 Zhenxin Sun
Zhenxin Sun Yun Heacock-Kang
Yun Heacock-Kang Ian A. McMillan
Ian A. McMillan Darlene Cabanas
Darlene Cabanas Jan Zarzycki-Siek
Jan Zarzycki-Siek
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 16 January 2023
Sec. Infectious Agents and Disease
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.1063287
This article is part of the Research Topic Burkholderia spp.-Transmission, Pathogenesis, Host-pathogen interaction, Prevention and Treatment View all 6 articles
Burkholderia pseudomallei (Bp), causing a highly fatal disease called melioidosis, is a facultative intracellular pathogen that attaches and invades a variety of cell types. We previously identified BP1026B_I0091 as a surface attachment protein (Sap1) and an essential virulence factor, contributing to Bp pathogenesis in vitro and in vivo. The expression of sap1 is regulated at different stages of Bp intracellular lifecycle by unidentified regulator(s). Here, we identified SapR (BP1026B_II1046) as a transcriptional regulator that activates sap1, using a high-throughput transposon mutagenesis screen in combination with Tn-Seq. Consistent with phenotypes of the Δsap1 mutant, the ΔsapR activator mutant exhibited a significant reduction in Bp attachment to the host cell, leading to subsequent decreased intracellular replication. RNA-Seq analysis further revealed that SapR regulates sap1. The regulation of sap1 by SapR was confirmed quantitatively by qRT-PCR, which also validated the RNA-Seq data. SapR globally regulates genes associated with the bacterial membrane in response to diverse environments, and some of the genes regulated by SapR are virulence factors that are required for Bp intracellular infection (e.g., type III and type VI secretion systems). This study has identified the complex SapR regulatory network and its importance as an activator of an essential Sap1 attachment factor.
Burkholderia pseudomallei (Bp) is a soil dwelling Gram-negative bacterium that causes the disease melioidosis with considerable morbidity and mortality in a broad range of hosts, including humans, cattle, horses, dolphins, goats, and koalas (Wiersinga et al., 2006; Currie, 2008). This pathogen demonstrates remarkable capabilities of surviving under hostile environments, particularly during interaction with the host immune system (Wiersinga et al., 2012). Bp infections can be acquired through three major routes: percutaneous inoculation via breaks in skin, inhalation of aerosolized water or soil, and ingestion of contaminated water (Dance, 2000; Wiersinga et al., 2006). Bp can disseminate to distant sites resulting in many possible manifestations associated with infection (Wiersinga et al., 2006). The most severe clinical manifestation is melioidosis septic shock where the eradication of Bp is difficult (Wiersinga et al., 2006). Bp has recently gained much interest from western countries despite having a high prevalence in Southeast Asia and Northern Australia (Inglis et al., 2006; Currie et al., 2008; Limmathurotsakul et al., 2016). Bp has been classified as a Tier 1 select agent by the United States. Centers for Disease Control and Prevention (CDC) due to its potential to pose a severe threat to public health and safety (Dance and Alibek, 2005; Gilad et al., 2007).
Burkholderia pseudomallei harbors two circular chromosomes totaling 7.25 mega bases, which contain a large number of genes encoding a wide array of virulence factors (Holden et al., 2004; Wiersinga et al., 2006) to promote successful infection of various hosts and survival within phagocytic and nonphagocytic cells (Jones et al., 1996; Harley et al., 1998). Amongst these virulence factors, adhesins (Balder et al., 2010) and surface attachment factors (BPSL0097/BP1026B_I0091 and BPSS1860/BP1026B_II1992; Heacock-Kang et al., 2021), the Bsa type III secretion system cluster 3 (T3SS3; Sun et al., 2010), a modulator of host cell tubulin (BPSS1818/BP1026B_II1948; Heacock-Kang et al., 2021), host cell autophagy evasion factor (BPSS0015/BP1026B_II0016; Heacock-Kang et al., 2021), the type VI secretion system cluster 5 (T6SS-5; Shalom et al., 2007; Burtnick et al., 2011; Chen et al., 2011), the cytotoxin Burkholderia lethal factor 1 (Cruz-Migoni et al., 2011), and capsular polysaccharide I (Reckseidler et al., 2001) facilitate Bp intracellular infection in the host. Many of these virulence factors are associated with the bacterial membrane. Bp initiates its intracellular life cycle by attaching and entering host cells, followed by rapid escape from endocytic vesicles through T3SS3 expression (Warawa and Woods, 2005; Burtnick et al., 2008). Once in the cytosol, Bp moves freely using BimA-facilitated polymerization of host cell actin (French et al., 2011; Benanti et al., 2015). Upon contact with host cell membranes, Bp induces T6SS-5 mediated membrane fusion with adjacent cells, resulting in multinucleated giant cell (MNGC) formation (Schwarz et al., 2014; Toesca et al., 2014). As recently demonstrated, Bp undergoes dynamic gene expression changes while transiting through host cells (Heacock-Kang et al., 2021). Up to 1,953 genes showed differential gene expression patterns in a stage specific manner during Bp intracellular infection indicating complex regulatory functions (Heacock-Kang et al., 2021; McMillan et al., 2021). The Bp genome is annotated to encode a wide spectrum of regulatory proteins, such as two-component sensor-regulator systems, alternative sigma factors, and different classes of regulatory proteins (Holden et al., 2004). Many of the virulence factors in Bp are controlled by the two-component or quorum sensing systems, indicating that Bp regulates its virulence systems by closely monitoring environmental cues (Jones et al., 1997; Ulrich et al., 2004; Valade et al., 2004; Horton et al., 2013; Schaefers, 2020). Bacterial gene regulation in response to environmental cues is vital for a successful transition from environmental living to intracellular infection (Ernst et al., 1999).
BP1026B_I0091/BPSL0097 (sap1) encodes a surface attachment protein that is expressed within the host cell endocytic vesicles during Bp infection (Heacock-Kang et al., 2021). Sap1 is a virulence factor required for the complete pathogenesis of Bp infecting cell lines and BALB/c mice, in particular contributing to the initial attachment stage of the infection (Heacock-Kang et al., 2021). Although sap1 has been characterized as a significant virulence determinant with a unique gene expression pattern during Bp infection, the transcriptional control of sap1 has yet to be identified. In this study, we used a high-throughput transposon mutagenesis screen coupled with Tn-Seq to identify potential regulators of sap1. We identified BP1026B_II1046 as a transcriptional regulator that activates the sap1 gene and designated it as SapR. Furthermore, we used RNA-Seq to characterize the SapR associated transcriptional network, providing insights into its role during Bp infection.
All manipulations of wildtype Bp were carried out in CDC approved and registered BSL-3 facilities at the University of Hawaiʻi at Mānoa (UHM) John A. Burns School of Medicine (JABSOM) and experiments with the select agent were performed in accordance with the recommended BSL-3 practices (Richmond and McKinney, 2007). Select-agent excluded Bp 1026b ∆asd strain and Bp82 strain (Propst et al., 2010) were manipulated in the BSL-2 laboratory at the UHM. The derivative of Escherichia coli EPmax10B (Bio-Rad) containing lacIq and pir [Lab ID: E1869 (Norris et al., 2009)] were routinely used as a cloning strain. The E. coli conjugal and suicidal strain HPS1-pir-Δasd-mob-Km-Δpro (Lab ID: E463 (Kang et al., 2009)) was used for the mobilization of plasmid containing oriT-origin into B. pseudomallei 1026b ∆asd [B0011 (Norris et al., 2011)] strain.
Growth of E. coli Δasd strains was carried out as previously described with 100 μg/ml of diaminopimelic acid (DAP; Sigma) supplied (Norris et al., 2009). Luria-Bertani (LB) medium (Difco) was generally used to culture all the bacteria. Wildtype Bp strain 1026b was cultured in LB while the B0011 strain (Norris et al., 2009) was cultured in LB with 200 μg/ml diaminopimelic acid (LB + DAP200) or in minimal glucose (MG) media (1 × M9 minimal medium plus 20 mM glucose) containing 1 mM lysine, 1 mM methionine, 1 mM threonine, and 200 μg/ml DAP (3AA + DAP200). Diaminopimelic acid was prepared in 1 M NaOH as a 100 mg/ml stock and was used when necessary as described previously (Barrett et al., 2008). Bp82 (Propst et al., 2010) is a ΔpurM derivative of Bp 1026b, and is an adenine and thiamine auxotroph. To culture Bp82, LB was supplemented with 0.6 mM adenine (LB + Ade) and M9-glucose medium was supplemented with both 0.6 mM adenine and 0.0005% thiamine (Propst et al., 2010). Merodiploids arising from chromosomal integration of the suicidal plasmid are resolved by utilization of the counter selectable marker pheS and sacB. 0.2% (w/v) p-chlorophenylalanine (cPhe; dl-4-chlorophenylalanine; Acros Organics) and 15% sucrose were used in Bp, respectively. LB + 1,000 μg/ml kanamycin was used for purifying the 1026b::T24 insertion mutants. All bacterial growth was carried out at 37°C and shaken cultures were maintained at 225 rpm.
Murine macrophage cell RAW264.7 was cultured in a humidified CO2 incubator at 37°C with 5% CO2. Dulbecco’s modified Eagle’s medium (DMEM) containing 4,500 mg/l glucose with 4 mM L-glutamine was used for culturing cell lines. All culture media were supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS; HyClone). Gibco 100× antibiotic/antimycotic (AntiAnti) was added at a 1 × working concentration (containing 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 250 ng/ml of amphotericin B) to prevent contamination by various bacteria and fungi. RAW264.7 cells were maintained at 60–80% confluency and were passaged by scrapping cells from the Corning™ T-75 flask. The passage numbers of cell lines were carefully recorded. CellBIND multi-well plates were used for cell infection assays.
Oligonucleotides were synthesized through Integrated DNA Technology. Unless otherwise indicated, all restriction enzymes, deoxynucleoside triphosphates, T4 DNA polymerase, T4 polynucleotide kinase, and T4 DNA ligase were purchased from New England Biolabs and used according to the manufacturer’s suggestion. Plasmids and DNA gel bands were isolated using the Zyppy plasmid miniprep kit and Zymoclean gel DNA recovery kit, respectively (Zymo Research Corporation). Escherichia coli competent cells were prepared as previously described (Kang et al., 2007). DNA ligations, restriction endonuclease digestions, and agarose gel electrophoresis were performed according to standard techniques, and all other molecular techniques were carried out according to methods described previously by Sambrook and Russell (2001).
Prokaryotes promoter predictor from Berkeley Drosophila Genome Project (Reese, 2001)1 was used to predict sap1 transcription start site. The transcript start sites of Sap1 was then experimentally mapped using a previously described method by direct sequencing of SMART™ RACE (rapid amplification of cDNA end) products (Tabansky and Nurminsky, 2003). All reagents required for promoter mapping were supplied in the SMART RACE cDNA Amplification kit (Clontech, BD Biosciences). Briefly, Bp wildtype 1026b strain was cultured in 3 ml LB broth and harvested in exponential phase for RNA extraction. Total RNA was prepared using the TRIzol® reagent (Invitrogen, Carlsbad, CA, United States), and isolated with an on-column DNase I treatment, using the Qiagen RNeasy plus mini kit. One μg of total RNA was used to generate RACE cDNA using the SMART RACE cDNA Amplification kit (Clontech, BD Biosciences) and following the manufacturer’s instructions. cDNA ends were amplified with SMART primer (5′-AAGCAGTGGTATCAACGCAGAGTACGCGGG-3′) and gene-specific primer (5′-GTCTTTCGACAGACATCC-3′) at a final concentration of 2 μM. The diluted cDNA was used as a template in a PCR with a second nested gene-specific primer (5′-CAGTTGAATCTCCTGCTGC-3′) and the SMART RACE primer (5′-AAGCAGTGGTATCAACGCAGAGT-3′). PCR products were purified from 2% agarose gel slices and sequenced with a third nested gene-specific primer (5′-AGCGATGCATCGCCGTTC-3′) at the University of Hawaii genomic core facility.
The engineering of Psap1-lacZ reporter strain was performed as described below. About 200 bp from up and down stream flanking the mapped transcriptional start sit was amplified from Bp genomic DNA with primers 5′-GAATGCCGGGTACCGAATGCGCCTG-3′ and 5′-ATGTCGTGTCTCGAATTCGCTCGGGTC-3′. A 400 bp PCR product was digested with EcoRI and KpnI and cloned into mini-Tn7-Km-lacZ vector digested with the same restriction enzymes. A reporter strain was constructed with a single-copy integration of a Psap1-lacZ fusion at a neutral glmS site in the chromosome of B0011 strain using the well-established mini-Tn7 system (Koch et al., 2001; Choi et al., 2005; Kang et al., 2009) with the aid of a nonreplicating helper plasmid pTNS3 encoding the transposase(Kang et al., 2009). Briefly, plasmids mini-Tn7-Km-Psap1-lacZ and pTNS3 was isolated using Zyppy Plasmid Kit. These two plasmids were mixed at an equal molar ratio and transformed into B0011 strain via electroporation. To make electro-competent bacterial cells, B0011 was grown to exponential phase in LB + DAP200 broth and 1 ml bacterial culture was harvested by centrifugation and washed four times with sterile water to remove salts. After electroporation, B0011 cells were immediately resuspended in 2 ml fresh LB + DAP200 and recovered in a 37°C incubator shaking at 225 RPM for 2 h. Cells were plated on LB + DAP200 + Km500 to select for the successful integration of Psap1-lacZ fusion. The B. pseudomallei chromosomes contain three insertion sites downstream of three glmS genes (Choi et al., 2008). Psap1-lacZ fusion was PCR confirmed to insert once at the glmS2 site with primers 5′-ATTAGCTTACGACGCTACACCC-3′ and 5′-ACACGACGCAAGAGCGGAATC-3′. The insertion of mini-Tn7 in bacterial chromosome is quite stable and the selective maintenance is not required (Norris et al., 2010). The reporter strain was purified three times before saving away in the -80°C freezer stocks due to the intrinsic stickiness of Bp cells.
