 Li Yin Tan
Li Yin Tan Thamil Vaani Komarasamy
Thamil Vaani Komarasamy William James3
William James3 Vinod R. M. T. Balasubramaniam
Vinod R. M. T. Balasubramaniam- 1Infection and Immunity Research Strength, Jeffrey Cheah School of Medicine and Health Sciences, Monash University Malaysia, Bandar Sunway, Malaysia
- 2Greenslopes Private Hospital, Greenslopes, QLD, Australia
- 3Sir William Dunn School of Pathology, University of Oxford, Oxford, United Kingdom
Zika virus (ZIKV) is a mosquito-borne, single-stranded RNA virus belonging to the genus Flavivirus. Although ZIKV infection is usually known to exhibit mild clinical symptoms, intrauterine ZIKV infections have been associated with severe neurological manifestations, including microcephaly and Guillain Barre syndrome (GBS). Therefore, it is imperative to understand the mechanisms of ZIKV entry into the central nervous system (CNS) and its effect on brain cells. Several routes of neuro-invasion have been identified, among which blood–brain barrier (BBB) disruption is the commonest mode of access. The molecular receptors involved in viral entry remain unknown; with various proposed molecular ZIKV-host interactions including potential non-receptor mediated cellular entry. As ZIKV invade neuronal cells, they trigger neurotoxic mechanisms via cell-autonomous and non-cell autonomous pathways, resulting in neurogenesis dysfunction, viral replication, and cell death, all of which eventually lead to microcephaly. Together, our understanding of the biological mechanisms of ZIKV exposure would aid in the development of anti-ZIKV therapies targeting host cellular and/or viral components to combat ZIKV infection and its neurological manifestations. In this present work, we review the current understanding of ZIKV entry mechanisms into the CNS and its implications on the brain. We also highlight the status of the drug repurposing approach for the development of potential antiviral drugs against ZIKV.
Introduction
Zika virus (ZIKV) is a mosquito-vectored flavivirus, consisting of three structural proteins [capsid (C), pre-membrane/membrane (prM/M), and envelope (E)], seven non-structural (NS) proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) and a single-stranded RNA genome of positive polarity (Yun and Lee, 2017). ZIKV has been detected in saliva, tears, urine, semen, brain, female genital tract, and testes (Morrison and Diamond, 2017). ZIKV along with the other members of the Flaviviridae family, including West Nile virus (WNV), dengue virus (DENV), hepatitis C virus (HCV), and Japanese encephalitis virus (JEV) possess significant neuroinvasive characteristics and are identified as neurotropic. In recent years, ZIKV received global attention due to the association with more severe neurological manifestations, such as Guillain-Barre syndrome (GBS) in adults (Oehler et al., 2014; Cao-Lormeau et al., 2016; Watrin et al., 2016) as well as microcephaly in infants (Brasil et al., 2016; Schuler-Faccini et al., 2016). Recent studies have demonstrated the neuro-invasiveness, tropism, and virulence of ZIKV (Shao et al., 2016; Costa et al., 2017a; Zhang et al., 2019). Owing to these severe complications, an extensive understanding of ZIKV neuroinvasion mechanisms and the host molecules involved is vital for therapy development.
Receptors in Central Nervous System Targeted by ZIKV
The entry mechanism of ZIKV into human cells, particularly neural cells, remains poorly understood. Previous studies have demonstrated several ZIKV entry factors, including dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN), AXL receptor tyrosine kinase (AXL), TYRO3 protein tyrosine kinase (TYRO3) and though to a lesser extent, T-cell immunoglobulin and mucin domain 1 (TIM-1), mediate entry of ZIKV into human dermal fibroblasts, epidermal keratinocytes, and immature dendritic cells (Hamel et al., 2015; Table 1). It is of note that ZIKV infection in primary dermal fibroblast was significantly decreased by RNA inhibitor and neutralizing antibody to AXL (Hamel et al., 2015). Therefore, the AXL receptor appears to play a vital role as the viral entry receptor. High expression of AXL receptor is seen within cells of the developing central nervous system (CNS), such as endothelial cells, microglial, astrocytes, and radial glial cells (Table 1; Nowakowski et al., 2016). A study found that the susceptibility of ZIKV to endothelial cells positively correlated with the cell surface levels of AXL (Liu et al., 2016). Belonging to the group of tyrosine kinase receptors TYRO3, AXL, and MER (TAM) family, AXL acts in apoptotic cells clearance and innate immunity modulation (Rothlin et al., 2007; Lemke and Rothlin, 2008). It is said that ZIKV is attached indirectly to the AXL receptor, mediated by the natural ligand of AXL, growth arrest–specific gene 6 (Gas6) based on the exposure of phosphatidylserine (PS) on viral envelope surface. Astrocytes and microglial cells appear to be the major ZIKV targets, having remained strongly expressed in the developing human cortex even as gestation progress (Meertens et al., 2017).
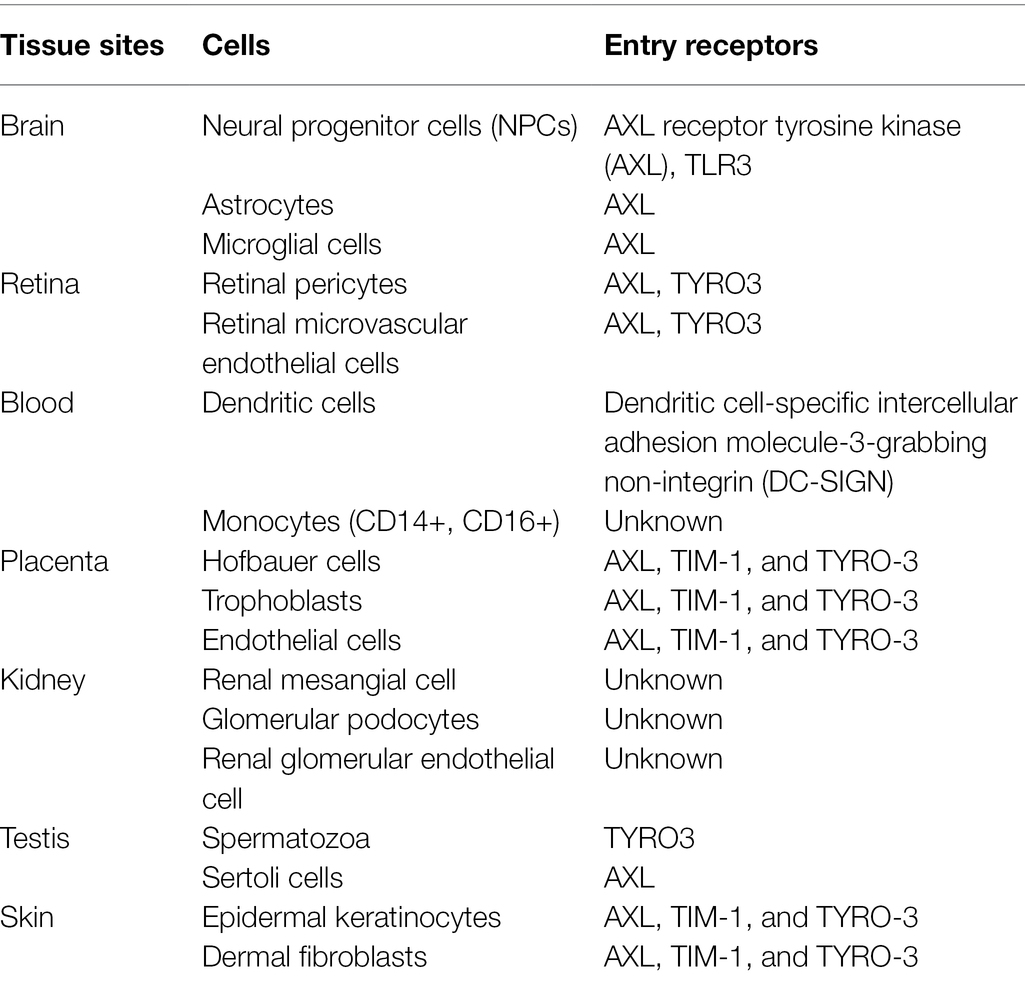
Table 1. Host cell entry receptors targeted by ZIKV (Lee et al., 2018; Lee and Shin, 2019).
However, recent findings demonstrated that ZIKV still showed effective invasion, infection, and replication in AXL-depleted cerebral organoids (Wells et al., 2016), neural progenitor cells (NPCs; Wells et al., 2016), and murine models (Wang et al., 2017b; Li et al., 2017c), suggesting that AXL may not serve as the exclusive receptor involved and that its dependency for viral entry may be cell-type or model specific. A further study reported the role of AXL in antagonizing ZIKV-induced activation of type I interferon (IFN) signaling, which facilitates ZIKV infection in astrocytes, instead of being a ZIKV entry receptor (Chen et al., 2018). While active investigations are still required on ZIKV target receptors, other receptor-independent mechanisms should be considered. Exosomes, secreted by most cell types, transport proteins, lipids, and nucleic acids between cells through systemic circulation or paracrine transmission, thereby exerting functional responses in target cells (Ramakrishnaiah et al., 2013). Several studies have shown exosome-mediated intercellular transmission of viruses (Narayanan et al., 2013; Ramakrishnaiah et al., 2013; Zhu et al., 2015). Tunneling nanotubes that connect a number of cell types, including immune and neuronal cells, are also shown to promote viral spread (Sowinski et al., 2008).
Tan et al. (2019) studied the mechanism underlying ZIKV infection in Vero, Huh-7, and induced pluripotent stem cell (iPSC)-derived human NPCs (hNPCs) using cell surface carbohydrates, sialic acid, and being the attachment receptor for several viruses. Although there was no direct involvement with ZIKV attachment, findings suggested that sialic acid could be an important mediator in the internalization of the ZIKV-receptor complex and its depletion significantly reduced infection in NPCs (Tan et al., 2019). Nevertheless, the underlying mechanism remains unclear and requires further research (Tan et al., 2019). Other factors, such as neural cell adhesion molecule 1 (NCAM1) and integrin αVβ5 have been postulated to act as cell-type-specific receptors for ZIKV entry (Srivastava et al., 2020; Wang et al., 2020).
The Mechanisms of ZIKV Crossing the BBB
The blood–brain barrier (BBB) comprises endothelial cells strongly adhered via tight junction proteins (TJP), which are associated with pericytes, astrocytes, and microglia (Abbott et al., 2010; Mustafa et al., 2019). The tight junctions (TJ) complexes of transmembrane proteins composed of structural proteins claudins and occludins found on plasma membranes of adjacent brain endothelial cells maintain the integrity of BBB. Several routes of invasion into the CNS have been demonstrated by neurotropic viruses: (Yun and Lee, 2017) transcellular (through cells) transport within endothelial cells through infection or transcytosis, and subsequent release of the virus into the CNS, (Morrison and Diamond, 2017) infected peripheral immune cells such as monocytes enter the CNS through Trojan horse strategy, (Oehler et al., 2014) paracellular (between cells) entry of virus following disruption of the BBB, and destabilization of TJ, (Cao-Lormeau et al., 2016) retrograde axonal transport of virus through peripheral nerves into the CNS, and (Watrin et al., 2016) blood-to-cerebral-spinal-fluid (CSF) translocation (Ayala-Nunez and Gaudin, 2020; Hsieh and St John, 2020; Chen and Li, 2021). Among these, BBB dysfunction is the most common neuroinvasion route for flavivirus due to indirect effects of systemic inflammatory cytokines such as tumor necrosis factor (TNF) and IFN or direct attachment to claudins (Neal, 2014).
It was demonstrated that ZIKV can infect and efficiently replicate in the human brain microvascular endothelial cells (HBMECs) and iPSC-derived BBB models without significantly altering the endothelial barrier integrity and permeability in vitro (Mladinich et al., 2017; Papa et al., 2017; Alimonti et al., 2018). Despite the efficient viral replication, the endothelium displayed tight BBB and its architecture was not extensively perturbed. However, actin cytoskeleton rearrangement was observed upon infection, suggesting possible changes in the morphology of the endothelium (Cle et al., 2020). These findings suggest that endothelial leakage and BBB disruption may not be essential for ZIKV to reach the brain, and ZIKV might utilize other mechanisms to invade the CNS (Figure 1).
