 Antonino Malacrinò
Antonino Malacrinò Ahmed Abdelfattah
Ahmed Abdelfattah Imen Belgacem
Imen Belgacem Leonardo Schena
Leonardo Schena- 1Dipartimento di Agraria, Università Mediterranea di Reggio Calabria, Reggio Calabria, Italy
- 2Institute of Environmental Biotechnology, Graz University of Technology, Graz, Austria
- 3Leibniz-Institute for Agricultural Engineering Potsdam (ATB) and University of Potsdam, Potsdam, Germany
- 4Agrocampus Ouest, INRAE, Université de Rennes, IGEPP, Le Rheu, France
Plant genotype is a crucial factor for the assembly of the plant-associated microbial communities. However, we still know little about the variation of diversity and structure of plant microbiomes across host species and genotypes. Here, we used six species of cereals (Avena sativa, Hordeum vulgare, Secale cereale, Triticum aestivum, Triticum polonicum, and Triticum turgidum) to test whether the plant fungal microbiome varies across species, and whether plant species use different mechanisms for microbiome assembly focusing on the plant ears. Using ITS2 amplicon metagenomics, we found that host species influences the diversity and structure of the seed-associated fungal communities. Then, we tested whether plant genotype influences the structure of seed fungal communities across different cultivars of T. aestivum (Aristato, Bologna, Rosia, and Vernia) and T. turgidum (Capeiti, Cappelli, Mazzancoio, Trinakria, and Timilia). We found that cultivar influences the seed fungal microbiome in both species. We found that in T. aestivum the seed fungal microbiota is more influenced by stochastic processes, while in T. turgidum selection plays a major role. Collectively, our results contribute to fill the knowledge gap on the wheat seed microbiome assembly and, together with other studies, might contribute to understand how we can manipulate this process to improve agriculture sustainability.
Introduction
Plant-associated microbial communities are well known for their impact on the ecology and evolution of their host (Trivedi et al., 2020; Malacrinò et al., 2022a). The structure of plant microbiomes largely differentiate within the same plant (e.g., between roots, leaves, fruits, flowers, seeds), and within the same compartment (e.g., different tissues within the same organ; Abdelfattah et al., 2016; Dastogeer et al., 2020; Trivedi et al., 2020), and it is influenced by several factors, including soil (Zarraonaindia et al., 2015; Malacrinò et al., 2021a), herbivores (French et al., 2021; Malacrinò et al., 2021a,b; Frew, 2022), pathogens (Ginnan et al., 2020; Wen et al., 2020; Ewing et al., 2021), and abiotic stresses (Vescio et al., 2021; Yu et al., 2022). Despite the great deal of research in this field, we still know little about the rules that govern the assembly of plant microbiomes. Plants, to a certain extent, are able to direct the assembly of their own microbiome. Indeed, plant genotype has been proven to influence, with different strengths, the structure of microbial communities associated with different plant species, including Boechera stricta (Wagner et al., 2016), Medicago trunculata (Brown et al., 2020), Glycine max (Liu et al., 2019), Olea europaea (Malacrinò et al., 2022b), and several others. This effect is thought to occur through changes in the plant metabolome (e.g., exudates, VOCs), and it can be modulated by plants to help coping with biotic and abiotic stresses (Liu et al., 2020; Tiziani et al., 2022). However, we are still not able to predict how microbiomes would vary across different plant genotypes.
Most of the research investigating the role of plant genotype on microbiome assembly has been done mostly focusing on roots, leaves, and fruits. However, given the functional importance that microbial vertical transmission can have across generations, it is essential to understand also whether plant genotype influences the composition of the seed microbiome. Few previous studies tested the variation of seed microbiomes across different plant genotypes. For example, Kim et al. (2020) found that host speciation and domestication shape seed bacterial and fungal communities in rice. Similarly, Wassermann et al. (2022) found different bacterial microbiota associated with different oilseed rape genotypes, with signatures of phylosymbiosis. In addition, while most of the studies constrain their observations to the bacterial and archeal communities (i.e., 16S rRNA gene amplicon metagenomics), it is also essential to focus on the plant-associated fungal communities. Recent research suggests that, within some taxonomical groups, the structure of plant microbiomes reconciliates with the host’s phylogeny (i.e., phylogenetically closer hosts associate with more similar microbial communities). This link, named phylosymbiosis, has been recently found for example in the apples (Abdelfattah et al., 2022) and chloridoid grasses (Van Bel et al., 2021). While we still do not know how common is phylosymbiosis among plants, it might be key to understand how different plant genotypes assemble their own microbial communities. This is particularly important considering that the vertical transmission of a portion of the plant microbiome has been found in several species, including oilseed rape (Wassermann et al., 2022), tomato (Bergna et al., 2018), wheat (Walsh et al., 2021), and oak (Abdelfattah et al., 2021).
In this study, we tested the influence of plant genotype on the seed fungal microbiome, using cereals as model. We characterized the microbiota of plant ears during the soft-dough phase to best capture the influence of plant genotype on the assembly of seed microbial communities, which might be hindered at a later stage by the plant senescence. First, we focused on the effect of plant species, testing whether the diversity and structure of the fungal microbiome would vary across different cereal species (Avena sativa, Hordeum vulgare, Secale cereale, Triticum aestivum, Triticum polonicum, and Triticum turgidum). While previous research would suggest that microbiome structure is driven by plant species, we also expect to detect a signature of phylosymbiosis (the reconciliation of microbiome structure with the host phylogeny). Second, we tested whether different cultivars within the species T. aestivum and T. turgidum would associate with different fungal communities. According to previous research on wheat (Donn et al., 2015; Azarbad et al., 2020; Yergeau et al., 2020) and other plant species (Wagner et al., 2016; Liu et al., 2019; Brown et al., 2020; Malacrinò et al., 2022b), we hypothesize to detect a strong genotype-depend signal on the structure of fungal communities within each group.
Materials and methods
Field experiment and sampling
The field experiment was set up during December 2015 in a common garden experiment (each genotype within a 5 × 5 m lot) located in Reggio Calabria, Italy (38°04′50.8”N 15°40′47.6″E). For this experiment we used non-sterilized seeds from six plant species: Avena sativa (cultivar Argentina), Hordeum vulgare (cultivar Pilastro), Secale cereale (cultivar Aspromonte), Triticum polonicum (cultivar Puglia), Triticum aestivum (four cultivars: Aristato, Bologna, Rosia, and Verna), and Triticum turgidum (five cultivars: Capeiti, Cappelli, Mazzancoio, Trinakria, and Timilia), with a total of 13 genotypes. Ears were harvested in May 2016 during their soft-dough phase. For each genotype, we collected ears (n = 5 for each sample) from 5 individual plants, for a total of 65 samples.
