 Lu Zhao
Lu Zhao Wen-Qiang Wang
Wen-Qiang Wang Sheng-Quan Xu
Sheng-Quan Xu De-Long Guan
De-Long Guan- 1College of Life Science, Shaanxi Normal University, Xi’an, China
- 2College of Life Sciences, Yan’an University, Yan’an, China
Gut bacteria exert effects on the health and fitness of their insect hosts. Grasshoppers are an important part of the grassland ecosystem and provide important ecosystem services. As the most valuable feature in grassland ecosystem, the compositions and potential influences of gut bacterial in herbivorous grasshoppers in the same ecological environment are essential but undetermined. To facilitate such studies, we collected nine species of grasshoppers (n = 110) from a rebuild grassland on the Loess Plateau in northern Shaanxi, China, which is a representative area of ecosystem restoration model. We characterized the composition and function of the gut bacteria. We found that 326 OTUs were exhibited in all grasshoppers in which Enterobacter, Pantoea, Bacillus, and Spiroplsma are dominant. Among them, 18 OTUs were shared across all nine species of grasshoppers. The predicted function showed that the majority function of those OTUs were involved in survival dependent processes including membrane transport, carbohydrate metabolism, amino acid metabolism, and DNA replication and repair. The composition of gut bacteria is specific to each grasshopper species, and the bacteria community is most various in Trilophidia annulata. These results highlight the gut bacterial community diversity in different grasshopper species. Our findings are necessary for better understanding the relationships between this important herbivorous insect and their microbiomes and have the potential contribution of evaluating the revegetation and ecosystem management in this area.
Introduction
Recently, the increasing interest in the research of insect microbiome has helped to reveal new insights into insect ecology (Muratore et al., 2020a). As an important part of the insect microbiome, gut bacteria played an essential role in the life course of their insect hosts, including nutrient absorption (Muratore et al., 2020b), behavioral changes (Dillon et al., 2000), improved reproduction (Kaltenpoth et al., 2009), and protection from environmental pathogens (Paniagua Voirol et al., 2018). In agricultural production, the interaction of gut bacteria with herbivores and pollinators can affect the yield of crops (Ray et al., 2007; Fierer et al., 2009). Gut bacterial communities and their insects are regarded as biogeochemical cycling mediators in the ecological environment (Ricci et al., 2012). Insect gut bacteria, for example, can regulate the decomposition of plant biomass, carbon cycling, and nitrogen fixation rates (Rajagopal, 2009).
Numerous studies of gut bacteria of insects have focused on several major insect orders, including Coleoptera (Scott et al., 2008; Kim et al., 2017), Orthoptera (Rogers et al., 2003; Dillon et al., 2005), Hymenoptera (Russell et al., 2009; Koch et al., 2013; Saraiva et al., 2015), Diptera (Behar et al., 2005; Dong et al., 2009; Wang et al., 2013), Lepidoptera (Ruokolainen et al., 2016), and Blattaria (Hongoh, 2010; Brune, 2014). Most of these studies mainly focused on the functional analysis of single species of gut bacteria of insects or compared the community structure and function of gut bacteria among different insect species. For instance, Shi et al. (2013) compared the composition and functionality of the gut bacteria in grasshopper, cutworm, and termite. Warnecke et al. (2007) had found that the gut bacteria of termite could assist the host in degrading lignocellulose, so as to make up for the deficiency of the termite. Engel and Moran (2013) found that the Gilliamella of the bees’ gut bacteria were able to encode pectin degrading enzymes that help the bees in breaking down the pollen wall. Sharon et al. (2010) found a close relationship between sexual selection and gut bacteria of Drosophila.
Grasshoppers belong to Orthoptera and are the main herbivores in the global grassland ecosystem (Muratore et al., 2020a). Due to their consumption of large amounts of plant biomass, grasshoppers are the most important primary consumers and play an important role in maintaining ecological balance (Aiello, 2005; Sun et al., 2015). Grasshoppers are also considered a key indicator of the stability of grassland ecosystems, especially in threatened habitats such as grasslands (Báldi and Kisbenedek, 1997; Muratore et al., 2020a). However, the exploration of the resident gut bacterial community of grasshopper is still limited. This study provided the characterization of the gut bacterial communities of grasshopper with similar feeding habits in the same specific ecological environment, which could help us to better understand the role that gut bacteria played on insect hosts.
In order to provide a broader environmental background, we chose the different species of herbivorous grasshoppers living in the Loess Plateau. The Loess Plateau (35.3′–39.17′N, 107.20′–111.04′E) of North Shaanxi Province in China is an area of approximately 92,521.4 km2, which is one of the most serious areas of ecological deterioration and soil erosion in the world (Fu et al., 2000). This region has a semi-arid continental climate. About 3 or 4 decades ago, due to deforestation, the forest had virtually disappeared. Therefore, the Chinese government implemented the policy of grain for green project in the 1990s to increase the rate of vegetation coverage (Shao et al., 2021). Until now, the existing plants are mainly composed of grasslands and shrubs (Qiu et al., 2001). Common plants in this area, including Stipa capillata, Artemisia giraldii, Bothriochloa ischaemum, and Artemisia gmelinii, provide a suitable living environment for grasshoppers (Fu et al., 2004; Gong et al., 2006). Common grasshoppers in this area include the genera of Oedaleus, Calliptamus, Atractomorpha, Epacromius, Trilophidia, and Oxya (Otte, 2005). These grasshoppers primarily feed on plants in the families of Gramineae and Asteraceae.
Nine species of grasshoppers in North Loess Plateau were collected in this study, including Heteropternis robusta Bey-Bienko, 1951; Acrida oxycephala Pallas, 1771; Epacromius coerulipes Ivanov, 1888; Eremippus mongolicus Ramme, 1952; Trilophidia annulata Thunberg, 1815; Oedaleus infernalis Saussure, 1884; Atractomorpha sinensis Bolívar, 1905; Aiolopus tamulus Fabricius, 1798; and Calliptamus abbreviatus Ikonnikov, 1913. We sequenced the 16S rRNA V3–V4 region of gut bacteria from nine grasshopper species to investigate (1) the composition of bacterial communities in different grasshoppers; (2) these gut bacteria function; (3) the comparison of gut bacteria communities across nine grasshopper species in the same area.
Materials and Methods
Sample Collection
A total of 110 adult (the fifth and sixth instar) individuals of grasshoppers were collected from Yuyang County and Yulin City of Shaanxi Province (Longitude: 109.73′ E, latitude: 38.35′ N). They are from different species: Heteropternis robusta (10 individuals), Acrida oxycephala (10 individuals), Epacromius coerulipes (10 individuals), Eremippus mongolicus (10 individuals), Trilophidia annulata (10 individuals), Oedaleus infernalis (30 individuals), Atractomorpha sinensis (10 individuals), and Calliptamus abbreviatus (10 individuals).