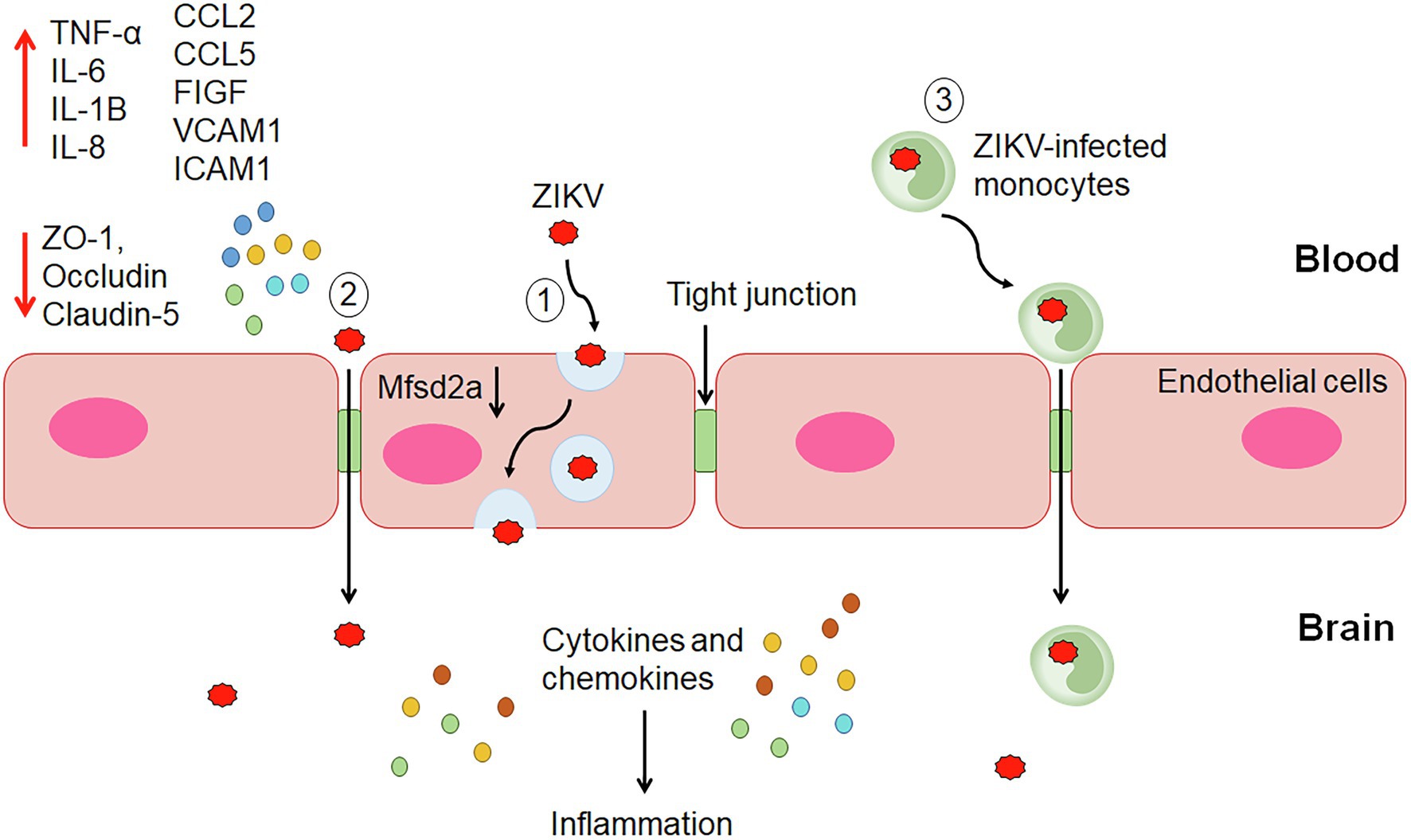
Figure 1. The proposed mechanisms of ZIKV invasion into the central nervous system (CNS). (1) Transcellular transport within endothelial cells of the BBB through infection or transcytosis mediated by ZIKV-induced degradation of Mfsd2a. (2) Paracellular trafficking of ZIKV across the blood–brain barrier (BBB) occurs through the upregulation of proinflammatory cytokines, chemokines, adhesion molecules and growth factors, and downregulation of tight junction proteins leading to alteration of the endothelial barrier integrity and permeability. (3) ZIKV-infected monocytes cross the BBB via the Trojan horse strategy. Once reach the CNS, ZIKV infects the brain cells, including astrocytes and microglial cells producing cytokines and chemokines leading to inflammation.
Several in vitro studies have demonstrated ZIKV infection and release from both apical and basolateral surfaces of brain endothelial cells without compromising the barrier permeability and integrity (Mladinich et al., 2017; Papa et al., 2017; Alimonti et al., 2018). ZIKV RNA was detected in the lower chamber of the transwell system despite maintaining the endothelial integrity. These studies support that ZIKV may cross the BBB through transcytosis, basolateral virus release, or paracytosis (Figure 1). A charge-based mechanism occurs whereby positively charged ZIKV particles foster attraction with the negatively charged membrane of brain endothelial cells for transcytosis (Nakayama et al., 2021). The secretion of type I and III IFNs and inflammatory cytokines did not affect microvascular endothelial cells (MECs) permeability but contributed to transinfection (Papa et al., 2017). These findings suggest potential drugs that block ZIKV replication or transcytosis can effectively prevent virus extravasation through MEC monolayer. Treatment with Chloroquine (inhibitor of cellular infection) and BFA (exocytosis inhibitor), inhibited virus RNA release and extravasation in a transwell system, indicating active virus replication is required for ZIKV to cross the BBB. On the other hand, treatment with Nystatin (inhibitor of caveolae-mediated transcytosis) impaired extravasation of ZIKV through HBMECs without affecting virus replication, suggesting utilization of transcytosis and/or basolateral release pathways by ZIKV to cross the BBB after infection and activation of endothelial cells (Papa et al., 2017).
The ZIKV transcytosis may be mediated by the major facilitator superfamily domain-containing protein 2 (Mfsd2a; Figure 1). Mfsd2a is selectively expressed on the CNS endothelial cells, and its genetic ablation resulted in increased transcytosis and leaky BBB (Ben-Zvi et al., 2014). It plays a critical role in transporting docosahexaenoic acid (DHA) into CNS (Chen and Li, 2021). The lipids transported by Mfsd2a mediate inhibition of caveolae vesicle formation in CNS endothelial cells and suppress transcytosis through which it maintains the integrity of the BBB. The deficiency of Msfd2a reduced the levels of DHA in the brain, which was accompanied by loss of neuronal cells, cognitive deficits, and microcephaly (Nguyen et al., 2014). ZIKV envelope (E) protein was found to interact with Mfsd2a. ZIKV E promotes polyubiquitination of Mfsd2a and mediates its proteasome-dependent degradation. ZIKV inhibited the levels of Mfsd2a in hBMECs and in neonatal mouse brains. In addition, ZIKV caused a reduction in the Mfsd2a-mediated DHA uptake and supplementation with DHA rescued ZIKV-indued abnormal brain development in mice (Zhou et al., 2019).
A study by Ayala-Nunez et al. (2019) proposed that ZIKV may use the Trojan horse strategy to cross the BBB by infecting the monocytes (Figure 1). The study found ZIKV-infected monocytes in the brain slices from the fetus with microcephaly. It was further demonstrated that ZIKV productively infected human primary monocytes and promoted viral dissemination to cerebral organoids. ZIKV manipulates the adhesive properties of monocytes, particularly CD14 and CD16 monocytes by increasing its expression of surface adhesion molecules (integrins, ICAM3, PECAM1, IQGAP1, catenin, myosins, actinin, KIF5B, vinculin, talin, and filamin A and B). This enables the ZIKV-infected monocytes to have a greater attachment to the blood vessel wall and transmigrate across the endothelium to infect neural cells (Ayala-Nunez et al., 2019). A study showed that HBMECs allowed transmigration of ZIKV-infected human monocyte THP-1 in a transwell system and subsequently infected the astrocytes in the basolateral compartment. As astrocytes play an important role in the maintenance of BBB through direct interaction with endothelial cells in the brains, ZIKV infection of these cells after crossing the endothelial layer may lead to inflammation and alteration of the barrier. In this context, modulation of inflammatory molecules, such as, C-C motif ligand-5 (CCL5/RANTES), C-X-C motif chemokine ligand 10 (CXCL10), and IFN-β was observed in astrocytes (Bramley et al., 2017; Cle et al., 2020). ZIKV infection of monocytes was also observed in macaques, and the infected cells were recruited to tissues, resulting in persistent viral infection (O’Connor et al., 2018). Hence, ZIKV-infected monocytes could represent a carrier for the Trojan horse strategy to invade the CNS.
The Trojan horse transmigration of ZIKV-infected monocytes across BBB depends on two receptors, chemokine receptor 7 (CCR7) and receptor for advanced glycation end (RAGE) expressed on ZIKV-infected monocytes. In a state of inflammation, the natural ligand of CCR7, chemokine ligand 19 (CCL19), and the danger-associated molecular pattern (DAMP) molecule, nuclear high mobility group box 1 (HMGB1) are upregulated. This receptor-ligand interaction induces dysregulation of the endothelium layer associated with disorganization of cadherins and actin fibers, thereby increasing the membrane’s permissiveness (de Carvalho et al., 2019). The process is mediated by the upregulation of C-X-C motif chemokine 12 (CXCL12) in monocyte during ZIKV infection, a key regulator of lymphocytes shifts across BBB into CNS parenchyma as it interacts with C-X-C chemokine receptor type 4 (CXCR4). The accompanying lymphocytes-induced inflammation in brain parenchyma further triggers subsequent downstream activation pathways resulting in increased BBB damage (Panganiban et al., 2020).
The paracellular pathway involving proteasomal degradation mechanism has been proposed as one of the pathways for BBB penetration (Figure 1). A study by Shao et al. showed that ZIKV infection induced altered vasculature and a leaky BBB in developing mouse brain (Shao et al., 2016). An in vitro study using transfected HBMECs (THBMEC) infected with ZIKV demonstrated vascular leakage enhancement by ZIKV through disruption in the cytoskeleton and tight junctional proteins arrangement. This occurs via the upregulation of genes that are involved in the production of proinflammatory cytokines, such as interleukin-6 (IL-6), TNF-α, cell adhesion molecules (CAMs), and growth factors (Ismail et al., 2018), which activates the RhoA/Rho-associated coiled-coil containing protein kinase (ROCK)/phosphorylated myosin light chain (pMLC) signaling effectors, thus intensifying stress fibers production (Chen and Li, 2021). Additionally, matrix metalloproteinases (MMP-2/MMP-9) and the proteasome are stimulated by the RhoA mechanism in the breakdown of TJP (Chen and Li, 2021). Downregulation of TJP zonula occludens-1 (ZO-1), occludin, and Claudin-5 by ZIKV (Leda et al., 2019), affecting their ability for transient phosphorylation and dephosphorylation (Siddiqui et al., 2015), was linked to an increase in trans endothelial permeability and BBB penetration (Argaw et al., 2009). The ZIKV-induced BBB disruption is likely to occur at later stages of the disease (Papa et al., 2017; Leda et al., 2019).
Zika virus infection of HBMECs and in vitro BBB model enhanced expression of type I and III IFNs and induced a significant increase in the release of cytokines [interleukin-1β (IL-1β), interleukin-8 (IL-8), IL-6, and TNF-α], chemokines [C-C motif ligand-2 (CCL2/MCP-1) and CCL5], and CAMs [vascular cell adhesion molecule 1 (VCAM1) and intercellular adhesion molecule 1 (ICAM1)]. ZIKV infection of pericytes showed upregulation of chemokines (CCL5 and CXCL10), cytokines [IL-6, IL-8, and interleukin-15 (IL-15)], as well as Toll-like receptor 3 (TLR3; Cle et al., 2020). The modulation of BBB proteins induced by ZIKV infection of endothelial cells and pericytes may trigger recruitment and docking of immune cells to the BBB and potentially lead to immune cell CNS infiltration and neuroinflammation (Papa et al., 2017; Cle et al., 2020). Increased levels of CAM were also observed in ZIKV-infected mouse models as well as in plasma from patients. In a mouse model, BBB permeability was not observed during the early time points, but subtle BBB alteration was detected at later time points, which could be associated with an inflammatory response triggered by viral replication (Papa et al., 2017). A study by Bramley et al. (2017) demonstrated that pre-treatment of a three-dimensional (3D) model of microvascular endothelial cells with TNF-α enhanced virus replication and disorganization of the junctional network, suggesting the potential role of inflammatory response in BBB disruption in vivo.
