 Hayley Cassidy
Hayley Cassidy Erley Lizarazo-Forero
Erley Lizarazo-Forero Leonard Schuele
Leonard Schuele Coretta Van Leer-Buter
Coretta Van Leer-Buter Hubert G. M. Niesters
Hubert G. M. Niesters- The University of Groningen, University Medical Centre Groningen, Department of Medical Microbiology and Infection Prevention, Division of Clinical Virology, Groningen, Netherlands
To explore an off-season enterovirus D68 (EV-D68) upsurge in the winter season of 2019/2020, we adapted a whole-genome sequencing approach for Nanopore Sequencing for 20 hospitalized patients with accompanying respiratory or neurological presentation. Applying phylodynamic and evolutionary analysis on Nextstrain and Datamonkey respectively, we report a highly diverse virus with an evolutionary rate of 3.05 × 10−3 substitutions per year (entire EV-D68 genome) and a positive episodic/diversifying selection with persistent yet undetected circulation likely driving evolution. While the predominant B3 subclade was identified in 19 patients, one A2 subclade was identified in an infant presenting with meningitis. Exploring single nucleotide variations using CLC Genomics Server showed high levels of non-synonymous mutations, particularly in the surface proteins, possibly highlighting growing problems with routine Sanger sequencing for typing enteroviruses. Surveillance and molecular approaches to enhance current knowledge of infectious pathogens capable of pandemic potential are paramount to early warning in health care facilities.
1. Introduction
Enterovirus D68 (EV-D68) has been increasingly recognized as an emerging virus, with outbreaks occurring primarily in children (Piralla et al., 2018; Kamau et al., 2019; Andrés et al., 2022). First reported in 1962 in California, EV-D68 was sporadically reported worldwide until 2013, with small outbreaks described in the Philippines (2009–2011; Imamura and Oshitani, 2015), Japan (2010; Hasegawa et al., 2011), United States of America (USA; 1970–2005; Khetsuriani et al., 2006) and in the Netherlands (2010; Rahamat-Langendoen et al., 2011). The largest known outbreak to date was reported in the USA in 2014, with over a thousand cases in children presenting with severe respiratory disease (Midgley et al., 2015). During the 2014 outbreak, a number of EV-D68 cases were linked to acute flaccid myelitis (AFM), presenting with asymmetric flaccid limb weakness and cranial nerve dysfunction (Messacar et al., 2015; Vogt and Crowe, 2018). In the same year, EV-D68 was also reported in Europe, Southeast Asia and Chile (Holm-Hansen et al., 2016). Since 2014, EV-D68 outbreaks have occurred in a biennial pattern between 2016 and 2018, typically in even years in temperate climates (Howson-Wells et al., 2022) and emerging most frequently in the USA, Europe, Argentina and Taiwan (Knoester et al., 2017; Hu and Chang, 2020). While EV-D68 typically peaks in late summer and autumn in temperate climates (Hodcroft et al., 2022), an off-season upsurge was observed across Europe during the 2019–2020 winter season (Midgley et al., 2020).
EV-D68 has been shown to have substantial genetic diversity (Kramer et al., 2018; Howson-Wells et al., 2022). Sequences can be separated into three main clades: A, B, and C, which can be further divided into A1–A2 and B1–B3 subclades (Hodcroft et al., 2022). The EV-D68 genome is approximately 7,500 nucleotides in length and encodes a single polyprotein which consists of four structural proteins (viral proteins 1–4) and seven non-structural proteins (2A–C and 3A–D; Eshaghi et al., 2017; Dyrdak et al., 2019). Generally, EV-D68 sequences have been investigated using the hypervariable viral protein 1 (VP1) gene, which is present on the viral capsid and has been used as a target for subtype differentiation (Harvala et al., 2018). Since 2014, subclades B1 and B3 have become dominant (Hodcroft et al., 2022). The emergence of the A2 subclade was additionally reported in East Asia in 2016 (Midgley et al., 2020). By 2018, clusters of subclades B3 and A2 appear to be predominating worldwide.
Whole-genome sequencing (WGS) has been used previously to investigate the diversity and evolution of EV-D68 during the 2014 and 2016 outbreaks (Dyrdak et al., 2019). Different subclades have been identified in respiratory and neurological samples, with varying rates of amino acid substitutions observed (Zhang et al., 2016; Wang et al., 2017; Hodcroft et al., 2022). In this study, we obtained near-complete EV-D68 sequences from patients presenting with respiratory or neurological disease at a regional university hospital in the Netherlands during the winter season of 2019–2020. Using WGS, we aimed to explore our patient cohort, along with the circulating clades responsible for the rise in cases with the intention of contributing to a greater understanding of potential evolutionarily changes within EV-D68.
2. Materials and methods
2.1. Patient selection
Patients were selected following a positive EV-D68 detection through our laboratory developed test (LDT) real-time reverse transcriptase PCR (RT-qPCR; Poelman et al., 2015). Sanger sequencing targeting the VP1 gene (approx. 326 bp; Nix et al., 2006) was performed on samples with a Ct value below 32. Sanger sequencing data was analyzed in BioNumerics v6.1, while patient information was extracted from the electronic patient database system.
2.2. Nucleic acid extraction
All samples were centrifuged at 6,000xg for 2 min. A total of 190 μL of supernatant was used as input for extraction on the easyMAG (bioMérieux, Inc., Marcy l’Etoile, France), which was eluted in 110 μL. Lysis buffer served as a negative control. A total of 70 μL of isolated nucleic acids were cleaned and concentrated to 40 μL using the RNA clean and concentrator kit-5 (Zymo Research, Irvine, USA), including an in-column DNase treatment using TurboDNase (Thermo Fisher Scientific, Waltham, USA), according to the manufacturer’s recommendations.
2.3. cDNA synthesis
Near full-length amplification was achieved using a one-step RT PCR and four overlapping fragments (approximately 2,000 bp) from primers designed by Dyrdak and colleagues (Dyrdak et al., 2019). Briefly, four separate reactions for each overlapping fragment contained; 1 μL of Superscript III RT/Platinum Taq HiFi Enzyme mix (Invitrogen, Stockholm, Sweden), 25 μL of 2× reaction mix, 2.5 μL of forward (0.5 μM) and reverse (0.5 μM) primers, 0.5 μL of random hexamers (1 ng/μL; Thermo Fisher Scientific) and 8.5 μL of RNase-free water. Finally, 10 μL of RNA template was added per fragment for each sample to attain a total reaction volume of 50 μL. We adjusted the PCR cycling conditions to account for the increased cDNA input required by Oxford Nanopore Technologies (ONT) ligation library preparation kit: 30 min at 50°C, 2 min at 94°C, ×33 (15 s at 94°C, 30s at 50°C, 2 min at 68°C), 5 min at 68°C, ∞ at 4°C. Each fragment was adjusted to 25 ng and pooled to achieve a total of 100 ng for each sample. cDNA fragments with <25 ng were re-amplified using the PCR reaction stated above with a modified PCR cycling condition to account for cDNA as an input: 1 min at 98°C, ×29 (10s at 98°C, 30s at 50°C, 3 min at 72°C), 5 min at 72°C, ∞ at 4°C.
2.4. Library preparation and sequencing
Sequencing libraries were generated using the Ligation Sequencing Kit (SQK-LSK109; ONT, Oxford, United Kingdom) and native barcoding expansion (EXP-NBD104; ONT). The One-pot protocol was followed for native barcoding for amplicons (Josh Quick, 2020)1. The libraries were pooled by equal mass and 25fM was loaded on FLO-MIN106 R9.4.1 flow cell on a MinION device for long-read sequencing (ONT).
