 Mariem Hanachi1,2
Mariem Hanachi1,2 Olfa Maghrebi3†
Olfa Maghrebi3† Haifa Bichiou4†
Haifa Bichiou4† Ferdaous Trabelsi5
Ferdaous Trabelsi5 Najla Maha Bouyahia6Fethi Zhioua6
Najla Maha Bouyahia6Fethi Zhioua6 Meriam Belghith3
Meriam Belghith3 Emna Harigua-Souiai7Meriem Baouendi1Lamia Guizani-Tabbane4
Emna Harigua-Souiai7Meriem Baouendi1Lamia Guizani-Tabbane4 Alia Benkahla1
Alia Benkahla1 Oussema Souiai1*
Oussema Souiai1*- 1Laboratory of Bioinformatics, bioMathematics and Biostatistics—LR16IPT09, Institut Pasteur de Tunis, Université de Tunis El Manar, Tunis, Tunisia
- 2Faculty of Science of Bizerte, University of Carthage, Tunis, Tunisia
- 3Laboratory of Transmission, Control, and Immunobiology of Infections—LR16 IPT02, Institut Pasteur de Tunis, Université de Tunis El Manar, Tunis, Tunisia
- 4Laboratory of Medical Parasitology, Biotechnology, and Biomolecules—LR16 IPT06, Institut Pasteur de Tunis, Université de Tunis El Manar, Tunis, Tunisia
- 5Service de Gynécologie et Obstétrique, Hôpital Régional de Zaghouan, Zaghouan, Tunisia
- 6Service de Gynécologie Obstétrique et Médecine de la Reproduction, Hôpital Aziza Othmana, Tunis, Tunisia
- 7Laboratory of Molecular Epidemiology and Experimental Pathology—LR16IPT04, Institut Pasteur de Tunis, Université de Tunis El Manar, Tunis, Tunisia
Microbiota colonization is a dynamic process that impacts the health status during an individual's lifetime. The composition of the gut microbiota of newborns is conditioned by multiple factors, including the delivery mode (DM). Nonetheless, the DM's influence remains uncertain and is still the subject of debate. In this context, the medical indication and the emergency of a cesarean delivery might have led to confounding conclusions regarding the composition and diversity of the neonatal microbiome. Herein, we used high-resolution shotgun sequencing to decipher the composition and dynamics of the gut microbiota composition of Tunisian newborns. Stool samples were collected from 5 elective cesarean section (ECS) and 5 vaginally delivered (VD) newborns at the following time points: Day 0, Day 15, and Day 30. The ECS and VD newborns showed the same level of bacterial richness and diversity. In addition, our data pointed to a shift in microbiota community composition during the first 2 weeks, regardless of the DM. Both ECS and VD showed a profile dominated by Proteobacteria, Actinobacteria, and Firmicutes. However, ECS showed an underrepresentation of Bacteroides and an enrichment of opportunistic pathogenic species of the ESKAPE group, starting from the second week. Besides revealing the intestinal microbiota of Tunisian newborns, this study provides novel insights into the microbiota perturbations caused by ECS.
Introduction
The bacterial colonization of the gut microbiota has a tremendous impact on health across the lifespan (Houghteling and Walker, 2015; Walker, 2017). Although suspected to be initiated in utero, microbiota colonization is a highly fluctuant and dynamic process during the first days of life (Perez-Muñoz et al., 2017; Walker, 2017; Sanidad and Zeng, 2020). This process occurs in successive stages, leading to a stable adult-like profile by the age of 3 years (Yatsunenko et al., 2012; Walker, 2017; Sanidad and Zeng, 2020). Pioneer bacteria that colonize the newborn's intestinal tract are essential for immune system maturation, gastrointestinal metabolism, and neurological development (Borre et al., 2014; Milani et al., 2017; Stiemsma and Michels, 2018; Sanidad and Zeng, 2020).
Most microbiome research has focused on Western countries, and only marginal analyses were interested in low-income African countries (Hamdi et al., 2021). To our knowledge, none concerned North Africa, despite the well-established effect of geographical localization and population habits on microbiome composition and diversity (Fallani et al., 2010; Yatsunenko et al., 2012). More specifically, Tunisia constitutes a particular case study, considering the dramatically high rate of (43%) cesarean section (CS) compared with thresholds established by the World Health Organization (WHO) (Unicef report, 2018).
Among all influencing factors, the delivery mode (DM) was extensively investigated and is still subject to various controversies and debates (Aagaard et al., 2016; Stinson et al., 2018). The first high throughput microbiome analyses focusing on this topic conducted by Dominguez-Bello et al. reported that the gut microbiota of the vaginally delivered (VD) newborns is similar to mother vaginal microbiota, while the microbiota of CS newborns resembles the maternal skin microbiota (Dominguez-Bello et al., 2010). Limited and altered diversity was subsequently described for CS delivered newborns (Rutayisire et al., 2016; Shi et al., 2018). These observations have been linked to a higher susceptibility to develop immune and metabolic disorders observed in CS infants (Darmasseelane et al., 2014; Sevelsted et al., 2015; Wampach et al., 2018; Busi et al., 2021). In spite of this established effect of DM on infant health, the consensus on the immature status of the CS delivery microbiome has been contradicted by some studies reporting no effects on newborn's microbiota neither in terms of composition nor in terms of functionality (Hu et al., 2013; Chu et al., 2017; Liu et al., 2019). It was suggested that the reported differences might be biased by confounding factors such as intrapartum antibiotic administration, differences in breastfeeding behaviors, gestational age, and like in our case of interest, the indication for CS (Aagaard et al., 2016; Chu et al., 2017; Stinson et al., 2018).
In fact, emergent and elective CS (ECS) differs in several aspects that may affect the maternal and newborn microbiota. First, ECS is undertaken without rupture of the fetal membranes, and newborns are delivered without passing through the birth canal. They are thereby not exposed to the maternal vaginal flora (Stinson et al., 2018). Second, emergent CS occurs after the start of labor and is characterized by a fluctuation in levels of endocrine, pro-inflammatory, and immune-mediating cytokines, in opposition to ECS (Malamitsi-Puchner et al., 2005; Francino, 2018; Stinson et al., 2018). Thus, ECS constitutes a unique opportunity to further assess the real impact of DM on the gut microbiota of newborns, as it prevents the contact of the newborn with the maternal vaginal flora during birth and the potential biases due to the medical indication of emergent CS.
Altogether, the arguments listed above raised our interest to first uncover the microbiota of Tunisian newborns and then to assess the impact of DM on the composition of early gut bacteria by minimizing confounding factors. A shotgun metagenomic approach was applied to better capture the microbial resolution in the ECS and VD newborns.
Materials and Methods
Ethics Statements
This study was approved by the Biomedical Ethics Committee of the Institute Pasteur of Tunis, under reference number 2018/10/I/LR161IPT09. Written informed consents were obtained from both parents.
Participants' Recruitment and Sample Collection
All participants originated from Zaghouan, a semirural region in Northwest Tunisia. The mothers had a non-declared pathology and did not receive antibiotics, anti-inflammatory drugs, or immunosuppressive therapy during the last trimester of pregnancy. Neonates were healthy, full-term born (>37 weeks), and did not receive any antibiotic treatment. Only newborns delivered by elective and unlabored cesarean were included in this study. No complications during the delivery were reported. Anonymized mother's health records with their corresponding metadata were compiled. Clinical metadata concerning the newborns were also collected including birth weight, gestational age, gender, weight, and feeding mode (Table 1).