As a matter of interest, cerebral organoid designs hold promising means in studying the 3D morphological and molecular characterization in ZIKV-induced microcephaly as it is able to recapitulate the human brain environment (Antonucci and Gehrke, 2019). However, this system has limited vascularization required for the prolonged culture period, presenting difficulty in discerning the necrotic core due to ZIKV or suboptimal preparation (Antonucci and Gehrke, 2019; Chen et al., 2019). The lack of BBB affects the ability to emulate the natural neuroimmune response toward ZIKV by infiltrating peripheral immune cells across the BBB (Antonucci and Gehrke, 2019). Therefore, co-culturing or other bioengineering techniques would improve the brain organoid model and provide a clear picture of the relationship between the CNS and peripheral circulatory system (Chen et al., 2019).
On the other hand, ZIKV infection and dissemination along peripheral nerves lack evidence and may not be the primary mechanism; however, it cannot be fully disregarded. Contrary to the BBB endothelial cells, the choroid plexus endothelial cells have fenestrated blood capillaries and are leaky, thereby could provide a pathway for the virus to spread out of the blood and enter into the choroid plexus cisternae. ZIKV can infect the pericytes around the fenestrated capillaries, forming a local amplification site and subsequently transcytosed across the blood-CSF layers (Kim et al., 2020).
The Implications of ZIKV Infection on the Brain
Similar to primary microcephaly, ZIKV-related microcephaly was reported to be the neurodevelopmental disruption during the first trimester when cortical neurogenesis is most active (Johansson et al., 2016). ZIKV causes microcephaly by affecting the NPCs through cell-autonomous and non-cell autonomous pathways.
Cell cycle perturbation by ZIKV, in particular the S-phase restriction, provides a favorable cellular environment for ZIKV replication as well as impairs the growth of hNPCs (Hammack et al., 2019). ZIKV causes host DNA breaks, activating the ataxia telangiectasia mutated (ATM)/Chk2 signaling cascade and inhibiting ataxia telangiectasia and rad3+ related (ATR)/Chk1 signaling cascade (Hammack et al., 2019). DNA damage response (DDR) is subsequently activated where DNA repair proteins H2A.X and 53BP1 are phosphorylated and cell cycle regulators proteins cell division cycle 25 A (CDC25A), cyclin A and cyclin E are degraded, resulting in G1/S transition arrest (Hammack et al., 2019). G0/G1 transition arrest was not shown to enhance ZIKV replication, implying a possible need for host DNA replication or damage repair factors for its replication (Hammack et al., 2019). Another in vitro study found that ZIKV envelope proteins restrict G2/M progression and induce apoptosis in neuroendocrine PC12 cells through upregulation of tumor suppressor protein, p53 and cyclin-dependent kinase (CDK) inhibitor (CDKi), p21Cip1/Waf1 leading to downregulation of G2/M phase regulator, cyclin B1, and augmentation of the proapoptotic pathway with the increased B-cell lymphoma protein 2 (Bcl-2)-associated X (Bax)/Bcl-2 ratio (Liu et al., 2018). In this context, the intrinsic cell death signaling mechanism is triggered by the activation of caspase-9 and caspase-3 (Liu et al., 2018). The attenuation of neural cell proliferation in the ventricular, subventricular, and intermediate zone of the fetal brain causes a significant decrease in intermediate progenitor cells, lateral ventricles size, and cortical surface area (Wu et al., 2016). Additionally, defective cell division due to ZIKV may precede the apoptotic program of neural stem cells; a cellular mechanism used to prevent genomic instability (Vitale et al., 2011; Souza et al., 2016). A number of mitotic dysfunctions were reported, such as extra centrosomes, multipolar spindles, partial segregation of spindle pole, chromosomal aneuploidy, and micronuclei production (Souza et al., 2016).
Recent findings revealed that ZIKV causes mitochondrial stress in human derived-iPSC astrocytes with alteration to both its structure and metabolism to supply the energy demand for viral replication through oxidative phosphorylation (OxPhos) pathway of ATP production (Ledur et al., 2020; Figure 2). Mitochondria being the putative avenue for reactive oxygen species (ROS) production were observed to have an increased level of oxidative stress in infected astrocytes, contributing to the DNA breaks and activation of DDR (Ledur et al., 2020; Figure 2). DDR plays an important role in inducing cell cycle arrest apoptosis (Devhare et al., 2017). The activation of DDR triggered phosphorylation of Histone H2AX (γH2AX) followed by assembly of 53BP1, both of which interact with other DNA repair proteins at the foci of DNA double-stranded break (Ledur et al., 2020; Figure 2). DDR plays an important role in inducing cell cycle arrest apoptosis (Devhare et al., 2017). Intermediate filaments, glial fibrillary acidic protein (GFAP), and Vimentin were elevated in infected astrocytes in vitro and in vivo, corroborating with reactive astrogliosis induced by ZIKV (Ledur et al., 2020). That said, mitochondrial dysfunction has been known to be associated with various neurological disorders, including neurodegenerative diseases, cerebral hypoxia, cerebral ischemia, and other brain injuries (Lopez-Domenech et al., 2016; Alimonti et al., 2018; Ludwig et al., 2018; Zhao et al., 2019). In the context of astrocytes, mitochondrial damage has been shown to cause apoptosis in motor neurons, and axon destruction in GBS (Madigan et al., 2017).
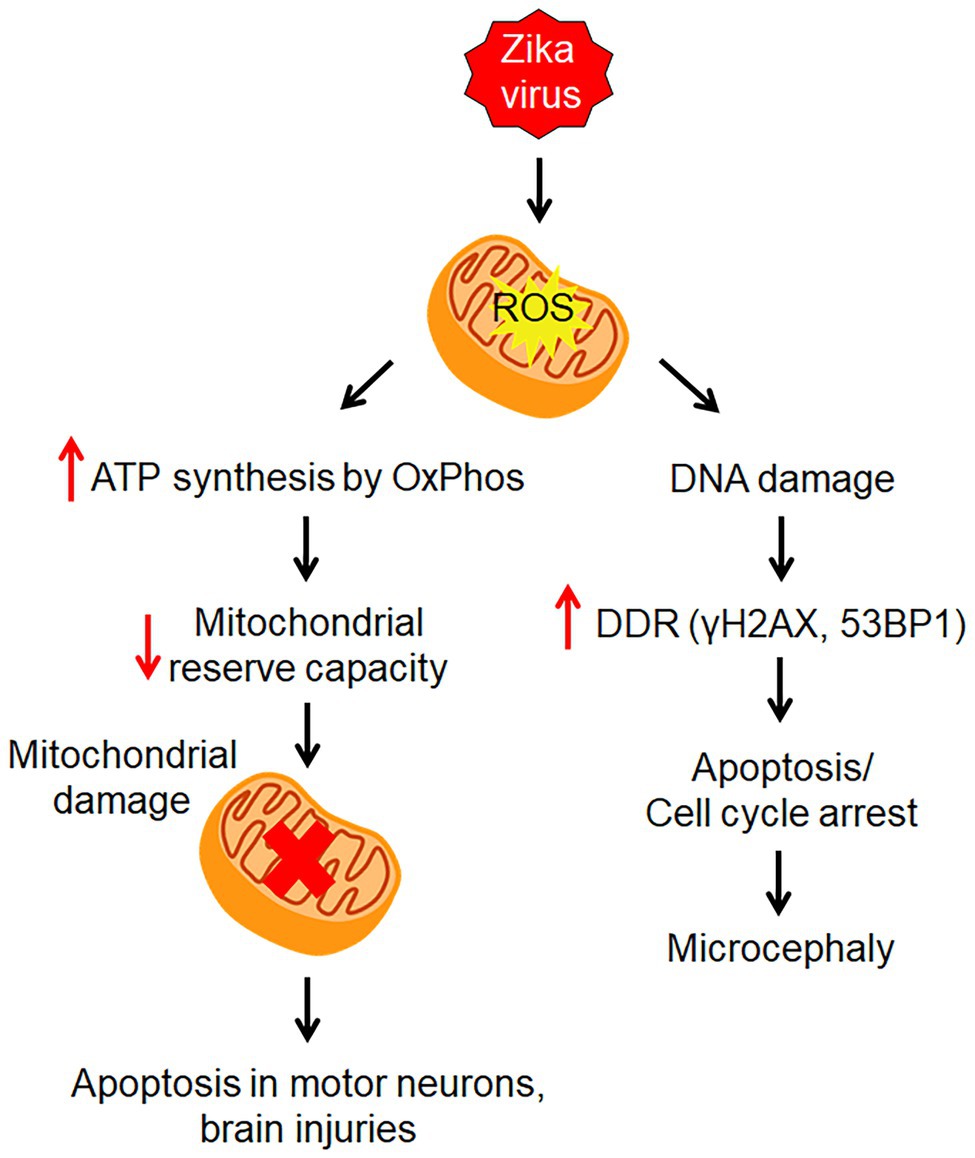
Figure 2. Zika virus-induced mitochondrial stress. ZIKV infection causes mitochondrial stress by altering its structure and metabolism to supply energy for viral replication through oxidative phosphorylation (OxPhos) pathway. This leads to an increase in reactive oxygen species (ROS) which causes DNA damage. Then the DNA damage response (DDR) is induced to activate repair pathways to monitor DNA damage.
Zika virus is capable of modulating the host endoplasmic reticulum (ER) structure for its replicative benefits (Ropidi et al., 2020; Figure 3). The viral NS4A exploits host reticulon 3.1A to promote ER membrane curvature for viral entry into ER, allowing replication to take place (Ropidi et al., 2020). The budded ZIKV RNA, along with other ZIKV proteins such as NS2B-NS3 and unprocessed C-prM-Env complexes are then assembled and cleaved in adjacent ER by ZIKV NS2A protein and NS2B-NS3 protease, respectively thereby producing new ZIKV virions (Ropidi et al., 2020). However, in the process of ZIKV infection, excessive formation of misfolded proteins overpowers the ER protein-folding capacity, causing ER stress and activation of unfolded protein response (UPR), which ultimately lead to apoptosis (Ropidi et al., 2020; Figure 3). In vitro and in vivo studies have shown an increase in ER stress proteins as well as expression of key molecules of the UPR, such as glucose regulatory protein 78 (GRP78), calreticulin, calnexin, and protein disulfide isomerase (PDI) in infected neural cells (Ropidi et al., 2020), resulting in neurogenesis inhibition and microcephaly (Gladwyn-Ng et al., 2018; Figure 3). In parallel, ZIKV-induced ER stress represses stress granules (SG) assembly, which functions to arrest global translation via eIF2α phosphorylation and SG proteins exploitation (Ropidi et al., 2020; Figure 3). Reticulophagy, a compensatory host innate defense response to engulf viral protein and damaged ER for lysosomal degradation, is inhibited by ZIKV NS2B-NS3 protease (NS2B/3; Ropidi et al., 2020; Figure 3). Besides intrinsic and extrinsic apoptotic cell death, the prolonged ER stress renders paraptosis-like death through extensive cytoplasmic vacuolization in ZIKV-infected cells (Ropidi et al., 2020; Figure 3).
