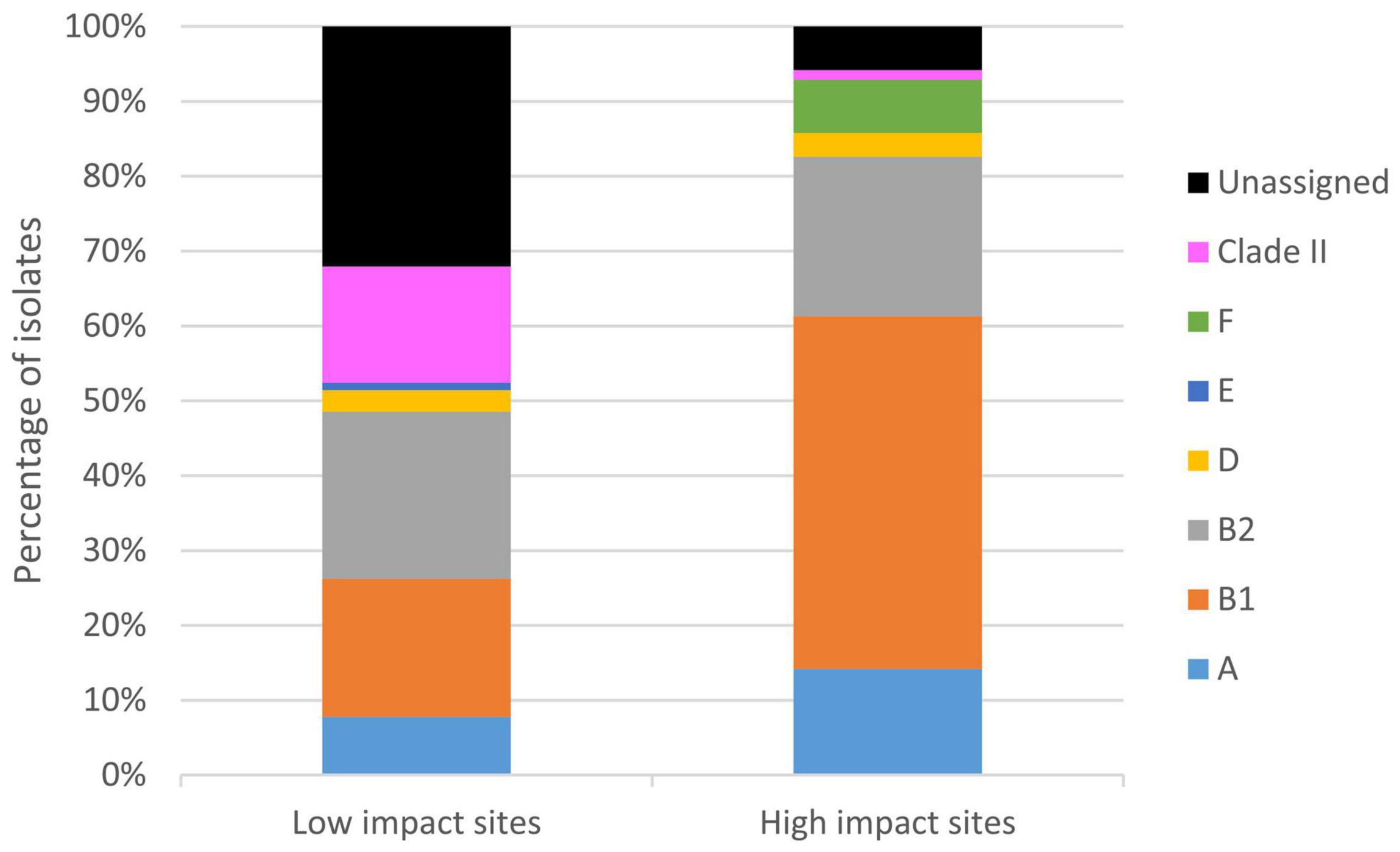
Figure 5. Distribution of E. coli phylogroups and clade II isolates in the TK watershed. An E. coli ST was assigned into one of the eight phylogroups according to information in Clermont et al. (2015, 2019) and EnteroBase (Alikhan et al., 2018). When information is not readily available, phylogroup was inferred for an ST by its genetic relatedness with STs of known phylogroup: two STs were assumed to belong to the same phylogroup only if they are SLVs or double locus variants (DLVs). No assignment was made if criteria were not met.
Novel STs (of E. coli and clade II) were retrieved at a higher frequency from the low human impact sites than sites with higher impact (68 vs. 40%, p < 0.01), contributed by 69 and 22% of isolates, respectively (p < 0.01). Considering only E. coli (as all clade II STs were novel), the percentage of new STs from the low human impact sites remained noticeably higher (61 vs. 38%, p = 0.02; contributed by 63 and 21% of isolates, respectively, p < 0.01).
On the other hand, E. coli isolates belonging to CCs of public health concern (CC10, CC28, and CC155) were mainly found at high human impact sites (n = 28, p < 0.01). Only one such isolate was detected in the low human impact sites. All isolates were then screened for the integrase gene of class 1 integron (intI1), which was proposed as a marker for anthropogenic pollution (Gillings et al., 2015). Notably, all intI1-positive isolates were E. coli. The percentage of intI1-positive E. coli was higher at the high human impact sites (12%) than at the low impact sites (2%), and the differences were statistically significant (p = 0.01).