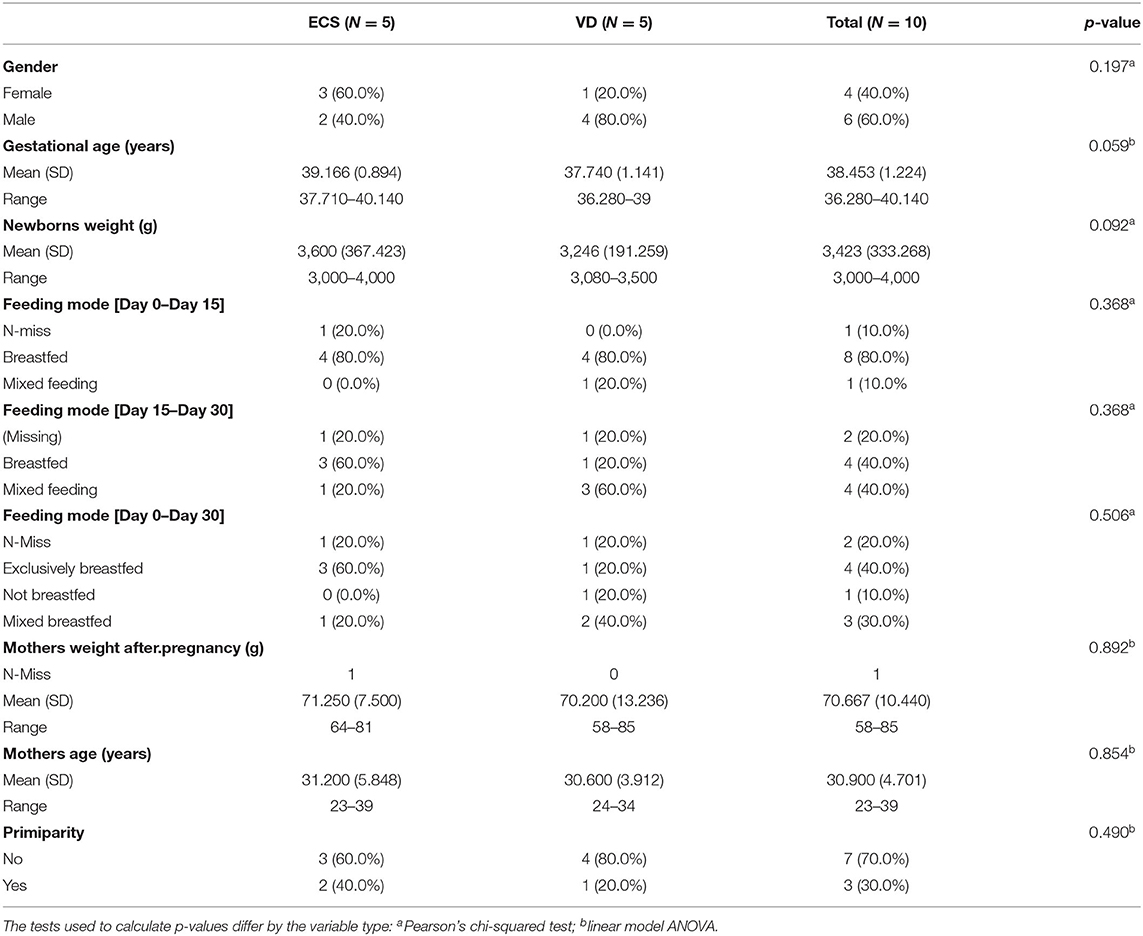
Table 1. Clinical characteristics of the newborns and mothers enrolled in this study.
A total of 10 participants were retained, and newborns' stool samples were collected at three time points. The neonatal meconium from VD and ECS newborns was collected during the first 24 h (Day 0) at the regional hospital of Zaghouan and at the Aziza Othmana hospital, located in Zaghouan and Tunis, respectively. The collection was renewed for 2 weeks (Day 15) and 1 month (Day 30) after birth. The samples were transported in ice bags to the laboratory and then stored at −80°C until DNA extraction.
DNA Extraction and Sequencing
Subsamples from each newborn stool (~200 mg) were used for DNA extraction. The solubilization and liquefaction of the samples were carried out using 1 ml of Tween 20 (10%). The solution was then vortexed horizontally for 20 min and centrifuged at 14,000 rpm for 10 min at 4°C. The obtained pellets were resuspended in a lysis buffer (100 mM NaCl, 25 mM EDTA, 0.5% SDS, 10 mM HEPES, and 1 mg/ml lysozyme) for chemical and enzymatic lysis. Then, bead-beating steps were proceeded using BeadBug and PowerBead Tubes, Garnet 0.70 mm (QIAGEN), with a setting of 3 × 20 s pulses, followed by incubation for 7 min at 95°C. After a centrifugation step, the supernatant was transferred into 2 ml tubes containing 30 μl proteinase K and 400 μl AL buffer (QIAamp Fast DNA Stool Mini Kit) and incubated at 70°C for 10 min. DNA was recovered using phenol-chloroform extraction and purified using the QIAamp DNA mini kit (QIAGEN). In brief, protein precipitation was performed using the phenol/chloroform method, followed by DNA precipitation using absolute ethanol. The supernatant was transferred to the QIAamp spin column, and DNA purification was performed according to the QIAamp Fast DNA Stool Mini Kit manufacturer's instructions. DNA samples were quantified using the Multiskan™ GO Microplate Spectrophotometer (Thermo Fisher™), and samples with >100 ng/μl DNA material proceeded to the paired-end (2 × 150 bp) metagenomic sequencing. Overall, we sequenced 33 samples: 9 participants at 3 time points, 1 participant at 2 time points (Day 0 and Day 30), and 4 technical replicates (2 at Day 0, 1 at Day 15, and 1 at Day 30).
Bioinformatics and Biostatistics Analyses
Quality control and preprocessing were undertaken on the raw data as follows: Trimmomatic was run to trim low-quality bases and short reads (SLIDINGWINDOW: 4:20, min length: 70) (Bolger et al., 2014). Following this step, we obtained on average 11 and 14 M reads for Day 15 and Day 30 samples. As expected, Day 0 samples harbored fewer sequences, 28.6 K reads on average (Supplementary Table 1). Then, human DNA and vector contaminants were screened and removed using FastQ Screen software (Wingett and Andrews, 2018). The taxonomic classification of cleaned metagenomic reads was performed using Kraken version 2.0.8 against the Minikraken Database (Wood et al., 2019). Then, Bracken version 2.5 was run on the Kraken taxonomic assignment outputs to increase the accuracy of the abundance estimates at species and genus levels (Lu et al., 2017).
The classification output consisted of a feature table containing 3,805 taxa composed of bacteria 3,489 operational taxonomic units (OTUs) as well as viruses (212 OTUs) and Archaea (103 OTUs). The statistical analysis started with filtering ambiguous and rare bacterial taxa (Supplementary Table 1). First, low-prevalent (<15% samples) and low abundant (<100 reads) phyla were removed. OTUs present in <15% of samples were also excluded. None of the filtered OTUs were specific to a mode of delivery or unique to collection day, thus not impacting the overall rates of abundance (data not shown). Ultimately, 2,609 OTUs belonging to bacteria remained in the taxonomy table.
Alpha diversity metrics (Shannon and Simpson indices and OTU richness) were analyzed using the microbiome R package (McMurdie and Holmes 2013).
