 Gao Feng
Gao Feng Yan Zeng2
Yan Zeng2 Yue-Qin Tang
Yue-Qin Tang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 06 January 2023
Sec. Systems Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.1098814
Microorganisms in anaerobic digestion (AD) are easily affected by ammonia, especially acetoclastic methanogens. Thus, in ammonia-suppressed AD systems, acetate degradation is reported to be carried out mainly by the cooperation of syntrophic acetate oxidizers and hydrogenotrophic methanogens. Previous studies have revealed ammonia inhibition on microbial flora by AD performance, but the effect mechanism of ammonia on microbial metabolism remains poorly understood. In this study, we constructed a mesophilic chemostat fed with acetate as the sole carbon source, gradually increased the total ammonia nitrogen (TAN) concentration from 1 g L−1 to 6 g L−1, and employed the 16S rRNA gene, metagenomics, and metatranscriptomics analysis to characterize the microbial community structure and metabolic behavior. The results showed that even at the TAN of 6 g L−1 (pH 7.5), the methanogenesis kept normal, the biogas production was approximately 92% of that at TAN of 1 g L−1 and the acetate degradation ratio reached 99%, suggesting the strong TAN tolerance of the microbial community enriched. 16S rRNA gene analysis suggested that the microbial community structure changed along with the TAN concentration. Methanothrix predominated in methanogens all the time, in which the dominant species was gradually replaced from M. soehngenii to M. harundinacea with the increased TAN. Dominant bacterial species also changed and Proteiniphilum showed a significant positive correlation with increased TAN. Meta-omics analysis showed that the absolute dominant microorganisms at TAN of 6 g L−1 were M. harundinacea and Proteiniphilum, both of which highly expressed genes for anti-oxidative stress. M. harundinacea and the second dominant methanogen Methanosarcina highly expressed both acetate cleavage and CO2 reduction pathways, suggesting the possibility that these two pathways contributed to methanogenesis together. Proteiniphilum and some other species in Firmicutes and Synergistetes were likely acetate oxidizers in the community as they highly expressed genes for syntrophic acetate oxidization, H2 generation, and electron transfer. These results suggested that Proteiniphilum as well as M. harundinacea have strong ammonia tolerance and played critical roles in acetate degradation under ammonia-suppressed conditions. The achievements of the study would contribute to the regulation and management of the AD process.
Anaerobic digestion (AD) is a core technology for organic waste treatment that generates methane as a renewable clean biofuel. However, AD is easily affected by various inhibitors, of which ammonia (NH3) and ammonium (NH4+) are the most prominent inhibitors (Hansen et al., 1998; Yenigün and Demirel, 2013). Although appropriate ammonium is beneficial for AD stability, undesirably high concentrations may be reached during the degradation of organic waste, especially those with high protein and urea content. It is generally agreed that the main factor leading to AD inhibition is ammonia, which causes proton imbalance and potassium loss and affects microbial metabolism (Gallert et al., 1998). McCarty and McKinney (1961) found that 0.15 g L−1 NH3 was completely inhibitory to AD. Another study showed that the total ammonia nitrogen (TAN) of around 1.7–1.8 g L−1 caused AD failure (Melbinger et al., 1971). Therefore, studying the effects of ammonia or TAN on AD microorganisms is crucial for developing countermeasures to reduce inhibition and maintain stable AD operation.
Volatile fatty acids (VFAs) are the most important intermediates in the AD process, especially acetate, which is the source of 70–80% of methane (Mackie and Bryant, 1981). As is well known, acetate can be converted to methane through two routes: acetate cleavage (Eq. 1) and acetate oxidation (Eqs 2, 3).
Because of the thermodynamically unfavorable energetics, acetate oxidation strongly depends on the removal of H2 by cooperating with partner hydrogenotrophic methanogens (Hattori, 2008). Such syntrophic cooperation is known to provide limited energy for the growth of two microbes, resulting in a low growth rate. However, previous studies have found that ammonia inhibits methanogens, especially acetate-consuming methanogens (Westerholm et al., 2012). Thus, under ammonia-inhibition conditions, syntrophic acetate oxidation is reported to become the main methanogenesis pathway (Schnürer and Nordberg, 2008). Jiang et al. (2018) used 14C radiolabeled acetate to investigate the link between TAN and the methanogenesis pathways. The results showed that the proportions of hydrogenotrophic methanogenesis in the high (11.1 g/kg) and low (0.2 g/kg) TAN concentration conditions were 68–75% and 9–23%, respectively, indicating the strong ammonia tolerance of syntrophic acetate oxidation methanogenesis pathway.
Therefore, studying the acetate degradation and methanogenesis pathways under ammonia stress is crucial for regulating AD and attenuating inhibition. Previous studies have revealed ammonia inhibition on microbial flora by AD performance; however, the effect mechanism of ammonia on microbial metabolism is not yet clear. Meanwhile, most previous studies were performed using batch cultures, which differed from the generally continuous state of the actual AD process. Therefore, in the present study, we constructed a mesophilic chemostat fed with acetate as the sole carbon source and gradually increased the TAN concentration for acclimation. We employed the 16S rRNA gene, metagenomics, and metatranscriptomics analysis to characterize the microbial community structure and metabolic behavior of syntrophs and methanogens supporting anaerobic acetate degradation. This study provides a basis for developing technology to improve ammonia tolerance and the operational stability of AD.
The acetate-degrading anaerobic chemostat was constructed using a continuous stirred tank reactor (CSTR) with a working volume of 1.8 l that was mixed using a magnetic stirrer at 300–400 rpm (Supplementary Figure S1). The reactor was immersed in a thermostat-controlled water bath to maintain a temperature of 37°C. The seed sludge from a swine manure treatment plant (Sichuan Province, China) was used to inoculate the reactor. Artificial wastewater containing acetate as the sole carbon source was continuously supplied to the CSTR by a peristaltic pump under an atmosphere of N2, and the effluent flowed out from a U-type tube (Supplementary Figure S1). The wastewater [total organic carbon (TOC) concentration of 8.0 g L−1] contained the following components per liter of distilled water: sodium acetate, 5.46 g; acetic acid, 16.0 g; KH2PO4, 0.3 g; KHCO3, 4.0 g; NH4Cl, 1.0 g; NaCl, 0.6 g; MgCl2·6H2O, 0.82 g; CaCl2·2H2O, 0.08 g; and cysteine-HCl·H2O, 0.1 g, supplemented with 10 ml trace element solution of DSMZ medium 318 (Deutsche Sammlung von Mikroorganismen und Zellkulturen, Germany) comprising 1.19 mg L−1 of Ni2+ and 0.34 mg L−1 of Co2+, and 10 ml vitamin solution of DSMZ medium 318 without vitamin B12.
The reactor was operated at a dilution rate of 0.05 d−1. After the system reached steady operation, additional NH4Cl was added to gradually increase TAN concentration from 1 g L−1 to 6 g L−1. Biogas production, gas composition, pH, concentrations of volatile suspended solids (VSS), suspended solids (SS), TOC, as well as VFAs were measured periodically using the protocols reported previously (Chen et al., 2020b). Microbial morphology at each stage was observed using a laser-scanning confocal fluorescence microscope (Olympus FV1000, Japan). During the steady operation period at each TAN concentration (N0 ~ N6), biomass was collected from the broth and used for DNA and RNA extraction.
To investigate the microbial community, 40 ml broth from the chemostat (days 103, 138, 187, 222, 250, 301, and 390) was collected in sterile DNase-free centrifuge tubes, centrifuged at 10,000 rpm at 4°C for 10 min, and rinsed thrice with sterile phosphate buffer saline (PBS) (10 mM, pH 7.5). Total DNA was extracted via the cyltrimethyl ammonium bromide (CTAB) method (Griffiths et al., 2000). DNA extracts were subjected to 16S rRNA gene amplicon sequencing. The V4-V5 hypervariable regions of both bacterial and archaeal 16S rRNA genes were amplified using universal primers 515F (5’-GTGCCAGCMGCCGCGGTAA-3′) and 909R (5’-CCCCGYCAATTCMTTTRAGT-3′). PCR product purification, Illumina sequencing, and data processing were conducted as described previously (Zheng et al., 2019). The raw reads of the 16S rRNA gene sequencing were deposited into the NCBI Sequence Read Archive (SRA) database with the accession number PRJNA524473.