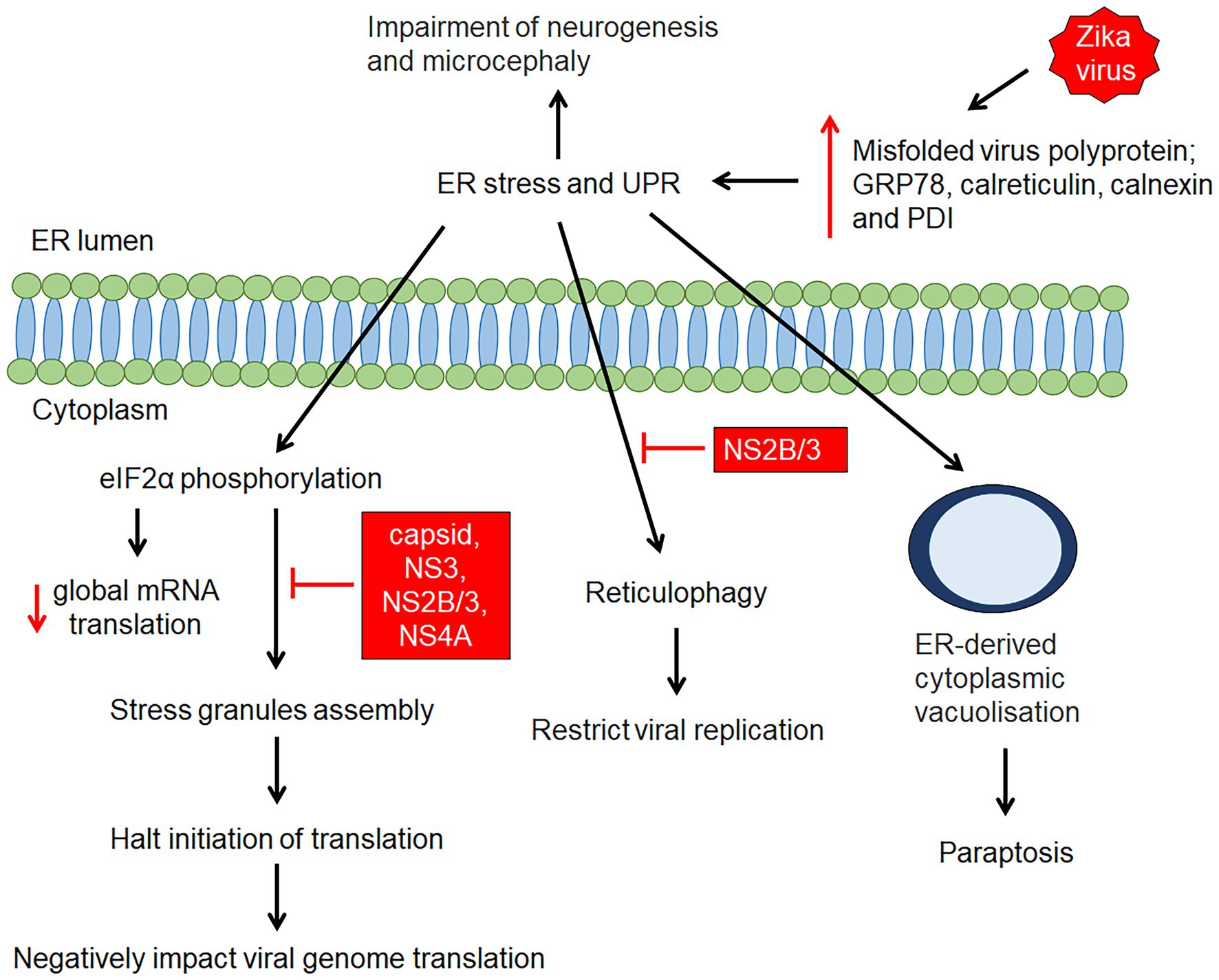
Figure 3. ZIKV-induced endoplasmic reticulum (ER) stress and unfolded protein response. ZIKV infection causes structural changes of the ER as a result of accumulation of misfolded/unfolded ZIKV proteins and remodelling of the ER structure for viral RNA replication. The accumulation of misfolded/unfolded ZIKV proteins and the increase in ER protein-folding capacity triggers ER stress and activates the unfolded protein response (UPR), resulting in elevation of GRP78, calnexin, calreticulin, and protein disulfide isomerase (PDI). This is followed by initiation of several mechanisms such as global protein translation, stress granule assembly, reticulophagy and cytoplasmic vacuolisation. Zika virus proteins (capsid, NS3, NS2B/3 and NS4A proteins) has shown to, suppress SGs assembly, while NS2B/3 has shown to inhibit reticulophagy which facilitate continuous viral replication.
Modulation of the host autophagy mechanism has been demonstrated by flaviviruses, such as DENV (Heaton and Randall, 2010), HCV (Sir et al., 2012), and ZIKV in skin fibroblast (Hamel et al., 2015) to promote viral replication. Recent findings showed that ZIKV induced the autophagosome-specific marker, cytosolic microtubule-associated light chain 3 (LC3), indicating autophagosomes are proviral and provide the avenue for ZIKV replication (Liang et al., 2016). ZIKV NS4A and NS4B have shown to inhibit the AKT-mammalian target of rapamycin (mTOR) signaling, resulting in reduced neurogenesis and increased autophagy in human fetal neural stem cells, therefore elevating viral replication (Liang et al., 2016). Although the precise molecular mechanism by ZIKV on autophagosomes is still elusive, various pathways, such as deregulating the host antiviral innate immunity (Ke and Chen, 2011), upregulating viral RNA translation (Dreux et al., 2009), and modulating host lipid metabolism for viral replication (Heaton and Randall, 2010) have been proposed and would benefit from further exploration.
From the non-cell autonomous pathway aspect, a recent study demonstrated that ZIKV induced neuronal apoptosis on adjacent healthy neurons while maintaining its replication in the infected neurons, potentially via ZIKV’s upregulation of pre-mRNA-processing-splicing factor 8 (Pprf8), an anti-apoptotic factor demonstrated in Picornavirus-infected neurons (Olmo et al., 2017). ZIKV-infected neurons undergo neuroinflammation process, secreting TNF-α, IL-1β, and glutamate (Olmo et al., 2017). These neurotoxic factors activate GluN2B-containing N-methyl-d-aspartate receptor (NMDAR), potentiating Ca2+ influx into the cell, which promotes excitotoxicity and eventually cell death (Olmo et al., 2017).
Cortical development is a highly complex process requiring tightly regulated and finely tuned sequential machinery. Hence, as vascular development is pivotal for organogenesis, the impairment of neurogenesis is demonstrated to be in part, due to vascular abnormalities (Garcez et al., 2018). Defective vasculature and decreased neuronal proliferation were concurrently apparent in the ZIKV-infected mice brain models, particularly in the ventricular zone, indicating the presence of a causal relationship between them (Garcez et al., 2018). These findings are further supported by the significant elevation in anti-angiogenic proteins, Ang1, Ang3, as well as endostatin and pigment epithelium-derived factor (PEDF), albeit to a smaller degree from proteomic analysis (Garcez et al., 2018).
Drug-Repurposing Strategy for the Treatment of ZIKV
Since the 2015 ZIKV outbreak and its association with congenital abnormalities, tremendous progress has been made in vaccine and antiviral research. More than 25 ZIKV vaccine candidates have been evaluated in nonclinical development and at least 12 are in clinical evaluations. These candidates include DNA, mRNA, viral-vectored, inactivated, and live attenuated vaccines (Barrett, 2018; Poland et al., 2019; Lee et al., 2021). Some of these candidates have demonstrated the ability to prevent viral transmission during pregnancy, as well as fetal malformations and demise in animal models (Richner et al., 2017; Shan et al., 2017; Li et al., 2018). Multiple candidates have been shown to be safe, well-tolerated and immunogenic in humans, with two candidates (VRC5288 and mRNA-1893) having advanced into phase 2 clinical trials (Barrett, 2018; Lee et al., 2021). In addition, therapeutic vaccination for ZIKV has been extensively explored as an alternative to vaccines. A total of 461 monoclonal antibodies (mAbs) that bind to E proteins have been identified, with 70 of them displaying moderate to high neutralizing activities (Wang et al., 2017c). Notably, administration of convalescent serum from a ZIKV-infected patient not only inhibited ZIKV replication but also prevented microcephaly in a mouse model (Wang et al., 2017a). While treatment with a human mAb, ZIKV-117 reduced vertical transmission and improved fetal outcome (Sapparapu et al., 2016). Another study showed that a cocktail of three neutralizing mAbs targeting different domains of the ZIKV E protein completely prevented viremia in non-human primates (NHPs; Magnani et al., 2017).
On the other hand, several approaches have been employed to identify drugs against ZIKV infection. The current search for ZIKV antivirals can be classified based on their mode of action, such as (i) host-directed antivirals, which focus on modulating host cellular processes used for viral life cycle or (ii) direct-acting antivirals, which target viral components. In order to expedite the development of effective antivirals against ZIKV, much focus has been given to drug repurposing or drug re-profiling (Table 2). Recommended therapies for ZIKV infection would require the ability to cross the placenta and BBB, have anti-ZIKV functionality primarily on fetal neural cells and be safe to use during pregnancy.
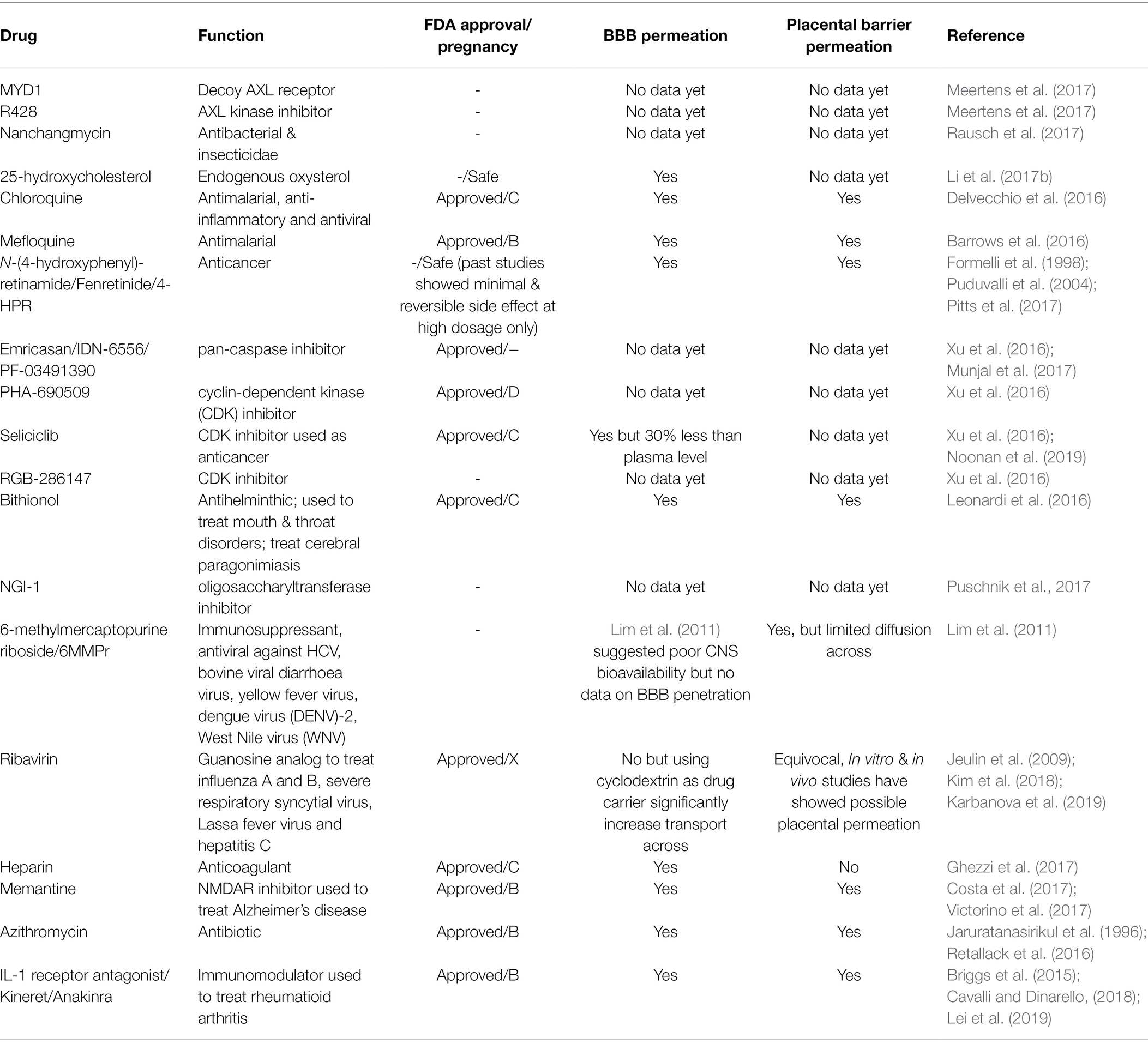
Table 2. Potential anti-ZIKV drugs focusing on studies done on neuronal cells.
