 Bing Fu
Bing Fu Junhui Ying2
Junhui Ying2 Ping Yu
Ping Yu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 16 January 2023
Sec. Systems Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.1111790
In this study, to construct the riboflavin-producing strain R1, five key genes, ribA, ribB, ribC, ribD, and ribE, were cloned and ligated to generate the plasmid pET-AE, which was overexpressed in Escherichia coli BL21. The R1 strain accumulated 182.65 ± 9.04 mg/l riboflavin. Subsequently, the R2 strain was constructed by the overexpression of zwf harboring the constructed plasmid pAC-Z in the R1 strain. Thus, the level of riboflavin in the R2 strain increased to 319.01 ± 20.65 mg/l (74.66% increase). To further enhance ribB transcript levels and riboflavin production, the FMN riboswitch was deleted from E. coli BL21 with CRISPR/Cas9 to generate the R3 strain. The R4 strain was constructed by cotransforming pET-AE and pAC-Z into the R3 strain. Compared to those of E. coli BL21, the ribB transcript levels of R2 and R4 improved 2.78 and 3.05-fold, respectively. The R4 strain accumulated 437.58 ± 14.36 mg/l riboflavin, increasing by 37.17% compared to the R2 strain. These results suggest that the deletion of the FMN riboswitch can improve the transcript level of ribB and facilitate riboflavin production. A riboflavin titer of 611.22 ± 11.25 mg/l was achieved under the optimal fermentation conditions. Ultimately, 1574.60 ± 109.32 mg/l riboflavin was produced through fed-batch fermentation with 40 g/l glucose. This study contributes to the industrial production of riboflavin by the recombinant E. coli BL21.
Riboflavin (RF), also known as vitamin B2, is a vitamin B complex essential to all life forms (Garcia-Angulo, 2017). Most microorganisms and plants can synthesize RF; however, vertebrates must obtain RF from their diets (Vitreschak et al., 2002). RF involves numerous reactions, and its derivatives flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) act as coenzymes for many flavoproteins in many metabolic pathways, such as carbohydrate, protein, vitamin, and fat metabolism (Northrop-Clewes and Thurnham, 2012; Saedisomeolia and Ashoori, 2018). In Escherichia coli, RF is successively converted to FMN and FAD by bifunctional RF kinase (RFK, EC 2.7.1.26)/FMN adenylyltransferase (FMNAT, EC 2.7.7.2), which is encoded by ribF. In rare cases, RF plays a cofactor role (Juarez et al., 2008). In addition, RF can provide protection against cancer, cardiovascular diseases, brain damage caused by ROS-induced damage in Alzheimer’s disease, lipid peroxidation, and reperfusion oxidative injury and maintain immunity (Powers, 2003; Mazur-Bialy and Pochec, 2016; Saedisomeolia and Ashoori, 2018; Zhao et al., 2018). RF is the second vitamin identified (Northrop-Clewes and Thurnham, 2012) and has been used in many fields, such as pharmaceuticals, cosmetics, and human and animal nutrition (Koizumi et al., 2000).
Riboflavin biosynthesis starts with the immediate precursor guanosine 5′-triphosphate (GTP) and D-ribulose 5-phosphate (Ru5P). This RF biosynthetic pathway (RBP) includes seven enzymatic reactions (Figure 1), which are mostly catalyzed by enzymes encoded by rib genes, namely, ribA (encoding GTP cyclohydrolase II, EC 3.5.4.25), riB (encoding 3,4-dihydroxy-2-butanone-4-phosphate synthase, EC 4.1.99.12), ribC (encoding RF synthase, EC 2.5.1.9), ribD (encoding bifunctional deaminase/reductase, EC 3.5.4.26), and ribE (encoding 6,7-dimethyl-8-ribityllumazine synthase, EC 2.5.1.78). However, the enzyme that catalyzes the dephosphorylation of 5-amino-6-(5-phospho-D-ribitylamino) uracil to form 5-amino-6-(1-D-ribitylamino) uracil is unknown. Some evidence has shown that YigB and YbjI in the haloacid dehalogenase superfamily can catalyze this elusive dephosphorylation step (Bacher et al., 2000; Haase et al., 2013; Garcia-Angulo, 2017). Escherichia coli rib genes are scattered across the genome, which distinguishes it from the classical RF-producing strain Bacillus subtilis with an entire operon (ribD-ribE-ribBA-ribH). The coexpression of the key genes of RBP enables E. coli to produce RF (Xu et al., 2015).
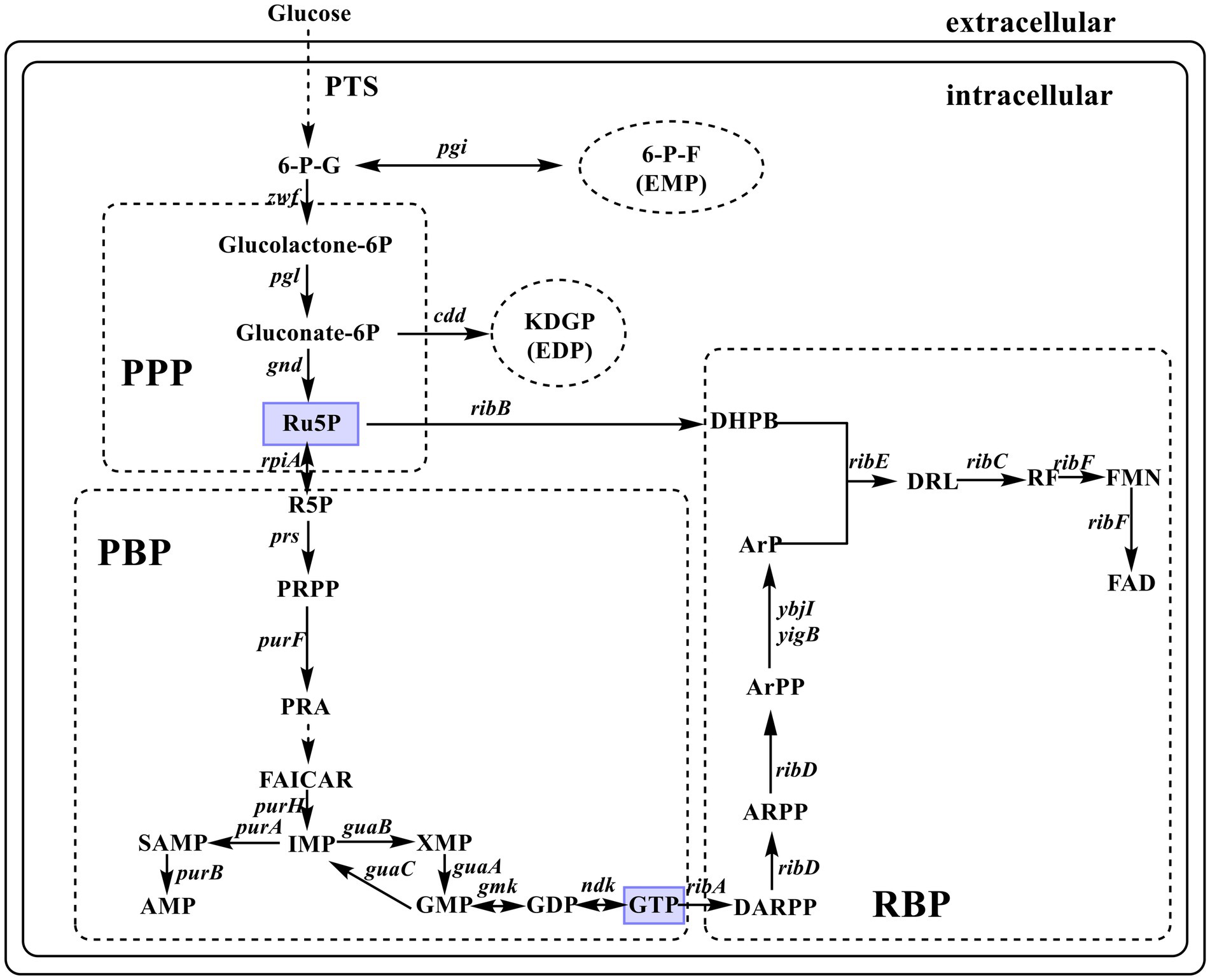
Figure 1. Diagrammatic representation of de novo biosynthesis of riboflavin (RF) in Escherichia coli. The enzymes encoded by the indicated genes are: zwf, glucose-6-phosphate-1-dehydrogenase; pgi, phosphoglucose isomerase; pgl, 6-phosphogluconolactonase; edd, phosphogluconate dehydratase; gnd, 6-phosphogluconate dehydrogenase; rpiA, ribose-5-phosphate isomerase A; prs, ribose-phosphate diphosphokinase; purF, amidophosphoribosyltransferase; purH, bifunctional AICAR transformylase/IMP cyclohydrolase; purA, adenylosuccinate synthase; purB, adenylosuccinate lyase; guaA, GMP synthase; guaB, IMP dehydrogenase; guaC, GMP reductase; gmk, guanylate kinase; ndk, nucleoside diphosphate kinase; ribA, GTP cyclohydrolase II; ribB, 3,4-dihydroxy-2-butanone-4-phosphate synthase; ribC, riboflavin synthase; ribD, bifunctional deaminase/reductase; ribE, 6,7-dimethyl-8-ribityllumazine synthase; ybjI and yigB, 5-amino-6-(5-phospho-D-ribitylamino) uracil phosphatase; ribF, bifunctional riboflavin kinase/FMN adenylyltransferase. PTS, phosphoenolpyruvate–carbohydrate phosphotransferase system; 6-G-P, D-glucose 6-phosphate; 6-P-F, D-fructose 6-phosphate; KDGP, 2-dehydro-3-deoxy-D-gluconate 6-phosphate; Ru5P, D-ribulose 5-phosphate; R5P, D-ribose 5-phosphate; PRPP, 5-phospho-α-D-ribose-1-diphosphate; PRA, 5-phosphoribosylamine; FAICAR, 5-formamido-1-(5-phospho-D-ribosyl)-imidazole-4-carboxamide; IMP, inosine 5′-monophosphate; SAMP, N6-(1,2-dicarboxyethyl) AMP; AMP, adenosine 5′-monophosphate; XMP, xanthosine 5′-monophosphate; GMP, guanosine 5′-monophosphate; GTP, guanosine 5′-triphosphate; DARPP, 2,5-diamino-6-(5-phospho-D-ribosylamino)pyrimidin-4(3H)-one; ARPP, 5-amino-6-(5′-phosphoribosylamino)uracil; ArPP, 5-amino-6-(5′-phospho-D-ribitylamino)uracil; ArP, 5-amino-6-(1-D-ribitylamino)uracil; DHPB, 3,4-dihydroxy-2-butanone-4-phosphate; DRL, 6,7-dimethyl-8-(1-D-ribityl)lumazine; FMN, flavin mononucleotide; FAD, flavin adenine dinucleotide; EDP, Entner–Doudoroff pathway; EMP, glycolytic pathway; PPP, pentose phosphate pathway; PBP, purine biosynthesis pathway; RBP, riboflavin biosynthetic pathway.