A phylogenetic tree was generated and used to estimate Faith phylogenetic diversity (standardized effect sizes = TRUE) using the function “phyloseq_phylo_div” from the R package PhyloMeasures (Tsirogiannis and Sandel, 2016) and metagMisc R package (available at: https://github.com/vmikk/metagMisc). The Friedman test was used to compare differences in alpha diversity between collection days (paired samples), followed by the Nemenyi post-hoc test using the PMCMRplus R package (available at https://cran.r-project.org/web/packages/PMCMRplus/index.html). When comparing alpha diversity between two unpaired groups, the non-parametric Wilcoxon was employed. Kruskal–Wallis test was used to assess statistical differences between more than two groups, followed by post-hoc Dunn's test (with Bonferroni correction). The clustering and heatmap were performed using the microbiomeutilities R package (Shetty and Lahti, 2020). The clustering based on taxa and samples was performed using Bray–Curtis dissimilarity distances.
Beta diversity was evaluated with the Bray–Curtis distance after data normalization using the Cumulative Sum Scaling (CSS) (Paulson et al., 2013). Visualization was performed using the Non-metric Multidimensional Scaling (NMDS) ordination method. Differences between the groups were tested using the permutational multivariate analysis of variance (PERMANOVA) test (number of the permutation set to 999). The linear discriminant analysis effect size (LEfSe) R package was used to identify the differentially abundant taxa between study groups on CSS normalized data (Segata et al., 2011) (parameters: kw_cutoff = 0.01, wilcoxon_cutoff = 0.01, and lda_cutoff = 3). PERMANOVA test was also run to determine covariate impact on newborns' gut microbial community based on Bray–Curtis distance (permutation set to 999).
Functional analysis was processed with SqueezeMeta Pipeline using the merged mode (Tamames and Puente-Sánchez, 2019). First, Megahit was used for the assembly (—min-count 1—k-list 27, 37, 47, 57, 67, 77, 87) (Li et al., 2015). We obtained 139,759 contigs ranging from 200 to 761,139 bp (Supplementary Table 1). Then, open reading frame (ORF)'s prediction and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway (KEGG database, release 95, July 2020) prediction were performed by Prodigal and Diamond, respectively (Hyatt et al., 2010; Buchfink et al., 2015). Coverage and abundance estimation for genes and contigs were performed using Bedtools and normalized using the reads per kilobase per million mapped reads (RPKM) method (Quinlan and Hall, 2010). The statistical downstream analyses were performed using the SQMtools and phyloseq R packages (McMurdie and Holmes, 2013; Puente-Sánchez et al., 2020).
Results
Characteristics of the Participants
Full-term newborns (5 from vaginal delivery and 5 from ECS delivery) were delivered at a mean gestational age of 38.5 weeks (SD ± 1.22). Samples were collected on the birthday (Day 0), Day 15, and Day 30 after birth. The characteristics of the mothers and newborns are listed in Table 1. Overall, 40% of newborns were exclusively breastfed, 10% exclusively received formula milk, and 30% received mixed feeding. Notably, 80% of neonates were breastfed until the second time point (Day 15). By Day 30, this percentage decreased to 40%. There were no significant differences in maternal age [31 years (SD ± 4.70)], mother's weight [70.76 kg (SD ± 10.44)], and birth weight [3.43 kg (SD ± 0.34)] between ECS and VD (Table 1).
Composition of Gut Microbiota of the Tunisian Newborns in the Early Stage of Life
We first aimed to determine the significant factors associated with microbiota structure. Therefore, we performed the PERMANOVA test individually for each covariate. The results showed that collection days explained most of the variance in population structure (Supplementary Table 2; p = 0.001, R2 = 0.210). Although not impacting the diversity, feeding mode was the second most significant factor explaining variation in neonatal gut microbiota (p = 0.033, R2 = 0.115; Supplementary Figure 1, Supplementary Table 2). The other clinical covariates (newborns' weight and gestational age) did not influence the community structure of the early microbiome (Supplementary Table 2).
We then scrutinized the early establishment of Tunisian newborns' gut microbiota by comparing the three sampling time points. This allowed us to identify the temporal patterns in terms of taxonomic diversity. A significant decrease in terms of Shannon evenness and Simpson diversity indices was observed when comparing Day 0 with Day 15 and Day 30 (Figure 1A, top left and top right panel). In addition, the number of observed OTUs and phylogenetic diversity significantly increases over time (Figure 1A, bottom left and bottom right panel). The beta-diversity analysis clearly and significantly distinguished the Day 0 samples from the other time points (Day 15 and Day 30) (p = 0.001, R2 = 0.210; Figure 1B). The functional annotation analysis corroborated this ascertainment (Figure 1C; p = 0.001, R2 = 0.688). This suggests that the functions potentially brought by bacteria at Day 0 are distinct from those ensured at Day 15 and Day 30. At the phylum level, a pattern of increasing abundance composed of Firmicutes, Proteobacteria, and Actinobacteria is observed (Figure 1D). Clustering at the genus level revealed that the abundance of Escherichia, Klebsiella from the Proteobacteria, Bifidobacterium from the Actinobacteria, and Streptococcus from the Firmicutes increases from Day 15 post-delivery (Figure 1E). The LEfSe analysis highlighted a significant increase for the above-mentioned genera and phyla (Supplementary Table 3).
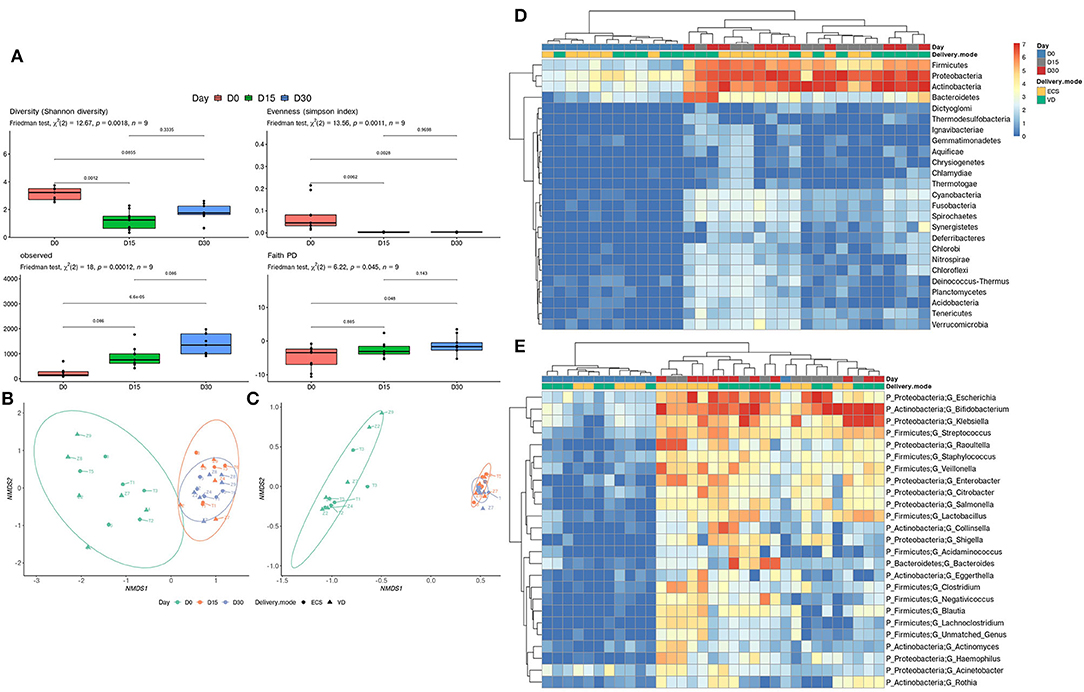
Figure 1. Development of early gut microbiota of Tunisian newborns. (A) Alpha diversity was measured by estimating the Shannon index (top right panel), Simpson index (top left panel), observed OTU richness (bottom left panel), and Faith phylogenetic diversity (bottom right panel). Significant differences between groups were determined using the Friedman test followed by a Nemenyi post-hoc test. Ordination plot with nonmetric multidimensional scaling (NMDS) based on the relative abundance of taxa (B) and pathways (C) in newborns' gut microbiota. Heatmap of relative abundance (log10 transformed) of the 25 most abundant taxa at the phylum (D) and genus level (E). The clustering was performed based on Bray–Curtis distance measure.