Host-Directed Antivirals
Attachment/Entry
MYD1, an engineered decoy AXL receptor, has a high-affinity binding to Gas6, preventing the interaction between ZIKV viral particles and the AXL receptor (Meertens et al., 2017). AXL kinase inhibitor R428 prevents phosphorylation of AXL, which activates the host innate immunity (Meertens et al., 2017). Both MYD1 and R428 inhibit ZIKV infection of human glial cells in a dose-dependent manner and are identified as potential antivirals through the AXL/Gas6 pathway inhibition (Meertens et al., 2017).
Nanchangmycin, an antibacterial and insecticidal polyether, potently inhibits the early entry stage in the ZIKV replication process and infection in primary cells of uterine, placenta, umbilical vein, BBB, and neuron-glial mixed midbrain at 0.1–0.4 μM with low cytopathic effects (Rausch et al., 2017).
25-hydroxycholesterol (25HC), an endogenous oxysterol formed via cholesterol oxidization, demonstrated antiviral activity by preventing ZIKV entry through internalization and fusion of ZIKV envelope with host endosomal membrane, not via ZIKV attachment (Li et al., 2017b). It decreased viral load and mortality in mice, inhibited ZIKV RNA shedding and symptoms in NHPs as well as protected developing human cortical organoids and embryonic mouse brain from ZIKV-associated neurological impairment and fetal microcephaly, respectively (Li et al., 2017b). 25HC displayed effectiveness in Vero cells at a concentration as low as 0.4 μM with no observable cytotoxicity at a concentration up to 10 μM. No adverse effects were seen on pregnant and neonatal mice at 50 mg/kg of 25HC (Li et al., 2017b).
Endosomal Fusion
Chloroquine, known for its antimalarial, anti-inflammatory, and antiviral activity, has been shown to protect Vero cells, HBMECs (in vitro BBB model), hNPCs, and mouse neurospheres from ZIKV infection (Delvecchio et al., 2016). It acts by increasing the endosomal pH, consequently preventing the fusion of ZIKV-host endosomal membrane, suppressing ZIKV-induced infections without exhibiting cytotoxicity (Delvecchio et al., 2016). Of note, chloroquine was said to be safe in pregnancy and able to penetrate the maternal-fetal placental barrier with a 4–30-fold higher concentration in the brain than in plasma (Delvecchio et al., 2016). These features are imperative in reducing the risk of infections and ZIKV-related microcephaly (Delvecchio et al., 2016).
Another antimalarial drug Mefloquine is proposed to work by preventing autophagy and interrupting the cellular lysosomal pH (Barrows et al., 2016). At a concentration of 10 μM, it was able to inhibit ZIKV infection to cervical HeLa cells, placental JEG3 cells, and primary human amnion epithelial cells (Barrows et al., 2016). Indeed, while further work is warranted to validate the beneficial anti-ZIKV effects of Mefloquine, the current data show promising anti-ZIKV effects, well-tolerated in pregnancy, as well as ability to cross to the placenta and the BBB (Barrows et al., 2016).
Translation/Transcription
N-(4-hydroxyphenyl)-retinamide (4-HPR or Fenretinide), known for its anticancer properties, is found to also exhibit anti-ZIKV activity, presumably facilitated by a host factor, in significantly reducing ZIKV RNA production without effect on viral polymerase or membrane-related replication complexes (Pitts et al., 2017). 4-HPR was shown to inhibit ZIKV in multiple mammalian cell lines culture as well as eliminate ZIKV viremia and brain viral load in vivo (Pitts et al., 2017).
Replication
Emricasan, a pan-caspase inhibitor (IDN-6556/PF-03491390), and PHA-690509, a CDKi, are able to suppress the caspase-3 pathway as well as improve hNPCs and astrocyte viability (Xu et al., 2016). Their effects are amplified even more when used in combination (Xu et al., 2016). Notably, the host cellular cyclin-dependent kinase (CDK) may serve as a valuable anti-ZIKV target given the close interconnection between CDKi, cell cycle regulation, and ZIKV proliferation (Xu et al., 2016). PHA-690509 alone is reported to partially protect hNPC against proliferation reduction and ZIKV replication (Xu et al., 2016). This inhibition effect on ZIKV production is corroborated by the identification of other CDKi, Seliciclib, and RGB-286147 with submicromolar half-maximal inhibitory concentration (IC50) at 24 and 27 nM, respectively (Xu et al., 2016). Bithionol, on the other hand, also blocks ZIKV-induced host caspases, possibly caspase-1, −3, −6, −7, and − 9, in Vero cells and human astrocytes (Leonardi et al., 2016). It is regarded highly as a potential therapy due to its ability to penetrate the placental and BBB and has been reported to be well-tolerated (Leonardi et al., 2016).
ER-Targeting Drugs
In various cell types, including hNPC, NGI-1 targets the ER-membrane multiprotein complex, and oligosaccharyltransferase (OST) complex, which is responsible for the catalysis of N-linked glycosylation of new proteins, by inhibiting viral RNA production and replication but independent of OST complex’s catalytic activity (Puschnik et al., 2017).
Nucleoside Biosynthesis
Azathioprine-derived thiopurine nucleoside analog, 6-methymercaptopurine riboside (6MMPr) was tested on epithelial and human neuronal cells, revealing efficient, dose-dependent inhibition of ZIKV infection, and lower cytotoxicity for neuronal cells (de Carvalho et al., 2017). It robustly blocks the de novo production pathway of purine, resulting in a smaller pool of nucleotides to be used for viral replication (de Carvalho et al., 2017). 6MMPr is reported to be well-tolerated during pregnancy and able to cross the placental barrier (de Carvalho et al., 2017). However, diffusion into the fetal vascular system did not reach a significant level (de Carvalho et al., 2017).
Ribavirin is a guanosine analog approved to treat influenza A and B, severe respiratory syncytial virus, Lassa fever virus, and hepatitis C (Kim et al., 2018). Similar to Favipiravir, Ribavirin showed a robust antiviral effect against ZIKV infection in hNPCs, as seen by the substantial dose-dependent decrease in mRNA expression of ZIKV E and NS5, with the greatest reduction at the 25 μg/ml (Kim et al., 2018). The study further demonstrated the extension of anti-ZIKV activity in human dermal fibroblasts, human lung adenocarcinoma cells, and Vero cells (Kim et al., 2018).
Cytopathic Effects Inhibition
Heparin, an anticoagulant, exhibited anti-apoptotic activity in hNPCs, potentially through inactivation of caspase-3 (Ghezzi et al., 2017). Its ultra-low molecular weight form is shown to infiltrate the BBB (Ghezzi et al., 2017). Unexpectedly, heparin only has a modest effect in curbing ZIKV infection and replication in hNPCs (Ghezzi et al., 2017). While heparin has no absolute contraindications during pregnancy, its inability to pass the placental barrier would necessitate a tailored drug delivery platform (Ghezzi et al., 2017).
Given that ZIKV can induce inflammation in infected neuronal cells and subsequent glutamate release to promote neurodegeneration of adjacent cells, Memantine which is a non-competitive NMDAR inhibitor used as Alzheimer’s disease therapy can inactivate NMDAR which are the primary ionotropic glutamate receptors in the brain, preventing high calcium influx and neurotoxicity at dosages 1, 10, and 30 μM (Costa et al., 2017b). The selective antagonism of Memantine toward overstimulated receptors presents lower cytotoxicity than other NMDAR antagonists and is thus classified as pregnancy class B by the US Food and Drug Administration (FDA; Costa et al., 2017b). Both in vitro and in vivo studies here demonstrated neuroprotective effects of memantine, Dizocilpine/MK-801, agmatine sulfate, and ifenprodil in ameliorating ZIKV-induced apoptosis without impacting the viral replication process (Costa et al., 2017b). Memantine was reported to be able to prevent microgliosis and overall brain injury, particularly in the cortical, striatal, and hippocampal regions (Costa et al., 2017b).
Unknown Mechanism
Antibiotic of macrolide-type Azithromycin, which is used to treat respiratory or sexually transmitted diseases, was reported to be able to cross the placental barrier and reach the fetal tissue, with concentrations of ~2.8 μM in placenta and 4–21 μM in the fetus (Retallack et al., 2016). Azithromycin was observed to decrease ZIKV infection, proliferation and cellular apoptosis in glial cells, and astrocytes (Retallack et al., 2016).
Placenta
As interleukin-1 (IL-1) receptors are expressed in microglial cells, IL-1 receptor antagonist (IRA) reduces fetal neuroinflammation by inhibiting fetal microglial activation (Lei et al., 2019). IRA indirectly prevents fetal neurocognitive abnormalities by preserving placental development despite existing ZIKV replication through its inhibition on ZIKV-induced placental proinflammatory cytokines IL-1β (Lei et al., 2019). This action reduces placental inflammation and increases trophoblast invasion and placental vascularity (Lei et al., 2019).
Direct-Acting Antivirals
NS2B–NS3 Protease
Zika virus utilizes its encoded NS3 protein and NS2B cofactor for proteolytic cleavage of its polyprotein in the viral maturation process. One such compound efficacious in ZIKV inhibition through this mechanism is Niclosamide (NIC), an FDA-approved anthelmintic therapy. Consistent with previous in vitro findings using glioblastoma SNB-19 cells (Xu et al., 2016), NIC is able to inhibit ZIKV production, decrease proinflammatory proteins such as CXCL10 and leukemia inhibitor factor (LIF), partially restore altered neuronal differentiation profile, and inhibit apoptosis in human induced neural stem cell (hiNSC), with pre- and/or concomitant NIC treatment (Cairns et al., 2018). Using humanized in vivo embryo, NIC showed neuroprotective effects by partially restoring morphological change of the brain and differentiation profile in hiNSCs (Cairns et al., 2018). Together with niclosamide, temoporfin, a photosensitizer used for squamous cell carcinoma and nitazoxanide, an anthelmintic drug, share a similar mechanism in inhibiting NS2B-NS3 proteins interactions (Li et al., 2017a). Both cause inactivation of ZIKV protease and attenuation of ZIKV polyprotein precursor processing, thus abolishing ZIKV replication in human placental epithelial cells (HPECs), iPSC, and iPSC-derived hNPCs (Li et al., 2017a). All except temoporfin possess good safety profiles for use in pregnant patients (Li et al., 2017a).
NS5 RdRp
Sofosbuvir, which is a uridine nucleotide analog and anti-HCV drug, uses its 2′-F radical to form a covalent bond with amino acid residue Asn612 (Sacramento et al., 2017). The formation of this covalent bond impairs the subsequent hydrogen bonds formed between nucleotides and ZIKV RNA polymerase, therefore acting as a transcription terminator which directly blocks ZIKV RNA polymerase (ZVRP) in various cell models (Sacramento et al., 2017). Interestingly, sequence analysis data found a higher mutation rate in Sofosbuvir-treated cells, inferring an additional anti-ZIKV effect where sofosbuvir initiates mutations on the ZIKV genetic profile, causing elevated A–G mutation levels and increasing ZIKV susceptibility to replication error (Sacramento et al., 2017).
Recently, a novel RdRp inhibitor Favipiravir (6-fluoro-3-hydroxy-2-pyrazinecarboxamide or T-705) has been shown to promote neuronal cell growth and ameliorate apoptosis (Kim et al., 2018). It upregulates the AKT phosphorylation in the phosphatidylinositol-3-kinase (PI3K)/AKT pathway essential for neurogenesis and increases the expression of anti-apoptotic mediator Bcl-2, whereas pro-apoptotic factor Bax is reduced (Kim et al., 2018). ZIKV E and NS5 gene expression levels were also significantly reduced in hNPCs at 1, 10, and 25 μM, suggesting an effective suppression of ZIKV infection (Kim et al., 2018). Though the mode of interaction has yet to be elucidated, probable hypotheses are either direct misincorporation into the developing viral RNA sequence or indirect inhibition of transcription via attachment to polymerase (Baz and Boivin, 2019).
