 Xinyue Xu
Xinyue Xu Baoyu Dong
Baoyu Dong Lijun Peng1
Lijun Peng1 Chao Gao
Chao Gao Zhiqun He
Zhiqun He Chuan Wang
Chuan Wang Jumei Zeng
Jumei Zeng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Microbiol. , 21 December 2022
Sec. Infectious Agents and Disease
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.1056608
This article is part of the Research Topic Mycobacterium spp.- Transmission, Pathogenesis, Host-pathogen interaction, Prevention and Treatment View all 10 articles
Mycobacterium tuberculosis possesses a dynamic cell envelope, which consists of a peptidoglycan layer, a mycolic acid layer, and an arabinogalactan polysaccharide. This envelope possesses a highly complex and unique structure representing a barrier that protects and assists the growth of M. tuberculosis and allows its adaptation to the host. It regulates the immune response of the host cells, causing their damage. Therefore, the cell envelope of M. tuberculosis is an attractive target for vaccine and drug development. The emergence of multidrug-resistant as well as extensively drug resistant tuberculosis and co-infection with HIV prevented an effective control of this disease. Thus, the discovery and development of new drugs is a major keystone for TB treatment and control. This review mainly summarizes the development of drug enzymes involved in the biosynthesis of the cell wall in M. tuberculosis, and other potential drug targets in this pathway, to provide more effective strategies for the development of new drugs.
Tuberculosis (TB) is an infectious disease caused by M. tuberculosis. In 2020, approximately 9.9 million people fell ill with TB globally (World Health, 2021). Since early 2020, the COVID-19 pandemic has had a dramatic impact on the outbreak and treatment of TB (Tadolini et al., 2020). The current standard treatment for drug-sensitive TB is a combination of four first-line drugs for 6 months. However, long-term use of multiple drugs may cause adverse drug reactions, which may lead to the interruption of anti-TB treatment, and even develop into multidrug-resistant TB (MDR-TB) and extensively drug-resistant TB (XDR-TB; Miotto et al., 2018). The emergence of multidrug-resistant TB and co-infection with HIV prevented an effective control of the disease (Bell and Noursadeghi, 2018). Therefore, the exploration of new drug targets and new anti-TB compounds is urgently needed.
The structure of the cell envelope of M. tuberculosis is very different from that of Gram-negative and Gram-positive bacteria. The mycolic acids (MA) of M. tuberculosis is covalently attached to the peptidoglycan (PG) layer and arabinogalactan (AG) polysaccharide,the whole complex is called mAGP (Kalscheuer et al., 2019). Mycobacterial outer membrane consists of two parts: the innermost leaflet contained mainly the mycolic acids, the outermost leaflet is composed of various glycolipids, trehalose monomycolate (TMM), trehalose 6,6′-dimycolate (TDM), phospholipids, porins, and waxes. (Alderwick et al., 2007; Jankute et al., 2015; Figure 1A). The lipids represents approximately 60% of the dry weight of the cell wall, and the virulence increase when the lipid contents (non-covalently exposed glycolipids, sulfoglycolipids and some phospholipids) increase; thus, the lipid content is closely related to the virulence of the bacteria. This unique cell wall enables the survival of M. tuberculosis in extremely harsh environments, as well as its survival under the treatment with chemotherapeutic agents (Daffe, 2015; Bhat et al., 2017). Changing the compounds entering pathway and/or increasing its entering amount are the challenges to developing inhibitors targeting the intracellular synthesis stage of bacterial cell wall. With the comprehensive understanding of the key enzymes and extensive studies in cell wall synthesis, the prospect of cell wall as a drug target is quite brilliant. Therefore, the biosynthesis pathways of the precursors of M. tuberculosis cell wall and their subsequent process of transport and polymerization are effective targets for anti-TB drug development. In this mini review, we summarized the potential drugs and drug targets existing in the biosynthetic pathways of mycobacteria cell envelope.