Enhancing the flux of the purine synthesis pathway by increasing the carbon flux through the pentose phosphate pathway (PPP) and regulating the supply of precursor metabolites can effectively increase RF production (Xu et al., 2015; Liu et al., 2016). The overexpression of zwf (encoding glucose-6-phosphate dehydrogenase, EC 1.1.1.49) can increase carbon flow through PPP (Schwechheimer et al., 2016; Sundara Sekar et al., 2017) to enhance the production of RF (Wang et al., 2011; Lu et al., 2012; Lin et al., 2014). A high rate of glucose utilization is important for production cost reduction.
The overexpression of key genes can enhance the yield of a target product, but it inevitably increases metabolic burden and may be counterproductive. Hence, introducing various types of genetic modification through genome engineering, including gene disruption, gene insertion, and point mutation, is essential to the enhancement of pathway efficiency and product yield (Li et al., 2015). CRISPR/Cas systems, comprising clustered regularly interspaced short palindromic repeats (CRISPR) and associated proteins, are innate immune systems in bacteria and archaea (Li et al., 2019; Liu et al., 2020). Bacterial CRISPR/Cas9, which belongs to the type-II CRISPR/Cas system, is widely used in genomic editing in various prokaryotes and eukaryotes (Jiang et al., 2015). The application of this system for gene editing is widely considered the greatest achievement in molecular biology after polymerase chain reaction (PCR) technology (Ledford, 2015).
The FMN riboswitch is the most widely distributed regulatory factor of RBP genes (Garcia-Angulo, 2017). It regulates the expression of genes in a cis fashion (Howe et al., 2016). In B. subtilis, the FMN riboswitch precedes the 5′ UTR rib operon mRNA, whereas in E. coli, it is present in the 5′ UTR of ribB mRNA (Pedrolli et al., 2015a). The FMN riboswitch comprises two distinct functional modules: an aptamer ligand-binding domain responsive to FMN and a downstream expression platform. The regulatory mechanism of the FMN riboswitch is clear. The binding of FMN to an aptamer domain induces a conformational change in the expression platform that regulates ribB expression typically through the attenuation of gene expression at the transcriptional and translational levels (Vitreschak et al., 2002; Pedrolli et al., 2015a,b; Howe et al., 2016; Figure 2). The knockout of the FMN riboswitch relieves FMN inhibition, allowing E. coli to accumulate RF (Pedrolli et al., 2015a). Owing to the absence of a counterpart in humans, the FMN riboswitch is believed to be a potentially attractive target for antibiotic development (Long et al., 2010; Howe et al., 2016).
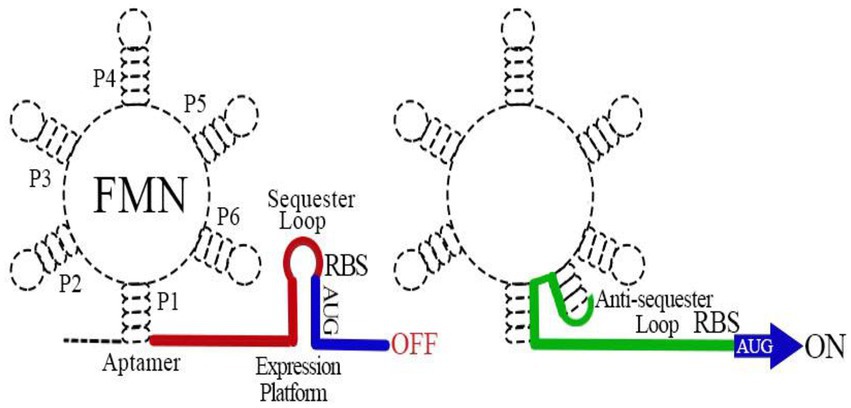
Figure 2. Schematic map of the flavin mononucleotide (FMN) riboswitch mechanism of action. The FMN riboswitch includes two main parts: the 5′ mRNA aptamer and the 3′ downstream expression platform. When FMN binds to the aptamer, the expression platform conforms to a sequester loop (Pritchett and Reddy, 2017) that inhibits ribB expression through early termination of transcription of the ribB and sequestration of the ribosome binding sequence to prevent translation of fully transcribed ribB mRNAs. In the absence of FMN, the aptamer ligand-binding domain adopts an alternative structural conformation that induces an anti-sequester loop (green) in the 3′ downstream expression platform, allowing the continuous expression of ribB.
In this study, to construct a genetically engineered E. coli strain that can produce RF, five key genes in the RBP of E. coli, ribA, ribB, ribC, ribD and ribE, were cloned and ligated to generate the plasmid pET-AE, which was overexpressed in E. coli BL21. The zwf harboring the constructed plasmid pAC-Z was further expressed to increase the carbon metabolism level and facilitate carbon flow through the PPP. The FMN inhibition of ribB was relieved by disrupting the FMN riboswitch in E. coli BL21 with CRISPR/Cas9. Through these efforts, the titer of riboflavin was enhanced to 1,574.60 ± 109.32 mg/l through fed-batch fermentation with 40 g/l glucose.
All strains and plasmids used in this study are listed in Table 1. E. coli DH5α was used as the host to propagate the plasmid DNA. E. coli BL21 was used as an expression host. The compatible plasmids pETDuet-1 and pACYCDuet-1 were used as expression plasmids. Luria-Bertani (LB) medium (10 g/l tryptone, 5 g/L yeast extract, and 10 g/l NaCl) was used for strain culture. M9Y medium (10/L glucose, 6.8 g/l Na2HPO4, 3.4 g/l KH2PO4, 0.5 g/l NaCl, 1.0 g/l NH4Cl, 5 g/l yeast extract, 2 mM MgSO4⋅7H2O, and 0.1 mM CaCl2; pH 7.2), LBG medium (LB medium with 10 g/l glucose; pH 7.2), and MSY media (10 g/l glucose, 3.8 g/l Na2HPO4, 1.5 g/l KH2PO4, 1.0 g/l (NH4)2SO4, 0.2 g/l MgSO4⋅7H2O, 5 g/l yeast extract, and 2% (v/v) trace element solution, pH 7.2) were used (Qiu et al., 2004), and the composition of trace element solution was as follows: 100 mg/l ZnSO4⋅7H2O, 30 mg/l MnCl2⋅4H2O, 300 mg/l H3BO3, 200 mg/l CoCl2⋅6H2O, 10 mg/l CuSO4⋅5H2O, 20 mg/l NiCl2⋅6H2O, 30 mg/l NaMoO4⋅2H2O, and 0.5 M HCl. Ampicillin (100 μg/ml), kanamycin (50 μg/ml), spectinomycin (50 μg/ml), chloramphenicol (25 μg/ml), and isopropylthio-β-galactoside (IPTG) were added if necessary. Unless otherwise stated, incubation was carried out at 37°C and 220 rpm, and the IPTG concentration was 1 mM.

Table 1. Strains and plasmids used in this study.
The FastPure plasmid mini kit, gel DNA extraction mini kit, bacterial DNA isolation mini kit, ClonExpress II one-step cloning kit, and FastPure cell/tissue total RNA isolation kit V2 were purchased from Vazyme Biotech (Nanjing, China). PrimeSTAR HS (premix), a competent cell preparation kit, and restriction enzymes (NdeI, XhoI, and DpnI) were purchased from Takara Biotech (Dalian, China). Primer synthesis and sequencing were performed by Shanghai Biotech (Shanghai, China).
All primers used in this study are listed in Table 2. The primers ribA-F/R, ribB-F/R, ribC-F/R, ribD-F/R, and ribE-F/R were used to obtain the gene fragments ribA, ribB, ribC, ribD, and ribE with the E. coli BL21 genome as the template. The artificial operon ribC–ribE–ribB–ribD–ribA was obtained by ligating the gel extraction products of the five genes through overlap-PCR with the primers ribC-F and ribA-R. After the plasmid pETDuet-1 was digested by NdeI and XhoI, the plasmid pET-AE was constructed by ligating the artificial operon ribC–ribE–ribB–ribD–ribA with the digested plasmid pETDuet-1 using a ClonExpress II one-step cloning kit (Figure 3A). The plasmid pAC-Z was obtained by amplifying the zwf gene from the E. coli BL21 genome with the primer zwf-F/R and conducting homologous combination with pACYCDuet-1, which was digested with NdeI and XhoI (Figure 3B).