Discussion
This study reported a relatively large number of clade II isolates from the less studied subtropical aquatic habitats, which, when present, constituted up to 60% of analyzed presumptive E. coli isolates per sample. In previous studies, clade II had been rarely observed despite thousands of host and non-host isolates resembling E. coli were screened, with only three fecal strains reported (Walk et al., 2009; Clermont et al., 2011; Lescat et al., 2013; Walk, 2015; Gangiredla et al., 2018). Two studies reported the presence of cryptic lineages (which included clade II) and E. coli in aquatic habitats, but each lineage’s proportions were not mentioned (Quero et al., 2015; Petit et al., 2017). Thus, little was known about the types of habitats where clade II may prevail, let alone other ecological characteristics. Clade II isolates were exclusively found in environmental matrices in this study, and none were from fecal samples. In contrast to most studies reporting that environmental cryptic clades belonged to clade V predominantly (see Supplementary File 6; Walk et al., 2009; Clermont et al., 2011; Deng et al., 2014; Vignaroli et al., 2015), here clade II was the most abundant from both freshwater and seawater habitats. Clade II accounted for all cryptic clades isolated at the primarily freshwater TK watershed and for 86% of cryptic isolates in the KLH mudflat (Figure 1).
We showed that periphyton and sediment were significant sources of clade II (Supplementary Table 1 and Supplementary Files 1, 6). Most clade II isolates (89%) were recovered from periphyton in the low human impact sites in the TK watershed. Despite a greater sampling effort, only two clade II isolates were recovered from the high human impact area downstream of the less disturbed area. Later, we obtained similar results in another study that was conducted at similar locations in the TK watershed (unpublished data). Cryptic clades (clade II = 25, clade IV = 3, clade V = 1) were also repeatedly isolated from coastal marine sediment over 8 months in the KLH mudflat, which was also a relatively undisturbed site, albeit having higher human impact level than the low impact sites in TK. Our high frequency of clade II isolation could be attributable to multiple factors. It is likely that pristine environments harbored more clade II, or that compared to temperate regions, subtropical environments generally sustained different proportions of cryptic lineages. Knowledge on the ecology of clade II will benefit from biogeography studies of the distribution of different cryptic clades in temperate, tropical, and subtropical aquatic environments and within diverse wildlife, which was less studied. Another pertinent possibility worth investigating is whether selective media have differential selectivity against different cryptic lineages. ChromAgar™ ECC was utilized in this study, whereas other studies utilized modified TEC, FC, modified FC, or RAPID’E. coli 2 Medium (Walk et al., 2007; Deng et al., 2014; Quero et al., 2015; Vignaroli et al., 2015; Petit et al., 2017). ChromAgar™ ECC was chosen in this study in accordance with the E. coli detection method developed by the Environmental Protection Department of Hong Kong for routine beach water quality assessment (Ho and Tam, 1997; Thoe et al., 2018).
Nonetheless, our observations that clade II prevail in subtropical aquatic environments with low human interference resonated another study where clades II, III, and V were abundant in river sediments that were less disturbed by human and agriculture activities (Petit et al., 2017). Such high frequencies of non-clade I cryptic clades could potentially confound the use of E. coli as fecal indicator bacteria. When they are abundant in sediments and associated vegetation, these cryptic clades could be suspended into overlaying water upon perturbations, for example, by storm events. Since current monitoring methods would misidentify them as E. coli unless MLST or PCR targeting cryptic clades (Clermont et al., 2011) were performed, this may cause a false conclusion of recent fecal pollution. Thus, it is imperative to determine their actual potential in interfering with E. coli-based water quality monitoring under different environmental perturbations or hydrology conditions.
While the observations above suggested that relatively undisturbed aquatic environments could be natural habitats of clade II, their presence there could have also possibly originated from recurring fecal input by wildlife, as the reference clade II strains originated from fecal materials of bird and yellow-backed duiker (Skurnik et al., 2006; Walk et al., 2009; Clermont et al., 2011; Gangiredla et al., 2018). Nonetheless, the absence of class 1 integrons in our clade II isolates was an indication that their habitat (be it wild animal host or external environment) was less disturbed by human activities. Integrons are genetic platforms capable of capturing and expressing gene cassettes that confer adaptive capabilities such as antimicrobial resistance. Class 1 integrons are the most clinically relevant, causing widespread dissemination of multidrug resistance (Cambray et al., 2010). Previously it was demonstrated that the prevalence of class 1 integrons in fecal E. coli was positively correlated with the exposure of their animal hosts to human activities (Skurnik et al., 2006) and that the integrase gene of class 1 integron could be a marker for anthropogenic pollution (Gillings et al., 2015).
Our hypothesis that the natural habitats of clade II were relatively pristine was supported by the AdaptML analysis (Figure 4), which inferred ecologically differentiated groups from phylogeny and ecological data (sample sources, which represented different types of habitats) using a hidden Markov model. Our dataset was heavily enriched with environmental isolates, while fecal isolates were underrepresented. Thus, to overcome the bias and for a more comprehensive representation of the habitats of E. coli and cryptic clades, reference strains from diverse sources were included in the analysis. The expanded dataset included strains representing the typical diversity of E. coli (Ochman and Selander, 1984) and representative genotypes of the cryptic clades. The analysis indicated that the ecological niche of clade II is distinct from typical host-associated E. coli and also cryptic clade I, V, and VI. Instead, most clade II genotypes shared habitats with the primarily environment-associated clades III and IV, and with E. coli that were mainly isolated from less disturbed freshwater habitats. The AdaptML prediction was conservative and inferred ecological differentiation based on our anticipated ecological categories (Hunt et al., 2008; Francisco et al., 2014). Therefore, these phylogenetically distinct groups may be further differentiated for other ecological parameters, as we focused on coarse dimensions such as host and environment types. Moreover, the use of merely seven housekeeping genes may miss out on ecological specializations that were not reflected in these genes. Sensitive and high-resolution approaches based on whole-genome data would further detail the ecological divergence between clade II and environmental E. coli, or even within clade II itself.