DNA extraction, library preparation, and sequencing
Samples were lyophilized and grind to a fine powder with stainless steel beads and a bead-beating homogenizer (Retsch GmbH, Haan, Germany). DNA was extracted from ~40 mg of each sample using the DNeasy Plant Mini Kit (QIAGEN, Venlo, Netherlands) according to the manufacturer’s instructions. DNA quality and concentration was then tested using a Nanodrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA, United States), and samples passing QC were stored at −80°C until further processing.
Libraries were prepared by amplifying the ITS2 region of the fungal rRNA using the primer pair ITS86f and ITS4 (Vancov and Keen, 2009). PCRs were conducted by mixing 12.5 μl of the KAPA HiFi Hot Start Ready Mix (KAPA Biosystems, Wilmington, MA) with 0.4 μM of each primer (modified to include Illumina adaptors), ~50 ng of DNA template, and nuclease-free water to a volume of 25 μl. Reactions were performed by setting the thermocycler (Mastercycler gradient, Eppendorf, Hamburg, Germany) for 3 min at 95°C, followed by 35 cycles of 20 s at 98°C, 15 s at 56°C, 30 s at 72°C, and by a final extension of 1 min at 72°C. A no-template control, in which nuclease-free water replaced the target DNA, was included in all PCR assays. Libraries were checked on agarose gel for successful amplification and purified with an Agencourt AMPure XP kit (Beckman Coulter Inc., Brea, CA, United States) using the supplier’s instructions. A second short-run PCR was performed in order to ligate the Illumina i7 and i5 barcodes (Nextera XT, Illumina, San Diego, CA, United States) setting the thermocycler for 3 min at 95°C, followed by 8 cycles of 30 s at 95°C, 30 s at 55°C, 30 s at 72°C, and by a final extension of 5 min at 72°C. Libraries were purified again as above, quantified using a Qubit spectrophotometer (Thermo Scientific, Waltham, MA, United States), normalized for even concentration using nuclease-free water, pooled together, and sequenced on an Illumina MiSeq (Illumina, San Diego, CA, United States) platform using the MiSeq Reagent Kit v3 300PE chemistry following the supplier’s protocol.
Data processing and analysis
Paired-end reads were processed using the DADA2 v1.22 (Callahan et al., 2016) pipeline implemented in R v4.1.2 (R Core Team, 2020) to remove low-quality data, identify Amplicon Sequence Variants (ASVs) and remove chimeras. Taxonomy was assigned using UNITE v8.3 database (Nilsson et al., 2019). Reads coming from amplification of plant DNA and singletons were then removed before further analyses.
Data analysis was performed in R v4.1.2 as well. Using the packages microbiome (Lahti and Shetty, 2017) and picante (Kembel et al., 2010) we estimated the diversity of the fungal community for each sample with three different indexes: Faith’s phylogenetic diversity, Shannon’s diversity, and Simpson’s dominance. Then, using the packages lme4 (Bates et al., 2014) and car (Fox and Weisberg, 2019), we tested the effect of plant genotype on the three different indexes by fitting three separate linear models specifying plant species as fixed factor. The package emmeans (Lenth, 2022) was used to infer pairwise contrasts (corrected using false discovery rate, FDR).
Similarly, we tested the effect of plant species and cultivar on the structure of seed fungal microbial communities using a multivariate approach with the tools implemented in the vegan package (Dixon, 2003). Distances between pairs of samples, in terms of community composition, were calculated using an unweighted Unifrac matrix. Tests were run using PERMANOVA (999 permutations), and visualization was performed using a NMDS procedure. First, we tested a model that includes both plant species and cultivar (nested within species; ~plant_species × plant_species/cultivar). Then, we tested the effect of cultivar within T. aestivum and T. turgidum, separately. Pairwise contrasts were inferred using the package RVAideMemoire (Hervé, 2022), correcting p-values for multiple comparisons (FDR).
When testing the influence of plant genotype on the structure of plant microbiota, results suggested that plant species explained a wide portion of the variation (~24.91%, see Results below). To further dissect this result, microbial data were further processed together with an ultrametric phylogenetic tree of Poaceae species obtained from TimeTree (Kumar et al., 2017). A Mantel test (9,999 permutations) was used to test the correlation between a Unifrac matrix of the distance between plant species calculated considering the composition of microbial communities (thus, averaged across replicated samples within the same host species) and a matrix of phylogenetic distance between plant species obtained using the function cophenetic.phylo() from the ape R package (Paradis and Schliep, 2019).
Results also showed a greater differentiation within T. aestivum compared to T. turgidum, suggesting that different mechanisms might contribute to the seed microbiome assembly within each group. We further dissected this by estimating the fungal taxa turnover (taxa replacement) and nestedness (gain/loss of taxa) by partitioning the beta diversity using the package betapart (Baselga, 2010). Differences between turnover and nestedness were tested, within each plant species, by fitting a linear model. For each cultivar of T. aestivum and T. turgidum, we tested whether the microbiome assembly fits a null model (Sloan et al., 2006), estimating the goodness of fit to the null model and the immigration coefficient using the package tyRa.1
Results
Data processing identified 242 fungal ASVs over our 65 samples (Supplementary Figure S1), mostly representing the genera Aureobasidium (16.19%), Cladosporium (15.87%), Alternaria (13.54%), Filobasidium (12.71%), Vishniacozyma (11.82%), Mycosphaerella (9.43%), and Stemphylium (7.51%).
We first tested whether there is variation in the diversity of fungal microbial communities between the different host plants. We found that host plant species drives an effect on the microbiome diversity, using Faith’s phylogenetic diversity index (F5, 56 = 7.84, p < 0.001; Figure 1A), Shannon’s diversity index (F5, 56 = 9.42, p < 0.001; Figure 1B), and Simpson’s dominance index (F5, 56 = 10.57, p < 0.001; Figure 1C). Post-hoc contrasts clarified that these effects are driven by A. sativa, which has a lower diversity (both Faith’s and Shannon’s indexes p < 0.001) and higher dominance (p < 0.001) compared to the other plant species, while no differences were found between the other host plants (p > 0.05).