2.5. Assay validation
An EV-D68 culture from the National Institute for Public Health and the Environment (RIVM) was used to validate the workflow prior to running the clinical samples (Supplementary materials and methods 1.1 and Supplementary Figure S1).
2.6. Data analysis
Sequencing reads were first base-called with Guppy v6.0.1 (ONT) with high accuracy mode enabling the “trim barcodes,” “barcode both ends” and “mid-read barcode filtering” option (Supplementary Table S1). Subsequent reads were uploaded onto the CLC Genomics Server 21.0.5 (CLC; Qiagen, Aarhus, Denmark). Only reads with >300 bp were kept and mapped against the human genome (hg19) to filter out human reads. To create a reference database, a total of 893 near-full length EV-D68 reference sequences (7,000–8,000 bp) were downloaded from the Virus Pathogen Resource (Supplementary materials and methods 1.2). The trimmed reads were mapped against this reference database using an 70% nucleotide identity and 80% length fraction on CLC to determine a best hit reference for each sample (Supplementary Table S2). Consensus sequences were extracted with a coverage cut-off of >30x (Karst et al., 2021), with confirmation using NCBI BLAST and uploaded onto the online enterovirus typing tool (v0.1) to obtain sub genogroups.2
2.7. Phylogenetic and evolutionary analysis
Patient consensus sequences and 893 reference genomes were aligned with MAFFT (v7.471; see Supplementary materials and methods 1.2). A time-scaled phylogenetic analysis using the augur pipeline (v7.0.2) and TimeTree v0.8.1 (Sagulenko et al., 2018) implemented in Nextstrain (Hadfield et al., 2018) was performed (Supplementary Table S3). A maximum likelihood tree was inferred using a GTR + gamma distribution, a strict molecular clock and a coalescent Skyline tree prior (Supplementary Table S3). The tree was rooted using GenBank accession number AY426531 (1962 Fermon strain). The time-scaled tree was then visualized using auspice (v0.8.0; Hadfield et al., 2018) and detailed with country of origin. To explore the temporal signal and heterochronous data, a root-to-tip regression analysis on TempEst (v2.7.0; Rambaut et al., 2016) was applied for the maximum likelihood tree without a molecular clock. To determine the type of selection pressure, a Mixed Effects Model of Evolution (MEME; Murrell et al., 2012) was applied on Datamonkey (v1.6.0) using two MAFFT alignments, one including the 824 EV-D68 references (see Supplementary materials and methods 1.2) and patient sequences, and the other with only patients sequences (CDS regions only; posterior probability [PP] value = 0.05; Supplementary Table S4). Finally, single nucleotide variation (SNV) was investigated within the patient sequences using a Fixed Ploidy algorithm with 90% variant probability and 90% minimum frequency on CLC, with 100x minimum coverage and a Q score of 30 (Supplementary Table S5).
2.8. Ethics statement
Oral consent for the use of clinical samples for research purposes is routinely obtained upon patient admission at the University Medical Center Groningen (UMCG), in accordance with the guidelines of the Medical Ethics Committee. All experiments were performed in accordance with the guidelines of the Declaration of Helsinki and all samples were anonymized. A waiver was obtained by the UMCG Ethics Committee: METc 2009.169. The sequencing data has been deposited in the Sequence Read Archive under the BioProject number: PRJNA865246.
3. Results
3.1. Off-season upsurge
From January 2010 to March 2020, a total of 157 EV-D68 detections were recorded at the UMCG, typically peaking between July and September (Figure 1), with the highest number of cases in July 2016 (n = 26 cases). Since 2014, there appears to be a general shift in the number of reported cases toward the late autumn months (Figure 1B). By the winter season of 2019–2020, a distinct rise in the number of cases (n = 20) can be observed, which is both off-season and in an odd year, with a peak in December 2019 (n = 8 detections; Figures 1A,B).
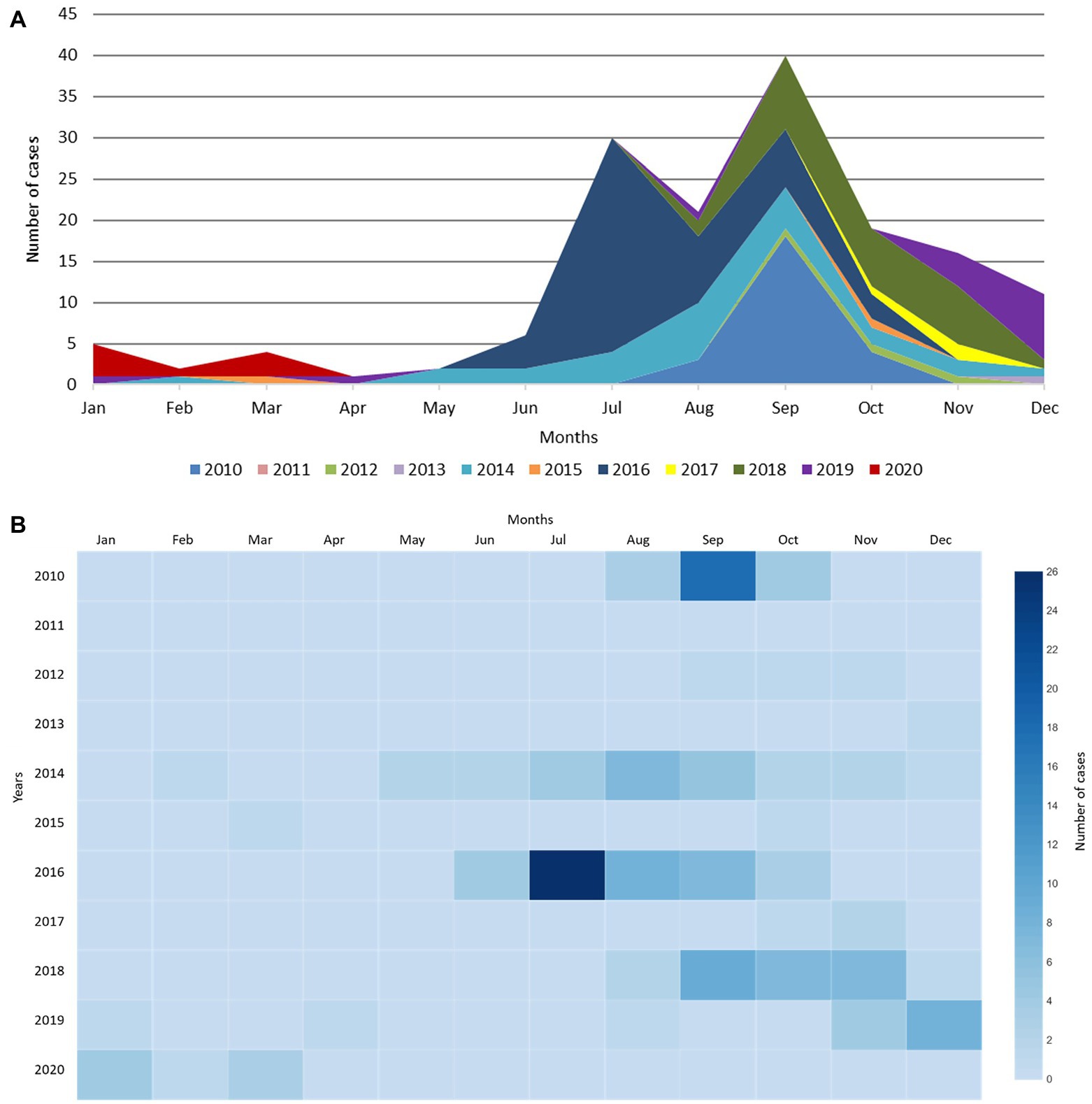
Figure 1. EV-D68 detection from January 2010 to March 2020 at the UMCG. (A) EV-D68 detection per month at the UMCG between 2010 and 2020. A distinct rise can be observed which does not follow the usual trend, shown in purple (2019) and red (2020). (B) Heatmap of EV-D68 detection per month at the UMCG between 2010 and 2020.