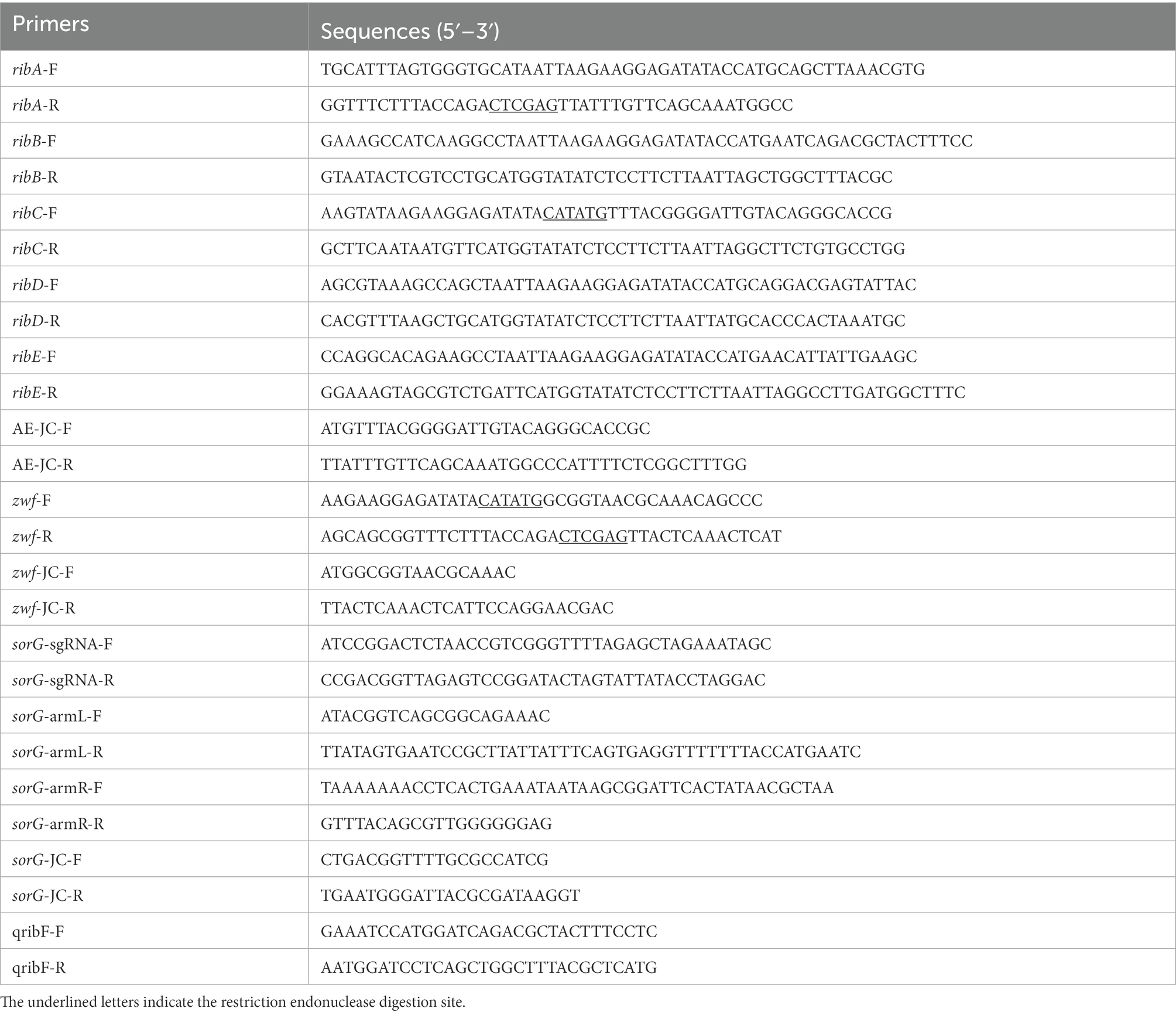
Table 2. Primers used in this study.
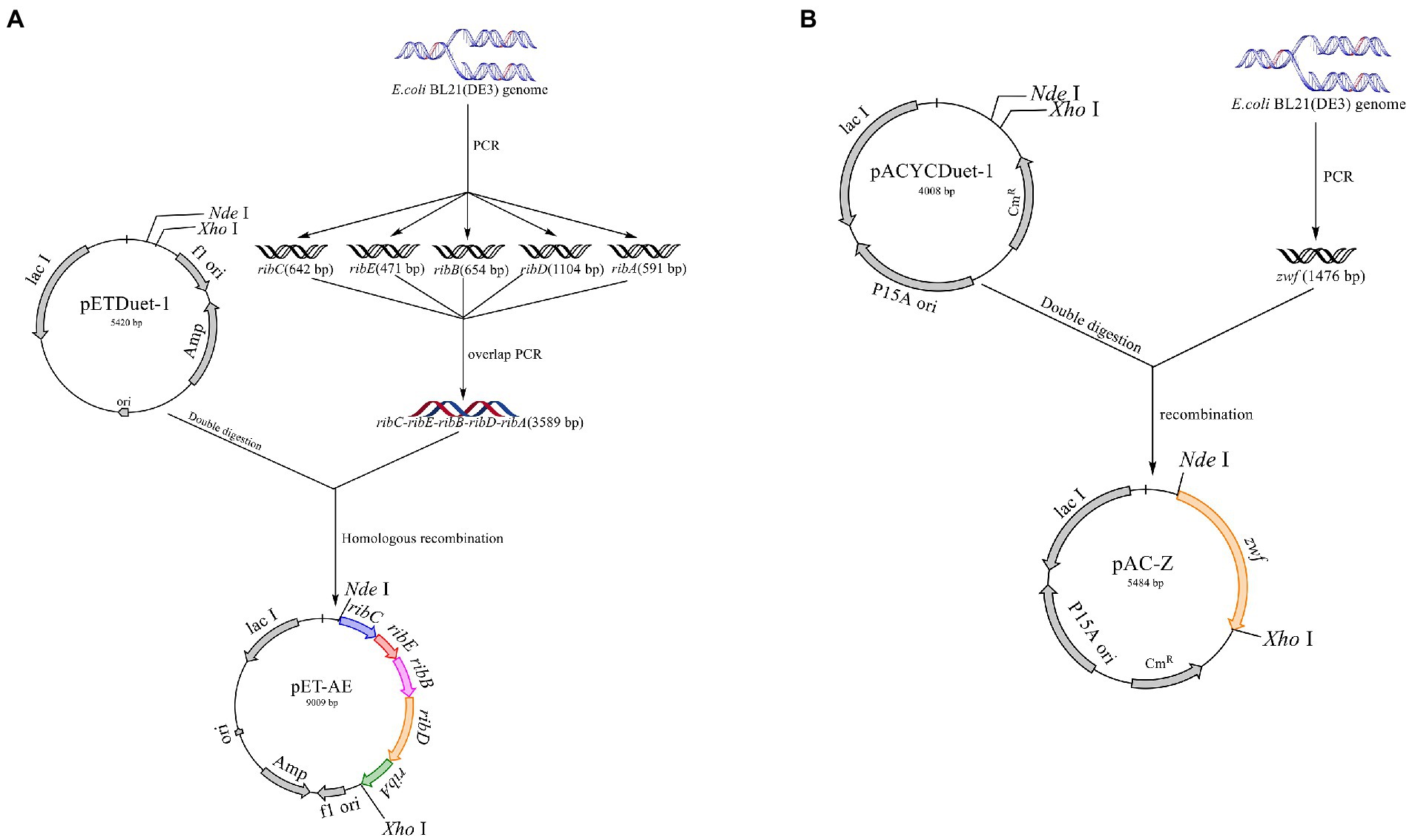
Figure 3. Schematic map of the construction of the plasmids pET-AE and pAC-Z. (A) Construction of the plasmid pET-AE; (B) the construction of the plasmid pAC-Z.
The plasmid pET-AE was transformed into E. coli BL21 competent cells to generate the R1 strain by the CaCl2-heat shock method (Fu et al., 2021). The R2 strain was constructed by introducing the plasmid pAC-Z into the R1 strain to increase the carbon metabolism level and facilitate carbon flow through the PPP.
A single colony was picked from agar plates and incubated in LB media containing appropriate antibiotics until the OD600 values reached 0.6. A final concentration of 2 mM IPTG was added to the cultures to induce the expression of the target proteins. After 6 h of incubation, the cells were collected through centrifugation at 12,000 rpm for 10 min. Phosphate buffer (PBS, pH 7.4) was used to resuspend the obtained cells, and ultrasonic treatment was performed by a sonicator (SCIENTZ-II D, China) to release the proteins (5 s sonication at 150 W, 10 s pause). The resultant crushing solution was centrifuged (12000 rpm, 10 min, 4°C). The precipitate was resuspended in PBS buffer (pH 7.4).
Approximately 10 μl of the precipitate and supernatant were subjected to SDS-PAGE analysis performed on a 12% running gel. Coomassie brilliant blue G-250 was used to stain the resolved proteins.
The CRISPR/Cas9 system is composed of pCas and pTargetF plasmids. Owing to its efficiency and simplicity, it is widely used in genomic editing (Liu et al., 2017; An et al., 2020).
The designed N20 sequence was evaluated with a web-based tool.1 sgRNA was obtained through reverse-PCR using primers with modified N20 at the 3′ end and pTargetF as the template. The PCR products were digested by DpnI at 37°C for 1 h to remove the template. The products were recovered and purified for the transformation of E. coli DH5α competent cells and incubated on an LB agar plate (50 μg/ml spectinomycin). Positive clones were screened out through sequencing.
pCas was transformed into E. coli BL21 competent cells. The positive clone was incubated in LB medium with kanamycin (50 μg/ml) until the OD600 reached 0.2. L-arabinose (30 mM) was added to the culture to induce the expression of Gam, Beta, and Exo. The cultures were collected for the preparation of electrocompetent cells after the OD600 reached 0.5. Donor DNA as the repair template which was used for homologous recombination was constructed as follows: approximately 500 bp of the upstream and downstream homologous arms were obtained through PCR, and E. coli DL21 genome DNA was used as template. The PCR products were ligated to generate donor DNA through overlap-PCR. The concentrations of sroG-sgRNA and donor DNA were measured with a microspectrophotometer (Thermo Scientific NanoDrop, United States). Approximately 400 ng of gRNA and 2 μg of donor DNA were added to E. coli BL21 electrocompetent cells, which harbored pCas, after electroporation at 2.5 kV in a 2 mm Gene Pulser cuvette (Bio-Rad, United States). The culture was separated on an LB agar plate (50 μg/ml spectinomycin and kanamycin) and incubated at 30°C overnight. Colony PCR and sequencing were carried out to screen the positive clones.
pCas contains a Cas9 protein from Streptococcus pyogenes MGAS5005 with its native promoter, the λ-Red gene under the control of the L-arabinose-inducible promoter (ParaB), temperature-sensitive replicon repA101ts for self-curing, and a sgRNA containing an N20 sequence targeting the pTarget pMB1 replicon (sgRNA-pMB1) under the control of the IPTG-inducible promoter (Ptrc; Jiang et al., 2015). Plasmid curing was performed as follows: the positive clone was incubated in LB medium containing 0.5 mM IPTG and 50 μg/ml spectinomycin and cultured overnight at 30°C. pCas was cured on LB medium overnight at 42°C. When a strain was subjected to further genomic editing, pCas curing was not performed.