As clade II strains were found in wildlife and aquatic habitats, they could resemble clade V that displayed traits of dual lifestyle in both host and external environments (Vignaroli et al., 2015), or represented environment-associated lineages with wildlife as spillover hosts, or consisted of at least two distinct ecotypes (host- vs. environment-associated). While our clade II isolates and the reference clade II strains formed a monophyletic cluster (Figure 1), the cluster’s relatively high genetic divergence and subclustering indicated possible ecological divergence. Moreover, the host-associated reference strains were generally more distantly related to our isolates of environmental origins. However, biogeographic effects cannot be dismissed (Cohan and Perry, 2007; Hunt et al., 2008). While our isolates were from Hong Kong, China, the host-associated clade II strains originated from Australia, Gabon, and United States (Walk et al., 2009; Clermont et al., 2011; Lescat et al., 2013; Walk, 2015; Gangiredla et al., 2018). We previously demonstrated that two groups of clade II genomes displayed signatures of functional divergence that were reflective of ecological differentiation to occupy gastrointestinal and external environments, respectively (Shen et al., 2020). The study included genomes of isolates in this study. Combined, results from our studies suggested that some of the clade II isolates in this study were closely related to the original clade II strains of fecal origins but were ecologically differentiated. Comparative genome sequence analysis will allow in-depth inquiries of the further phylogenetic and ecological differentiation within clade II.
The population structure of clade II was a mixture of clonal and divergent genotypes (Figure 2). Clade II STs were generally more distantly related, even for isolates from the same sample. More isolates need to be analyzed to verify the observed patterns. Although cryptic clades coexist with E. coli in aquatic environments, we observed limited recombination of the housekeeping genes between E. coli and cryptic clades II, IV, and V in this study (Table 6). The large FST value (> 0.5) between clade II vs. different E. coli populations also indicated insignificant gene flow (Supplementary File 5). The sharing of alleles, however, is not absent. For example, the phylogenetically distinct E. coli and clade II shared a common allele at the recA locus (Figure 2). Likewise, comparative genomics showed that core gene recombinations were mainly restricted to within enteric Escherichia (including clade I) or within environment-associated clades III–V. However, clade II was not included in the analysis (Luo et al., 2011).
In this study, all seven MLST loci were under purifying selection, as anticipated for housekeeping genes (Chaudhuri and Henderson, 2012). Nonetheless, for the same gene, we observed different selection strengths between phylogenetically distinct Escherichia (E. coli vs. cryptic clade II) and for populations isolated from environments of different conditions (host-associated vs. environmental; primarily freshwater vs. marine), signifying different evolutionary processes among these compared entities (Table 4). Different populations are likely subjected to different selection pressures specific to particular environmental and ecological conditions (Feil and Spratt, 2001). In general, marine habitats exerted stronger selection on the housekeeping genes of both E. coli and clade II than freshwater habitats (Table 4). At the KLH mudflat, stronger purifying selection was exerted on clade II than E. coli for most of the genes. At this point, whether this is an exemplification of one group being better at coping with the marine environment remained debatable. Similarly, the selection strength exerted on clade II in host vs. external environment was not probed as there were merely three fecal isolates with genetic information. These questions could be better answered when more isolates from different habitats are available and analyzed with approaches that are more sensitive in detecting response to selection.
Thus far, this work revealed previously undescribed genetic diversity of clade II (all STs and most of the alleles at the majority of MLST loci were novel) and uncovered some of their ecological characteristics and population genetics. Similarly, many new STs and alleles were also found within environmental E. coli. The discovery of these new genetic diversities attested to the biodiversity potential of the subtropical aquatic environment. The isolates may represent more genetic diversity than was demonstrated, as the Achtman MLST scheme adopted in this study has the lowest nucleotide diversity among the three E. coli MLST schemes (Kaas et al., 2012). Nonetheless, the Achtman MLST scheme was chosen for its phylogeny based on concatenated sequence data have the best congruency with the “true” phylogeny represented by whole-genome data (Sahl et al., 2012).
For E. coli, we noticed that while the environmental isolates contributed significantly more novel STs and alleles than the fecal counterparts, they displayed lower ST frequency and fewer alleles across all seven loci (Table 3). In environmental habitats, E. coli need to survive conditions that differ substantially from hosts, such as sunlight radiation, nutrient limitation, high salinity (in marine habitat), and predation (Van Elsas et al., 2011). Thus, environmental E. coli population structure depends on both origin and environmental survivability: rapid decay, persistence, or naturalization (Van Elsas et al., 2011; Berthe et al., 2013). Moreover, the cultivation method may miss out on injured or dormant cells that were viable but non-culturable. Consequently, environmental populations typically were less diverse than host-associated populations despite having more novel genotypes (McLellan, 2004; Walk et al., 2007; Perchec-Merien and Lewis, 2013). Our observations above not only indicated a discovery of new diversities that suggested adaptation to specific environments (Perchec-Merien and Lewis, 2013), but also exemplified natural selection favoring genotypes that survive better in the external environment (McLellan, 2004; Walk et al., 2007; Perchec-Merien and Lewis, 2013).