3.2. Off-season EV-D68 clinical characterization
The clinical characteristics of the 20 patients with an EV-D68 detection during the 2019/2020 off-season upsurge are shown in Table 1. For four patients (1–3 and 16), only birth date, gender and sample collection information were recorded. Sample 16 had additional diagnosis information.
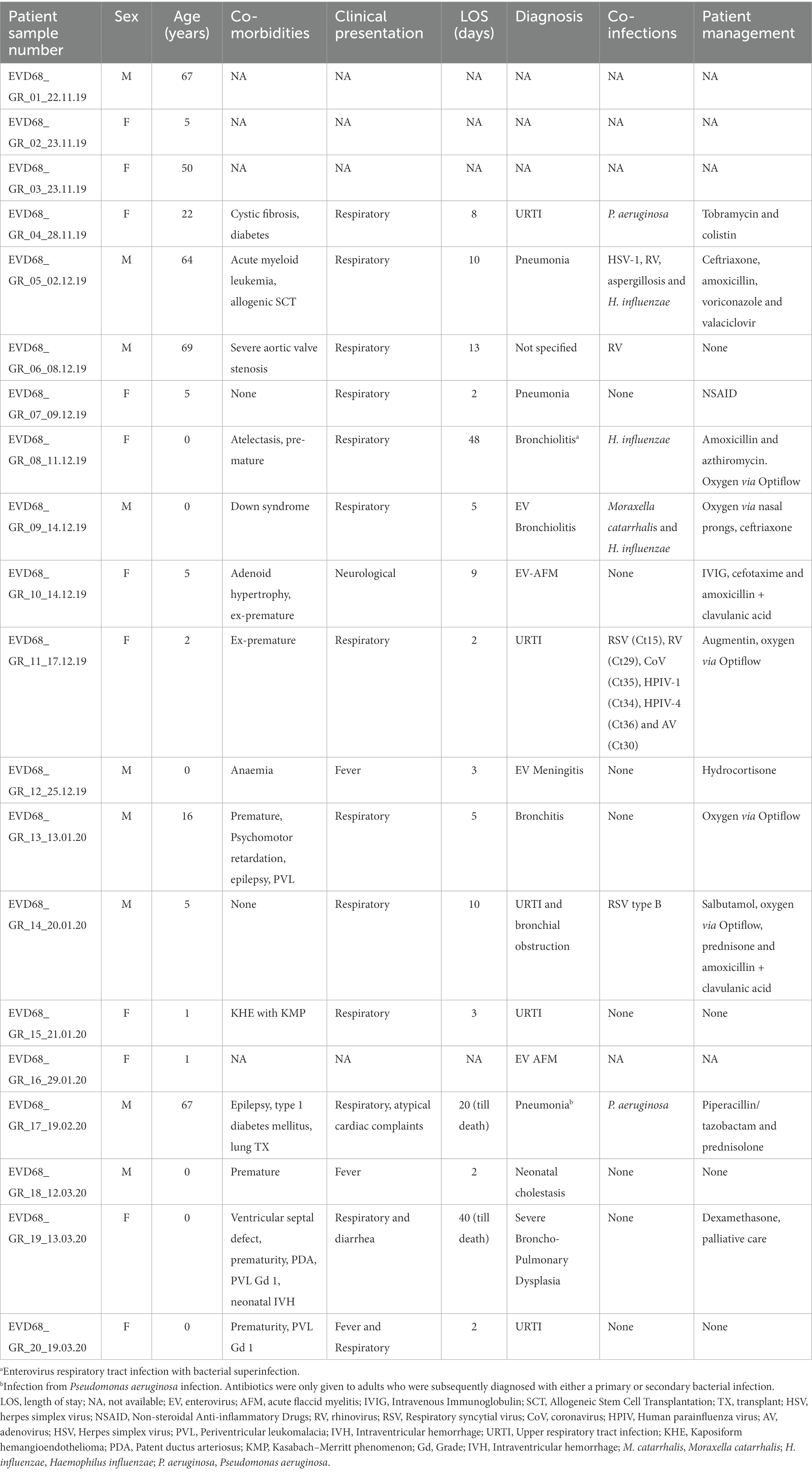
Table 1. Clinical characteristics of patients with an enterovirus D68 detection.
A total of 13 children (<16 years of age; median 1 year) and seven adults (median 64 years) were included in the study. Eight children (61%) were found to have an underlying medical condition, of which prematurity had the highest occurrence (n = 6; Table 1). All adults with available clinical information (n = 5) had at least one underlying condition. Females accounted for 55% of the patient population (n = 11).
Of the eight children with a respiratory infection, four had a short length of stay (LOS; 2–3 days), one had a medium LOS (4–7 days) and three had a long LOS (>7 days). According to the attending clinician, EV-D68 was found to be the causative agent in five of the eight children. Of the four adults with a respiratory infection, one had a medium LOS and three had a long LOS (one adult died after 20 days in hospital). According to the attending clinician, EV-D68 was found to be the causative agent in only one adult (Table 1). Of the three children with an EV-D68 neurological infection, one child (<1 year) was diagnosed with meningitis and two children (5 years and 1 year) were diagnosed with AFM. The 5-year-old child was given antibiotics and intravenous immune globulin (IVIG) treatment prior to their AFM diagnosis. Within this small off-season cohort, there was no EV-D68 neurological presentation reported in adults.
3.3. EV-D68 whole-genome sequencing
Seventeen respiratory, two cerebrospinal fluid (CSF), and one fecal sample were included for WGS. Four samples (20%) had an unsuccessful Sanger sequencing typing result previously. Twenty near-full genomes were recovered (genome size range 7,173–7,222 bp), indicating a robust approach (Table 2). All but one sample (patient number 5) had sufficient sequencing depth (59.4×–66,459.4×) for SNV calling and phylogenetic analysis. Supplementary Figure S2 illustrates an example of the genome coverage pattern achieved.
