 Chandrima Gain
Chandrima Gain Theodoros Kelesidis
Theodoros Kelesidis
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 13 January 2023
Sec. Virology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.1111930
This article is part of the Research Topic Molecular and Cellular Mechanisms Involved in Inflammation, Metabolism and Oxidative Stress Induced by Coronaviruses View all 8 articles
Coronaviruses can cause serious respiratory tract infections and may also impact other end organs such as the central nervous system, the lung and the heart. The coronavirus disease 2019 (COVID-19) has had a devastating impact on humanity. Understanding the mechanisms that contribute to the pathogenesis of coronavirus infections, will set the foundation for development of new treatments to attenuate the impact of infections with coronaviruses on host cells and tissues. During infection of host cells, coronaviruses trigger an imbalance between increased production of reactive oxygen species (ROS) and reduced antioxidant host responses that leads to increased redox stress. Subsequently, increased redox stress contributes to reduced antiviral host responses and increased virus-induced inflammation and apoptosis that ultimately drive cell and tissue damage and end organ disease. However, there is limited understanding how different coronaviruses including SARS-CoV-2, manipulate cellular machinery that drives redox responses. This review aims to elucidate the redox mechanisms involved in the replication of coronaviruses and associated inflammation, apoptotic pathways, autoimmunity, vascular dysfunction and tissue damage that collectively contribute to multiorgan damage.
The coronavirus disease 2019 (COVID-19) has had a devastating impact on humanity. Coronaviruses can cause serious respiratory tract infections and may impact other end organs such as the central nervous system. Coronaviruses are enveloped single-stranded positive-sense RNA viruses named after their crown-like appearance of their spike proteins on their surface (Singhal, 2020). To date, there has been seven human coronaviruses (HCoVs) identified: severe acute respiratory syndrome coronavirus (SARS-CoV-2), SARS-CoV, Middle East respiratory syndrome coronavirus (MERS-CoV), Human coronavirus 229E (HCoV-229E), HCoV-OC43, HCoV-NL63, and HKU-1. Four of them including HCoV-OC43, HCoV-NL63, HCoV-229E, and HKU-1, typically trigger only mild respiratory illnesses in humans. On the other hand, SARS-CoV-2, SARS and MERS are known to cause more severe illness, acute respiratory distress syndrome (ARDS) or multi-organ dysfunction, especially in aged people with comorbidities (Li et al., 2021a). Understanding the mechanisms that contribute to the pathogenesis of coronavirus infections, will set the foundation for development of new treatments to attenuate the impact of coronaviruses on host cells and tissues. However, there is limited understanding how different coronaviruses including SARS-CoV-2, manipulate cellular machinery to drive host cell responses.
Emerging evidence suggests that human diseases including viral infections often disrupt the host natural balance between increased production of reactive oxygen species (ROS) and reduced antioxidant host responses that collectively increases redox stress (Amini et al., 2022; Figure 1). ROS are free radical and nonradical byproducts of metabolic processes in organelles such as plasma and nuclear membranes, the mitochondria, peroxisomes and the endoplasmic reticulum (ER; Reshi et al., 2014). ROS are necessary for cellular processes like mitochondrial energy production, host defense, cellular signaling, and the regulation of gene expression. Mitochondria are the main location of production of ROS (mito-ROS) during energy production. Increased ROS during viral infections have not only detrimental impact on the cells and tissues but are also important for antiviral immune function (Yang et al., 2007; Finkel, 2011) during viral infections like influenza (To et al., 2014), respiratory syncytial virus (RSV; Fink et al., 2008) and rhinoviruses (Kaul et al., 2000; Fink et al., 2008).
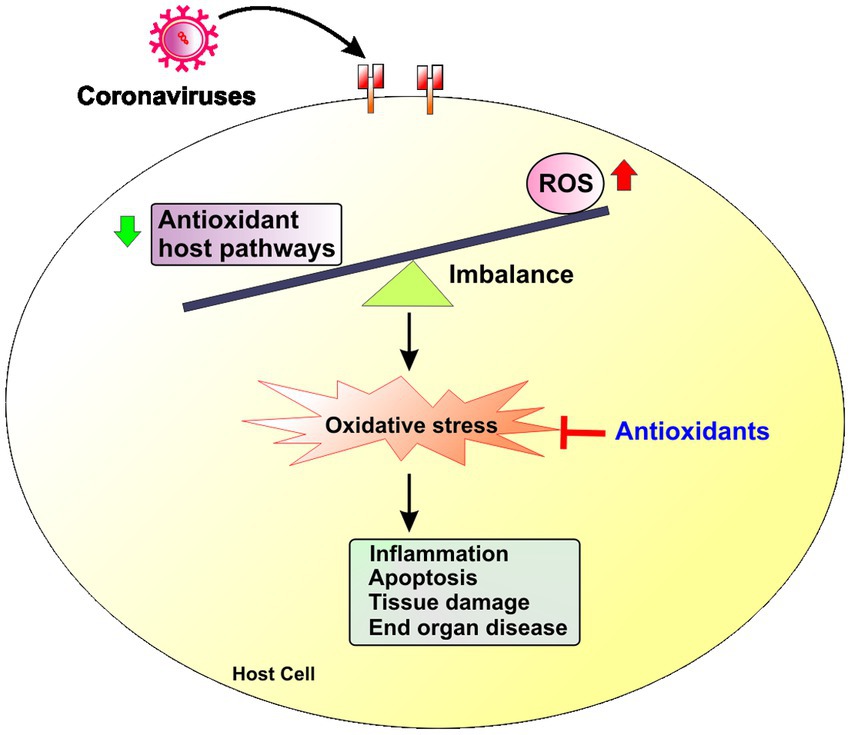
Figure 1. Redox imbalance in coronavirus infections. Coronavirus infection triggers an imbalance between increased production of reactive oxygen species (ROS) and reduced antioxidant host responses that leads to increased redox stress in the host cell. Increased redox stress induces inflammation, apoptosis and ultimately tissue damage and end organ disease.
However, an excess of ROS can damage cellular components including lipids, proteins, and DNA, alter immune functions, inflammatory responses and induce organ and tissue dysfunction (Preiser, 2012; Reshi et al., 2014; Labarrere and Kassab, 2022). Indeed, several studies have shown that oxidative stress contributes to the pathogenesis of respiratory viral infections (Khomich et al., 2018), influenza and RSV. Increased oxidative stress in severe COVID-19 contributes to inflammation, endothelial cell dysfunction, thrombosis that can lead to multiorgan damage (Li et al., 2021a; Alam and Czajkowsky, 2022). Oxidative stress, induced by coronavirus, also interferes with inflammatory pathways that may lead to more long-lasting tissue damage. However, there is limited understanding how different coronaviruses including SARS-CoV-2, manipulate cellular machinery that drives redox responses.
In this review, we summarize the scientific evidence regarding the cellular and molecular pathways modulated by oxidative stress that are implicated in the pathogenesis of coronavirus infections. We specifically review the role of redox pathways in major pathophysiological underpinnings that contribute to cell and tissue damage in coronavirus infection: (1) virus replication, (2) virus-associated inflammation, (3) virus-associated apoptosis, (4) redox-related end organ disease. We review the scientific evidence related to these redox pathways, separately for SARS-CoV-2 versus all the other coronaviruses [SARS-CoV, MERS, respiratory coronaviruses and other coronaviruses used to model SARS-CoV-2 infection such as the murine hepatitis virus (MHV)]. Finally, we discuss the relevance of these redox pathways with regards to acute severe COVID-19 and Post-Acute Sequelae of SARS-CoV-2 infection (PASC) and potential antioxidant treatments.
Several redox mechanisms can regulate both viral entry and cytosolic replication of coronaviruses (Figure 2; Table 1; Wang and Zhang, 1999; Kulisz et al., 2002; Halestrap et al., 2004; Mizutani et al., 2004; Emerling et al., 2005; Kefaloyianni et al., 2006; Doughan et al., 2008; Lucas et al., 2008; Cho et al., 2009; Garrido and Griendling, 2009; Hosakote et al., 2009; Jamaluddin et al., 2009; Wosniak et al., 2009; de Wilde et al., 2011; Kesic et al., 2011; Xia et al., 2011; Kosmider et al., 2012; Yamada et al., 2012; Kim et al., 2012b; Lee et al., 2013; Nguyen Dinh Cat et al., 2013; Komaravelli and Casola, 2014; Hyser and Estes, 2015; Kindrachuk et al., 2015; Komaravelli et al., 2015; Paszti-Gere et al., 2015; Shirihai et al., 2015; Simon et al., 2015; Demers-Lamarche et al., 2016; Kau et al., 2016; Morris et al., 2016; Zhang et al., 2016; Daiber et al., 2017; Trempolec et al., 2017; Khomich et al., 2018; Tu et al., 2019; Olagnier et al., 2020; Tao et al., 2020; Verdecchia et al., 2020; Herengt et al., 2021; Moghimi et al., 2021; Youn et al., 2021).
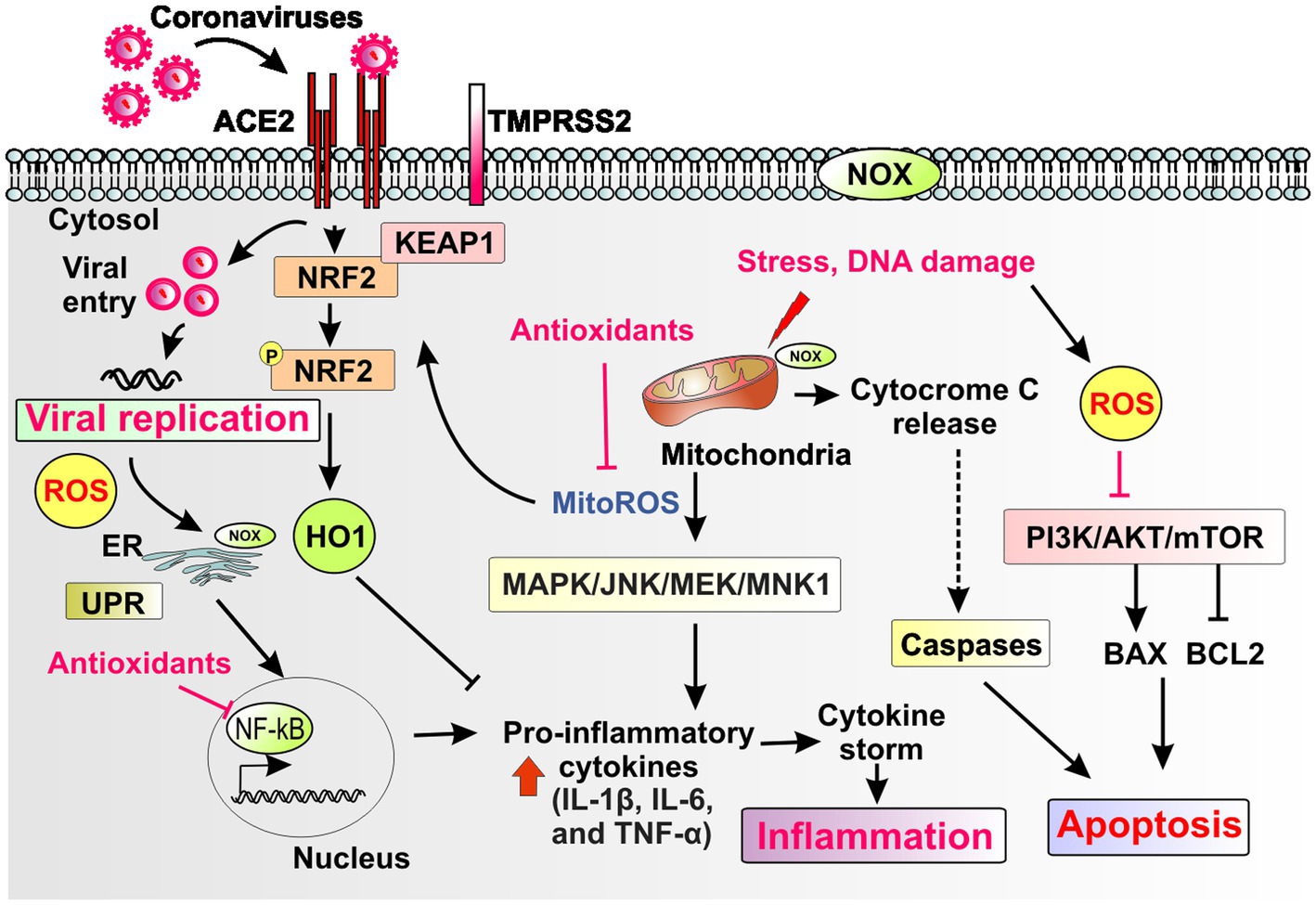
Figure 2. Schematic representation of redox pathways that contribute to viral replication, inflammation, and apoptosis during coronavirus infection. Coronaviruses bind to the ACE2 receptor and replicate through host proteases such as TMPRSS2 and by hijacking cytosolic cellular machinery such as the mitochondria and the endoplasmic reticulum (ER), which engages the unfolded protein response (UPR). The plasma membrane, the ER and mitochondria harbor different isoforms of the NADPH oxidase (NOX) enzyme. Coronaviruses induce cellular oxidative stress with generation of reactive oxygen species (ROS) and mitochondrial ROS (mito-ROS) and impairment of stress-inducible, antioxidant, anti-inflammatory and antiviral responses such as the Nrf2 pathway and other key downstream mediators such as Heme oxygenase-1 (HO-1). Mito-ROS induce downstream signaling pathways such as MAPK, JNK, MEK/MNK1 that induce both viral replication and proinflammatory pathways such as induction of cytokines (e.g., IL-1b, IL-6, and TNF-a). Mito-ROS, ROS and ER stress response induce the proinflammatory pathway NF-κB. ROS and mito-ROS also induce apoptosis through alterations in apoptotic pathways such as PI3K/AKT, mTOR and induction of mitochondrial apoptosis. Collectively, redox mediated pathways that drive viral replication, inflammation and apoptosis contribute to cell and tissue damage that drive end organ disease in coronavirus infection. Endogenous antioxidant host pathways and exogenous therapeutic antioxidants could attenuate redox mediated pathways that drive pathogenesis of coronavirus infections.
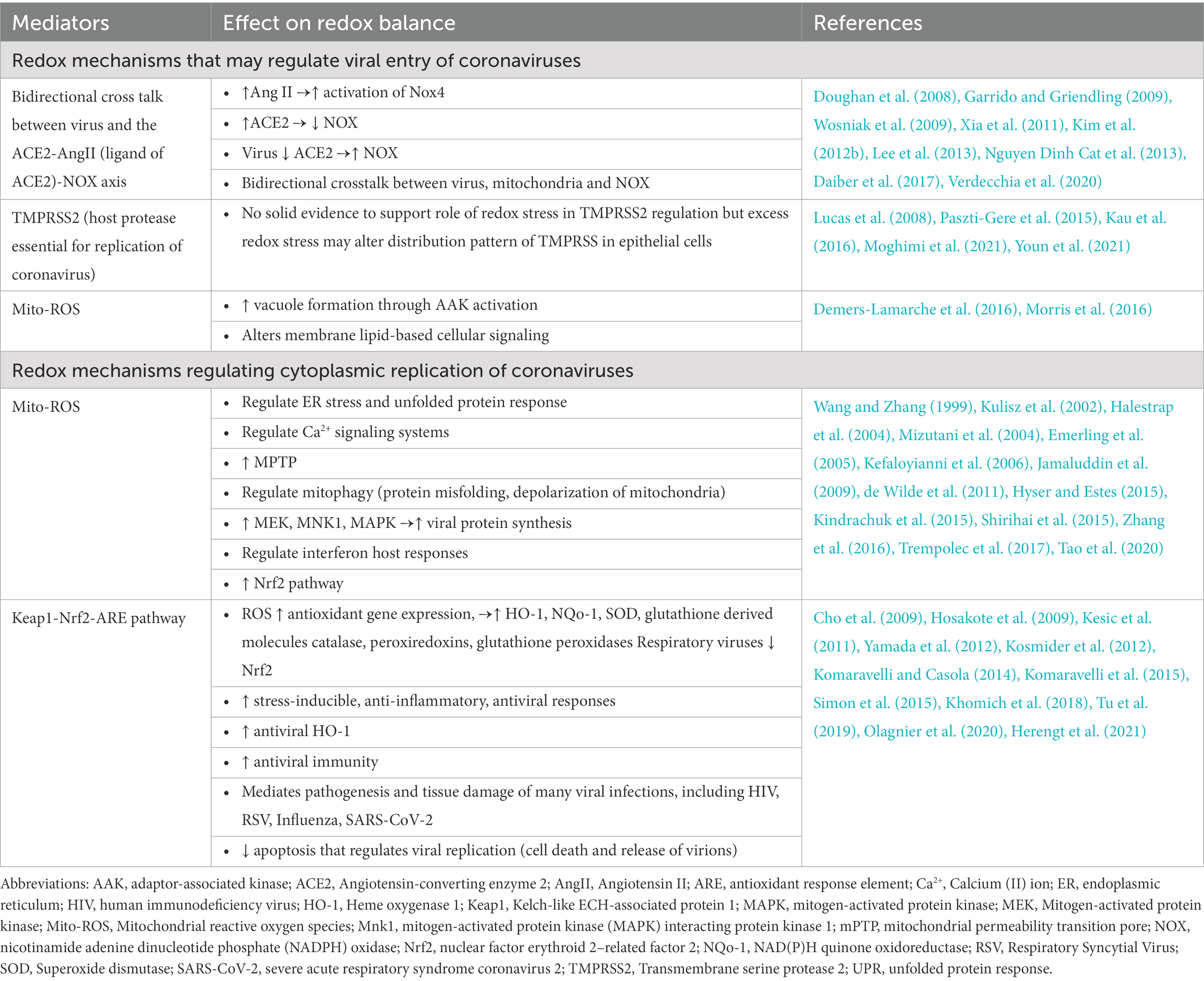
Table 1. Redox mechanisms that regulate replication of coronaviruses.