To analyze the metabolic characteristics of the community under high ammonia stress (TAN concentration of 6 g L−1), sludge samples for metagenomics and metatranscriptomics sequencing were collected on days 390 and 393. Total DNA and RNA were extracted via the CTAB method (Griffiths et al., 2000). The metagenome DNA was sequenced on an Illumina HiSeq 3,000 platform (Illumina). The obtained raw data were trimmed via Trimmomatic v0.36 (Bolger et al., 2014) with a quality cutoff of 30, a sliding window of 6 bp, and a minimum length cutoff of 100 bp. The clean reads from two metagenomes corresponding were co-assembled via SPAdes v.3.5.0 (Bankevich et al., 2012), binned using MetaBAT (Kang et al., 2015), checked for completeness and contamination using CheckM (Parks et al., 2015), and calculated relative abundance using BBMap (Bushnell, 2014). Phylogenomic trees were built with PhyloPhlAn v0.99 (“-u” option) (Segata et al., 2013). Genes were then annotated using Prokka (Seemann, 2014) and manual curation was performed as described previously (Chen et al., 2020a).
For metatranscriptomics sequencing, total RNA was treated with DNase to remove the residual DNA using an RNase-free DNase set (Qiagen, Hilden, Germany). Ribosomal RNA (rRNA) was removed from the DNase-treated RNA via the Ribo-Zero rRNA Removal Kits (Illumina, San Diego, CA, United States). RNAseq libraries were created using the TruSeq RNA sample prep kit (Illumina, San Diego, CA, United States) with the standard protocol. The sample libraries were sequenced on an Illumina HiSeq 3,000 sequencer. The metatranscriptomics sequences were trimmed as the DNA-trimming step described above and mapped to metagenomic bins using the Bowtie2 aligner (Langmead and Salzberg, 2012). The expression levels of given genes from each bin were calculated separately as reads per kilobase transcript per million reads (RPKM) mapped to the bin averaged from the duplicate samples. In the heatmap illustration, the gene expression levels were further normalized to the median gene expression levels of each bin (RPKM-NM) averaged from the duplicate samples (Nobu et al., 2017). The raw reads of the metagenomics and metatranscriptomics sequencing are accessible at http://bigd.big.ac.cn/gsa, accession number: CRA008529.
The reactor operated continuously for more than 400 days and the methanogenesis kept stable during each stage with different TAN concentrations (N0 ~ N6 stages). The pH was stable at about 7.5 at all stages. From N0 to N2 stages, the biogas production did not decrease, and the acetate fed was completely degraded. From N3, the biogas production decreased slightly, but at the N6 stage, it was about 92% of that at N0 ~ N2 (Figure 1). In biogas, CH4 accounted for 53–59%, CO2 accounted for 41–47%, and the partial pressure of H2 was less than 1 Pa. Acetate built up in the initial period of N5 and N6, but with the extension of the running time, it was degraded. The acetate degradation ratio was high of 99% at the N6 stage. The SS and VSS concentrations increased slightly with increased TAN which was around 1.69 and 0.98 g L−1, respectively, at stage N6 (Figure 1). These results suggested that the methanogenesis was not repressed obviously even at a TAN of 6 g L−1 (free ammonia nitrogen concentration of 0.23 g L−1).
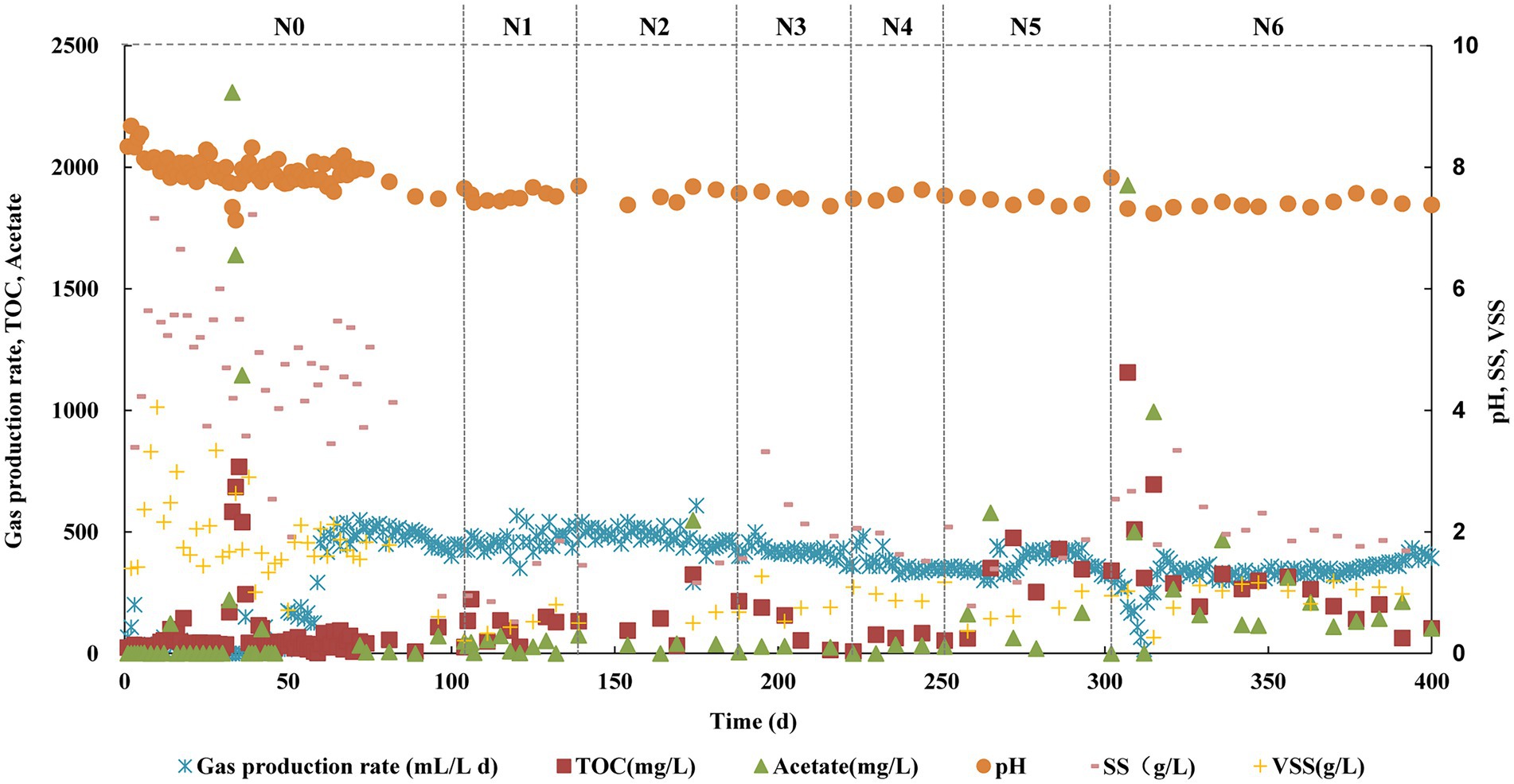
Figure 1. Performance of the anaerobic chemostat fed with acetate as the sole carbon source under different TAN concentrations.
We performed fluorescence observation of the microbial community at each stage. F420, the unique key coenzyme in the hydrogenotrophic methanogenesis pathway, can make cells fluoresce blue-green under UV light at 420 mm wavelength (Braks et al., 1994). During the N0 to N4 stages, the microbial morphology was mainly tubular, and almost no fluorescence cell was detected (Supplementary Figure S2). When the TAN concentration reached 5 g L−1, spherical cells were clearly observed, and a strong fluorescence was detected (Supplementary Figure S2). This result indicated that with the increase of TAN concentration, cells able to produce methane through the hydrogenotrophic pathway probably increased, which also suggested that acetate oxidation probably occurred in the reactor.
The microbial community analysis was carried out using the sludge samples on the last day of each operation stage (N0 ~ N6). Based on 16S ribosomal RNA gene analysis, the structure of the microbial community varied greatly with increased TAN concentration (Supplementary Figure S3). Compared with bacteria, the archaeal populations were significantly inhibited, and the relative abundance decreased to 3.54% during the stage of N3 and N4 (Supplementary Figure S4). As the TAN concentration continued to be increased, the relative abundance of archaea gradually recovered to 18.35%. To further reveal the impact of TAN on microbial communities, the dominant bacterial and archaeal (>1% relative abundance at any stage) communities at the operational taxonomic units (OTU) level were analyzed and Spearman’s correlation coefficients were calculated to illustrate the correlation between microbial community structure and TAN concentration (Figure 2). The dominant bacterial phyla included Bacteroidetes, Synergistetes, Firmicutes, and Spirochaetae (Figure 2A; Supplementary Figure S6A). OTUs in Bacteroidetes prevailed in high TAN conditions, in which the relative abundance of Petrimonas (OTU230) and unclassified Lentimicrobiaceae (OTU281) were significantly positively correlated with TAN concentration and Proteiniphilum (OTU214) was extremely significantly correlated, suggesting the high TAN tolerance of these OTUs. In the Synergistetes phylum, the Thermovirga (OTU244) predominated in the N2 stage (61%), but the relative abundance continued to decrease in the later stages. The most abundant OTU in Firmicutes was Ruminiclostridium (OTU97), which was negatively correlated with TAN concentration, while syntrophic butyrate (Syntrophomonas [OTU233]) and acetate (Tepidanaerobacter syntrophicus [OTU176])-oxidizing bacteria were positively correlated with TAN. The main genera in Spirochaetae were unclassified Spirochaetaceae (OTU4) and Sphaerochaeta (OTU275, 202, 220, and 222), their responses to TAN stress are diametrically opposite. Unclassified Spirochaetaceae (OTU4) was strongly inhibited by TAN, whereas Sphaerochaeta was not.