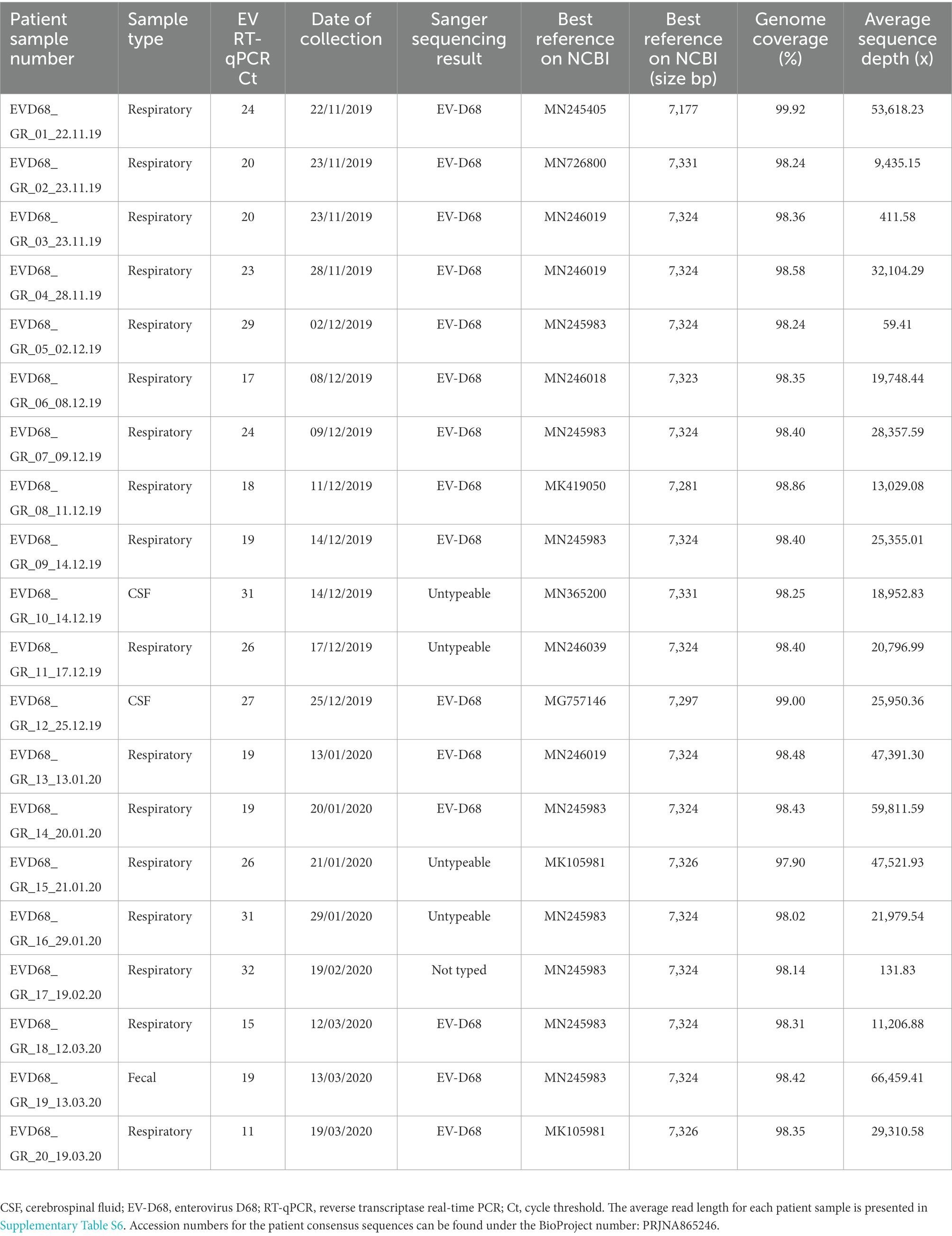
Table 2. Metatable of sample information.
3.4. Phylogenetic analysis
Phylodynamic analysis indicated that 18 sequences from the off-season upsurge clustered within three distinct subgroups within the B3 lineage, with a most recent common ancestor (MRCA) estimated to have occurred in late 2018, following a period of high genetic diversity in mid-2017 (Figure 2; Supplementary Figures S4, S5). Clusters 1 and 2 had the highest similarity to strains from the USA collected in 2018, with 99% (98.9%–99.0%) and 98.9% (98.85%–99.1%) identities, respectively. Cluster 3 had highest similarity to strains from the Netherlands (99.4%) and Sweden (98.87%), collected in 2019. Patient 12 (Cluster 4) clustered within the A2 lineage with a USA strain collected in 2017 (97.90% identity). To initially estimate the evolutionary changes of EV-D68, the temporal signal was explored using a root-to-tip regression analysis (Supplementary Figure S3; Supplementary Table S7). A strong association was observed between genetic distances and collection dates, highlighting heterogeneity between the EV-D68 sequences (R2 = 0.9544). Applying a time-scaled tree using the Nextstrain pipeline, we estimated the evolutionary rate for the entire EV-D68 genome to have 3.05 × 10−3 substitutions per site per year.
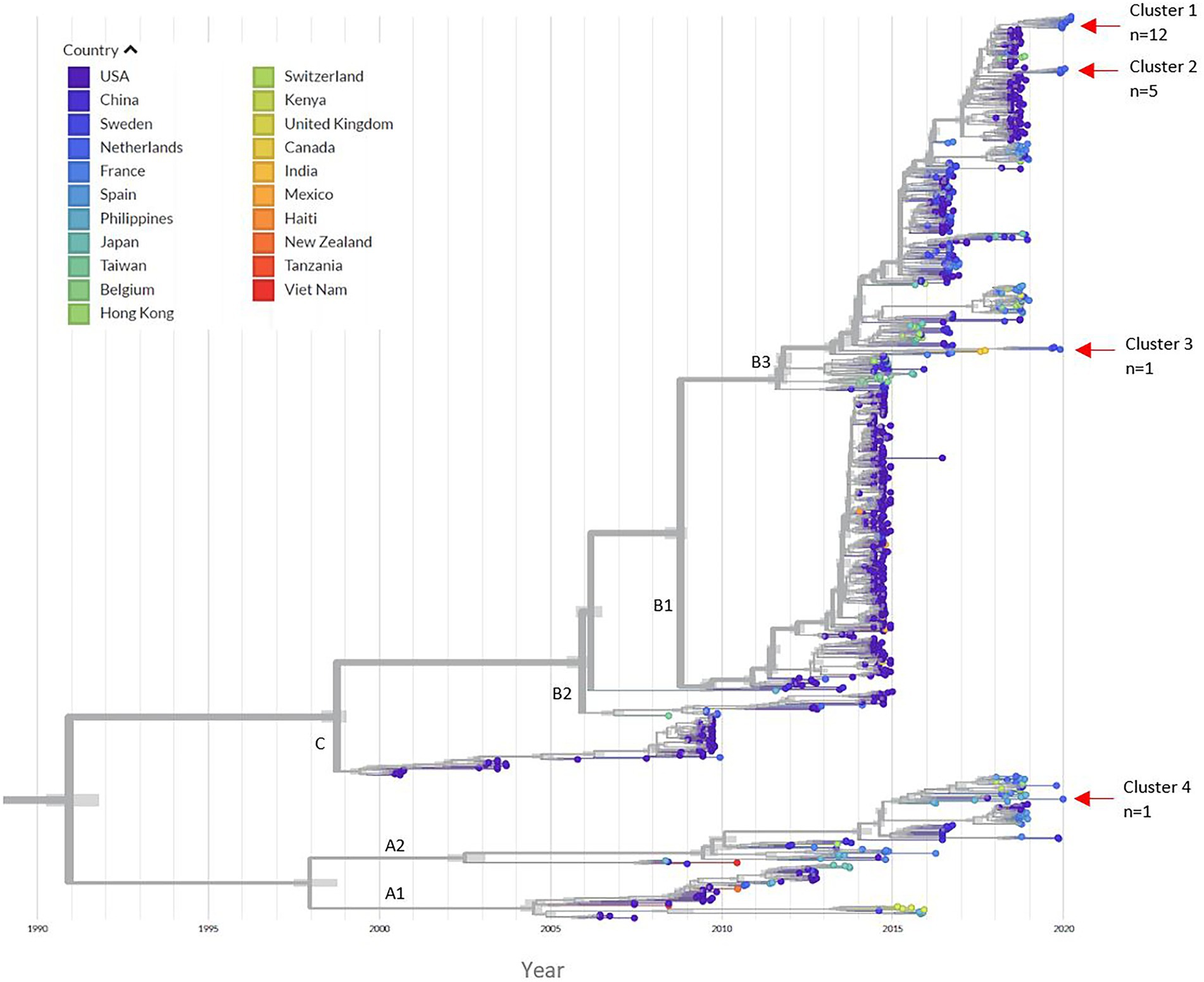
Figure 2. Time-scaled phylogenetic tree. The left panel indicates the corresponding countries to each EV-D68 sequence in the tree. The red arrows indicate the location of the four different clusters of our 2019/2020 patient samples. From the 892 references downloaded; 21 sequences were manually removed due to poor quality after the initial alignment (Supplementary Table S8). A total of 872 sequences were run through the Nextstrain pipeline. An additional 29 references were removed after a second alignment. The Nextstrain pipeline automatically removed problematic sequences during tree refinement (n = 12 references) and uploading to auspice (n = 7 references), this included EVD68_GR_05_02.12.19 (Supplementary Table S8). A total of 824 EV-D68 references were included in the phylogenetic analysis. Supplementary Figures S4, S5 illustrate zoomed in images of the clusters. The time-scaled tree had an estimated evolution rate of 3.05 × 10−3.