The spike S proteins on the surface of coronaviruses are responsible to their attachment to host receptors in airway epithelial cells such as the angiotensin-converting enzyme 2 (ACE2) receptors that interact with host cell proteases, such as transmembrane protease serine 2 (TMPRSS2; Hamming et al., 2004; Irigoyen et al., 2016; Lukassen et al., 2020; Xu et al., 2020). While many coronaviruses utilize peptidases, such as ACE2, dipeptidyl peptidase 4, aminopeptidase N, as their cellular receptors, SARS-CoV, SARS-CoV-2 and HCoV-NL63 utilize ACE2 as their receptors thus disrupting the renin-angiotensin system (Verdecchia et al., 2020).
ACE2, a peptidase that exists on the cell surfaces of most organs (Hamming et al., 2004), is one of the most crucial key players in induction of redox stress (Shatizadeh Malekshahi et al., 2022). Angiotensin II (AngII), the ligand of ACE2, is a potent activator of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and an inducer of ROS production in the vasculature, kidney and brain (Garrido and Griendling, 2009). Typically, ACE2 helps avert NAPDH oxidase activity by converting Ang II into angiotensin 1–7, thereby reducing ROS levels; Ang II stimulates NAPDH oxidase. ACE2 overexpression has been shown to reduce ROS, and ACE2 deficiency has been shown to induce oxidative stress (Xia et al., 2011; Pena Silva et al., 2012). The complex cross-talk between ACE2 and redox pathways is further emphasized by a possible bidirectional redox regulation of ACE2 levels. High ACE2 activity may reduce redox stress but vice versa high redox stress may regulate ACE2 activity. In vitro studies showed that NOX-driven ROS may reduce ACE2 in vascular smooth muscle cells (Lavrentyev and Malik, 2009). Consistent with this evidence, independent in vitro studies demonstrated that Ang II-induced activation of mitochondrial Nox4 is an important endogenous source of ROS and is related to cell survival in kidney epithelial cells (Kim et al., 2012b). The crosstalk between NOX and ACE2 has also been shown in vivo in mouse models of disease and increased levels of ACE2 are generally associated with reduced oxidative stress in mammalian cells (Xia et al., 2011).
Angiotensin II is often upregulated in viral infections (Doughan et al., 2008; Wosniak et al., 2009; Lee et al., 2013; Daiber et al., 2017). However, when cells are infected with coronavirus, there is a reduction of ACE2 receptors on the cell surface and this results in an increase of Ang II which binds to ACE1 and increases ROS levels through NADPH oxidase (Nguyen Dinh Cat et al., 2013). Experimental studies have demonstrated that in vitro exposure to S protein induces excessive oxidative stress in endothelial cells, which is mediated specifically by activation of NADPH oxidase isoform 2 (NOX2), but not NOX1 or NOX4 (Youn et al., 2021). However, it is unclear if there is bidirectional link between ACE2 levels and increased redox cellular pathways in the setting of SARS-CoV-2-induced ACE2 downregulation in airway epithelial cells.
TMPRSS2 is expressed in both the cytoplasm as well as in the cell membrane in epithelial cells (Lucas et al., 2008). In vitro studies with porcine intestinal epithelial cells have shown that acute excessive oxidative stress induces altered distribution pattern of TMPRSS2 and relocalized transmembrane serine protease activity that may contribute to weakening of epithelial barrier integrity (Paszti-Gere et al., 2015). However, a small study of COVID-19 patients and uninfected controls showed that measures of oxidative stress in sperm epithelial cells were not associated with levels of TMPRSS2 (Moghimi et al., 2021). Similarly, another experimental study showed that cigarette smoking extract (CSE) that is an established trigger of oxidative stress (Kau et al., 2016) had no effect on ACE2 and TMPRSS2 expression in endothelial cells (Youn et al., 2021). Overall, there is no solid evidence to support a role of increased redox stress in regulation of TMPRSS2.
Other than redox-dependent regulation of membrane receptors for coronaviruses, mito-ROS are also instigators of aberrant vacuole formation (Demers-Lamarche et al., 2016) by activation of adaptor-associated kinase 1 (AAK1), a regulator of endocytosis (Chen et al., 2006) that has been targeted therapeutically in SARS-CoV-2 infection with baricitinib (Stebbing et al., 2020). Mito-ROS can also induce alterations in membrane lipid rafts and lipid-based cellular signaling changing their properties (Morris et al., 2016) and these membrane changes may also impact viral entry of coronaviruses. Thus, redox mechanisms may regulate entry of coronaviruses in mammalian cells but these mechanisms need to be further studied specifically in airway epithelial cells and in vivo.
Viral infections may alter the mitochondrial dynamics leading to excessive mito-ROS generation, mitochondrial biogenesis, and altered mitochondrial β-oxidation (Elesela and Lukacs, 2021). Mitochondria are targeted by coronavirus (Shi et al., 2014). Coronaviruses may directly induce production of mito-ROS in cells. Non-structured viral proteins, such as coronavirus 3a protein directly activate NLRP3 inflammasome in macrophages, which is mediated by increased mito-ROS level (Zhou et al., 2011; Chen et al., 2019). Finally, redox pathways also regulate cellular machinery that propagates replication of coronaviruses through multiple pathways.
First, Mito-ROS regulate the endoplasmic reticulum stress and the unfolded protein response (UPR) that contribute to replication of coronaviruses (de Wilde et al., 2011; Hyser and Estes, 2015; Kindrachuk et al., 2015; Zhang et al., 2016) and associated Ca2+ signaling systems. Second, mito-ROS induce the mitochondrial permeability transition pore (mPTP) that is a proviral factor for replication of coronaviruses. Indeed, by blocking the mPTP, cyclosporin A impacts coronavirus replication (Halestrap et al., 2004). Mitochondria-targeted antioxidants inhibit mPTP, mito-ROS (Halestrap et al., 2004), and ROS (Dikalova et al., 2010; Dikalov et al., 2014). Third, mito-ROS regulate mitophagy that regulates replication of coronaviruses. Protein misfolding mitochondrial depolarization and ROS activate mitophagy (Shirihai et al., 2015). Viral proteins like SARS-CoV ORF-9 (Shi et al., 2014) interact with mitophagic machinery such as LC3 and Beclin1 (Zhang et al., 2018). Therapeutic targeting of aberrant autophagy through Beclin1 reduces MERS infection (Gassen et al., 2019). Fifth, mito-ROS trigger MEK (Zhang et al., 2016), MNK1 (Wang and Zhang, 1999) and MAPK signaling pathways (Kulisz et al., 2002; Emerling et al., 2005; Trempolec et al., 2017) that propagate viral protein synthesis and SARS-Co-V replication (Mizutani et al., 2004; Kefaloyianni et al., 2006; Jamaluddin et al., 2009). Sixth, ROS regulate cytoplasmic interferon host antiviral responses during coronavirus infection. ROS promotes MHV replication by downregulating interferon host responses during MHV infection (Tao et al., 2020). Lastly, preclinical studies suggest that mito-ROS may contribute to viral reservoirs and replication of SARS-CoV-2 in macrophages, but this has not been clearly demonstrated in vivo (Codo et al., 2020). Thus, mito-ROS induce multiple proviral cytoplasmic pathways.
The primary transcription factor regulating the antioxidant response is the nuclear factor E2-related factor 2 (Nrf2), which regulates the Kelch-like ECH-associated protein 1 (Keap1)-Nrf2-antioxidant response elements (ARE) pathway (Khomich et al., 2018). Under normal circumstances, the Keap1-Nrf2-ARE pathway is activated by the oxidative stress resulting from ROS production. Nrf2, which is usually bound to Keap1 by ubiquitination or degraded by Keap1 in the absence of oxidative stress, is translocated to the nucleus when oxidative stress modifies the conformational structure of Keap1 and prevents it from binding Nrf2 (Komaravelli and Casola, 2014; Han et al., 2021). Mito-ROS activate Nrf2 through protein kinases, and induce production of antioxidant proteins and genes involved in mitochondrial quality control (Kasai et al., 2020). The activation of Nrf2 results in the upregulation of antioxidant gene expression as Nrf2 binds to antioxidant response element (ARE) sites, leading to the expression of key players of the antioxidant response, including heme oxygenase-1 (HO-1), NADPH quinone oxidoreductase 1 (NQO-1), superoxide dismutases (SOD), and glutathione derived molecules catalase, peroxiredoxins, and glutathione peroxidases which collectively attenuate oxidative stress (Khomich et al., 2018; Tu et al., 2019).
Several studies have found that respiratory viruses downregulate the expression of antioxidant genes by inhibiting Nrf2, preventing it from mobilizing to the nucleus and binding to ARE sites (Komaravelli and Casola, 2014). The Nrf2 pathway that mediates pathogenesis and tissue damage of several viral infections including HIV, RSV (Cho et al., 2009; Hosakote et al., 2009; Komaravelli et al., 2015), influenza (Kesic et al., 2011; Kosmider et al., 2012; Yamada et al., 2012; Simon et al., 2015), and SARS-CoV-2 (Olagnier et al., 2020). Induction of the Nrf2 pathway and key downstream mediators such as Heme oxygenase-1 (HO-1) triggers stress-inducible, anti-inflammatory, and antiviral responses present in most human cells (Espinoza et al., 2017). NRF2 has antiviral properties but, it remains unclear which genes mediate these effects and how they exert antiviral effect (Herengt et al., 2021).
Emerging evidence has increased our understanding of the role of Nrf2 activation in SARS-CoV-2 infection. In vitro experiments with Vero hTMPRSS2 cells, Calu-3 and primary human airway epithelial cell lines and using gene silencing of Keap1 and Nrf2 agonists 4-octyl-itaconate (4-OI) and dimethyl fumarate (DMF), it was shown that the Nrf2 pathway has a critical role in inhibiting SARS-CoV-2 replication, in addition to limiting the host inflammatory response. SARS-CoV-2 reduced in vitro basal levels of HO-1 and NQO-1 in lung cells. Notably, considering Nrf2’s known role in inhibiting anti-viral IFN responses, it was shown that the antiviral effect of Nrf2 is independent of interferon responses (Olagnier et al., 2020). Mechanistic preclinical studies showed that Nrf2 activation reduced SARS-CoV-2 replication by inducing the metabolite biliverdin, whereas SARS-CoV-2 altered the NRF2 axis through the cross-talk between the nonstructural viral protein NSP14 and the NAD-dependent deacetylase Sirtuin 1 (SIRT1; Olagnier et al., 2020; Zhang et al., 2022).
Experimental studies have also shown that downregulation of antioxidant genes by SARS-CoV-2 and SARS-CoV-1 is combined with an upregulation of oxidative stress genes like myeloperoxidase (MPO), calprotectin (S100A8 and S100A9), sulfiredoxin-1 (SRXN1), glutamate cysteine ligase modifier subunit (GCLM), sestrin2 (SESN2), and thioredoxin-1 (TXN; Saheb Sharif-Askari et al., 2021). The results of these studies have revealed key aspects of SARS-CoV-2 infection: such as downregulation of host’s antioxidant pathway as an important role in viral replication, and possible utility of activators of antioxidant pathways as specific therapeutic targets.
Many viruses alter apoptosis or programmed cell death of the infected cell as a mechanism of increased production of virus progeny, cell killing and virus spread (Roulston et al., 1999). Apoptosis is the programmed cell death that involves the activation of proteases called caspases and a cascade of events that link apoptosis-initiating stimuli to final death of the cell. ROS (Pierce et al., 1991; Kasahara et al., 1997) and mitochondria play pivotal roles in induction of apoptosis under both physiologic and pathologic conditions. Increased mito-ROS induce apoptosis and cell death (Orrenius et al., 2007). Excessive ROS can activate pro-apoptotic Bcl-2 family proteins by increasing mitochondrial permeability to drive the mitochondrial membrane potential, release cytochrome c, mtDNA (Santos et al., 2003), and pro-apoptotic caspase-3 and-9. This leads to the activation of intrinsic or mitochondrial driven cell death by apoptosis (Green and Llambi, 2015). Coronaviruses impact apoptosis through several pathways. Notably, mitochondrial apoptosis is directly and uniquely induced by SARS-CoV (Pfefferle et al., 2011) triggering viral replication (Supinski et al., 2009; Maiti et al., 2017). SARS-CoV-2 infection also downregulates the Nrf2 pathway (Olagnier et al., 2020; Zhang et al., 2022) which has antiapoptotic cellular effect (Niture and Jaiswal, 2012; Khan et al., 2018). Thus, coronaviruses induce apoptosis through multiple pathways, either directly (Pfefferle et al., 2011), or indirectly by inducing production of mito-ROS and downregulating antiapoptotic pathways such as Nrf2 and the virus-induced alteration of mitochondrial apoptosis contributes to increased replication of coronaviruses (Supinski et al., 2009; Pfefferle et al., 2011; Maiti et al., 2017).
The complement system is a major host defense mechanism against viral replication. Several viruses hijack the complement system for cellular entry and spread (Agrawal et al., 2017). The role of the complement system in the pathogenesis of coronavirus infections is complex and contradictory (Santiesteban-Lores et al., 2021). During SARS-CoV-2 infection, the complement system is a host defense mechanism against viral replication in asymptomatic or mild cases (Santiesteban-Lores et al., 2021). However, complement activation has also potent proinflammatory effect and can increase local and systemic damage in severe COVID-19 (Santiesteban-Lores et al., 2021). As outlined above, coronavirus induce production of mito-ROS during infection. Mito-ROS induce the “complement–metabolism–inflammasome axis”(Arbore and Kemper, 2016). MERS-CoV can also directly induce the complement system (Chen et al., 2010). Collectively, limited evidence suggests that complement activation through redox pathways may have a more important role in cell and tissue damage in severe coronavirus infections rather than a major regulatory role in replication of coronaviruses.
Mitophagy, the cellular process that clears excess or damaged mitochondria, has a key role in function of mitochondria and mammalian cells and regulates severeal physiological and pathological processes, including apoptosis, immunity and inflammation. Emerging evidence suggests that several viruses hijack mitophagy to enable viral replication and escape host immune responses (Li et al., 2022). SARS-CoV can encode open reading frame-9b (ORF-9b), which is localized in mitochondria and induces mitochondrial elongation which further triggers mitophagy and coronavirus replication (Shi et al., 2014). Preclinical studies have shown that SARS-CoV-2 directly causes mitochondrial dysfunction and mitophagy impairment (Shang et al., 2021). Notably, defects in autophagy and mitophagy processes may regulate host response to coronavirus infection (Pacheco et al., 2021). Coronaviruses also induce production of mito-ROS that have an established complex crosstalk with mitophagy (Schofield and Schafer, 2021). Overall, further evidence is needed to clearly link the role of aberrant redox pathways and mitophagy in the regulation of replication of coronaviruses.