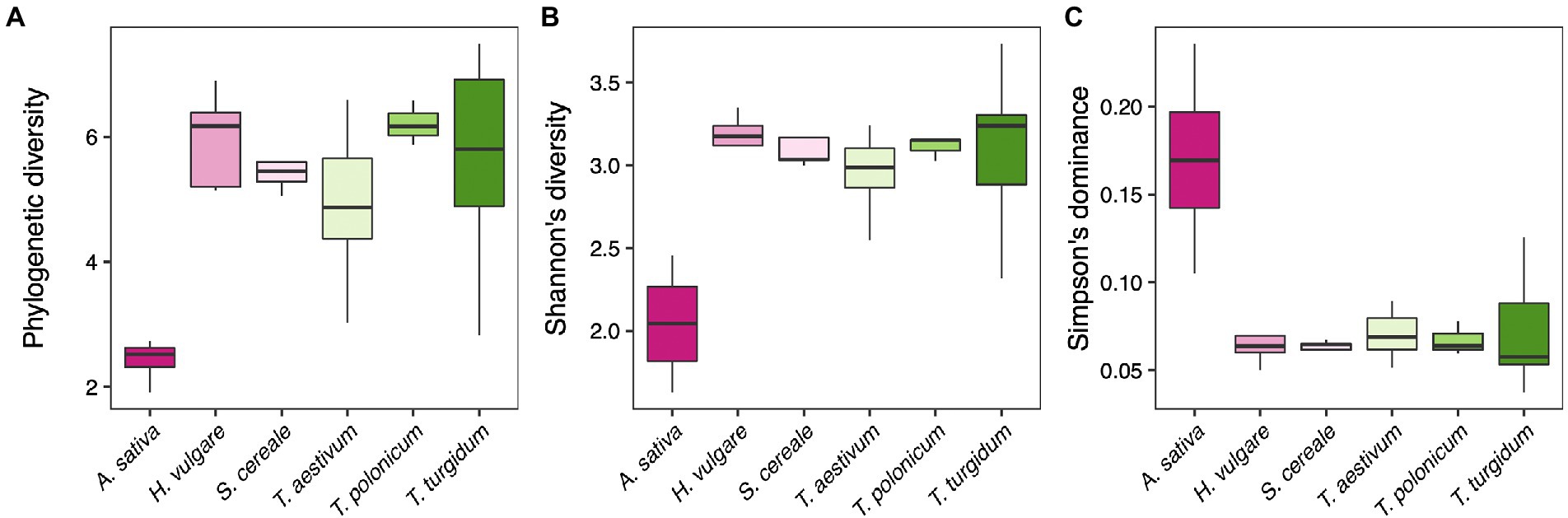
Figure 1. (A) Faith’s phylogenetic diversity, (B) Shannon’s diversity, and (C) Simpson’s dominance index of fungal communities for each host plant species.
When focusing on the structure of the fungal microbiome, PERMANOVA suggests both an effect driven by host plant species (F5, 49 = 4.10, p < 0.001) and cultivar (F7, 49 = 1.83, p < 0.001; nested within species), although species explained a higher proportion of the variance (24.91%) compared to cultivar (15.61%). Post-hoc contrasts show that the structure of the fungal microbiome of A. sativa was different compared to all the other plant species (p < 0.05), and the one of H. vulgare was different from the microbiome of T. aestivum, while no other differences were recorded (Supplementary Table S1).
We then tested whether we could detect a signal of phylosymbiosis, the convergence between host phylogeny and the structure of their microbial communities. Mantel’s correlation shows a weak non-significant correlation between the structure of the fungal microbiota and the host phylogenetic distance (r = 0.31, p = 0.21), although we found overlap across many species when comparing the plant phylogeny with the hierarchical clustering of fungal communities based on Unifrac distances, excluding H. vulgare and T. polonicum (Figure 2).
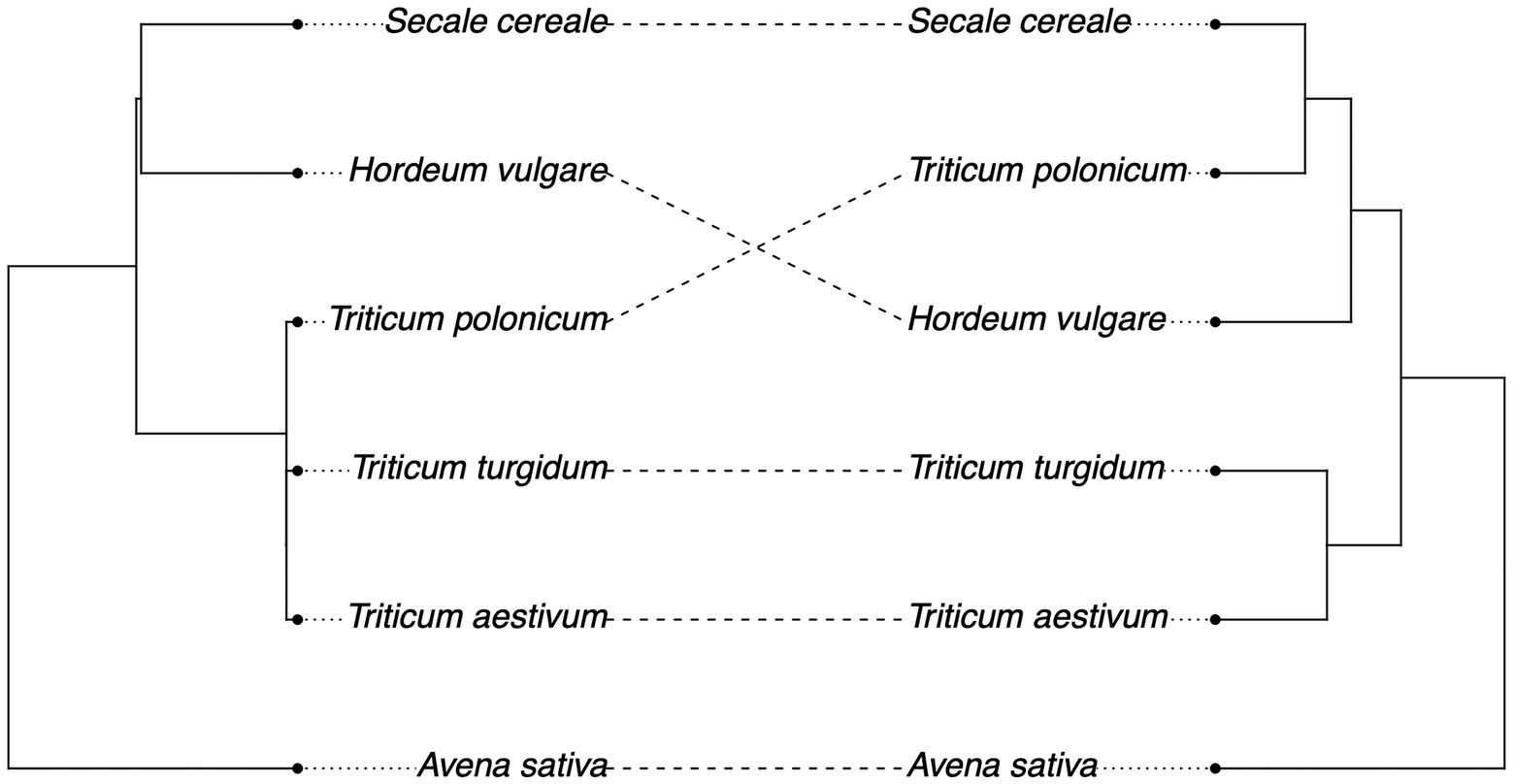
Figure 2. Weak convergence of plant phylogeny (left) and fungal microbiome hierarchical clustering (right) based on Unifrac distances (Mantel’s test: r = 0.31, p = 0.21).