This study observed a largely clonal population structure for E. coli (Figure 2), corroborating the global E. coli phylogeny (Tenaillon et al., 2010; Chaudhuri and Henderson, 2012). Clonality was implied by the presence of extensive linkage disequilibrium despite the significant role of recombination in generating allelic and ST variations at all population levels (Table 5). Strong linkage arose probably due to natural selection imposed by specific environmental conditions (Walk et al., 2007; Perchec-Merien and Lewis, 2013). Clonality was also indicated by isolates sharing identical STs or forming CCs despite diverse geographical and temporal origins (Figure 2). Similar observations were reported elsewhere (Maynard Smith et al., 1993; Wirth et al., 2006; Walk et al., 2007; Perchec-Merien and Lewis, 2013). The lack of geographical and temporal differentiation was further confirmed by little genetic differentiation between different E. coli populations, as reflected by low FST values (Supplementary File 5). All these observations are testaments to the clonality of E. coli. Intra-phylogroup recombination was more frequent while inter-phylogroup recombination was limited (Figure 3 and Table 6). These observations corroborated Walk et al. (2007), albeit utilizing different sets of housekeeping genes. Analysis with single-nucleotide polymorphisms also demonstrated that inter-phylogroup recombination was primarily between A and B1, A and E, and B2 and D, with ecological niche partitioning among plausible explanations (Leopold et al., 2011).
AdaptML analysis iterated the broad ecological niche of E. coli, with its genotypes distributed over all four ecologically distinct groups (Figure 4, Supplementary Figure 4, and Supplementary File 3). While group 1 consisted mainly of typical host-associated E. coli, group 2 appeared to lead a more generalist lifestyle. This group contained roughly equal portions of isolates from both host and environmental sources from various locations, reflecting a possibility that these genotypes not only were adapted to life within hosts but may also persist in the external environments, especially marine environments. Group 4 appeared to harbor characteristics that enabled survival in environments with occasional salinity fluctuation due to seawater intrusion. Finally, group 3 notably harbored many novel E. coli genotypes and grouped with most clade II genotypes. These novel genotypes were mainly from the TK freshwater sites where many clade II were isolated, which remained largely undisturbed by human activities. Thus, this group of E. coli may represent genotypes that were more suited to live in relatively pristine environments. Further research is warranted to confirm the ecological distinctness of these inferred groups and the underlying functional divergence that resulted in such differentiation. It is also investigation-worthy to determine if genotypes of groups 3 and 4 contained naturalized E. coli.
In this Anthropocene epoch, human activities have significantly altered the dynamics of microbes (and their gene repertoire) dissemination (Zhu et al., 2018). The TK watershed is an aquatic continuum with differential human impacts where human activities/land use shaped the population structures of E. coli and the distribution of clade II. The clade II isolates were the main contributor to the moderate genetic difference between populations (containing both E. coli and clade II) from the two differentially impacted areas. Little genetic differentiation was present when clade II isolates were removed, exemplifying the effect they could bring to E. coli population genetics study when present in high frequency and not properly distinguished.
Nonetheless, by looking at both E. coli and clade II, we were able to investigate the partitioning of these two Escherichia in relation to human activities. The low impact area harbored E. coli populations with a significantly higher proportion of novel genotypes than the high impact area. Moreover, clade II isolates were conspicuously abundant in sediment and periphyton of the low impact area (Supplementary Table 1 and Supplementary File 1). The differential composition of E. coli phylogroups between the two areas was not elaborated as the phylogroups for many isolates from the low impact sites (slightly over one-third) were unassigned (Figure 5). Nonetheless, the presence of this many isolates with unassigned phylogroups is a manifestation of the newly discovered diversity of E. coli from these sites, which were genetically more distant to known sequence variants in the public MLST database. All these observations pointed to less human fecal pollution at these sites. E. coli that were present likely originated from wild animal feces, and some could even represent genotypes that are more adapted to external environments.
Shifting downstream to the high human impact area, the abundance of clade II isolates and novel E. coli genotypes reduced substantially, and was contrasted by a significant increase of E. coli belonging to globally successful CCs of public health concern, such as CC10 and CC155. ST155 (primary founder of CC155, the largest CC in our study) has been suggested to be responsible for the spread of extended-spectrum β-lactamase gene from animals to humans, causing widespread resistance to β-lactam antibiotics (Skurnik et al., 2015). The second-largest CC in our dataset, CC10, was associated with multiple E. coli pathotypes, found in a wide range of hosts and environmental matrices, and frequently antibiotic-resistant (Reid et al., 2018). The concentration of these genotypes around the high human impact area suggested the directionality of the flow of these isolates from human activities. As selection was neutral or purifying in the slow-evolving housekeeping genes that encode essential functions (Zheng et al., 2004), detecting genes that respond quickly and positively would better depict how bacteria respond to selection pressures in habitats. Examples are mobile genetic elements such as prophages and integrons, which have been vital for habitat adaptation (Touchon et al., 2020). Class 1 integrons enabled E. coli to better adapt to antibiotic pressure with very low fitness cost and were not stably maintained without selection pressure (Díaz-Mejía et al., 2008; Lacotte et al., 2017). Thus, when these E. coli are introduced into receiving water contaminated with antibiotics, class 1 integrons are retained as the selection pressure calls for its maintenance and expression. Sources of antibiotic contamination include the discharge of antibiotic residues from sewage outfall, antibiotics runoff from animal farming on surrounding lands, and administration of antibiotics in aquaculture. The presence of class I integrons was influenced by human activities as shown in other studies (Gillings et al., 2015; Petit et al., 2017; Uyaguari-Díaz et al., 2018; Touchon et al., 2020). intI1-positive E. coli were more abundant at the high human impact sites that were within the vicinity of the beach. These sites received the highest level of human disturbance: direct body contact (beach activities), sewage effluent discharge, and possible leakage from failing sewage infrastructure or cross-connected sewage and stormwater networks. Meanwhile, as mentioned earlier, clade II isolates, which were mainly found in low impact sites, were all intI1-negative.