Several redox mechanisms regulate inflammation during infection with coronaviruses (Figure 2; Table 2; Shono et al., 1996; Wesselborg et al., 1997; Chua et al., 1998; Canty et al., 1999; Tenjinbaru et al., 1999; Wang and Zhang, 1999; Cooke and Davidge, 2002; Pearlstein et al., 2002; Takada et al., 2003; Mizutani et al., 2004; Desouki et al., 2005; Mukherjee et al., 2005; Kefaloyianni et al., 2006; Xie and Shaikh, 2006; Schrader et al., 2007; Doughan et al., 2008; Nanduri et al., 2008; Cho et al., 2009; Hosakote et al., 2009; Jamaluddin et al., 2009; Martinon et al., 2009; Wosniak et al., 2009; Dikalova et al., 2010; Bulua et al., 2011; Kesic et al., 2011; Kosmider et al., 2012; Yamada et al., 2012; Lee et al., 2013; Nakajima and Kitamura, 2013; Nguyen Dinh Cat et al., 2013; Komaravelli and Casola, 2014; Zinovkin et al., 2014; Komaravelli et al., 2015; Simon et al., 2015; Sun et al., 2016; Zhang et al., 2016; Daiber et al., 2017; Espinoza et al., 2017; Khomich et al., 2018; Tu et al., 2019; Valle et al., 2019; Connors and Levy, 2020; Mahmud-Al-Rafat et al., 2020; Olagnier et al., 2020; Herengt et al., 2021; Saheb Sharif-Askari et al., 2021; Toro et al., 2022).
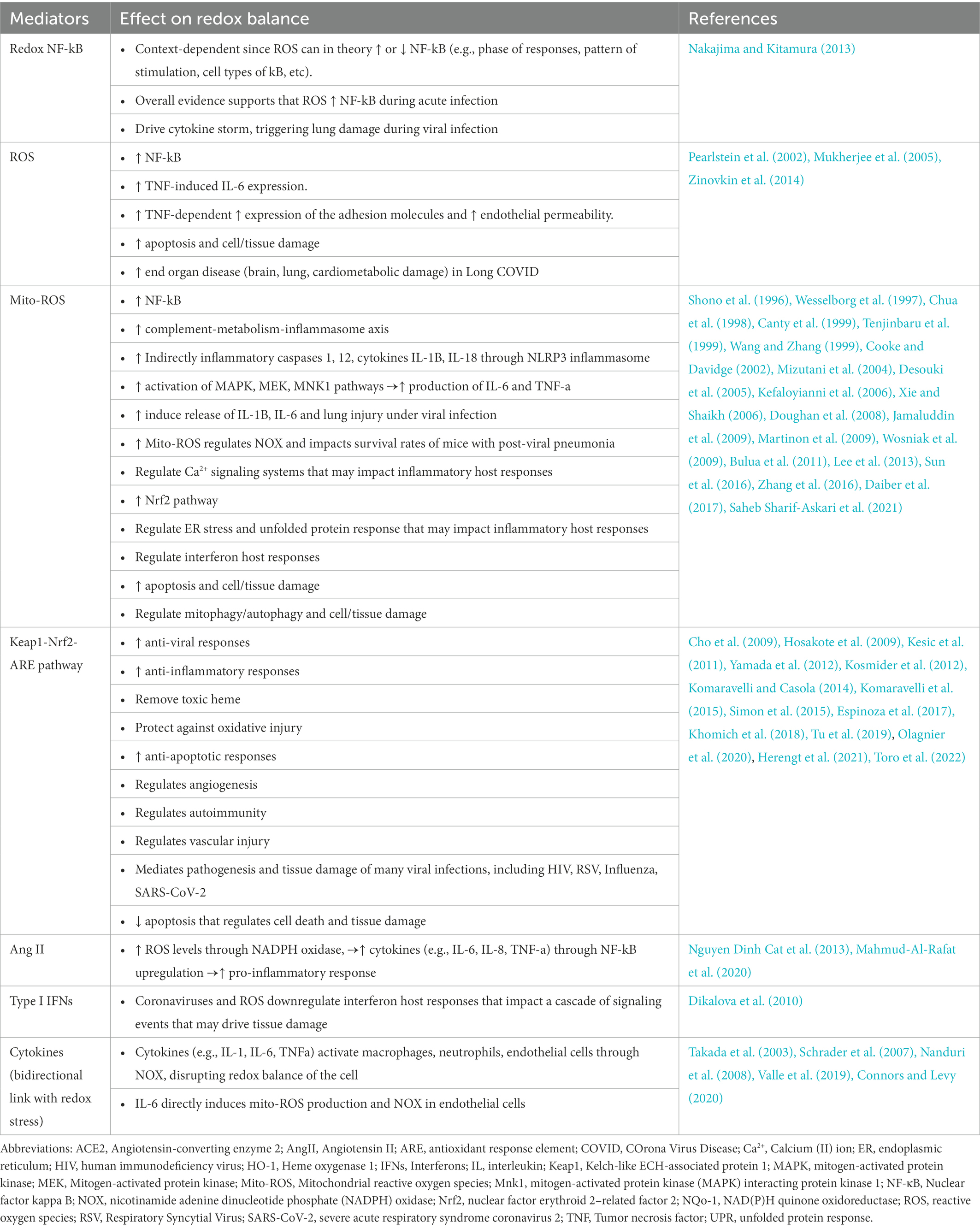
Table 2. Redox mechanisms that regulate cell and tissue damage during infection with coronaviruses.
Nuclear factor-κB (NF-κB) is a redox-sensitive transcription factor that is regulated by ROS through the classical IkB kinase (IKK)-dependent canonical pathway (Liu et al., 2017) and coordinates innate and adaptive immunity, inflammation, and apoptosis (Piette et al., 1997). The redox regulation of the NF-κB pathway has been reviewed elsewhere and varies between different mammalian cells and in the setting of cancer (Gloire et al., 2006). Although it is established that cytokines and lipopolysaccharides induce proinflammatory activation of NF-κB (Schreck and Baeuerle, 1991), ROS may also reduce NF-κB activity (Nakajima and Kitamura, 2013). Oxidative stress in the early phase may induce activation of NF-κB in epithelial cells (Wesselborg et al., 1997; Tenjinbaru et al., 1999; Thevenod et al., 2000) and endothelial cells (Shono et al., 1996; Chua et al., 1998; Canty et al., 1999; Cooke and Davidge, 2002) which are targets of coronaviruses. Redox stress in epithelial cells in the late phase may also inhibit basal and inducible activation of NF-κB (Xie and Shaikh, 2006; Yang et al., 2007; Nakajima and Kitamura, 2013). The regulation of NF-κB by ROS is dependent not only on the phase of responses and the pattern of stimulation, but also depends on specific cell types (Nakajima and Kitamura, 2013). However, most of the evidence regarding redox regulation of the NF-κB pathway is not based on airway epithelial cells, the main target of SARS-CoV-2, and heterogeneous redox stimuli have been utilized in several experimental studies, often in supraphysiological concentrations. Thus, it is not well defined how ROS regulate activity of NF-κB in a bidirectional fashion in airway epithelial cells (Nakajima and Kitamura, 2013).
Overall, cumulative evidence suggests that there is context-dependent regulation of NF-κB by ROS (Nakajima and Kitamura, 2013). Preclinical studies have shown that ROS trigger NF-κB activation in airway epithelial cells (Jany et al., 1995; Ito et al., 2004). In contrast, inhibition of cytokine-triggered NF-κB activation under pre-exposure to ROS has been described in distal airway alveolar epithelial cells (Korn et al., 2001; Reynaert et al., 2006). The oxidative stress– unfolded protein response (UPR) pathway and redox ER responses play a key role in the bidirectional control of NF-κB (Nakajima and Kitamura, 2013). Thus, the opposite, bidirectional effects of redox stimuli on NF-κB seem to depend on the phase of response, the context, the type of cells and the specific redox stimuli. Overall, this bidirectional crosstalk is not well characterized specifically in coronavirus infections.
Viruses may hijack cellular signaling pathways and transcription factors and control them to their own advantage. In particular, the NF-κB pathway appears to be an attractive target for common human viral pathogens (Santoro et al., 2003). Distinct viral proteins encoded by viruses such as HCV, rotavirus, EBV, HBV, HTLV-1, and HIV-1 activate NF-κB by interacting with cellular signaling pathways including calcium-or redox-regulated signals or through ER stress mechanisms. Accumulation of viral dsRNA activates PKR, which in turn stimulates IKK. However, most of the evidence regarding virus-induced regulation of the NF-κB pathway is based on chronic viral infections or infections with DNA viruses (Santoro et al., 2003). There is limited evidence regarding the direct impact of coronaviruses on this pathway.
Evidence has suggested that proteins of SARS-CoV-2 can directly or indirectly impact NF-kB activation. In vitro studies showed that the spike protein of SARS-CoV induces a strong cytokine response through the NF-kB pathway (Dosch et al., 2009). It was also shown that SARS-CoV nucleocapsid protein activated NF-kB in Vero E6 cells in a dose dependent manner (Liao et al., 2005). ORF7a protein of SARS-CoV-2 mediates activation of NF-kB and induced proinflammatory expression of cytokines (Su et al., 2021). Similarly, Nsp5 in SARS-CoV-2 activated NF-kB pathway through upregulation of SUMOylation of mitochondrial antiviral-signaling proteins (Li et al., 2021b). Notably, studies show that the NF-κB signal pathway is a central pathway involved in induction of pro-inflammatory cytokines and chemokines in respiratory virus infection, including SARS-CoV-2-triggered COVID-19 (Kircheis et al., 2020; Hariharan et al., 2021; Kandasamy, 2021). Thus, the pharmacological inactivation of the NF-κB signaling pathway can represent a potential therapeutic target to treat severe COVID-19 (Kircheis et al., 2020; Hariharan et al., 2021; Kandasamy, 2021).
As outlined above, coronavirus induce production of mito-ROS during infection. Mito-ROS have been shown to inhibit interferons and induce aberrant alterations of lipids, membranes, proteins and ultimately tissue damage. Mito-ROS induce inflammasome activation (Dashdorj et al., 2013; Han et al., 2018) and the “complement–metabolism–inflammasome axis”(Arbore and Kemper, 2016), Mito-ROS indirectly regulate inflammatory caspases 1 and 12, as well as the cytokines IL-1β and IL-18 in macrophages through the NLRP3 inflammasome (Martinon et al., 2009). Mito-ROS induce NFκB (Imai et al., 2008) which drives a cytokine storm, triggering lung damage during viral infection. Mito-ROS also induce activate MAPK pathways and promote production of IL-6 and TNF-α (Wang and Zhang, 1999; Mizutani et al., 2004; Kefaloyianni et al., 2006; Jamaluddin et al., 2009; Bulua et al., 2011; Zhang et al., 2016). Mito-ROS directly induce release of IL-1β (Dashdorj et al., 2013; Han et al., 2018), IL-6 (Lowes et al., 2008, 2013; Bulua et al., 2011; Li et al., 2019). Consistent with this evidence it has been shown that Mito-ROS induce inflammatory response and lung injury in mouse models of viral infections (Hu et al., 2019a; Hu et al., 2019b). Thus, mito-ROS may regulate redox cytoplasmic proinflammatory responses in respiratory viral infections.
Heme oxygenase 1 (HO-1), a downstream protein of the Nrf2 pathway, contributes to anti-inflammatory and antiviral responses, removes toxic heme, protects against oxidative injury and also regulates apoptosis, inflammation and angiogenesis (Espinoza et al., 2017). While the exact mechanism by which SARS-CoV-2 affects HO-1 and, conversely, how HO-1 exerts its antiviral effects against SARS-CoV-2 is still being studied, there is an established association between HO-1 and a reduction of tissue damage through its anti-inflammatory and antioxidative functions throughout the body (Toro et al., 2022). This makes HO-1 an important target for developing novel COVID-19 therapeutics.
During SAS-CoV-2 infection, the reduction of ACE2 on the cell surface leads to increase of Ang II and NOX (Nguyen Dinh Cat et al., 2013). Bidirectional crosstalk between mitochondria and NOX, markedly affects redox responses to angiotensin II, the ligand of ACE2 that is upregulated in viral infections (Doughan et al., 2008; Wosniak et al., 2009; Lee et al., 2013; Daiber et al., 2017). Indeed, therapeutic targeting of NOX, triggered by mito-ROS (Desouki et al., 2005), increased the survival of mice with post-influenza pneumonia (Sun et al., 2016). Thus, as a result of increased NOX, NF-κβ activation there is activation of the pro-inflammatory response and release of cytokines like IL-6, IL-8, and TNFα (Mahmud-Al-Rafat et al., 2020). Pro-inflammatory cytokines like IL-1, IL-6, and TNFα activate macrophages, neutrophils, and endothelial cells through NADPH oxidase, resulting in a greater production of superoxide and H2O2 (Takada et al., 2003; Nanduri et al., 2008; Connors and Levy, 2020).
As outlined above, coronavirus induce production of mito-ROS which trigger the “complement–metabolism–inflammasome axis”(Arbore and Kemper, 2016). MERS-CoV can also directly induce the complement system (Chen et al., 2010). The complement activation has also potent proinflammatory effect and can increase local and systemic damage in severe COVID-19 (Santiesteban-Lores et al., 2021). Preclinical in vitro studies have shown controversial data regarding the role of the complement system in binding coronaviruses (Santiesteban-Lores et al., 2021). Experimental studies with animals have shown that complement activation induces a systemic pro-inflammatory response during experimental infection with SARS-CoV and MERS that drives disease progression (Gralinski et al., 2018; Jiang et al., 2018). Small human cohorts also show that complement activation is associated with disease progression of SARS (Wang et al., 2005). Collectively, limited and often controversial evidence suggests that complement activation through redox pathways may have an important role in cell and tissue damage in severe coronavirus infections.
Other than activation of proinflammatory NF-kB, mito-ROS and NOX pathways and downregulation of anti-inflammatory ACE2 and Nrf2 pathways, different coronaviruses may also directly induce other proinflammatory effects. MERS-CoV can induce the complement system and increase inflammatory response, pyroptosis and eventually lung tissue damage (Chen et al., 2010). MERS-CoV infected macrophages increase pro-inflammatory cytokines and chemokines (Pruijssers and Denison, 2019). SARS-CoV and MERS-CoV may also attenuate levels of endogenous Type I IFNs that are immunomodulatory (Dikalova et al., 2010). Finally, mouse hepatitis virus (MHV) directly upregulated interleukin signaling such as IL-27 during acute encephalomyelitis.
As described above, coronaviruses induce apoptosis through multiple pathways, either directly (Pfefferle et al., 2011), or indirectly by inducing production of mito-ROS and downregulating antiapoptotic pathways such as Nrf2. Excessive ROS generation can lead to loss of mitochondrial function and apoptosis of lung epithelial cells (Sun et al., 2013). Increased mito-ROS also directly contribute to acute injury in lung tissue in mouse models of viral infections (Hu et al., 2019a,b). Indeed, increased apoptosis of epithelial cells is associated with lung injury in COVID-19 (Hussman, 2020). Studies have also shown that CD4 and CD8 T cells in patients with COVID-19 are more likely to get affected by apoptosis (Nieto-Torres et al., 2015). Thus, increased apoptosis during coronavirus infection contributes to increased tissue damage and pathogenesis of coronavirus infections.
As outlined above, coronaviruses induce production of mito-ROS that have an established complex crosstalk with mitophagy (Schofield and Schafer, 2021). Mitochondrial ROS and damage-associated molecular patterns (DAMPs) activate inflammasomes to induce inflammatory responses and tissue injury. Emerging evidence suggests that mitophagy protects against the hyperinflammation induced by ROS and DAMPs and regulates inflammatory responses in several diseases (Zhao et al., 2015). Thus, by inducing production of mito-ROS, mitochondrial dysfunction and mitophagy impairment, SARS-CoV-2 may contribute to inflammation and tissue damage (Shang et al., 2021).
Other than regulation of viral replication, inflammation and apoptosis, redox pathways may also contribute to regulation of other pathways that contribute to tissue damage such as autoimmunity and vascular dysfunction. Oxidative stress plays a central in autoimmune diseases (Ramani et al., 2020). Specifically, the antioxidant pathway Nrf2 has also a key role in regulation of autoimmunity (Freeborn and Rockwell, 2021). Given the possible role of autoimmunity in pathogenesis of COVID-19 and Long COVID, further understanding of the contribution of dysregulation redox pathways in development of autoimmunity during coronavirus infections is needed (Liu et al., 2021; Saad et al., 2021).
ROS induce levels of the adhesion molecules and increase permeability in endothelial cells (Mukherjee et al., 2005; Zinovkin et al., 2014). ROS also contribute to TNF-induced IL-6 expression and NF-κB activation (Pearlstein et al., 2002). Notably, IL-6 directly induces mito-ROS production and NOX in endothelial cells (Schrader et al., 2007; Valle et al., 2019) and impact NO bioavailability and endothelial function (Saura et al., 2006). SARS-CoV-2 S-protein binds to ACE2 and subsequently triggers reduction in ACE2 levels that cleaves ATII. High ATII level further leads to oxidative stress and endothelial dysfunction (Chernyak et al., 2020) and induces ROS production via NOX in endothelial cells. Thus, increased redox stress induced by SARS-CoV-2 may impact not only vascular permeability and vasodilation but also vascular inflammation.