Random mutagenesis was performed in the B0011 Psap1-lacZ fusion strain using the pBT20-gat plasmid containing the Himar1 mariner transposon and its transposase-encoding tnp gene (Kulasekara et al., 2005). Escherichia coli strain E463 (HPS1-pir-Δasd-mob-Km-Δpro) was used as the conjugal donor to introduce transposon plasmid pBT20-gat into B0011 Psap1-lacZ fusion strain. Briefly, the donor and recipient were grown up to mid-exponential phase and the cells were harvested separately by centrifugation at 9000 × g for 1 min at room temperature and washed twice with 1 ml of fresh LB. The pellets were combined at approximately equal cell densities and gently resuspended in 50 ul of LB. The mixture was spotted onto a LB + DAP200 plate for conjugation at 37°C. After 4 h of conjugation, the bacteria were harvested and washed twice with 1 ×M9 buffer, then plated on 10×150 mm agar plates containing MG + DAP 200 μg/ml + 3AA (1 mM of methionine, lysine, and threonine) + 60 μg/ml X-gal + 0.4% glyphosate. Without adding amino acid proline, the donor strain is unable to grow. The recipient strain cannot grow with the presence of glyphosate unless it has pBT20-gat transposon inserted into the chromosomes. The plates were incubated at room temperature for at least 24 h to allow the color to fully develop after colonies were visible following incubation at 37o C (Supplementary Figure S1). The white and dark blue colonies were picked and patched on the same agar to confirm β-galactosidase activity. A total of 230,000 transposon mutant colonies, approximately 40 × coverage of genome, were screened for the β-galactosidase activity. 205 colonies with higher (dark blue) and 220 colonies with lower (white) β-galactosidase activity were pooled separately for transposon junctions identification through Illumina® deep-sequencing (Tn-seq).
Genomic DNA (gDNA) of dark blue and white colony pools was purified with phenol-chloroform extraction and quantified via Nanodrop. Shearing buffer (40% glycerol + 10 mM Tris pH 8.0 + 1 mM EDTA) was used to resuspend 10 μg of gDNA sample to a total volume of 800 μl followed by nebulization. Briefly, the samples were each nebulized for 5 min at 35 p.s.i. of N2 and followed by adding 30 mg/ml of dextran sulfate dissolved in water to each sample to a final concentration of 0.2 μg/ml. One tenth the volume of 3 M sodium acetate was added and vortexed to mix well. To precipitate DNA, an equal volume of ice cold isopropanol was added to the sample and freeze at –80o C for at least 30 min. The samples were centrifuged at 4°C at full speed for 20 min and washed with old 70% ethanol two times. Excessive ethanol was further removed by heated speed vacuum and DNA pellets were resuspended in 16 μl Qiagen EB buffer for Nanodrop quantification. The sheared DNA was end repaired with the NEB Next End Repair Kit according to the manufacturer’s protocol and purified with Qiagen QIAquick PCR Purification Kit following the protocol. Each sample was further divided into four samples and 3′ A-tailing was carried out with Taq polymerase at 70°C for 20 min. The adapters were designed following previously described method with modification for sequence specific to our transposon (Gallagher et al., 2011). Purified DNA samples were digested with XbaI overnight at 37°C to separate the two genome-transposon junctions. Two % TAE agarose gel was used to visualize the DNA samples and the DNA smears between 200 and 400 bp were excised and purified via QIAquick Gel Extraction Kit and eluted with 30 μl of EB buffer. Circularization of adaptor ligated gDNA and exonuclease digestion were performed as previously described using Ampligase heat stable ligase (Gallagher et al., 2011). The transposon ends were amplified with PCR and Tn-seq libraries were built with the NEBNext DNA library prep kit for Ilumina® sequencing based on manufacturer’s protocols. Library samples were cleaned up with MinElute PCR purification Kit and eluted with EB buffer.
The sample was sequenced at the Genetics Core and Confocal Microscopy Facility at University of Hawaiʻi at Mānoa. The dark blue and the white colony sample pools received 3.2 and 4.5 million reads, respectively. Reads were mapped onto the Bp genome with Bowtie (Langmead et al., 2009) using the Galaxy suite (Blankenberg et al., 2001; Giardine et al., 2005; Goecks et al., 2010) to identify the transposon junctions.
The well-established mini-Tn7 integration system was used to construct the 1026b sapR::T24 complement strain (Norris et al., 2010) in BSL-3 laboratory. Briefly, sapR and its predicted native promoter was cloned into mini-Tn7-gat plasmid, yielding mini-Tn7-gat-sapR. The helper plasmid pTNS3 encoding transposase was electroporated with mini-Tn7-gat-sapR into the 1026b sapR::T24 mutant. After recovering in LB for 2 h, cells were harvested by centrifugation in a bio-safety cabinet and washed twice with 1x M9 minimal salts buffer to remove trace nutrient from LB. Mutants contains the mini-Tn7 insertion were selected for on MG + 0.4% GS plate. Colonies were purified on the same medium three times and insertion at one of the three glmS sites in the chromosome was verified by PCR as previously described (Kang et al., 2009). This extensive purification procedure is very important and highly recommended, because GS is bacteriostatic, rather than bactericidal, at this effective concentration. 1026b sapR::T24 mutant, sapR::T24 complement, and the wildtype strain were used in the RT-PCR validation study and RNA-seq analysis.
We used our previously published primers and probe for sap1 in this real-time PCR experiment (Heacock-Kang et al., 2021). The primers and probe were designed using Integrated DNA Technologies Primer Quest software2 following the criteria (Son et al., 2007) that were mentioned below. Briefly, the amplicon size should range from 60 to 100 bp and the primer melting temperatures were designed for 62°C, when more one primer pair were designed a melting temperature difference of less than 4°C for each pair would be recommended. The probe melting temperatures were designed to be 5–10°C higher than those for the corresponding primer pairs. To eliminate the possibility of nonspecific binding, primer and probe sequences were also subjected to BLASTN analysis against the B. pseudomallei wildtype 1026b genome, and the primers were also ensured to had no significant complementarity at the 3′ end and the probe to had no significant complementarity at the 5′ end to nonspecific locations on Bp 1026b genome.
The same preparation of RNA as described above for high throughput RNA-Seq experiment was used for real-time RT-PCR. Three μg of RNA was used for cDNA synthesis with random hexamer and SuperScript III (Invitrogen) per manufacturer’s protocol. To ensure no genomic DNA contamination was carried over from the RNA extraction, a control lacking reverse transcriptase was included for cDNA synthesis. The final cDNA product was dissolved in 1,200 μl RNase and DNase free water. In order to yield more accurate normalization analysis (Vandesompele et al., 2002) and control for variations between runs, three housekeeping genes (Heacock-Kang et al., 2021) (BPSL0602, BPSL2502, and BPSS2061 express consistently across all conditions) were chosen to be amplified at the same time with sap1 on one 96-well plate. The primers set to amplify sap1 are forward_5′-CATCGTTGCATTTTCGG-3′, reverse_ 5′-GGAATCCAGATTGTCTTTCG-3′, and probe_/56-FAM/TTCGTCAGTTGAATCTCCTGC/3BHQ_1/. Briefly, master mixtures were made and aliquoted into sub mixtures for each gene assayed. The final volume of each real-time PCR mixture is 25 μl containing 10 μl of cDNA, 12.5 μl of iQ Supermix (Bio- Rad), 120 nM of each primer, and 12 nM probe. Real-time PCR was performed with an iCycler iQ (Bio-Rad) using the following protocol: denaturation at 95°C for 10 min and then 55 cycles of amplification and quantification at 95°C for 20 s and at 65°C for 45 s. For each gene, reactions with amplification efficiency variant within 5% are subjected to data analysis. Real-time PCR fold changes were calculated using the amplification plot method and the available macro functions for data analyses of real-time PCR (DART-PCR; Peirson et al., 2003).
Deletion mutation of sapR gene in Bp82 was made by allelic-replacement method based on the concept of the double homologous recombination as previously described (Barrett et al., 2008) with minor modification. Bp82 was grown in LB + Ade to exponential phase and harvested by centrifugation. Electro-competent Bp82 cells were made as described above (Barrett et al., 2008). Briefly, cells were washed four times with sterile ddH2O for electroporation with the allelic-replacement and suicidal vector pEX18Km-pheS-ΔsapR-gat-FRT2. After 2–3 h recovery in fresh LB + Ade, the bacterial cells were plated onto MG + 0.4% GS Ade/Thi agar plate. Merodiploids arising from chromosomal integration of the suicide plasmid are resolved by utilization of the counter selectable marker pheS. Colonies were purified on MG + 0.4% GS + 0.2% cPhe + Ade/Thi agar plates to resolve the merodiploid state. Plates were incubated at 37°C for 48 h, colonies were patched with toothpicks onto both MG + 0.4% GS + 0.2% cPhe + Ade/Thi and LB + Ade + Km 500 μg/ml plates. Mutants unable to grow in the presence of kanamycin were purified on MG + 0.4% GS Ade/Thi. Single colonies were purified on MG + 0.4% GS Ade/Thi media. The same purification procedure was repeated three times ensuring no wildtype background carrying over. Further screening and confirmation of mutants were performed by PCR using oligonucleotides pairs 5′-TGTCGATGCCGTCGAAG-3′/5′-CACATCCTGATGTACAAGCGGATCA-3′ (upstream junction) and 5′-GGCATCGAGAGTGTCATAC-3′/5′-CGTTGATGGGCTTGACCTCGATC-3′ (downstream junction) to amplify the chromosome and vector junctions. Primers 5′-CTGTCGATGCCGTCGAAG-3′ and 5′-GGCATCGAGAGTGTCATAC-3′ annealed to the upstream region of the chromosome outside of the region cloned for allelic replacement and the downstream region, respectively.
An unmarked strain was required to be constructed before engineering the complement strain. The gat-FRT2 marker excision was performed using a previously described protocol (Choi et al., 2008) with minor modifications. Briefly, ΔsapR mutant was electroporated with 200 ng of pFlpe4, a curable Flp recombinase-expressing plasmid. Hundred μl aliquot of 1 ml recovery culture was plated on LB + Ade + Km500 + 0.2% l-rhamnose plate and incubated at 30°C for 48 h or until colonies were grown up. Single colonies were picked and patched on the same media to incubate at 30°C to ensure full excision. Fully-grown patches were streaked out on LB + Ade plate for single colony isolation and pFlpe4 plasmid was cured by incubating at 42°C. The successful excision of gat marker was confirmed by PCR and no growth on MG + 0.4% GS Ade/Thi plate. The well-established mini-Tn7 integration system was used to construct the Bp82 ΔsapR complement strain (Norris et al., 2010) in BSL-2 laboratory as described above. Bp82 ΔsapR mutant and ΔsapR/ comp were used in the mammalian cell infection assay.
Bp82, Bp82 Δsap1, Bp82 ΔsapR, and Bp82 ΔsapR complement strains were grown up overnight in LB + Ade broth in a shaking incubator at 37°C. The bacteria were harvested and diluted 100 times in 250 μl fresh LB + Ade and placed in a 96-well plate. This experiment was conducted in triplicate. Growth analysis was carried out in the BioTek EL × 808IU plate reader at 37°C with constant shaking and OD600 measurement was recorded every 30 min. Data was analyzed and plotted in Prism.
pBLAC is a derivative of pMLBAD, a low-copy broad-host-range plasmid (Lefebre and Valvano, 2002) that can replicate in Bp (Barrett et al., 2008). The sapR gene is placed immediately downstream of a lac operator and the expression of sapR can be induced by IPTG. pBLAC was constructed by replacing the E. coli araBAD arabinose-inducible promoter and araC, transcriptional regulator gene, of pMLBAD (Lefebre and Valvano, 2002) with lac promoter and lacI-encoded Lac repressor of mini-Tn7-lacZ that has been routinely used in our lab for Bp wildtype 1026b strain gene induction experiments. pBLAC was designed to be used for sapR induction study in Bp82 strains and its usage can be extended to Burkholderia thailandensis (Bt), the experimental surrogate of Bp that is manipulated in a BSL-1 laboratory. New vector pBLAC carries the backbone sequence derived from broad-host-range vector pMLBAD, including mob, gene required for conjugal transfer of plasmid; rep, replication protein gene; ori, origin of replication; the dhfr, dihydrofolate reductase gene encoding trimethoprim resistance; rrnB, transcriptional terminator; MCS, multiple cloning site. SapR cloned into the MCS was transcribed from the lac promoter and regulated by Lac repressor. Trimethoprim (Tp) 150 μg/ml was used in media to maintain the plasmid in Bp82 strains.