DM Impacts the Distribution of Key Taxa in Newborns' Gut Microbiota
To address the impact of DM on newborns' gut microbiota, we carried out various taxonomic and functional diversity analyzes. Alpha and beta-diversity showed no significant difference neither in terms of diversity nor in bacterial community structure between ECS and VD groups (p = 0.331, R2 = 0.034) (Figures 2A,C). Similarly, we did not observe differences in community function between ECS and VD (Figure 2B; p-value = 0.75, R2 = 0.011).
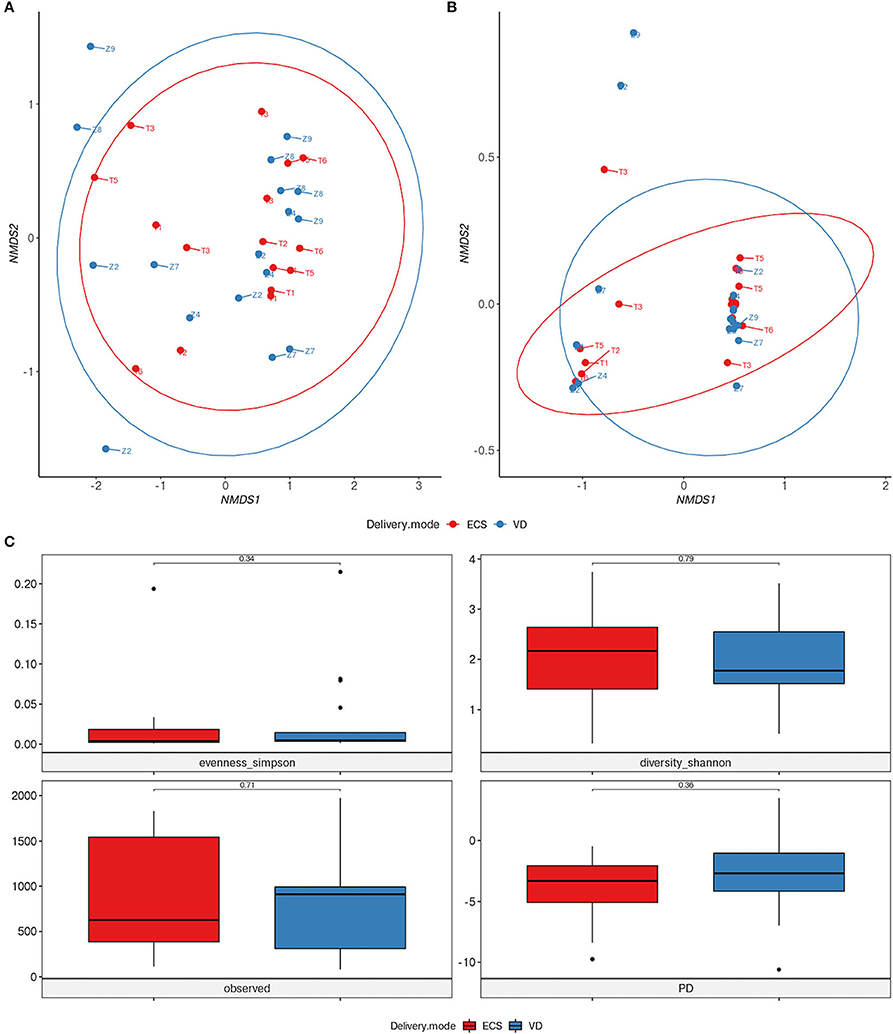
Figure 2. Impact of vaginal and elective cesarean on the diversity of the early life gut microbiome. NMDS ordination plot based on the relative abundance of taxa (A) and pathways (B) in newborns' gut microbiota. Samples are marked according to delivery mode (DM). (C) Alpha diversity was measured by Shannon and Simpson indices (panel top), observed OTU richness (panel bottom left), and Faith phylogenetic diversity (panel bottom right). Significant differences between groups were determined using a Mann–Whitney U-test.
Although the ECS and VD microbiota shares a similar community structure, marginal taxonomic differences do exist. To further characterize these differences, LEfSe analysis was performed to detect specific differentially abundant features at various taxonomic ranks in ECS and VD (Supplementary Figure 2). In the ECS group, species belonging to Actinomyces, Pseudopropionibacterium, and Citrobacter genera were overrepresented compared with the VD group. Interestingly, the microbiota of ECS newborns was enriched in the genus Clostridium. Among these genera, the Clostridium perfringens, an opportunistic pathogenic species, was detected as overrepresented. In addition, the ECS showed an overrepresentation of Enterobacter species, a highly virulent and antibiotic-resistant member of the ESKAPE bacterial group.
The Bacteroidetes phylum, including species belonging to the Bacteroides genus (Bacteroides helcogenes; Bacteroides heparinolyticus), and Parabacteroides were identified as overrepresented in the VD group (Supplementary Figure 2). The ECS group also showed an enrichment in the Bifidobacterium dentium, while the Bifidobacterium pseudocatenulatum was overrepresented in the VD group.
Taken together, these results indicate that although the mode of delivery does not apparently impact the overall diversity, there is nevertheless a significant difference at phylum and genus level between the two groups. These differences are mirrored by the enrichment of opportunistic pathogenic species in ECS and Bacteroidetes in VD.
The Establishment of Gut Microbiota in ECS and VD Newborns Exhibits Different Dynamics
We then sought to understand how the composition and developmental trajectory of the early gut microbiota occurs in ECS and VD. A transition that occurs within the first 2 weeks of life was observed in both ECS (p = 0.002, R2 = 0.246) and VD (p = 0.002, R2 = 0.269; Figure 3).
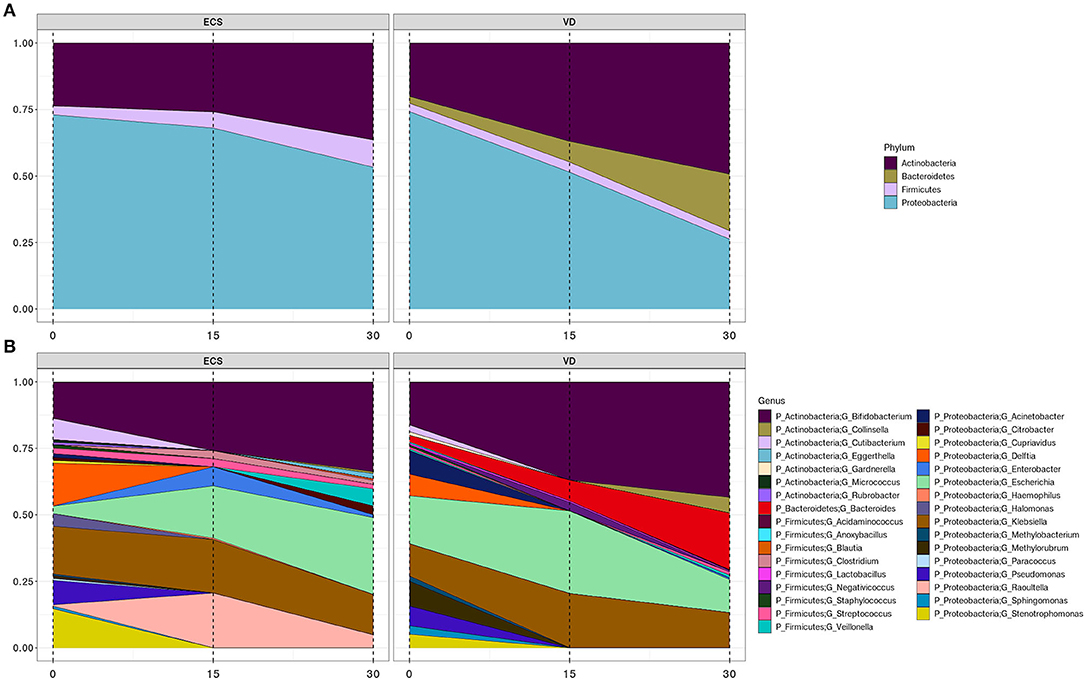
Figure 3. Development trajectory of the newborn's gut microbiota according to DM. Area plots showing the longitudinal changes of >2% relative abundance across all samples of bacterial phyla (A) and their corresponding genera (B) in elective cesarean section (ECS) and vaginally delivered (VD).