We then tested whether different cultivars within the groups T. aestivum and T. turgidum associate with a different fungal microbiome. Results suggest an effect driven by cultivar in both T. aestivum (F3, 16 = 1.93, p = 0.005; Figure 3A) and T. turgidum (F4, 20 = 1.71, p = 0.02; Figure 3B). Post-hoc contrasts within the T. aestivum group show reciprocal differences between the cultivar Bologna, Rosia, and Verna (p = 0.034), while no differences were recorded between the cultivar Aristato and the others (p > 0.05). When testing pairwise differences within the T. turgidum group, results did not show any difference between the cultivars (p > 0.05), suggesting that the overall effect driven by cultivar (F4, 20 = 1.71, p = 0.02) is marginal.
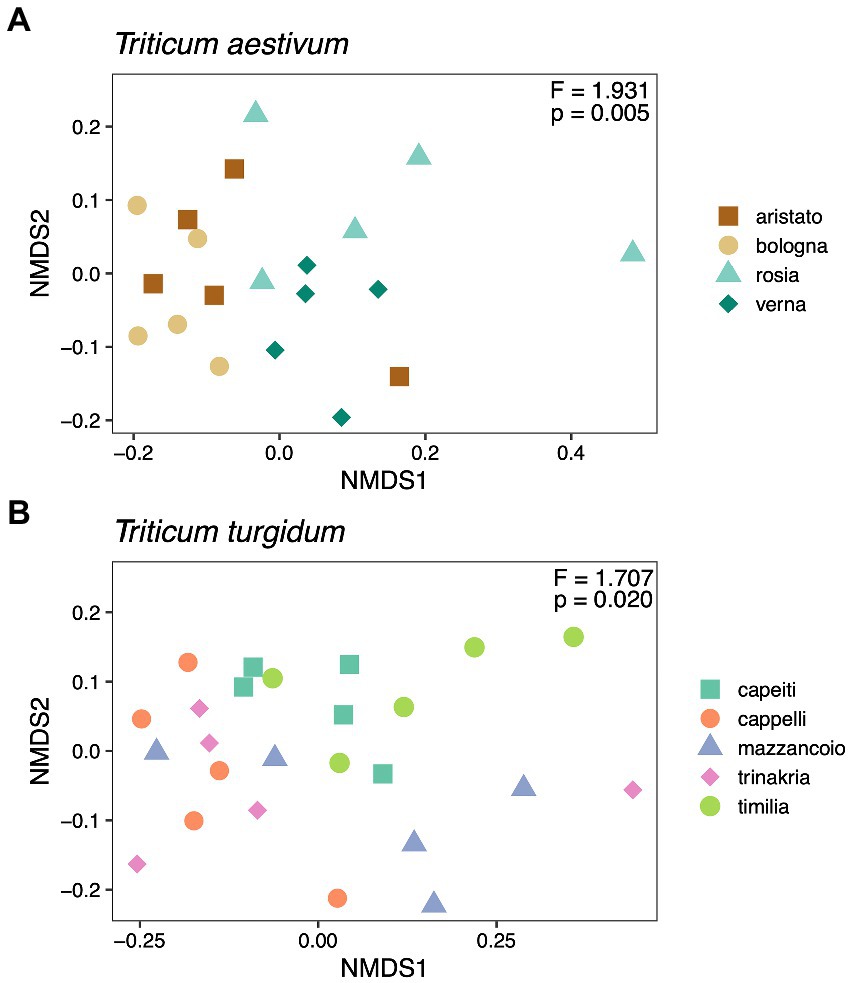
Figure 3. Non-metric Multidimensional Scaling (NMDS) ordination of cultivars within the Triticum aestivum (A) and Triticum turgidum (B) groups, calculated on a Unifrac distance matrix. Each panel shows also results from a PERMANOVA analysis (999 permutations, Unifrac distance matrix) testing the effect of cultivar within each group.
The strongest distinction between cultivars within the group T. aestivum compared to T. turgidum might suggest that the fungal microbiome of different cultivars assembles by different mechanisms. We tested this idea by estimating the fungal taxa turnover and nestedness within both groups T. aestivum and T. turgidum. In both T. aestivum and T. turgidum turnover and nestedness contribute to explain the structure of the fungal microbiome (Figure 4A). In T. aestivum, the contribution of taxa turnover was higher than nestedness (F1, 378 = 36.31, p < 0.001), while for T. turgidum we found the opposite pattern (F1, 598 = 5.71, p = 0.017). This suggests that in T. aestivum there might be a higher replacement of taxa between cultivars, probably driven by stochastic processes, while in T. turgidum the fungal community assembles by gain/loss of fungal taxa, probably driven by selection. To test for this idea, we fit our data to a neutral model (Sloan et al., 2006), and we estimated the ecological drift (as goodness of fit to the neutral model) and the dispersal (as immigration coefficient from the model). Results show that cultivars of T. aestivum tend to have higher R2 and dispersal values, while cultivars of T. turgidum show the opposite pattern (Figure 4B). This suggests that the assembly of seed fungal communities in T. aestivum is more driven by neutral processes, while in T. turgidum is more driven by selection.