RAW264.7 macrophage cells were cultured in the T-75 corning flask to confluence and washed with pre-warmed sterile 1 x PBS twice. Then, cells were scraped off from the flask and seeded in 250 μl of DMEM without AntiAnti at 7 × 104 cells per well into 48-well Corning CellBIND culture plates. Cells were allowed to attach overnight before the infection study. Bp82 strain grown in LB medium + Ade overnight at 37°C was used to infect RAW264.7 macrophages at MOI of 10:1 in the 48-well CellBIND plates. Bacteria were diluted in 1 × PBS for plating and numeration to obtain the initial infecting bacteria number. At 1 h post infection, DMEM containing unattached bacteria were removed and the cell monolayers were washed three times with pre-warmed sterile 1 × PBS. Monolayers were lysed with 500 μl 1 × PBS + 0.2% Triton X-100, and serial diluted in 1 × PBS. Fifty μl of 100 (undiluted lysate), 10−1, 10−2, 10−3 dilutions were plated onto LB + Ade 12-well agar plates and colonies were enumerated after 48 h incubation at 37°C as the attached bacteria number. The attachment efficiency was determined by dividing the attached number by the initial infecting bacteria number. The experiment was carried out in triplicate and the numbers represent the average of all three replicates with the error bars representing the SEM. The student t-test was used to determine the statistical significance between attachment efficiencies of the different strains.
Intracellular replication assay was carried out as previously described (Norris et al., 2011) with slight modifications. Briefly, RAW264.7 murine macrophage cells were infected with Bp82, mutant and complement strains at an MOI of 10:1 in an aminoglycoside protection assay. RAW264.7 cells were cultured and seeded into a 48-well Corning CellBIND culture plates as described above. After allowing the infection to progress for 1 h, the medium containing bacteria was removed, and the monolayers were washed three times with 1 × PBS to remove any unattached bacteria. Fresh DMEM + Ade with 1,000 μg/ml each of amikacin and kanamycin was added to the monolayers to kill any non-internalized bacteria and inhibit extracellular bacterial growth for the remainder of the assay. During the assay, medium was removed from the wells at 2, 8, and 24 hpi (hours post infection), and the infected cell monolayers were washed, lysed and diluted as described above. Bacteria colonies were numerated after 48 h incubation at 37°C, and data was calculated and plotted in Prism. All the assays were performed in triplicate, and the SEM were calculated for each.
Burkholderia pseudomallei wildtype strain1026b and sapR::T24 insertion mutant, were grown overnight in 3 ml LB broth, subcultured to grow to mid exponential phase for RNA extraction. Total RNA from 12 ml culture of each strain was isolated using TRIzol® reagent and purify through QIAGEN RNeasy plus mini kit according to the manufacturer’s instructions. Sequencing was performed by the Tufts University Core Facility Genomics on the Illumina HiSeq 2500 platform after rRNA depletion and library generation. More than 20 million reads with > 96% alignment rate to Bp genome were generated for each triplicate sample from each strain. The fastq files were uploaded to Rockhopper tool (McClure et al., 2013) designed specifically for analysis of bacterial RNA-Seq data. The normalized expression values generated in the Rockhopper analysis, in which the sapR mutant was compared to the wild type control, were used for the differential gene expression analysis. Genes with a false discovery rate (q-value) of < 0.01 were considered to be significantly differentially expressed and were designated for further analysis. A subset of these significantly differentially expressed genes with log2FC ≤ −1 and ≥ 1 were used for Clusters of Orthologous Groups of proteins (COG) functional analysis and subcellular localization analysis for their gene products. The subcellular localization was computationally predicted through PSORTb V3.0 (Yu et al., 2010). The list of predicted gene across the whole genome was downloaded from Burkholderia.com to use in the analysis. Graphs were generated in Prism. Differentially regulated gene clusters (q-value < 0.01) identified from RNA-Seq were mapped along two Bp chromosomes using WoPPER web server (Puccio et al., 2017) to visualize their physical localizations.
To find regulators of sap1 using high-throughput transposon mutagenesis and deep sequencing, we must first determine the approximate sap1 regulatory region by identifying its transcriptional start site. Using the neural network based promoter prediction server of the Berkeley Drosophila Genome Project (BDGP; Reese, 2001), we found two possible transcriptional start sites. The first possible transcriptional start site is 182 bp before the sap1 start codon and has a confidence value of 0.81. The second possible transcriptional start site is 281 bp before the sap1 start codon and has a confidence value of 1.00. Transcription of sap1 could occur from one or both of these possible transcriptional start sites. To experimentally determine if one or both of these transcriptional start sites is used, we employed the well-established non-radioactive SMART RACE method (Tabansky and Nurminsky, 2003). As shown in Figure 1A, one SMART product approximately 500 bp in size was obtained. Sanger sequencing determined that the sap1 transcriptional start site is 281 bp upstream of its start codon, confirming the high confidence prediction from BDGP (Figures 1B,C). From this information, we determined the promoter of sap1 (Psap1) and identified the −10 and − 35 elements (Figure 1C). The sap1 promoter region encompassing 200 bps flanking either side of the predicted sap1 transcriptional start site was utilized to identify potential regulators of sap1 (Figure 1C).
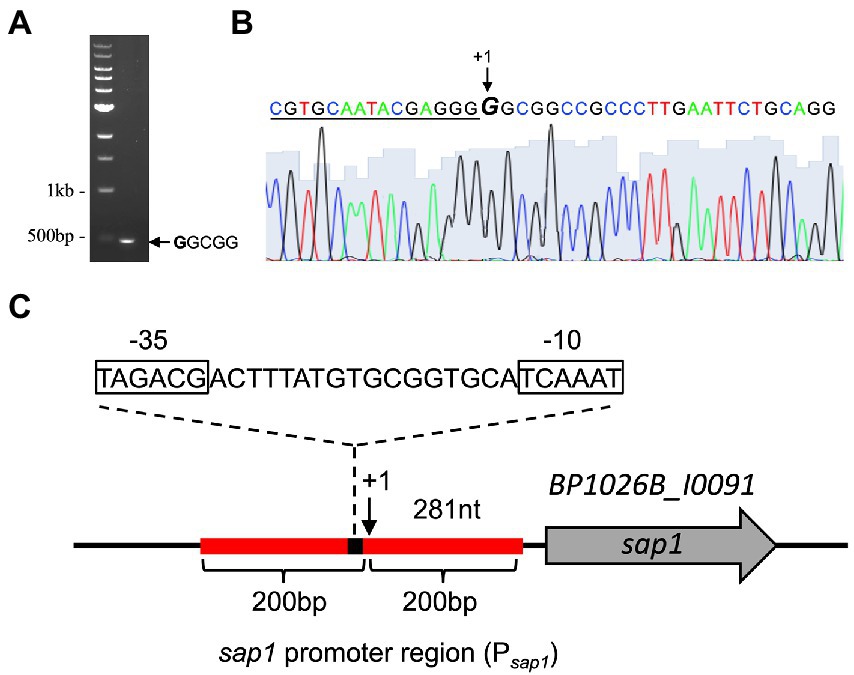
Figure 1. SMART RACE mapping identifies the transcriptional start site for sap1. (A) One SMART product was observed after PCR amplification of the cDNA with SMART primer and a second nested gene-specific primer. (B) The sap1 transcriptional start site (indicated by + 1 at the GGCGG sequence) was identified by sequencing the purified PCR product with a third nested gene-specific primer. The underlined nucleotides indicate the SMART primer sequence. (C) Schematic representation of sap1 locus. The sap1 transcript start site is at 281 bp upstream of its start codon; the sap1 promoter region, 200 bp flanking the predicted transcriptional start site (indicated by red line), was used to identify potential regulators of sap1; the sequences of predicted − 10 and − 35 elements were shown.
We designed a high-throughput strategy to identify the regulator(s) for sap1 using transposon mutagenesis of the select-agent exempt strain Bp 1026b Δasd (B0011) and deep sequencing (Figure 2; Gallagher et al., 2011; Norris et al., 2011). A reporter strain was constructed with a single-copy integration of a Psap1-lacZ fusion at a neutral site on the chromosome of B0011 using the mini-Tn7 system (Koch et al., 2001; Choi et al., 2005; Kang et al., 2009). Random mutagenesis was then performed on this B0011 reporter fusion strain, utilizing the mariner transposon on pBT20-gat (Kulasekara et al., 2005). To ensure comprehensive coverage of both chromosomes, a total of 230,000 transposon mutant colonies, approximately 40 × coverage of Bp genome, were screened for variations in β-galactosidase activity. Colonies with increased β-galactosidase activity would have dark blue color indicating that the transposon insertions disrupted genes that repress expression from the sap1 promoter (Figure 2B). On the other hand, if a gene that activates expression from the sap1 promoter was mutated, lower β-galactosidase activity would be observed, and colonies would be less blue than the basal level (Figure 2B). Several hundreds of both dark blue and white colonies were patched to ensure a consistent β-galactosidase activity pattern. Eventually, 205 dark blue colonies and 220 white colonies were pooled separately and the transposon junctions were identified by Tn-seq49. From the pool of white colonies and through this analysis, we identified BP1026B_II1046, a hypothetical protein with a DNA binding motif. BP1026B_III046 shows similarities to MurR/RipR DNA binding transcriptional regulators (COG1737) with an N-terminal helix-turn-helix like HTH_6 domain (Pfam: PF01418) and a C-terminal a SIS (Sugar ISomerase) domain (cd05013), which is found in many phosphosugar isomerases and phosphosugar binding proteins (Sørensen and Hove-Jensen, 1996; Finn et al., 2016; Lu et al., 2020). In addition, both Phyre2 (Kelley et al., 2015) and I-TASSER (Zhang, 2009; Roy et al., 2012; Yang and Zhang, 2015) predicted that BP1026B_II1046 was homologous to Vibrio vulnificus NanR further indicating that BP1026B_II1046 is a regulator. We, therefore, designated BP1026B_II1046 as sapR for the remainder of this study.
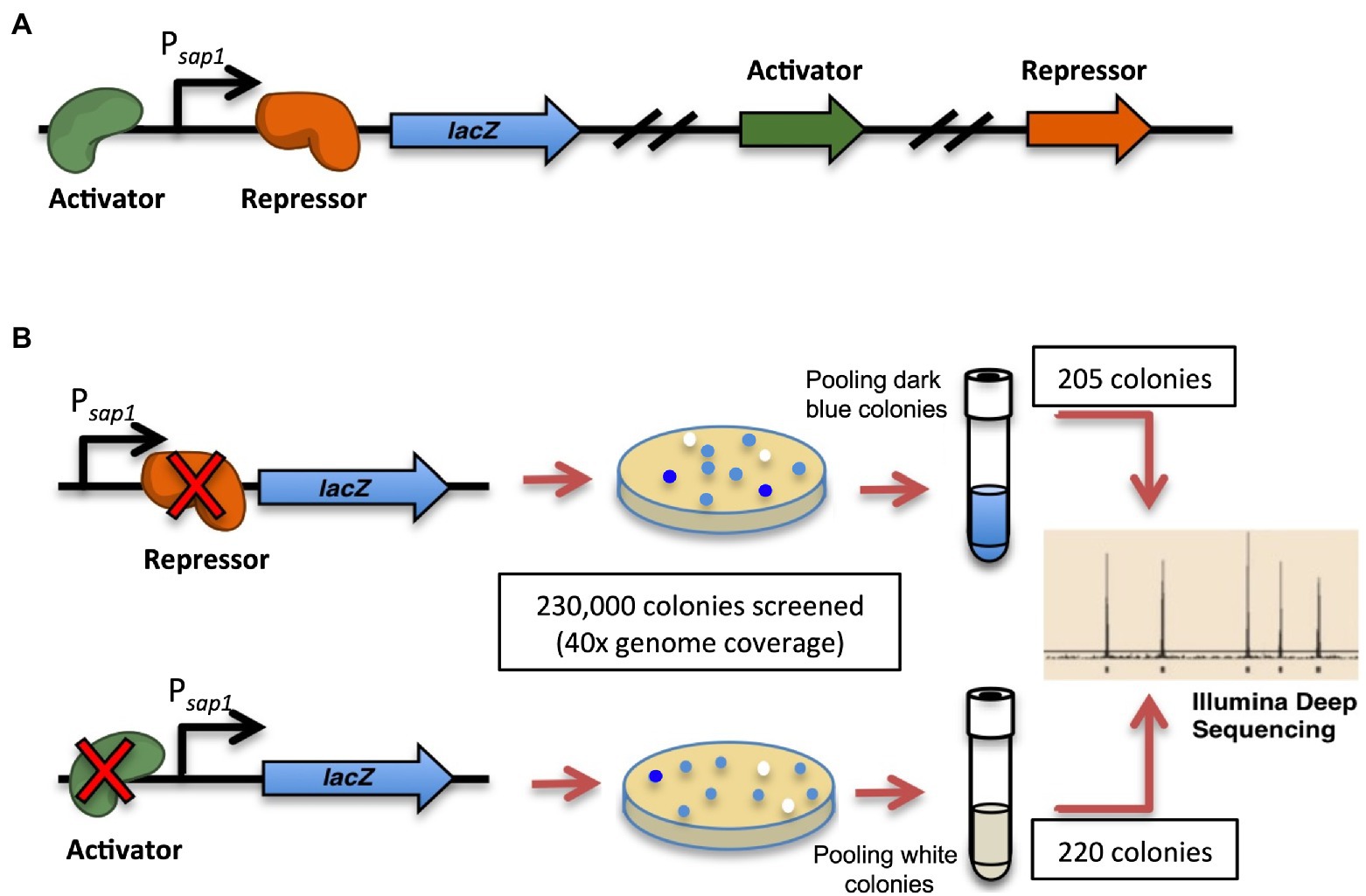
Figure 2. Strategy to identify potential sap1 regulator(s): high-throughput transposon mutagenesis and Tn-Seq. (A) A reporter strain was generated with a single-copy integration of a Psap1-lacZ fusion at a neutral site on the chromosome of B0011 strain. (B) Random mutagenesis was performed on the reporter strain using mariner transposon pBT20-gat. A total of 230,000 transposon mutant colonies, approximately 40 × coverage of genome, were screened for their β-galactosidase activities. Colonies with higher (205) and lower (220) expression of β-galactosidase activity were pooled separately and the transposon junctions were identified by Tn-Seq.