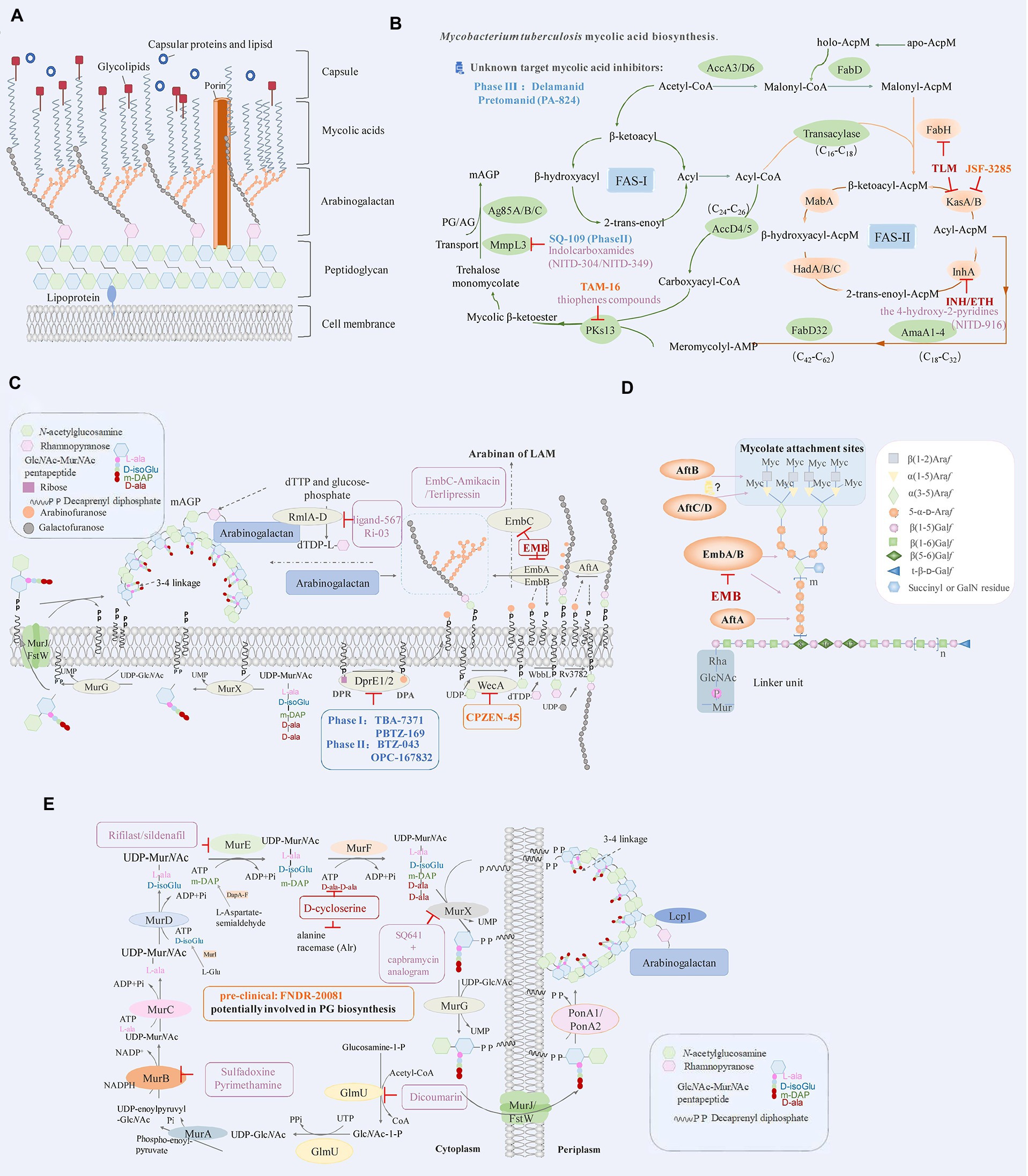
Figure 1. (A) Schematic representation of Mycobacterium tuberculosis cell envelope. (B) Mycobacterium tuberculosis mycolic acid biosynthesis. The key enzymes and drug targets of mycolic acids biosynthesis are shown as indicated. FAS-I: fatty acid synthase types I, FAS-II: fatty acid synthase types I. (C) Drugs targeting the decaprenylphosphoryl arabinose biosynthesis pathway. DPA, the precursor of arabinan, is synthesized by the epimerization of DPR by DprE1 and DprE2. The key enzymes and drug targets of decaprenylphosphoryl arabinose biosynthesis are shown as indicated. (D) Structure of mycobacterial arabinogalactan and roles of key enzymes that are responsible for its biosynthesis. (E) The biosynthesis pathway of peptidoglycan in M. tuberculosis and roles of key enzymes that are responsible for its biosynthesis. The key enzymes and drug targets of peptidoglycan biosynthesis are shown as indicated. The diagram represents the drugs (red) that are effective against M. tuberculosis, drugs in clinical trials (blue), pre-clinical (orange), and compounds in the discovery stage with promising development (purple).