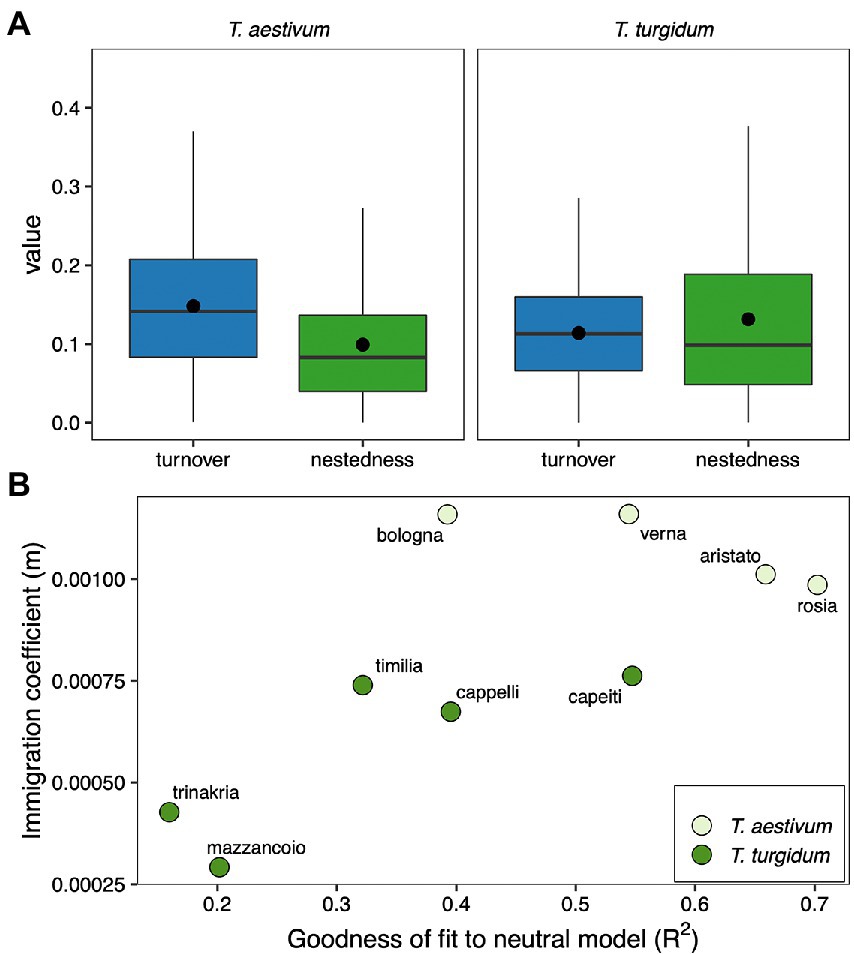
Figure 4. (A) Fungal ASVs turnover and nestedness within the Triticum aestivum (left) and Triticum turgidum (right) groups (dots represent the group mean, clarifying that nestedness is higher than turnover in T. turgidum). The term “value” on the y-axis represents either the turnover or nestedness values. (B) Scatterplot of the goodness of fit to a neutral model (R2) and immigration coefficient of each cultivar of T. aestivum (light green) and T. turgidum (dark green).
Discussion
In this study we analyzed the fungal microbial communities associated with ears of six cereal species, and we found variation of the diversity and structure of the microbiome across species, but weak evidence of co-diversification of plant species and microbiota composition. We then focused on the fungal microbiome of different cultivars within the species T. aestivum and T. turgidum, and we found differences in the microbiota composition between cultivars, with a stronger effect on T. aestivum compared to T. turgidum. This difference posed the question about the existence of different mechanisms within each plant species when assembling their seed microbiome. Further analyses suggested that in T. aestivum the seed fungal microbiome assembly might be driven by the genotype-specific association with fungal taxa, while in T. turgidum this assembly might be more driven by stochastic events.
Differences in the diversity and structure of microbial communities between different plant species have been previously reported (Trivedi et al., 2020). In cereals, previous research found evidence of differentiation of the bacterial microbiota between plant species (Kinnunen-Grubb et al., 2020; Wipf and Coleman-Derr, 2021; Gholizadeh et al., 2022), while reporting little or no variation in the fungal community (Hassani et al., 2020; Sun et al., 2020; Tkacz et al., 2020; Abdullaeva et al., 2021) at different plant compartments. However, to the best of our knowledge, only Abdullaeva et al. (2021) tested this idea on the seed microbiome of different cereal species, suggesting that plant species is a strong driver of the bacterial seed microbiome structure. Similarly, our results show differences in the diversity and structure of fungal microbiome between the different cereal species, although these differences were mostly driven by A. sativa and H. vulgare. When further testing for the co-diversification of plant species and their fungal microbiome, we found a weak support for phylosymbiosis. This is not surprising, also considering that previous studies focusing on the effect of wheat domestication on the plant microbiome found little or no effect of plant species on the fungal microbiome (Hassani et al., 2020; Sun et al., 2020; Tkacz et al., 2020; Abdullaeva et al., 2021), albeit not directly testing for phylosymbiosis.
When further digging into the variation of the seed microbiome between different cultivars, we found that the structure of fungal communities associated with different varieties of T. aestivum and T. turgidum were shaped by plant genotype. A genotype-driven effect on plant microbiome has been previously shown for several plant species (Wagner et al., 2016; Liu et al., 2019; Brown et al., 2020; Wassermann et al., 2022; Malacrinò et al., 2022b), but also among wheat varieties (Kavamura et al., 2020). Indeed, previous research found that wheat genotype can shape the diversity and structure of microbial communities in bulk soil (Yergeau et al., 2020), rhizosphere (Donn et al., 2015; Mahoney et al., 2017; Azarbad et al., 2020; Kavamura et al., 2020; Rossmann et al., 2020; Simonin et al., 2020; Wolińska et al., 2020), roots (Azarbad et al., 2020; Cui et al., 2022), and leaves (Sapkota et al., 2017; Azarbad et al., 2020; Žiarovská et al., 2020). However, these previous studies mainly focused on T. aestivum and on the bacterial communities at each compartment. Latz et al. (2021) focused instead on the fungal microbiome of seeds, leaves, and roots of 12 cultivars of T. aestivum, suggesting that genotype is one of the major drivers of the fungal communities. Our results show that such genotype-driven effect occurs in the seed fungal microbiome of T. aestivum and T. turgidum, with a stronger effect in T. aestivum.