All coronaviruses have the potential to induce tissue damage and end organ disease through viral replication, increased inflammation and apoptosis, induction of ROS and reduction of cytoprotective pathways such as the Nrf2 and HO-1 pathways. Increased redox stress is known instigator of lung dysfunction (Kellner et al., 2017), cardiovascular disease (Dubois-Deruy et al., 2020), central nervous system dysfunction such as neurodegeneration and neuropsychiatric disease (Reiter, 1998; Patel, 2016; Salim, 2017) and the metabolic syndrome (Ando and Fujita, 2009; Roberts and Sindhu, 2009; Carrier, 2017) which are all manifestations of both acute severe COVID-19 and post-acute sequelae of SARS-CoV-2 infection (often called Long COVID syndrome; Figure 3; Nalbandian et al., 2021). Coronaviruses differ in their potential to induce end organ damage (Table 3; Bonavia et al., 1997; De Albuquerque et al., 2006; de Wilde et al., 2013; Josset et al., 2013; Zhao et al., 2015; Li et al., 2016; Agostini et al., 2018; Coperchini et al., 2020; Huang et al., 2020; Petersen et al., 2020; Wang et al., 2020; Yi et al., 2020; Caldera-Crespo et al., 2021; Paidas et al., 2021; Tian et al., 2021; Jansen et al., 2022). Among the various human coronaviruses, end organ damage is observed in MERS, SARS-CoV-1, and SARS-CoV-2. These coronaviruses demonstrate a more severe pathology than HCoV-229E, HCoV-OC43, HCoV-NL63, and HCoV-HKU1 in terms of their fatality and systemic effects on multiple organ systems. Multiple animal models have been used to uncover the ways coronaviruses lead to the end organ damage that presents in patient autopsies. The MHV mice model is the most studied model among the coronaviruses, and it has served as a useful proxy in understanding SARS-CoV-2 (Paidas et al., 2022); MHV is known to enteric and respiratory disease, hepatitis, encephalitis, and chronic demyelination and is useful in studying infection of the liver and brain (Weiss and Navas-Martin, 2005). Despite MHV-1 utilizing a different receptor than either MERS or the SARS coronaviruses (carcinoembryonic antigen-related cell adhesion molecule 1 instead of dipeptidylpeptidase 4 and angiotensin-converting enzyme 2), end organ damage in the MHV-1 model has been acclaimed as an appropriate model for MERS, SARS-CoV-1, and SARS-CoV-2 (De Albuquerque et al., 2006; Agostini et al., 2018; Caldera-Crespo et al., 2021; Paidas et al., 2021; Tian et al., 2021). Herein, we briefly summarize redox pathways that regulate damage of the lung and the brain, the two main target organs for end organ disease in acute COVID-19 and Long COVID.
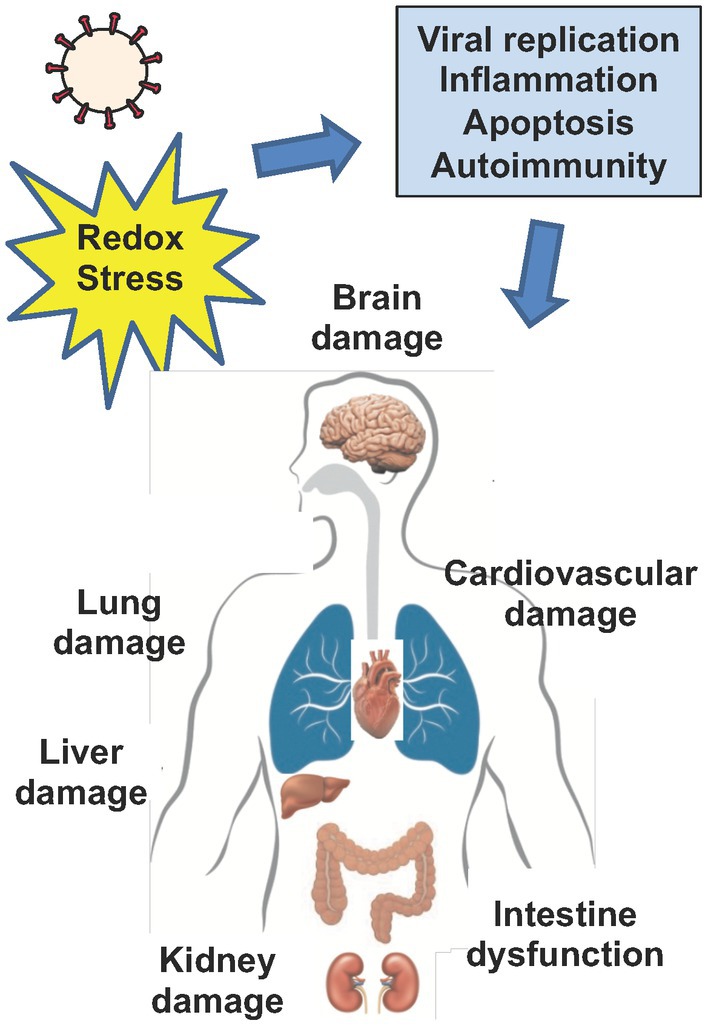
Figure 3. Viral infection and end organ damage. Increased redox stress drives viral replication, inflammation, apoptosis, vascular dysfunction and autoimmunity, in both acute infection with coronaviruses and in the setting of Post-Acute Sequelae of SARS-CoV-2 (PASC or Long COVID). Collectively, redox mediated pathways that drive viral replication, inflammation, apoptosis, autoimmunity, and vascular dysfunction contribute to cell and tissue damage that drives end organ disease in coronavirus infection. Cells enriched in mitochondria such as neurons, endothelial and epithelial cells may be particularly susceptible to increased redox stress driven by coronaviruses. Ultimately, increased redox stress during acute infection with coronaviruses and in the setting of PASC can directly or indirectly drive end organ disease such as brain, lung, liver, kidney and cardiovascular damage and induce intestinal dysfunction.
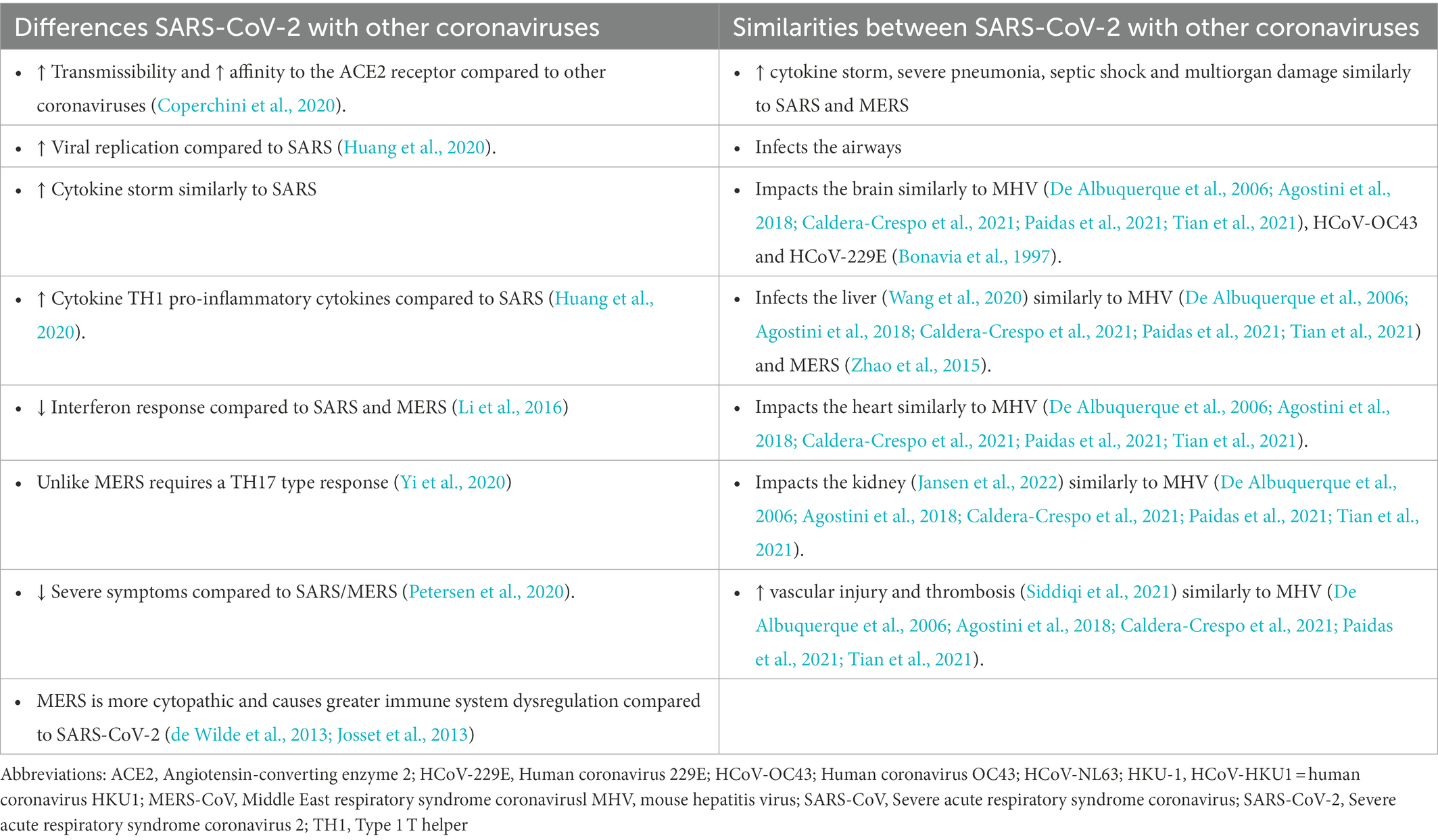
Table 3. Comparison of coronaviruses with regards to impact on end organ disease.
The excessive generation of oxygen radicals under pathological conditions such as acute lung injury (ALI) and its most severe form acute respiratory distress syndrome (ARDS) leads to increased endothelial permeability. Increased redox stress leads to increased permeability of lung blood vessels, increased infiltration of immune cells and increased accumulation of fluids in the alveolar system (Kellner et al., 2017). Mitochondria, NADPH oxidase (NOX), xanthine oxidase (Shasby et al., 1985; Barnard and Matalon, 1992), and eNOS are the major contributors of ROS in cells of vasculature during active metabolism that also contribute to the pathogenesis of ALI (Gross et al., 2015). Imbalance of antioxidant enzymes such as superoxide dismutase (SOD; Ndengele et al., 2005; Cai et al., 2014), catalase (Flick et al., 1988; Kozower et al., 2003) and glutathione peroxidase (GPx; Aggarwal et al., 2012; Kim et al., 2012a) and Nrf2 (Zhu et al., 2013; Peng et al., 2016) also contribute to pathogenesis of ALI and ARDS. Similarly, to MERS and SARS, severe SARS-CoV-2 infection presents with high levels of pro-inflammatory cytokines like IL-6, and can lead to ARDS, which is associated with acute renal injury, acute respiratory injury, and septic shock (Chen et al., 2020). COVID-19-related ARDS has a high prevalence and is different to ARDS due to other etiologies (Park et al., 2009).
SARS-CoV-2 directly impacts several of established instigators that contribute to pathogenesis of ALI/ARDS including mitochondrial function (Srinivasan et al., 2021), NOX (Damiano et al., 2020; Violi et al., 2020; de Oliveira and Nunes, 2021), xanthine oxidase (Pratomo et al., 2021; Al-Kuraishy et al., 2022), eNOS (Guimaraes et al., 2021), glutathione peroxidase (Labarrere and Kassab, 2022) and Nrf2 (Olagnier et al., 2020; Zhang et al., 2022). To date, there is no treatment for ARDS in COVID-19 disease (Jafari-Oori et al., 2021).
The brain is highly susceptible to oxidative stress due to enrichment for lipids, mitochondria, calcium, glutamate and increased redox stimuli (Cobley et al., 2018). Brain damage induced by oxidative stress may negatively impact normal functions of central nervous system and may contribute to the pathogenesis of neurodegenerative disorders such as Alzheimer and Parkinson disease and in the pathogenesis of neuropsychiatric disorders, including anxiety and depression (Salim, 2017). For these, increased oxidative stress through mitochondrial dysfunction, increased inflammation and energy imbalance has also been hypothesized to contribute to pathogenesis of neurocognitive dysfunction in Long COVID (Paul et al., 2021; Jarrott et al., 2022).
Multiple trials underway have tested antioxidants as therapeutic agents in COVID-19.1 Several therapies targeting redox imbalance already have been used for the treatment of COVID-19 including inhaled NO (Lotz et al., 2021), ubiquinol (Fukuda et al., 2016), combination of NADH and CoQ10 (Castro-Marrero et al., 2015), N-acetyl cysteine, mitochondria-targeted antioxidant MitoQ (Codo et al., 2020; Petcherski et al., 2022) and Nrf2 agonists (Zinovkin and Grebenchikov, 2020). Other potential antioxidant treatments that have been considered include, ubiquinol, nicotinamide, glutathione (and glutathione donors), cysteamine, sulforaphane, melatonin vitamin C, vitamin D, vitamin E, melatonin plus pentoxifylline and selenium. However, most of the proposed antioxidant treatments have either not been directly tested in humans in the setting of randomized control clinical trials or due to several methodological issues of heterogeneous studies, the data were inconclusive (Table 4). Many ongoing clinical trials regarding the use of antioxidants in treatment of COVID-19 have not been published. Notably, oral antioxidants have not produced dramatic improvements in conditions associated with redox imbalance (Barcelos et al., 2020). No single antioxidant can scavenge all the various ROS and reactive nitrogen species (RNS). Further validation with animal models and clinical trials are necessary to reveal therapeutic potential of combination therapies of antivirals, antioxidant and anti-inflammatory treatments.
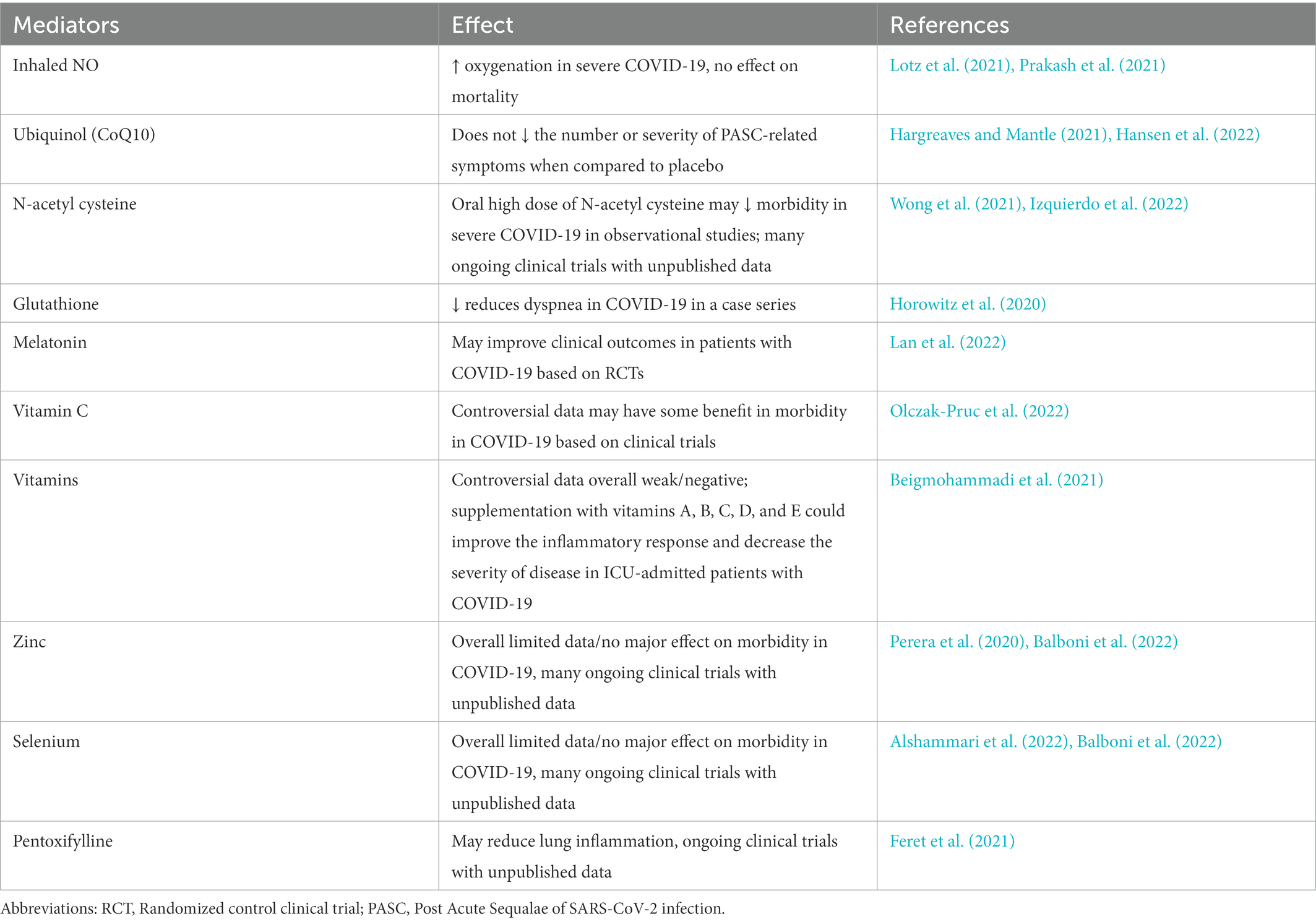
Table 4. Antioxidant treatments that have been tested in humans for treatment of coronavirus infections.