We used qRT-PCR to verify SapR regulatory activity on sap1 (Table 1). RNA was isolated from Bp 1026b wildtype strain and a sapR mutant strain from the Bp 1026b T24 transposon mutant library (Borlee et al., 2017). In the absence of SapR, the sap1 gene was down-regulated with a log2FC (fold change) of −0.78 compared to wildtype Bp 1026b, consistent with the lower β-galactosidase activity in our initial transposon mutagenesis screening (Table 1). We performed beta-gal assay (Son et al., 2008) in the Psap1-lacZ reporter strains of Bp82 and Bp82 ΔsapR mutant to further quantify the regulatory activity of SapR. The beta-galactosidase activity was significantly reduced in the ΔsapR mutant, and the reduction level agreed with our RNA-seq and RT-PCR results (Supplementary Figure S2). Additionally, from our previous Bp single cell ‘TRANSITomic’ analysis that revealed dynamic gene-expression flux as Bp transit through RAW264.7 macrophage at defined stages: vacuole entry; cytoplasmic escape and replication; and membrane protrusion (Heacock-Kang et al., 2021), we identified that both sap1 and sapR were up-regulated in the endocytic vesicle. This finding suggests a coordinated expression of sap1 and sapR during intracellular infection (Supplementary Figure S4). Together these data indicate that SapR is an activator of the sap1 gene.

Table 1. Summary of experimental data indicating that SapR positively regulates sap1.
To further validate the SapR regulatory function of sap1, attachment assays that measure the efficiency of bacterial adherence to the host cell surface were carried out, using the select-agent excluded Bp82 strain derived from wildtype strain 1026b. RAW264.7 murine macrophages were infected with strains Bp82, Bp82 ΔsapR, Bp82 ΔsapR-attTn7::sapR complement (Bp82 ΔsapR/comp), and Bp82 Δsap1 at an MOI of 10:1. The ΔsapR mutant was significantly defective in attachment to RAW264.7 monolayers, showing 48.16% of Bp82 attachment ability (Figure 3A). This is comparable to the attachment defect of the Δsap1 mutant exhibiting 46.72% wildtype attachment (Figure 3A). This result shows that the ΔsapR mutant has a similar phenotype to the Δsap1 mutant during host cell infection and suggests that SapR and Sap1 act in a similar pathway. The attachment defect of the ΔsapR mutant was restored by single copy complementation of the sapR gene.
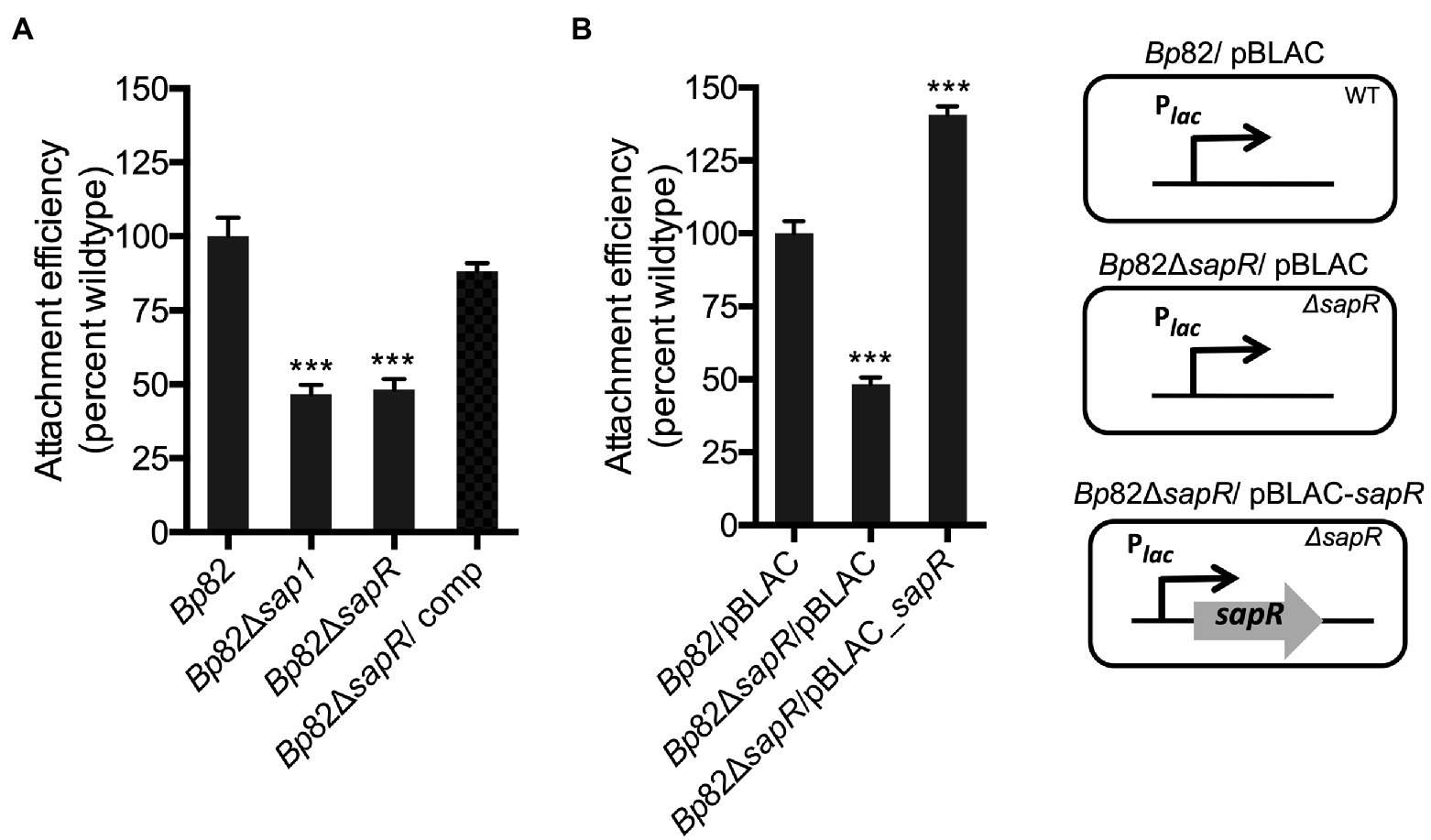
Figure 3. SapR is required to activate sap1 for attaching to host cells. Attachment assays on RAW264.7 macrophage monolayers were performed to further validate SapR’s role as an activator for sap1. (A) The Bp82 ΔsapR mutant exhibited significant attachment deficiency to RAW264.7 cells, comparing to the Bp82 strain. This deficiency is comparable to the defect in attachment of the Bp82 Δsap1 strain. Complement with a single copy of sapR restored the impaired attachment to wildtype level. (B) Bp82 ΔsapR mutant/pBLAC displayed drastic defect in its attachment ability. Whereas the constitutive expression of sapR in ΔsapR mutant/pBLAC-sapR by inducing the culture with 1 mM IPTG increased bacterial attachment significantly. Data in bar graphs represent means ± s.e.m. and analyzed via unpaired t-test. ***p < 0.001.
In addition, we over expressed SapR in trans to compare the attachment efficiencies with Bp82 and Bp82 ΔsapR mutant. RAW264.7 murine macrophages were infected with Bp82/pBLAC, Bp82 ΔsapR/pBLAC, and the over-expressed sapR strain Bp82 ΔsapR/pBLAC-sapR all under IPTG induction (Figure 3B). The strain ΔsapR /pBLAC-sapR that over expresses SapR demonstrated a significantly increased ability to attachment to RAW264.7 cells, at approximately 140% of Bp82 (Figure 3B). This observation that increased expression of sapR leads to increased attachment provides further evidence that SapR is an activator of sap1. Taken together, these data suggest that both SapR and Sap1 have roles in the host cell attachment process.
To investigate if the attachment deficiencies of the ΔsapR mutant lead to subsequent decreased in intracellular replication, an intracellular replication assay was carried out in RAW264.7 macrophage cells. RAW264.7 cells were infected with an MOI of 10:1 with Bp82, Bp82 ΔsapR, Bp82 ΔsapR-attTn7::sapR complement (Bp82 ΔsapR/comp), and Bp82 Δsap1 strains (Figure 4A). At 2 h post infection (hpi), no significant difference was observed initially amongst all the strains indicating a similar number of bacteria were uptake by macrophage cells. As the infection progressed, the intracellular replication capability of Bp82 ΔsapR was significantly impaired, replicating at 45% of Bp82 at 8 hpi and 48% of Bp82 at 24 hpi (Figure 4B). The defect of the ΔsapR mutant was restored by single copy complementation of the sapR gene (Figures 4A,B). These results are similar in the Bp82 Δsap1 mutant showing defects at 8 h and 24 hpi, replicating at 35 and 18 percent of Bp82 (Figure 4B). In vitro growth analysis showed that these mutants have similar growth patterns to wildtype and complement strains (Figure 4C). Taken together, the results indicate that the defect in intracellular replication of the Bp82 ΔsapR mutant is due to impaired pathogenesis rather than defect in in vitro fitness.
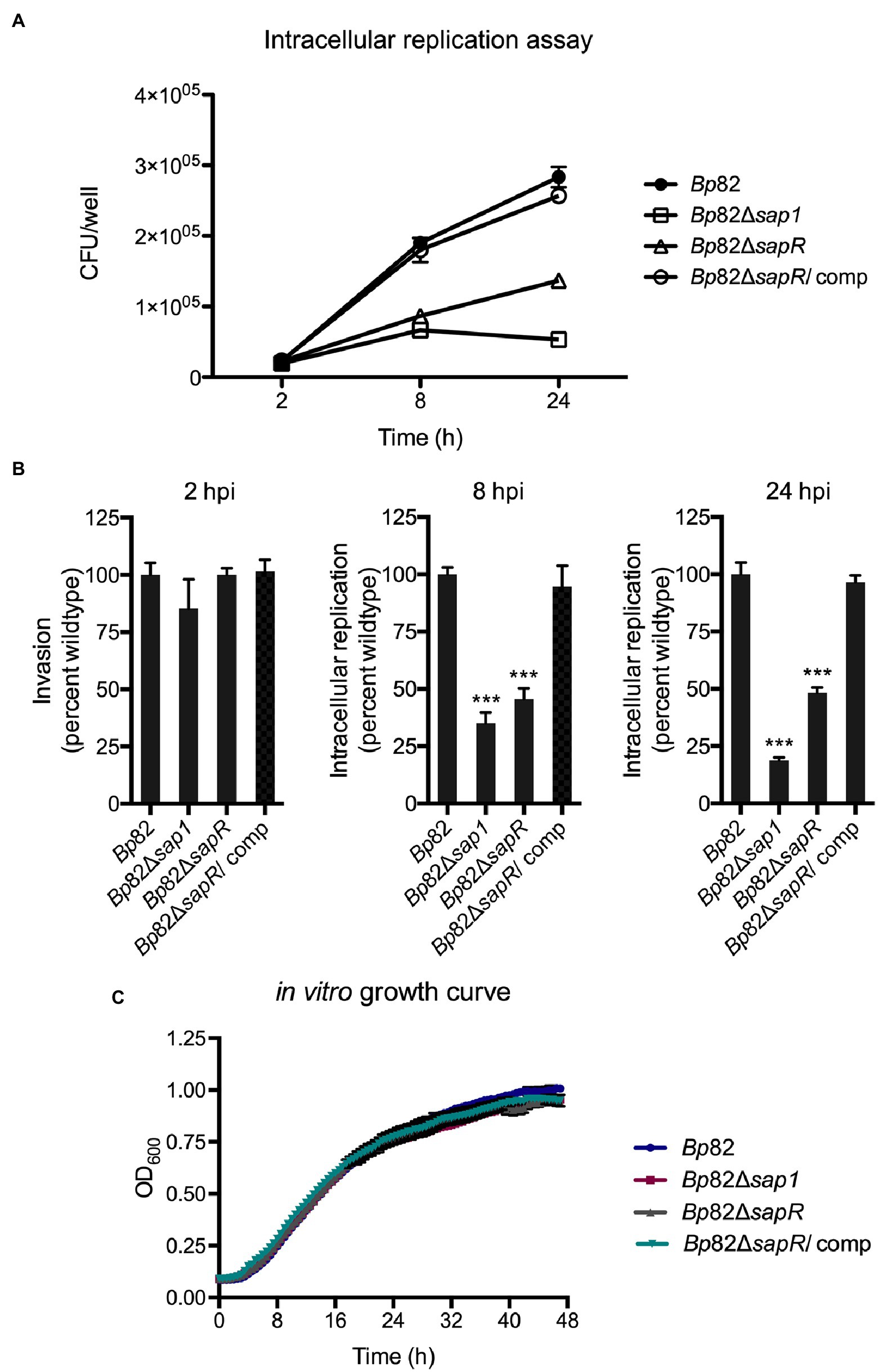
Figure 4. The ΔsapR mutant is highly defective during intracellular replication. (A) Bp82, Δsap1, ΔsapR, and ΔsapR/comp strains were analyzed for their ability to replicate in RAW264.7 murine macrophages at initial MOI of 10:1. CFU/well of each strain was numerated at 2, 8, and 24 h post infection (hpi). (B) The percent wildtype were calculated and the averages of triplicates were presented with error bars representing SEM. The Δsap1 and ΔsapR mutants displayed significant defect in replication at 8 and 24 hpi compared to the Bp82 strain. Data in bar graphs represent means ± s.e.m. and analyzed via unpaired t-test. ***p < 0.001. (C) In vitro growth analysis of Bp82, Δsap1, ΔsapR, and ΔsapR/comp in LB + Ade liquid media. Mutants show no in vitro growth defects compared to wildtype strain while growing under the same conditions.