Mycolic acids (MA) are important components of the cell wall in M. tuberculosis (Asselineau and Lederer, 1950); its biosynthetic pathway mainly involves FAS-I (fatty acid synthase type I) and FAS-II (fatty acid synthase type II; Figure 1B). The FAS-I utilizes acetyl-CoA and malonyl-CoA to generate a butyryl-S-enzyme complex; then the butyryl group receives two carbon units from malonyl-CoA. Each round of elongation lengthens the chain by two carbons and finally synthesizes 16- or 18-carbon-containing fatty acids (Jankute et al., 2015). The FAS-II uses ACP transacylase (FabD, Rv2243; Kremer et al., 2001), β-ketoacyl-ACP synthesis III (FabH, Rv0533c; Lai and Cronan, 2003), β-ketoacyl-ACP reductase (MabA/fabG1, Rv1483; Parish et al., 2007), β-hydroxy acyl-ACP dehydratase (HadABC, Rv0635/Rv0636/Rv0637; Sacco et al., 2007; Slama et al., 2016; Farjallah et al., 2021), 2-trans-enoyl-ACP reductase (InhA, Rv1484; Prasad et al., 2021) and β-ketoacyl-ACP synthetase (KasA/B, Rv2245/ Rv2246; Lee and Engels, 2013; Vilcheze et al., 2014) for the extension of the substrate. The acyl-CoA and meroacyl-ACP are synthesized after several cycles. Meromycolyl-AMP and 2-carboxyl-acyl-CoA are generated with the catalysis of FabD32 (Rv3801c) ligase (Portevin et al., 2005; Gavalda et al., 2009) and AccD4/AccD5 (Rv3799c/Rv3280) enzymes (Portevin et al., 2005; Oh et al., 2006). The condensation of meromycolate and C-26 is manipulated by polyketide synthase 13 (Pks13, Rv3800c; Figure 1B; Portevin et al., 2004; Gavalda et al., 2009). Mycolic acids biosynthesis in M. tuberculosis occurs through the concerted action of more than 20 enzymes that are components of different multi-enzyme complexes. Therefore, this pathway represents an important reservoir of novel targets for the development of new drugs to cure TB, especially in the context of the emergence of drug resistance.
Isoniazid (INH), with the enoyl-AcpM reductase InhA being the primary target, can attack many targets in M. tuberculosis after oxidation by KatG (Rv1908c) (catalase-peroxidase). The isonicotinic acyl–NADH complex is created, which blocks the metabolic pathway of mycolic acids in the cell wall by inhibiting 2-trans-enoyl-ACP reductase (InhA) active site, and finally leads M. tuberculosis death (Vilcheze and Jacobs Jr., 2019). Ethionamide (ETH) is similar in structure to INH and is also a prodrug. It is activated by the enzyme ethA (Rv3854c, a monooxygenase), and binds NAD+ to form an ETH-NAD adduct which inhibits the same target site as INH (Vilcheze and Jacobs Jr., 2014).