There is limited understanding how different coronaviruses including SARS-CoV-2, manipulate cellular redox machinery to drive viral replication and associated host cell responses including inflammation, apoptosis and associated end organ disease. The crosstalk between NOX and ACE2 as well mito-ROS may impact viral entry of coronaviruses while mito-ROS may also induce multiple proviral cytoplasmic pathways. Experimental studies have also shown that coronaviruses induce downregulation of antioxidant genes such as Nrf2 in combination with an upregulation of oxidative stress genes like myeloperoxidase that may contribute to both increased viral replication and inflammation. Coronaviruses may induce several redox sensitive proinflammatory pathways such NF-kB, mito-ROS and NOX pathways and downregulate anti-inflammatory ACE2 and Nrf2 pathways. Coronaviruses may further trigger cell damage through activation of redox sensitive pyroptosis and apoptosis. Finally, other than regulation of viral replication, inflammation and apoptosis, redox pathways may also contribute to regulation of other pathways that contribute to tissue damage such as autoimmunity and vascular dysfunction. Thus, coronaviruses have the potential to induce tissue damage and end organ disease through viral replication, increased inflammation and apoptosis, induction of ROS and reduction of cytoprotective pathways such as the Nrf2 and HO-1 pathways. Coronaviruses differ in their potential to induce end organ damage. Among the various human coronaviruses, end organ damage is observed in MERS, SARS-CoV-1, and SARS-CoV-2. Increased redox stress is known instigator of lung dysfunction (Kellner et al., 2017), cardiovascular disease (Dubois-Deruy et al., 2020), central nervous system dysfunction such as neurodegeneration and neuropsychiatric disease (Reiter, 1998; Patel, 2016; Salim, 2017) and the metabolic syndrome (Ando and Fujita, 2009; Roberts and Sindhu, 2009; Carrier, 2017) which are all manifestations of both acute severe COVID-19 and Long COVID syndrome (Nalbandian et al., 2021). Given the complexity of the pathogenesis of coronavirus infections and that oral antioxidants have not produced dramatic improvements in conditions associated with redox imbalance, further validation with animal models and clinical trials are necessary to reveal therapeutic potential of combination therapies of antivirals, antioxidant and anti-inflammatory treatments. Understanding the mechanisms that contribute to the pathogenesis of coronavirus infections, will set the foundation for development of new treatments for coronavirus infections.
CG, SiS, TA, and TK wrote the manuscript, reviewed the literature, and collected the information. SaS revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported in part by National Institute of Health grant R01AG059501 (TK), National Institute of Health grant R01AG059502 04S1 (TK), and California HIV/AIDS Research Program grant OS17-LA-002 (TK).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aggarwal, S., Dimitropoulou, C., Lu, Q., Black, S. M., and Sharma, S. (2012). Glutathione supplementation attenuates lipopolysaccharide-induced mitochondrial dysfunction and apoptosis in a mouse model of acute lung injury. Front. Physiol. 3:161. doi: 10.3389/fphys.2012.00161
Agostini, M. L., Andres, E. L., Sims, A. C., Graham, R. L., Sheahan, T. P., Lu, X., et al. (2018). Coronavirus susceptibility to the antiviral Remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. MBio 9:2. doi: 10.1128/mBio.00221-18
Agrawal, P., Nawadkar, R., Ojha, H., Kumar, J., and Sahu, A. (2017). Complement evasion strategies of viruses: an overview. Front. Microbiol. 8:1117. doi: 10.3389/fmicb.2017.01117
Alam, M. S., and Czajkowsky, D. M. (2022). SARS-CoV-2 infection and oxidative stress: pathophysiological insight into thrombosis and therapeutic opportunities. Cytokine Growth Factor Rev. 63, 44–57. doi: 10.1016/j.cytogfr.2021.11.001
Al-Kuraishy, H. M., Al-Gareeb, A. I., Al-Niemi, M. S., Aljowaie, R. M., Almutairi, S. M., Alexiou, A., et al. (2022). The prospective effect of allopurinol on the oxidative stress index and endothelial dysfunction in Covid-19. Inflammation 45, 1651–1667. doi: 10.1007/s10753-022-01648-7
Alshammari, M. K., Fatima, W., Alraya, R. A., Khuzaim Alzahrani, A., Kamal, M., Alshammari, R. S., et al. (2022). Selenium and COVID-19: A spotlight on the clinical trials, inventive compositions, and patent literature. J. Infect. Public Health 15, 1225–1233. doi: 10.1016/j.jiph.2022.09.011
Amini, M. A., Karimi, J., Talebi, S. S., and Piri, H. (2022). The association of COVID-19 and reactive oxygen species modulator 1 (ROMO1) with oxidative stress. Chonnam Med. J. 58, 1–5. doi: 10.4068/cmj.2022.58.1.1
Ando, K., and Fujita, T. (2009). Metabolic syndrome and oxidative stress. Free Radic. Biol. Med. 47, 213–218. doi: 10.1016/j.freeradbiomed.2009.04.030
Arbore, G., and Kemper, C. (2016). A novel "complement-metabolism-inflammasome axis" as a key regulator of immune cell effector function. Eur. J. Immunol. 46, 1563–1573. doi: 10.1002/eji.201546131
Balboni, E., Zagnoli, F., Filippini, T., Fairweather-Tait, S. J., and Vinceti, M. (2022). Zinc and selenium supplementation in COVID-19 prevention and treatment: a systematic review of the experimental studies. J. Trace Elem. Med. Biol. 71:126956. doi: 10.1016/j.jtemb.2022.126956
Barcelos, I., Shadiack, E., Ganetzky, R. D., and Falk, M. J. (2020). Mitochondrial medicine therapies: rationale, evidence, and dosing guidelines. Curr. Opin. Pediatr. 32, 707–718. doi: 10.1097/MOP.0000000000000954
Barnard, M. L., and Matalon, S. (1992). Mechanisms of extracellular reactive oxygen species injury to the pulmonary microvasculature. J. Appl. Physiol. 72, 1724–1729. doi: 10.1152/jappl.1992.72.5.1724
Beigmohammadi, M. T., Bitarafan, S., Hoseindokht, A., Abdollahi, A., Amoozadeh, L., and Soltani, D. (2021). The effect of supplementation with vitamins A, B, C, D, and E on disease severity and inflammatory responses in patients with COVID-19: a randomized clinical trial. Trials 22:802. doi: 10.1186/s13063-021-05795-4
Bonavia, A., Arbour, N., Yong, V. W., and Talbot, P. J. (1997). Infection of primary cultures of human neural cells by human coronaviruses 229E and OC43. J. Virol. 71, 800–806. doi: 10.1128/jvi.71.1.800-806.1997
Bulua, A. C., Simon, A., Maddipati, R., Pelletier, M., Park, H., Kim, K. Y., et al. (2011). Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 208, 519–533. doi: 10.1084/jem.20102049
Cai, L., Yi, F., Dai, Z., Huang, X., Zhao, Y. D., Mirza, M. K., et al. (2014). Loss of caveolin-1 and adiponectin induces severe inflammatory lung injury following LPS challenge through excessive oxidative/nitrative stress. Am. J. Phys. Lung Cell. Mol. Phys. 306, L566–L573. doi: 10.1152/ajplung.00182.2013
Caldera-Crespo, L. A., Paidas, M. J., Roy, S., Schulman, C. I., Kenyon, N. S., Daunert, S., et al. (2021). Experimental models of COVID-19. Front. Cell. Infect. Microbiol. 11:792584. doi: 10.3389/fcimb.2021.792584
Canty, T. G. Jr., Boyle, E. M. Jr., Farr, A., Morgan, E. N., Verrier, E. D., and Pohlman, T. H. (1999). Oxidative stress induces NF-kappaB nuclear translocation without degradation of IkappaBalpha. Circulation 100, II361–II364.
Carrier, A. (2017). Metabolic syndrome and oxidative stress: A complex relationship. Antioxid. Redox Signal. 26, 429–431. doi: 10.1089/ars.2016.6929
Castro-Marrero, J., Cordero, M. D., Segundo, M. J., Saez-Francas, N., Calvo, N., Roman-Malo, L., et al. (2015). Does oral coenzyme Q10 plus NADH supplementation improve fatigue and biochemical parameters in chronic fatigue syndrome? Antioxid. Redox Signal. 22, 679–685. doi: 10.1089/ars.2014.6181
Chen, Z., Krmar, R. T., Dada, L., Efendiev, R., Leibiger, I. B., Pedemonte, C. H., et al. (2006). Phosphorylation of adaptor protein-2 mu2 is essential for Na+, K+-ATPase endocytosis in response to either G protein-coupled receptor or reactive oxygen species. Am. J. Respir. Cell Mol. Biol. 35, 127–132. doi: 10.1165/rcmb.2006-0044OC
Chen, J., Lau, Y. F., Lamirande, E. W., Paddock, C. D., Bartlett, J. H., Zaki, S. R., et al. (2010). Cellular immune responses to severe acute respiratory syndrome coronavirus (SARS-CoV) infection in senescent BALB/c mice: CD4+ T cells are important in control of SARS-CoV infection. J. Virol. 84, 1289–1301. doi: 10.1128/JVI.01281-09
Chen, I. Y., Moriyama, M., Chang, M. F., and Ichinohe, T. (2019). Severe acute respiratory syndrome coronavirus Viroporin 3a activates the NLRP3 Inflammasome. Front. Microbiol. 10:50. doi: 10.3389/fmicb.2019.00050
Chen, N., Zhou, M., Dong, X., Qu, J., Gong, F., Han, Y., et al. (2020). Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 395, 507–513. doi: 10.1016/S0140-6736(20)30211-7
Chernyak, B. V., Popova, E. N., Prikhodko, A. S., Grebenchikov, O. A., Zinovkina, L. A., and Zinovkin, R. A. (2020). COVID-19 and oxidative stress. Biochemistry (Mosc) 85, 1543–1553. doi: 10.1134/S0006297920120068
Cho, H. Y., Imani, F., Miller-Degraff, L., Walters, D., Melendi, G. A., Yamamoto, M., et al. (2009). Antiviral activity of Nrf2 in a murine model of respiratory syncytial virus disease. Am. J. Respir. Crit. Care Med. 179, 138–150. doi: 10.1164/rccm.200804-535OC
Chua, C. C., Hamdy, R. C., and Chua, B. H. (1998). Upregulation of vascular endothelial growth factor by H2O2 in rat heart endothelial cells. Free Radic. Biol. Med. 25, 891–897. doi: 10.1016/S0891-5849(98)00115-4
Cobley, J. N., Fiorello, M. L., and Bailey, D. M. (2018). 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 15, 490–503. doi: 10.1016/j.redox.2018.01.008
Codo, A. C., Davanzo, G. G., Monteiro, L. B., De Souza, G. F., Muraro, S. P., Virgilio-Da-Silva, J. V., et al. (2020). Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1alpha/glycolysis-dependent axis. Cell Metab. 32:e435, 437–446.e5. doi: 10.1016/j.cmet.2020.07.007
Connors, J. M., and Levy, J. H. (2020). COVID-19 and its implications for thrombosis and anticoagulation. Blood 135, 2033–2040. doi: 10.1182/blood.2020006000
Cooke, C. L., and Davidge, S. T. (2002). Peroxynitrite increases iNOS through NF-kappaB and decreases prostacyclin synthase in endothelial cells. Am. J. Phys. Cell Physiol. 282, C395–C402. doi: 10.1152/ajpcell.00295.2001
Coperchini, F., Chiovato, L., Croce, L., Magri, F., and Rotondi, M. (2020). The cytokine storm in COVID-19: an overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 53, 25–32. doi: 10.1016/j.cytogfr.2020.05.003
Daiber, A., Di Lisa, F., Oelze, M., Kroller-Schon, S., Steven, S., Schulz, E., et al. (2017). Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 174, 1670–1689. doi: 10.1111/bph.13403
Damiano, S., Sozio, C., La Rosa, G., and Santillo, M. (2020). NOX-dependent signaling dysregulation in severe COVID-19: clues to effective treatments. Front. Cell. Infect. Microbiol. 10:608435. doi: 10.3389/fcimb.2020.608435
Dashdorj, A., Jyothi, K. R., Lim, S., Jo, A., Nguyen, M. N., Ha, J., et al. (2013). Mitochondria-targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines. BMC Med. 11:178. doi: 10.1186/1741-7015-11-178
De Albuquerque, N., Baig, E., Ma, X., Zhang, J., He, W., Rowe, A., et al. (2006). Murine hepatitis virus strain 1 produces a clinically relevant model of severe acute respiratory syndrome in A/J mice. J. Virol. 80, 10382–10394. doi: 10.1128/JVI.00747-06
De Oliveira, A. A., and Nunes, K. P. (2021). Crosstalk of TLR4, vascular NADPH oxidase, and COVID-19 in diabetes: what are the potential implications? Vasc. Pharmacol. 139:106879. doi: 10.1016/j.vph.2021.106879
De Wilde, A. H., Raj, V. S., Oudshoorn, D., Bestebroer, T. M., Van Nieuwkoop, S., Limpens, R., et al. (2013). MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-alpha treatment. J. Gen. Virol. 94, 1749–1760. doi: 10.1099/vir.0.052910-0
De Wilde, A. H., Zevenhoven-Dobbe, J. C., Van Der Meer, Y., Thiel, V., Narayanan, K., Makino, S., et al. (2011). Cyclosporin A inhibits the replication of diverse coronaviruses. J. Gen. Virol. 92, 2542–2548. doi: 10.1099/vir.0.034983-0
Demers-Lamarche, J., Guillebaud, G., Tlili, M., Todkar, K., Belanger, N., Grondin, M., et al. (2016). Loss of mitochondrial function impairs lysosomes. J. Biol. Chem. 291, 10263–10276. doi: 10.1074/jbc.M115.695825
Desouki, M. M., Kulawiec, M., Bansal, S., Das, G. M., and Singh, K. K. (2005). Cross talk between mitochondria and superoxide generating NADPH oxidase in breast and ovarian tumors. Cancer Biol. Ther. 4, 1367–1373. doi: 10.4161/cbt.4.12.2233
Dikalov, S. I., Nazarewicz, R. R., Bikineyeva, A., Hilenski, L., Lassegue, B., Griendling, K. K., et al. (2014). Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid. Redox Signal. 20, 281–294. doi: 10.1089/ars.2012.4918
Dikalova, A. E., Bikineyeva, A. T., Budzyn, K., Nazarewicz, R. R., Mccann, L., Lewis, W., et al. (2010). Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 107, 106–116. doi: 10.1161/CIRCRESAHA.109.214601
Dosch, S. F., Mahajan, S. D., and Collins, A. R. (2009). SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-kappaB pathway in human monocyte macrophages in vitro. Virus Res. 142, 19–27. doi: 10.1016/j.virusres.2009.01.005
Doughan, A. K., Harrison, D. G., and Dikalov, S. I. (2008). Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 102, 488–496. doi: 10.1161/CIRCRESAHA.107.162800
Dubois-Deruy, E., Peugnet, V., Turkieh, A., and Pinet, F. (2020). Oxidative stress in cardiovascular diseases. Antioxidants 9, 864–879. doi: 10.3390/antiox9090864
Elesela, S., and Lukacs, N. W. (2021). Role of mitochondria in viral infections. Life 11, 232–247. doi: 10.3390/life11030232
Emerling, B. M., Platanias, L. C., Black, E., Nebreda, A. R., Davis, R. J., and Chandel, N. S. (2005). Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol. Cell. Biol. 25, 4853–4862. doi: 10.1128/MCB.25.12.4853-4862.2005
Espinoza, J. A., Gonzalez, P. A., and Kalergis, A. M. (2017). Modulation of antiviral immunity by Heme Oxygenase-1. Am. J. Pathol. 187, 487–493. doi: 10.1016/j.ajpath.2016.11.011
Feret, W., Nalewajska, M., Wojczynski, L., Witkiewicz, W., Klos, P., Dziedziejko, V., et al. (2021). Pentoxifylline as a potential adjuvant therapy for COVID-19: impeding the burden of the cytokine storm. J. Clin. Med. 10, 5305–5313. doi: 10.3390/jcm10225305
Fink, K., Duval, A., Martel, A., Soucy-Faulkner, A., and Grandvaux, N. (2008). Dual role of NOX2 in respiratory syncytial virus-and Sendai virus-induced activation of NF-kappaB in airway epithelial cells. J. Immunol. 180, 6911–6922. doi: 10.4049/jimmunol.180.10.6911
Finkel, T. (2011). Signal transduction by reactive oxygen species. J. Cell Biol. 194, 7–15. doi: 10.1083/jcb.201102095
Flick, M. R., Milligan, S. A., Hoeffel, J. M., and Goldstein, I. M. (1988). Catalase prevents increased lung vascular permeability during air emboli in unanesthetized sheep. J. Appl. Physiol. 64, 929–935. doi: 10.1152/jappl.1988.64.3.929
Freeborn, R. A., and Rockwell, C. E. (2021). The role of Nrf2 in autoimmunity and infectious disease: therapeutic possibilities. Adv. Pharmacol. 91, 61–110. doi: 10.1016/bs.apha.2020.10.003
Fukuda, S., Nojima, J., Kajimoto, O., Yamaguti, K., Nakatomi, Y., Kuratsune, H., et al. (2016). Ubiquinol-10 supplementation improves autonomic nervous function and cognitive function in chronic fatigue syndrome. Biofactors 42, 431–440. doi: 10.1002/biof.1293
Garrido, A. M., and Griendling, K. K. (2009). NADPH oxidases and angiotensin II receptor signaling. Mol. Cell. Endocrinol. 302, 148–158. doi: 10.1016/j.mce.2008.11.003
Gassen, N. C., Niemeyer, D., Muth, D., Corman, V. M., Martinelli, S., Gassen, A., et al. (2019). SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-coronavirus infection. Nat. Commun. 10:5770. doi: 10.1038/s41467-019-13659-4
Gloire, G., Legrand-Poels, S., and Piette, J. (2006). NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem. Pharmacol. 72, 1493–1505. doi: 10.1016/j.bcp.2006.04.011
Gralinski, L. E., Sheahan, T. P., Morrison, T. E., Menachery, V. D., Jensen, K., Leist, S. R., et al. (2018). Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. MBio 9, e01753–e01718. doi: 10.1128/mBio.01753-18
Green, D. R., and Llambi, F. (2015). Cell death signaling. Cold Spring Harb. Perspect. Biol. 7:7. doi: 10.1101/cshperspect.a006080
Gross, C. M., Rafikov, R., Kumar, S., Aggarwal, S., Ham, P. B. 3rd, Meadows, M. L., et al. (2015). Endothelial nitric oxide synthase deficient mice are protected from lipopolysaccharide induced acute lung injury. PLoS One 10:e0119918. doi: 10.1371/journal.pone.0119918
Guimaraes, L. M. F., Rossini, C. V. T., and Lameu, C. (2021). Implications of SARS-Cov-2 infection on eNOS and iNOS activity: consequences for the respiratory and vascular systems. Nitric Oxide 111-112, 64–71. doi: 10.1016/j.niox.2021.04.003
Halestrap, A. P., Clarke, S. J., and Javadov, S. A. (2004). Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc. Res. 61, 372–385. doi: 10.1016/S0008-6363(03)00533-9
Hamming, I., Timens, W., Bulthuis, M. L., Lely, A. T., Navis, G., and Van Goor, H. (2004). Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 203, 631–637. doi: 10.1002/path.1570
Han, M., Lee, D., Lee, S. H., and Kim, T. H. (2021). Oxidative stress and antioxidant pathway in allergic rhinitis. Antioxidants 10, 1266–1281. doi: 10.3390/antiox10081266
Han, Y., Xu, X., Tang, C., Gao, P., Chen, X., Xiong, X., et al. (2018). Reactive oxygen species promote tubular injury in diabetic nephropathy: the role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol. 16, 32–46. doi: 10.1016/j.redox.2018.02.013
Hansen, K. S., Mogensen, T. H., Agergaard, J., Schiottz-Christensen, B., Ostergaard, L., Vibholm, L. K., et al. (2022). High-dose coenzyme Q10 therapy versus placebo in patients with post COVID-19 condition: A randomized, phase 2, crossover trial. Lancet Reg. Health Eur.,:100539, doi: 10.1016/j.lanepe.2022.100539 [Epub ahead of print].