The R3 strain was generated by deleting the FMN riboswitch from primitive E. coli BL21 with the CRISPR/Cas9 system according to the above method. The R4 strain was constructed by cotransforming pET-AE and pAC-Z into the R3 strain.
Given that the binding of FMN to the FMN riboswitch represses ribB expression, the effect of the FMN riboswitch deletion on ribB transcript levels was detected through quantitative real-time PCR (qPCR). The E. coli BL21, R2, R3, and R4 strains were incubated in LB medium at 37°C. When the OD600 reached 0.6, 2 mM IPTG was added to the LB medium to induce the transcription of ribB. The cells were harvested when O600 reached 1.0. Total RNA was extracted from the E. coli cells with a bacterial total RNA isolation kit. After cDNA was synthesized with an M5 Super plus qPCR RT kit (Mei5bio, Beijing, China) with the total RNA as the template, qPCR was performed on a LightCycler480 (Roche, Basel, Switzerland) with 2 × RealStar Green Fast Mixture (GenStar, China).
Differences in ribB gene transcript levels between the engineered strains and wild-type E. coli BL21 were measured in triplicate, normalized to the 16S internal control, and calculated according to the 2−∆∆CT method (Livak and Schmittgen, 2001).
Batch shake-flask culture was used to compare the RF production capacities of different strains. The engineered R4 strain was used to optimize the fermentation conditions (medium, glucose concentration, incubation temperature, and IPTG concentration). The R4 strain was inoculated into 10 ml of LB medium and cultured overnight as a preseed culture. Preseed cultures (0.5 ml) were added to 50 ml of LB and cultured to mid-exponential growth phase at 220 rpm as the seed cultures. One percent of each seed culture (v/v) was transferred to LB (or M9Y or MSY) medium to produce RF. When the OD600 reached 0.6, IPTG was added to induce the expression of target genes for producing RF. If necessary, appropriate antibiotics were added to the medium. Time-course analysis of RF production in a 1 L shake flask was also performed according to the above procedure.
OD600 was measured with a PGENERAL New Century T6 spectrophotometer (Beijing, China). The concentration of glucose was measured by the 3,5-dinitrosalicylic acid method (Hu et al., 2008). The concentration of RF was determined by high-performance liquid chromatography (HPLC; Agilent 1,260 Infinity, CA, United States). Bacterial cultures were diluted before centrifugation because RF is poorly soluble under neutral or acidic conditions and form crystals. The supernatants were filtered (0.22 μm inorganic membrane) and analyzed under the following conditions (Pedrolli et al., 2015a): mobile phase, 18% (v/v) methanol–20 mM formic acid–20 mM ammonium formate (pH 3.7); flow rate, 0.8 ml/min; injection volume, 10 μl; column, ZORBAX SB-C18 (4.6 × 150 mm; Agilent, CA, United States); and RF detection at 445 nm.
All experiments were performed in triplicate. The results are presented as the mean ± SD of three independent experiments. SPSS 17.0 software was used for statistical analysis. Data were graphed using Origin 8.5 software. The plasmid construction map was generated with ChemDraw Professional 2017 software.
For the construction of the RF-producing strain R1, the plasmid pET-AE was transformed into E. coli BL21 competent cells. The R2 strain with enhanced glucose utilization ability was obtained by transforming the plasmid pAC-Z into the R1 strain.
The expression of the target proteins (RibA, RibB, RibC, RibD, RibE, and Zwf) in the recombinant strain R2 was verified through SDS-PAGE. The electrophoresis results are shown in Figure 4A. The apparent molecular weights of the six proteins were 22, 23.4, 23, 40, 16.2, and 52 kDa, respectively, which were consistent with the expected values, indicating that the target proteins were correctly expressed in the recombinant strain R2.
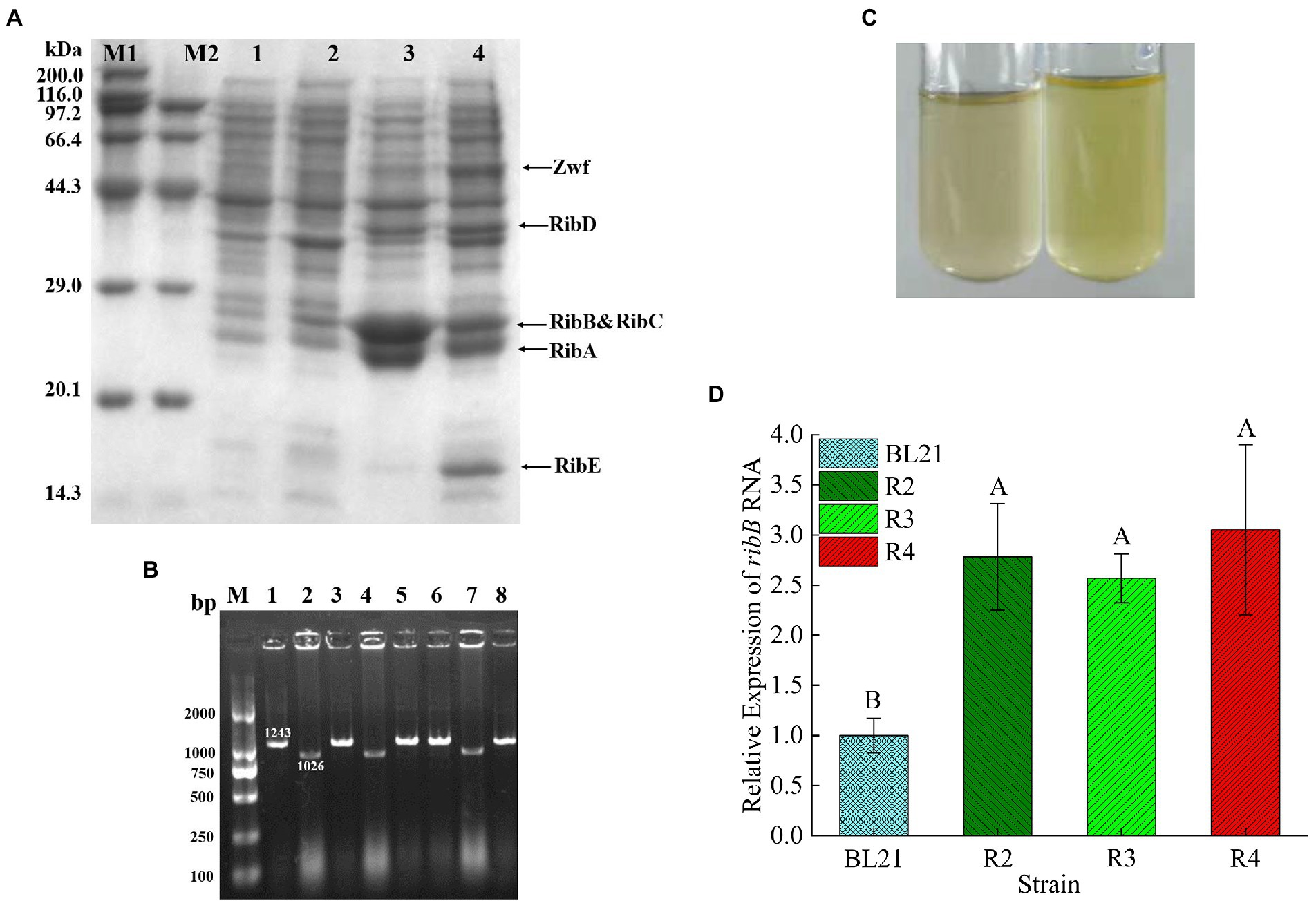
Figure 4. (A) SDS-PAGE analysis of the recombinant E. coli. Lane M: protein molecular weight marker; Lane 1: precipitated proteins from E. coli BL21/pETDuet-1&pACYCDuet-1 after IPTG induction; Lane 2: proteins in supernatant from E. coli BL21/pETDuet-1&pACYCDuet-1 after IPTG induction; Lane 3: precipitated proteins from strain R2 after IPTG induction; Lane 4: proteins in supernatant from strain R2 after IPTG induction. (B) Colony PCR analysis of E. coli BL21▵sroG. The length of the positive band was 1,026 bp, while the negative band was 1,243 bp. (C) Comparison of the supernatant color after E. coli was incubated overnight in 5 ml of LB medium (left: E. coli BL21; right: R3). (D) Comparison of ribB transcript levels of different strains by qPCR. p < 0.01. Letters indicate significant differences.
The RF production ability of the recombinant strains was measured through HPLC, and the OD600 was measured with a spectrophotometer. The key fermentation parameters (OD600 and RF titer) in the strains R1 and R2 are listed in Table 3. After 72 h of incubation, the engineered strain R1 produced 182.65 ± 9.04 mg/l RF, and the OD600 reached 7.18 ± 0.36. The engineered strain R2 produced 319.01 ± 20.65 mg/l RF, and the OD600 reached 8.76 ± 0.12, showing 74.66 and 22.01% increases compared to the R1 strain, respectively. These results suggested that the overexpression of the key genes in E. coli BL21 conferred on the strain the ability to produce RF and that the overexpression of zwf on this basis further enhanced RF production and promoted the growth of the recombinant E. coli BL21.
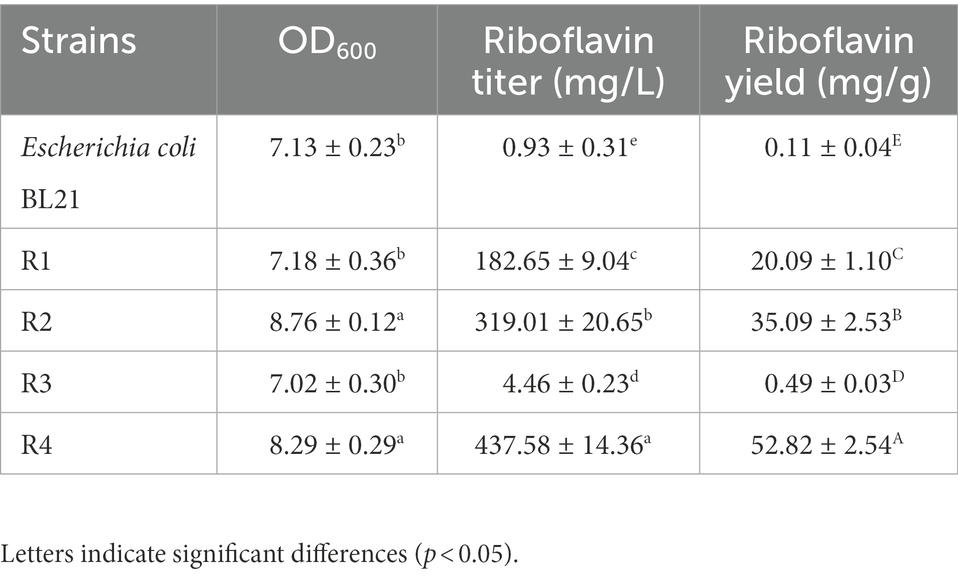
Table 3. Riboflavin production in various strains.