Aside adduct-forming compounds, there are many inhibitors that directly bind to InhA occupying the fatty-acyl binding site and even bi-substrate inhibitors, that bind simultaneously in both FA and cofactor-binding sites. These directly binding small molecule inhibitors include, arylamides (He et al., 2007), triclosan, diphenyl ethers derivatives (Freundlich et al., 2009; Kamsri et al., 2014), pyrrolidine carboxamide analogs (Kouassi et al., 2015), imidazopiperidine (Wall et al., 2007), and 4-hydroxy-2-pyridones (Figure 1B; Manjunatha et al., 2015; Guardia et al., 2016). The isoniazid-NAD truncated adducts are bi-substrate inhibitors of InhA, the hydrophobic substituents of isoniazid-NAD truncated adducts would be recognized by the fatty-acyl binding site of InhA, and the nicotinamide moiety interacts with the cofactor capsule (Delaine et al., 2010). These inhibitors directly bind to the InhA target without prior activation, avoiding the issues due to clinical drug resistance related to KatG (Prasad et al., 2021). A drug directly targeting InhA in a different location than the activated INH and not requiring activation by KatG may have bactericidal and sterilizing properties superior to those of INH. Since most INH resistance is mediated by mutations in katG, there should also be little or no cross-resistance between a direct inhibitor of InhA and INH. However, these compounds have some limitations. The triclosan, which has been shown associated with the low oral bioavailability (Vosatka et al., 2018), the neurodevelopment impairment (Alfhili et al., 2021), gestational diabetes (Ouyang et al., 2018) and the decline of reproductive function in male mice (Priyanka et al., 2020). Future novel agent should be orally administered, well-tolerated, and with a low propensity to generate resistance.
The β-ketoacyl-AcpM synthase (KasA) of M. tuberculosis is an essential enzyme in the mycobacterial fatty acid biosynthesis (FAS-II) pathway and is a potentially promising target for antibacterial drug development. Inhibitors of KasA have been previously reported, and among them, the most significant inhibitor is thiolactomycin (TLM; Figure 