Hargreaves, I. R., and Mantle, D. (2021). COVID-19, coenzyme Q10 and selenium. Adv. Exp. Med. Biol. 1327, 161–168. doi: 10.1007/978-3-030-71697-4_13
Hariharan, A., Hakeem, A. R., Radhakrishnan, S., Reddy, M. S., and Rela, M. (2021). The role and therapeutic potential of NF-kappa-B pathway in severe COVID-19 patients. Inflammopharmacology 29, 91–100. doi: 10.1007/s10787-020-00773-9
Herengt, A., Thyrsted, J., and Holm, C. K. (2021). NRF2 in viral infection. Antioxidants 10, 1491–1688. doi: 10.3390/antiox10091491
Horowitz, R. I., Freeman, P. R., and Bruzzese, J. (2020). Efficacy of glutathione therapy in relieving dyspnea associated with COVID-19 pneumonia: A report of 2 cases. Respir. Med. Case. Rep. 30:101063. doi: 10.1016/j.rmcr.2020.101063
Hosakote, Y. M., Liu, T., Castro, S. M., Garofalo, R. P., and Casola, A. (2009). Respiratory syncytial virus induces oxidative stress by modulating antioxidant enzymes. Am. J. Respir. Cell Mol. Biol. 41, 348–357. doi: 10.1165/rcmb.2008-0330OC
Hu, M., Bogoyevitch, M. A., and Jans, D. A. (2019a). Subversion of host cell mitochondria by RSV to favor virus production is dependent on inhibition of mitochondrial complex I and ROS generation. Cells 8, 1417–1433. doi: 10.3390/cells8111417
Hu, M., Schulze, K. E., Ghildyal, R., Henstridge, D. C., Kolanowski, J. L., New, E. J., et al. (2019b). Respiratory syncytial virus co-opts host mitochondrial function to favour infectious virus production. elife 8, 1–30. doi: 10.7554/eLife.42448
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506. doi: 10.1016/S0140-6736(20)30183-5
Hussman, J. P. (2020). Cellular and molecular pathways of COVID-19 and potential points of therapeutic intervention. Front. Pharmacol. 11:1169. doi: 10.3389/fphar.2020.01169
Hyser, J. M., and Estes, M. K. (2015). Pathophysiological consequences of calcium-conducting Viroporins. Annu. Rev. Virol. 2, 473–496. doi: 10.1146/annurev-virology-100114-054846
Imai, Y., Kuba, K., Neely, G. G., Yaghubian-Malhami, R., Perkmann, T., Van Loo, G., et al. (2008). Identification of oxidative stress and toll-like receptor 4 signaling as a key pathway of acute lung injury. Cells 133, 235–249. doi: 10.1016/j.cell.2008.02.043
Irigoyen, N., Firth, A. E., Jones, J. D., Chung, B. Y., Siddell, S. G., and Brierley, I. (2016). High-resolution analysis of coronavirus gene expression by RNA sequencing and ribosome profiling. PLoS Pathog. 12:e1005473. doi: 10.1371/journal.ppat.1005473
Ito, K., Hanazawa, T., Tomita, K., Barnes, P. J., and Adcock, I. M. (2004). Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: role of tyrosine nitration. Biochem. Biophys. Res. Commun. 315, 240–245. doi: 10.1016/j.bbrc.2004.01.046
Izquierdo, J. L., Soriano, J. B., Gonzalez, Y., Lumbreras, S., Ancochea, J., Echeverry, C., et al. (2022). Use of N-acetylcysteine at high doses as an oral treatment for patients hospitalized with COVID-19. Sci. Prog. 105:003685042210745. doi: 10.1177/00368504221074574
Jafari-Oori, M., Ghasemifard, F., Ebadi, A., Karimi, L., Rahimi-Bashar, F., Jamialahmadi, T., et al. (2021). Acute respiratory distress syndrome and COVID-19: A scoping review and meta-analysis. Adv. Exp. Med. Biol. 1321, 211–228. doi: 10.1007/978-3-030-59261-5_18
Jamaluddin, M., Tian, B., Boldogh, I., Garofalo, R. P., and Brasier, A. R. (2009). Respiratory syncytial virus infection induces a reactive oxygen species-MSK1-phospho-Ser-276 RelA pathway required for cytokine expression. J. Virol. 83, 10605–10615. doi: 10.1128/JVI.01090-09
Jansen, J., Reimer, K. C., Nagai, J. S., Varghese, F. S., Overheul, G. J., De Beer, M., et al. (2022). SARS-CoV-2 infects the human kidney and drives fibrosis in kidney organoids. Cell Stem Cell 29:e218, 217–231.e8. doi: 10.1016/j.stem.2021.12.010
Jany, B., Betz, R., and Schreck, R. (1995). Activation of the transcription factor NF-kappa B in human tracheobronchial epithelial cells by inflammatory stimuli. Eur. Respir. J. 8, 387–391. doi: 10.1183/09031936.95.08030387
Jarrott, B., Head, R., Pringle, K. G., Lumbers, E. R., and Martin, J. H. (2022). "LONG COVID"-A hypothesis for understanding the biological basis and pharmacological treatment strategy. Pharmacol. Res. Perspect. 10:e00911. doi: 10.1002/prp2.911
Jiang, Y., Zhao, G., Song, N., Li, P., Chen, Y., Guo, Y., et al. (2018). Blockade of the C5a-C5aR axis alleviates lung damage in hDPP4-transgenic mice infected with MERS-CoV. Emerg. Microbes Infect. 7, 1–12. doi: 10.1038/s41426-018-0063-8
Josset, L., Menachery, V. D., Gralinski, L. E., Agnihothram, S., Sova, P., Carter, V. S., et al. (2013). Cell host response to infection with novel human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus. MBio 4, e00165–e00113. doi: 10.1128/mBio.00165-13
Kandasamy, M. (2021). NF-kappaB signalling as a pharmacological target in COVID-19: potential roles for IKKbeta inhibitors. Naunyn Schmiedeberg's Arch. Pharmacol. 394, 561–567. doi: 10.1007/s00210-020-02035-5
Kasahara, Y., Iwai, K., Yachie, A., Ohta, K., Konno, A., Seki, H., et al. (1997). Involvement of reactive oxygen intermediates in spontaneous and CD95 (Fas/APO-1)-mediated apoptosis of neutrophils. Blood 89, 1748–1753. doi: 10.1182/blood.V89.5.1748
Kasai, S., Shimizu, S., Tatara, Y., Mimura, J., and Itoh, K. (2020). Regulation of Nrf2 by mitochondrial reactive oxygen species in physiology and pathology. Biomol. Ther. 10, 320–341. doi: 10.3390/biom10020320
Kau, H. C., Wu, S. B., Tsai, C. C., Liu, C. J., and Wei, Y. H. (2016). Cigarette smoke extract-induced oxidative stress and fibrosis-related genes expression in orbital fibroblasts from patients with Graves' Ophthalmopathy. Oxidative Med. Cell. Longev. 2016, 1–10. doi: 10.1155/2016/4676289
Kaul, P., Biagioli, M. C., Singh, I., and Turner, R. B. (2000). Rhinovirus-induced oxidative stress and interleukin-8 elaboration involves p47-phox but is independent of attachment to intercellular adhesion molecule-1 and viral replication. J. Infect. Dis. 181, 1885–1890. doi: 10.1086/315504
Kefaloyianni, E., Gaitanaki, C., and Beis, I. (2006). ERK1/2 and p38-MAPK signalling pathways, through MSK1, are involved in NF-kappaB transactivation during oxidative stress in skeletal myoblasts. Cell. Signal. 18, 2238–2251. doi: 10.1016/j.cellsig.2006.05.004
Kellner, M., Noonepalle, S., Lu, Q., Srivastava, A., Zemskov, E., and Black, S. M. (2017). ROS signaling in the pathogenesis of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS). Adv. Exp. Med. Biol. 967, 105–137. doi: 10.1007/978-3-319-63245-2_8
Kesic, M. J., Simmons, S. O., Bauer, R., and Jaspers, I. (2011). Nrf2 expression modifies influenza A entry and replication in nasal epithelial cells. Free Radic. Biol. Med. 51, 444–453. doi: 10.1016/j.freeradbiomed.2011.04.027
Khan, N. M., Ahmad, I., and Haqqi, T. M. (2018). Nrf2/ARE pathway attenuates oxidative and apoptotic response in human osteoarthritis chondrocytes by activating ERK1/2/ELK1-P70S6K-P90RSK signaling axis. Free Radic. Biol. Med. 116, 159–171. doi: 10.1016/j.freeradbiomed.2018.01.013
Khomich, O. A., Kochetkov, S. N., Bartosch, B., and Ivanov, A. V. (2018). Redox biology of respiratory viral infections. Viruses 10, 392–419. doi: 10.3390/v10080392
Kim, S. M., Kim, Y. G., Jeong, K. H., Lee, S. H., Lee, T. W., Ihm, C. G., et al. (2012b). Angiotensin II-induced mitochondrial Nox4 is a major endogenous source of oxidative stress in kidney tubular cells. PLoS One 7:e39739. doi: 10.1371/journal.pone.0039739
Kim, K. S., Suh, G. J., Kwon, W. Y., Kwak, Y. H., Lee, K., Lee, H. J., et al. (2012a). Antioxidant effects of selenium on lung injury in paraquat intoxicated rats. Clin. Toxicol. 50, 749–753. doi: 10.3109/15563650.2012.708418
Kindrachuk, J., Ork, B., Hart, B. J., Mazur, S., Holbrook, M. R., Frieman, M. B., et al. (2015). Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob. Agents Chemother. 59, 1088–1099. doi: 10.1128/AAC.03659-14
Kircheis, R., Haasbach, E., Lueftenegger, D., Heyken, W. T., Ocker, M., and Planz, O. (2020). NF-kappaB pathway as a potential target for treatment of critical stage COVID-19 patients. Front. Immunol. 11:598444. doi: 10.3389/fimmu.2020.598444
Komaravelli, N., and Casola, A. (2014). Respiratory viral infections and subversion of cellular antioxidant defenses. J. Pharmacogenomics Pharmacoproteomics 5, 1000141–1000161. doi: 10.4172/2153-0645.1000141
Komaravelli, N., Tian, B., Ivanciuc, T., Mautemps, N., Brasier, A. R., Garofalo, R. P., et al. (2015). Respiratory syncytial virus infection down-regulates antioxidant enzyme expression by triggering deacetylation-proteasomal degradation of Nrf2. Free Radic. Biol. Med. 88, 391–403. doi: 10.1016/j.freeradbiomed.2015.05.043
Korn, S. H., Wouters, E. F., Vos, N., and Janssen-Heininger, Y. M. (2001). Cytokine-induced activation of nuclear factor-kappa B is inhibited by hydrogen peroxide through oxidative inactivation of IkappaB kinase. J. Biol. Chem. 276, 35693–35700. doi: 10.1074/jbc.M104321200
Kosmider, B., Messier, E. M., Janssen, W. J., Nahreini, P., Wang, J., Hartshorn, K. L., et al. (2012). Nrf2 protects human alveolar epithelial cells against injury induced by influenza A virus. Respir. Res. 13:43. doi: 10.1186/1465-9921-13-43
Kozower, B. D., Christofidou-Solomidou, M., Sweitzer, T. D., Muro, S., Buerk, D. G., Solomides, C. C., et al. (2003). Immunotargeting of catalase to the pulmonary endothelium alleviates oxidative stress and reduces acute lung transplantation injury. Nat. Biotechnol. 21, 392–398. doi: 10.1038/nbt806
Kulisz, A., Chen, N., Chandel, N. S., Shao, Z., and Schumacker, P. T. (2002). Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am. J. Phys. Lung Cell. Mol. Phys. 282, L1324–L1329. doi: 10.1152/ajplung.00326.2001
Labarrere, C. A., and Kassab, G. S. (2022). Glutathione deficiency in the pathogenesis of SARS-CoV-2 infection and its effects upon the host immune response in severe COVID-19 disease. Front. Microbiol. 13:979719. doi: 10.3389/fmicb.2022.979719
Lan, S. H., Lee, H. Z., Chao, C. M., Chang, S. P., Lu, L. C., and Lai, C. C. (2022). Efficacy of melatonin in the treatment of patients with COVID-19: A systematic review and meta-analysis of randomized controlled trials. J. Med. Virol. 94, 2102–2107. doi: 10.1002/jmv.27595
Lavrentyev, E. N., and Malik, K. U. (2009). High glucose-induced Nox1-derived superoxides downregulate PKC-betaII, which subsequently decreases ACE2 expression and ANG(1-7) formation in rat VSMCs. Am. J. Physiol. Heart Circ. Physiol. 296, H106–H118. doi: 10.1152/ajpheart.00239.2008
Lee, D. Y., Wauquier, F., Eid, A. A., Roman, L. J., Ghosh-Choudhury, G., Khazim, K., et al. (2013). Nox4 NADPH oxidase mediates peroxynitrite-dependent uncoupling of endothelial nitric-oxide synthase and fibronectin expression in response to angiotensin II: role of mitochondrial reactive oxygen species. J. Biol. Chem. 288, 28668–28686. doi: 10.1074/jbc.M113.470971