3.5. Evolution analysis
SNV’s (non-synonymous mutations only) were explored within all 20 patient samples, with the 1962 Fermon stain (accession number AY426531) used as a reference to allow comparisons with other studies (Dyrdak et al., 2019). Non-synonymous mutations were scattered across the genome, both in structural and non-structural segments of the polyprotein, except in the 3B gene (essential for replication and comprises of 0.88% of genome). We observed a total of 836 non-synonymous mutations (in relation to the Fermon strain), with VP1 containing the highest number of variants (n = 257; Figure 3; Supplementary Table S9). All variants detected were unique per genome position, and therefore we can report one EV-D68 infection per patient.
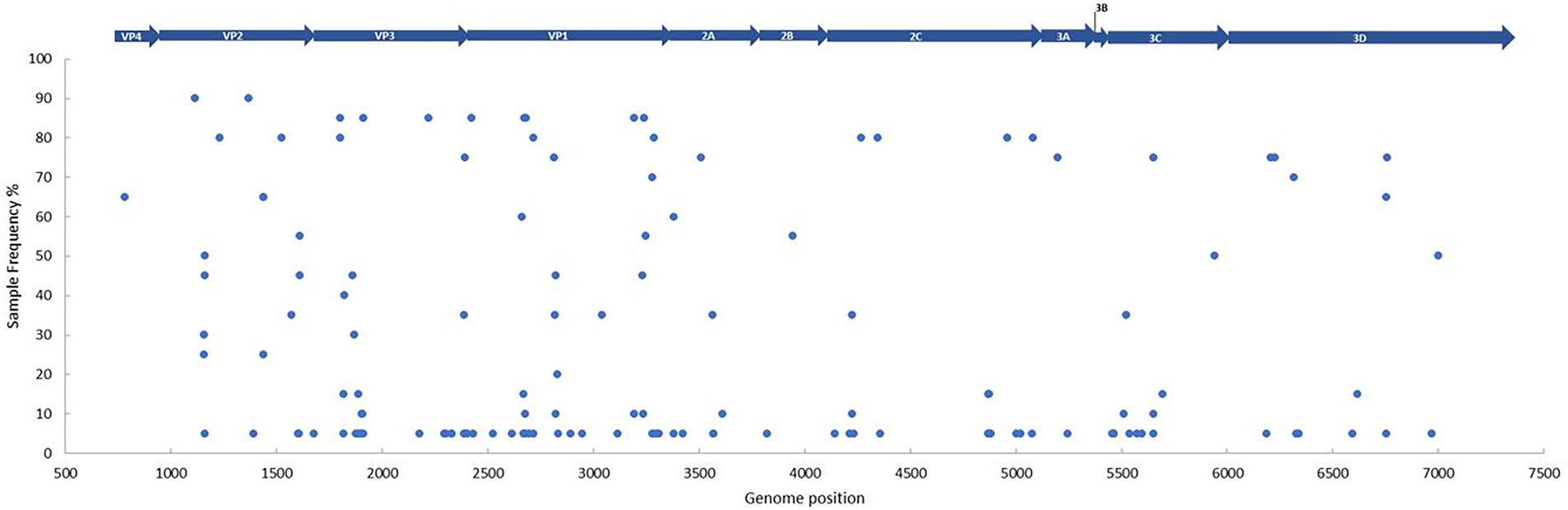
Figure 3. The frequency and distribution of single nucleotide variants per genome position between the patient samples (n = 20) and the Fermon strain (accession number AY426531). Every blue dot represents a non-synonymous change on the sequence, with the gene position depicted above. Sample frequency denotes the percentage of samples in the data set with the non-synonymous change. A Fixed Ploidy algorithm was applied on CLC Genomics Server v21.0.5 using the following parameters: 90% variant probability, 90% minimum frequency, min 100x coverage, max 100,000x coverage, Q-score threshold = 30.
Overall, 44 multiple and 92 single nucleotide variants were observed, with particularly high activity within the BC loop region (position 2,659–2,698) in the VP1 gene. The following frame-shift mutations in the VP1 gene were of note: one deletion (removal of GA [His651] in position 2,679–2,680), two replacements (replacement of GGG to A [Gly649] in position 2,677–2,679 and C to TGA [Ser647] in position 2,670) and one insertion (TA [ser647] in position 2,669–2,670). All non-synonymous mutations for each sample, along with their corresponding regions are detailed in Supplementary Table S9. Although no shared variants were observed amongst all the off-season sequences, 18 did share variants in two regions (with 90% frequency) present in the VP2 gene (Figure 3; Supplementary Table S9). No obvious variant patterns were observed in patients with CSF collection (Supplementary Table S9) or neurological presentation (Supplementary Figure S6).
To determine any potential changes in the type of selection pressure during our off-season upsurge, two MEME models were used to explore the distribution of variation at the codon level within the EV-D68 population (n = 824) and the patient sequences (n = 19; Figure 4). Exploring the whole population (shown in blue), we found evidence of episodic or diversifying selection at 54 codon sites (PP value = 0.05 [occurring 95%]), scattered throughout the genome, with high clustering within the viral capsid genes. Exploring within the patient population (shown in red), we found evidence of episodic or diversifying selection at 10 codon sites, with the highest clustering in the 3C gene, followed by the capsid genes.
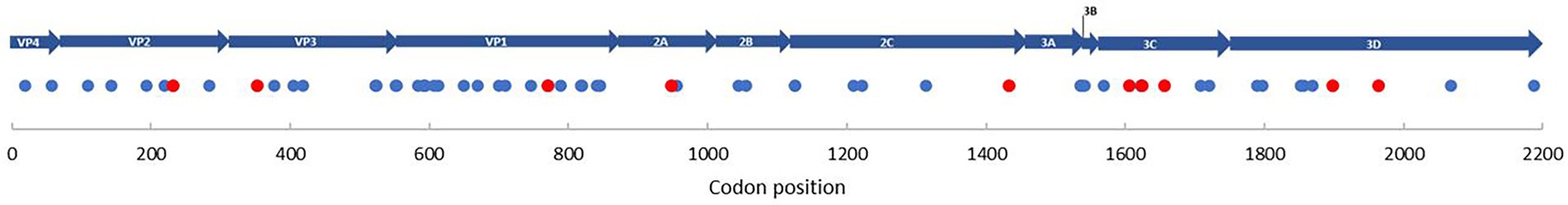
Figure 4. Selection pressure on individual codon sites. Blue dots represent the selection pressure on individual codon sites for all reference genomes (n = 824), along with EV-D68 patients (n = 19; 1962–2020). Red dots represent the selection pressure on individual codon sites for only EV-D68 patients (n = 19). MEME was used to determine diversifying selection (PP value < 0.05). Individual sites are specifically indicated in Supplementary Figure S10.