We have shown that SapR controls the expression of sap1 and that there is a functional correlation between these two proteins. To further understand the role of SapR in the context of Bp pathophysiology, we sought to determine the regulation network of SapR. We compared the transcriptome profile from Bp 1026b wildtype and 1026b sapR::T24 mutant strains growing under the same conditions using RNA-Seq analysis. The sap1 gene was differentially expressed with a log2FC of −0.56 and q-value (the false discovery rate) of 9.56E-93, further confirming that SapR is a positive regulator of sap1 (Table 1). To gain a better understanding of the SapR regulatory network, we did a further investigation on the role of SapR regulating membrane associate components which was summarized in Table 2. This table listed interesting findings of differentially expressed membrane associate components that are either known to associate with Bp pathogenesis or newly identified membrane-associated clusters from WoPPER analysis in our study.
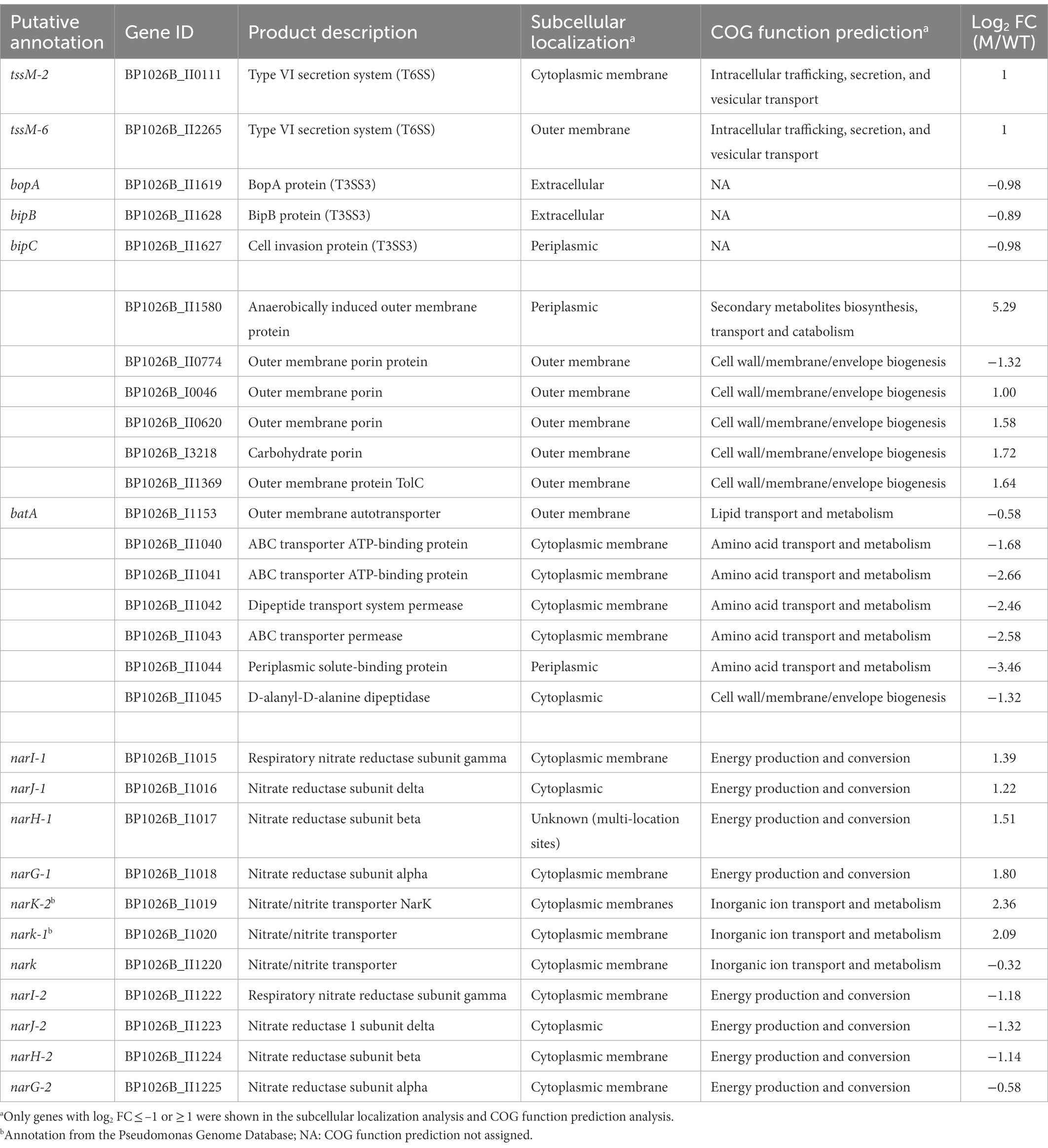
Table 2. Genes regulated by SapR encoding products closely associated with cell envelope.
In total, 483 genes were regulated by SapR using a statistical cut off of q-value < 0.01 and a log2FC cut off of ≤ −1 or ≥ 1 (Supplementary Table S1; Figure 5A). Among these 483 differentially expressed genes (DEGs), there are 67 predicted RNA transcripts, and 416 annotated open reading frames. Of the 416 protein coding genes, 56% are assigned a subcellular location and 44% are unknown (Figure 5B). Some of these SapR-regulated proteins are associated with the cytoplasmic membrane (14%), periplasmic space (3%), outer membrane (3%) and extracellular space (2%; Figure 5B). Cytoplasmic membrane associated proteins showed a large enrichment to 24% when increasing the stringency to a log2FC cut off of ≤ −2 or ≥ 2 (Figure 5C). BP1026B_I1015, BP1026B_I1020 and BP1026B_II1220, BP1026B_II1225 associated with cell envelope for nitrogen metabolism were regulated by SapR with the mean of log2FC > 1 and < −1 (Table 2). SapR also regulated known virulence factors associated with pathogenesis. SapR regulates components of type III and type VI secretion systems (T3SS3 and T6SS). BP1026B_II0111 (tssM-2) and BP1026B_II2265 (tssM-6) encoding an inner membrane protein TssM essential for the function of a sub-complex of the T6SS. Both genes were significantly up-regulated in the sapR mutant with log2FC of 1 (Table 2).
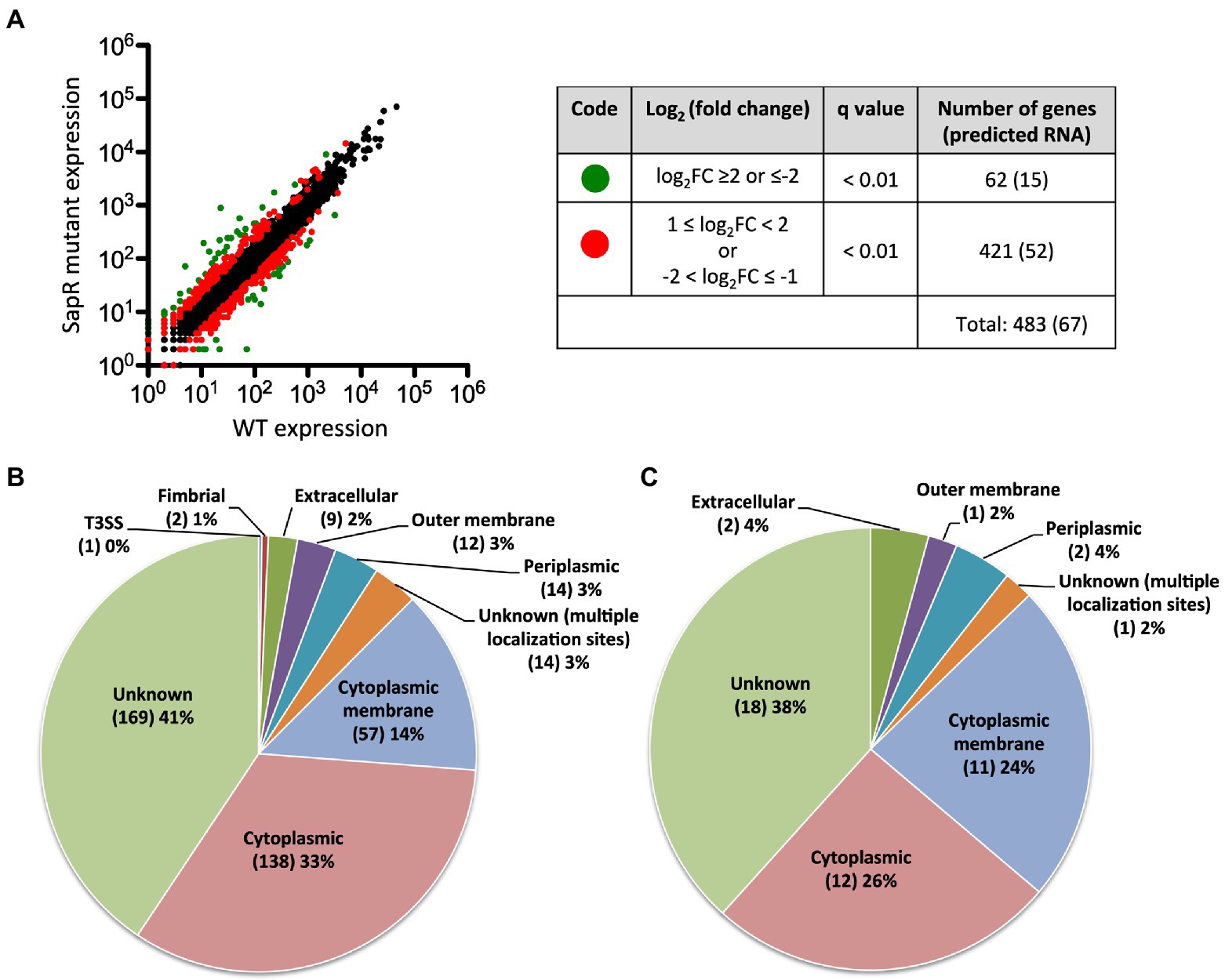
Figure 5. SapR differentially regulates genes in many subcellular locations. (A) Expression values generated from Rockhopper of all detected genes were used to create the plot. A total of 483 DEGs are highlighted. Sixty-two genes with a q-value < 0.01 and a log2FC ≤ −2 or ≥ 2 are highlighted in green, and 421 genes with 1 ≤ log2FC < 2 or − 2 < log2FC ≤ −1 are highlighted in red. Numbers of predicted RNAs shown in parentheses were not included in this analysis. (B) The 416 DEGs with a cut off of a log2FC ≤ −1 or ≥ 1 were further analyzed based on their assigned subcellular localization from Burkholderia.com. (C) A large enrichment was showed in proteins associated with cytoplasmic membrane while increasing the stringency to cut off of a log2FC ≤ −2 or ≥ 2. Number of genes in each category was presented in parenthesis followed by its percentage.