In conclusion, this study unearthed the vast genetic diversity of cryptic clades and E. coli embedded in the freshwater and marine habitats in Hong Kong, attesting to the less tapped biodiversity potential of these subtropical environments. Contrary to previous reports, clade II was the most abundant cryptic lineage co-isolated with E. coli, especially in relatively pristine subtropical aquatic environments. Clade II and E. coli showed limited recombination and significant genetic divergence. Our works provided preliminary indications that clade II was ecologically differentiated from typical E. coli. Some members may even represent novel environmental Escherichia clades closely related to the original clade II strains of fecal origins. Many questions remained about the ecological and clinical significance of clade II and their further phylogenetic and ecological differentiation. Nonetheless, discovering the habitats where clade II maintained relatively large population sizes and the availability of genome-based data would pave the ways to answer these questions. E. coli of diverse origins exhibited clonality amidst divergent STs, echoing other studies in that recombination in housekeeping genes was insufficient to disrupt phylogenetic signals of the largely clonal E. coli. Notably, environmental E. coli were less diverse than fecal isolates despite contributing many new alleles and STs. Finally, we demonstrated that human activities influenced E. coli and clade II distribution in a small aquatic continuum. Moving from relatively pristine sites toward areas with higher human disturbance, the abundance of clade II isolates and new E. coli genotypes reduces, while E. coli bearing class I integrons and belonging to CCs of public health concern accumulates. Altogether, this work exemplified that sampling of relatively pristine subtropical environments could lead to the discovery of more E. coli and cryptic clade diversity, which is imperative for understanding the extent of their genetic and functional variations in relation to diverse habitat conditions.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, OM025257–OM030212.
Author Contributions
XK: experiments, bioinformatics, analysis and interpretation, original draft, and review and editing. ZS: bioinformatics, analysis and interpretation, and original draft. CW, YY, HL, and SC: experiments, analysis and interpretation, and review and editing. JK: review and editing, and supervision. SL: review and editing, and supervision and funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the University Grants Committee Theme-based Research Scheme (T21-602/16-R) and the General Research Fund (16301018) of the Hong Kong Research Grants Council.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Samuel C. S. Cheng for his assistance in experimental setups and field sampling coordination.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.811755/full#supplementary-material
Footnotes
References
Alikhan, N. F., Zhou, Z., Sergeant, M. J., and Achtman, M. (2018). A genomic overview of the population structure of Salmonella. PloS Genet. 14:e1007261. doi: 10.1371/journal.pgen.1007261
Bermúdez, M., and Hazen, T. C. (1988). Phenotypic and genotypic comparison of Escherichia coli from pristine tropical waters. Appl. Environ. Microbiol. 54, 979–983. doi: 10.1128/aem.54.4.979-983.1988
Berthe, T., Ratajczak, M., Clermont, O., Denamur, E., and Petit, F. (2013). Evidence for coexistence of distinct Escherichia coli populations in various aquatic environments and their survival in estuary water. Appl. Environ. Microbiol. 79, 4684–4693. doi: 10.1128/AEM.00698-13
Bruen, T. C., Philippe, H., and Bryant, D. (2006). A simple and robust statistical test for detecting the presence of recombination. Genetics 172, 2665–2681. doi: 10.1534/genetics.105.048975
Chaudhuri, R. R., and Henderson, I. R. (2012). The evolution of the Escherichia coli phylogeny. Infect. Genet. Evol. 12, 214–226. doi: 10.1016/j.meegid.2012.01.005
Chun, J., Oren, A., Ventosa, A., Christensen, H., Arahal, D. R., da Costa, M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. doi: 10.1099/ijsem.0.002516
Cianciotto, N. P. (2006). Legionella: State of the Art 30 Years After its Recognition. Washington, DC: ASM Press.