1B), which can inhibit KasA, KasB, and FabH (Choi et al., 2000; Kremer et al., 2000). Many M. tuberculosis beta-ketoacyl-ACP synthase KasA inhibitors lack sufficient potency and/or pharmacokinetic properties. JSF-3285 (Table 1) is an indazole targeting KasA, as demonstrated by the article published in 2020 by Rutgers University (Inoyama et al., 2020). JSF-3285 is a promising preclinical candidate for TB. This study plans to identify and develop novel, small molecule inhibitors of KasA that can be used in combination with other agents to improve the therapeutic effect and to treat drug-resistant forms of TB.
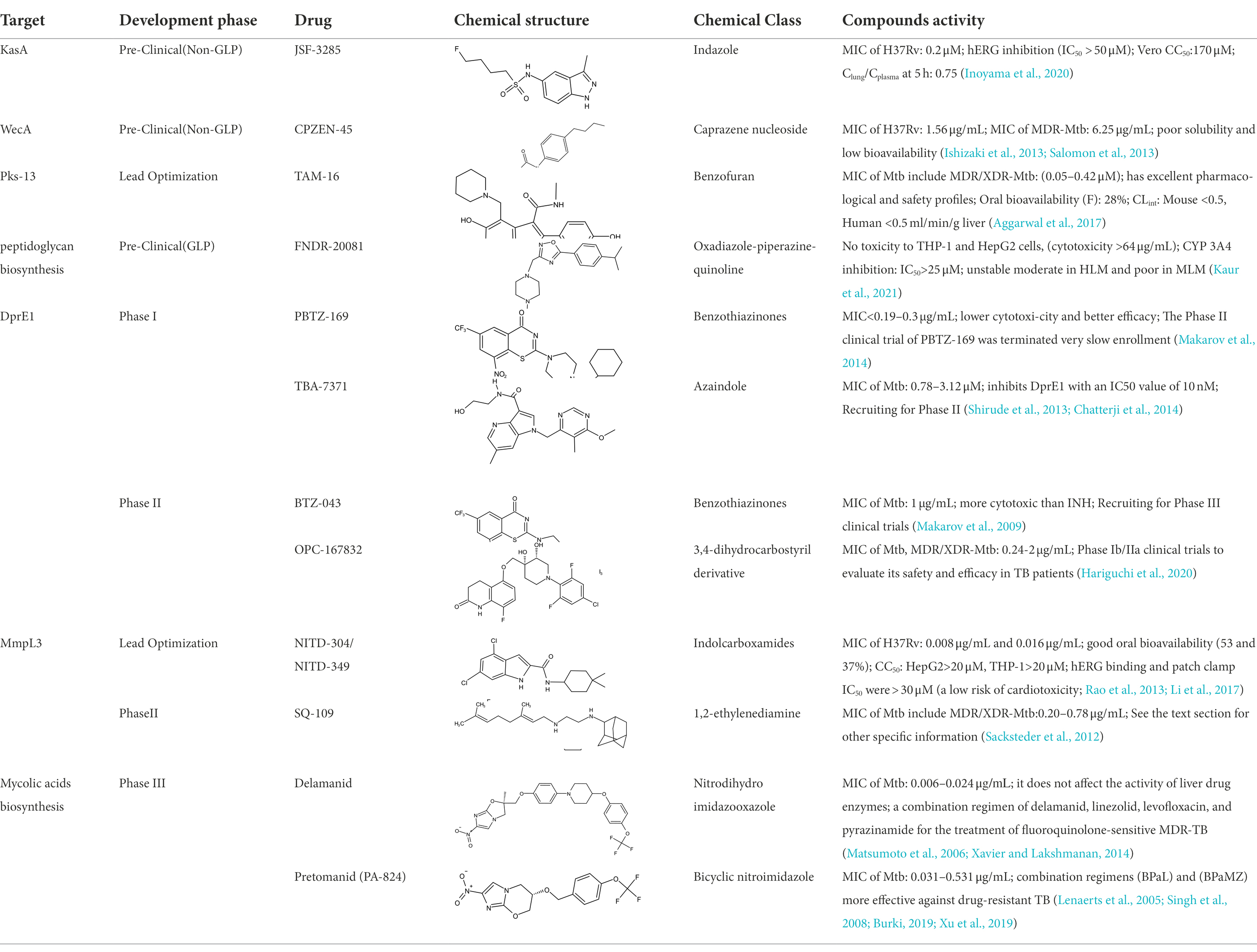
Table 1. Molecules in various stages of drug discovery, their targets, chemical structure, efficacy against Mycobacterium tuberculosis and some other information.
Polyketide synthase 13 (PKs13) is an essential enzyme that forms mycolic acids, and it is required for the formation of the cell wall of M. tuberculosis. PKs13 is involved in the last step of the mycolic acids biosynthesis pathway and has been widely studied as a drug target for TB (Figure 1B; Yu, M. et al., 2018). Aggarwal et al. (2017) used structure-guided methods to develop a lead molecule that targets the thioesterase activity of Pks13 (Figure 1B; Table 1). In another study, a series of thiophenes that kill M. tuberculosis were identified by targeting the N-terminal ACPN domain of Pks13. Wilson et al. (2013) revealed that the compounds work by blocking the interaction of ACPN with the FabD32 protein, which transfers the meromycolyl chain and is essential for the growth of mycobacteria. It can also inhibit the activity of PKs13 by blocking the TE domain of PKs13 (Ioerger et al., 2013; Zhang, W. et al., 2021). These results confirm Pks13 as a target for drugs against M. tuberculosis and highlight its potential in the development of new TB drugs that interfere with the critical pathway of mycolic acids synthesis. It further proves that structure-guided drug development is an effective method for producing new agents, although with limited application in the process of developing new antimycobacterial compounds.