Li, W., Qiao, J., You, Q., Zong, S., Peng, Q., Liu, Y., et al. (2021b). SARS-CoV-2 Nsp5 activates NF-kappaB pathway by upregulating SUMOylation of MAVS. Front. Immunol. 12:750969. doi: 10.3389/fimmu.2021.750969
Li, R., Ren, T., and Zeng, J. (2019). Mitochondrial coenzyme Q protects sepsis-induced acute lung injury by activating PI3K/Akt/GSK-3beta/mTOR pathway in rats. Biomed. Res. Int. 2019, 1–9. doi: 10.1155/2019/5240898
Li, K., Wohlford-Lenane, C., Perlman, S., Zhao, J., Jewell, A. K., Reznikov, L. R., et al. (2016). Middle East respiratory syndrome coronavirus causes multiple organ damage and lethal disease in mice transgenic for human dipeptidyl peptidase 4. J. Infect. Dis. 213, 712–722. doi: 10.1093/infdis/jiv499
Li, Y., Wu, K., Zeng, S., Zou, L., Li, X., Xu, C., et al. (2022). The role of Mitophagy in viral infection. Cells 11, 711–723. doi: 10.3390/cells11040711
Li, M., Zhu, D., Yang, J., Yan, L., Xiong, Z., Lu, J., et al. (2021a). Clinical treatment experience in severe and critical COVID-19. Mediat. Inflamm. 2021:9924542. doi: 10.1155/2021/9924542
Liao, Q. J., Ye, L. B., Timani, K. A., Zeng, Y. C., She, Y. L., Ye, L., et al. (2005). Activation of NF-kappaB by the full-length nucleocapsid protein of the SARS coronavirus. Acta Biochim. Biophys. Sin. Shanghai 37, 607–612. doi: 10.1111/j.1745-7270.2005.00082.x
Liu, Y., Sawalha, A. H., and Lu, Q. (2021). COVID-19 and autoimmune diseases. Curr. Opin. Rheumatol. 33, 155–162. doi: 10.1097/BOR.0000000000000776
Liu, T., Zhang, L., Joo, D., and Sun, S. C. (2017). NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2, 1–9. doi: 10.1038/sigtrans.2017.23
Lotz, C., Muellenbach, R. M., Meybohm, P., Mutlak, H., Lepper, P. M., Rolfes, C. B., et al. (2021). Effects of inhaled nitric oxide in COVID-19-induced ARDS - is it worthwhile? Acta Anaesthesiol. Scand. 65, 629–632. doi: 10.1111/aas.13757
Lowes, D. A., Thottakam, B. M., Webster, N. R., Murphy, M. P., and Galley, H. F. (2008). The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide-peptidoglycan model of sepsis. Free Radic. Biol. Med. 45, 1559–1565. doi: 10.1016/j.freeradbiomed.2008.09.003
Lowes, D. A., Webster, N. R., Murphy, M. P., and Galley, H. F. (2013). Antioxidants that protect mitochondria reduce interleukin-6 and oxidative stress, improve mitochondrial function, and reduce biochemical markers of organ dysfunction in a rat model of acute sepsis. Br. J. Anaesth. 110, 472–480. doi: 10.1093/bja/aes577
Lucas, J. M., True, L., Hawley, S., Matsumura, M., Morrissey, C., Vessella, R., et al. (2008). The androgen-regulated type II serine protease TMPRSS2 is differentially expressed and mislocalized in prostate adenocarcinoma. J. Pathol. 215, 118–125. doi: 10.1002/path.2330
Lukassen, S., Chua, R. L., Trefzer, T., Kahn, N. C., Schneider, M. A., Muley, T., et al. (2020). SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 39:e105114. doi: 10.15252/embj.2020105114
Mahmud-Al-Rafat, A., Muzammal Haque Asim, M., Taylor-Robinson, A. W., Majumder, A., Muktadir, A., Muktadir, H., et al. (2020). A combinational approach to restore cytokine balance and to inhibit virus growth may promote patient recovery in severe COVID-19 cases. Cytokine 136:155228. doi: 10.1016/j.cyto.2020.155228
Maiti, A. K., Saha, N. C., More, S. S., Panigrahi, A. K., and Paul, G. (2017). Neuroprotective efficacy of mitochondrial antioxidant MitoQ in suppressing peroxynitrite-mediated mitochondrial dysfunction inflicted by lead toxicity in the rat brain. Neurotox. Res. 31, 358–372. doi: 10.1007/s12640-016-9692-7
Martinon, F., Mayor, A., and Tschopp, J. (2009). The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27, 229–265. doi: 10.1146/annurev.immunol.021908.132715
Mizutani, T., Fukushi, S., Saijo, M., Kurane, I., and Morikawa, S. (2004). Phosphorylation of p38 MAPK and its downstream targets in SARS coronavirus-infected cells. Biochem. Biophys. Res. Commun. 319, 1228–1234. doi: 10.1016/j.bbrc.2004.05.107
Moghimi, N., Eslami Farsani, B., Ghadipasha, M., Mahmoudiasl, G. R., Piryaei, A., Aliaghaei, A., et al. (2021). COVID-19 disrupts spermatogenesis through the oxidative stress pathway following induction of apoptosis. Apoptosis 26, 415–430. doi: 10.1007/s10495-021-01680-2
Morris, G., Walder, K., Puri, B. K., Berk, M., and Maes, M. (2016). The deleterious effects of oxidative and nitrosative stress on palmitoylation, membrane lipid rafts and lipid-based cellular signalling: New drug targets in neuroimmune disorders. Mol. Neurobiol. 53, 4638–4658. doi: 10.1007/s12035-015-9392-y
Mukherjee, T. K., Mukhopadhyay, S., and Hoidal, J. R. (2005). The role of reactive oxygen species in TNFalpha-dependent expression of the receptor for advanced glycation end products in human umbilical vein endothelial cells. Biochim. Biophys. Acta 1744, 213–223. doi: 10.1016/j.bbamcr.2005.03.007
Nakajima, S., and Kitamura, M. (2013). Bidirectional regulation of NF-kappaB by reactive oxygen species: a role of unfolded protein response. Free Radic. Biol. Med. 65, 162–174. doi: 10.1016/j.freeradbiomed.2013.06.020
Nalbandian, A., Sehgal, K., Gupta, A., Madhavan, M. V., Mcgroder, C., Stevens, J. S., et al. (2021). Post-acute COVID-19 syndrome. Nat. Med. 27, 601–615. doi: 10.1038/s41591-021-01283-z
Nanduri, J., Yuan, G., Kumar, G. K., Semenza, G. L., and Prabhakar, N. R. (2008). Transcriptional responses to intermittent hypoxia. Respir. Physiol. Neurobiol. 164, 277–281. doi: 10.1016/j.resp.2008.07.006
Ndengele, M. M., Muscoli, C., Wang, Z. Q., Doyle, T. M., Matuschak, G. M., and Salvemini, D. (2005). Superoxide potentiates NF-kappaB activation and modulates endotoxin-induced cytokine production in alveolar macrophages. Shock 23, 186–193. doi: 10.1097/01.shk.0000144130.36771.d6
Nguyen Dinh Cat, A., Montezano, A. C., Burger, D., and Touyz, R. M. (2013). Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal. 19, 1110–1120. doi: 10.1089/ars.2012.4641
Nieto-Torres, J. L., Verdia-Baguena, C., Jimenez-Guardeno, J. M., Regla-Nava, J. A., Castano-Rodriguez, C., Fernandez-Delgado, R., et al. (2015). Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 485, 330–339. doi: 10.1016/j.virol.2015.08.010
Niture, S. K., and Jaiswal, A. K. (2012). Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 287, 9873–9886. doi: 10.1074/jbc.M111.312694
Olagnier, D., Farahani, E., Thyrsted, J., Blay-Cadanet, J., Herengt, A., Idorn, M., et al. (2020). SARS-CoV2-mediated suppression of NRF2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat. Commun. 11:4938. doi: 10.1038/s41467-020-18764-3
Olczak-Pruc, M., Swieczkowski, D., Ladny, J. R., Pruc, M., Juarez-Vela, R., Rafique, Z., et al. (2022). Vitamin C supplementation for the treatment of COVID-19: A systematic review and meta-analysis. Nutrients 14, 4217–4231. doi: 10.3390/nu14194217
Orrenius, S., Gogvadze, V., and Zhivotovsky, B. (2007). Mitochondrial oxidative stress: implications for cell death. Annu. Rev. Pharmacol. Toxicol. 47, 143–183. doi: 10.1146/annurev.pharmtox.47.120505.105122
Pacheco, Y., Valeyre, D., El Jammal, T., Vallee, M., Chevalier, F., Lamartine, J., et al. (2021). Autophagy and Mitophagy-related pathways at the crossroads of genetic pathways involved in familial sarcoidosis and host-pathogen interactions induced by coronaviruses. Cells 10, 1995–2024. doi: 10.3390/cells10081995
Paidas, M. J., Mohamed, A. B., Norenberg, M. D., Saad, A., Barry, A. F., Colon, C., et al. (2021). Multi-organ histopathological changes in a mouse hepatitis virus model of COVID-19. Viruses 13. doi: 10.3390/v13091703
Paidas, M. J., Sampath, N., Schindler, E. A., Cosio, D. S., Ndubizu, C. O., Shamaladevi, N., et al. (2022). Mechanism of multi-organ injury in experimental COVID-19 and its inhibition by a small molecule peptide. Front. Pharmacol. 13:864798. doi: 10.3389/fphar.2022.864798
Park, H. S., Kim, S. R., and Lee, Y. C. (2009). Impact of oxidative stress on lung diseases. Respirology 14, 27–38. doi: 10.1111/j.1440-1843.2008.01447.x
Paszti-Gere, E., Barna, R. F., Kovago, C., Szauder, I., Ujhelyi, G., Jakab, C., et al. (2015). Changes in the distribution of type II transmembrane serine protease, TMPRSS2 and in paracellular permeability in IPEC-J2 cells exposed to oxidative stress. Inflammation 38, 775–783. doi: 10.1007/s10753-014-9988-9
Patel, M. (2016). Targeting oxidative stress in central nervous system disorders. Trends Pharmacol. Sci. 37, 768–778. doi: 10.1016/j.tips.2016.06.007
Paul, B. D., Lemle, M. D., Komaroff, A. L., and Snyder, S. H. (2021). Redox imbalance links COVID-19 and myalgic encephalomyelitis/chronic fatigue syndrome. Proc. Natl. Acad. Sci. U. S. A. 118, 1–10. doi: 10.1073/pnas.2024358118
Pearlstein, D. P., Ali, M. H., Mungai, P. T., Hynes, K. L., Gewertz, B. L., and Schumacker, P. T. (2002). Role of mitochondrial oxidant generation in endothelial cell responses to hypoxia. Arterioscler. Thromb. Vasc. Biol. 22, 566–573. doi: 10.1161/01.ATV.0000012262.76205.6A
Pena Silva, R. A., Chu, Y., Miller, J. D., Mitchell, I. J., Penninger, J. M., Faraci, F. M., et al. (2012). Impact of ACE2 deficiency and oxidative stress on cerebrovascular function with aging. Stroke 43, 3358–3363. doi: 10.1161/STROKEAHA.112.667063
Peng, S., Hang, N., Liu, W., Guo, W., Jiang, C., Yang, X., et al. (2016). Andrographolide sulfonate ameliorates lipopolysaccharide-induced acute lung injury in mice by down-regulating MAPK and NF-kappaB pathways. Acta Pharm. Sin. B 6, 205–211. doi: 10.1016/j.apsb.2016.02.002
Perera, M., El Khoury, J., Chinni, V., Bolton, D., Qu, L., Johnson, P., et al. (2020). Randomised controlled trial for high-dose intravenous zinc as adjunctive therapy in SARS-CoV-2 (COVID-19) positive critically ill patients: trial protocol. BMJ Open 10:e040580. doi: 10.1136/bmjopen-2020-040580
Petcherski, A., Sharma, M., Daskou, M., Satta, S., Vasilopoulos, H., Hugo, C., et al. (2022). Mitoquinone mesylate targets SARS-CoV-2 and associated lung inflammation through host pathways. bioRxiv
Petersen, E., Koopmans, M., Go, U., Hamer, D. H., Petrosillo, N., Castelli, F., et al. (2020). Comparing SARS-CoV-2 with SARS-CoV and influenza pandemics. Lancet Infect. Dis. 20, e238–e244. doi: 10.1016/S1473-3099(20)30484-9
Pfefferle, S., Schopf, J., Kogl, M., Friedel, C. C., Muller, M. A., Carbajo-Lozoya, J., et al. (2011). The SARS-coronavirus-host interactome: identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathog. 7:e1002331. doi: 10.1371/journal.ppat.1002331
Pierce, G. B., Parchment, R. E., and Lewellyn, A. L. (1991). Hydrogen peroxide as a mediator of programmed cell death in the blastocyst. Differentiation 46, 181–186. doi: 10.1111/j.1432-0436.1991.tb00880.x
Piette, J., Piret, B., Bonizzi, G., Schoonbroodt, S., Merville, M. P., Legrand-Poels, S., et al. (1997). Multiple redox regulation in NF-kappaB transcription factor activation. Biol. Chem. 378, 1237–1245.