Isolation of the Guts From the Grasshoppers
We divided all the grasshoppers into three groups (Group A, Group B, and Group C). Group A includes five species belonging to different subfamilies, which are A. sinensis (Pyrgomorphinae), C. abbreviatus (Calliptaminae), E. mongolicus (Gomphocerinae), A. oxycephala (Acridinae), and O. infernalis (Oedipodinae). The five species of group B are collected from the E. coerulipes, A. tamulus, H. robusta, O. infernalis, and T. annulate, and they belong to Oedipodinae. Group C includes three samples collected from the fore-gut and mid-gut of O. infernalis (Supplementary Table 1). Dissection was performed on each grasshopper by removing the entire gut before extracting their bacteria. All samples were firstly quick-frozen in liquid nitrogen for 2–3 min, and then they were disinfected with 70% alcohol for 1 min. The gut of grasshoppers was extracted by sterile scissors. All the anatomical procedures were performed on clean bench library preparation.
DNA Extraction and PCR Amplification of 16S rRNA
DNA was extracted from grasshoppers using the E.Z.N.A.® Soil DNA Kit (Omega Bio-Tek, Norcross, GA, United States) according to the manufactures’ manual. The V3–V4 region of the bacteria 16S ribosomal RNA gene was amplified by PCR using primers 341F (5′-barcode- CCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′), where barcode is an eight-base sequence unique to each sample. PCR reactions were performed in a triplicate 20-μl mixture containing 4 μl of 5 × FastPfu Buffer, 2 μl of 2.5 mM dNTPs, 0.8 μl of each primer (5 μM), 0.4 μl of FastPfu Polymerase, and 10 ng of template DNA at 95°C for 2 min, followed by 25 cycles at 95°C for 30 s, 55°C for 30 s, 72°C for 30 s and a final extension at 72°C for 5 min. The PCR amplicon products were extracted from 2% agarose gels and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, United States) according to the manufacturer’s instructions and quantified using QuantiFluor™ -ST (Promega, Madison, WI, United States).
16S rRNA Sequencing
Purified PCR products were quantified by Qubit®3.0 (Life Invitrogen) and every 24 amplicons whose barcodes were different were mixed equally. The mixed DNA product was used to construct Illumina Pair-End library following Illumina’s genomic DNA library preparation procedure. Then, the amplicon library was paired-end sequenced (2 × 250 bp) on an Illumina HiSeq 2000 platform (McHardy et al., 2013) according to the standard protocols.
Overall, for each species, over 30 Mb raw data were retrieved and saved in two separated files which contain the forward and reversed reads. These raw data were initially conducted by the Trimmomatic software (Bolger et al., 2014) to perform the quality check. In this step, low-quality reads including the adaptor contaminated reads, along with the reads that have quality score Q20 less than 20, or length less than 50 bp, were all removed and those retained were restored as clean reads.
Data Analysis
To perform the statistical analysis of these clean reads, we used the QIIME software package environment (version 2.01) (Bolyen et al., 2019). First of all, the forward and reverse reads were imported and merged using the Join_paired_end.py script, then the reads were demultiplexed by different types of barcodes using the split_libraries_fastq.py script. The generated demultiplexed reads were saved in one single fasta format file named seq.fna. This file was compared with the downloaded core_set_aligned.fasta.inputed file1 to identify and remove the chimeric reads. After these steps, we obtained the organized sequences, which are suitable for OTU classification. We use the UPARSE pipeline (version 7.1)2 (Edgar, 2013) to classify all these sequences into a OTU table with the threshold of 97%. The OTUs were picked by both the opened and the closed methods using the SILVA (SSU126) 16S rRNA database and Greengenes 13.8 (DeSantis et al., 2006) as reference. The open-picked and closed-picked OTUs were used for different analysis purposes. The open-picked OTUs comprise new bacteria information that lack annotation, and they are more sufficient in generating more accurate OTUs which are better for further alpha and beta analysis and phylogenetic affiliation of each 16S rRNA genes. The RDP Classifier3 software (Cole et al., 2005) is used to conduct this analysis and the confidence threshold is 70%. The closed-picked OTUs only contain those with KEGG annotation, which were later used in PICRUSt analysis.
Functional Predictions
PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) (Douglas et al., 2018) was used to predict the function of bacterial communities based on 16S rRNA sequencing data. For this analysis, the OTUs were obtained from the Greengenes 13.8 (DeSantis et al., 2006) reference database. The mapped OTUs were further characterized and annotated using the KEGG database.
Results
Sequencing and Diversity of Gut Bacteria Community
On sequencing of the 16S rRNA V3–V4 region of nine species of grasshoppers, respectively, a total of 461,372 high-quality sequences were obtained after basic processes and a series of quality filtering. The average length of reads was 427 bp. These high-quality sequences which were clustered into OTUs were at 97% similarity threshold. The taxonomic classification of OTUs was performed using RDP Classifier (see text footnote 3). A total of 326 OTUs were identified from all gut bacteria of grasshoppers (Supplementary Tables 2, 3).
Rarefaction curve can be used to determine whether the current sequencing depth of each sample is sufficient to reflect the microbial diversity contained in the community sample. In this study, the Shannon index rarefaction curves in all individuals reached the plateau (Supplementary Figure 1), indicating that most of the bacterial diversity in all grasshoppers was captured in the current sequencing. Other undiscovered rare species will not affect our conclusion based on diversity-index.
The Composition of the Gut Bacteria Community
According to the results of species annotation and statistical analysis, nearly all reads were assignable to 20 phyla, 210 genera. The distribution of dominant phyla and genera in nine species of grasshoppers was as follows: except for A. tamulus and T. annulata, the main phyla of the other seven species of grasshoppers were Proteobacteria. At the genus level, the most dominant genus in A. oxycephala, C. abbreviate, E. mongolicus, H. robusta, and O. infernalis was Enterobacter, and the proportion was 31.50, 30.01, 64.16, 60.58, and 76.25%, respectively. The dominant genus in A. sinensis and E. coerulipes was Pantoea; the proportion was 45.64 and 72.18%, respectively. The dominant genus of T. annulata and A. tamulus was Bacillus; the proportion was 28.22 and 30.97%, respectively. More than 1% of the other genera were Halomonas, Enterococcus, and Lactococcus (Figure 1A).