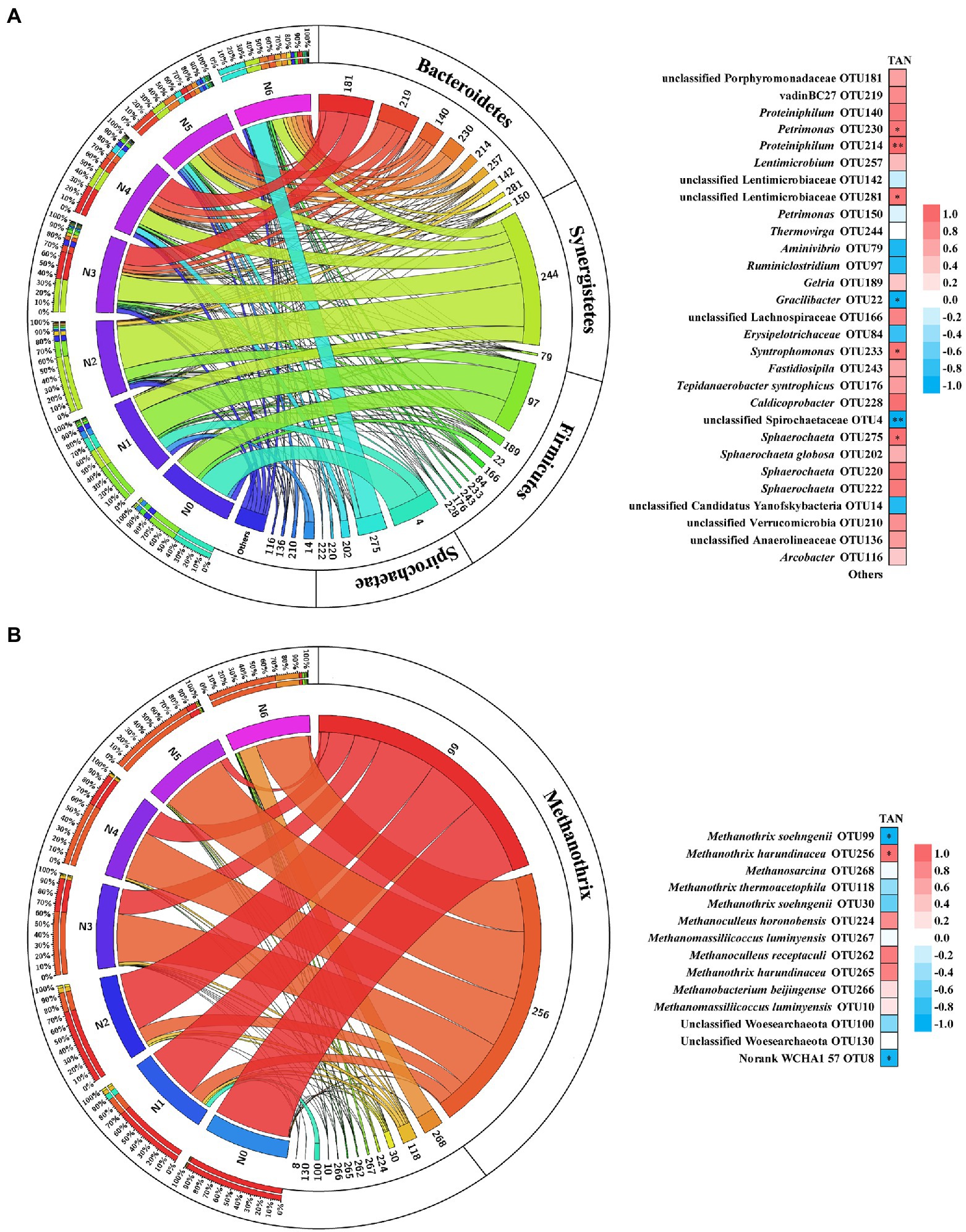
Figure 2. Relative abundance in different stages and the Spearman’s correlation with TAN concentration of bacterial (A) and archaeal (B) OTUs. Ribbon width represents the relative abundance. Red represents a positive correlation; blue represents a negative correlation; significant correlations (value of p ≤ 0.05) are labeled by *; extremely significant correlations (value of p ≤ 0.01) are labeled by **.
Regarding the archaeal community, the OTUs were determined at the species level by phylogenetic analysis (Supplementary Figure S5). Among them, Methanothrix was the most abundant methanogen across all the stages, the genus mainly included M. soehngenii (OTU99) and M. harundinacea (OTU256), which showed totally opposite trends (Figure 2B). With the increase of TAN concentration, the dominant methanogen in the chemostat was gradually replaced from M. soehngenii (OTU99) to M. harundinacea (OTU256), suggesting that M. harundinacea was more tolerant to TAN suppression. Moreover, the diversity of methanogens increased significantly with the increase in TAN concentration. Methanosarcina (OTU 268) was only detected in the N6 stage, and its relative abundance accounted for 23.5% of total archaeal species (Figure 2B; Supplementary Figure S6B).
To profile the metabolic capability of TAN-tolerant bacteria and methanogens, metagenomics analysis was performed on microbial community samples taken from the chemostat at the N6 stage on two different dates. Overall, 34 Gbp metagenomic clean sequences were obtained. Illumina paired-end reads from the two duplicate samples were co-assembled, and binning the assembled contigs of metagenomes yielded 89 bins. Among these, 45 bacterial and 6 archaeal bins which have at least 72% genome completeness and < 6.5% contamination as estimated by CheckM (Parks et al., 2015) were analyzed (Supplementary Figures S7, S8; Supplementary Table S1). To further reveal the metabolic behavior of the microbial populations, 56.6 million metatranscriptomic reads (4.5 Gbp) were produced and mapped to the metagenomic bins. The percentages of metatranscriptomic reads mapped to dominant genomes were, respectively, analyzed at two sampling time points, and the two transcriptomics analyses displayed similar results (Figure 3). Based on mapping meta-omic reads to the obtained bins, the bacterial populations retrieved accounted for 86 and 60% of the metagenomic and metatranscriptomic reads, respectively (Supplementary Figure S9). Among all bacterial phyla, Bacteroidetes represented major fractions of the 16S rRNA (40.85%), metagenomes (49.59%), and metatranscriptomes (39.15%) at the N6 stage (Figure 3; Supplementary Figures S6A). The major genera in Bacteroidetes were Proteiniphilum (Bin76, Bin79, and Bin41), unclassified Bacteroidetes (Bin83 and Bin32), and Paludibacter (Bin 89; (Figure 3). In addition, as observed in the 16S rRNA gene analysis, populations associated with known syntrophic fatty acid-oxidizing bacteria (Tepidanaerobacter Bin10 and Syntrophomonas Bin24) were detected in Firmicutes (Figures 2A, 3).