Others
A study using a mice model revealed an engineered AH peptide with D-enantiomer (AH-D) confers protection against ZIKV by stimulating viral liposome rupture (Jackman et al., 2018). Therapeutic administration of AH-D is reported to be able to abrogate ZIKV-related infection, apoptosis, and replication in neuronal and other systemic cells (Jackman et al., 2018). This finding is corroborated by a decrease in mortality and morbidity even when AH-D is given prophylactically (Jackman et al., 2018). Additionally, it can infiltrate the intact BBB without altering BBB permeability to reduce ZIKV burden and neuroinflammation in CNS through reduction of proinflammatory mediators, thus preventing BBB injury and neurodegeneration (Jackman et al., 2018).
Conclusion
Following the sudden increase in the number of new-borns with microcephaly during the Brazillian outbreak of ZIKV, numerous studies have been conducted and they verify the causal relationship between ZIKV infection and neurological disorders. ZIKV possesses the ability to cross the placental barrier and BBB, targeting brain cells, particularly in the developing brain. The virus crosses the BBB through (i) transcellular pathway, (ii) monocyte transmigration through Trojan horse pathway, (iii) vascular leakage enhancement, and (iv) disruption of the choroid plexus epithelial layer. Upon crossing the BBB and entering the brain, ZIKV infects various cells in the developing brain, causing neuropathogenesis through (i) cell cycle perturbation, (ii) mitochondrial dysfunction, (iii) ER stress and UTP, (iv) modulation of host autophagy, and (v) neuronal apoptosis. Despite major advances in this field, many important questions remain answered: (i) Why only a small number of fetuses born to infected mothers develop microcephaly? What are the long-term outcomes of infected neonates without detectable abnormalities at birth? (ii) Is an intact BBB permissive to ZIKV infection? Can ZIKV target astrocytes in the adult brain? Will there be any long-term effects in adults infected with ZIKV? and (iii) Since the innate immune responses and mitochondria functions are connected, how does mitochondrial dysfunction upon ZIKV infection affect the cellular immune responses? Therefore, more studies are required to fully understand the mechanisms of cross-talk between ZIKV and the human brain, as well as the host factors involved in utero transmission and neuropathogenesis. In addition, systematic and long-term follow-ups are imperative to determine the unknown long-term neuropathological and behavioral consequences in both new-borns and adults.
Importantly, despite its severe implications and potential future outbreaks, there are still no vaccine or antiviral drugs available against ZIKV. The development of new therapeutic approaches should be one of the priorities for future research. The drug-repurposing strategy offers a promising avenue to identify potential antiviral drugs against ZIKV within a shorter development timeline and known safety. This approach is also particularly appealing for mosquito-borne viruses, which receive less attention from the affluent regions and pharmaceutical companies. A number of host-targeting and virus-targeting agents with potential inhibitory activity against ZIKV have been identified. However, the majority of these drugs have only been tested in vitro. It is also critical to evaluate their efficacy in appropriate animal models to better predict their clinical outcome. In addition, the ability of these drugs to prevent ZIKV CNS invasion needs to be evaluated. Importantly, ZIKV drugs should be clinically safe for use in pregnant women and fetuses.
Author Contributions
LT and VB: conceptualization. LT and TK: methodology and writing—original draft preparation. LT, TK, WJ, and VB: writing, review, and editing. VB and WJ: supervision. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the reviewer for critical comments and suggestions. We would also like to thank the school and administration staff of Jeffrey Cheah School of Medicine and Health Sciences, Monash University Malaysia for their continuous support to this project and lab members of Infectious Disease Laboratory for critically reading this review.
References
Abbott, N. J., Patabendige, A. A., Dolman, D. E., Yusof, S. R., and Begley, D. J. (2010). Structure and function of the blood-brain barrier. Neurobiol. Dis. 37, 13–25. doi: 10.1016/j.nbd.2009.07.030
Alimonti, J. B., Ribecco-Lutkiewicz, M., Sodja, C., Jezierski, A., Stanimirovic, D. B., Liu, Q., et al. (2018). Zika virus crosses an in vitro human blood brain barrier model. Fluid. Barrier. CNS. 15:15. doi: 10.1186/s12987-018-0100-y
Antonucci, J., and Gehrke, L. (2019). Cerebral organoid models for neurotropic viruses. ACS Infect. Dis. 5, 1976–1979. doi: 10.1021/acsinfecdis.9b00339
Argaw, A. T., Gurfein, B. T., Zhang, Y., Zameer, A., and John, G. R. (2009). VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc. Natl. Acad. Sci. U. S. A. 106, 1977–1982. doi: 10.1073/pnas.0808698106
Ayala-Nunez, N. V., Follain, G., Delalande, F., Hirschler, A., Partiot, E., Hale, G. L., et al. (2019). Zika virus enhances monocyte adhesion and transmigration favoring viral dissemination to neural cells. Nat. Commun. 10:4430. doi: 10.1038/s41467-019-12408-x
Ayala-Nunez, N. V., and Gaudin, R. (2020). A viral journey to the brain: current considerations and future developments. PLoS Pathog. 16:e1008434. doi: 10.1371/journal.ppat.1008434
Barrett, A. D. T. (2018). Current status of Zika vaccine development: Zika vaccines advance into clinical evaluation. NPJ Vaccines 3:24. doi: 10.1038/s41541-018-0061-9
Barrows, N. J., Campos, R. K., Powell, S. T., Prasanth, K. R., Schott-Lerner, G., Soto-Acosta, R., et al. (2016). A screen of FDA-approved drugs for inhibitors of zika virus infection. Cell Host Microbe 20, 259–270. doi: 10.1016/j.chom.2016.07.004
Baz, M., and Boivin, G. (2019). Antiviral agents in development for zika virus infections. Pharmaceuticals 12:101. doi: 10.3390/ph12030101
Ben-Zvi, A., Lacoste, B., Kur, E., Andreone, B. J., Mayshar, Y., Yan, H., et al. (2014). Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature 509, 507–511. doi: 10.1038/nature13324
Bramley, J. C., Drummond, C. G., Lennemann, N. J., Good, C. A., Kim, K. S., and Coyne, C. B. (2017). A three-dimensional cell culture system to model RNA virus infections at the blood-brain barrier. mSphere 2, e00206–e00217. doi: 10.1128/mSphere.00206-17
Brasil, P., Calvet, G. A., Siqueira, A. M., Wakimoto, M., de Sequeira, P. C., Nobre, A., et al. (2016). Zika virus outbreak in Rio de Janeiro, Brazil: clinical characterization, epidemiological and virological aspects. PLoS Negl. Trop. Dis. 10:e0004636. doi: 10.1371/journal.pntd.0004636
Briggs, G. G., Freeman, R. K., and Yaffe, S. J. (2015). Drugs in Pregnancy and Lactation: A Reference Guide to Fetal and Neonatal Risk. Philadelphia: Wolters Kluwer Health.
Cairns, D. M., Boorgu, D. S. S. K., Levin, M., and Kaplan, D. L. (2018). Niclosamide rescues microcephaly in a humanized in vivo model of Zika infection using human induced neural stem cells. Biol. Open. 7:bio031807. doi: 10.1242/bio.031807
Cao-Lormeau, V.-M., Blake, A., Mons, S., Lastère, S., Roche, C., Vanhomwegen, J., et al. (2016). Guillain-Barré syndrome outbreak associated with Zika virus infection in French Polynesia: a case-control study. Lancet 387, 1531–1539. doi: 10.1016/S0140-6736(16)00562-6
Cavalli, G., and Dinarello, C. A. (2018). Anakinra therapy for non-cancer inflammatory diseases. Front. Pharmacol. 9:1157. doi: 10.3389/fphar.2018.01157
Chen, Z., and Li, G. (2021). Immune response and blood-brain barrier dysfunction during viral neuroinvasion. Innate Immun. 27, 109–117. doi: 10.1177/1753425920954281
Chen, H. I., Song, H., and Ming, G.-l. (2019). Applications of human brain organoids to clinical problems. Dev. Dyn. 248, 53–64. doi: 10.1002/dvdy.24662
Chen, J., Yang, Y.-f., Yang, Y., Zou, P., Chen, J., He, Y., et al. (2018). AXL promotes Zika virus infection in astrocytes by antagonizing type I interferon signalling. Nat. Microbiol. 3, 302–309. doi: 10.1038/s41564-017-0092-4
Cle, M., Desmetz, C., Barthelemy, J., Martin, M. F., Constant, O., Maarifi, G., et al. (2020). Zika virus infection promotes local inflammation, cell adhesion molecule upregulation, and leukocyte recruitment at the blood-brain barrier. MBio 11, e01183–e01120. doi: 10.1128/mBio.01183-20
Costa, V. V., Del Sarto, J. L., Rocha, R. F., Silva, F. R., Doria, J. G., Olmo, I. G., et al. (2017a). N-methyl-d-aspartate (NMDA) receptor blockade prevents neuronal death induced by zika virus infection. MBio 8, e00350–e00317. doi: 10.1128/mBio.00350-17
Costa, V. V., Del Sarto, J. L., Rocha, R. F., Silva, F. R., Doria, J. G., Olmo, I. G., et al. (2017b). Aspartate (NMDA) receptor blockade prevents neuronal death induced by zika virus infection. MBio 8, e00350–e00317. doi: 10.1128/mBio.00350-17
de Carvalho, G. C., Borget, M. Y., Bernier, S., Garneau, D., da Silva Duarte, A. J., and Dumais, N. (2019). RAGE and CCR7 mediate the transmigration of Zika-infected monocytes through the blood-brain barrier. Immunobiology 224, 792–803. doi: 10.1016/j.imbio.2019.08.007
de Carvalho, O. V., Felix, D. M., de Mendonca, L. R., de Araujo, C., de Oliveira Franca, R. F., Cordeiro, M. T., et al. (2017). The thiopurine nucleoside analogue 6-methylmercaptopurine riboside (6MMPr) effectively blocks Zika virus replication. Int. J. Antimicrob. Agents 50, 718–725. doi: 10.1016/j.ijantimicag.2017.08.016
Delvecchio, R., Higa, L. M., Pezzuto, P., Valadão, A. L., Garcez, P. P., Monteiro, F. L., et al. (2016). Chloroquine, an endocytosis blocking agent, inhibits zika virus infection in different cell models. Viruses 8:322. doi: 10.3390/v8120322
Devhare, P., Meyer, K., Steele, R., Ray, R. B., and Ray, R. (2017). Zika virus infection dysregulates human neural stem cell growth and inhibits differentiation into neuroprogenitor cells. Cell Death Dis. 8:e3106. doi: 10.1038/cddis.2017.517
Dreux, M., Gastaminza, P., Wieland, S. F., and Chisari, F. V. (2009). The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. U. S. A. 106, 14046–14051. doi: 10.1073/pnas.0907344106
Formelli, F., De Palo, G., Costa, A., and Veronesi, U. (1998). Human transplacental passage of the retinoid fenretinide (4HPR). Eur. J. Cancer 34, 428–429. doi: 10.1016/S0959-8049(97)10013-2
Garcez, P. P., Stolp, H. B., Sravanam, S., Christoff, R. R., Ferreira, J. C. C. G., Dias, A. A., et al. (2018). Zika virus impairs the development of blood vessels in a mouse model of congenital infection. Sci. Rep. 8:12774. doi: 10.1038/s41598-018-31149-3
Ghezzi, S., Cooper, L., Rubio, A., Pagani, I., Capobianchi, M. R., Ippolito, G., et al. (2017). Heparin prevents Zika virus induced-cytopathic effects in human neural progenitor cells. Antivir. Res. 140, 13–17. doi: 10.1016/j.antiviral.2016.12.023
Gladwyn-Ng, I., Cordon-Barris, L., Alfano, C., Creppe, C., Couderc, T., Morelli, G., et al. (2018). Stress-induced unfolded protein response contributes to Zika virus-associated microcephaly. Nat. Neurosci. 21, 63–71. doi: 10.1038/s41593-017-0038-4
Hamel, R., Dejarnac, O., Wichit, S., Ekchariyawat, P., Neyret, A., Luplertlop, N., et al. (2015). Biology of Zika virus infection in human skin cells. J. Virol. 89:8880. doi: 10.1128/JVI.00354-15
Hammack, C., Ogden, S. C., Madden, J. C., Medina, A., Xu, C., Phillips, E., et al. (2019). Zika virus infection induces DNA damage response in human neural progenitors that enhances viral replication. J. Virol. 93, e00638–e00619. doi: 10.1128/JVI.00638-19
Heaton, N. S., and Randall, G. (2010). Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 8, 422–432. doi: 10.1016/j.chom.2010.10.006
Hsieh, J. T., and St John, A. L. (2020). Japanese encephalitis virus and its mechanisms of neuroinvasion. PLoS Pathog. 16:e1008260. doi: 10.1371/journal.ppat.1008260
Ismail, A., Mahboob, T., Chandramathi, S., and Sekaran, S. (2018). Zika virus modulates blood-brain barrier of brain microvascular endothelial cells. Trop. Biomed. 36, 888–897.