To relieve FMN inhibition, a total of 223 bp of nucleotides located upstream of the RBS sequence of ribB were deleted. sgRNA was obtained through reverse-PCR using the primer sroG-sgRNA-F/R and designated sroG-sgRNA. The positive clones were screened out through sequencing after plasmid extraction. The primers sroG-armL-F/R and sroG-armR-F/R were used to amplify the upstream and downstream homologous arms, sroG-Up and sroG-Down (with length of approximately 500 bp), with E. coli BL21 genome DNA as the template. The primers sroG-armL-F and sroG-armR-R were used to obtain approximately 1-kb-long donor DNA through overlap-PCR. After cotransformation with sroG-sgRNA and donor DNA, the positive clones with 1,026 bp bands were screened out through colony PCR, whereas the negative clones had 1,243 bp bands (Figure 4B). The sequencing results further confirmed the successful knockout of the FMN riboswitch. Before proceeding to the next step, strain R3 was successively subjected to IPTG induction and 42°C culture to cure the sroG-sgRNA and pCas. After incubation in 5 ml of LB medium for 24 h, the color of the supernatant turned yellow, indicating that the deletion strain R3 had derepressed the inhibition of RF biosynthesis by FMN compared to the wild-type E. coli BL21 (Figure 4C).
Northern blot analysis showed that the deletion of the FMN riboswitch in E. coli CmpX13 led to the constitutive synthesis of ribB mRNA (Pedrolli et al., 2015a). To compare the effects of FMN riboswitch deletion and ribB overexpression on ribB transcript levels, the ribB transcript levels of E. coli BL21, R2, R3, and R4 were quantified through qPCR. As shown in Figure 4D and Table 4, compared to those of E. coli BL21, the ribB transcript levels of R2, R3, and R4 improved 2.78, 2.57, and 3.05-fold, respectively. These results suggested that the deletion of the FMN riboswitch could derepress the effect of FMN on ribB transcription. However, knockout of the FMN riboswitch on the basis of the overexpression of ribB resulted in only a slight increase in the transcript level of ribB (an increase of 9.7%). This result is consistent with that reported by Pedrolli et al. (2015a).
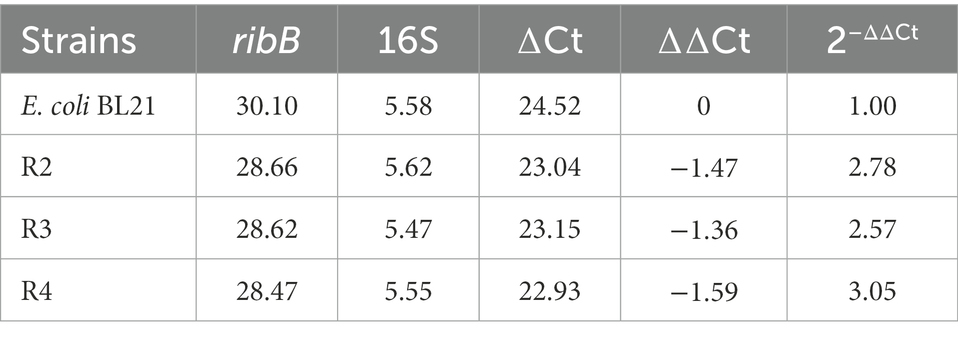
Table 4. RibB gene transcript level assays of different strains by qPCR.
After 72 h of incubation in M9Y medium, the FMN riboswitch deletion strain R3 accumulated 4.46 ± 0.23 mg/l RF, increasing by 3.8-fold compared to the control strain E. coli BL21 (0.93 ± 0.31 mg/l). In addition, no significant difference was observed in OD600 values between the R3 strain and E. coli BL21 (Table 3), suggesting that the deletion of the FMN riboswitch did not affect the growth of the strain. The engineered strain R4 produced 437.58 ± 14.36 mg/l RF, and the OD600 value of the cells reached 8.29 ± 0.29. Compared to the engineered strain R2, the RF titer of the engineered strain R4 increased by 0.37-fold (Table 3). This result showed that the deletion of the FMN riboswitch on the basis of the overexpression of key genes was effective in enhancing the RF production capacity of the engineered strain.
Riboflavin (RF) production can be further improved by optimizing fermentation conditions, such as culture medium, glucose concentration, culture temperature, and IPTG concentration (Figure 5). The engineered strain R4 was chosen for the optimization of the culture conditions. M9Y, MSY, and LB culture media were used to screen the optimum medium for RF production in this study. As shown in Figure 5A, MSY was the most suitable medium for the fermentation of the R4 strain for RF production, and the titer of RF in MSY medium after incubation for 72 h was 505.01 ± 45.88 mg/l. A variety of metal elements (e.g., Co2+, Mn2+, etc.) in the trace element solution in MSY may be cofactors for key enzymes in the de novo synthesis of RF. Therefore, MSY medium was effective in enhancing RF production in the engineered strain R4. Thus, MSY was selected as the fermentation medium, and a glucose concentration gradient (10, 20, 30, and 40 g/l) was set up to screen the optimal glucose concentration. As shown in Figure 5B, the optimum glucose concentration was 20 g/l, and the titer of RF reached 537.88 ± 10.26 mg/l. A temperature gradient (28, 30, 37, and 42°C) was used to determine the optimum fermentation temperature. MSY containing 20 g/l glucose was used as the fermentation medium. As shown in Figure 5C, the highest RF titer was obtained at 37°C, which was consistent with the optimum growth temperature for E. coli. The titer of RF reached 537.88 ± 10.26 mg/l in the engineered strain at 37°C. Since strain R4 contained compatible pET-AE and pAC-Z plasmids under the control of the T7-Lac promoter, an induction concentration gradient of IPTG (0.1, 0.25, 0.5, 1.0, 1.5, and 2.0 mM) was set up for the identification of the most suitable IPTG induction concentration. The medium was MSY (20 g/l glucose), and the temperature was 37°C. As shown in Figure 5D, the highest RF titer was obtained at 2 mM IPTG. The RF titer reached 611.22 ± 11.25 mg/l. Taken together, the optimum fermentation conditions of the engineered strain R4 for the production of RF were as follows: medium, MSY; glucose, 20 g/l; temperature, 37°C; and IPTG concentration, 2 mM.
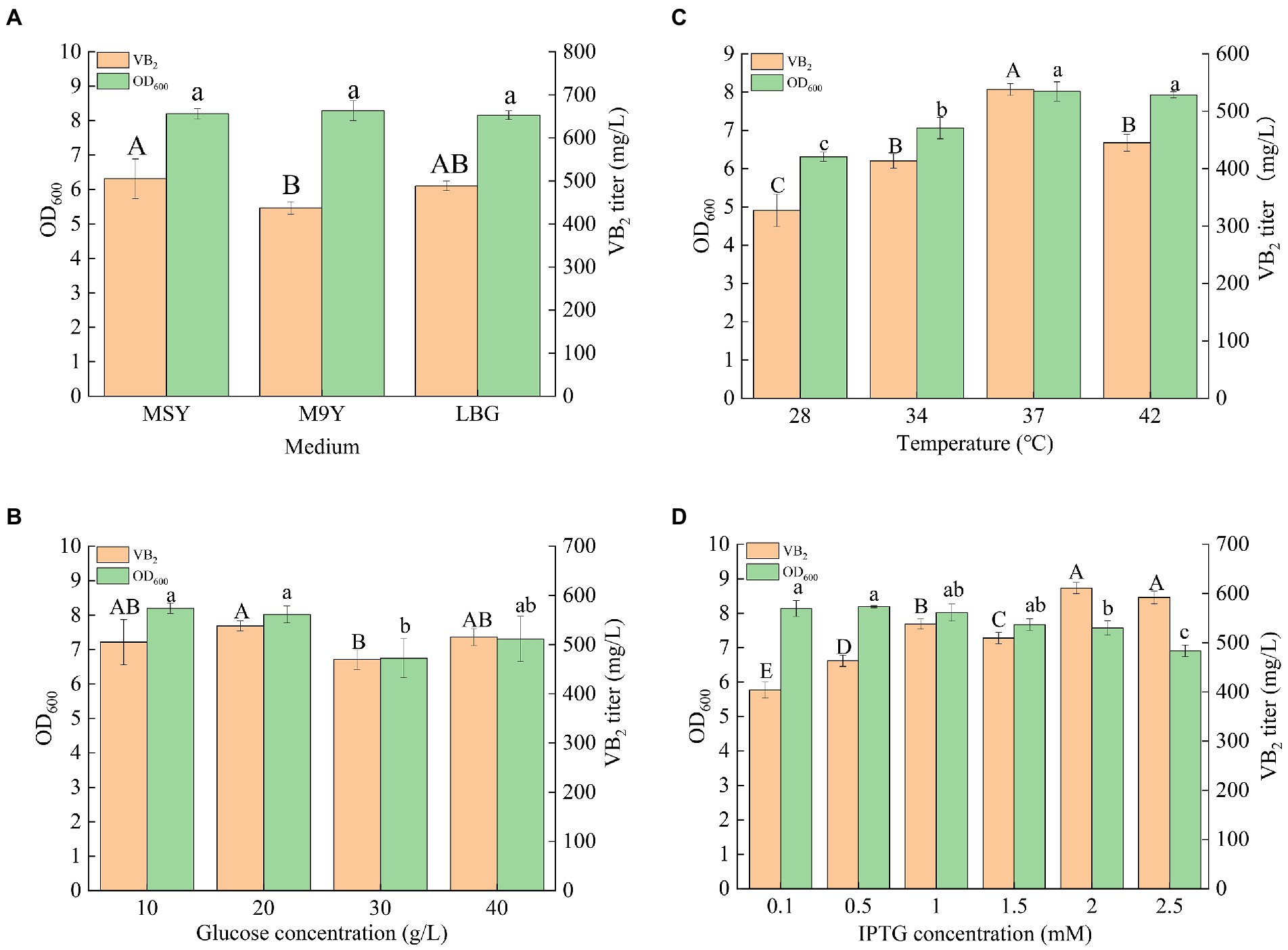
Figure 5. Results of the culture condition optimization of the engineered strain R4: (A) fermentation results in different culture media; (B) fermentation results with MSY medium containing different concentrations of glucose; (C) fermentation results at different temperatures; (D) fermentation results at different IPTG concentrations (p < 0.05). Letters indicate significant differences.