MmpL3 (Rv0206c) is an essential inner membrane protein in M. tuberculosis (Domenech et al., 2005). MmpL3 is responsible for the transport of mycolic acids in the form of trehalose monomycolate (TMM), the precursor of trehalose dimycolate (TDM) and mycolates bound to arabinogalactan that together forms the mycomembrane (Figure 1B). MmpL3 is critical for mycobacterial replication and viability (Belisle et al., 1997; Bhatt et al., 2005). MmpL3 has a periplasmic pore domain and a 12-helix transmembrane domain (Zhang et al., 2019), the structural data will greatly advance the development of MmpL3 inhibitors. Zhang et al. (2019) determined the crystal structure of the compound-MmpL3 and proved the direct interaction between the compound and MmpL3. Those compounds including the ethylenediamine derivative SQ109 (Table 1; Sacksteder et al., 2012; Tahlan et al., 2012; Li et al., 2014), indole-2-carboxamide ICA38 (Rao et al., 2013), adamantylurea AU1235 (Grzegorzewicz et al., 2012; McNeil et al., 2020). SQ109 is currently under Sequella’s US investigational new drug (IND) and completed three Phase 1 studies in the U.S. and two Phase 2 studies in drug-sensitive TB patients in Africa, in addition to the Phase 2b-3 study in Russia (Bukhdruker et al., 2020). SQ-109 appears to be safe and well-tolerated in human studies, with mild to moderate, dose-dependent gastrointestinal discomfort being the most frequently observed adverse events (Heinrich et al., 2015).
The new anti-TB drugs Delamanid (Table 1) producing nitric oxide, block the synthesis of mycolic acids, further damaging the stability of cell membranes (Matsumoto et al., 2006; Xavier and Lakshmanan, 2014). Preclinical studies showed that Delamanid is not genotoxic or potentially carcinogenic, and it has been approved for marketing by the State Food and Drug Administration in 2018 Clinical trials with a treatment plan containing Delamanid are underway in the United States because of the current increase in TB cases among AIDS patients (Nguyen et al., 2020). Pretomanid (PA-824), a bicyclic nitroimidazoles, is a novel anti-tuberculosis agent (Lenaerts et al., 2005). PA824 is a pro-drug that operates directly as a NO donor, it has many attractive characteristics as a potential TB therapy, most notably its novel mechanism of action. PA824 involves a dual-mode function, disrupting mycolic acids synthesis pathway for aerobic bacteria (Table 1; Tyagi et al., 2005; Manjunatha et al., 2009) and respiratory poisoning for anaerobic bacteria (Singh et al., 2008; Bahuguna and Rawat, 2020; Priyanka et al., 2020). PA-824 is converted by deazaflavin nitroreductase to nitrous oxide (NO) and other toxic products, that accumulate within bacteria and obstruct normal electron flow and homeostasis, which abrogate M. tuberculosis growth. The TB Alliance’s new drug application (NDA) for the novel TB drug candidate pretomanid has been accepted for review by the United States Food and Drug Administration (FDA). The application was submitted for the use of pretomanid as part of a new regimen, in combination with bedaquiline and linezolid, in the treatment of extensively drug-resistant (XDR) TB, treatment intolerant multidrug-resistant (MDR) TB, and treatment non-responsive MDR-TB (Conradie et al., 2020), the effectiveness of PA824 in treating MDR/XDR TB with combinations of first-line TB medications has been confirmed. In conclusion, the key enzymes in the mycolic acids synthesis pathway can represent an important approach in the discovery of anti-TB drugs.
LAM and AG are the two major mycobacterial cell wall (lipo) polysaccharides, which contain a structurally similar arabinan domain that is highly branched. AG is synthesized on a decaprenyl phosphate (C50-P) lipid carrier and then attached to peptidoglycan and mycolic acids (Jankute et al., 2012). The phosphatidyl-myo-inositol mannosidase (PIMs), lipomannan (LM) and LAM are essential for the survival and pathogenicity of M. tuberculosis (Torrelles et al., 2009); the enzymes involved in the biosynthesis of AG and LAM could be the ideal targets for anti-TB drug discovery.
Arabinosyltransferase (Emb) is a key enzyme responsible for the biosynthesis of AG and LAM. EmbC is mainly responsible for the synthesis of LAM and associates with ethambutol (EMB) resistance (Zhang et al., 2020). Ethambutol competitively inhibits the substrate to bind EmbB and EmbC subunits. M. tuberculosis has difficulties in developing drug resistance without affecting its complex cell wall construction (Figure 1C). The above research enabled the optimization in the use of ethambutol and the development of new drugs targeting Emb proteins (Jankute et al., 2012; Tan et al., 2020a). Singh et al. (2019) analyzed the US Food and Drug Administration (FDA) library for EmbC, and selected drugs with higher binding affinity to EmbC, such as Terlipressin and Amikacin. The analysis of the size and shape of minimal energy landscape area shows greater stability of the EmbC-Terlipressin complex than the other drugs. It demonstrates the EmbC binding potential of the drug Terlipressin and Amikacin (Figure 1C), further underlining the urgency to develop new and improved treatments, with the reconsideration of existing drugs representing a potential route shortcut.