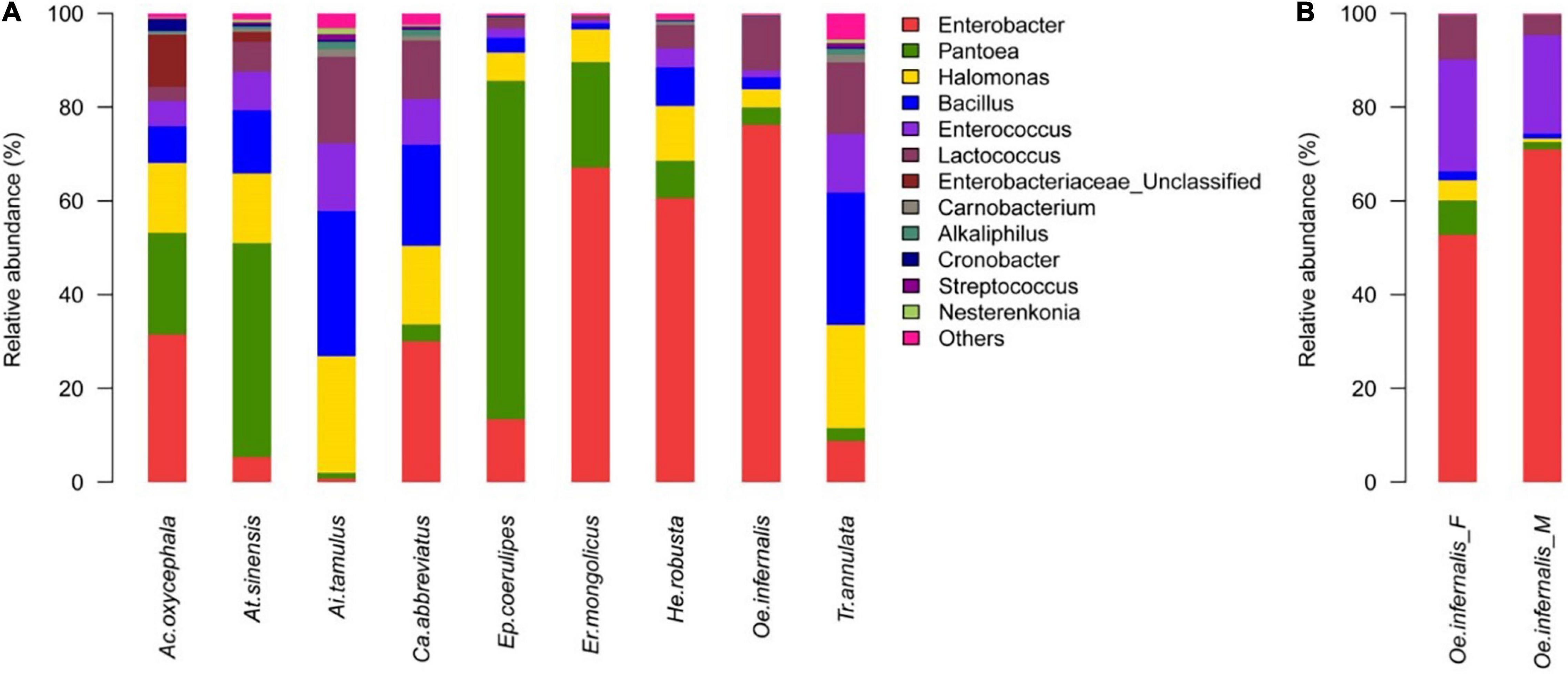
Figure 1. Genus-Level Gut Bacteria Composition of Grasshoppers. (A) Relative contribution of the gut bacteria in Ac. oxycephala, At. sinensis, Ai. tamulus, Ca. abbreviatus, Ep. coerulipes, Er. mongolicus, He. robusta, Oe. infernalis, and Tr. annulata. (B) Relative contribution of the gut microbiota in fore-gut and mid-gut of Oe. infernalis.
Moreover, we found that the distribution of gut bacteria in for-gut and mid-gut of O. infernalis were different. The dominant genus was Enterobacter: the proportion was 52.78 and 71.01%, respectively (Figure 1B). This could be related to the function of different intestinal segments in host insects. The for-gut absorbs and stores food, while the mid-gut secretes digestive enzymes to digest food and absorb nutrients. The gut bacteria of insects interact with its host to perform these functions.
The Comparative Analysis of Gut Bacteria Communities Among the Nine Grasshopper Species
We divided all the grasshoppers into three groups: Group A, Group B, and Group C (see Methods section “Isolation of the Guts From the Grasshoppers”). Then, we described the common and unique gut bacteria of nine grasshopper species in groups A, B, and C. A total of 140 OTUs were identified from group A, of which 24 OTUs were shared by them and the highest percentage of shared OTUs belonged to Bacillus. The number of unique OTUs in C. abbreviatus, A. sinensis, A. oxycephala, O. infernalis, and E. mongolicus was 50, 12, 2, 1, and 1, respectively (Figure 2A). A total of 300 OTUs were identified from group B, of which 26 OTUs were shared by them and the highest percentage of shared OTUs belonged to Bacillus and Enterobacter. The 136, 36, 12, 4, and 1 OTUs were uniquely identified with T. annulata, A. tamulus, H. robusta, E. coerulipes, and O. infernalis. The number of unique OTUs of T. annulata was significantly higher than that of other grasshopper species (Figure 2B). A total of 64 OTUs were identified from group C, of which 26 OTUs were shared by them and the highest percentage of shared OTUs belonged to Bacillus and Enterobacter. The 19 and 1 OTUs were uniquely identified of fore-gut and mid-gut in O. infernalis (Figure 2C). A total of 18 OTUs were shared across all grasshopper species, and the highest percentage of shared OTUs was Bacillus, Enterobacter and Enterococcus, respectively (Supplementary Table 4).