For fed-batch cultivation, 1% (v/v) seed cultures were added to 250 ml of MSY with 40 g/l glucose in a 1 l shake flask (10 g/l glucose was added to the initial medium and feed medium separately), and the mixture was shaken at 37°C and 220 rpm. 3-(N-morpholino) propanesulfonate (MOPS, 50 mM) was added to the cultures to maintain a neutral pH. IPTG at a final concentration of 2 mM induced the expression of the target genes. Ammonia was used to adjust the pH to 7.2 during fermentation. As shown in Figure 6, with the addition of glucose, the RF titer and the OD600 values of the cells increased quickly. The RF titer was the highest after induction with IPTG for 96 h (1574.60 ± 109.32 mg/l with a yield of 12.00 ± 0.83 mg/g glucose). Meanwhile, the OD600 value of the cells reached a maximum of 13.9 ± 0.87. Afterward, these values began to decline due to the depletion of glucose and the accumulation of byproducts.
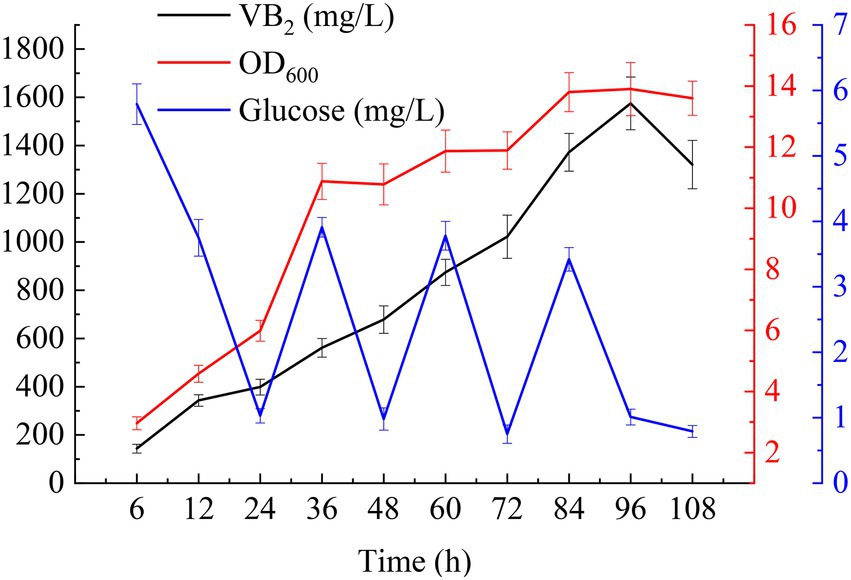
Figure 6. Time-course profiles of key fermentation parameters: glucose concentration, OD600, and RF titer in MSY medium during fed-batch cultivations.
Owing to its wide range of functions in the human body, RF is one of the six main indicators for assessing human body status according to WHO regulations. Given that the human body cannot synthesize RF, humans must consume RF from food, particularly milk, eggs, meat, cheese, and grains. Deficiency in the RF can cause ariboflavinosis (Northrop-Clewes and Thurnham, 2012).
Current strategies for RF production include total chemical synthesis, chemical semisynthesis, and microbial fermentation. Microbial fermentation can be further divided into two types: traditional microbial fermentation and engineered bacterial fermentation. Currently, B. subtilis and Aphis gossypii are the main strains used in the industrial production of RF (Revuelta et al., 2017). As a common workhorse in fundamental biological research, E. coli is used in producing various substances due to its numerous advantages: fast growth rate, clear genetic background, simple genetic manipulation, and low culture requirements. Unlike wild-type E. coli, E. coli BL21 can accumulate RF under normal culture conditions because of the His115Leu mutation of RibF (Wang et al., 2015). In this study, the titer of RF in the constructed strain R3 was 3.48-fold higher than that in E. coli CmpX13 (Pedrolli et al., 2015a). These results demonstrate the potential of E. coli BL21 as an RF-producing bacterium.
The overexpression of key genes, the knockout of metabolic branches for the enhancement of the supply of direct precursor substances GTP and Ru5P, deregulation of key enzymes, enhancement of energy generation, reduction of consumption for metabolic maintenance, and optimization of culture media are common strategies for metabolic engineering (Fordjour et al., 2019).
The de novo biosynthesis pathway of RF consists of three main pathways: PPP, purine biosynthesis pathway (PBP), and RBP (Figure 1). Metabolic engineering modifications in these pathways are the focus of research, as follows. (1) Metabolic engineering in the PPP phase. The coexpression of the mutant genes zwf (A243T) and gnd(S361F) from Corynebacterium glutamicum and native pgl in E. coli MG1655 can enhance RF production (Lin et al., 2014). The coexpression of the mutant genes zwf(A243T) and gnd(S361F) from C. glutamicum in B. subtilis also enhances RF production (Wang et al., 2011). Deletion of the pfkA, edd, and eda genes enhances the flux toward PPP (Liu et al., 2016). The knockout of pgi can promote metabolic flux toward PPP, which in turn increases RF production (Lin et al., 2014). This can markedly reduce the translation level of the glucose transporter IICBGlc (encoded by ptsG) simultaneously (Kimata et al., 2001). Unexpectedly, in this study, pgi was disrupted in the engineered strain R4, and the titer of RF decreased by 23% when cultured in M9Y medium without glycine. The disruption of pgi decreased the energy supplementation and RF production, and the reduction in OD600 verified this effect. (2) Metabolic engineering in the PBP phase. The co-overexpression of purF, purM, purN, purH, and purD, the key genes in the PBP, increased the ability of B. subtilis PK-P to produce RF by 31% (Shi et al., 2009). The purine repressor PurR (encoded by purR) tightly regulates the transcription of genes involved in the biosynthesis of inosine 5′-monophosphate (IMP), guanosine 5′-monophosphate (GMP), and adenosine 5′-monophosphate (AMP) from 5-phospho-α-D-ribose-1-diphosphate (PRPP; Cho et al., 2011). The deregulation of the purine pathway by disruption of purR improves purine nucleotide supply and RF production in B. subtilis (Shi et al., 2014). However, in E. coli, although the knockout of purR increased the expression level of purine synthesis genes, the production of GTP, the direct precursor of RF synthesis, did not increase, similar to the production of RF (Liu et al., 2021). This difference may exist due to the regulation of different genes in the two different microorganisms by PurR. As one of the direct precursors of RF synthesis, GTP is synthesized by PBP. Coexpression of ndk and gmk can enhance GTP supply and the production of RF in E. coli (Xu et al., 2015). PRS and PurF are considered key enzymes in the de novo and salvage synthesis of purine nucleotides (Shimaoka et al., 2007; Shi et al., 2009). PRS is inhibited by adenosine diphosphate (ADP), and the prsD128A mutation severely affects the affinity of ADP for the PRS binding site (Shimaoka et al., 2007), whereas PurF is inhibited by AMP and GMP. purF(K326Q, P410W, T304E) mutations desensitize these inhibitors (Jimenez et al., 2005; Shimaoka et al., 2007). The overexpression of the mutant prs and purF indeed enhances GTP supplementation, and thus the yield of the target products in different microorganisms (Shimaoka et al., 2007; Xu et al., 2015). Unexpectedly, in this study, the overexpression of mutant prs and purF did not enhance RF production. The main reason for this may be the excessive metabolic burden. Constitutive expression on chromosomes by gene editing techniques may be a better option. (3) Metabolic engineering in the RBP phase. The coexpression of RBP genes in different microorganisms (E. coli, B. subtilis, Pichia pastoris, and C. famata) contributed to the efficient production of RF (Perkins et al., 1999; Marx et al., 2008; Dmytruk et al., 2011; Lin et al., 2014). In the present study, the deletion of the FMN riboswitch derepresses the transcription of ribB. Compared to strains that overexpress only rib genes and zwf, the strain that further knocked out the FMN riboswitch showed a 9.71% increase in ribB transcript level but a 37.17% increase in RF production. In E. coli, rib genes are scattered at different locations on the chromosome, and ribB contains an FMN-regulated riboswitch in the upstream 5′ UTR, while no similar regulatory sequences are found in front of other RBP genes, suggesting that ribB should be a key gene in RF synthesis (Pedrolli et al., 2015a). These results suggest that RF production can be enhanced by knocking out the FMN riboswitch. To further enhance the titer of RF, reducing the conversion ratio from RF to FMN and FAD is necessary, given that ribF is an essential gene for E. coli growth (Baba et al., 2006). Thus, ideal options include reducing the expression level of ribF or introducing a mutant. Lin et al. (2014) replaced the native RBS of ribF with a weaker RBS. This modulation reduced the expression of ribF and contributed to RF overproduction. Hu et al. (2021) increased the production of RF by knocking down the ribF, purA, and guaC genes of E. coli by using synthetic regulatory small RNA. In Leuconostoc citreum, the expression of ribF was downregulated by a CRISPR interference system, and production was improved (Son et al., 2020).