The rhamnose-GlcNAc disaccharide (RmlA-D, Rv0334/Rv3464/Rv3465/Rv3266c) linker is fundamental for the structural integrity of the mycobacterial cell wall (Li et al., 2006; Qu et al., 2021). M. tuberculosis encode four RmlA-D enzymes involved in the synthesis of the donor dTDP-L-rhamnose beginning with dTTP and glucose-phosphate (Dhaked et al., 2019). Van et al., used a biological layer interference method to identify inhibitory lead compounds that bind to RmlB and RmlC and determined that Ri03 affects the viability of streptococcus and mycobacteria (Figure 1C). Thus, it can be used as a lead compound in the development of a new class of antibiotics against the biosynthesis of dTDP-L-rhamnose in pathogenic bacteria (van der Beek et al., 2019). Ravichandran et al. (2020) used the Super Natural-II database with AutoDock4.0 to perform a virtual screening of the RmlD protein and found a potential inhibitor of RmlD, thus inhibiting the synthesis of the cell wall in M. tuberculosis. The above studies prove that RmlB-D are potential new anti-TB drug targets, although no mature drug is available yet, and much research still needs to be performed.
DprE1 (Rv3790) and DprE2 (Rv3791) are key enzymes in the biosynthesis of arabinose donors (DPA; Figure 1C; Bhutani et al., 2015). Benzothiazolinones (BTZ) target DprE1, and BTZ043 (Table 1; phase II drug) is the most effective among these inhibitors, showing high potency (MIC of 1 ng/ml) against M. tuberculosis H37Rv, multidrug-resistant and extensively resistant M. tuberculosis isolates (Makarov et al., 2009; Trefzer et al., 2010; Zhang et al., 2018). PBTZ169 (Table 1; phase I drug) was optimized from BTZ043 by pharmacochemistry and exhibits an increased efficacy and safety (Makarov et al., 2014; Shi et al., 2018). Subsequently, many compounds with different scaffold structures inhibit enzymes in covalent or non-covalent way (Shirude et al., 2013). For example, TBA7371 (Azaindole derivative), which is a non-covalent inhibitor of DprE1 inhibitors, completed phase I clinical trials (Table 1; Chatterji et al., 2014; Zhang et al., 2018; Robertson et al., 2021; Figure 1C). Another phenotypic screening effort around the carbostyril core results in the discovery of OPC-167832. OPC-167832 in regimens combined with delamanid showed superior efficacy to a standard RHZE regimen (rifampicin + INH + pyrazinamide + ethambutol) in mice. However, trials need to compare the proportion of subjects with favorable outcomes in each experimental treatment arm with OPC-167832, delamanid and bedaquiline versus patients receiving a standard RHZE regimen (Table 1; Hariguchi et al., 2020). These results confirm DprE1 as a target for drug against M. tuberculosis, since it interferes with the critical pathway of mycolic acids synthesis.
The AftA (Rv3792), AftB (Rv3805c), and AftC (Rv2673) genes encode for the enzyme arabinofuranosyltransferase (ArafT), which is responsible for the polymerization of the arabinofuranyl (Araf) residues of DPA into the arabinose components AG and LAM (Figure 1D; Giri et al., 2019). Kolly et al. (2015) explored the transcription of the GtrA (Rv3789) protein and its adjacent genes and found that AftA prime the transfer of the first arabinose residue to the galactose chain. AftA is co-expressed with GtrA, DprE1 and DprE2. AftC in M. tuberculosis is an α-1,3 arabinosyltransferase involved in the branching of α-1,5 linear arabinan of both l LAM and AG (Zhang et al., 2011). AftD (Rv0236c) is the largest glycosyltransferase in the genome of M. tuberculosis. Glycan array analysis shows that AftD binds to complex arabinose glycans (Figure 1D; Tan et al., 2020b). All the identified arabinosyltransferases are essential in the growth of M. tuberculosis and are potential targets of new anti-TB drugs.