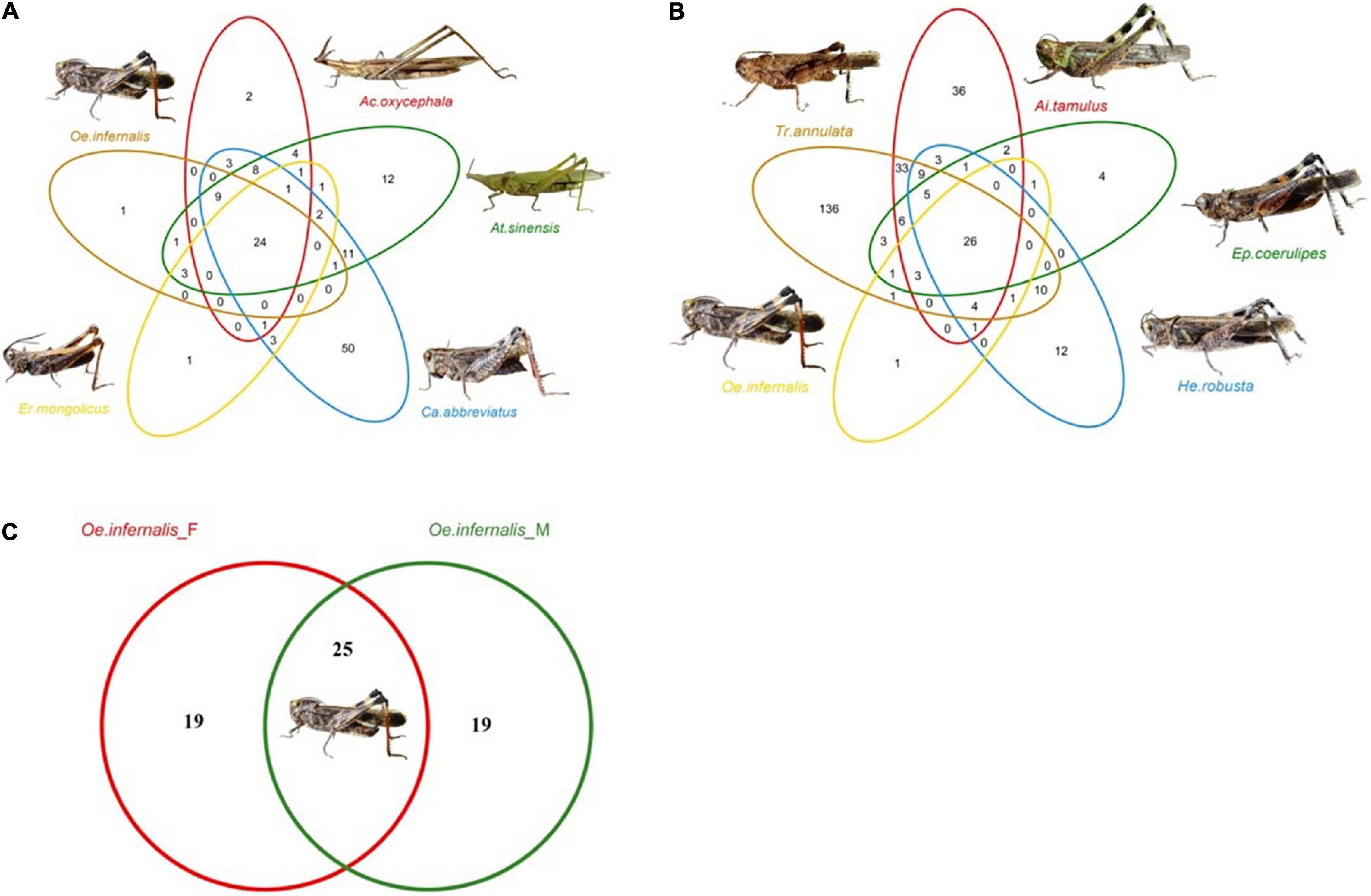
Figure 2. Venn diagrams show the common and unique OTUs of three groups. (A) The gut bacteria of five families, namely At. sinensis (Pyrgomorphinae), Ca. abbreviatus (Calliptaminae), Er. mongolicus (Gomphocerinae), Ac. oxycephala (Acridinae) and Oe. infernalis (Oedipodinae). (B) The gut bacteria of Ep. coerulipes, Ai. tamulus, He. robusta, Oe. infernalis, and Tr. annulata and they belonged to Oedipodinae. (C) The gut bacteria of fore-gut and mid-gut of Oe. infernalis.
We calculated the Shannon, Simpson, and the Chao index of different grasshoppers to analyze the alpha diversity among the samples. The results showed that the alpha diversity of T. annulata was the highest, and the alpha diversity of E. coerulipes was the lowest (Table 1). Beta diversity of bacterial communities was assessed using non-metric multidimensional scaling (NMDS). NMDS ordination plots revealed clear differences in the community composition at the species level (Supplementary Figure 2). A heat map of representative genus indicated that all the grasshoppers were clustered into two categories according to the similarity of the gut bacteria composition of grasshoppers. The first category includes C. abbreviatus, A. tamulus, and T. annulata. The second category includes O. infernalis, E. mongolicus, H. robusta, A. oxycephala, A. sinensis, and E. coerulipes. The grasshoppers of the same family did not come together, indicating the differences between the gut bacterial communities of grasshoppers from the same family (Figure 3).
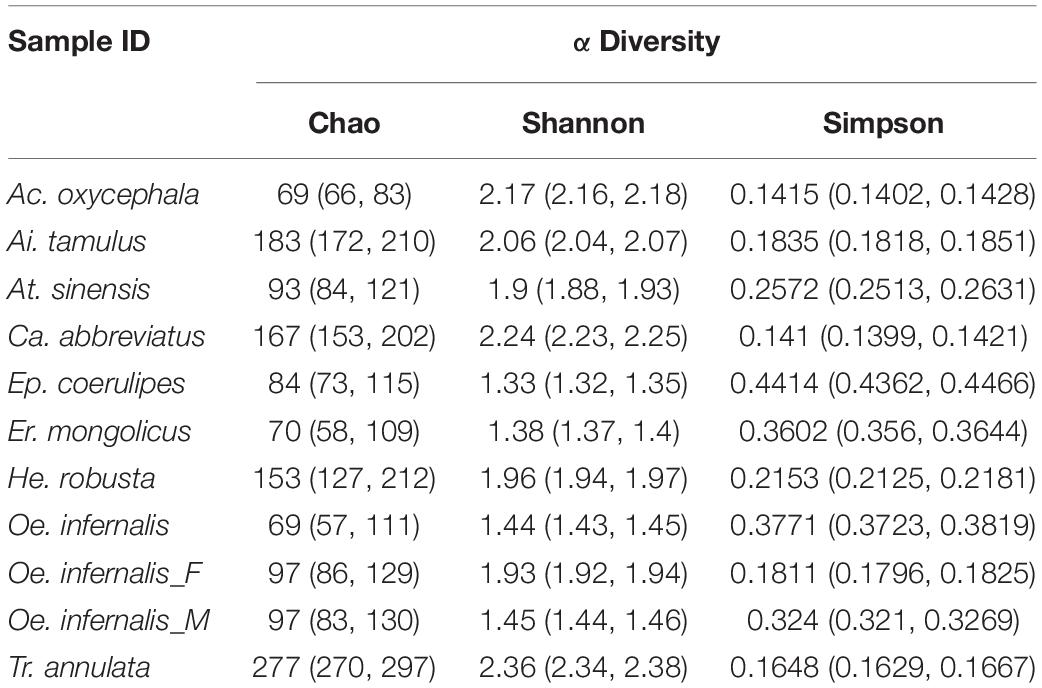
Table 1. Diversity Index of Gut Bacteria in Each Grasshopper.