While the gut microbiota of ECS neonates was colonized by Actinobacteria, Proteobacteria, and Firmicutes, the microbiota of the VD group also included the Bacteroidetes phylum. The Bacteroidetes were represented exclusively by the genus Bacteroides, while the genus Raoultella was strictly observed in ECS neonates (Figures 3A,B, Supplementary Table 4). We also observed a gradual increase of Actinobacteria at the expense of Proteobacteria (Figure 3A). This trend appears to be a consequence of the progressive growth of the genus Bifidobacterium, at a lower rate in ECS, and the decline of the genera Delftia, Enterobacter, Sphingomonas, and Stenotrophomonas (Figure 3B, Supplementary Table 3). With a lower relative abundance, the Firmicutes remain constant in the ECS while increasing in the VD, possibly due to expansion of the Veillonella genus (Figures 3A,B, Supplementary Table 4). At the genus level, the most striking difference between the two groups is the presence of Lactobacillus and Collinsella in VD.
Some genera present at birth do not persist in the remaining time points. This was observed for Cutibacterium, Acinetobacter, Delftia, Pseudomonas, Sphingomonas, Stenotrophomonas, and Methylobacterium, regardless of DM. While focusing on ECS newborns' meconium, we noticed that genera Citrobacter, Cupriavidus, Halomonas, and Paracoccus are undetected from Day 15. Similarly, Gardnerella and Methylorubrum are solely present in VD newborns' meconium during the first 2 weeks.
Again, LEfSe analysis was used to detect overrepresented bacterial taxa between ECS and VD at each sampling time point. We found that few distinctive taxa signatures were detected in the neonatal microbiota at Day 0 (Figure 4A). While the Clostridiale order and the Citrobacter genus were overrepresented in ECS, the Methylobacter genus was detected in VD.
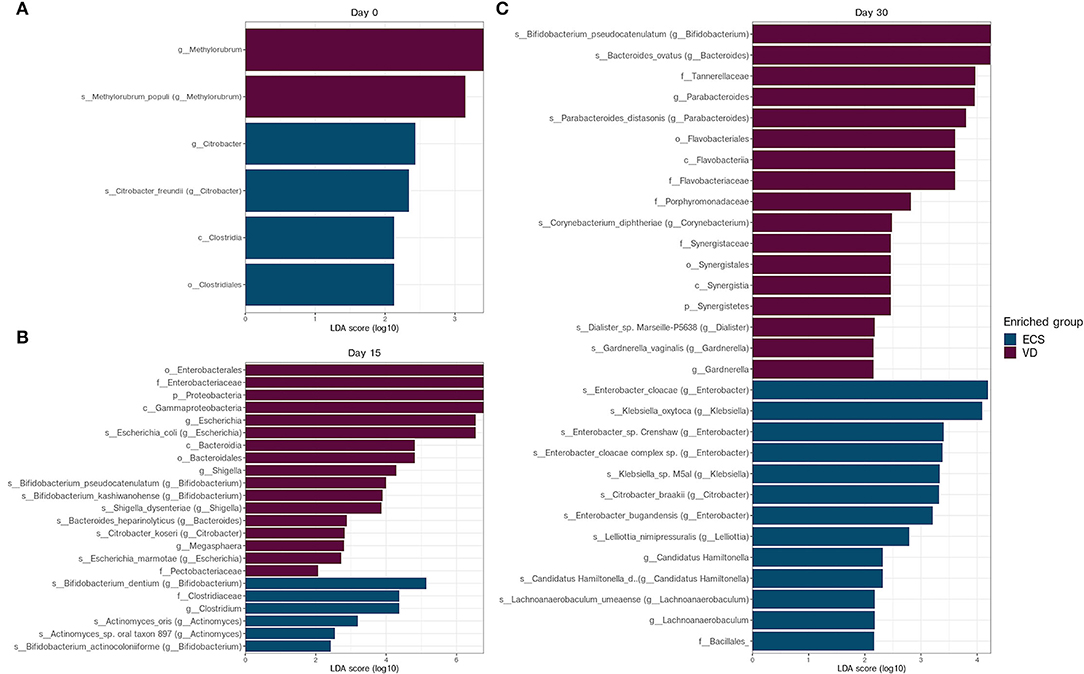
Figure 4. Differentially abundant taxa between ECS and VD newborns at each collection data. Linear discriminant analysis effect size (LEfSe) analysis of metagenomic sequences from ECS and VD newborn stool samples at Day 0 (A), Day 15 (B), and Day 30 (C). The blue shaded bars indicate the taxa enriched in the microbiome of VD newborns. The purple shaded bars indicate the taxa enriched in the microbiome of ECS newborns. The prefixes “p,” “c,” “o,” “f,” “g,” “s,” and “s” indicate the annotation levels of phylum, class, order, family, genus, species, and strain, respectively.
On Day 15, Proteobacteria, especially Escherichia and Shigella, were overrepresented in VD newborns compared with the ECS group (Figure 4B). The ECS group had a higher relative abundance of Clostridium and Clostridiaceae. In addition, the ECS group was enriched in species belonging to the Bifidobacterium genus, namely B. dentium and Bifidobacterium actinocoloniform (Figure 4B).
On Day 30, LEfSe analysis revealed an overrepresentation of species belonging to Bacteroides, Parabacteroides, and Gardnerella genera, respectively, represented by Bacteroides ovatus, Parabacteroides distasonis, and Gardnerella vaginalis, respectively, for the VD group (Figure 4C). The ECS samples showed an enrichment of Klebsiella oxytoca and various Enterobacter species (Figure 4C). Given the prevalence of these opportunistic pathogens in the gut of ECS neonates, we extended this analysis and compared the relative abundance of the ESKAPE group. We found that the ECS group had a significantly higher relative abundance at Day 15 and Day 30 (Supplementary Figure 4).
The above-mentioned results indicate that a shift occurs during the same timeframe in ECS and VD. However, ECS showed a disturbed establishment of the Bacteroides genus and an increased relative abundance of ESKAPE bacteria members from the second week.
Discussion
This longitudinal study was designed to provide an unprecedented insight into the Tunisian newborn's gut microbiota using high-resolution shotgun sequencing. In our context of low biomass, this technique demonstrated its ability to better accurately assign reads to species level compared to 16s sequencing as stated by Peterson et al. (2021). It assessed the effect of DM, more precisely the ESC, on its composition during the first month of life. In fact, ECS represents a public health concern as this procedure is often over-practiced (Jenabi et al., 2020). Considering the establishment of newborn microbiota, we herein highlighted major differences between the day of birth and the other two time points in terms of alpha diversity. We observed the highest diversity at Day 0 compared with the other two time points. This trend is possibly due to the rapid influx of microbes composing the pioneering microbiome, originating from maternal and environmental origins, as previously suggested (Wampach et al., 2017; Ferretti et al., 2018). The subsequent decrease in diversity could be attributed to anterior loss or replacement of poorly adapted bacteria in the gastrointestinal tract (Ferretti et al., 2018). Similar to previous findings summarized by Milani et al. the Tunisian gut microbiota is initially colonized mainly by Proteobacteria, Actinobacteria, Firmicutes, and Bacteroidetes (Milani et al., 2017). At the genus level, this study indicates that the initial gut microbiota is dynamic and undergoes several changes in terms of composition starting from the second week onwards. This shift observed on Day 15 concerned particularly the genera Klebsiella, Escherichia, Lactobacillus, and Staphylococcus. These findings shade previous descriptions that suggested initial colonization by Firmicutes members such as Streptococcus, Staphylococcus, and Enterobacteriaceae (Rautava et al., 2012).