The coutilization of carbon sources to reduce production costs has become an important research topic in metabolic engineering (Wu et al., 2016). As the most abundant renewable carbohydrate source on Earth, the efficient use of lignocellulose has attracted the attention of researchers. Although many aspects, such as lignin pretreatment and hydrolysis, are still not well addressed, the coutilization of the hydrolysates of lignocellulose, such as glucose, xylose, and arabinose, has been extensively studied in different organisms (Karhumaa et al., 2006; Jørgensen et al., 2007; Xin et al., 2016). Nevertheless, most industrial microorganisms preferentially utilize glucose over xylose owing to the regulatory phenomenon of carbon catabolite repression (CCR; Gawand et al., 2013). CCR was first studied in microorganisms such as E. coli, B. subtilis, and Salmonella typhimurium during the 1940s, and it emerged as an important gene-regulatory mechanism in bacteria, controlling the expression of approximately 10% of total bacterial genes (Choudhury and Saini, 2017). The accumulation of dephosphorylated EIIAGlc is the main cause of CCR in E. coli (Yuan et al., 2020), and the disruption of ptsG can relieve CCR, enabling the simultaneous utilization of mixed sugars (Ma et al., 2017; Yuan et al., 2020). In addition to being involved in glucose transport, in some rhizosphere microorganisms (e.g., B. cereus C1L), ptsG is also involved in root colonization and the production of beneficial metabolites to induce plant systemic disease resistance (Lin et al., 2020). However, the deletion of ptsG normally impairs the growth of strains (Chen et al., 2021). In many PTS-strains, this problem is addressed by the coexpression of galactose permease (galP) and glucokinase (glK), which are responsible for glucose transport and phosphorylation (Gawand et al., 2013; Li et al., 2020; Chen et al., 2021). In this study, ptsG was disrupted. Although the growth of the bacterium was restored by introducing galP and glK, the RF production (86.82 ± 3.22 mg/l) was obviously reduced compared to the strain with pgi deletion in R4 (165.55 ± 5.89 mg/l) after 24 h incubation. The main reason for this may be that the overexpression of these two genes consumed a considerable amount of energy.
In conclusion, the production of RF was enhanced in the recombinant E. coli BL21 strain by combinatorial strategies with key gene overexpression and FMN riboswitch deletion. The overexpression of the key genes enables the E. coli BL21 strain to produce a high yield of RF. The knockout of the FMN riboswitch further increases ribB transcript levels and RF production. To the best of our knowledge, this is the first use of the knockout of the FMN riboswitch in the construction of an engineered E. coli strain for RF production. The engineered strain R4 with the coexpression of five key genes and zwf and the deletion of the FMN riboswitch could accumulate 611.22 ± 11.25 mg/l RF under optimal fermentation conditions. Ultimately, 1,574.60 ± 109.32 mg/l RF was obtained through fed-batch fermentation with 40 g/l glucose. This study provides a basis for the production of RF in the recombinant E. coli BL21 in the future.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
PY conceptualized and designed the experiments. BF, JY, QC, and QZ conducted the experiments. JL and ZZ analyzed the data. PY wrote the manuscript. All authors contributed to the article and approved the submitted version.
This study was financed by the Natural Science Foundation of Zhejiang Province, China (No. LY21C200006).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
An, J. H., Zhang, W. L., Jing, X. R., Nie, Y., and Xu, Y. (2020). Reconstitution of TCA cycle involving L-isoleucine dioxygenase for hydroxylation of L-isoleucine in Escherichia coli using CRISPR-Cas9. 3 Biotech. 10, 167–110. doi: 10.1007/s13205-020-2160-3
Baba, T., Ara, T., Hasegawa, M., Takai, Y., Okumura, Y., Baba, M., et al. (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 1–11. doi: 10.1038/msb4100050
Bacher, A., Eberhardt, S., Fischer, M., Kis, K., and Richter, G. (2000). Biosynthesis of vitamin b2 (riboflavin). Annu. Rev. Nutr. 20, 153–167. doi: 10.1146/annurev.nutr.20.1.153
Chen, M. L., Ma, C. W., Chen, L., and Zeng, A. P. (2021). Integrated laboratory evolution and rational engineering of GalP/Glk-dependent Escherichia coli for higher yield and productivity of L-tryptophan biosynthesis. Metab. Eng. Commun. 12, e00167–e00112. doi: 10.1016/j.mec.2021.e00167
Cho, B. K., Federowicz, S. A., Embree, M., Park, Y. S., Kim, D., and Palsson, B. O. (2011). The PurR regulon in Escherichia coli K-12 MG1655. Nucleic Acids Res. 39, 6456–6464. doi: 10.1093/nar/gkr307
Choudhury, D., and Saini, S. (2017). “Sugar Co-utilization in Microorganisms,” in Current Developments in Biotechnology and Bioengineering. eds. P. Gunasekaran, S. Noronha and A. Pandey (Elsevier), 243–268.
Dmytruk, K. V., Yatsyshyn, V. Y., Sybirna, N. O., Fedorovych, D. V., and Sibirny, A. A. (2011). Metabolic engineering and classic selection of the yeast Candida famata (Candida flareri) for construction of strains with enhanced riboflavin production. Metab. Eng. 13, 82–88. doi: 10.1016/j.ymben.2010.10.005
Fordjour, E., Adipah, F. K., Zhou, S., Du, G., and Zhou, J. (2019). Metabolic engineering of Escherichia coli BL21 (DE3) for de novo production of L-DOPA from D-glucose. Microb. Cell Factories 18:74. doi: 10.1186/s12934-019-1122-0
Fu, B., Ren, Q., Ma, J., Chen, Q., Zhang, Q., and Yu, P. (2021). Enhancing the production of physiologically active vitamin D3 by engineering the hydroxylase CYP105A1 and the electron transport chain. World J. Microbiol. Biotechnol. 38, 14–10. doi: 10.1007/s11274-021-03193-1
Garcia-Angulo, V. A. (2017). Overlapping riboflavin supply pathways in bacteria. Crit. Rev. Microbiol. 43, 196–209. doi: 10.1080/1040841X.2016.1192578
Gawand, P., Hyland, P., Ekins, A., Martin, V. J. J., and Mahadevan, R. (2013). Novel approach to engineer strains for simultaneous sugar utilization. Metab. Eng. 20, 63–72. doi: 10.1016/j.ymben.2013.08.003
Haase, I., Sarge, S., Illarionov, B., Laudert, D., Hohmann, H. P., Bacher, A., et al. (2013). Enzymes from the haloacid dehalogenase (HAD) superfamily catalyse the elusive dephosphorylation step of riboflavin biosynthesis. Chembiochem 14, 2272–2275. doi: 10.1002/cbic.201300544
Howe, J. A., Xiao, L., Fischmann, T. O., Wang, H., Tang, H., Villafania, A., et al. (2016). Atomic resolution mechanistic studies of ribocil: a highly selective unnatural ligand mimic of the E. coli FMN riboswitch. RNA Biol. 13, 946–954. doi: 10.1080/15476286.2016.1216304
Hu, R., Lin, L., Liu, T., Ouyang, P., He, B., and Liu, S. (2008). Reducing sugar content in hemicellulose hydrolysate by DNS method: a revisit. J. Biobased Materials Bioenergy 2, 156–161. doi: 10.1166/jbmb.2008.306
Hu, W., Liu, S., Wang, Z., and Chen, T. (2021). Improving riboflavin production by knocking down ribF, purA and guaC genes using synthetic regulatory small RNA. J. Biotechnol. 336, 25–29. doi: 10.1016/j.jbiotec.2021.05.007
Jiang, Y., Chen, B., Duan, C., Sun, B., Yang, J., and Yang, S. (2015). Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 81, 2506–2514. doi: 10.1128/AEM.04023-14
Jimenez, A., Santos, M. A., Pompejus, M., and Revuelta, J. L. (2005). Metabolic engineering of the purine pathway for riboflavin production in Ashbya gossypii. Appl. Environ. Microbiol. 71, 5743–5751. doi: 10.1128/AEM.71.10.5743-5751.2005
Jørgensen, H., Kristensen, J. B., and Felby, C. (2007). Enzymatic conversion of lignocellulose into fermentable sugars: challenges and opportunities. Biofuels Bioprod. Biorefin. 1, 119–134. doi: 10.1002/bbb.4
Juarez, O., Nilges, M. J., Gillespie, P., Cotton, J., and Barquera, B. (2008). Riboflavin is an active redox cofactor in the Na+−pumping NADH: quinone oxidoreductase (Na+-NQR) from vibrio cholerae. J. Biol. Chem. 283, 33162–33167. doi: 10.1074/jbc.M806913200
Karhumaa, K., Wiedemann, B., Hahn-Hagerdal, B., Boles, E., and Gorwa-Grauslund, M. F. (2006). Co-utilization of L-arabinose and D-xylose by laboratory and industrial Saccharomyces cerevisiae strains. Microb. Cell Factories 5, 1–11. doi: 10.1186/1475-2859-5-18
Kimata, K., Tanaka, Y., Inada, T., and Aiba, H. (2001). Expression of the glucose transporter gene, ptsG, is regulated at the mRNA degradation step in response to glycolytic flux in Escherichia coli. EMBO J. 20, 3587–3595. doi: 10.1093/emboj/20.13.3587
Koizumi, S., Yonetani, Y., Maruyama, A., and Teshiba, S. (2000). Production of riboflavin by metabolically engineered Corynebacterium ammoniagenes. Appl. Microbiol. Biotechnol. 53, 674–679. doi: 10.1007/s002539900295
Li, Y. F., Lin, Z. Q., Huang, C., Zhang, Y., Wang, Z., Tang, Y. J., et al. (2015). Metabolic engineering of Escherichia coli using CRISPR-Cas9 meditated genome editing. Metab. Eng. 31, 13–21. doi: 10.1016/j.ymben.2015.06.006