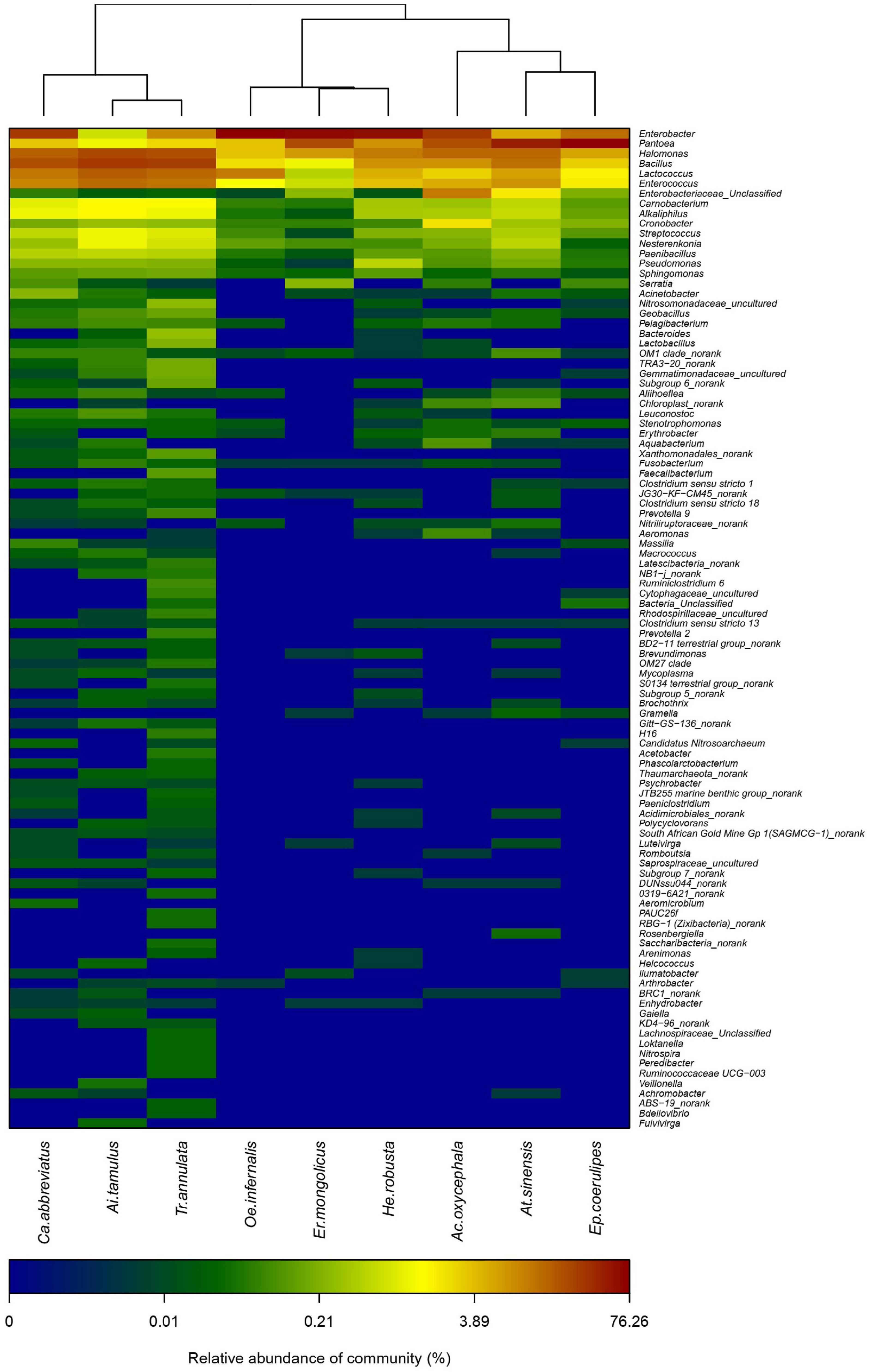
Figure 3. Heatmap showing the relative abundance of OTUs and clustering across nine species.
Prediction and Differences in the Gut Bacteria Pathways
Based on 16S rRNA sequence and KEGG database, PICRUSt was conducted to predict the metabolic pathways involved in the gut bacteria of grasshoppers. Using this method, 41 KEGG pathways were found involved by all gut bacteria in the nine grasshopper species (Figure 4A). The 10 dominant KEGG pathways found in all gut bacteria and the common 18 OTUs in nine species of grasshoppers were identical, the majority of which belonged to membrane transport, carbohydrate metabolism, amino acid metabolism, replication and repair, poor characterization, cellular processes and signal, energy metabolism, translation, metabolism of cofactors and vitamins, and cell motility (Figures 4B,C). In addition, T. annulata had a higher proportion of three KEGG pathways (membrane transport, carbohydrate metabolism, and amino acid metabolism) than other grasshoppers.
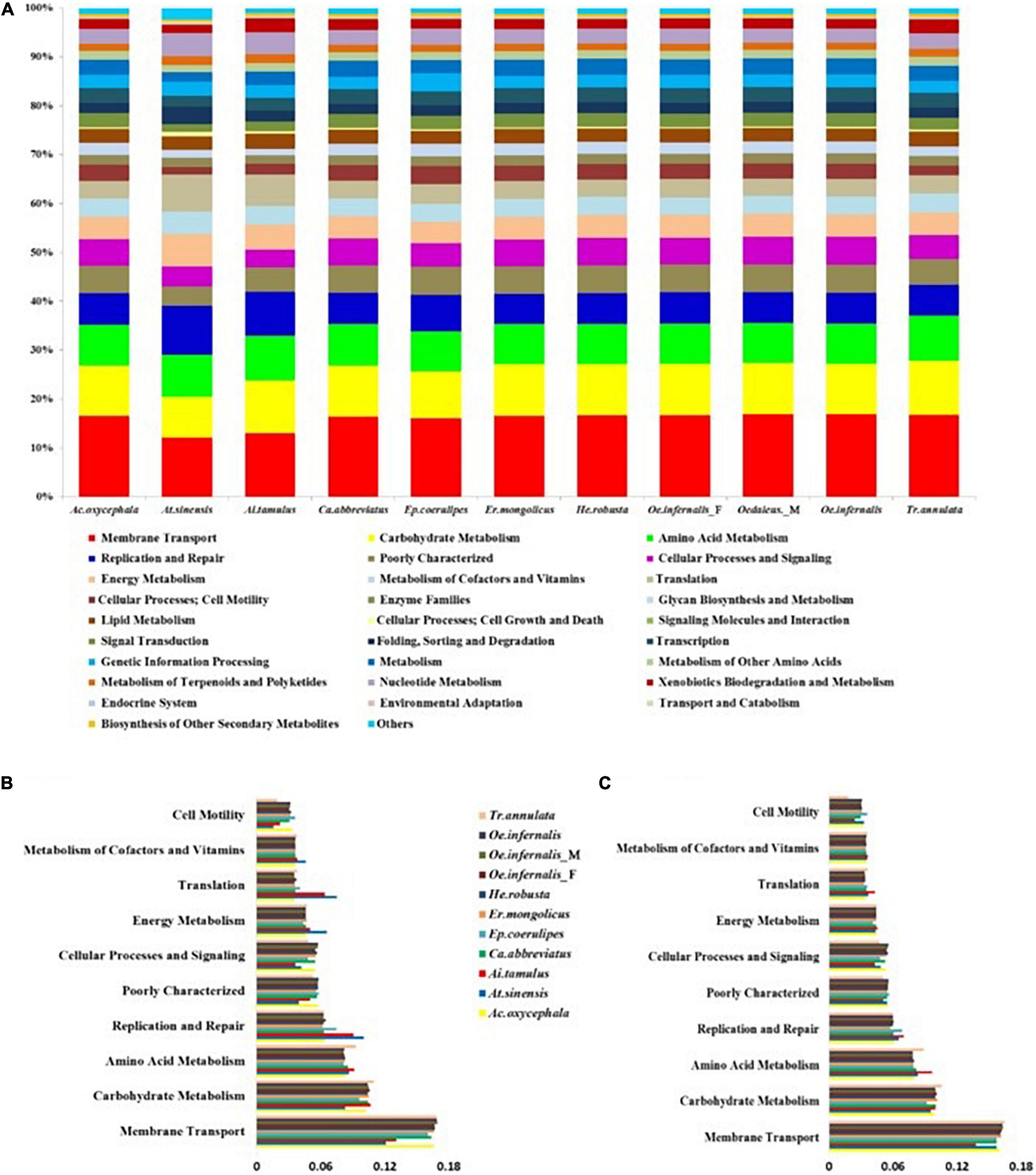
Figure 4. The predicted functions of gut bacteria in grasshoppers using PICRUSt. (A) Functional prediction of gut bacteria in all grasshoppers. (B) Histogram showing the 10 dominant KEGG pathways of the common 18 OTUs. (C) Histogram showing the 10 dominant KEGG pathways of gut bacteria in all grasshoppers.
Discussion
Grasshoppers, one of the most important primary consumers, have enormous ecological and economic value. Herbivorous grasshoppers rely on their gut bacteria to extract nutrients from indigestible plant tissue (Muratore et al., 2020a). However, when compared to the study of agricultural pest prevention and control, there are few studies on gut bacteria in grasshoppers. In the current study, we investigated the composition and function of the gut bacterial communities of nine grasshopper species in the Loess Plateau in northern Shaanxi, China. Firmicutes and Proteobacteria dominated the gut bacteria of nine grasshopper species. At the genus level, the detailed composition of these grasshoppers varied greatly. The highest proportion of the genera in Firmicutes was Bacillus, Enterococcus, and Lactococcus, respectively, and the highest proportion of the genera in Proteobacteria was Enterobacter, Pantoea, Halomonas, respectively. Several other herbivores have been found to have a similar composition of gut bacteria. For example, Enterobacter, Bacillus, and Pantoea are also found in the herbivorous Hooded crane (Zhao et al., 2017). The predominance of the gut bacteria of the herbivorous Spodoptera also includes Enterococcus and Pantoea (Chen et al., 2016). Enterobacter was also found in the silkworm and other herbivorous insects (Liang et al., 2015). Enterobacter has the ability to produce 1, 4-β-xylosidase, which is involved in the degradation of the plant cell wall (Suen et al., 2010). Bacillus can produce the endoglycanase enzyme required for the degradation of cellulose, which has the effect of degrading cellulose (Wakarchuk et al., 1994). Lactococcus can secrete expressive proteins (antigens or antibodies) that can interact directly with the substrate or the immune system of the intestinal mucosa (Hanniffy et al., 2007).