Early life microbiome colonization is influenced by numerous factors, and a particular emphasis has been placed on the impact of the mode of delivery. A considerable amount of literature has been devoted to this topic and has established that the gut microbiota of CS newborns is characterized by a reduced diversity and an altered composition (Dominguez-Bello et al., 2010; Shi et al., 2018; Stinson et al., 2018; Shao et al., 2019).
When considering the medical indication and the urgency of cesarean deliveries, previous analyses reported that infants born by ECS had the lowest richness and diversity compared with infants born by urgent cesarean delivery and those vaginally delivered (Azad et al., 2013). However, no significant difference was detected in terms of diversity between ECS and VD in the first days of life (Liu et al., 2015). Our results further support the latter observation, extending the observed trend to the first month of life. In addition, our results show a significant change in the composition of the microbiota during the first 2 weeks for the VD and ECS groups. These results are in agreement with those presented previously by Kim et al. The authors had also highlighted that this transition is progressive in VD, whereas it is abrupt for those delivered by CS (Kim et al., 2020).
Low species-specific diversity is commonly associated with a higher susceptibility to develop immune and metabolic diseases (Milani et al., 2017). Therefore, we sought to identify key taxa, highlighting this aspect. Interestingly, the Bacteroides genus was absent in ECS newborns, consistent with previous findings (Jakobsson et al., 2014; Shao et al., 2019). Bacteroides are acquired through exposure to maternal vaginal flora, thus explaining the low-Bacteroides profile for CS (Shao et al., 2019). Recently, Mitchell et al. challenged this dogma by demonstrating that Bacteroides were present in the first week and absent during the second week in both emergent and ECS newborns (Mitchell et al., 2020). The delayed establishment of Bacteroides in the gut microbiota of CS neonates was associated with health issues, notably allergic diseases, and milk oligosaccharides' breakdown (Marcobal et al., 2011; Jakobsson et al., 2014).
In the same context, previous studies considered the Bifidobacterium genus as the most abundant during the first days of life, with a lower prevalence in CS (Tanaka and Nakayama, 2017). In this study, we confirmed the high level of Bifidobacterium in VD compared with CS. In this frame, Yassour et al. suggested that the lack of Bacteroides is balanced by the abundance of Bifidobacterium (Yassour et al., 2016). Our differential analysis showed an enrichment of B. dentium and Bifidobacterium actinocoloniiforme in CS, while B. pseudocatenulatum and Bifidobacterium kashiwanohense are overrepresented in the VD group. Other studies described a delay in the transmission of maternal strains of Bifidobacterium (e.g., Bifidobacterium longum, Bifidobacterium breve, and B. pseudocatenulatum) in newborns delivered by cesarean (Shao et al., 2019). Moreover, Saturio et al. (2021) reported a higher abundance of B. pseudocatenulatum in the VD and B. dentium species in the CS.
Similar to Bifidobacterium, Lactobacillus is also considered a major beneficial bacterium for the health of the human host (Rastall, 2004). Its low prevalence was associated with increased susceptibility to allergic diseases (Wang et al., 2020). In fact, Lactobacillus is typically present in the maternal vaginal flora (Tannock et al., 1990; Chu et al., 2017) and is reported as enriched in VD newborns compared with CS newborns (Dominguez-Bello et al., 2010; Kim et al., 2020). However, it was reported that the presence of the Lactobacillus genus is not dependent on the urgency of CS (Liu et al., 2015). Our results showed that Lactobacillus is barely observable in VD, while undetectable in ECS.
Another striking result was the detection of an enrichment of opportunistic pathogens in ECS newborns. Among these pathogenic species, we particularly identified ESKAPE pathogen members, responsible for several nosocomial infections (Santajit and Indrawattana, 2016). Other highly opportunistic pathogens, namely Klebsiella oxytoca, Enterobacter cloacae, and Clostridium perfringens, were also retrieved as overrepresented in ECS (Shao et al., 2019). Using lower resolution methods, prior work showed a similar pathogen prevalence between ECS and VD, thus confirming our results (Liu et al., 2015; Stokholm et al., 2016). The prevalence of the latter pathogens in CS, and to a lesser extent in VD, was associated with Bacteroides alteration and non-breastfeeding practices (Shao et al., 2019). Thus, the observed low-Bacteroides profile could partially explain the dominance of ESKAPE bacteria in ECS at Day 15 and Day 30. Regarding breastfeeding practices, we could not assess its protective effect due to the small size of the groups.
In conclusion, this study provides the first overview of the ethnically homogenous and still unresolved microbiota of Tunisian newborns. Using a higher resolution Shotgun sequencing approach, we complemented previous findings regarding the impact of ECS on the initial colonization of the gut microbiota.
Data Availability Statement
The datasets presented in this study can be found in SRA repository at: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA762668.
Ethics Statement
The studies involving human participants were reviewed and approved by Comité d'éthique bio-médicale de l'institut Pasteur de Tunis. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
OS and AB conceived this study and were in charge of overall direction and planning. MH, OM, HB, FZ, MBa, LG-T, FT, and OM assisted in the implementation of collection and DNA extraction protocols. MH, OS, LG-T, AB, MBe, and EH-S contributed to the interpretation of the results and provided critical feedback, and helped shape the research, analysis, and manuscript. MH, OS, AB, and LG-T analyzed the data and wrote the manuscript. All authors contributed with valuable discussions and editions and approving the final version of the manuscript.
Funding
This project was partly funded by H3ABioNet, which was supported by the National Institutes of Health Common Fund (Grant No. U41HG006941). Financial support was also afforded by the foundation Mérieux. We would like to acknowledge the European project PHINDaccess: Strengthening Omics data analysis capacities in pathogen-host interaction (Grant Agreement ID: 811034).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.780568/full#supplementary-material
Supplementary Figure 1. Impact of feeding mode on newborn's gut microbiota. Alpha diversity was measured by estimating the Shannon index (panel top right), Simpson index (panel top left), observed OTU richness (panel bottom left), and Faith phylogenetic diversity (panel bottom right). Significant differences between groups were determined using the Kruskal–Wallis test, followed by the Dunn test with Bonferroni correction.
Supplementary Figure 2. Differentially abundant taxa between elective cesarean section (ECS) and vaginally delivered (VD) newborns detected by linear discriminant analysis effect size (LEfSe) analysis. The blue shaded bars indicate the taxa enriched in the microbiome of VD newborns. The purple shaded bars indicate the taxa enriched in the microbiome of ECS newborns. The prefixes “p,” “c,” “o,” “f,” “g,” “s,” and “s” indicate the annotation levels of phylum, class, order, family, genus, species, and strain, respectively.
Supplementary Figure 3. Bacterial diversity transition in ECS and VD newborns. Nonmetric Multidimensional Scaling (NMDS) ordination plot based on the relative abundance of taxa in VD (A) and ECS (B).