BP1026B_II1619 (BopA), an effector of T3SS3 responsible for phagosome escape (Gong et al., 2011), and two translocators of T3SS3, BP1026B_II1628 (BipB) involved in stimulating cell-to-cell fusion and BP1026B_II1627 (BipC) bound to host F-actin to facilitate internalization of Bp into host cells (Kang et al., 2016; Vander Broek et al., 2017), were down-regulated in the sapR mutant. In addition, SapR regulates many outer membrane porins (BP1026B_II0620, BP1026B_II0774, BP1026B_II1369, BP1026B_I0046 and BP1026B_I3218) generally responsible for cellular permeability and antibiotic resistance (Choi and Lee, 2019). Furthermore, an autotransporter BP1026B_I1153 (batA; Lafontaine et al., 2019) was down-regulated in the sapR mutant. We also observed altered expression of genes near the sapR gene, namely BP1026B_II1040–BP1026B_II1045, amongst the highest down-regulated genes in the SapR mutant (Table 2; Supplementary Figure S3). These include a dipeptidase, dipeptide and ABC transport systems, and a periplasmic binding protein that could be involved in peptidoglycan metabolism. The sapR mutant was complemented by inserting a copy of sapR back into the genome via the mini-Tn7 system. This demonstrates that the transposon mutation of sapR is unlikely to cause polar effects on the adjacent genes. This evidence strongly suggests that SapR regulates many proteins associated with the cell envelope that are also tied to Bp pathogenesis.
Among the 483 DEGs, 249 were up regulated and 234 were down regulated (Figure 6; Supplementary Table S1). Approximately 50% of DEGs are classified as hypothetical proteins, proteins of unknown function, proteins with no COG prediction, or hypothetical RNA transcripts (Figure 6). The rest of DEGs encode core functions such as lipid, inorganic ion, carbohydrate, or amino acid transport and metabolism, energy production and conversion, secondary metabolites biosynthesis, and cell wall/membrane/envelope biogenesis (Figure 6). Among those functional categories, lipid transport and metabolism and energy production and conversion account for the largest proportions of the analyzed DEGs (Figure 6). This suggests that SapR controls many pathways of unknown function and several known pathways including lipid metabolism and energy utilization.
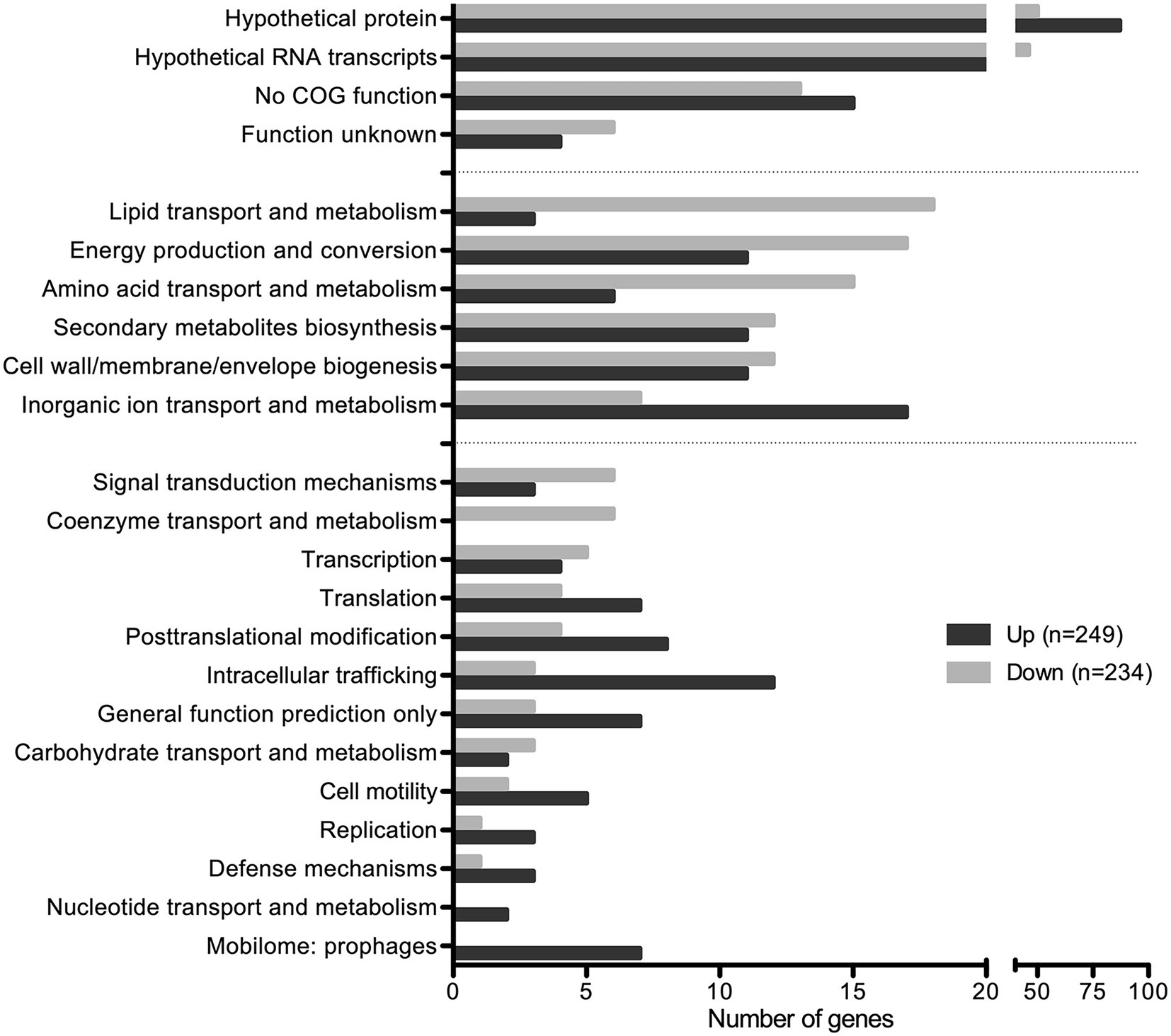
Figure 6. SapR disproportionally regulates hypothetical proteins. Differentially expressed genes (483) with statistical analysis (q-value < 0.01) combined with a cut off of log2 FC ≤ −1 or ≥ 1 were analyzed by the Clusters of Orthologous Genes (COG) functional predictions. Hypothetical proteins, proteins of unknown function, proteins with no COG prediction, or hypothetical RNA transcripts account for approximately 50% of DEGs. Among the rest, the top functional categories (29%) regulated by SapR are lipid transport and metabolism, energy production and conversion, amino acid transport and metabolism, cell wall/membrane/envelope biogenesis, secondary metabolites biosynthesis, transport and catabolism, and inorganic ion transport and metabolism.
To understand what genomic regions are activated and repressed by SapR, we analyzed the RNA-seq data set using WoPPER (Puccio et al., 2017). WoPPER is a web tool based on Position RElated genomics Data Analysis (PREDA) with the integration of gene expression data and the physical position of genes along the genome to identify differentially expressed chromosomal regions in bacteria (Puccio et al., 2017). WoPPER analysis identified 72 gene clusters up-regulated and 73 gene clusters down-regulated in the sapR mutant strain (Supplementary Figure S5). This analysis highlights the broad reach that SapR has on gene expression throughout the entire genome and indicates that SapR has the ability to activate and repress transcription. This analysis also identifies two nitrogen metabolism gene clusters that are differently regulated by SapR (Supplementary Figure S5; Table 2; Supplementary Table S2). One of these gene clusters (BP1026B_I1015–BP1026B_I1020) is located on chromosome I (Cluster ID: 21) and up-regulated, while the second (BP1026B_II1220, BP1026B_II1222-BP1026B_II1225) is located on chromosome II (Cluster ID: 30) and down-regulated in the sapR mutant. These nitrate reduction gene clusters share 69–79% sequence homology (Mangalea et al., 2017) but have different expression patterns controlled by SapR indicating possible distinct roles for each. This evidence suggests that Bp may exploit different nitrogen metabolism operons to survive in diverse environments.
Once being exposed to various environments, the bacterial cell surface is the front line for sensing environmental cues followed by transducing signals inside the cell to alter gene expression and adapt to new conditions. Bacteria have evolved sophisticated regulatory strategies to modulate their cell surface components. Many membrane-associated Bp virulence factors, including T6SS-5 and T3SS3, are controlled by transcriptional regulators (Sun et al., 2010; Chen et al., 2011; Lennings et al., 2018). Sap1 was recently identified to be a critical Bp attachment factor for intracellular infection (Heacock-Kang et al., 2021), but there is no information on how it is regulated. To identify possible regulators of the sap1 gene, we first mapped the transcriptional start site of sap1 and defined its promoter region, which was used for the high-throughput random transposon mutagenesis screen. BP1026B_II1046 (SapR) was identified as a potential regulator for sap1. SapR models with 100.0% confidence to V. vulnificus NanR that responses specifically to ManNAc-6P the catabolic intermediate of sialic acid present on the mucus layer of human intestines (Roy et al., 2010; Hwang et al., 2013; Kelley et al., 2015). Through the present manuscript we show that SapR has a role in pathogenesis consistent with how NanR regulation is critical for V. vulnificus pathogenesis (Hwang et al., 2013). We identified a functional correlation between Sap1 and SapR, where SapR is also important for attachment and infection in RAW264.7 murine macrophage cells. The ΔsapR mutant showed impaired attachment efficiency comparable to Δsap1 mutant. Its attachment defect was restored and increased to 140% of wildtype when SapR was constitutively expressed on a plasmid. The involvement of SapR during infection was also confirmed by the fact that ΔsapR mutant had a significant defect during intracellular replication in macrophage cells. The expression of sapR is up-regulated within the endocytic vesicle during intracellular infection, the same intracellular niche that sap1 is expressed (Heacock-Kang et al., 2021; Supplementary Figure S4). This indicates that sap1 and sapR expression are highly coordinated during Bp intracellular infection.
We utilized an RNA-seq approach and identified that SapR regulates genes locally and globally within the Bp genome. In addition to showing that SapR regulates genes on both chromosomes, 23% of the SapR regulated proteins are localized to the cellular envelope. BP1026B_II1045, the neighboring gene to sapR, was highly down-regulated in ΔsapR mutant and encodes D-alanyl-D-alanine dipeptidase sharing 33% amino acid identity with Enterococcus faecium VanX responsible for vancomycin resistance (Lessard and Walsh, 1999). Vancomycin is an important drug for the treatment of Gram-positive bacterial infections, but is rarely used to treat Gram-negative bacterial infections due to the limited permeability of the outer membrane (Lessard and Walsh, 1999). However, vanX homologs in Gram-negative bacteria have been reported in multiple physiological roles including peptidoglycan metabolism that may contribute to preventing activation of the host immune system (Hilbert et al., 1999). SapR also regulates other genes involved in a putative dipeptide transport system for cell survival under starvation conditions (Lessard and Walsh, 1999) including a periplasmic solute-binding protein (BP1026B_II1044), an ABC transporter permease (BP1026B_II1043), a dipeptide transport system permease (BP1026B_II1042), and two ABC transporter ATP-binding proteins (BP1026B_II1041, BP1026B_II1040). Modifying peptidoglycan under stress to reduce intracellular defense stimulation has been exploited by intracellular pathogens to promote adaption to eukaryotic intracellular niche (Chaput et al., 2006; Rico-Pérez et al., 2016; García-Del Portillo, 2020). These data suggest that SapR up-regulates this gene cluster within the endocytic vesicle and it could be a critical step for Bp adaptation to intracellular survival. SapR also regulates other membrane-associated genes encoding outer membrane porins (BP1026B_II0620, BP1026B_II0774, BP1026B_II1369, BP1026B_I0046 and BP1026B_I3218), the autotransporter BP1026B_I1153 (BatA), anaerobically induced outer membrane protein BP1026B_II1580 and two TssM proteins BP1026B_II0111 (tssM-2) and BP1026B_II2265 (tssM-6). BatA localized on the bacterial surface is protective in vaccination against lethal aerosol infection with Bp and Burkholderia mallei (Lafontaine et al., 2019). TssM protein is a three-transmembrane spanning segment inner membrane protein, directly interacting with TssL by its N-terminal domain (Ma et al., 2009; Zoued et al., 2014). TssM is an essential protein for the function of a sub-complex of the T6SS comprised of four cell envelope proteins TssL, TssM, TagL, and TssJ (Aschtgen et al., 2010; Burtnick et al., 2011; Lennings et al., 2018). Although tssM-2 and tssM-6 belong to T6SS systems that are not generally believed to be important for complete pathogenesis in infecting mammalian cells, our data showing these components are regulated by SapR could provide genomic clues for the identification of its additional functions. In addition, SapR regulates BP1026B_II1369, which encodes an outer membrane channel forming protein TolC. This channel is involved in the expulsion of diverse molecules from the cell including protein toxins and antibacterial drugs (Koronakis et al., 2004). TolC is ubiquitous among Gram-negative bacteria, and has been tied to bacterial virulence and pathogenesis in Enterobacter, Borrelia, Salmonella, Vibrio, Legionella, Francisella (Masi and Pagès, 2013). Taken together these data suggest that SapR regulates virulence factors associated with the cell membrane and plays a role in dynamic modification of Bp membrane components for adaptation to changes of its residing environment.