The functional contribution of the insect’s gut bacteria to its behavior and survival is indisputable (Dillon et al., 2000). Among grasshoppers, the highest proportion of the common 18 OTUs was Bacillus, Enterobacter, and Enterococcus. We used the PICRUSt to infer the function of all gut bacteria and the common 18 OTUs of nine grasshopper species based on 16S rRNA sequencing. It was found that the dominant functions of all gut bacteria were the same as the common 18 OTUs of all individuals, and these functions are membrane transport, carbohydrate metabolism, amino acid metabolism, etc., which are closely related to food transportation, digestion, and nutrient absorption. The results revealed that the common 18 OTUs of gut bacteria in nine grasshopper species are the basic components of gut bacteria in grasshoppers. However, due to the limitation of PICRUSt, the result of functional predictions should be cautious (Corby-Harris et al., 2014). For example, PICRUSt could only predict the OTUs that were matched with the available database and could not predict novel and unstudied OTUs (Langille et al., 2013). Many practical functions of gut bacteria of grasshopper are still worthy of being discovered and studied.
We found that although these grasshoppers have same ecological environments (such as climate, temperature, etc.); the composition of gut bacterial communities varies greatly across species. This indicates that the host species was an important factor for the gut bacteria community structure. This is consistent with previous results (Yang et al., 2016). The diversity of gut bacteria communities in T. annulata is greater than in the other eight species, as is the number of OUTs in T. annulata. T. annulata is a widely distributed insect in Asia, from east of Japan, west to Pakistan, and India (Bai et al., 2016; Guan and Xu, 2016). T. annulate has a more varied diet than other grasshopper species, so it may need more abundant gut bacteria to adapt the diversification of environment. A better understanding of these important host–microbiome interactions will provide insights into both host ecology and bacterial functions, potentially leading to better conservation of these species and their habitats (Muratore et al., 2020a).
Conclusion
Here we demonstrated the composition and function of the gut bacterial communities of nine grasshopper species in the Loess Plateau in northern Shaanxi, China. The dominant bacteria genera of all the samples are Enterobacter, Pantoea, and Halomonas. The differences in the composition of gut bacterial communities of the nine grasshopper species may be affected by the host species and the dietary diversity. Despite the fact that our sample was not representative of age, sex, or physiological status, all of them are factors affecting the gut bacteria composition of insects. Our study provides a meaningful reference for the future study of gut bacteria in grasshoppers.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
LZ, W-QW, D-LG, and S-QX conceived the study and designed the experiments. LZ and D-LG performed the experiments, analyzed the data, and wrote the manuscript. D-LG, S-QX, and W-QW revised the manuscript. S-QX and D-LG are the co-corresponding authors. All authors read and approved the final manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China (Nos. 31760101 and 31872273) and the Fundamental Research Funds for the Central Universities (Nos. GK201903063, GK201902010, GK201702010, and GK201701006).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Sheng-jiang Tan from Cambridge Inst Med Res, Keith Peters Bldg, Hills Rd, Cambridge CB2 0XY, England, for conception of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.806927/full#supplementary-material
Footnotes
- ^ http://greengenes.lbl.gov/Download/Sequence_Data
- ^ http://drive5.com/uparse/
- ^ http://rdp.cme.msu.edu/
References
Aiello, A. (2005). Field guide to grasshoppers, katydids, and crickets of the United States. Library J. 130, 120–120.