Supplementary Figure 4. Relative abundance of ESKAPE members in ECS and VD newborns at each collection time point. Significance in the differences between groups was determined using the Mann–Whitney U-test.
References
Aagaard, K., Stewart, C. J., and Chu, D. (2016). Una destinatio, viae diversae: Does exposure to the vaginal microbiota confer health benefits to the infant, and does lack of exposure confer disease risk? EMBO Rep. 17, 1679–1684. doi: 10.15252/embr.201643483
Azad, M. B., Konya, T., Maughan, H., Guttman, D. S., Field, C. J., Chari, R. S., et al. (2013). Gut microbiota of healthy Canadian infants: profiles by mode of delivery and infant diet at 4 months. CMAJ 185, 385–394. doi: 10.1503/cmaj.121189
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Borre, Y. E., O'Keeffe, G. W., Clarke, G., Stanton, C., Dinan, T. G., and Cryan, J. F. (2014). Microbiota and neurodevelopmental windows: implications for brain disorders. Trends Mol. Med. 20, 509–518. doi: 10.1016/j.molmed.2014.05.002
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Busi, S. B., de Nies, L., Habier, J., Wampach, L., Fritz, J. V., Heintz-Buschart, A., et al. (2021). Persistence of birth mode-dependent effects on gut microbiome composition, immune system stimulation and antimicrobial resistance during the first year of life. ISME Commun. 1, 8. doi: 10.1038/s43705-021-00003-5
Chu, D. M., Ma, J., Prince, A. L., Antony, K. M., Seferovic, M. D., and Aagaard, K. M. (2017). Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat. Med. 23, 314–326. doi: 10.1038/nm.4272
Darmasseelane, K., Hyde, M. J., Santhakumaran, S., Gale, C., and Modi, N. (2014). Mode of delivery and offspring body mass index, overweight and obesity in adult life: a systematic review and meta-analysis. PLoS ONE 9, e87896. doi: 10.1371/journal.pone.0087896
Dominguez-Bello, M. G., Costello, E. K., Contreras, M., Magris, M., Hidalgo, G., Fierer, N., et al. (2010). Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Nat. Acad. Sci. U.S.A. 107, 11971–11975. doi: 10.1073/pnas.1002601107
Fallani, M., Young, D., Scott, J., Norin, E., Amarri, S., Adam, R., et al. (2010). Intestinal microbiota of 6-week-old infants across Europe: geographic influence beyond delivery mode, breast-feeding, and antibiotics. J. Pediatr. Gastroenterol. Nutr. 51, 77–84. doi: 10.1097/MPG.0b013e3181d1b11e
Ferretti, P., Pasolli, E., Tett, A., Asnicar, F., Gorfer, V., Fedi, S., et al. (2018). Mother-to-infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell Host Microbe 24, 133–145. doi: 10.1016/j.chom.2018.06.005
Francino, M. P. (2018). Birth mode-related differences in gut microbiota colonization and immune system development. Ann Nutr Metab. 73, 12–16. doi: 10.1159/000490842
Hamdi, Y., Zass, L., Othman, H., Radouani, F., Allali, I., Hanachi, M., et al. (2021). Human OMICs and computational biology research in Africa: current challenges and prospects. Omics 25, 213–233. doi: 10.1089/omi.2021.0004
Houghteling, P. D., and Walker, W. A. (2015). Why is initial bacterial colonization of the intestine important to the infant's and child's health?. J. Pediatr. Gastroenterol. Nutr. 60, 294. doi: 10.1097/MPG.0000000000000597
Hu, J., Nomura, Y., Bashir, A., Fernandez-Hernandez, H., Itzkowitz, S., Pei, Z., et al. (2013). Diversified microbiota of meconium is affected by maternal diabetes status. PLoS ONE 8, e78257. doi: 10.1371/journal.pone.0078257
Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119. doi: 10.1186/1471-2105-11-119
Jakobsson, H. E., Abrahamsson, T. R., Jenmalm, M. C., Harris, K., Quince, C., Jernberg, C., et al. (2014). Decreased gut microbiota diversity, delayed bacteroidetes colonisation and reduced Th1 responses in infants delivered by caesarean section. Gut 63, 559–566. doi: 10.1136/gutjnl-2012-303249
Jenabi, E., Khazaei, S., Bashirian, S., Aghababaei, S., and Matinnia, N. (2020). Reasons for elective cesarean section on maternal request: a systematic review. J. Mater. Fetal Neonatal Med. 33, 3867–3872. doi: 10.1080/14767058.2019.1587407
Kim, G., Bae, J., Kim, M. J., Kwon, H., Park, G., Kim, S. J., et al. (2020). Delayed establishment of gut microbiota in infants delivered by cesarean section. Front. Microbiol. 11, 2099. doi: 10.3389/fmicb.2020.02099
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Liu, C. J., Liang, X., Niu, Z. Y., Jin, Q., Zeng, X. Q., Wang, W. X., et al. (2019). Is the delivery mode a critical factor for the microbial communities in the meconium? EBioMedicine 49, 354–363. doi: 10.1016/j.ebiom.2019.10.045
Liu, D., Yu, J., Li, L., Ai, Q., Feng, J., Song, C., et al. (2015). Bacterial community structure associated with elective cesarean section versus vaginal delivery in Chinese newborns. J. Pediatr. Gastroenterol. Nutr. 60, 240–246. doi: 10.1097/MPG.0000000000000606
Lu, J., Breitwieser, F. P., Thielen, P., and Salzberg, S. L. (2017). Bracken: estimating species abundance in metagenomics data. PeerJ Comp. Sci. 3, e104. doi: 10.7717/peerj-cs.104
Malamitsi-Puchner, A., Protonotariou, E., Boutsikou, T., Makrakis, E., Sarandakou, A., and Creatsas, G. (2005). The influence of the mode of delivery on circulating cytokine concentrations in the perinatal period. Early Hum. Dev. 81, 387–392. doi: 10.1016/j.earlhumdev.2004.10.017
Marcobal, A., Barboza, M., Sonnenburg, E. D., Pudlo, N., Martens, E. C., Desai, P., et al. (2011). Bacteroides in the infant gut consume milk oligosaccharides via mucus-utilization pathways. Cell Host Microbe 10, 507–514. doi: 10.1016/j.chom.2011.10.007
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217. doi: 10.1371/journal.pone.0061217
Milani, C., Duranti, S., Bottacini, F., Casey, E., Turroni, F., Mahony, J., et al. (2017). The first microbial colonizers of the human gut: composition, activities, and health implications of the infant gut microbiota. Microbiol. Mol. Biol. Rev. 81, e00036–e00017. doi: 10.1128/MMBR.00036-17
Mitchell, C. M., Mazzoni, C., Hogstrom, L., Bryant, A., Bergerat, A., Cher, A., et al. (2020). Delivery mode affects stability of early infant gut microbiota. Cell Rep. Med. 1, 100156. doi: 10.1016/j.xcrm.2020.100156
Paulson, J. N., Stine, O. C., Bravo, H. C., and Pop, M. (2013). Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 10, 1200–1202. doi: 10.1038/nmeth.2658
Perez-Muñoz, M. E., Arrieta, M. C., Ramer-Tait, A. E., and Walter, J. (2017). A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: implications for research on the pioneer infant microbiome. microbiome 5, 48. doi: 10.1186/s40168-017-0268-4
Peterson, D., Bonham, K. S., Rowland, S., Pattanayak, C. W., and Klepac-Ceraj, V. (2021). Comparative analysis of 16S rRNA gene and metagenome sequencing in pediatric gut microbiomes. Front. Microbiol. 12, 1651. doi: 10.3389/fmicb.2021.670336