Clermont, O., Dixit, O. V., Vangchhia, B., Condamine, B., Dion, S., Bridier-Nahmias, A., et al. (2019). Characterization and rapid identification of phylogroup G in Escherichia coli, a lineage with high virulence and antibiotic resistance potential. Environ. Microbiol. 21, 3107–3117. doi: 10.1111/1462-2920.14713
Clermont, O., Gordon, D., and Denamur, E. (2015). Guide to the various phylogenetic classification schemes for Escherichia coli and the correspondence among schemes. Microbiology 161, 980–988. doi: 10.1099/mic.0.000063
Clermont, O., Gordon, D. M., Brisse, S., Walk, S. T., and Denamur, E. (2011). Characterization of the cryptic Escherichia lineages: rapid identification and prevalence. Environ. Microbiol. 13, 2468–2477. doi: 10.1111/j.1462-2920.2011.02519.x
Cohan, F. M., and Kopac, S. M. (2011). Microbial genomics: E. coli relatives out of doors and out of body. Curr. Biol. 21, R587–R589. doi: 10.1016/j.cub.2011.06.011
Cohan, F. M., and Perry, E. B. (2007). A systematics for discovering the fundamental units of bacterial diversity. Curr. Biol. 17, R373–R386. doi: 10.1016/j.cub.2007.03.032
Coleman, M. L., and Chisholm, S. W. (2010). Ecosystem-specific selection pressures revealed through comparative population genomics. Proc. Natl. Acad. Sci. U.S.A. 107, 18634–18639. doi: 10.1073/pnas.1009480107
Deng, D., Zhang, N., Mustapha, A., Xu, D., Wuliji, T., Farley, M., et al. (2014). Differentiating enteric Escherichia coli from environmental bacteria through the putative glucosyltransferase gene (ycjM). Water Res. 61, 224–231. doi: 10.1016/j.watres.2014.05.015
Díaz-Mejía, J. J., Amabile-Cuevas, C. F., Rosas, I., and Souza, V. (2008). An analysis of the evolutionary relationships of integron integrases, with emphasis on the prevalence of class 1 integrons in Escherichia coli isolates from clinical and environmental origins. Microbiology 154, 94–102. doi: 10.1099/mic.0.2007/008649-0
Dusek, N., Hewitt, A. J., Schmidt, K. N., and Bergholz, P. W. (2018). Landscape-scale factors affecting the prevalence of Escherichia coli in surface soil include land cover type, edge interactions, and soil pH. Appl. Environ. Microbiol. 84, e02714–e02717. doi: 10.1128/AEM.02714-17
Excoffier, L., Laval, G., and Schneider, S. (2005). Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol. Bioinform. 1:117693430500100003.
Feil, E. J., and Spratt, B. G. (2001). Recombination and the population structures of bacterial pathogens. Annu. Rev. Microbiol. 55, 561–590. doi: 10.1146/annurev.micro.55.1.561
Francisco, A. P., Bugalho, M., Ramirez, M., and Carriço, J. A. (2009). Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinformatics 10:152. doi: 10.1186/1471-2105-10-152
Francisco, J. C., Cohan, F. M., and Krizanc, D. (2014). Accuracy and efficiency of algorithms for the demarcation of bacterial ecotypes from DNA sequence data. Int. J. Bioinform. Res. Appl. 10, 409–425. doi: 10.1504/IJBRA.2014.062992
Fraser, C., Hanage, W. P., and Spratt, B. G. (2007). Recombination and the nature of bacterial speciation. Science 315, 476–480. doi: 10.1126/science.1127573
Gangiredla, J., Mammel, M. K., Barnaba, T. J., Tartera, C., Gebru, S. T., Patel, I. R., et al. (2018). Draft genome sequences of Escherichia albertii, Escherichia fergusonii, and strains belonging to six cryptic lineages of Escherichia spp. Genome Announc. 6, e00271–18. doi: 10.1128/genomeA.00271-18
Gillings, M. R., Gaze, W. H., Pruden, A., Smalla, K., Tiedje, J. M., and Zhu, Y. G. (2015). Using the class 1 integron-integrase gene as a proxy for anthropogenic pollution. ISME J. 9, 1269–1279. doi: 10.1038/ismej.2014.226
Gonzalez-Alba, J. M., Baquero, F., Cantón, R., and Galán, J. C. (2019). Stratified reconstruction of ancestral Escherichia coli diversification. BMC Genomics 20:936. doi: 10.1186/s12864-019-6346-1
Gordon, D. M., and Cowling, A. (2003). The distribution and genetic structure of Escherichia coli in Australian vertebrates: host and geographic effects. Microbiology 149, 3575–3586. doi: 10.1099/mic.0.26486-0
Haubold, B., and Hudson, R. R. (2000). LIAN 3.0: detecting linkage disequilibrium in multilocus data. Bioinformatics 16, 847–849. doi: 10.1093/bioinformatics/16.9.847
Ho, B. S. W., and Tam, T. Y. (1997). Enumeration of E. coli in environmental waters and wastewater using a chromogenic medium. Water Sci. Technol. 35, 409–413.
Hunt, D. E., David, L. A., Gevers, D., Preheim, S. P., Alm, E. J., and Polz, M. F. (2008). Resource partitioning and sympatric differentiation among closely related bacterioplankton. Science 320, 1081–1085. doi: 10.1126/science.1157890
Huson, D. H. (1998). SplitsTree: analysing and visualizing evolutionary data. Bioinformatics 14, 68–73. doi: 10.1093/bioinformatics/14.1.68
Huson, D. H., and Bryant, D. (2005). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Ingle, D. J., Clermont, O., Skurnik, D., Denamur, E., Walk, S. T., and Gordon, D. M. (2011). Biofilm formation, thermal niche and virulence characteristics of Escherichia spp. Appl. Environ. Microbiol. 77, 2695–2700. doi: 10.1128/AEM.02401-10
Ishii, S., and Sadowsky, M. J. (2008). Escherichia coli in the environment: implications for water quality and human health. Microbes Environ. 23, 101–108. doi: 10.1264/jsme2.23.101
Jolley, K. A., Feil, E. J., Chan, M. S., and Maiden, M. C. J. (2001). Sequence type analysis and recombinational tests (START). Bioinformatics 17, 1230–1231.