Jackman, J., Costa, V., Park, S., Real, A., Park, J., Leal Cardozo, P., et al. (2018). Therapeutic treatment of Zika virus infection using a brain-penetrating antiviral peptide. Nat. Mater. 17, 971–977. doi: 10.1038/s41563-018-0194-2
Jaruratanasirikul, S., Hortiwakul, R., Tantisarasart, T., Phuenpathom, N., and Tussanasunthornwong, S. (1996). Distribution of azithromycin into brain tissue, cerebrospinal fluid, and aqueous humor of the eye. Antimicrob. Agents Chemother. 40, 825–826. doi: 10.1128/AAC.40.3.825
Jeulin, H., Venard, V., Carapito, D., Finance, C., and Kedzierewicz, F. (2009). Effective ribavirin concentration in mice brain using cyclodextrin as a drug carrier: evaluation in a measles encephalitis model. Antivir. Res. 81, 261–266. doi: 10.1016/j.antiviral.2008.12.006
Johansson, M. A., Mier-y-Teran-Romero, L., Reefhuis, J., Gilboa, S. M., and Hills, S. L. (2016). Zika and the risk of microcephaly. N. Engl. J. Med. 375, 1–4. doi: 10.1056/NEJMp1605367
Karbanova, S., Cerveny, L., Jiraskova, L., Karahoda, R., Ceckova, M., Ptackova, Z., et al. (2019). Transport of ribavirin across the rat and human placental barrier: roles of nucleoside and ATP-binding cassette drug efflux transporters. Biochem. Pharmacol. 163, 60–70. doi: 10.1016/j.bcp.2019.01.024
Ke, P. Y., and Chen, S. S. (2011). Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Invest. 121, 37–56. doi: 10.1172/JCI41474
Kim, J., Alejandro, B., Hetman, M., Hattab, E. M., Joiner, J., Schroten, H., et al. (2020). Zika virus infects pericytes in the choroid plexus and enters the central nervous system through the blood-cerebrospinal fluid barrier. PLoS Pathog. 16:e1008204. doi: 10.1371/journal.ppat.1008204
Kim, J.-A., Seong, R.-K., Kumar, M., and Shin, O. S. (2018). Favipiravir and ribavirin inhibit replication of Asian and African strains of Zika virus in different cell models. Viruses 10:72. doi: 10.3390/v10020072
Leda, A. R., Bertrand, L., Andras, I. E., El-Hage, N., Nair, M., and Toborek, M. (2019). Selective disruption of the blood–brain barrier by Zika virus. Front. Microbiol. 10:2158. doi: 10.3389/fmicb.2019.02158
Ledur, P. F., Karmirian, K., Pedrosa, C. S. G., Souza, L. R. Q., Assis-de-Lemos, G., Martins, T. M., et al. (2020). Zika virus infection leads to mitochondrial failure, oxidative stress and DNA damage in human iPSC-derived astrocytes. Sci. Rep. 10:1218. doi: 10.1038/s41598-020-57914-x
Lee, I., Bos, S., Li, G., Wang, S., Gadea, G., Desprès, P., et al. (2018). Probing molecular insights into Zika virus−host interactions. Viruses 10:233. doi: 10.3390/v10050233
Lee, L. J., Komarasamy, T. V., Adnan, N. A. A., James, W., and Balasubramaniam, R. M. T. V. (2021). Hide and seek: the interplay between zika virus and the host immune response. Front. Immunol. 12:750365. doi: 10.3389/fimmu.2021.750365
Lee, J. K., and Shin, O. S. (2019). Advances in Zika virus-host cell interaction: current knowledge and future perspectives. Int. J. Mol. Sci. 20:1101. doi: 10.3390/ijms20051101
Lei, J., Vermillion, M. S., Jia, B., Xie, H., Xie, L., McLane, M. W., et al. (2019). IL-1 receptor antagonist therapy mitigates placental dysfunction and perinatal injury following Zika virus infection. JCI Insight 4:e122678. doi: 10.1172/jci.insight.122678
Lemke, G., and Rothlin, C. V. (2008). Immunobiology of the TAM receptors. Nat. Rev. Immunol. 8, 327–336. doi: 10.1038/nri2303
Leonardi, W., Zilbermintz, L., Cheng, L. W., Zozaya, J., Tran, S. H., Elliott, J. H., et al. (2016). Bithionol blocks pathogenicity of bacterial toxins, ricin, and Zika virus. Sci. Rep. 6:34475. doi: 10.1038/srep34475
Li, Z., Brecher, M., Deng, Y.-Q., Zhang, J., Sakamuru, S., Liu, B., et al. (2017a). Existing drugs as broad-spectrum and potent inhibitors for Zika virus by targeting NS2B-NS3 interaction. Cell Res. 27, 1046–1064. doi: 10.1038/cr.2017.88
Li, C., Deng, Y.-Q., Wang, S., Ma, F., Aliyari, R., Huang, X.-Y., et al. (2017b). 25-hydroxycholesterol protects host against zika virus infection and its associated microcephaly in a mouse model. Immunity 46, 446–456. doi: 10.1016/j.immuni.2017.02.012
Li, X. F., Dong, H. L., Wang, H. J., Huang, X. Y., Qiu, Y. F., Ji, X., et al. (2018). Development of a chimeric Zika vaccine using a licensed live-attenuated flavivirus vaccine as backbone. Nat. Commun. 9:673. doi: 10.1038/s41467-018-02975-w
Li, F., Wang, P.-R., Qu, L.-B., Yi, C.-H., Zhang, F.-C., Tang, X.-P., et al. (2017c). AXL is not essential for Zika virus infection in the mouse brain. Emerg. Microbe. Infect. 6:e16. doi: 10.1038/emi.2017.10
Liang, Q., Luo, Z., Zeng, J., Chen, W., Foo, S.-S., Lee, S.-A., et al. (2016). Zika virus NS4A and NS4B proteins deregulate Akt-mTOR signaling in human fetal neural stem cells to inhibit neurogenesis and induce autophagy. Cell Stem Cell 19, 663–671. doi: 10.1016/j.stem.2016.07.019
Lim, P. Y., Keating, J. A., Hoover, S., Striker, R., and Bernard, K. A. (2011). A thiopurine drug inhibits West Nile virus production in cell culture, but not in mice. PLoS One 6:e26697. doi: 10.1371/journal.pone.0026697
Liu, S., DeLalio, L. J., Isakson, B. E., and Wang, T. T. (2016). AXL-mediated productive infection of human endothelial cells by Zika virus. Circ. Res. 119, 1183–1189. doi: 10.1161/CIRCRESAHA.116.309866
Liu, J., Li, Q., Li, X., Qiu, Z., Li, A., Liang, W., et al. (2018). Zika virus envelope protein induces G2/M cell cycle arrest and apoptosis via an intrinsic cell death signaling pathway in neuroendocrine PC12 cells. Int. J. Biol. Sci. 14, 1099–1108. doi: 10.7150/ijbs.26400
Lopez-Domenech, G., Higgs, N. F., Vaccaro, V., Ros, H., Arancibia-Carcamo, I. L., MacAskill, A. F., et al. (2016). Loss of dendritic complexity precedes neurodegeneration in a mouse model with disrupted mitochondrial distribution in mature dendrites. Cell Rep. 17, 317–327. doi: 10.1016/j.celrep.2016.09.004
Ludwig, P. E., Thankam, F. G., Patil, A. A., Chamczuk, A. J., and Agrawal, D. K. (2018). Brain injury and neural stem cells. Neural Regen. Res. 13, 7–18. doi: 10.4103/1673-5374.224361
Madigan, C. A., Cambier, C. J., Kelly-Scumpia, K. M., Scumpia, P. O., Cheng, T. Y., Zailaa, J., et al. (2017). A macrophage response to mycobacterium leprae phenolic glycolipid initiates nerve damage in leprosy. Cell 170, 973–985.e10. doi: 10.1016/j.cell.2017.07.030
Magnani, D. M., Rogers, T. F., Beutler, N., Ricciardi, M. J., Bailey, V. K., Gonzalez-Nieto, L., et al. (2017). Neutralizing human monoclonal antibodies prevent Zika virus infection in macaques. Sci. Transl. Med. 9:eaan8184. doi: 10.1126/scitranslmed.aan8184
Meertens, L., Labeau, A., Dejarnac, O., Cipriani, S., Sinigaglia, L., Bonnet-Madin, L., et al. (2017). Axl mediates ZIKA virus entry in human glial cells and modulates innate immune responses. Cell Rep. 18, 324–333. doi: 10.1016/j.celrep.2016.12.045
Mladinich, M. C., Schwedes, J., and Mackow, E. R. (2017). Zika virus persistently infects and is basolaterally released from primary human brain microvascular endothelial cells. MBio 8, e00952–e00917. doi: 10.1128/mBio.00952-17
Morrison, T. E., and Diamond, M. S. (2017). Animal models of Zika virus infection, pathogenesis, and immunity. J. Virol. 91, e00009–e00017. doi: 10.1128/JVI.00009-17
Munjal, A., Khandia, R., Dhama, K., Sachan, S., Karthik, K., Tiwari, R., et al. (2017). Advances in developing therapies to combat Zika virus. Curr. Knowled. Fut. Perspect. Front. Microbiol. 8:1469. doi: 10.3389/fmicb.2017.01469
Mustafa, Y. M., Meuren, L. M., Coelho, S. V. A., and de Arruda, L. B. (2019). Pathways exploited by flaviviruses to counteract the blood-brain barrier and invade the central nervous system. Front. Microbiol. 10:525. doi: 10.3389/fmicb.2019.00525
Nakayama, E., Kato, F., Tajima, S., Ogawa, S., Yan, K., Takahashi, K., et al. (2021). Neuroinvasiveness of the MR766 strain of Zika virus in IFNAR−/− mice maps to prM residues conserved amongst African genotype viruses. PLoS Pathog. 17:e1009788. doi: 10.1371/journal.ppat.1009788
Narayanan, A., Iordanskiy, S., Das, R., Van Duyne, R., Santos, S., Jaworski, E., et al. (2013). Exosomes derived from HIV-1-infected cells contain trans-activation response element RNA. J. Biol. Chem. 288, 20014–20033. doi: 10.1074/jbc.M112.438895
Neal, J. W. (2014). Flaviviruses are neurotropic, but how do they invade the CNS? J. Inf. Secur. 69, 203–215. doi: 10.1016/j.jinf.2014.05.010
Nguyen, L. N., Ma, D., Shui, G., Wong, P., Cazenave-Gassiot, A., Zhang, X., et al. (2014). Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 509, 503–506. doi: 10.1038/nature13241
Noonan, J. J., Jarzabek, M., Lincoln, F. A., Cavanagh, B. L., Pariag, A. R., Juric, V., et al. (2019). Implementing patient-derived xenografts to assess the effectiveness of cyclin-dependent kinase inhibitors in glioblastoma. Cancer 11:2005. doi: 10.3390/cancers11122005
Nowakowski, T. J., Pollen, A. A., Di Lullo, E., Sandoval-Espinosa, C., Bershteyn, M., and Kriegstein, A. R. (2016). Expression analysis highlights AXL as a candidate zika virus entry receptor in neural stem cells. Cell Stem Cell 18, 591–596. doi: 10.1016/j.stem.2016.03.012
O’Connor, M. A., Tisoncik-Go, J., Lewis, T. B., Miller, C. J., Bratt, D., Moats, C. R., et al. (2018). Early cellular innate immune responses drive Zika viral persistence and tissue tropism in pigtail macaques. Nat. Commun. 9:3371. doi: 10.1038/s41467-018-05826-w
Oehler, E., Watrin, L., Larre, P., Leparc-Goffart, I., Lastere, S., Valour, F., et al. (2014). Zika virus infection complicated by Guillain-Barre syndrome–case report, French Polynesia, December 2013. Euro. Surveill. 19:20720. doi: 10.2807/1560-7917.es2014.19.9.20720