4. Discussion
In this study, we provide clinical and phylogenetic information from hospitalized patients from an off-season EV-D68 upsurge in the winter season of 2019–2020. Using a modified WGS approach, we were able to obtain near-complete EV-D68 genomes from a wide range of Ct values, including Ct values >30 and sample material, including CSF. The summer/autumn seasonal circulation is well recognized for all enteroviruses in temperate climates, with a biennial recurrence of EV-D68 observed in North America and Europe (Elrick et al., 2021; Fall et al., 2022). Interestingly, our data shows a general shift towards the later months, with the upsurge emerging in an odd year (Figure 1). This off-season occurrence was also observed by other European countries (Midgley et al., 2020). Although the seasonality of enteroviruses is likely determined by environmental factors such as temperature and air humidity (Fares, 2013; Pons-Salort et al., 2018), the biennial occurrence of EV-D68 is thus far not understood. While factors including herd immunity, human behavior and circulation of other (entero-) viruses may play a part, the role and occurrence of viral variants requires more investigation.
The majority of patients in our study had underlying conditions, with prematurity highest among children and typically multiple comorbidities in adults, echoing previous studies (Lau et al., 2016; Fall et al., 2022). Overall, we observed 13 cases (65%) of respiratory illness and three cases (15%) of neurological illness (all in children; Table 1), two of which had AFM and one had EV-D68 meningitis. In our small cohort, we found a relatively high number of cases requiring admission to hospital, with 75% of patients admitted for over 2 days. This most likely reflects the tertiary-care character of our hospital. A similar ratio of severe illness was additionally observed within Europe during the same time period (Midgley et al., 2020). Interestingly, we identified one A2 subclade from a child (<1 year) with meningitis. Although detection of EV-D68 in CSF is rare (Fares, 2013), to the best of our knowledge, we report the first detection of an A2 subclade associated meningitis in CSF. EV-D68 was also detected from one fecal sample from a patient presenting with respiratory and diarrhea illness. Although EV-D68 detection from fecal samples is uncommon, it could provide an alternative for widespread population surveillance studies (Corpuz et al., 2020; Tedcastle et al., 2022). It is important to note that the true burden and circulation within the community is difficult to estimate, given that community surveillance is not performed and subtype differentiation is typically only performed for severer cases following clinical consultation.
Genotyping enteroviruses is important for understanding changes in epidemiology, which could aid outbreak preparedness and patient management (Harvala et al., 2018). WGS has been previously used to explore viral dynamics, particularly during outbreak scenarios and especially during the SARS-CoV-2 pandemic (Oude Munnink et al., 2021). We adapted a previously applied EV-D68 WGS method for ONT platforms to characterize our upsurge samples, including previously untypeable samples.
Using the Nextstrain pipeline, we could perform more real-time tracking of EV-D68 evolution and spread overtime (Figure 1). A high genetic diversity can be observed among the available EV-D68 sequences, indicating diversification and persistence even in low circulation years, similarly reported by Hodcroft et al. (2022). Interestingly, there appears to be continuous emergence in conjuncture with clade replacement prior to 2018, with the rise of the predominating B3 subclade. In our study, 18 samples clustered with the B3 subclade within three different branches, suggesting introduction at several different timepoints in the Netherlands, followed by local spread. Subclade B3 exhibits high levels of geographical circulation (and mixing) since its emergence in 2016 (Wang et al., 2017), having been found in several countries globally (Midgley et al., 2020). The detection of the A2 subclade additionally suggests an ongoing low-level (and potentially undetected) transmission in Europe. Given their increasing observation between 2014 and 2020 (Lau et al., 2016; Midgley et al., 2020; Hodcroft et al., 2022), it could render this finding potentially important for monitoring. Moreover, although the majority of sequences from our study appear to cluster with USA strains, as some countries deposit more sequences than others, it could also result in a geographical bias. Our time-scaled tree generated an estimated evolution rate of 3.05 × 10−3 for the near-complete genome, which is slightly lower than 3.8 × 10−3 in 2018 (Dyrdak et al., 2019), but higher than another estimate of 2.99 × 10−3 from 2017 (Eshaghi et al., 2017). Some patient sequences (patient 1, 4,12) appeared to have higher genetic diversity than average for the sampling date (Supplementary Figure S3; Supplementary Table S7), suggesting a possible higher level of viral evolution within these patients
The generation of high-quality near-complete genomes potentially allows the investigation of inter-and intra-host relations of SNV (Dyrdak et al., 2019; Cassidy et al., 2022). As seen in the SARS-CoV-2 pandemic, variants may explain changes in transmissibility and pathogenicity (Yang et al., 2022). We report a high level of variation within our study, particularly in the VP1 and 3C (transcribing the EV-D68-encoded protease) genes, however in this small cohort, any link between SNV and clinical outcome would not be statistically relevant. Moreover, coverage bias may have been introduced in our study with some samples generating a low volume of reads, while other samples obtaining sequencing depths higher than 100,000× in some regions. This coverage bias was particularly prominent in regions with overlapping primer fragments (Supplementary Figure S2). As a result, we applied a coverage cut-off of 100,000× to reduce the chances of sequencing or PCR errors which could be called wrongly due to the sheer number of reads. Additionally, although continuous improvements in ONT error rate have been made over the recent years (Wick et al., 2021), the overall high error rate compared to Illumina sequencing could also be a limitation. Variants with the highest frequency (in 90% of patient sequences) were subsequently revealed to be highly conserved within the B3 and A2 subclades (>90% in our reference database).
The high number of variants within the hypervariable VP1 gene could highlight the impact of immune pressure and its continual influence to shape this region (Opanda et al., 2014; Eshaghi et al., 2017). Other studies have identified rapidly evolving neutralizing epitopes in the VP1 gene, particularly in the BC and DE loops, with higher amino acid substitution rates than the rest of the genome (Hodcroft et al., 2022). With the emergence of new EV-D68 strains, given the high genetic diversity observed over time (Figures 2, 3), coupled with declining immunity (Kramer et al., 2018), it could indicate a growing susceptible population. At the same time, the requirement of re-amplification in some sample fragments could increase the chances for PCR driven mutations (Salk et al., 2018).
Finally, we show evidence of high levels of episodic positive diversifying selection events within the EV-D68 population and within our patient dataset scattered along the genome, particularly in the 3C and VP1 genes (referred to as “hotspot” regions; Muslin et al., 2019). As our patient samples are the most recently deposited (near-full) EV-D68 sequences, it could highlight the areas which are most likely being selected in the future. Interestingly, the 3C gene has a central role in inhibiting antiviral immunity (Xiang et al., 2015). It has been speculated that the now dominant B3 subclade has arose from diversifying selection and likely represents a newly emerging strain, as opposed to evolving from the B1 or B2 clades (Wang et al., 2017; Hodcroft et al., 2022). Given the strong selection pressure observed (in this case positive diversifying selection) of subclade B3, along with a potentially increasing naïve population, it could have driven its predominance since 2018.
5. Conclusion
We report an off-season EV-D68 upsurge likely caused by highly diverse subclades and associated with severe respiratory and neurological disease, particularly in children and immunocompromised adults. It could be that the positive episodic or diversifying selection, along with persistent yet undetected circulation may be driving EV-D68 evolution, however at this point this is still a working hypothesis. Furthermore, high levels of non-synonymous mutations, particularly in the surface proteins, could suggest monitoring closely for potential challenges with routine Sanger sequencing. Applying WGS revealed an dynamic picture of EV-D68, highlighting evolutionary patterns. With the re-emergence of EV-D68 following the COVID-19 lockdown and poliovirus recently detected in non-epidemic countries, it is important to closely monitor this virus which may become a threat to public health.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA900246.