Li, M., Liu, C., Yang, J., Nian, R., Xian, M., Li, F., et al. (2020). Common problems associated with the microbial productions of aromatic compounds and corresponding metabolic engineering strategies. Biotechnol. Adv. 41:107548. doi: 10.1016/j.biotechadv.2020.107548
Li, Y., Yan, F., Wu, H., Li, G., Han, Y., Ma, Q., et al. (2019). Multiple-step chromosomal integration of divided segments from a large DNA fragment via CRISPR/Cas9 in Escherichia coli. J. Ind. Microbiol. Biotechnol. 46, 81–90. doi: 10.1007/s10295-018-2114-5
Lin, C. H., Lu, C. Y., Tseng, A. T., Huang, C. J., Lin, Y. J., and Chen, C. Y. (2020). The ptsG gene encoding the major glucose transporter of Bacillus cereus C1L participates in root colonization and beneficial metabolite production to induce plant systemic disease resistance. Mol. Plant-Microbe Interact. 33, 256–271. doi: 10.1094/MPMI-06-19-0165-R
Lin, Z., Xu, Z., Li, Y., Wang, Z., Chen, T., and Zhao, X. (2014). Metabolic engineering of Escherichia coli for the production of riboflavin. Microb. Cell Factories 13, 104–112. doi: 10.1186/s12934-014-0104-5
Liu, S., Hu, W. Y., Wang, Z. W., and Chen, T. (2021). Rational engineering of Escherichia coli for high-level production of riboflavin. J. Agric. Food Chem. 69, 12241–12249. doi: 10.1021/acs.jafc.1c04471
Liu, S., Kang, P., Cui, Z. Z., Wang, Z., and Chen, T. (2016). Increased riboflavin production by knockout of 6-phosphofructokinase I and blocking the Entner-Doudoroff pathway in Escherichia coli. Biotechnol. Lett. 38, 1307–1314. doi: 10.1007/s10529-016-2104-5
Liu, C., Zhang, L., Liu, H., and Cheng, K. (2017). Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J. Control. Release 266, 17–26. doi: 10.1016/j.jconrel.2017.09.012
Liu, L., Zhao, D., Ye, L., Zhan, T., Xiong, B., Hu, M., et al. (2020). A programmable CRISPR/Cas9-based phage defense system for Escherichia coli BL21 (DE3). Microb. Cell Factories 19:136. doi: 10.1186/s12934-020-01393-2
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T)) method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Long, Q., Ji, L., Wang, H., and Xie, J. (2010). Riboflavin biosynthetic and regulatory factors as potential novel anti-infective drug targets. Chem. Biol. Drug Des. 75, 339–347. doi: 10.1111/j.1747-0285.2010.00946.x
Lu, J., Tang, J., Liu, Y., Zhu, X., Zhang, T., and Zhang, X. (2012). Combinatorial modulation of galP and glk gene expression for improved alternative glucose utilization. Appl. Microbiol. Biotechnol. 93, 2455–2462. doi: 10.1007/s00253-011-3752-y
Ma, Q., Zhang, Q., Xu, Q., Zhang, C., Li, Y., Fan, X., et al. (2017). Systems metabolic engineering strategies for the production of amino acids. Synth. Syst. Biotechnol. 2, 87–96. doi: 10.1016/j.synbio.2017.07.003
Marx, H., Mattanovich, D., and Sauer, M. (2008). Overexpression of the riboflavin biosynthetic pathway in Pichia pastoris. Microb. Cell Factories 7:23. doi: 10.1186/1475-2859-7-23
Mazur-Bialy, A. I., and Pochec, E. (2016). Riboflavin reduces pro-inflammatory activation of adipocyte-macrophage co-culture. Potential application of vitamin B2 enrichment for attenuation of insulin resistance and metabolic syndrome development. Molecules 21, 1–15. doi: 10.3390/molecules21121724
Northrop-Clewes, C. A., and Thurnham, D. I. (2012). The discovery and characterization of riboflavin. Ann. Nutr. Metab. 61, 224–230. doi: 10.1159/000343111
Pedrolli, D. B., Kuhm, C., Sevin, D. C., Vockenhuber, M. P., Sauer, U., Suess, B., et al. (2015b). A dual control mechanism synchronizes riboflavin and sulphur metabolism in Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 112, 14054–14059. doi: 10.1073/pnas.1515024112
Pedrolli, D. B., Langer, S., Hobl, B., Schwarz, J., Hashimoto, M., and Mack, M. (2015a). The ribB FMN riboswitch from Escherichia coli operates at the transcriptional and translational level and regulates riboflavin biosynthesis. FEBS J. 282, 3230–3242. doi: 10.1111/febs.13226
Perkins, J., Sloma, A., Hermann, T., Theriault, K., Zachgo, E., Erdenberger, T., et al. (1999). Genetic engineering of Bacillus subtilis for the commercial production of riboflavin. J. Ind. Microbiol. Biotechnol. 22, 8–18. doi: 10.1038/sj.jim.2900587
Powers, H. (2003). Riboflavin (vitamin B2) and health. Am. J. Clin. Nutr. 77, 1352–1360. doi: 10.1556/AAlim.32.2003.2.9
Pritchett, D., and Reddy, A. B. (2017). No FAD, no CRY: redox and circadian rhythms. Trends Biochem. Sci. 42, 497–499. doi: 10.1016/j.tibs.2017.05.007
Qiu, Y., Ouyang, S., Shen, Z., Wu, Q., and Chen, G. (2004). Metabolic engineering for the production of copolyesters consisting of 3-hydroxybutyrate and 3-hydroxyhexanoate by Aeromonas hydrophila. Macromol. Biosci. 4, 255–261. doi: 10.1002/mabi.200300090
Revuelta, J. L., Ledesma-Amaro, R., Lozano-Martinez, P., Diaz-Fernandez, D., Buey, R. M., and Jimenez, A. (2017). Bioproduction of riboflavin: a bright yellow history. J. Ind. Microbiol. Biotechnol. 44, 659–665. doi: 10.1007/s10295-016-1842-7
Saedisomeolia, A., and Ashoori, M. (2018). Riboflavin in human health: a review of current evidences. Adv. Food Nutr. Res. 83, 57–81. doi: 10.1016/bs.afnr.2017.11.002
Schwechheimer, S. K., Park, E. Y., Revuelta, J. L., Becker, J., and Wittmann, C. (2016). Biotechnology of riboflavin. Appl. Microbiol. Biotechnol. 100, 2107–2119. doi: 10.1007/s00253-015-7256-z
Shi, S., Shen, Z., Chen, X., Chen, T., and Zhao, X. (2009). Increased production of riboflavin by metabolic engineering of the purine pathway in Bacillus subtilis. Biochem. Eng. J. 46, 28–33. doi: 10.1016/j.bej.2009.04.008
Shi, T., Wang, Y., Wang, Z., Wang, G., Liu, D., Fu, J., et al. (2014). Deregulation of purine pathway in Bacillus subtilis. Microb. Cell Factories 13, 1–16. doi: 10.1186/s12934-014-0101-8
Shimaoka, M., Takenaka, Y., Kurahashi, O., Kawasaki, H., and Matsui, H. (2007). Effect of amplification of desensitized purF and prs on inosine accumulation in Escherichia coli. J. Biosci. Bioeng. 103, 255–261. doi: 10.1263/jbb.103.255
Son, J., Jang, S. H., Cha, J. W., and Jeong, K. J. (2020). Development of CRISPR interference (CRISPRi) platform for metabolic engineering of Leuconostoc citreum and its application for engineering riboflavin biosynthesis. Int. J. Mol. Sci. 21:5614. doi: 10.3390/ijms21165614
Sundara Sekar, B., Seol, E., and Park, S. (2017). Co-production of hydrogen and ethanol from glucose in Escherichia coli by activation of pentose-phosphate pathway through deletion of phosphoglucose isomerase (pgi) and overexpression of glucose-6-phosphate dehydrogenase (zwf) and 6-phosphogluconate dehydrogenase (gnd). Biotechnol. Biofuels 10:85. doi: 10.1186/s13068-017-0768-2
Vitreschak, A. G., Rodionov, D. A., Mironov, A. A., and Gelfand, M. S. (2002). Regulation of riboflavin biosynthesis and transport genes in bacteria by transcriptional and translational attenuation. Nucleic Acids Res. 30, 3141–3151. doi: 10.1093/nar/gkf433
Wang, Z., Chen, T., Ma, X., Shen, Z., and Zhao, X. (2011). Enhancement of riboflavin production with Bacillus subtilis by expression and site-directed mutagenesis of zwf and gnd gene from Corynebacterium glutamicum. Bioresour. Technol. 102, 3934–3940. doi: 10.1016/j.biortech.2010.11.120
Wang, X., Wang, Q., and Qi, Q. (2015). Identification of riboflavin: revealing different metabolic characteristics between Escherichia coli BL21 (DE3) and MG1655. FEMS Microbiol. Lett. 362, 1–8. doi: 10.1093/femsle/fnv071
Wu, Y., Shen, X., Yuan, Q., and Yan, Y. (2016). Metabolic engineering strategies for co-utilization of carbon sources in microbes. Bioengineering (Basel) 3, 1–10. doi: 10.3390/bioengineering3010010
Xin, B., Wang, Y., Tao, F., Li, L., Ma, C., and Xu, P. (2016). Co-utilization of glycerol and lignocellulosic hydrolysates enhances anaerobic 1,3-propanediol production by Clostridium diolis. Sci. Rep. 6, 1–10. doi: 10.1038/srep19044
Xu, Z., Lin, Z., Wang, Z., and Chen, T. (2015). Improvement of the riboflavin production by engineering the precursor biosynthesis pathways in Escherichia coli. Chin. J. Chem. Eng. 23, 1834–1839. doi: 10.1016/j.cjche.2015.08.013
Yuan, X., Tu, S., Lin, J., Yang, L., Shen, H., and Wu, M. (2020). Combination of the CRP mutation and ptsG deletion in Escherichia coli to efficiently synthesize xylitol from corncob hydrolysates. Appl. Microbiol. Biotechnol. 104, 2039–2050. doi: 10.1007/s00253-019-10324-0
Keywords: metabolic engineering, riboflavin, FMN riboswitch, CRISPR/Cas9, Escherichia coli BL21
Citation: Fu B, Ying J, Chen Q, Zhang Q, Lu J, Zhu Z and Yu P (2023) Enhancing the biosynthesis of riboflavin in the recombinant Escherichia coli BL21 strain by metabolic engineering. Front. Microbiol. 13:1111790. doi: 10.3389/fmicb.2022.1111790
Edited by:
Hyun-Seob Song, University of Nebraska-Lincoln, United StatesReviewed by:
Liya Liang, Dalian University of Technology, ChinaCopyright © 2023 Fu, Ying, Chen, Zhang, Lu, Zhu and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Yu,  eXVwOTIwMkBob3RtYWlsLmNvbQ==
eXVwOTIwMkBob3RtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.