These results might suggest different mechanisms in the seed fungal microbiome assembly in T. aestivum and T. turgidum. When testing this hypothesis, we found that in T. aestivum the fungal community has a higher rate of taxa replacement between cultivars (high turnover), a higher fit to a null assembly model, and higher taxa immigration rates, suggesting that neutral processes are the main driver of seed microbiome assembly. On the other hand, in T. turgidum, we found a higher rate of taxa gain/loss (nestedness), a lower fit to a null assembly model, and lower taxa immigration rates, suggesting that selection of fungal taxa drives the seed fungal community assembly. This finds support in higher differentiation of fungal communities between cultivars of T. aestivum compared to T. turgidum, suggesting that in T. aestivum the seed microbiome is under the influence of stochastic processes, generating a wider differentiation between cultivars, while in T. turgidum selection of the fungal taxa plays a major role, making fungal microbiome assembly more conservative within this group and generating less differences between cultivars. A previous study tested the effect of selection, drift, and dispersal on the assembly of bacterial and fungal communities of rhizosphere, roots, and leaves of T. aestivum and its wild ancestors (Hassani et al., 2020), suggesting that in T. aestivum the assembly of microbial communities is driven by neutral processes, while in its wild ancestors selection played a major role. Thus, we can speculate that the domestication process somehow influenced the way T. aestivum cultivars assemble their microbiome, while this did not happen in T. turgidum, and future studies can focus on understanding the reason behind these differences and their functional consequences. For example, the fact that selection of the seed microbiome is more relaxed in T. aestivum compared to T. turgidum might have consequences on the ability of exerting protection against fungal pathogens (Hassani et al., 2020; Tkacz et al., 2020). In addition, future research can overcome a possible caveat of our study, which is the limited spatial and temporal variability we considered, and a wider study on multiple years and locations might help to further dig into the processes/mechanisms behind wheat microbiome assembly.
Collectively, our results show that the seed fungal microbiome is variable across cereal species, but also among different genotype of T. aestivum and T. turgidum, where it is assembled using different mechanisms. While here we are limited on the extent we can infer about microbiome assembly mechanisms and their functional consequences, future studies are needed to finely understand how plants assemble their own microbial communities, how this influence their fitness, ecology, and evolution, and whether we can manipulate the outcome of plant-microbe interactions to improve the agricultural sustainability, ecosystem restoration and natural resources protection efforts.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/sra, PRJNA848675.
Author contributions
AA and LS conceived the study. IB performed the experiments and collected the data. AM and AA analyzed the data. AM wrote the first draft of the manuscript. All authors commented on previous versions of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by The Italian Ministry of Education, University and Research (MIUR) with “PON Ricerca e competitività 2007–2013” Innovazione di prodotto e di processo nelle filiera dei prodotti da forno e dolciari (PON03PE_00090_01).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1075399/full#supplementary-material
Footnotes
References
Abdelfattah, A., Tack, A. J. M., Wasserman, B., Liu, J., Berg, G., Norelli, J., et al. (2022). Evidence for host-microbiome co-evolution in apple. New Phytol. 234, 2088–2100. doi: 10.1111/nph.17820
Abdelfattah, A., Wisniewski, M., Droby, S., and Schena, L. (2016). Spatial and compositional variation in the fungal communities of organic and conventionally grown apple fruit at the consumer point-of-purchase. Hortic. Res. 3:16047. doi: 10.1038/hortres.2016.47
Abdelfattah, A., Wisniewski, M., Schena, L., and Tack, A. J. M. (2021). Experimental evidence of microbial inheritance in plants and transmission routes from seed to phyllosphere and root. Environ. Microbiol. 23, 2199–2214. doi: 10.1111/1462-2920.15392
Abdullaeva, Y., Ambika Manirajan, B., Honermeier, B., Schnell, S., and Cardinale, M. (2021). Domestication affects the composition, diversity, and co-occurrence of the cereal seed microbiota. J. Adv. Res. 31, 75–86. doi: 10.1016/j.jare.2020.12.008
Azarbad, H., Tremblay, J., Giard-Laliberté, C., Bainard, L. D., and Yergeau, E. (2020). Four decades of soil water stress history together with host genotype constrain the response of the wheat microbiome to soil moisture. FEMS Microbiol. Ecol. 96:fiaa098. doi: 10.1093/femsec/fiaa098
Baselga, A. (2010). Partitioning the turnover and nestedness components of beta diversity. Glob. Ecol. Biogeogr. 19, 134–143. doi: 10.1111/j.1466-8238.2009.00490.x
Bates, D., Mächler, M., Bolker, B., and Walker, S. (2014). Fitting linear mixed-effects models using lme4. Journal of Statistical Software 67, 1–48. doi: 10.18637/jss.v067.i01
Bergna, A., Cernava, T., Rändler, M., Grosch, R., Zachow, C., and Berg, G. (2018). Tomato seeds preferably transmit plant beneficial endophytes. Phytobiomes J. 2, 183–193. doi: 10.1094/PBIOMES-06-18-0029-R
Brown, S. P., Grillo, M. A., Podowski, J. C., and Heath, K. D. (2020). Soil origin and plant genotype structure distinct microbiome compartments in the model legume Medicago truncatula. Microbiome 8:139. doi: 10.1186/s40168-020-00915-9