Prakash, A., Kaur, S., Kaur, C., Prabha, P. K., Bhatacharya, A., Sarma, P., et al. (2021). Efficacy and safety of inhaled nitric oxide in the treatment of severe/critical COVID-19 patients: A systematic review. Indian J. Pharm. 53, 236–243. doi: 10.4103/ijp.ijp_382_21
Pratomo, I. P., Noor, D. R., Kusmardi, K., Rukmana, A., Paramita, R. I., Erlina, L., et al. (2021). Xanthine oxidase-induced inflammatory responses in respiratory epithelial cells: A review in immunopathology of COVID-19. Int. J. Inflamm. 2021, 1–10. doi: 10.1155/2021/1653392
Preiser, J. C. (2012). Oxidative stress. JPEN J. Parenter. Enteral Nutr. 36, 147–154. doi: 10.1177/0148607111434963
Pruijssers, A. J., and Denison, M. R. (2019). Nucleoside analogues for the treatment of coronavirus infections. Curr. Opin. Virol. 35, 57–62. doi: 10.1016/j.coviro.2019.04.002
Ramani, S., Pathak, A., Dalal, V., Paul, A., and Biswas, S. (2020). Oxidative stress in autoimmune diseases: an under dealt malice. Curr. Protein Pept. Sci. 21, 611–621. doi: 10.2174/1389203721666200214111816
Reiter, R. J. (1998). Oxidative damage in the central nervous system: protection by melatonin. Prog. Neurobiol. 56, 359–384. doi: 10.1016/S0301-0082(98)00052-5
Reshi, M. L., Su, Y. C., and Hong, J. R. (2014). RNA viruses: ROS-mediated cell death. Int. J. Cell Biol. 2014:467452. doi: 10.1155/2014/467452
Reynaert, N. L., Van Der Vliet, A., Guala, A. S., Mcgovern, T., Hristova, M., Pantano, C., et al. (2006). Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc. Natl. Acad. Sci. U. S. A. 103, 13086–13091. doi: 10.1073/pnas.0603290103
Roberts, C. K., and Sindhu, K. K. (2009). Oxidative stress and metabolic syndrome. Life Sci. 84, 705–712. doi: 10.1016/j.lfs.2009.02.026
Roulston, A., Marcellus, R. C., and Branton, P. E. (1999). Viruses and apoptosis. Annu. Rev. Microbiol. 53, 577–628. doi: 10.1146/annurev.micro.53.1.577
Saad, M. A., Alfishawy, M., Nassar, M., Mohamed, M., Esene, I. N., and Elbendary, A. (2021). COVID-19 and autoimmune diseases: A systematic review of reported cases. Curr. Rheumatol. Rev. 17, 193–204. doi: 10.2174/1573397116666201029155856
Saheb Sharif-Askari, N., Saheb Sharif-Askari, F., Mdkhana, B., Hussain Alsayed, H. A., Alsafar, H., Alrais, Z. F., et al. (2021). Upregulation of oxidative stress gene markers during SARS-COV-2 viral infection. Free Radic. Biol. Med. 172, 688–698. doi: 10.1016/j.freeradbiomed.2021.06.018
Salim, S. (2017). Oxidative stress and the central nervous system. J. Pharmacol. Exp. Ther. 360, 201–205. doi: 10.1124/jpet.116.237503
Santiesteban-Lores, L. E., Amamura, T. A., Da Silva, T. F., Midon, L. M., Carneiro, M. C., Isaac, L., et al. (2021). A double edged-sword - the complement system during SARS-CoV-2 infection. Life Sci. 272:119245. doi: 10.1016/j.lfs.2021.119245
Santoro, M. G., Rossi, A., and Amici, C. (2003). NF-kappaB and virus infection: who controls whom. EMBO J. 22, 2552–2560. doi: 10.1093/emboj/cdg267
Santos, J. H., Hunakova, L., Chen, Y., Bortner, C., and Van Houten, B. (2003). Cell sorting experiments link persistent mitochondrial DNA damage with loss of mitochondrial membrane potential and apoptotic cell death. J. Biol. Chem. 278, 1728–1734. doi: 10.1074/jbc.M208752200
Saura, M., Zaragoza, C., Bao, C., Herranz, B., Rodriguez-Puyol, M., and Lowenstein, C. J. (2006). Stat3 mediates interleukin-6 [correction of interelukin-6] inhibition of human endothelial nitric-oxide synthase expression. J. Biol. Chem. 281, 30057–30062. doi: 10.1074/jbc.M606279200
Schofield, J. H., and Schafer, Z. T. (2021). Mitochondrial reactive oxygen species and mitophagy: A complex and nuanced relationship. Antioxid. Redox Signal. 34, 517–530. doi: 10.1089/ars.2020.8058
Schrader, L. I., Kinzenbaw, D. A., Johnson, A. W., Faraci, F. M., and Didion, S. P. (2007). IL-6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler. Thromb. Vasc. Biol. 27, 2576–2581. doi: 10.1161/ATVBAHA.107.153080
Schreck, R., and Baeuerle, P. A. (1991). A role for oxygen radicals as second messengers. Trends Cell Biol. 1, 39–42. doi: 10.1016/0962-8924(91)90072-H
Shang, C., Liu, Z., Zhu, Y., Lu, J., Ge, C., Zhang, C., et al. (2021). SARS-CoV-2 causes mitochondrial dysfunction and mitophagy impairment. Front. Microbiol. 12:780768. doi: 10.3389/fmicb.2021.780768
Shasby, D. M., Lind, S. E., Shasby, S. S., Goldsmith, J. C., and Hunninghake, G. W. (1985). Reversible oxidant-induced increases in albumin transfer across cultured endothelium: alterations in cell shape and calcium homeostasis. Blood 65, 605–614. doi: 10.1182/blood.V65.3.605.605
Shatizadeh Malekshahi, S., Yavarian, J., and Shafiei-Jandaghi, N. Z. (2022). Usage of peptidases by SARS-CoV-2 and several human coronaviruses as receptors: A mysterious story. Biotechnol. Appl. Biochem. 69, 124–128. doi: 10.1002/bab.2087
Shi, C. S., Qi, H. Y., Boularan, C., Huang, N. N., Abu-Asab, M., Shelhamer, J. H., et al. (2014). SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 193, 3080–3089. doi: 10.4049/jimmunol.1303196
Shirihai, O. S., Song, M., and Dorn, G. W. 2nd (2015). How mitochondrial dynamism orchestrates mitophagy. Circ. Res. 116, 1835–1849. doi: 10.1161/CIRCRESAHA.116.306374
Shono, T., Ono, M., Izumi, H., Jimi, S. I., Matsushima, K., Okamoto, T., et al. (1996). Involvement of the transcription factor NF-kappaB in tubular morphogenesis of human microvascular endothelial cells by oxidative stress. Mol. Cell. Biol. 16, 4231–4239. doi: 10.1128/MCB.16.8.4231
Siddiqi, H. K., Libby, P., and Ridker, P. M. (2021). COVID-19 - A vascular disease. Trends Cardiovasc. Med. 31, 1–5. doi: 10.1016/j.tcm.2020.10.005
Simon, P. F., Mccorrister, S., Hu, P., Chong, P., Silaghi, A., Westmacott, G., et al. (2015). Highly pathogenic H5N1 and novel H7N9 influenza A viruses induce More profound proteomic host responses than seasonal and pandemic H1N1 strains. J. Proteome Res. 14, 4511–4523. doi: 10.1021/acs.jproteome.5b00196
Singhal, T. (2020). A review of coronavirus Disease-2019 (COVID-19). Indian J. Pediatr. 87, 281–286. doi: 10.1007/s12098-020-03263-6
Srinivasan, K., Pandey, A. K., Livingston, A., and Venkatesh, S. (2021). Roles of host mitochondria in the development of COVID-19 pathology: could mitochondria be a potential therapeutic target? Mol. Biomed. 2:38. doi: 10.1186/s43556-021-00060-1
Stebbing, J., Phelan, A., Griffin, I., Tucker, C., Oechsle, O., Smith, D., et al. (2020). COVID-19: combining antiviral and anti-inflammatory treatments. Lancet Infect. Dis. 20, 400–402. doi: 10.1016/S1473-3099(20)30132-8
Su, C. M., Wang, L., and Yoo, D. (2021). Activation of NF-kappaB and induction of proinflammatory cytokine expressions mediated by ORF7a protein of SARS-CoV-2. Sci. Rep. 11:13464. doi: 10.1038/s41598-021-92941-2
Sun, S., Sursal, T., Adibnia, Y., Zhao, C., Zheng, Y., Li, H., et al. (2013). Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLoS One 8:e59989. doi: 10.1371/journal.pone.0059989
Sun, K., Yajjala, V. K., Bauer, C., Talmon, G. A., Fischer, K. J., Kielian, T., et al. (2016). Nox2-derived oxidative stress results in inefficacy of antibiotics against post-influenza S. aureus pneumonia. J. Exp. Med. 213, 1851–1864. doi: 10.1084/jem.20150514
Supinski, G. S., Murphy, M. P., and Callahan, L. A. (2009). MitoQ administration prevents endotoxin-induced cardiac dysfunction. Am. J. Phys. Regul. Integr. Comp. Phys. 297, R1095–R1102. doi: 10.1152/ajpregu.90902.2008
Takada, Y., Mukhopadhyay, A., Kundu, G. C., Mahabeleshwar, G. H., Singh, S., and Aggarwal, B. B. (2003). Hydrogen peroxide activates NF-kappa B through tyrosine phosphorylation of I kappa B alpha and serine phosphorylation of p65: evidence for the involvement of I kappa B alpha kinase and Syk protein-tyrosine kinase. J. Biol. Chem. 278, 24233–24241. doi: 10.1074/jbc.M212389200
Tao, L., Lemoff, A., Wang, G., Zarek, C., Lowe, A., Yan, N., et al. (2020). Reactive oxygen species oxidize STING and suppress interferon production. elife 9, 1–23. doi: 10.7554/eLife.57837
Tenjinbaru, K., Furuno, T., Hirashima, N., and Nakanishi, M. (1999). Nuclear translocation of green fluorescent protein-nuclear factor kappaB with a distinct lag time in living cells. FEBS Lett. 444, 1–4. doi: 10.1016/S0014-5793(99)00002-2
Thevenod, F., Friedmann, J. M., Katsen, A. D., and Hauser, I. A. (2000). Up-regulation of multidrug resistance P-glycoprotein via nuclear factor-kappaB activation protects kidney proximal tubule cells from cadmium-and reactive oxygen species-induced apoptosis. J. Biol. Chem. 275, 1887–1896. doi: 10.1074/jbc.275.3.1887
Tian, J., Middleton, B., and Kaufman, D. L. (2021). GABA(A)-receptor agonists limit pneumonitis and death in murine coronavirus-infected mice. Viruses 13, 966–978. doi: 10.3390/v13060966
To, E. E., Broughton, B. R., Hendricks, K. S., Vlahos, R., and Selemidis, S. (2014). Influenza A virus and TLR7 activation potentiate NOX2 oxidase-dependent ROS production in macrophages. Free Radic. Res. 48, 940–947. doi: 10.3109/10715762.2014.927579
Toro, A., Ruiz, M. S., Lage-Vickers, S., Sanchis, P., Sabater, A., Pascual, G., et al. (2022). A journey into the clinical relevance of Heme oxygenase 1 for human inflammatory disease and viral clearance: why does it matter on the COVID-19 scene? Antioxidants 11, 276–298. doi: 10.3390/antiox11020276
Trempolec, N., Munoz, J. P., Slobodnyuk, K., Marin, S., Cascante, M., Zorzano, A., et al. (2017). Induction of oxidative metabolism by the p38alpha/MK2 pathway. Sci. Rep. 7:11367. doi: 10.1038/s41598-017-11309-7
Tu, W., Wang, H., Li, S., Liu, Q., and Sha, H. (2019). The anti-inflammatory and anti-oxidant mechanisms of the Keap1/Nrf2/ARE signaling pathway in chronic diseases. Aging Dis. 10, 637–651. doi: 10.14336/AD.2018.0513
Valle, M. L., Dworshak, J., Sharma, A., Ibrahim, A. S., Al-Shabrawey, M., and Sharma, S. (2019). Inhibition of interleukin-6 trans-signaling prevents inflammation and endothelial barrier disruption in retinal endothelial cells. Exp. Eye Res. 178, 27–36. doi: 10.1016/j.exer.2018.09.009
Verdecchia, P., Cavallini, C., Spanevello, A., and Angeli, F. (2020). The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 76, 14–20. doi: 10.1016/j.ejim.2020.04.037
Violi, F., Oliva, A., Cangemi, R., Ceccarelli, G., Pignatelli, P., Carnevale, R., et al. (2020). Nox2 activation in Covid-19. Redox Biol. 36:101655. doi: 10.1016/j.redox.2020.101655
Wang, F. S., Chu, F. L., Jin, L., Li, Y. G., Zhang, Z., Xu, D., et al. (2005). Acquired but reversible loss of erythrocyte complement receptor 1 (CR1, CD35) and its longitudinal alteration in patients with severe acute respiratory syndrome. Clin. Exp. Immunol. 139, 112–119. doi: 10.1111/j.1365-2249.2005.02681.x
Wang, Y., Liu, S., Liu, H., Li, W., Lin, F., Jiang, L., et al. (2020). SARS-CoV-2 infection of the liver directly contributes to hepatic impairment in patients with COVID-19. J. Hepatol. 73, 807–816. doi: 10.1016/j.jhep.2020.05.002
Wang, Y., and Zhang, X. (1999). The nucleocapsid protein of coronavirus mouse hepatitis virus interacts with the cellular heterogeneous nuclear ribonucleoprotein A1 in vitro and in vivo. Virology 265, 96–109. doi: 10.1006/viro.1999.0025
Weiss, S. R., and Navas-Martin, S. (2005). Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 69, 635–664. doi: 10.1128/MMBR.69.4.635-664.2005
Wesselborg, S., Bauer, M. K., Vogt, M., Schmitz, M. L., and Schulze-Osthoff, K. (1997). Activation of transcription factor NF-kappaB and p38 mitogen-activated protein kinase is mediated by distinct and separate stress effector pathways. J. Biol. Chem. 272, 12422–12429. doi: 10.1074/jbc.272.19.12422
Wong, K. K., Lee, S. W. H., and Kua, K. P. (2021). N-acetylcysteine as adjuvant therapy for COVID-19 - A perspective on the current state of the evidence. J. Inflamm. Res. 14, 2993–3013. doi: 10.2147/JIR.S306849
Wosniak, J. Jr., Santos, C. X., Kowaltowski, A. J., and Laurindo, F. R. (2009). Cross-talk between mitochondria and NADPH oxidase: effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid. Redox Signal. 11, 1265–1278. doi: 10.1089/ars.2009.2392
Xia, H., Suda, S., Bindom, S., Feng, Y., Gurley, S. B., Seth, D., et al. (2011). ACE2-mediated reduction of oxidative stress in the central nervous system is associated with improvement of autonomic function. PLoS One 6:e22682. doi: 10.1371/journal.pone.0022682
Xie, J., and Shaikh, Z. A. (2006). Cadmium-induced apoptosis in rat kidney epithelial cells involves decrease in nuclear factor-kappa B activity. Toxicol. Sci. 91, 299–308. doi: 10.1093/toxsci/kfj131
Xu, H., Zhong, L., Deng, J., Peng, J., Dan, H., Zeng, X., et al. (2020). High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 12:8. doi: 10.1038/s41368-020-0074-x
Yamada, Y., Limmon, G. V., Zheng, D., Li, N., Li, L., Yin, L., et al. (2012). Major shifts in the spatio-temporal distribution of lung antioxidant enzymes during influenza pneumonia. PLoS One 7:e31494. doi: 10.1371/journal.pone.0031494
Yang, P. M., Chen, H. C., Tsai, J. S., and Lin, L. Y. (2007). Cadmium induces Ca2+−dependent necrotic cell death through calpain-triggered mitochondrial depolarization and reactive oxygen species-mediated inhibition of nuclear factor-kappaB activity. Chem. Res. Toxicol. 20, 406–415. doi: 10.1021/tx060144c
Yi, Y., Lagniton, P. N. P., Ye, S., Li, E., and Xu, R. H. (2020). COVID-19: what has been learned and to be learned about the novel coronavirus disease. Int. J. Biol. Sci. 16, 1753–1766. doi: 10.7150/ijbs.45134
Youn, J. Y., Zhang, Y., Wu, Y., Cannesson, M., and Cai, H. (2021). Therapeutic application of estrogen for COVID-19: attenuation of SARS-CoV-2 spike protein and IL-6 stimulated, ACE2-dependent NOX2 activation, ROS production and MCP-1 upregulation in endothelial cells. Redox Biol. 46:102099. doi: 10.1016/j.redox.2021.102099
Zhang, L., Qin, Y., and Chen, M. (2018). Viral strategies for triggering and manipulating mitophagy. Autophagy 14, 1665–1673. doi: 10.1080/15548627.2018.1466014
Zhang, J., Wang, X., Vikash, V., Ye, Q., Wu, D., Liu, Y., et al. (2016). ROS and ROS-mediated cellular signaling. Oxidative Med. Cell. Longev. 2016, 1–18. doi: 10.1155/2016/4350965
Zhang, S., Wang, J., Wang, L., Aliyari, S., and Cheng, G. (2022). SARS-CoV-2 virus NSP14 impairs NRF2/HMOX1 activation by targeting Sirtuin 1. Cell. Mol. Immunol. 19, 872–882. doi: 10.1038/s41423-022-00887-w
Zhao, G., Jiang, Y., Qiu, H., Gao, T., Zeng, Y., Guo, Y., et al. (2015). Multi-organ damage in human dipeptidyl peptidase 4 transgenic mice infected with Middle East respiratory syndrome-coronavirus. PLoS One 10:e0145561. doi: 10.1371/journal.pone.0145561
Zhou, R., Yazdi, A. S., Menu, P., and Tschopp, J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225. doi: 10.1038/nature09663
Zhu, T., Wang, D. X., Zhang, W., Liao, X. Q., Guan, X., Bo, H., et al. (2013). Andrographolide protects against LPS-induced acute lung injury by inactivation of NF-kappaB. PLoS One 8:e56407. doi: 10.1371/journal.pone.0056407
Zinovkin, R. A., and Grebenchikov, O. A. (2020). Transcription factor Nrf2 as a potential therapeutic target for prevention of cytokine storm in COVID-19 patients. Biochemistry (Mosc) 85, 833–837. doi: 10.1134/S0006297920070111
Keywords: SARS-CoV-2, coronavirus, inflammation, oxidative stress, tissue damage, apoptosis
Citation: Gain C, Song S, Angtuaco T, Satta S and Kelesidis T (2023) The role of oxidative stress in the pathogenesis of infections with coronaviruses. Front. Microbiol. 13:1111930. doi: 10.3389/fmicb.2022.1111930
Edited by:
Wenjun Song, Guangzhou National Laboratory, ChinaReviewed by:
Paola Checconi, San Raffaele Telematic University, ItalyCopyright © 2023 Gain, Song, Angtuaco, Satta and Kelesidis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Theodoros Kelesidis, ✉ dGtlbGVzaWRpc0BtZWRuZXQudWNsYS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.