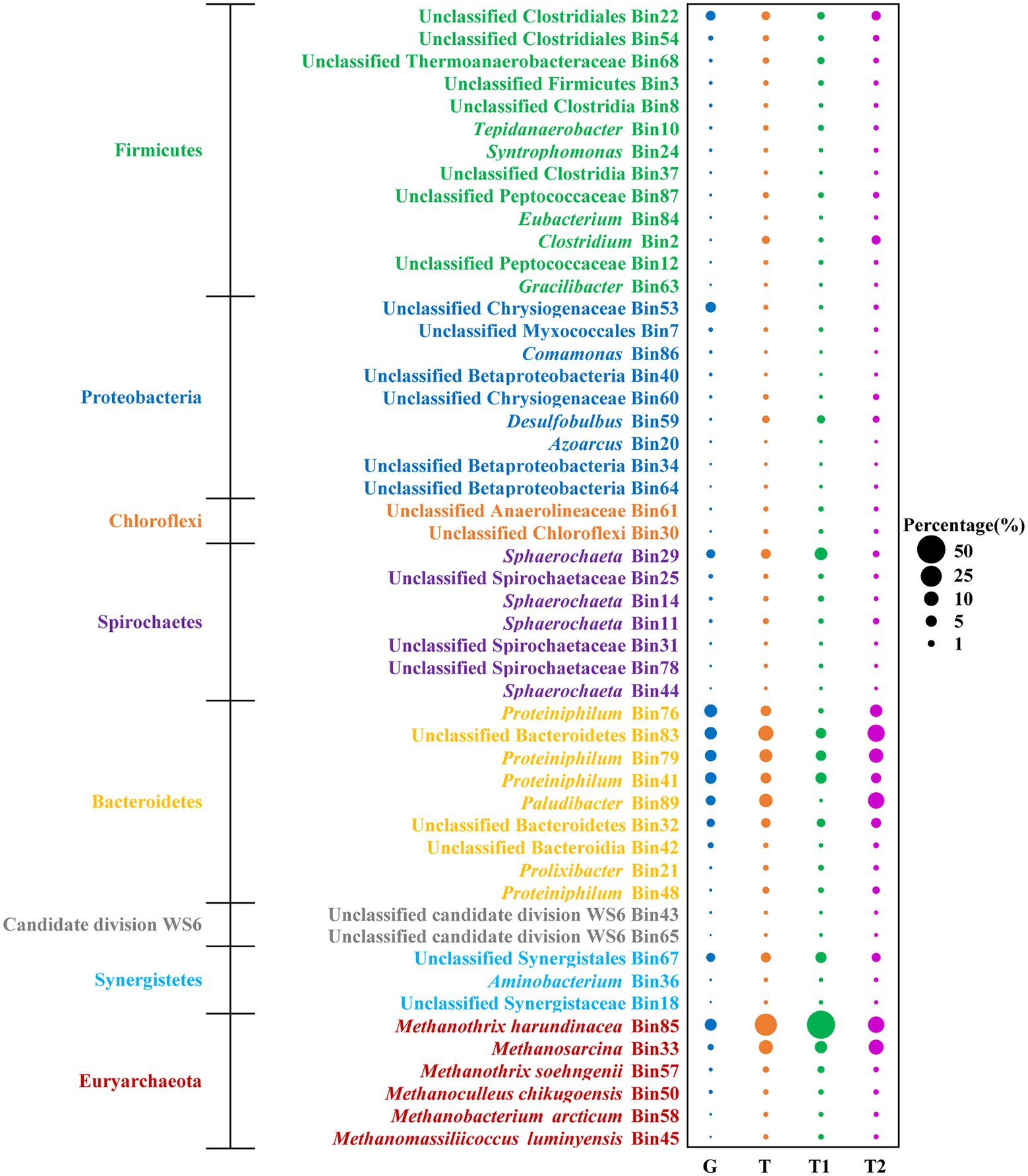
Figure 3. Metagenomic abundance (G) and percentage of metatranscriptomic reads (T) of bacterial and archaeal bins. The corresponding abundance of genomic bins in the N6 stage are estimated from their metagenomic coverage calculated as the percentage of metagenomics (MG) reads mapped to each genome relative to the total reads mapped to all constructed bacterial and archaeal genomes. The estimated activity of these genomes is shown as the percentage of metatranscriptomic (MT) reads mapped to each genome relative to the total reads mapped to all constructed bacterial and archaeal genomes. G, total MG reads; T, total MT reads; T1, MT reads of sampling time point 1; T2, MT reads of sampling time point 2.
As for the methanogen, M. harundinacea (Bin85) was the most abundant, accounting for 9.10 and 28.07% of the metagenomic and metatranscriptomic reads, respectively (Figure 3). The second dominant methanogen was Methanosarcina (Bin33) with 1.95% metagenomic abundance and 8.98% metatranscriptomic abundance. It is worth mentioning that M. soehngenii (Bin57) only accounted for 0.58 and 0.65% of the metagenomic and metatranscriptomic reads, consistent with the results of 16S rRNA gene analysis (Figures 2B, 3).
The community hosted diverse methanogens, including M. harundinacea (Bin85), M. soehngenii (Bin57), Methanosarcina (Bin33), Methanoculleus chikugoensis (Bin50), Methanobacterium arcticum (Bin58), and Methanomassiliicoccus luminyensis (Bin45) (Supplementary Figure S8; Supplementary Table S1). These methanogens have distinct substrate spectrums for methane production, including the acetate cleavage pathway, CO2 reduction pathway, and methyl cleavage pathway (Figure 4A; Supplementary Table S2). During methanogenesis, methanogens synthesize ATP via a transmembrane Na+/H+ gradient formed by an electron transfer chain (Figure 4B; Supplementary Table S3). To further understand the metabolic profile of methanogens, we analyzed the methanogenic and energy metabolic pathways of target bins by comparison with known pathways in the UniProt database, then mapped the metatranscriptomic reads to bins and calculate RPKM-NM to analyze the expression level of target pathways (Figure 4C).
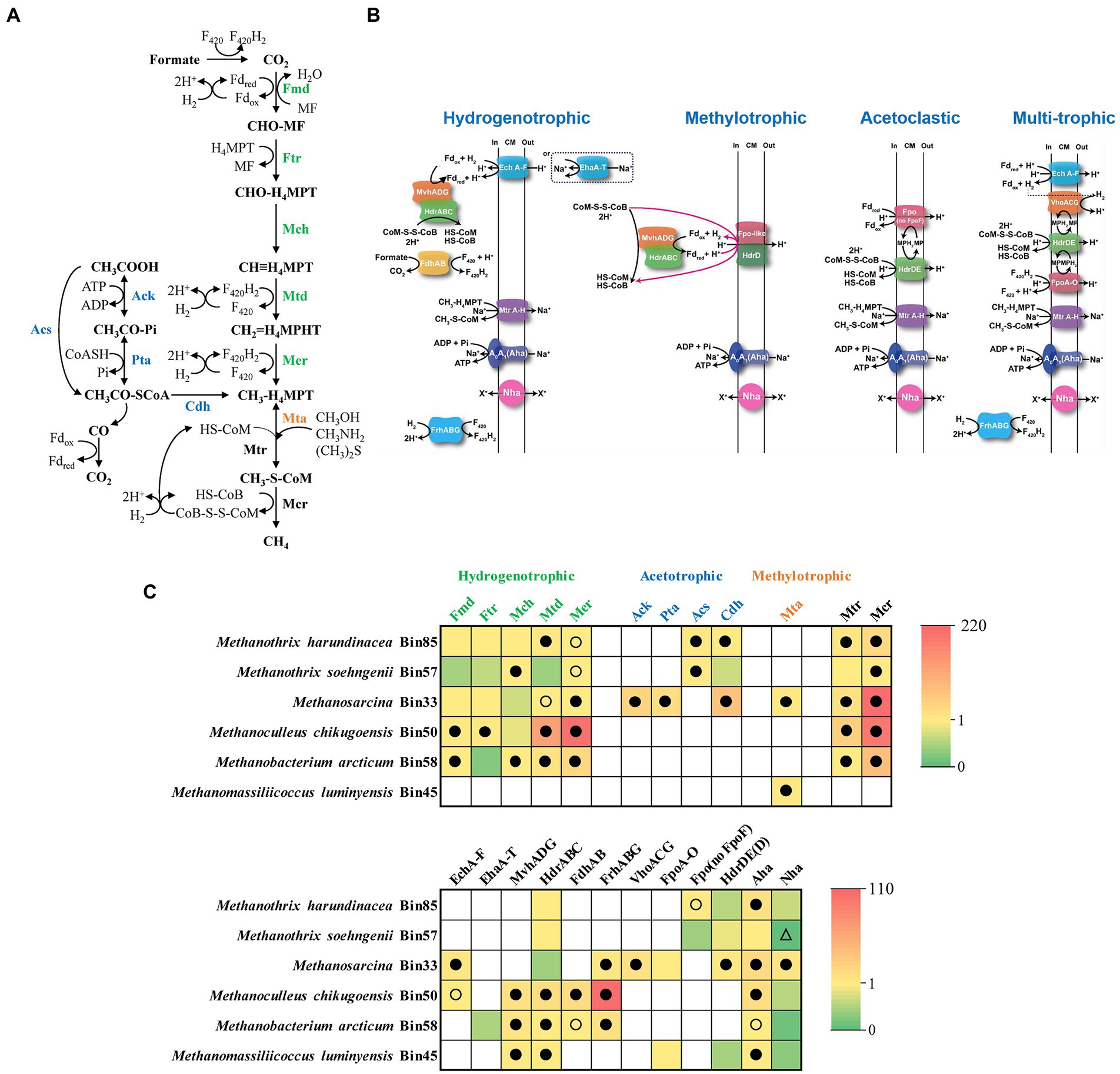
Figure 4. Metabolic pathways of methanogenesis (A), methanogenic electron transfer (B), and the gene expression level (C) of methanogens in the N6 stage. The gene expression levels are calculated as reads per kilobase of transcript per million reads mapped to individual bins (RPKM) normalized to the median gene expression for the corresponding bin (RPKM-NM) averaged from duplicate samples. Pathways containing genes with RPKM-NM of 0 and greater than the octile and quartile are marked (open triangle, filled and open dots, respectively). Enzyme abbreviations and their corresponding genes are elaborated in Supplementary Tables S2, S3.