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Cui, M.-H., Chen, X.-Y., Yin, F.-X., Xia, G.-M., Yi, Y., Zhang, Y.-B., et al. (2022). Hybridization affects the structure and function of root microbiome by altering gene expression in roots of wheat introgression line under saline-alkali stress. Sci. Total Environ. 835:155467. doi: 10.1016/j.scitotenv.2022.155467
Dastogeer, K. M. G., Tumpa, F. H., Sultana, A., Akter, M. A., and Chakraborty, A. (2020). Plant microbiome–an account of the factors that shape community composition and diversity. Current Plant Biology 23:100161. doi: 10.1016/j.cpb.2020.100161
Dixon, P. (2003). VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x
Donn, S., Kirkegaard, J. A., Perera, G., Richardson, A. E., and Watt, M. (2015). Evolution of bacterial communities in the wheat crop rhizosphere. Environ. Microbiol. 17, 610–621. doi: 10.1111/1462-2920.12452
Ewing, C. J., Slot, J., Benítez, M.-S., Rosa, C., Malacrinò, A., Bennett, A., et al. (2021). The foliar microbiome suggests that fungal and bacterial agents may be involved in the beech leaf disease pathosystem. Phytobiomes J. 5, 335–349. doi: 10.1094/PBIOMES-12-20-0088-R
Fox, J., and Weisberg, S. (2019). An R Companion to Applied Regression. 3rd edn. Thousand Oaks, CA: Sage. Available at: https://socialsciences.mcmaster.ca/jfox/Books/Companion
French, E., Kaplan, I., and Enders, L. (2021). Foliar aphid herbivory alters the tomato rhizosphere microbiome, but initial soil community determines the legacy effects. Front. Sustain. Food Syst. 5:629684. doi: 10.3389/fsufs.2021.629684
Frew, A. (2022). Root herbivory reduces species richness and alters community structure of root-colonising arbuscular mycorrhizal fungi. Soil Biol. Biochem. 171:108723. doi: 10.1016/j.soilbio.2022.108723
Gholizadeh, S., Mohammadi, S. A., and Salekdeh, G. H. (2022). Changes in root microbiome during wheat evolution. BMC Microbiol. 22:64. doi: 10.1186/s12866-022-02467-4
Ginnan, N. A., Dang, T., Bodaghi, S., Ruegger, P. M., McCollum, G., England, G., et al. (2020). Disease-induced microbial shifts in citrus indicate microbiome-derived responses to huanglongbing across the disease severity spectrum. Phytobiomes J. 4, 375–387. doi: 10.1094/PBIOMES-04-20-0027-R
Hassani, M. A., Özkurt, E., Franzenburg, S., and Stukenbrock, E. H. (2020). Ecological assembly processes of the bacterial and fungal microbiota of wild and domesticated wheat species. Phytobiomes J. 4, 217–224. doi: 10.1094/PBIOMES-01-20-0001-SC
Hervé, M. (2022). RVAideMemoire: Testing and Plotting Procedures for Biostatistics. R package version 0.9-81-2. Available at: https://cran.r-project.org/package=RVAideMemoire
Kavamura, V. N., Robinson, R. J., Hughes, D., Clark, I., Rossmann, M., and Melo, I. S.De, (2020). Wheat dwarfing influences selection of the rhizosphere microbiome. Sci. Rep. 10,:1452, doi: 10.1038/s41598-020-58402-y
Kembel, S. W., Cowan, P. D., Helmus, M. R., Cornwell, W. K., Morlon, H., Ackerly, D. D., et al. (2010). Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26, 1463–1464. doi: 10.1093/bioinformatics/btq166
Kim, H., Lee, K. K., Jeon, J., Harris, W. A., and Lee, Y.-H. (2020). Domestication of Oryza species eco-evolutionarily shapes bacterial and fungal communities in rice seed. Microbiome 8:20. doi: 10.1186/s40168-020-00805-0
Kinnunen-Grubb, M., Sapkota, R., Vignola, M., Nunes, I. M., and Nicolaisen, M. (2020). Breeding selection imposed a differential selective pressure on the wheat root-associated microbiome. FEMS Microbiol. Ecol. 96:196. doi: 10.1093/femsec/fiaa196
Kumar, S., Stecher, G., Suleski, M., and Hedges, S. B. (2017). TimeTree: a resource for timelines, Timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819. doi: 10.1093/molbev/msx116
Lahti, L., and Shetty, S. (2017). Tools for microbiome analysis in R package. Bioconductor. doi: 10.18129/B9.bioc.microbiome
Latz, M. A. C., Kerrn, M. H., Sørensen, H., Collinge, D. B., Jensen, B., Brown, J. K. M., et al. (2021). Succession of the fungal endophytic microbiome of wheat is dependent on tissue-specific interactions between host genotype and environment. Sci. Total Environ. 759:143804. doi: 10.1016/j.scitotenv.2020.143804
Lenth, R. V. (2022). Emmeans: Estimated Marginal Means, aka Least-Squares Means. R package version 1.7.4-1. Available at: https://github.com/rvlenth/emmeans
Liu, H., Brettell, L. E., Qiu, Z., and Singh, B. K. (2020). Microbiome-mediated stress resistance in plants. Trends Plant Sci. 25, 733–743. doi: 10.1016/j.tplants.2020.03.014
Liu, F., Hewezi, T., Lebeis, S. L., Pantalone, V., Grewal, P. S., and Staton, M. E. (2019). Soil indigenous microbiome and plant genotypes cooperatively modify soybean rhizosphere microbiome assembly. BMC Microbiol. 19:201. doi: 10.1186/s12866-019-1572-x
Mahoney, A. K., Yin, C., and Hulbert, S. H. (2017). Community structure, species variation, and potential functions of rhizosphere-associated bacteria of different winter wheat (Triticum aestivum) cultivars. Front. Plant Sci. 8:132. doi: 10.3389/fpls.2017.00132
Malacrinò, A., Abdelfattah, A., Berg, G., Benitez, M.-S., Bennett, A. E., Böttner, L., et al. (2022a). Exploring microbiomes for plant disease management. Biol. Control 169:104890. doi: 10.1016/j.biocontrol.2022.104890
Malacrinò, A., Karley, A. J., Schena, L., and Bennett, A. E. (2021a). Soil microbial diversity impacts plant microbiota more than herbivory. Phytobiomes J. 5, 408–417. doi: 10.1094/PBIOMES-02-21-0011-R
Malacrinò, A., Mosca, S., Li Destri Nicosia, M. G., Agosteo, G. E., and Schena, L. (2022b). Plant genotype shapes the bacterial microbiome of fruits, leaves, and soil in olive plants. Plants (Basel) 11:613. doi: 10.3390/plants11050613
Malacrinò, A., Wang, M., Caul, S., Karley, A. J., and Bennett, A. E. (2021b). Herbivory shapes the rhizosphere bacterial microbiota in potato plants. Environ. Microbiol. Rep. 13, 805–811. doi: 10.1111/1758-2229.12998