Author contributions
HC: study design, laboratory and data analysis, writing-original draft preparation, and writing-reviewing and editing. EL-F: data analysis and writing-reviewing and editing. LS: study design, laboratory and data analysis, and writing-reviewing and editing. CL-B and HGMN: conceptualization, editing, and supervision. All authors contributed to the article and approved the submitted version.
Funding
Hayley Cassidy and Leonard Schuele received funding from the European Union’s Horizon 2020 research and innovation program, under the Marie Sklodowska-Curie grant agreement 713660 (MSCA-COFUND-2015-DP “Pronkjewail”). Erley Lizarazo-Forero was funded by Quality Control Molecular Diagnostics (QCMD, Glasgow, Scotland) under an unrestricted grant.
Acknowledgments
Special thanks to the routine Clinical Virology department at the University Medical Center Groningen for performing diagnostics.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1088770/full#supplementary-material
Footnotes
References
Andrés, C., Vila, J., Creus-Costa, A., Piñana, M., González-Sánchez, A., Esperalba, J., et al. (2022). Enterovirus D68 in hospitalized children, Barcelona, Spain, 2014–2021. Emerg. Infect. Dis. 28, 1327–1331. doi: 10.3201/eid2807.220264
Cassidy, H., Schuele, L., Lizarazo-Forero, E., Couto, N., Rossen, J. W. A., Friedrich, A. W., et al. (2022). Exploring a prolonged enterovirus C104 infection in a severely ill patient using nanopore sequencing. Virus Evol. 8:veab109. doi: 10.1093/ve/veab109
Corpuz, M. V. A., Buonerba, A., Vigliotta, G., Zarra, T., Ballesteros, F. Jr., Campiglia, P., et al. (2020). Viruses in wastewater: occurrence, abundance and detection methods. Sci. Total Environ. 745:140910. doi: 10.1016/j.scitotenv.2020.140910
Dyrdak, R., Mastafa, M., Hodcroft, E. B., Neher, R. A., and Albert, J. (2019). Intra-and interpatient evolution of enterovirus D68 analyzed by whole-genome deep sequencing. Virus Evol. 5:vez007. doi: 10.1093/ve/vez007
Elrick, M. J., Pekosz, A., and Duggal, P. (2021). Enterovirus D68 molecular and cellular biology and pathogenesis. J. Biol. Chem. 296:100317. doi: 10.1016/j.jbc.2021.100317
Eshaghi, A., Duvvuri, V. R., Isabel, S., Banh, P., Li, A., Peci, A., et al. (2017). Global distribution and evolutionary history of enterovirus D68, with emphasis on the 2014 outbreak in Ontario, Canada. Front Microbiol. 8:257. doi: 10.3389/fmicb.2017.00257
Fall, A., Gallagher, N., Morris, C. P., Norton, J. M., Pekosz, A., Klein, E., et al. (2022). Circulation of enterovirus D68 during period of increased influenza-like illness, Maryland, USA, 2021. Emerg. Infect. Dis. 28, 1525–1527. doi: 10.3201/eid2807.212603
Fall, A., Kenmoe, S., Ebogo-Belobo, J. T., Mbaga, D. S., Bowo-Ngandji, A., Foe-Essomba, J. R., et al. (2022). Global prevalence and case fatality rate of enterovirus D68 infections, a systematic review and meta-analysis. PLoS Negl. Trop. Dis. 16:e0010073. doi: 10.1371/journal.pntd.0010073
Fares, A. (2013). Factors influencing the seasonal patterns of infectious diseases. Int. J. Prev. Med. 4, 128–132.
Hadfield, J., Megill, C., Bell, S. M., Huddleston, J., Potter, B., Callender, C., et al. (2018). Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123. doi: 10.1093/bioinformatics/bty407
Harvala, H., Broberg, E., Benschop, K., Berginc, N., Ladhani, S., Susi, P., et al. (2018). Recommendations for enterovirus diagnostics and characterisation within and beyond Europe. J. Clin. Virol. 101, 11–17. doi: 10.1016/j.jcv.2018.01.008
Hasegawa, S., Hirano, R., Okamoto-Nakagawa, R., Ichiyama, T., and Shirabe, K. (2011). Enterovirus 68 infection in children with asthma attacks: virus-induced asthma in Japanese children. Allergy 66, 1618–1620. doi: 10.1111/j.1398-9995.2011.02725.x
Hodcroft, E. B., Dyrdak, R., Andrés, C., Egli, A., Reist, J., Martínez, G., et al. (2022). Evolution, geographic spreading, and demographic distribution of enterovirus D68. PLoS Pathog. 18:e1010515. doi: 10.1371/journal.ppat.1010515
Holm-Hansen, C. C., Midgley, S. E., and Fischer, T. K. (2016). Global emergence of enterovirus D68: a systematic review. Lancet Infect. Dis. 16, e64–e75. doi: 10.1016/S1473-3099(15)00543-5
Howson-Wells, H. C., Tsoleridis, T., Zainuddin, I., Tarr, A. W., Irving, W. L., Ball, J. K., et al. (2022). Enterovirus D68 epidemic, UK, 2018, was caused by subclades B3 and D1, predominantly in children and adults, respectively, with both subclades exhibiting extensive genetic diversity. Microb. Genom. 8:mgen 000825. doi: 10.1099/mgen.0.000825
Hu, Y. L., and Chang, L. Y. (2020). Current status of enterovirus D68 worldwide and in Taiwan. Pediatr. Neonatol. 61, 9–15. doi: 10.1016/j.pedneo.2019.09.007
Imamura, T., and Oshitani, H. (2015). Global reemergence of enterovirus D68 as an important pathogen for acute respiratory infections. Rev. Med. Virol. 25, 102–114. doi: 10.1002/rmv.1820
Josh Quick (2020). One-pot native barcoding of amplicons v2. Protocols.Io. doi: 10.17504/protocols.io.bdp8i5rw
Kamau, E., Harvala, H., Blomqvist, S., Nguyen, D., Horby, P., Pebody, R., et al. (2019). Increase in enterovirus D68 infections in young children, United Kingdom, 2006-2016. Emerg. Infect. Dis. 25, 1200–1203. doi: 10.3201/eid2506.181759
Karst, S. M., Ziels, R. M., Kirkegaard, R. H., Sørensen, E. A., McDonald, D., Zhu, Q., et al. (2021). High-accuracy long-read amplicon sequences using unique molecular identifiers with Nanopore or Pac bio sequencing. Nat. Methods 18, 165–169. doi: 10.1038/s41592-020-01041-y
Khetsuriani, N., Lamonte-Fowlkes, A., Oberst, S., and Pallansch, M. A. (2006). Centers for Disease Control and Prevention. Enterovirus surveillance--United States, 1970-2005. MMWR Surveill. Summ. 55, 1–20.