Specifically, M. harundinacea (Bin85) and Methanosarcina (Bin33) expressed acetate utilization pathway highly (top octile of expressed genes in the corresponding bin) and M. soehngenii (Bin57) expressed at a lower level (Figures 4A,C; Supplementary Table S2). The M. harundinacea and Methanosarcina correspondingly highly expressed (top quartile or octile) genes for acetoclastic methanogenesis mediated by Fpo (no FpoF) and H2-cycling (via EchA-F and VhoACG; Figures 4B,C; Supplementary Table S3; Welte and Deppenmeier, 2014).
Bins of hydrogenotrophic methanogens, such as M. chikugoensis (Bin50), M. arcticum (Bin58), and Methanosarcina (Bin33), encode and express genes for CO2-reducing methanogenesis (Figures 4A,C; Supplementary Table S2). The Methanoculleus highly expressed (top quartile or octile) genes for the oxidation of H2 (MvhADG-HdrABC and FrhABG) and formate (FdhAB; Figures 4B,D; Supplementary Table S3). Another H2-oxidizing population, M. luminyensis (Bin45), highly expressed genes for coupling of H2 oxidation with the reduction of methanol and dimethylamine to methane (Figure 4; Supplementary Tables S2, S3). Interestingly, Methanothrix is a well-known obligate acetotrophic methanogen (Hattori, 2008), but we found the expression of the CO2-reducing methanogenesis pathway was high in Methanothrix, especially in Bin85 (Figures 4A,C; Supplementary Table S2; Holmes et al., 2017). However, Methanothrix did not express the key genes for utilizing H2 (Figures 4B,C), it may directly use extracellular electrons for reducing CO2 to CH4 (Rotaru et al., 2014; Zhao et al., 2020). Thus, the acetate-fed chemostat was likely to produce methane not only via acetoclastic pathway but also possible via the CO2 reduction pathway under high TAN-suppressed conditions.
As discussed above, in the acetate-fed and TAN-suppressed methanogenic system, besides directly decomposing acetate into methane, methanogens can also use the products (electron, H2, and CO2) of syntrophic acetate oxidizers to generate methane. There are currently two acetate oxidation pathways reported (Zhu et al., 2020), the reversed Wood–Ljungdahl pathway and the glycine cleavage pathway, which are different in the process from CH3-CO-S-CoA to CH2 = THF (Figure 5A). To systematically analyze the potential acetate-oxidizing bacteria in the system, 45 bacterial bins were annotated and then compared with the known acetate-oxidizing pathways in the UniProt database (Supplementary Figure S10; Supplementary Table S4). We screened out 20 bacterial bins with complete acetate oxidation pathway or metagenomic abundance greater than 1%, then mapped the metatranscriptomic reads to bins and calculated RPKM-NM to analyze the expression levels of genes related to acetate oxidation, H2/formate metabolism, and electron transport (Figure 5).
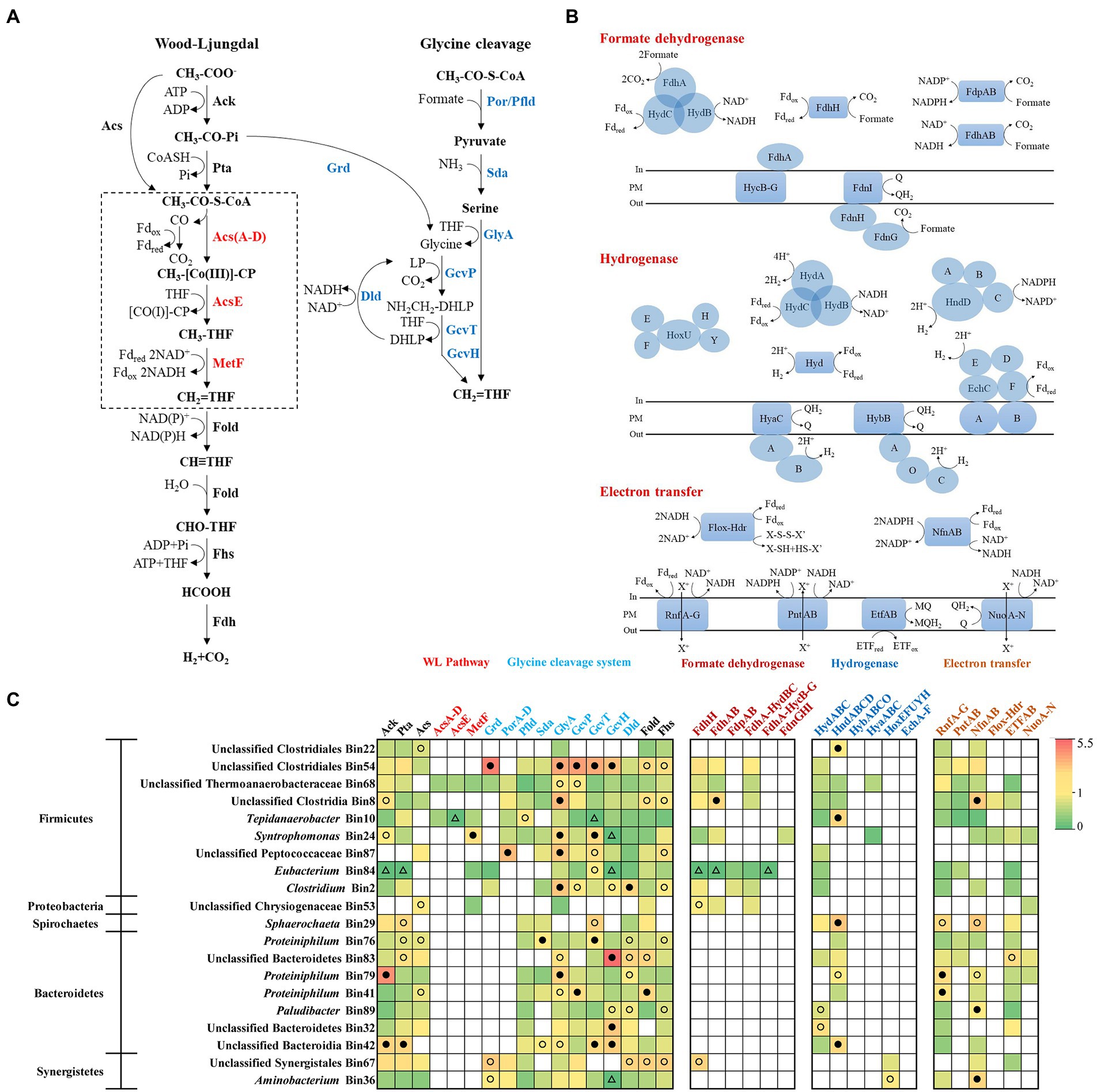
Figure 5. Metabolic pathways of acetate oxidation (A), H2/formate metabolism and electron transfer (B), and the gene expression level (C) of syntrophs which may syntrophically degrade acetate in the N6 stage. The gene expression levels are calculated as reads per kilobase of transcript per million reads mapped to individual bins (RPKM) normalized to the median gene expression for the corresponding bin (RPKM-NM) averaged from duplicate samples. Pathways containing genes with RPKM-NM of 0 and greater than the octile and quartile are marked (open triangle, filled and open dots, respectively). Enzyme abbreviations and their corresponding genes are elaborated in Supplementary Tables S4, S5.
Within the acetate-degrading community, unclassified Thermoanaerobacteraceae (Bin68) encode all genes for both reversed Wood–Ljungdahl and glycine cleavage pathways with high expression of GlyA and GcvP (top quartile of expressed genes in the corresponding bin; Figures 5A,C; Rui et al., 2011). A population (Bin10) related to a known acetate-degrading genus, Tepidanaerobacter, expressed conversion of acetate to formate through the reversed Wood–Ljungdahl pathway (Figures 5A,C; Westerholm et al., 2011). Another population (Bin54) belongs to Clostridiales expressed genes for acetate degradation (Grd, GlyA, GcvP, GcvT, and GcvH) at high levels (top octile; Figure 5C; Westerholm et al., 2018). Notably, Proteiniphilum (Bin76, Bin79, and Bin41) had the highest abundance in both metagenomic and metatranscriptomic reads (Figure 3) and highly expressed genes of glycine cleavage pathway (top quartile or octile; Figures 5A,C); thus, these populations are likely novel acetate degraders.