Kaas, R. S., Friis, C., Ussery, D. W., and Aarestrup, F. M. (2012). Estimating variation within the genes and inferring the phylogeny of 186 sequenced diverse Escherichia coli genomes. BMC Genomics 13:577. doi: 10.1186/1471-2164-13-577
Koeppel, A., Perry, E. B., Sikorski, J., Krizanc, D., Warner, A., Ward, D. M., et al. (2008). Identifying the fundamental units of bacterial diversity: a paradigm shift to incorporate ecology into bacterial systematics. Proc. Natl. Acad. Sci. U.S.A. 105, 2504–2509. doi: 10.1073/pnas.0712205105
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lacotte, Y., Ploy, M. C., and Raherison, S. (2017). Class 1 integrons are low-cost structures in Escherichia coli. ISME J. 11:1535. doi: 10.1038/ismej.2017.38
Leopold, S. R., Sawyer, S. A., Whittam, T. S., and Tarr, P. I. (2011). Obscured phylogeny and possible recombinational dormancy in Escherichia coli. BMC Evol. Biol. 11:183. doi: 10.1186/1471-2148-11-183
Lescat, M., Clermont, O., Woerther, P. L., Glodt, J., Dion, S., Skurnik, D., et al. (2013). Commensal Escherichia coli strains in Guiana reveal a high genetic diversity with host-dependant population structure. Environ. Microbiol. Rep. 5, 49–57. doi: 10.1111/j.1758-2229.2012.00374.x
Letunic, I., and Bork, P. (2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Luo, C., Walk, S. T., Gordon, D. M., Feldgarden, M., Tiedje, J. M., and Konstantinidis, K. T. (2011). Genome sequencing of environmental Escherichia coli expands understanding of the ecology and speciation of the model bacterial species. Proc. Natl. Acad. Sci. U.S.A. 108, 7200–7205. doi: 10.1073/pnas.1015622108
Maiden, M. C., Bygraves, J. A., Feil, E., Morelli, G., Russell, J. E., Urwin, R., et al. (1998). Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U.S.A. 95, 3140–3145. doi: 10.1073/pnas.95.6.3140
Maynard Smith, J., Smith, N. H., O’Rourke, M., and Spratt, B. G. (1993). How clonal are bacteria? Proc. Natl. Acad. Sci. U.S.A. 90, 4384–4388.
McLellan, S. L. (2004). Genetic diversity of Escherichia coli isolated from urban rivers and beach water. Appl. Environ. Microbiol. 70, 4658–4665. doi: 10.1128/AEM.70.8.4658-4665.2004
Nascimento, M., Sousa, A., Ramirez, M., Francisco, A. P., Carriço, J. A., and Vaz, C. (2016). PHYLOViZ 2.0: providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics 33, 128–129. doi: 10.1093/bioinformatics/btw582
Nei, M., and Kumar, S. (2000). Molecular Evolution and Phylogenetics. Oxford: Oxford university press.
Ochman, H., and Selander, R. K. (1984). Standard reference strains of Escherichia coli from natural populations. J. Bacteriol. 157, 690–693.
Perchec-Merien, A. M., and Lewis, G. D. (2013). Naturalized Escherichia coli from New Zealand wetland and stream environments. FEMS Microbiol. Ecol. 83, 494–503.
Petit, F., Clermont, O., Delannoy, S., Servais, P., Gourmelon, M., Fach, P., et al. (2017). Change in the structure of Escherichia coli population and the pattern of virulence genes along a rural aquatic continuum. Front. Microbiol. 8:609. doi: 10.3389/fmicb.2017.00609
Quero, G. M., Fasolato, L., Vignaroli, C., and Luna, G. M. (2015). Understanding the association of Escherichia coli with diverse macroalgae in the lagoon of Venice. Sci. Rep. 5:10969. doi: 10.1038/srep10969
Ratajczak, M., Laroche, E., Berthe, T., Clermont, O., Pawlak, B., Denamur, E., et al. (2010). Influence of hydrological conditions on the Escherichia coli population structure in the water of a creek on a rural watershed. BMC Microbiol. 10:222. doi: 10.1186/1471-2180-10-222
Reid, C. J., DeMaere, M. Z., and Djordjevic, S. P. (2018). Australian porcine clonal complex 10 (CC10) Escherichia coli belong to multiple sublineages of a highly diverse global CC10 phylogeny. Microb. Genom. 5:e000225. doi: 10.1099/mgen.0.000225
Reid, S. D., Herbelin, C. J., Bumbaugh, A. C., Selander, R. K., and Whittam, T. S. (2000). Parallel evolution of virulence in pathogenic Escherichia coli. Nature 406:64.