Nilsson, R. H., Larsson, K.-H., Taylor, A. F. S., Bengtsson-Palme, J., Jeppesen, T. S., Schigel, D., et al. (2019). The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 47, D259–D264. doi: 10.1093/nar/gky1022
Paradis, E., and Schliep, K. (2019). Ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. doi: 10.1093/bioinformatics/bty633
R Core Team. (2020). R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. Available at: https://www.R-project.org/
Rossmann, M., Pérez-Jaramillo, J. E., Kavamura, V. N., Chiaramonte, J. B., Dumack, K., Fiore-Donno, A. M., et al. (2020). Multitrophic interactions in the rhizosphere microbiome of wheat: from bacteria and fungi to protists. FEMS Microbiol. Ecol. 96:032. doi: 10.1093/femsec/fiaa032
Sapkota, R., Jørgensen, L. N., and Nicolaisen, M. (2017). Spatiotemporal variation and networks in the mycobiome of the wheat canopy. Front. Plant Sci. 8:1357. doi: 10.3389/fpls.2017.01357
Simonin, M., Dasilva, C., Terzi, V., Ngonkeu, E. L. M., Diouf, D., Kane, A., et al. (2020). Influence of plant genotype and soil on the wheat rhizosphere microbiome: evidences for a core microbiome across eight African and European soils. FEMS Microbiol. Ecol. 96:067. doi: 10.1093/femsec/fiaa067
Sloan, W. T., Lunn, M., Woodcock, S., Head, I. M., Nee, S., and Curtis, T. P. (2006). Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 8, 732–740. doi: 10.1111/j.1462-2920.2005.00956.x
Sun, X., Kosman, E., and Sharon, A. (2020). Stem endophytic mycobiota in wild and domesticated wheat: structural differences and hidden resources for wheat improvement. J. Fungi (Basel) 6:180. doi: 10.3390/jof6030180
Tiziani, R., Miras-Moreno, B., Malacrinò, A., Vescio, R., Lucini, L., Mimmo, T., et al. (2022). Drought, heat, and their combination impact the root exudation patterns and rhizosphere microbiome in maize roots. Environ. Exp. Bot. 203:105071. doi: 10.1016/j.envexpbot.2022.105071
Tkacz, A., Pini, F., Turner, T. R., Bestion, E., Simmonds, J., Howell, P., et al. (2020). Agricultural selection of wheat has been shaped by plant-microbe interactions. Front. Microbiol. 11:132. doi: 10.3389/fmicb.2020.00132
Trivedi, P., Leach, J. E., Tringe, S. G., Sa, T., and Singh, B. K. (2020). Plant–microbiome interactions: from community assembly to plant health. Nat. Rev. Microbiol. 18, 607–621. doi: 10.1038/s41579-020-0412-1
Van Bel, M., Fisher, A. E., Ball, L., Columbus, J. T., and Berlemont, R. (2021). Phylosymbiosis in the rhizosphere microbiome extends to nitrogen cycle functional potential. Microorganisms (Basel) 9:2476. doi: 10.3390/microorganisms9122476
Vancov, T., and Keen, B. (2009). Amplification of soil fungal community DNA using the ITS86F and ITS4 primers. FEMS Microbiol. Lett. 296, 91–96. doi: 10.1111/j.1574-6968.2009.01621.x
Vescio, R., Malacrinò, A., Bennett, A. E., and Sorgonà, A. (2021). Single and combined abiotic stressors affect maize rhizosphere bacterial microbiota. Rhizosphere 17:100318. doi: 10.1016/j.rhisph.2021.100318
Wagner, M. R., Lundberg, D. S., del Rio, T. G., Tringe, S. G., Dangl, J. L., and Mitchell-Olds, T. (2016). Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat. Commun. 7:12151. doi: 10.1038/ncomms12151
Walsh, C. M., Becker-Uncapher, I., Carlson, M., and Fierer, N. (2021). Variable influences of soil and seed-associated bacterial communities on the assembly of seedling microbiomes. ISME J. 15, 2748–2762. doi: 10.1038/s41396-021-00967-1
Wassermann, B., Abdelfattah, A., Wicaksono, W. A., Kusstatscher, P., Müller, H., Cernava, T., et al. (2022). The Brassica napus seed microbiota is cultivar-specific and transmitted via paternal breeding lines. Microb. Biotechnol. 15, 2379–2390. doi: 10.1111/1751-7915.14077
Wen, T., Zhao, M., Liu, T., Huang, Q., Yuan, J., and Shen, Q. (2020). High abundance of Ralstonia solanacearum changed tomato rhizosphere microbiome and metabolome. BMC Plant Biol. 20:166. doi: 10.1186/s12870-020-02365-9
Wipf, H. M. L., and Coleman-Derr, D. (2021). Evaluating domestication and ploidy effects on the assembly of the wheat bacterial microbiome. PLoS One 16:e0248030. doi: 10.1371/journal.pone.0248030
Wolińska, A., Kuźniar, A., and Gałązka, A. (2020). Biodiversity in the rhizosphere of selected winter wheat (Triticum aestivum L.) cultivars—genetic and catabolic fingerprinting. Agronomy (Basel) 10:953. doi: 10.3390/agronomy10070953
Yergeau, É., Quiza, L., and Tremblay, J. (2020). Microbial indicators are better predictors of wheat yield and quality than N fertilization. FEMS Microbiol. Ecol. 96:205. doi: 10.1093/femsec/fiz205
Yu, T., Cheng, L., Liu, Q., Wang, S., Zhou, Y., Zhong, H., et al. (2022). Effects of waterlogging on soybean Rhizosphere bacterial community using V4, LoopSeq, and PacBio 16S rRNA sequence. Microbiol Spectr 10:e0201121. doi: 10.1128/spectrum.02011-21
Zarraonaindia, I., Owens, S. M., Weisenhorn, P., West, K., Hampton-Marcell, J., Lax, S., et al. (2015). The soil microbiome influences grapevine-associated microbiota. MBio 6, e02527–e02514. doi: 10.1128/mBio.02527-14
Keywords: wheat, ears, cultivar, metabarcoding, ITS, phylosymbiosis
Citation: Malacrinò A, Abdelfattah A, Belgacem I and Schena L (2023) Plant genotype influence the structure of cereal seed fungal microbiome. Front. Microbiol. 13:1075399. doi: 10.3389/fmicb.2022.1075399
Edited by:
Mamadou L. Fall, Agriculture and Agri-Food Canada (AAFC), CanadaReviewed by:
Inmaculada Larena, Instituto Nacional de Investigación y Tecnología Agroalimentaria (INIA), SpainJames T. Tambong, Agriculture and Agri-Food Canada (AAFC), Canada
Copyright © 2023 Malacrinò, Abdelfattah, Belgacem and Schena. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahmed Abdelfattah, ✉ YWFiZGVsZmF0dGFoQGF0Yi1wb3RzZGFtLmRl