Multiple genes/gene clusters encoding formate dehydrogenase and hydrogenase are detected in the aforementioned syntrophs for formate and H2 generation. These enzymes are necessary for the reoxidation of reducing equivalents (i.e., menaquinone, NADH, NADPH, and reduced ferredoxin [Fd]) generated during acetate oxidation (Figures 5B; Supplementary Table S5). For formate metabolism, all syntrophic metabolizers in Firmicutes expressed Fd-dependent formate dehydrogenases (FdhH), NADH-dependent formate dehydrogenases (FdhAB), and putative electron-confurcating formate dehydrogenases (FdhA-HydBC) except for Tepidanaerobacter (Bin10) and unclassified Peptococcaceae (Bin87; Figures 5B,C; Supplementary Figure S11). Among them, unclassified Clostridia Bin8 highly expressed FdhAB (top octile; Figure 5C). For H2 generation, all syntrophic metabolizers in Firmicutes (except for Syntrophomonas) expressed cytoplasmic [FeFe]-type electron-confurcating hydrogenases (HydABC) (Figures 5B,C; Supplementary Figure S11). These hydrogenases catalyze the thermodynamically favorable production of H2 from Fdred to drive the unfavorable production of H2 from NADH (Buckel and Thauer, 2013). We detected high expression of [FeFe]-type NADP+-dependent hydrogenases (HndABCD) in unclassified Clostridiales (Bin22), Tepidanaerobacter (Bin10), Sphaerochaeta (Bin29), Proteiniphilum (Bin79), and unclassified Bacteroidia (Bin42) (top octile or quartile; Figure 5C; de Luca et al., 1998).
To support the thermodynamically challenging H2 and formate generation, the syntrophs also expressed energy-conserving electron transfer enzymes. The Proteiniphilum populations (Bin79 and Bin41) highly expressed (top octile) the membrane-bound Rhodobacter nitrogen fixation complex (RnfA-G), which can either extrude cation and transfer an electron from Fdred to NAD+ to gain energy or transport cation inward and transfer electron from NADH to Fdox to drive electron-confurcating hydrogen or formate generation (Figures 5B,C; Biegel et al., 2011). In addition, Proteiniphilum (Bin79) also highly expressed (top quartile) the NADP+-Fdred oxidoreductase (NfnAB), which transfers electrons from NADPH to NAD+ and Fdox via electron disproportionation (Figures 5B,C; Wang et al., 2010).
Since the ROS-detoxification mechanism supported the adaptation of Proteiniphilum species (Wu et al., 2021) and ammonia caused oxidative stress in methanogens (Kato et al., 2014; Li et al., 2022), we also examined the occurrence of antioxidant genes in target bins (Figure 6; Supplementary Table S6). All the populations of Proteiniphilum highly expressed genes (top octile of expressed genes in the corresponding bin) encoding superoxide dismutase (Sod) and peroxiredoxins (Prx), respectively, which belong to energy-free ROS scavenger (Supplementary Table S6; Martins et al., 2019). For methanogens, in addition to detecting highly expressed Sod and Prx, we also identified the expression of energy-dependent ROS scavengers (superoxide reductase, Sor; rubrerythrin, Rbr) (Figure 6; Supplementary Table S6), which require additional energy sources with F420H2 as the ultimate electron donor (Martins et al., 2019). Methanothrix harundinacea (Bin85) and Methanosarcina (Bin33) which represented major fractions of the metagenomes and metatranscriptomes in methanogens highly expressed Sor and Rbr (top octile; Figures 3, 6). Thus, the predominant bacterial genus Proteiniphilum and methanogens in the TAN-suppressed system possessed mechanisms of anti-oxidative stress thereby providing a selective advantage under suppressive conditions.

Figure 6. Gene expression level for anti-oxidative stress in syntrophs and methanogens. The gene expression levels are calculated as reads per kilobase of transcript per million reads mapped to individual bins (RPKM) normalized to the median gene expression for the corresponding bin (RPKM-NM) averaged from duplicate samples. Pathways containing genes with RPKM-NM of 0 and greater than the octile and quartile are marked (open triangle, filled and open dots, respectively). Enzyme abbreviations and their corresponding genes are elaborated in Supplementary Table S6.
It was generally reported that TAN concentrations of around 1.7–1.8 g L−1 were completely inhibitory to AD under unacclimated inoculation conditions, and the TAN level could increase up to 5 g L−1 with acclimation (Albertson, 1961; Melbinger et al., 1971). In this study, the continuous anaerobic digestion reactor fed with acetate as the sole carbon source was operated stably at a high TAN concentration (6 g L−1) after a long-term continuous acclimation with progressively increasing TAN. During acclimation, both bacterial and archaeal communities underwent significant species turnover (Figure 2; Supplementary Figure S3). Thus, we evaluated why under such strong TAN inhibition, Methanothrix harundinacea was able to dominate methanogens, and the only dominant bacteria were Proteiniphilum species.
As for methanogens, the only dominant genus was Methanothrix during the acclimation, although multitrophic Methanosarcina appeared at the N6 stage, Methanothrix still dominated. This result suggests that acetate cleavage was probably the main way of methanogenesis under high TAN stress, which to some extent contradicts the previous studies that the methanogenic pathway was transformed from acetotrophic to hydrogenotrophic under ammonia stress (Schnürer and Nordberg, 2008; Yenigün and Demirel, 2013; Gao et al., 2015; Jiang et al., 2018). In addition, based on the fluorescence of F420 observed in methanogens and the increasing relative abundance of bacteria (Supplementary Figures S2, S4), it is reasonable to speculate that a part of bacteria carried out acetate oxidative metabolism in the high TAN stages. To clarify this contradiction, further validation of the methanogenesis pathway with meta-omics was conducted.
Methanothrix harundinacea (Bin85) and Methanosarcina (Bin33) predominated in methanogens at the N6 stage (Figure 3). Through meta-omics analysis, we found that M. harundinacea highly expressed both CO2 reduction and acetate cleavage pathways, and the expression of acetate cleavage pathway was higher (Figure 4C), consistent with existing research results (Holmes et al., 2017). However, we did not detect the key genes responsible for utilizing H2 in M. harundinacea (Figure 4C), suggesting the possibility that M. harundinacea uses extracellular electrons directly to reduce CO2 (Rotaru et al., 2014; Zhao et al., 2020). It has been found that Methanothrix can reduce CO2 to methane by directly accepting electrons from Geobacter in an AD reactor supplied with granular activated carbon (Yang et al., 2019). It is possible that M. harundinacea is similar to multitrophic Methanosarcina, which may gain more energy through multiple pathways to combat ammonia stress (Hao et al., 2015). Though M. soehngenii is reported also be able to receive extracellular electrons for reducing CO2 to CH4 (Yang et al., 2019), possibly due to its much lower ammonia resistance (much lower expression of Sor and Rbr; Figure 6), it lost competitiveness compared with M. harundinacea. It has been reported that M. soehngenii can be completely inhibited at a TAN concentration of 560 mg L−1 in pure culture (Sprott and Patel, 1986). The reason why previous studies reported that the methanogenesis pathway was transformed from acetotrophic to hydrogenotrophic under ammonia stress is probably that there was no strong ammonia-tolerant M. harundinacea in their systems or the lack of acclimatization to ammonia (Zhang et al., 2014; Jiang et al., 2018).
Furthermore, the key genes for both energy-free and energy-dependent anti-oxidative stress mechanisms were detected in methanogens, which may play roles in resistance to the oxidative stress caused by ammonia, thereby beneficial to adapt to a high ammonia condition (Wu et al., 2021). In our study, only the M. harundinacea and Methanosarcina highly expressed energy-dependent anti-oxidative stress genes (sor and rbr), which made them predominant methanogens at the N6 stage. The dominant Methanothrix and Methanosarcina yielded methane possibly through both acetate cleavage and CO2 reduction, and the CO2 reduction pathway might provide energy for anti-oxidative stress metabolism, thereby achieving ammonia-inhibition resistance. However, as the expression of genes in a pathway does not mean the pathway has metabolic activity, further studies are needed in the future to obtain direct evidence that the CO2 reduction pathway of the two dominant methanogens has activity. In addition, further studies using pure cultures of these methanogens should be conducted to obtain physiological and biochemical evidence of their ammonia resistance.