Knoester, M., Schölvinck, E. H., Poelman, R., Smit, S., Vermont, C. L., Niesters, H. G., et al. (2017). Upsurge of enterovirus D68, the Netherlands, 2016. Emerg. Infect. Dis. 23, 140–143. doi: 10.3201/eid2301.161313
Kramer, R., Sabatier, M., Wirth, T., Pichon, M., Lina, B., Schuffenecker, I., et al. (2018). Molecular diversity and biennial circulation of enterovirus D68: a systematic screening study in Lyon, France, 2010 to 2016. Euro Surveill. 23:1700711. doi: 10.2807/1560-7917.ES.2018.23.37.1700711
Lau, S. K., Yip, C. C., Zhao, P. S., Chow, W. N., To KK, Wu, A. K., et al. (2016). Enterovirus D68 infections associated with severe respiratory illness in elderly patients and emergence of a novel clade in Hong Kong. Sci. Rep. 6:25147. doi: 10.1038/srep25147
Messacar, K., Schreiner, T. L., Maloney, J. A., Wallace, A., Ludke, J., Oberste, M. S., et al. (2015). A cluster of acute flaccid paralysis and cranial nerve dysfunction temporally associated with an outbreak of enterovirus D68 in children in Colorado, USA. Lancet 385, 1662–1671. doi: 10.1016/S0140-6736(14)62457-0
Midgley, S. E., Benschop, K., Dyrdak, R., Mirand, A., Bailly, J. L., Bierbaum, S., et al. (2020). Co-circulation of multiple enterovirus D68 subclades, including a novel B3 cluster, across Europe in a season of expected low prevalence, 2019/20. Euro Surveill. 25:1900749. doi: 10.2807/1560-7917.ES.2020.25.2.1900749
Midgley, C. M., Watson, J. T., Nix, W. A., Curns, A. T., Rogers, S. L., Brown, B. A., et al. (2015). Severe respiratory illness associated with a nationwide outbreak of enterovirus D68 in the USA (2014): a descriptive epidemiological investigation. Lancet Respir. Med. 3, 879–887. doi: 10.1016/S2213-2600(15)00335-5
Murrell, B., Wertheim, J. O., Moola, S., Weighill, T., Scheffler, K., and Kosakovsky Pond, S. L. (2012). Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 8:e1002764. doi: 10.1371/journal.pgen.1002764
Muslin, C., Mac Kain, A., Bessaud, M., Blondel, B., and Delpeyroux, F. (2019). Recombination in enteroviruses, a multi-step modular evolutionary process. Viruses 11:859. doi: 10.3390/v11090859
Nix, W. A., Oberste, M. S., and Pallansch, M. A. (2006). Sensitive, seminested PCR amplification of VP1 sequences for direct identification of all enterovirus serotypes from original clinical specimens. J. Clin. Microbiol. 44, 2698–2704. doi: 10.1128/JCM.00542-06
Opanda, S. M., Wamunyokoli, F., Khamadi, S., Coldren, R., and Bulimo, W. D. (2014). Genetic diversity of human enterovirus 68 strains isolated in Kenya using the hypervariable 3′-end of VP1 gene. PLoS One 9:e102866. doi: 10.1371/journal.pone.0102866
Oude Munnink, B. B., Worp, N., Nieuwenhuijse, D. F., Sikkema, R. S., Haagmans, B., Fouchier, R. A. M., et al. (2021). The next phase of SARS-CoV-2 surveillance: real-time molecular epidemiology. Nat. Med. 27, 1518–1524. doi: 10.1038/s41591-021-01472-w
Piralla, A., Principi, N., Ruggiero, L., Girello, A., Giardina, F., De Sando, E., et al. (2018). Enterovirus-D68 (EV-D68) in pediatric patients with respiratory infection: the circulation of a new B3 clade in Italy. J. Clin. Virol. 99–100, 91–96. doi: 10.1016/j.jcv.2018.01.005
Poelman, R., Schölvinck, E. H., Borger, R., Niesters, H. G., and van Leer-Buter, C. (2015). The emergence of enterovirus D68 in a Dutch university medical center and the necessity for routinely screening for respiratory viruses. J. Clin. Virol. 62, 1–5. doi: 10.1016/j.jcv.2014.11.011
Pons-Salort, M., Oberste, M. S., Pallansch, M. A., Abedi, G. R., Takahashi, S., Grenfell, B. T., et al. (2018). The seasonality of nonpolio enteroviruses in the United States: patterns and drivers. Proc. Natl. Acad. Sci. U. S. A. 115, 3078–3083. doi: 10.1073/pnas.1721159115
Rahamat-Langendoen, J., Riezebos-Brilman, A., Borger, R., van der Heide, R., Brandenburg, A., Schölvinck, E., et al. (2011). Upsurge of human enterovirus 68 infections in patients with severe respiratory tract infections. J. Clin. Virol. 52, 103–106. doi: 10.1016/j.jcv.2011.06.019
Rambaut, A., Lam, T. T., Max Carvalho, L., and Pybus, O. G. (2016). Exploring the temporal structure of heterochronous sequences using temp Est (formerly path-O-gen). Virus Evol 2:vew 007. doi: 10.1093/ve/vew007
Sagulenko, P., Puller, V., and Neher, R. A. (2018). Tree time: maximum-likelihood phylodynamic analysis. Virus Evol. 4:vex 042. doi: 10.1093/ve/vex042
Salk, J. J., Schmitt, M. W., and Loeb, L. A. (2018). Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat. Rev. Genet. 19, 269–285. doi: 10.1038/nrg.2017.117.Epub2018
Tedcastle, A., Wilton, T., Pegg, E., Klapsa, D., Bujaki, E., Mate, R., et al. (2022). Detection of enterovirus D68 in wastewater samples from the UK between July and November 2021. Viruses 14:143. doi: 10.3390/v14010143
Vogt, M. R., and Crowe, J. E. Jr. (2018). Current understanding of humoral immunity to enterovirus D68. J. Pediatr. Infect Dis Soc. 7, S49–S53. doi: 10.1093/jpids/piy124
Wang, G., Zhuge, J., Huang, W., Nolan, S. M., Gilrane, V. L., Yin, C., et al. (2017). Enterovirus D68 subclade B3 strain circulating and causing an outbreak in the United States in 2016. Sci. Rep. 7:1242. doi: 10.1038/s41598-017-01349-4
Wick, R. R., Judd, L. M., Cerdeira, L. T., Hawkey, J., Méric, G., Vezina, B., et al. (2021). Trycycler: consensus long-read assemblies for bacterial genomes. Genome Biol. 22:266. doi: 10.1186/s13059-021-02483-z
Xiang, Z., Liu, L., Lei, X., Zhou, Z., He, B., and Wang, J. (2015). 3C protease of enterovirus D68 inhibits cellular defense mediated by interferon regulatory factor 7. J. Virol. 90, 1613–1621. doi: 10.1128/JVI.02395-15
Yang, Z., Zhang, S., Tang, Y. P., Zhang, S., Xu, D. Q., Yue, S. J., et al. (2022). Clinical characteristics, transmissibility, pathogenicity, susceptible populations, and re-infectivity of prominent COVID-19 variants. Aging Dis. 13, 402–422. doi: 10.14336/AD.2021.1210
Keywords: enterovirus D68, respiratory infection, neurological infection, whole-genome sequencing, long-read sequencing
Citation: Cassidy H, Lizarazo-Forero E, Schuele L, Van Leer-Buter C and Niesters HGM (2023) Off-season circulation and characterization of enterovirus D68 with respiratory and neurological presentation using whole-genome sequencing. Front. Microbiol. 13:1088770. doi: 10.3389/fmicb.2022.1088770
Edited by:
Jun Hang, Walter Reed Army Institute of Research, United StatesReviewed by:
Katja Wolthers, University of Amsterdam, NetherlandsAvram Levy, University of Western Australia, Australia
Copyright © 2023 Cassidy, Lizarazo-Forero, Schuele, Van Leer-Buter and Niesters. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hubert G. M. Niesters,✉ aC5nLm0ubmllc3RlcnNAdW1jZy5ubA==