Bai, Y., Dong, J. J., Guan, D. L., Xie, J. Y., and Xu, S. Q. (2016). Geographic variation in wing size and shape of the grasshopper Trilophidia annulata (Orthoptera: Oedipodidae): morphological trait variations follow an ecogeographical rule. Sci. Rep. 6:32680. doi: 10.1038/srep32680
Báldi, A., and Kisbenedek, T. (1997). Orthopteran assemblages as indicators of grassland naturalness in Hungary. Environment 66, 121–129. doi: 10.1016/s0167-8809(97)00068-6
Behar, A., Yuval, B., and Jurkevitch, E. (2005). Enterobacteria-mediated nitrogen fixation in natural populations of the fruit fly Ceratitis capitata. Mol. Ecol. 14, 2637–2643. doi: 10.1111/j.1365-294X.2005.02615.x
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Brune, A. (2014). Symbiotic digestion of lignocellulose in termite guts. Nat. Rev. Microbiol. 12, 168–180. doi: 10.1038/nrmicro3182
Chen, B., Teh, B. S., Sun, C., Hu, S., Lu, X., Boland, W., et al. (2016). Biodiversity and Activity of the Gut Microbiota across the Life History of the Insect Herbivore Spodoptera littoralis. Sci. Rep. 6:29505. doi: 10.1038/srep29505
Cole, J. R., Chai, B., Farris, R. J., Wang, Q., Kulam, S. A., McGarrell, D. M., et al. (2005). The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res. 33, D294–D296. doi: 10.1093/nar/gki038
Corby-Harris, V., Maes, P., and Anderson, K. E. (2014). The bacterial communities associated with honey bee (Apis mellifera) foragers. PLoS One 9:e95056. doi: 10.1371/journal.pone.0095056
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/aem.03006-05
Dillon, R. J., Vennard, C. T., Buckling, A., and Charnley, A. K. (2005). Diversity of locust gut bacteria protects against pathogen invasion. Ecol. Lett. 8, 1291–1298. doi: 10.1111/j.1461-0248.2005.00828.x
Dillon, R. J., Vennard, C. T., and Charnley, A. K. (2000). Exploitation of gut bacteria in the locust. Nature 403:851. doi: 10.1038/35002669
Dong, Y., Manfredini, F., and Dimopoulos, G. (2009). Implication of the mosquito midgut microbiota in the defense against malaria parasites. PLoS Pathog. 5:e1000423. doi: 10.1371/journal.ppat.1000423
Douglas, G. M., Beiko, R. G., and Langille, M. G. I. (2018). Predicting the Functional Potential of the Microbiome from Marker Genes Using PICRUSt. Methods Mol. Biol. 1849, 169–177. doi: 10.1007/978-1-4939-8728-3_11
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Engel, P., and Moran, N. A. (2013). Functional and evolutionary insights into the simple yet specific gut microbiota of the honey bee from metagenomic analysis. Gut. Microbes 4, 60–65. doi: 10.4161/gmic.22517
Fierer, N., Strickland, M. S., Liptzin, D., Bradford, M. A., and Cleveland, C. C. (2009). Global patterns in belowground communities. Ecol. Lett. 12, 1238–1249. doi: 10.1111/j.1461-0248.2009.01360.x
Fu, B. J., Chen, L. D., Ma, K. M., Zhou, H. F., and Wang, J. (2000). The relationships between land use and soil conditions in the hilly area of the loess plateau in northern Shaanxi, China. Catena 39, 69–78. doi: 10.1016/s0341-8162(99)00084-3
Fu, B. J., Meng, Q. H., Qiu, Y., Zhao, W. W., Zhang, Q. J., and Davidson, D. A. (2004). Effects of land use on soil erosion and nitrogen loss in the hilly area of the Loess Plateau. China. Land Degrad. Dev. 15, 87–96. doi: 10.1002/ldr.572
Gong, J., Chen, L., Fu, B., Huang, Y., Huang, Z., and Peng, H. (2006). Effect of land use on soil nutrients in the loess hilly area of the Loess Plateau. China. Land Degrad. Dev. 17, 453–465. doi: 10.1002/ldr.701
Guan, D. L., and Xu, S. Q. (2016). Complete mitochondrial genome of the geophilous grasshopper Trilophidia annulata (Acrididae: Oedipodinae: Trilophidia). Mitochondrial DNA A DNA Mapp. Seq. Anal. 27, 3143–3144. doi: 10.3109/19401736.2015.1007304
Hanniffy, S. B., Carter, A. T., Hitchin, E., and Wells, J. M. (2007). Mucosal delivery of a pneumococcal vaccine using Lactococcus lactis affords protection against respiratory infection. J. Infect. Dis. 195, 185–193. doi: 10.1086/509807
Hongoh, Y. (2010). Diversity and genomes of uncultured microbial symbionts in the termite gut. Biosci. Biotechnol. Biochem. 74, 1145–1151. doi: 10.1271/bbb.100094
Kaltenpoth, M., Winter, S. A., and Kleinhammer, A. (2009). Localization and transmission route of Coriobacterium glomerans, the endosymbiont of pyrrhocorid bugs. FEMS Microbiol. Ecol. 69, 373–383. doi: 10.1111/j.1574-6941.2009.00722.x
Kim, J. M., Choi, M. Y., Kim, J. W., Lee, S. A., Ahn, J. H., Song, J., et al. (2017). Effects of diet type, developmental stage, and gut compartment in the gut bacterial communities of two Cerambycidae species (Coleoptera). J. Microbiol. 55, 21–30. doi: 10.1007/s12275-017-6561-x
Koch, H., Abrol, D. P., Li, J., and Schmid-Hempel, P. (2013). Diversity and evolutionary patterns of bacterial gut associates of corbiculate bees. Mol. Ecol. 22, 2028–2044. doi: 10.1111/mec.12209
Langille, M. G., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Liang, X., Fu, Y., and Liu, H. (2015). Isolation and characterization of enzyme-producing bacteria of the silkworm larval gut in bioregenerative life support system. Acta Astronautica 116, 247–253. doi: 10.1016/j.actaastro.2015.07.010
McHardy, I. H., Goudarzi, M., Tong, M., Ruegger, P. M., Schwager, E., Weger, J. R., et al. (2013). Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome 1:17. doi: 10.1186/2049-2618-1-17
Muratore, M., Prather, C., and Sun, Y. (2020a). The gut bacterial communities across six grasshopper species from a coastal tallgrass prairie. PLoS One 15:e0228406. doi: 10.1371/journal.pone.0228406