As for the bacteria, Petrimonas, Proteiniphilum, unclassified Lentimicrobiaceae, Syntrophomonas, and Sphaerochaeta showed a significantly positive correlation trend with TAN concentration (Figure 2). As discussed above, methanogens need electrons from syntrophic acetate-oxidizing bacteria for CO2 reduction. According to the metagenomic analysis, we found several bins with acetate-oxidizing potential in Firmicutes, such as unclassified Thermoanaerobacteraceae (Bin68), unclassified Clostridiales (Bin54), and Tepidanaerobacter (Bin10), etc., consistent with previous studies (Hattori, 2008; Westerholm et al., 2018). Interestingly, Proteiniphilum, the only dominant bacterial genus belonging to Bacteroidetes, also fully expressed the acetate oxidation pathway (Figure 5C). At present, it is believed that the function of Proteiniphilum species is only hydrolysis and fermentation, its acetate oxidation ability is a new discovery that has not been found in previous studies (Hahnke et al., 2016; Wu et al., 2021). It is reasonable to assume that Proteiniphilum represents a broad range of metabolic capabilities including hydrolysis and fermentation and acetate oxidation to gain more energy, so it can dominate in inhibitory environments. We found a complete glycine cleavage pathway in the genomes of isolated P. acetatigenes and P. saccharofermentans strains (data not shown). Isolates of P. acetatigenes were found able to reduce CO2 with H2 to produce acetate (Kim et al., 2020), suggesting the expression of the glycine cleavage pathway. Some syntrophic acetate oxidizers including Thermacetogenium phaeum, Clostridium ultunense, etc., are also able to act as reductive acetogens (Hattori, 2008). Therefore, based on our analysis results and these reports, Proteiniphilum in our acetate-fed chemostat largely acted as syntrophic acetate oxidizers. Moreover, Proteiniphilum highly expressed the genes for energy-free anti-oxidative stress, making it more adaptable to ammonia stress, consistent with previous studies (Wu et al., 2021). Multiple studies have shown that Proteiniphilum can prevail in inhibitory conditions, such as high ammonia, high salt, and high oxygen (He et al., 2017; Lee et al., 2019; Wu et al., 2021). To further determine the acetate oxidation capacity of Proteiniphilum, further studies using pure monoculture of Proteiniphilum or co-culture with methanogens can be performed in the future.
This study revealed the microbial community composition and methanogenesis metabolic networks in the acetate-fed chemostat under high TAN conditions. Through gradual acclimation, the chemostat was able to operate stably at a TAN concentration of 6 g L−1. Methanothrix predominated in methanogens all the time, in which the dominant species was gradually replaced from M. soehngenii to M. harundinacea with the increasing TAN, suggesting a stronger TAN tolerance of M. harundinacea. Furthermore, we found that the genes involved in the CO2-reducing methanogenesis pathway in M. harundinacea had high expression levels, suggesting the possibility that Methanothrix might directly use extracellular electrons from syntrophic bacteria for reducing CO2 to CH4. During acclimation, the bacterial community underwent significant species turnover. We found diverse potential acetate-oxidizing bacteria under high TAN conditions, including the only dominant bacteria, Proteiniphilum, which highly expressed genes for acetate degradation. Thus, Proteiniphilum populations in the chemostat were possibly novel acetate degraders. The findings of this study provide a basis for improving ammonia tolerance and operational stability of AD.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA524473; http://bigd.big.ac.cn/gsa, CRA008529.
GF operated reactor, analyzed data, and wrote the manuscript. YZ and H-ZW operated reactor and analyzed data. Y-TC analyzed the data. Y-QT directed experiments, data analysis, and manuscript revision. All authors contributed to the article and approved the submitted version.
This study was financially supported by the National Key R&D Program of China (2022YFE0108500) and the National Natural Science Foundation of China (No. 51678378).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1098814/full#supplementary-material
Albertson, O. E. (1961). Ammonia nitrogen and the anaerobic environment. J. Water Pollut. Control Fed. 33, 978–995.
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Biegel, E., Schmidt, S., González, J. M., and Müller, V. (2011). Biochemistry, evolution and physiological function of the Rnf complex, a novel ion-motive electron transport complex in prokaryotes. Cell. Mol. Life Sci. 68, 613–634. doi: 10.1007/s00018-010-0555-8
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Braks, I. J., Hoppert, M., Roge, S., and Mayer, F. (1994). Structural aspects and immunolocalization of the F420-reducing and non-F420-reducing hydrogenases from Methanobacterium thermoautotrophicum Marburg. J. Bacteriol. 176, 7677–7687. doi: 10.1128/jb.176.24.7677-7687.1994
Buckel, W., and Thauer, R. K. (2013). Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochim. Biophys. Acta 1827, 94–113. doi: 10.1016/j.bbabio.2012.07.002
Bushnell, B. (2014). BBMap: A Fast, Accurate, Splice-Aware Aligner. Berkeley, CA: Lawrence Berkeley National Laboratory.
Chen, Y.-T., Zeng, Y., Li, J., Zhao, X.-Y., Yi, Y., Gou, M., et al. (2020a). Novel syntrophic isovalerate-degrading bacteria and their energetic cooperation with methanogens in methanogenic chemostats. Environ. Sci. Technol. 54, 9618–9628. doi: 10.1021/acs.est.0c01840
Chen, Y.-T., Zeng, Y., Wang, H.-Z., Zheng, D., Kamagata, Y., Narihiro, T., et al. (2020b). Different interspecies electron transfer patterns during mesophilic and thermophilic syntrophic propionate degradation in chemostats. Microb. Ecol. 80, 120–132. doi: 10.1007/s00248-020-01485-x
de Luca, G., de Philip, P., Rousset, M., Belaich, J. P., and Dermoun, Z. (1998). The NADP-reducing hydrogenase of Desulfovibrio fructosovorans: evidence for a native complex with hydrogen-dependent methyl-viologen-reducing activity. Biochem. Biophys. Res. Commun. 248, 591–596. doi: 10.1006/bbrc.1998.9022
Gallert, C., Bauer, S., and Winter, J. (1998). Effect of ammonia on the anaerobic degradation of protein by a mesophilic and thermophilic biowaste population. Appl. Microbiol. Biotechnol. 50, 495–501. doi: 10.1007/s002530051326
Gao, S., Zhao, M., Chen, Y., Yu, M., and Ruan, W. (2015). Tolerance response to in situ ammonia stress in a pilot-scale anaerobic digestion reactor for alleviating ammonia inhibition. Bioresour. Technol. 198, 372–379. doi: 10.1016/j.biortech.2015.09.044
Griffiths, R. I., Whiteley, A. S., O'Donnell, A. G., and Bailey, M. J. (2000). Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA-and rRNA-based microbial community composition. Appl. Environ. Microbiol. 66, 5488–5491. doi: 10.1128/aem.66.12.5488-5491.2000
Hahnke, S., Langer, T., Koeck, D. E., and Klocke, M. (2016). Description of Proteiniphilum saccharofermentans sp nov., Petrimonas mucosa sp. nov., and Fermentimonas caenicola gen. Nov., sp nov., isolated from mesophilic laboratory-scale biogas reactors, and emended description of the genus Proteiniphilum. Int. J. Syst. Evol. Microbiol. 66, 1466–1475. doi: 10.1099/ijsem.0.000902
Hansen, K. H., Angelidaki, I., and Ahring, B. K. (1998). Anaerobic digestion of swine manure: inhibition by ammonia. Water Res. 32, 5–12. doi: 10.1016/s0043-1354(97)00201-7
Hao, L., Lue, F., Mazeas, L., Quemener, E. D.-L., Madigou, C., Guenne, A., et al. (2015). Stable isotope probing of acetate fed anaerobic batch incubations shows a partial resistance of acetoclastic methanogenesis catalyzed by Methanosarcina to sudden increase of ammonia level. Water Res. 69, 90–99. doi: 10.1016/j.watres.2014.11.010
Hattori, S. (2008). Syntrophic acetate-oxidizing microbes in methanogenic environments. Microbes Environ. 23, 118–127. doi: 10.1264/jsme2.23.118
He, Q., Li, L., and Peng, X. (2017). Early warning indicators and microbial mechanisms for process failure due to organic overloading in food waste digesters. J. Environ. Eng. 143:04017077. doi: 10.1061/(asce)ee.1943-7870.0001280
Holmes, D. E., Shrestha, P. M., Walker, D. J. F., Dang, Y., Nevin, K. P., Woodard, T. L., et al. (2017). Metatranscriptomic evidence for direct interspecies electron transfer between Geobacter and Methanothrix species in methanogenic rice paddy soils. Appl. Environ. Microbiol. 83:e00223-17. doi: 10.1128/AEM.00223-17
Jiang, Y., Banks, C., Zhang, Y., Heaven, S., and Longhurst, P. (2018). Quantifying the percentage of methane formation via acetoclastic and syntrophic acetate oxidation pathways in anaerobic digesters. Waste Manag. 71, 749–756. doi: 10.1016/j.wasman.2017.04.005
Kang, D. D., Froula, J., Egan, R., and Wang, Z. (2015). MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3:e1165. doi: 10.7717/peerj.1165
Kato, S., Sasaki, K., Watanabe, K., Yumoto, I., and Kamagata, Y. (2014). Physiological and transcriptomic analyses of the thermophilic, aceticlastic methanogen Methanosaeta thermophila responding to ammonia stress. Microbes Environ. 29, 162–167. doi: 10.1264/jsme2.ME14021
Kim, S., Lindner, S. N., Aslan, S., Yishai, O., Wenk, S., Schann, K., et al. (2020). Growth of E. coli on formate and methanol via the reductive glycine pathway. Nat. Chem. Biol. 16, 538–545. doi: 10.1038/s41589-020-0473-5
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lee, J., Koo, T., Yulisa, A., and Hwang, S. (2019). Magnetite as an enhancer in methanogenic degradation of volatile fatty acids under ammonia-stressed condition. J. Environ. Manag. 241, 418–426. doi: 10.1016/j.jenvman.2019.04.038
Li, J., Ran, X., Zhou, M., Wang, K., Wang, H., and Wang, Y. (2022). Oxidative stress and antioxidant mechanisms of obligate anaerobes involved in biological waste treatment processes: a review. Sci. Total Environ. 838:156454. doi: 10.1016/j.scitotenv.2022.156454
Mackie, R. I., and Bryant, M. P. (1981). Metabolic-activity of fatty acid-oxidizing bacteria and the contribution of acetate, propionate, butyrate, and CO2 to methanogenesis in cattle waste at 40-degrees-c and 60-degrees-c. Appl. Environ. Microbiol. 41, 1363–1373. doi: 10.1128/aem.41.6.1363-1373.1981
Martins, M. C., Romão, C. V., Folgosa, F., Borges, P. T., Frazão, C., and Teixeira, M. (2019). How superoxide reductases and flavodiiron proteins combat oxidative stress in anaerobes. Free Radic. Biol. Med. 140, 36–60. doi: 10.1016/j.freeradbiomed.2019.01.051
McCarty, P. L., and McKinney, R. E. (1961). Salt toxicity in anaerobic digestion. J. Water Pollut. Control Fed. 33, 399–415.