Through WoPPER analysis, the narGHJI-1/narK-1 complex (BP1026B_I1015 - BP1026B_I1020; ChI_Cluster ID 21) was shown to be up-regulated in the sapR mutant that encodes a nitrate reductase system, responsible for nitrate/nitrite transport and nitrate reduction in the absence of oxygen (Moreno-Vivián et al., 1999; Mangalea et al., 2017; Supplementary Figure S5). The narX/narL two-component regulatory system upstream of the narGHJI operon used in sensing and regulating nitrate metabolism in response to exogenous nitrate (Mangalea et al., 2017) was not influenced by SapR. BP1026B_II1220 and BP1026B_II1222-BP1026B_II1225 encode another nitrate/nitrite transporter (narK) and nitrate reductase complex (narGHJI-2) on the second chromosome (ChII_Cluster ID 30; Supplementary Figure S5), respectively. Interestingly, narGHJI-2 and BP1026B_II1220 were significantly down-regulated in the sapR mutant suggesting that this gene cluster is activated by SapR. This evidence indicates that Bp may exploit different nitrogen metabolism operons to survive in diverse environments.
In summary, this study has identified SapR as a positive transcriptional regulator for Sap1 and shown that SapR regulates membrane-associated virulence factors, peptidoglycan, lipid, and nitrogen metabolism. Future investigation on the genome wide binding sites of SapR will aid in a better understanding of the molecular mechanism of SapR regulation. Overall, our data presented here contribute to valuable insights into the complex regulation network during Bp intracellular infection.
The data presented in the study are deposited in the GEO repository, accession number GSE220579.
ZS, YH-K, and TH designed the experiments. ZS, YH-K, DC, and JZ-S conducted the experiments. ZS, YH-K, IM, and TH analyzed the data and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This project was supported by the United States National Institutes of Health (NIH)/National Institute of Allergy and Infectious Diseases (NIAID) grant number R21AI123913 awarded to TH.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1063287/full#supplementary-material
Aschtgen, M. S., Thomas, M. S., and Cascales, E. (2010). Anchoring the type VI secretion system to the peptidoglycan: TssL, TagL, TagP. What else? Virulence 1, 535–540. doi: 10.4161/viru.1.6.13732
Balder, R., Lipski, S., Lazarus, J. J., Grose, W., Wooten, R. M., Hogan, R. J., et al. (2010). Identification of Burkholderia mallei and Burkholderia pseudomallei adhesins for human respiratory epithelial cells. BMC Microbiol. 10:250. doi: 10.1186/1471-2180-10-250
Barrett, A. R., Kang, Y., Inamasu, K. S., Son, M. S., Vukovich, J. M., and Hoang, T. T. (2008). Genetics tools for allelic-replacement in Burkholderia species. Appl. Environ. Microbiol. 74, 4498–4508. doi: 10.1128/AEM.00531-08
Benanti, E. L., Nguyen, C. M., and Welch, M. D. (2015). Virulent Burkholderia species mimic host actin polymerases to drive actin-based motility. Cells 161, 348–360. doi: 10.1016/j.cell.2015.02.044
Blankenberg, D., Von Kuster, G., Coraor, N., Ananda, G., Lazarus, R., Mangan, M.,, et al. (2001). Current Protocols in Molecular Biology. New York, NY: John Wiley & Sons, Inc.
Borlee, G. I., Plumley, B. A., Martin, K. H., Somprasong, N., Mangalea, M. R., Islam, M. N., et al. (2017). Genome-scale analysis of the genes that contribute to Burkholderia pseudomallei biofilm formation identifies a crucial exopolysaccharide biosynthesis gene cluster. PLoS Negl. Trop. Dis. 11:e0005689. doi: 10.1371/journal.pntd.0005689
Burtnick, M. N., Brett, P. J., Harding, S. V., Ngugi, S. A., Ribot, W. J., Chantratita, N., et al. (2011). The cluster 1 type VI secretion system is a major virulence determinant in Burkholderia pseudomallei. Infect. Immun. 79, 1512–1525. doi: 10.1128/IAI.01218-10
Burtnick, M. N., Brett, P. J., Nair, V., Warawa, J. M., Woods, D. E., and Gherardini, F. C. (2008). Burkholderia pseudomallei type III secretion system mutants exhibit delayed vacuolar escape phenotypes in RAW 264.7 murine macrophages. Infect. Immun. 76, 2991–3000. doi: 10.1128/IAI.00263-08
Chaput, C., Ecobichon, C., Cayet, N., Girardin, S. E., Werts, C., Guadagnini, S., et al. (2006). Role of AmiA in the morphological transition of Helicobacter pylori and in immune escape. PLoS Pathog. 2:e97. doi: 10.1371/journal.ppat.0020097
Chen, Y., Wong, J., Sun, G. W., Liu, Y., Tan, G. Y. G., and Gan, Y. H. (2011). Regulation of type VI secretion system during Burkholderia pseudomallei infection. Infect. Immun. 79, 3064–3073. doi: 10.1128/IAI.05148-11
Choi, K.-H., Gaynor, J. B., White, K. G., Lopez, C., Bosio, C. M., Karkhoff-Schweizer, R. A. R., et al. (2005). A Tn7-based broad-range bacterial cloning and expression system. Nat. Methods 2, 443–448. doi: 10.1038/nmeth765
Choi, U., and Lee, C. R. (2019). Distinct roles of outer membrane Porins in antibiotic resistance and membrane integrity in Escherichia coli. Front. Microbiol. 10:953. doi: 10.3389/fmicb.2019.00953
Choi, K.-H., Mima, T., Casart, Y., Rholl, D., Kumar, A., Beacham, I. R., et al. (2008). Genetic tools for select-agent-compliant manipulation of Burkholderia pseudomallei. Appl. Environ. Microbiol. 74, 1064–1075. doi: 10.1128/AEM.02430-07
Cruz-Migoni, A., Hautbergue, G. M., Artymiuk, P. J., Baker, P. J., Bokori-Brown, M., Chang, C. T., et al. (2011). A Burkholderia pseudomallei toxin inhibits helicase activity of translation factor eIF4A. Science 334, 821–824. doi: 10.1126/science.1211915
Currie, B. J. (2008). Advances and remaining uncertainties in the epidemiology of Burkholderia pseudomallei and melioidosis. Trans. R. Soc. Trop. Med. Hyg. 102, 225–227. doi: 10.1016/j.trstmh.2007.11.005
Currie, B. J., Dance, D. A., and Cheng, A. C. (2008). The global distribution of Burkholderia pseudomallei and melioidosis: an update. Trans. R. Soc. Trop. Med. Hyg. 102, S1–S4. doi: 10.1016/S0035-9203(08)70002-6
Dance, D. A. (2000). Ecology of Burkholderia pseudomallei and the interactions between environmental Burkholderia spp. and human–animal hosts. Acta Trop. 74, 159–168. doi: 10.1016/S0001-706X(99)00066-2
Dance, D.A., and Alibek, F.a. (eds.) (2005). Bioterrorism and Infectious Agents. (pp. 99–145). Springer Science+Business Media Inc., New York, NY.
Ernst, R. K., Guina, T., and Miller, S. I. (1999). How intracellular bacteria survive: surface modifications that promote resistance to host innate immune responses. J. Infect. Dis. 179, S326–S330. doi: 10.1086/513850
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., Mitchell, A. L., et al. (2016). The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285. doi: 10.1093/nar/gkv1344
French, C. T., Toesca, I. J., Wu, T. H., Teslaa, T., Beaty, S. M., Wong, W., et al. (2011). Dissection of the Burkholderia intracellular life cycle using a photothermal nanoblade. Proc. Natl. Acad. Sci. 108, 12095–12100. doi: 10.1073/pnas.1107183108
Gallagher, L. A., Shendure, J., and Manoil, C. (2011). Genome-scale identification of resistance functions in Pseudomonas aeruginosa using Tn-seq. MBio 2, e00315–e00310. doi: 10.1128/mBio.00315-10
García-Del Portillo, F. (2020). Building peptidoglycan inside eukaryotic cells: a view from symbiotic and pathogenic bacteria. Mol. Microbiol. 113, 613–626. doi: 10.1111/mmi.14452
Giardine, B., Riemer, C., Hardison, R. C., Burhans, R., Elnitski, L., Shah, P., et al. (2005). Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15, 1451–1455. doi: 10.1101/gr.4086505
Gilad, J., Harary, I., Dushnitsky, T., Schwartz, D., and Amsalem, Y. (2007). Burkholderia mallei and Burkholderia pseudomallei as bioterrorism agents: national aspects of emergency preparedness. IMAJ 9, 499–503.
Goecks, J., Nekrutenko, A., Taylor, J., and The Galaxy, T. (2010). Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11:R86. doi: 10.1186/gb-2010-11-8-r86
Gong, L., Cullinane, M., Treerat, P., Ramm, G., Prescott, M., Adler, B., et al. (2011). The Burkholderia pseudomallei type III secretion system and BopA are required for evasion of LC3-associated phagocytosis. PLoS One 6:e17852. doi: 10.1371/journal.pone.0017852
Harley, V. S., Dance, D. A., Tovey, G., McCrossan, M. V., and Drasar, B. S. (1998). An ultrastructural study of the phagocytosis of Burkholderia pseudomallei. Microbios 94, 35–45.
Heacock-Kang, Y., McMillan, I. A., Norris, M. H., Sun, Z., Zarzycki-Siek, J., Bluhm, A. P., et al. (2021). The Burkholderia pseudomallei intracellular 'TRANSITome'. Nat. Commun. 12:1907. doi: 10.1038/s41467-021-22169-1
Hilbert, F., García-del Portillo, F., and Groisman, E. A. (1999). A periplasmic D-alanyl-D-alanine dipeptidase in the gram-negative bacterium Salmonella enterica. J. Bacteriol. 181, 2158–2165. doi: 10.1128/JB.181.7.2158-2165.1999
Holden, M. T., Titball, R. W., Peacock, S. J., Cerdeño-Tárraga, A. M., Atkins, T., Crossman, L. C., et al. (2004). Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei. Proc. Natl. Acad. Sci. U. S. A. 101, 14240–14245. doi: 10.1073/pnas.0403302101
Horton, R. E., Grant, G. D., Matthews, B., Batzloff, M., Owen, S. J., Kyan, S., et al. (2013). Quorum sensing negatively regulates multinucleate cell formation during intracellular growth of Burkholderia pseudomallei in macrophage-like cells. PLoS One 8:e63394. doi: 10.1371/journal.pone.0063394
Hwang, J., Kim, B. S., Jang, S. Y., Lim, J. G., You, D. J., Jung, H. S., et al. (2013). Structural insights into the regulation of sialic acid catabolism by the Vibrio vulnificus transcriptional repressor NanR. Proc. Natl. Acad. Sci. U. S. A. 110, E2829–E2837. doi: 10.1073/pnas.1302859110
Inglis, T. J., Rolim, D. B., and Sousa Ade, Q. (2006). Melioidosis in the Americas. Am. J. Trop. Med. Hyg. 75, 947–954. doi: 10.4269/ajtmh.2006.75.947
Jones, A. L., Beveridge, T. J., and Woods, D. E. (1996). Intracellular survival of Burkholderia pseudomallei. J. Bacteriol. 64, 782–790. doi: 10.1128/iai.64.3.782-790.1996
Jones, A. L., DeShazer, D., and Woods, D. E. (1997). Identification and characterization of a two-component regulatory system involved in invasion of eukaryotic cells and heavy-metal resistance in Burkholderia pseudomallei. Infect. Immun. 65, 4972–4977. doi: 10.1128/iai.65.12.4972-4977.1997
Kang, Y., Norris, M. H., Barrett, A. R., Wilcox, B. A., and Hoang, T. T. (2009). Engineering of tellurite-resistant genetic tools for single-copy chromosomal analysis of Burkholderia spp. and characterization of the Burkholderia thailandensis betBA operon. Appl. Environ. Microbiol. 75, 4015–4027. doi: 10.1128/AEM.02733-08
Kang, Y., Son, M. S., and Hoang, T. T. (2007). One step engineering of T7-expression strains for protein production: increasing the host-range of the T7-expression system. Protein Expr. Purif. 55, 325–333. doi: 10.1016/j.pep.2007.06.014
Kang, W. T., Vellasamy, K. M., Rajamani, L., Beuerman, R. W., and Vadivelu, J. (2016). Burkholderia pseudomallei type III secreted protein BipC: role in actin modulation and translocation activities required for the bacterial intracellular lifecycle. PeerJ 4:e2532. doi: 10.7717/peerj.2532
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. (2015). The Phyre2 web portal for protein modelling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Koch, B., Jensen, L. E., and Nybroe, O. (2001). A panel of Tn7-based vectors for insertion of the gfp marker gene or for delivery of cloned DNA into gram-negative bacteria at a neutral chromosomal site. J. Microbiol. Meth. 45, 187–195. doi: 10.1016/S0167-7012(01)00246-9