Muratore, M., Sun, Y., and Prather, C. (2020b). Environmental Nutrients Alter Bacterial and Fungal Gut Microbiomes in the Common Meadow Katydid. Orchelimum vulgare. Front. Microbiol. 11:557980. doi: 10.3389/fmicb.2020.557980
Paniagua Voirol, L. R., Frago, E., Kaltenpoth, M., Hilker, M., and Fatouros, N. E. (2018). Bacterial Symbionts in Lepidoptera: Their Diversity. Transmission, and Impact on the Host. Front. Microbiol. 9:556. doi: 10.3389/fmicb.2018.00556
Qiu, Y., Fu, B. J., Wang, J., and Chen, L. D. (2001). Spatial variability of soil moisture content and its relation to environmental indices in a semi-arid gully catchment of the Loess Plateau. China. J. Arid Environ. 49, 723–750. doi: 10.1006/jare.2001.0828
Rajagopal, R. (2009). Beneficial interactions between insects and gut bacteria. Indian J. Microbiol. 49, 114–119. doi: 10.1007/s12088-009-0023-z
Ray, J. L., Andersen, H. K., Young, S., Nielsen, K. M., and O’Callaghan, M. (2007). An assessment of the potential of herbivorous insect gut bacteria to develop competence for natural transformation. Environ. Biosafety Res. 6, 135–147. doi: 10.1051/ebr:2007032
Ricci, I., Valzano, M., Ulissi, U., Epis, S., Cappelli, A., and Favia, G. (2012). Symbiotic control of mosquito borne disease. Pathog. Glob. Health 106, 380–385. doi: 10.1179/2047773212y.0000000051
Rogers, S. M., Matheson, T., Despland, E., Dodgson, T., Burrows, M., and Simpson, S. J. (2003). Mechanosensory-induced behavioural gregarization in the desert locust Schistocerca gregaria. J. Exp. Biol. 206, 3991–4002. doi: 10.1242/jeb.00648
Ruokolainen, L., Ikonen, S., Makkonen, H., and Hanski, I. (2016). Larval growth rate is associated with the composition of the gut microbiota in the Glanville fritillary butterfly. Oecologia 181, 895–903. doi: 10.1007/s00442-016-3603-8
Russell, J. A., Moreau, C. S., Goldman-Huertas, B., Fujiwara, M., Lohman, D. J., and Pierce, N. E. (2009). Bacterial gut symbionts are tightly linked with the evolution of herbivory in ants. Proc. Natl. Acad. Sci. U. S. A. 106, 21236–21241. doi: 10.1073/pnas.0907926106
Saraiva, M. A., Zemolin, A. P., Franco, J. L., Boldo, J. T., Stefenon, V. M., Triplett, E. W., et al. (2015). Relationship between honeybee nutrition and their microbial communities. Antonie Van Leeuwenhoek 107, 921–933. doi: 10.1007/s10482-015-0384-8
Scott, J. J., Oh, D. C., Yuceer, M. C., Klepzig, K. D., Clardy, J., and Currie, C. R. (2008). Bacterial protection of beetle-fungus mutualism. Science 322:63. doi: 10.1126/science.1160423
Shao, L. M., Ren, J. D., Lü, F., Zhang, H., and He, P. J. (2021). [Experimental Influence of Food Waste Fermentation Broth on the Soil Quality in a Loess Hilly Area]. Huan Jing Ke Xue 42, 4500–4509. doi: 10.13227/j.hjkx.202012234
Sharon, G., Segal, D., Ringo, J. M., Hefetz, A., Zilber-Rosenberg, I., and Rosenberg, E. (2010). Commensal bacteria play a role in mating preference of Drosophila melanogaster. Proc. Natl. Acad. Sci. U. S. A. 107, 20051–20056. doi: 10.1073/pnas.1009906107
Shi, W., Xie, S., Chen, X., Sun, S., Zhou, X., Liu, L., et al. (2013). Comparative genomic analysis of the microbiome [corrected] of herbivorous insects reveals eco-environmental adaptations: biotechnology applications. PLoS Genet. 9:e1003131. doi: 10.1371/journal.pgen.1003131
Suen, G., Scott, J. J., Aylward, F. O., Adams, S. M., Tringe, S. G., Pinto-Tomás, A. A., et al. (2010). An insect herbivore microbiome with high plant biomass-degrading capacity. PLoS Genet. 6:e1001129. doi: 10.1371/journal.pgen.1001129
Sun, T., Liu, Z. Y., Qin, L. P., and Long, R. J. (2015). Grasshopper (Orthoptera: Acrididae) Community Composition in the Rangeland of the Northern Slopes of the Qilian Mountains in Northwestern China. J Insect Sci. 1:171.
Wakarchuk, W. W., Campbell, R. L., Sung, W. L., Davoodi, J., and Yaguchi, M. (1994). Mutational and crystallographic analyses of the active site residues of the Bacillus circulans xylanase. Protein Sci. 3, 467–475. doi: 10.1002/pro.5560030312
Wang, J., Weiss, B. L., and Aksoy, S. (2013). Tsetse fly microbiota: form and function. Front. Cell Infect. Microbiol. 3:69. doi: 10.3389/fcimb.2013.00069
Warnecke, F., Luginbühl, P., Ivanova, N., Ghassemian, M., Richardson, T. H., Stege, J. T., et al. (2007). Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450, 560–565. doi: 10.1038/nature06269
Yang, Y., Deng, Y., and Cao, L. (2016). Characterising the interspecific variations and convergence of gut microbiota in Anseriformes herbivores at wintering areas. Sci. Rep. 6:32655. doi: 10.1038/srep32655
Keywords: gut bacteria, grasshopper, comparison, ecological environment, Loess Plateau
Citation: Zhao L, Wang W-Q, Xu S-Q and Guan D-L (2022) The Comparison of Gut Bacteria Communities and the Functions Among the Sympatric Grasshopper Species From the Loess Plateau. Front. Microbiol. 13:806927. doi: 10.3389/fmicb.2022.806927
Received: 01 November 2021; Accepted: 22 February 2022;
Published: 05 April 2022.
Edited by:
Hongjie Li, Ningbo University, ChinaReviewed by:
Zhijun Zhou, Hebei University, ChinaGuan-Hong Wang, Chinese Academy of Sciences (CAS), China
Zhihang Zhuo, Hainan University, China
Copyright © 2022 Zhao, Wang, Xu and Guan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sheng-Quan Xu, eHVzaGVuZ3F1YW5Ac25udS5lZHUuY24=; De-Long Guan, Z3VhbmRlbG9uZ0Bzbm51LmVkdS5jbg==