Melbinger, N., Donnellon, J., and Zablatzky, H. R. (1971). Toxic effects of ammonia nitrogen in high-rate digestion [with discussion]. J. Water Pollut. Control Fed. 43, 1658–1670.
Nobu, M. K., Narihiro, T., Liu, M., Kuroda, K., Mei, R., and Liu, W. T. (2017). Thermodynamically diverse syntrophic aromatic compound catabolism. Environ. Microbiol. 19, 4576–4586. doi: 10.1111/1462-2920.13922
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.7287/peerj.preprints.554v1
Rotaru, A.-E., Shrestha, P. M., Liu, F., Shrestha, M., Shrestha, D., Embree, M., et al. (2014). A new model for electron flow during anaerobic digestion: direct interspecies electron transfer to Methanosaeta for the reduction of carbon dioxide to methane. Energy Environ. Sci. 7, 408–415. doi: 10.1039/c3ee42189a
Rui, J., Qiu, Q., and Lu, Y. (2011). Syntrophic acetate oxidation under thermophilic methanogenic condition in Chinese paddy field soil. FEMS Microbiol. Ecol. 77, 264–273. doi: 10.1111/j.1574-6941.2011.01104.x
Schnürer, A., and Nordberg, Å. (2008). Ammonia, a selective agent for methane production by syntrophic acetate oxidation at mesophilic temperature. Water Sci. Technol. 57, 735–740. doi: 10.2166/wst.2008.097
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Segata, N., Börnigen, D., Morgan, X. C., and Huttenhower, C. (2013). PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 4, 2304–2311. doi: 10.1038/ncomms3304
Sprott, G. D., and Patel, G. B. (1986). Ammonia toxicity in pure cultures of methanogenic bacteria. Syst. Appl. Microbiol. 7, 358–363. doi: 10.1016/s0723-2020(86)80034-0
Wang, S., Huang, H., Moll, J., and Thauer, R. K. (2010). NADP+ reduction with reduced ferredoxin and NADP+ reduction with NADH are coupled via an electron-bifurcating enzyme complex in Clostridium kluyveri. J. Bacteriol. 192, 5115–5123. doi: 10.1128/jb.00612-10
Welte, C., and Deppenmeier, U. (2014). Bioenergetics and anaerobic respiratory chains of aceticlastic methanogens. Biochim. Biophys. Acta 1837, 1130–1147. doi: 10.1016/j.bbabio.2013.12.002
Westerholm, M., Leven, L., and Schnurer, A. (2012). Bioaugmentation of syntrophic acetate-oxidizing culture in biogas reactors exposed to increasing levels of ammonia. Appl. Environ. Microbiol. 78, 7619–7625. doi: 10.1128/aem.01637-12
Westerholm, M., Mueller, B., Singh, A., Karlsson Lindsjo, O., and Schnurer, A. (2018). Detection of novel syntrophic acetate-oxidizing bacteria from biogas processes by continuous acetate enrichment approaches. Microb. Biotechnol. 11, 680–693. doi: 10.1111/1751-7915.13035
Westerholm, M., Roos, S., and Schnurer, A. (2011). Tepidanaerobacter acetatoxydans sp. nov., an anaerobic, syntrophic acetate-oxidizing bacterium isolated from two ammonium-enriched mesophilic methanogenic processes. Syst. Appl. Microbiol. 34, 260–266. doi: 10.1016/j.syapm.2010.11.018
Wu, Z., Nguyen, D., Lam, T. Y., Zhuang, H., Shrestha, S., Raskin, L., et al. (2021). Synergistic association between cytochrome bd-encoded Proteiniphilum and reactive oxygen species (ROS)-scavenging methanogens in microaerobic-anaerobic digestion of lignocellulosic biomass. Water Res. 190:116721. doi: 10.1016/j.watres.2020.116721
Yang, P., Tan, G. A., Aslam, M., Kim, J., and Lee, P. H. (2019). Metatranscriptomic evidence for classical and RuBisCO-mediated CO2 reduction to methane facilitated by direct interspecies electron transfer in a methanogenic system. Sci. Rep. 9:4116. doi: 10.1038/s41598-019-40830-0
Yenigün, O., and Demirel, B. (2013). Ammonia inhibition in anaerobic digestion: a review. Process Biochem. 48, 901–911. doi: 10.1016/j.procbio.2013.04.012
Zhang, C., Yuan, Q., and Lu, Y. (2014). Inhibitory effects of ammonia on methanogen mcrA transcripts in anaerobic digester sludge. FEMS Microbiol. Ecol. 87, 368–377. doi: 10.1111/1574-6941.12229
Zhao, Z., Wang, J., Li, Y., Zhu, T., Yu, Q., Wang, T., et al. (2020). Why do DIETers like drinking: metagenomic analysis for methane and energy metabolism during anaerobic digestion with ethanol. Water Res. 171:115425. doi: 10.1016/j.watres.2019.115425
Zheng, D., Wang, H.-Z., Gou, M., Nobu, M. K., Narihiro, T., Hu, B., et al. (2019). Identification of novel potential acetate-oxidizing bacteria in thermophilic methanogenic chemostats by DNA stable isotope probing. Appl. Microbiol. Biotechnol. 103, 8631–8645. doi: 10.1007/s00253-019-10078-9
Keywords: anaerobic digestion, ammonia inhibition, microbial community, syntrophic acetate oxidation, methanogenic pathways, energy conservation, anti-oxidative stress, meta-omics
Citation: Feng G, Zeng Y, Wang H-Z, Chen Y-T and Tang Y-Q (2023) Proteiniphilum and Methanothrix harundinacea became dominant acetate utilizers in a methanogenic reactor operated under strong ammonia stress. Front. Microbiol. 13:1098814. doi: 10.3389/fmicb.2022.1098814
Edited by:
Biswarup Sen, Tianjin University, ChinaReviewed by:
Fanghua Liu, Guangdong Institute of Eco-environmental and Soil Sciences (CAS), ChinaCopyright © 2023 Feng, Zeng, Wang, Chen and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yue-Qin Tang,  dGFuZ3lxQHNjdS5lZHUuY24=
dGFuZ3lxQHNjdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.