Olmo, I. G., Carvalho, T. G., Costa, V. V., Alves-Silva, J., Ferrari, C. Z., Izidoro-Toledo, T. C., et al. (2017). Zika virus promotes neuronal cell death in a non-cell autonomous manner by triggering the release of neurotoxic factors. Front. Immunol. 8:1016. doi: 10.3389/fimmu.2017.01016
Panganiban, A. T., Blair, R. V., Hattler, J. B., Bohannon, D. G., Bonaldo, M. C., Schouest, B., et al. (2020). A Zika virus primary isolate induces neuroinflammation, compromises the blood-brain barrier and upregulates CXCL12 in adult macaques. Brain Pathol. 30, 1017–1027. doi: 10.1111/bpa.12873
Papa, M. P., Meuren, L. M., Coelho, S. V. A., Lucas, C. G. O., Mustafá, Y. M., Lemos Matassoli, F., et al. (2017). Zika virus infects, activates, and crosses brain microvascular endothelial cells, without barrier disruption. Front. Microbiol. 8:2557. doi: 10.3389/fmicb.2017.02557
Pitts, J. D., Li, P.-C., de Wispelaere, M., and Yang, P. L. (2017). Antiviral activity of N-(4-hydroxyphenyl) retinamide (4-HPR) against Zika virus. Antivir. Res. 147, 124–130. doi: 10.1016/j.antiviral.2017.10.014
Poland, G. A., Ovsyannikova, I. G., and Kennedy, R. B. (2019). Zika vaccine development: current status. Mayo Clin. Proc. 94, 2572–2586. doi: 10.1016/j.mayocp.2019.05.016
Puduvalli, V. K., Yung, W. K., Hess, K. R., Kuhn, J. G., Groves, M. D., Levin, V. A., et al. (2004). Phase II study of fenretinide (NSC 374551) in adults with recurrent malignant gliomas: a north American brain tumor consortium study. J. Clin. Oncol. 22, 4282–4289. doi: 10.1200/JCO.2004.09.096
Puschnik, A. S., Marceau, C. D., Ooi, Y. S., Majzoub, K., Rinis, N., Contessa, J. N., et al. (2017). A small-molecule oligosaccharyltransferase inhibitor with pan-flaviviral activity. Cell Rep. 21, 3032–3039. doi: 10.1016/j.celrep.2017.11.054
Ramakrishnaiah, V., Thumann, C., Fofana, I., Habersetzer, F., Pan, Q., de Ruiter, P. E., et al. (2013). Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. U. S. A. 110, 13109–13113. doi: 10.1073/pnas.1221899110
Rausch, K., Hackett, B. A., Weinbren, N. L., Reeder, S. M., Sadovsky, Y., Hunter, C. A., et al. (2017). Screening bioactives reveals nanchangmycin as a broad spectrum antiviral active against zika virus. Cell Rep. 18, 804–815. doi: 10.1016/j.celrep.2016.12.068
Retallack, H., Di Lullo, E., Arias, C., Knopp, K. A., Laurie, M. T., Sandoval-Espinosa, C., et al. (2016). Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. U. S. A. 113, 14408–14413. doi: 10.1073/pnas.1618029113
Richner, J. M., Jagger, B. W., Shan, C., Fontes, C. R., Dowd, K. A., Cao, B., et al. (2017). Vaccine mediated protection against Zika virus-induced congenital disease. Cell 170, 273–283.e12. doi: 10.1016/j.cell.2017.06.040
Ropidi, I., Khazali, A. S., Nor Rashid, N., and Yusof, R. (2020). Endoplasmic reticulum: a focal point of Zika virus infection. J. Biomed. Sci. 27:27. doi: 10.1186/s12929-020-0618-6
Rothlin, C. V., Ghosh, S., Zuniga, E. I., Oldstone, M. B., and Lemke, G. (2007). TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 131, 1124–1136. doi: 10.1016/j.cell.2007.10.034
Sacramento, C. Q., de Melo, G. R., de Freitas, C. S., Rocha, N., Hoelz, L. V. B., Miranda, M., et al. (2017). The clinically approved antiviral drug sofosbuvir inhibits Zika virus replication. Sci. Rep. 7:40920. doi: 10.1038/srep46772
Sapparapu, G., Fernandez, E., Kose, N., Bin, C., Fox, J. M., Bombardi, R. G., et al. (2016). Neutralizing human antibodies prevent Zika virus replication and fetal disease in mice. Nature 540, 443–447. doi: 10.1038/nature20564
Schuler-Faccini, L., Ribeiro, E. M., Feitosa, I. M., Horovitz, D. D., Cavalcanti, D. P., Pessoa, A., et al. (2016). Possible association between Zika virus infection and microcephaly—Brazil, 2015. Morb. Mortal. Wkly Rep. 65, 59–62. doi: 10.15585/mmwr.mm6503e2
Shan, C., Muruato, A. E., Jagger, B. W., Richner, J., Nunes, B. T. D., Medeiros, D. B. A., et al. (2017). A single-dose live-attenuated vaccine prevents Zika virus pregnancy transmission and testis damage. Nat. Commun. 8:676. doi: 10.1038/s41467-017-00737-8
Shao, Q., Herrlinger, S., Yang, S. L., Lai, F., Moore, J. M., Brindley, M. A., et al. (2016). Zika virus infection disrupts neurovascular development and results in postnatal microcephaly with brain damage. Development 143, 4127–4136. doi: 10.1242/dev.143768
Siddiqui, M. R., Mayanil, C. S., Kim, K. S., and Tomita, T. (2015). Angiopoietin-1 regulates brain endothelial permeability through PTPN-2 mediated tyrosine dephosphorylation of occludin. PLoS One 10:e0130857. doi: 10.1371/journal.pone.0130857
Sir, D., Kuo, C.-f., Tian, Y., Liu, H. M., Huang, E. J., Jung, J. U., et al. (2012). Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 287, 18036–18043. doi: 10.1074/jbc.M111.320085
Souza, B. S. F., Sampaio, G. L. A., Pereira, C. S., Campos, G. S., Sardi, S. I., Freitas, L. A. R., et al. (2016). Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells. Sci. Rep. 6:39775. doi: 10.1038/srep39775
Sowinski, S., Jolly, C., Berninghausen, O., Purbhoo, M., Chauveau, A., Köhler, K., et al. (2008). Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell Biol. 10, 211–219. doi: 10.1038/ncb1682
Srivastava, M., Zhang, Y., Chen, J., Sirohi, D., Miller, A., Zhang, Y., et al. (2020). Chemical proteomics tracks virus entry and uncovers NCAM1 as Zika virus receptor. Nat. Commun. 11:3896. doi: 10.1038/s41467-020-17638-y
Tan, C. W., Huan Hor, C. H., Kwek, S. S., Tee, H. K., Sam, I. C., Goh, E. L. K., et al. (2019). Cell surface α2,3-linked sialic acid facilitates Zika virus internalization. Emerg. Microbe. Infect. 8, 426–437. doi: 10.1080/22221751.2019.1590130
Victorino, D. B., Bederman, I. R., and Costa, A. C. S. (2017). Pharmacokinetic properties of memantine after a single intraperitoneal administration and multiple oral doses in euploid mice and in the Ts65Dn mouse model of down’s syndrome. Basic Clin. Pharmacol. Toxicol. 121, 382–389. doi: 10.1111/bcpt.12816
Vitale, I., Galluzzi, L., Castedo, M., and Kroemer, G. (2011). Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 12, 385–392. doi: 10.1038/nrm3115
Wang, S., Hong, S., Deng, Y. Q., Ye, Q., Zhao, L. Z., Zhang, F. C., et al. (2017a). Transfer of convalescent serum to pregnant mice prevents Zika virus infection and microcephaly in offspring. Cell Res. 27, 158–160. doi: 10.1038/cr.2016.144
Wang, Z.-Y., Wang, Z., Zhen, Z.-D., Feng, K.-H., Guo, J., Gao, N., et al. (2017b). Axl is not an indispensable factor for Zika virus infection in mice. J. Gen. Virol. 98, 2061–2068. doi: 10.1099/jgv.0.000886
Wang, Q., Yan, J., and Gao, G. F. (2017c). Monoclonal antibodies against Zika virus: therapeutics and their implications for vaccine design. J. Virol. 91, e01049–e01017. doi: 10.1128/JVI.01049-17
Wang, S., Zhang, Q., Tiwari, S. K., Lichinchi, G., Yau, E. H., Hui, H., et al. (2020). Integrin alphavbeta5 internalizes Zika virus during neural stem cells infection and provides a promising target for antiviral therapy. Cell Rep. 30, 969–983.e4. doi: 10.1016/j.celrep.2019.11.020
Watrin, L., Ghawché, F., Larre, P., Neau, J.-P., Mathis, S., and Fournier, E. (2016). Guillain–Barré syndrome (42 cases) occurring during a Zika virus outbreak in French Polynesia. Medicine 95:e3257. doi: 10.1097/MD.0000000000003257
Wells, M. F., Salick, M. R., Wiskow, O., Ho, D. J., Worringer, K. A., Ihry, R. J., et al. (2016). Genetic ablation of AXL does not protect human neural progenitor cells and cerebral organoids from Zika virus infection. Cell Stem Cell 19, 703–708. doi: 10.1016/j.stem.2016.11.011
Wu, K.-Y., Zuo, G.-L., Li, X.-F., Ye, Q., Deng, Y.-Q., Huang, X.-Y., et al. (2016). Vertical transmission of Zika virus targeting the radial glial cells affects cortex development of offspring mice. Cell Res. 26, 645–654. doi: 10.1038/cr.2016.58
Xu, M., Lee, E. M., Wen, Z., Cheng, Y., Huang, W.-K., Qian, X., et al. (2016). Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat. Med. 22, 1101–1107. doi: 10.1038/nm.4184
Yun, S. I., and Lee, Y. M. (2017). Zika virus: an emerging flavivirus. J. Microbiol. 55, 204–219. doi: 10.1007/s12275-017-7063-6
Zhang, W., Tan, Y. W., Yam, W. K., Tu, H., Qiu, L., Tan, E. K., et al. (2019). In utero infection of Zika virus leads to abnormal central nervous system development in mice. Sci. Rep. 9:7298. doi: 10.1038/s41598-019-43303-6
Zhao, X. Y., Lu, M. H., Yuan, D. J., Xu, D. E., Yao, P. P., Ji, W. L., et al. (2019). Mitochondrial dysfunction in neural injury. Front. Neurosci. 13:30. doi: 10.3389/fnins.2019.00030
Zhou, J., Chi, X., Cheng, M., Huang, X., Liu, X., Fan, J., et al. (2019). Zika virus degrades the omega-3 fatty acid transporter Mfsd2a in brain microvascular endothelial cells and impairs lipid homeostasis. Sci. Adv. 5:eaax7142. doi: 10.1126/sciadv.aax7142
Keywords: zika virus, blood-brain barrier, transcytosis, Trojan horse, inflammatory response, endoplasmic reticulum stress, autophagy, drug repurposing
Citation: Tan LY, Komarasamy TV, James W and Balasubramaniam VRMT (2022) Host Molecules Regulating Neural Invasion of Zika Virus and Drug Repurposing Strategy. Front. Microbiol. 13:743147. doi: 10.3389/fmicb.2022.743147
Edited by:
Linqi Zhang, Tsinghua University, ChinaReviewed by:
Jing An, Capital Medical University, ChinaElisa Vicenzi, San Raffaele Hospital (IRCCS), Italy
Copyright © 2022 Tan, Komarasamy, James and Balasubramaniam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vinod R. M. T. Balasubramaniam, dmlub2QuYmFsYXN1YnJhbWFuaWFtQG1vbmFzaC5lZHU=
†These authors have contributed equally to this work