Koronakis, V., Eswaran, J., and Hughes, C. (2004). Structure and function of TolC: the bacterial exit duct for proteins and drugs. Annu. Rev. Biochem. 73, 467–489. doi: 10.1146/annurev.biochem.73.011303.074104
Kulasekara, H. D., Ventre, I., Kulasekara, B. R., Lazdunski, A., Filloux, A., and Lory, S. (2005). A novel two-component system controls the expression of Pseudomonas aeruginosa fimbrial cup genes. Mol. Microbiol. 55, 368–380. doi: 10.1111/j.1365-2958.2004.04402.x
Lafontaine, E. R., Chen, Z., Huertas-Diaz, M. C., Dyke, J. S., Jelesijevic, T. P., Michel, F.,, et al. (2019). The autotransporter protein BatA is a protective antigen against lethal aerosol infection with Burkholderia mallei and Burkholderia pseudomallei. Vaccine X 1:100002. doi: 10.1016/j.jvacx.2018.100002
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Lefebre, M. D., and Valvano, M. A. (2002). Construction and evaluation of plasmid vectors optimized for constitutive and regulated gene expression in Burkholderia cepacia complex isolates. Appl. Environ. Microbiol. 68, 5956–5964. doi: 10.1128/AEM.68.12.5956-5964.2002
Lennings, J., West, T. E., and Schwarz, S. (2018). The Burkholderia type VI secretion system 5: composition, regulation and role in virulence. Front. Microbiol. 9:3339. doi: 10.3389/fmicb.2018.03339
Lessard, I. A., and Walsh, C. T. (1999). VanX, a bacterial D-alanyl-D-alanine dipeptidase: resistance, immunity, or survival function? Proc. Natl. Acad. Sci. U. S. A. 96, 11028–11032. doi: 10.1073/pnas.96.20.11028
Limmathurotsakul, D., Golding, N., Dance, D. A. B., Messina, J. P., Pigott, D. M., Moyes, C. L., et al. (2016). Predicted global distribution of Burkholderia pseudomallei and burden of melioidosis. Nat. Microbiol. 1:15008. doi: 10.1038/nmicrobiol.2015.8
Lu, S., Wang, J., Chitsaz, F., Derbyshire, M. K., Geer, R. C., Gonzales, N. R., et al. (2020). CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 48, D265–D268. doi: 10.1093/nar/gkz991
Ma, L. S., Lin, J. S., and Lai, E. M. (2009). An IcmF family protein, ImpLM, is an integral inner membrane protein interacting with ImpKL, and its walker a motif is required for type VI secretion system-mediated Hcp secretion in Agrobacterium tumefaciens. J. Bacteriol. 191, 4316–4329. doi: 10.1128/JB.00029-09
Mangalea, M. R., Plumley, B. A., and Borlee, B. R. (2017). Nitrate sensing and metabolism inhibit biofilm formation in the opportunistic pathogen Burkholderia pseudomallei by reducing the intracellular concentration of c-di-GMP. Front. Microbiol. 8:1353. doi: 10.3389/fmicb.2017.01353
Masi, M., and Pagès, J. M. (2013). Structure, function and regulation of outer membrane proteins involved in drug transport in Enterobactericeae: the OmpF/C–TolC case. Open Microbiol. J 7, 22–33. doi: 10.2174/1874285801307010022
McClure, R., Balasubramanian, D., Sun, Y., Bobrovskyy, M., Sumby, P., Genco, C. A., et al. (2013). Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res. 41:e140. doi: 10.1093/nar/gkt444
McMillan, I. A., Norris, M. H., Zarzycki-Siek, J., Heacock-Kang, Y., Sun, Z., Borlee, B. R., et al. (2021). Identification of a PadR-type regulator essential for intracellular pathogenesis of Burkholderia pseudomallei. Sci. Rep. 11:10405. doi: 10.1038/s41598-021-89852-7
Moreno-Vivián, C., Cabello, P., Martínez-Luque, M., Blasco, R., and Castillo, F. (1999). Prokaryotic nitrate reduction: molecular properties and functional distinction among bacterial nitrate reductases. J. Bacteriol. 181, 6573–6584. doi: 10.1128/JB.181.21.6573-6584.1999
Norris, M. H., Kang, Y., Lu, D., Wilcox, B. A., and Hoang, T. T. (2009). Glyphosate resistance as a novel select-agent-compliant, non-antibiotic selectable-marker in chromosomal mutagenesis of the essential genes asd and dapB of Burkholderia pseudomallei. Appl. Environ. Microbiol. 75, 6062–6075. doi: 10.1128/AEM.00820-09
Norris, M. H., Kang, Y., Wilcox, B., and Hoang, T. T. (2010). Stable, site-specific fluorescent tagging constructs optimized for Burkholderia species. Appl. Environ. Microbiol. 76, 7635–7640. doi: 10.1128/AEM.01188-10
Norris, M. H., Propst, K. L., Kang, Y., Dow, S. W., Schweizer, H. P., and Hoang, T. T. (2011). The Burkholderia pseudomallei Δasd mutant exhibits attenuated intracellular infectivity and imparts protection against acute inhalation melioidosis in mice. Infect. Immun. 79, 4010–4018. doi: 10.1128/IAI.05044-11
Peirson, S. N., Butler, J. N., and Foster, R. G. (2003). Experimental validation of novel and conventional approaches to quantitative real-time PCR data analysis. Nucleic Acids Res. 31, 73e–773e. doi: 10.1093/nar/gng073
Propst, K. L., Mima, T., Choi, K.-H., Dow, S. W., and Schweizer, H. P. (2010). A Burkholderia pseudomallei ΔpurM mutant is avirulent in immunocompetent and immunodeficient animals: candidate strain for exclusion from select-agent lists. Infect. Immun. 78, 3136–3143. doi: 10.1128/IAI.01313-09
Puccio, S., Grillo, G., Licciulli, F., Severgnini, M., Liuni, S., Bicciato, S., et al. (2017). WoPPER: web server for position Related data analysis of gene expression in prokaryotes. Nucleic Acids Res. 45, W109–W115. doi: 10.1093/nar/gkx329
Reckseidler, S. L., DeShazer, D., Sokol, P. A., and Woods, D. E. (2001). Detection of bacterial virulence genes by subtractive hybridization: identification of capsular polysaccharide of Burkholderia pseudomallei as a major virulence determinant. Infect. Immun. 69, 34–44. doi: 10.1128/IAI.69.1.34-44.2001
Reese, M. G. (2001). Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput. Chem. 26, 51–56. doi: 10.1016/S0097-8485(01)00099-7
Richmond, J.Y., and McKinney, R.W. (2007). Biosafety in Microbiological and Biomedical Laboratories, (5th Edn.). Centers for Disease Control and Prevention: Atlanta, GA.
Rico-Pérez, G., Pezza, A., Pucciarelli, M. G., de Pedro, M. A., Soncini, F. C., and García-del Portillo, F. (2016). A novel peptidoglycan DL-endopeptidase induced by salmonella inside eukaryotic cells contributes to virulence. Mol. Microbiol. 99, 546–556. doi: 10.1111/mmi.13248
Roy, A., Kucukural, A., and Zhang, Y. (2010). I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738. doi: 10.1038/nprot.2010.5
Roy, A., Yang, J., and Zhang, Y. (2012). COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 40, W471–W477. doi: 10.1093/nar/gks372
Sambrook, J., and Russell, D.W. (2001). Molecular cloning: A laboratory manual, (2nd Edn.). Cold Spring Harbor Laboratory Press, Cold Spring Harbor: New York, NY.
Schaefers, M. M. (2020). Regulation of virulence by two-component systems in pathogenic Burkholderia. Infect. Immun. 88:e00927–19. doi: 10.1128/IAI.00927-19
Schwarz, S., Singh, P., Robertson, J. D., LeRoux, M., Skerrett, S. J., Goodlett, D. R., et al. (2014). VgrG-5 is a Burkholderia type VI secretion system-exported protein required for multinucleated giant cell formation and virulence. Infect. Immun. 82, 1445–1452. doi: 10.1128/IAI.01368-13
Shalom, G., Shaw, J. G., and Thomas, M. S. (2007). In vivo expression technology identifies a type VI secretion system locus in Burkholderia pseudomallei that is induced upon invasion of macrophage. Microbiology 153, 2689–2699. doi: 10.1099/mic.0.2007/006585-0
Son, M. S., Matthews, W. J., Kang, Y., Nguyen, D. T., and Hoang, T. T. (2007). In vivo evidence of Pseudomonas aeruginosa nutrient acquisition and pathogenesis in the lungs of cystic fibrosis patients. Infect. Immun. 75, 5313–5324. doi: 10.1128/IAI.01807-06
Son, M. S., Nguyen, D. T., Kang, Y., and Hoang, T. T. (2008). Engineering of FRT-lacZ fusion constructs: induction of the Pseudomonas aeruginosa fadAB1 operon by medium and long chain-length fatty acids. Plasmid 59, 111–118. doi: 10.1016/j.plasmid.2007.12.002
Sørensen, K. I., and Hove-Jensen, B. (1996). Ribose catabolism of Escherichia coli: characterization of the rpiB gene encoding ribose phosphate isomerase B and of the rpiR gene, which is involved in regulation of rpiB expression. J. Bacteriol. 178, 1003–1011. doi: 10.1128/jb.178.4.1003-1011.1996
Sun, G. W., Chen, Y., Liu, Y., Tan, G. Y. G., Ong, C., Tan, P., et al. (2010). Identification of a regulatory cascade controlling type III secretion system 3 gene expression in Burkholderia pseudomallei. Mol. Microbiol. 76, 677–689. doi: 10.1111/j.1365-2958.2010.07124.x
Tabansky, I., and Nurminsky, D. I. (2003). Mapping of transcription start sites by direct sequencing of SMART RACE products. BioTechniques 34, 485–486. doi: 10.2144/03343bm06
Toesca, I. J., French, C. T., and Miller, J. F. (2014). The type VI secretion system spike protein VgrG5 mediates membrane fusion during intercellular spread by pseudomallei group Burkholderia species. Infect. Immun. 82, 1436–1444. doi: 10.1128/IAI.01367-13
Ulrich, R. L., DeShazer, D., Brueggemann, E. E., Hines, H. B., Oyston, P. C., and Jeddeloh, J. A. (2004). Role of quorum sensing in the pathogenicity of Burkholderia pseudomallei. J. Med. Microbiol. 53, 1053–1064. doi: 10.1099/jmm.0.45661-0
Valade, E., Thibault, F. M., Gauthier, Y. P., Palencia, M., Popoff, M. Y., and Vidal, D. R. (2004). The PmlI-PmlR quorum sensing system in Burkholderia pseudomallei plays a key role in virulence and modulates production of the MprA protease. J. Bacteriol. 186, 2288–2294. doi: 10.1128/JB.186.8.2288-2294.2004
Vander Broek, C. W., Zainal Abidin, N., and Stevens, J. M. (2017). BipC, a predicted Burkholderia pseudomallei type 3 secretion system Translocator protein with actin binding activity. Front. Cell. Infect. Microbiol. 7:333. doi: 10.3389/fcimb.2017.00333
Vandesompele, J., de Preter, K., Pattyn, F., Poppe, B., van Roy, N., de Paepe, A., et al. (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, research0034.1–research0034.12. doi: 10.1186/gb-2002-3-7-research0034
Warawa, J., and Woods, D. E. (2005). Type III secretion system cluster 3 is required for maximal virulence of Burkholderia pseudomallei in hamster infection model. FEMS Microbiol. Lett. 242, 101–108. doi: 10.1016/j.femsle.2004.10.045
Wiersinga, W. J., Currie, B. J., and Peacock, S. J. (2012). Melioidosis. N. Engl. J. Med. 367, 1035–1044. doi: 10.1056/NEJMra1204699
Wiersinga, W. J., van der Poll, T., White, N. J., Day, N. P., and Peacock, S. J. (2006). Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat. Rev. Microbiol. 4, 272–282. doi: 10.1038/nrmicro1385
Yang, J., and Zhang, Y. (2015). I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 43, W174–W181. doi: 10.1093/nar/gkv342
Yu, N. Y., Wagner, J. R., Laird, M. R., Melli, G., Rey, S., Lo, R., et al. (2010). PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615. doi: 10.1093/bioinformatics/btq249
Zhang, Y. (2009). I-TASSER: fully automated protein structure prediction in CASP8. Proteins 77, 100–113. doi: 10.1002/prot.22588
Keywords: Burkholderia pseudomallei, transposon mutagenesis, Tn-Seq, RNA-Seq, bacterial cell envelope, gene regulation, virulence, single-cell transcriptomic
Citation: Sun Z, Heacock-Kang Y, McMillan IA, Cabanas D, Zarzycki-Siek J and Hoang TT (2023) A virulence activator of a surface attachment protein in Burkholderia pseudomallei acts as a global regulator of other membrane-associated virulence factors. Front. Microbiol. 13:1063287. doi: 10.3389/fmicb.2022.1063287
Edited by:
Sorujsiri Chareonsudjai, Khon Kaen University, ThailandReviewed by:
Christopher French, Northern Arizona University, United StatesCopyright © 2023 Sun, Heacock-Kang, McMillan, Cabanas, Zarzycki-Siek and Hoang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tung T. Hoang,  dG9uZ2hAaGF3YWlpLmVkdQ==
dG9uZ2hAaGF3YWlpLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.