Puente-Sánchez, F., García-García, N., and Tamames, J. (2020). SQMtools: automated processing and visual analysis of'omics data with R and anvi'o. BMC Bioinform. 21, 358. doi: 10.1186/s12859-020-03703-2
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Rastall, R. A. (2004). Bacteria in the gut: friends and foes and how to alter the balance. J Nutr. 134, 2022S−2026S. doi: 10.1093/jn/134.8.2022S
Rautava, S., Luoto, R., Salminen, S., and Isolauri, E. (2012). Microbial contact during pregnancy, intestinal colonization and human disease. Nat. Rev. Gastroenterol. Hepatol. 9, 565–576. doi: 10.1038/nrgastro.2012.144
Rutayisire, E., Huang, K., Liu, Y., and Tao, F. (2016). The mode of delivery affects the diversity and colonization pattern of the gut microbiota during the first year of infants' life: a systematic review. BMC Gastroenterol. 16, 86. doi: 10.1186/s12876-016-0498-0
Sanidad, K. Z., and Zeng, M. Y. (2020). Neonatal gut microbiome and immunity. Curr. Opin. Microbiol. 56, 30–37. doi: 10.1016/j.mib.2020.05.011
Santajit, S., and Indrawattana, N. (2016). Mechanisms of antimicrobial resistance in ESKAPE pathogens. Biomed Res. Int. 2016, 2475067. doi: 10.1155/2016/2475067
Saturio, S., Nogacka, A. M., Suárez, M., Fernández, N., Mantecón, L., Mancabelli, L., et al. (2021). Early-life development of the bifidobacterial community in the infant gut. Int. J. Mol. Sci. 22, 3382. doi: 10.3390/ijms22073382
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. doi: 10.1186/gb-2011-12-6-r60
Sevelsted, A., Stokholm, J., Bønnelykke, K., and Bisgaard, H. (2015). Cesarean section and chronic immune disorders. Pediatrics 135, e92–e98. doi: 10.1542/peds.2014-0596
Shao, Y., Forster, S. C., Tsaliki, E., Vervier, K., Strang, A., Simpson, N., et al. (2019). Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature 574, 117–121. doi: 10.1038/s41586-019-1560-1
Shetty, S. A., and Lahti, L. (2020). microbiomeutilities: Utilities for Microbiome Analytics. Available online at: https://microsud.github.io/microbiomeutilities/
Shi, Y. C., Guo, H., Chen, J., Sun, G., Ren, R. R., Guo, M. Z., et al. (2018). Initial meconium microbiome in Chinese neonates delivered naturally or by cesarean section. Sci. Rep. 8, 3255. doi: 10.1038/s41598-018-21657-7
Stiemsma, L. T., and Michels, K. B. (2018). The role of the microbiome in the developmental origins of health and disease. Pediatrics 141, e20172437. doi: 10.1542/peds.2017-2437
Stinson, L. F., Payne, M. S., and Keelan, J. A. (2018). A critical review of the bacterial baptism hypothesis and the impact of cesarean delivery on the infant microbiome. Front. Med. 5, 135. doi: 10.3389/fmed.2018.00135
Stokholm, J., Thorsen, J., Chawes, B. L., Schjørring, S., Krogfelt, K. A., Bønnelykke, K., et al. (2016). Cesarean section changes neonatal gut colonization. J. Allergy Clin. Immunol. 138, 881–889. doi: 10.1016/j.jaci.2016.01.028
Tamames, J., and Puente-Sánchez, F. (2019). SqueezeMeta, a highly portable, fully automatic metagenomic analysis pipeline. Front. Microbiol. 9, 3349. doi: 10.3389/fmicb.2018.03349
Tanaka, M., and Nakayama, J. (2017). Development of the gut microbiota in infancy and its impact on health in later life. Allergol. Int. 66, 515–522. doi: 10.1016/j.alit.2017.07.010
Tannock, G. W., Fuller, R., Smith, S. L., and Hall, M. A. (1990). Plasmid profiling of members of the family Enterobacteriaceae, Lactobacilli, and Bifidobacteria to study the transmission of bacteria from mother to infant. J. Clin. Microbiol. 28, 1225–1228. doi: 10.1128/jcm.28.6.1225-1228.1990
Tsirogiannis, C., and Sandel, B. (2016). PhyloMeasures: a package for computing phylogenetic biodiversity measures and their statistical moments. Ecography 39, 709–714. doi: 10.1111/ecog.01814
Unicef report (2018). UNICEF Data Warehouse, 2018. Available online at: https://data.unicef.org/resources/data_explorer/unicef_f/?ag=UNICEF&df=GLOBAL_DATAFLOW&ver=1.0&dq=.MNCH_CSEC..&startPeriod=2015&endPeriod=2018
Walker, W. A. (2017). The importance of appropriate initial bacterial colonization of the intestine in newborn, child, and adult health. Pediatr. Res. 82, 387–395. doi: 10.1038/pr.2017.111
Wampach, L., Heintz-Buschart, A., Fritz, J. V., Ramiro-Garcia, J., Habier, J., Herold, M., et al. (2018). Birth mode is associated with earliest strain-conferred gut microbiome functions and immunostimulatory potential. Nat. Commun. 9, 5091. doi: 10.1038/s41467-018-07631-x
Wampach, L., Heintz-Buschart, A., Hogan, A., Muller, E. E., Narayanasamy, S., Laczny, C. C., et al. (2017). Colonization and succession within the human gut microbiome by archaea, bacteria, and microeukaryotes during the first year of life. Front. Microbiol. 8, 738. doi: 10.3389/fmicb.2017.00738
Wang, S., Egan, M., Ryan, C. A., Boyaval, P., Dempsey, E. M., Ross, R. P., et al. (2020). A good start in life is important—perinatal factors dictate early microbiota development and longer term maturation. FEMS Microbiol. Rev. 44, 763–781. doi: 10.1093/femsre/fuaa030
Wingett, S. W., and Andrews, S. (2018). FastQ Screen: a tool for multi-genome mapping and quality control. F1000Research 7, 1338. doi: 10.12688/f1000research.15931.1
Wood, D. E., Lu, J., and Langmead, B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257. doi: 10.1186/s13059-019-1891-0
Yassour, M., Vatanen, T., Siljander, H., Hämäläinen, A. M., Härkönen, T., Ryhänen, S. J., et al. (2016). Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci. Transl. Med. 8, 343ra81. doi: 10.1126/scitranslmed.aad0917
Keywords: shotgun metagenome sequencing, newborns, elective cesarean deliveries, ESKAPE bacteria, Tunisia, microbiome
Citation: Hanachi M, Maghrebi O, Bichiou H, Trabelsi F, Bouyahia NM, Zhioua F, Belghith M, Harigua-Souiai E, Baouendi M, Guizani-Tabbane L, Benkahla A and Souiai O (2022) Longitudinal and Comparative Analysis of Gut Microbiota of Tunisian Newborns According to Delivery Mode. Front. Microbiol. 13:780568. doi: 10.3389/fmicb.2022.780568
Received: 21 September 2021; Accepted: 15 March 2022;
Published: 25 April 2022.
Edited by:
David W. Ussery, University of Arkansas for Medical Sciences, United StatesReviewed by:
Roshonda Jones, Independent Researcher, Oakland, United StatesFabrice Armougom, Institut de Recherche Pour le Développement (IRD), France
Copyright © 2022 Hanachi, Maghrebi, Bichiou, Trabelsi, Bouyahia, Zhioua, Belghith, Harigua-Souiai, Baouendi, Guizani-Tabbane, Benkahla and Souiai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oussema Souiai, b3Vzc2VtYS5zb3VpYWlAcGFzdGV1ci50bg==
†These authors have contributed equally to this work