Riley, L. W. (2014). Pandemic lineages of extraintestinal pathogenic Escherichia coli. Clin. Microbiol. Infect. 20, 380–390. doi: 10.1111/1469-0691.12646
Rochelle-Newall, E., Nguyen, T. M. H., Le, T. P. Q., Sengtaheuanghoung, O., and Ribolzi, O. (2015). A short review of fecal indicator bacteria in tropical aquatic ecosystems: knowledge gaps and future directions. Front. Microbiol. 6:308. doi: 10.3389/fmicb.2015.00308
Sahl, J. W., Matalka, M. N., and Rasko, D. A. (2012). Phylomark, a tool to identify conserved phylogenetic markers from whole-genome alignments. Appl. Environ. Microbiol. 78, 4884–4892. doi: 10.1128/AEM.00929-12
Schloter, M., Lebuhn, M., Heulin, T., and Hartmann, A. (2000). Ecology and evolution of bacterial microdiversity. FEMS Microbiol. Rev. 24, 647–660. doi: 10.1111/j.1574-6976.2000.tb00564.x
Shen, Z. Y., Koh, X. P., Yu, Y. P., and Lau, S. C. (2020). Genetic variation and preliminary indications of divergent niche adaptation in cryptic clade II of Escherichia. Microorganisms 8:1713. doi: 10.3390/microorganisms8111713
Skurnik, D., Clermont, O., Guillard, T., Launay, A., Danilchanka, O., Pons, S., et al. (2015). Emergence of antimicrobial-resistant Escherichia coli of animal origin spreading in humans. Mol. Biol. Evol. 33, 898–914. doi: 10.1093/molbev/msv280
Skurnik, D., Ruimy, R., Andremont, A., Amorin, C., Rouquet, P., Picard, B., et al. (2006). Effect of human vicinity on antimicrobial resistance and integrons in animal faecal Escherichia coli. J. Antimicrob. Chemother. 57, 1215–1219. doi: 10.1093/jac/dkl122
Spratt, B. G. (1999). Multilocus sequence typing: molecular typing of bacterial pathogens in an era of rapid DNA sequencing and the internet. Curr. Opin. Microbiol. 2, 312–316. doi: 10.1016/S1369-5274(99)80054-X
Stoesser, N., Sheppard, A. E., Pankhurst, L., De Maio, N., Moore, C. E., Sebra, R., et al. (2016). Evolutionary history of the global emergence of the Escherichia coli epidemic clone ST131. mBio 7:e02162. doi: 10.1128/mBio.02162-15
Tenaillon, O., Skurnik, D., Picard, B., and Denamur, E. (2010). The population genetics of commensal Escherichia coli. Nat. Rev. Microbiol. 8:207.
Thoe, W., Lee, O. H., Leung, K. F., Lee, T., Ashbolt, N. J., Yang, R. R., et al. (2018). Twenty five years of beach monitoring in Hong Kong: a re-examination of the beach water quality classification scheme from a comparative and global perspective. Mar. Pollut. Bull. 131, 793–803. doi: 10.1016/j.marpolbul.2018.05.002
Touchon, M., Perrin, A., De Sousa, J. A. M., Vangchhia, B., Burn, S., O’Brien, C. L., et al. (2020). Phylogenetic background and habitat drive the genetic diversification of Escherichia coli. PLoS Genet. 16:e1008866. doi: 10.1371/journal.pgen.1008866
Uyaguari-Díaz, M. I., Croxen, M. A., Luo, Z., Cronin, K. I., Chan, M., Baticados, W. N., et al. (2018). Human activity determines the presence of integron-associated and antibiotic resistance genes in Southwestern British Columbia. Front. Microbiol. 9:852. doi: 10.3389/fmicb.2018.00852
Van Elsas, J. D., Semenov, A. V., Costa, R., and Trevors, J. T. (2011). Survival of Escherichia coli in the environment: fundamental and public health aspects. ISME J. 5:173. doi: 10.1038/ismej.2010.80
Vignaroli, C., Di Sante, L., Magi, G., Luna, G. M., Di Cesare, A., Pasquaroli, S., et al. (2015). Adhesion of marine cryptic Escherichia isolates to human intestinal epithelial cells. ISME J. 9:508. doi: 10.1038/ismej.2014.164
Walk, S. T., Alm, E. W., Calhoun, L. M., Mladonicky, J. M., and Whittam, T. S. (2007). Genetic diversity and population structure of Escherichia coli isolated from freshwater beaches. Environ. Microbiol. 9, 2274–2288. doi: 10.1111/j.1462-2920.2007.01341.x
Walk, S. T., Alm, E. W., Gordon, D. M., Ram, J. L., Toranzos, G. A., Tiedje, J. M., et al. (2009). Cryptic lineages of the genus Escherichia. Appl. Environ. Microbiol. 75, 6534–6544.
Waters, N. R., Abram, F., Brennan, F., Holmes, A., and Pritchard, L. (2020). Easy phylotyping of Escherichia coli via the EzClermont web app and command-line tool. Access Microbiol. 2:acmi000143. doi: 10.1099/acmi.0.000143
Wirth, T., Falush, D., Lan, R., Colles, F., Mensa, P., Wieler, L. H., et al. (2006). Sex and virulence in Escherichia coli: an evolutionary perspective. Mol. Microbiol. 60, 1136–1151.
Yang, H. H., Vinopal, R. T., Grasso, D., and Smets, B. F. (2004). High diversity among environmental Escherichia coli isolates from a bovine feedlot. Appl. Environ. Microbiol. 70, 1528–1536. doi: 10.1128/AEM.70.3.1528-1536.2004
Zheng, Y., Roberts, R. J., and Kasif, S. (2004). Segmentally variable genes: a new perspective on adaptation. PLoS Biol. 2:e81. doi: 10.1371/journal.pbio.0020081
Keywords: Escherichia, genetic diversity, subtropical, aquatic environment, cryptic clades, environmental E. coli, ecological differentiation, human impact
Citation: Koh XP, Shen Z, Woo CF, Yu Y, Lun HI, Cheung SW, Kwan JKC and Lau SCK (2022) Genetic and Ecological Diversity of Escherichia coli and Cryptic Escherichia Clades in Subtropical Aquatic Environments. Front. Microbiol. 13:811755. doi: 10.3389/fmicb.2022.811755
Received: 09 November 2021; Accepted: 12 January 2022;
Published: 17 February 2022.
Edited by:
Joshua A. Steele, Southern California Coastal Water Research Project, United StatesReviewed by:
Satoshi Ishii, University of Minnesota Twin Cities, United StatesVolker Siegfried Brozel, South Dakota State University, United States
Copyright © 2022 Koh, Shen, Woo, Yu, Lun, Cheung, Kwan and Lau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stanley Chun Kwan Lau, c2NrbGF1QHVzdC5oaw==