The biosynthesis of PG is a highly coordinated process composed of three stages of sequential reactions. In the first stage, UDP-GlcNAc is synthesized by the acetylation and uridylation of essential enzyme GlmU (Rv1018c) beginning with the fructose-1-phosphate (Figure 1E). In the second stage, the synthesis of UDP-MurNAc-pentapeptide is catalyzed by the MurA-F (Rv1315/ Rv0482/Rv2152c/Rv2155c/Rv2158c/Rv2157c) ligase pathway (Rani and Khan, 2016). Starting from UDP-GlcNAc, the enzyme MurA adds phosphoenol pyruvate to form UDP-enoylpyr uvyl-GlcNAc, which is in turn transformed in UDP-MurNAc upon reduction of the enoylpyruvyl moiety to a lactoyl ether moiety with NADPH as an electron hydrogen donor catalyzed by MurB (Figure 1E; Squeglia et al., 2018). Besides, the MurX (Rv2156c) transfers the nucleotide glycopentapeptide to the decallyl phosphate to form the first membrane-bound peptidoglycan precursor (lipid I; Siricilla et al., 2014). Subsequently, the β (1 → 4) bond is formed between GlcNAc of UDP-GlcNAc and MurNAc/Glyc of lipid I by the MurG (Rv2153c) enzyme, thus forming lipid II (Zhang, L. et al., 2021). An enzyme with transglycosylase and transpeptidase activity is necessary for the final stage of peptidoglycan synthesis (Raghavendra et al., 2018). Finally, the Lcp1 enzyme links the arabinogalactan complex to the peptidoglycan (Alderwick et al., 2015). Mur ligases enable the development of cell walls through the cytoplasmic to periplasmic biosynthesis (Sangshetti et al., 2017; Figure 1E). Thus, the Mur ligases may be an ideal target for the discovery of new antibiotics/anti-TB drugs.
GlmU is a bifunctional enzyme with glucosamine-1-phosphate acetyltransferase activity and N-acetylglucosamine-1-phosphate uridine transferase activity considered as a potential drug target (Agarwal et al., 2021). Kolly et al. (2015); Han et al. (2019) screened many compounds from different natural products against M. tuberculosis by DTNB colorimetry. The results showed that dicoumarin has a good inhibitory effect on GlmU acetyltransferase (Figure 1E). MurA-F represent important transferase/ligase in the steps of peptidoglycan biosynthesis. The amide ligases MurC, MurD, MurE and MurF are characterized by the same catalytic function because they possess similar amino acid regions and preserved comparable structural properties (Rani and Khan, 2016). Kumar et al. (2020) analyzed two peptide compound libraries (Asinex and ChemDiv) based on the structure of MurA, and they found that four compounds have acceptable pharmacokinetic properties. After screening FDA-approved drugs from two repositories, they found that sulfadoxine and pyrimethamine have stable interaction with MurB, while rifilast and sildenafil have the most reliable interaction with MurE (Figure 1E; Rani et al., 2020). D-cycloserine is a structural analog of D-alanine, it can competitively inhibit two essential enzymes in the synthesis of peptidoglycan: alanine racemase (Alr) and D-Ala: D-Ala ligase, which are involved in pentapeptide core formation (Figure 1E; Prosser and de Carvalho, 2013). Cycloserine is a broad-spectrum antibiotic introduced in 1952 and was recommended by the World Health Organization (WHO) to be administered to MDR-TB patients. Due to fewer patients with drug-resistant disease have been reported than for some other second-line antituberculosis drugs (Caminero et al., 2010; Yu, X. et al., 2018). A capbramycin analogram kills nonreplicating M. tuberculosis at low concentrations, with strong synergistic effects with SQ641 (a MurX inhibitor; Figure 1E; Siricilla et al., 2015).
β-lactam (Bla) antibiotics inhibit the transpeptidase activity of penicillin-binding proteins (PBPs) to block the cross-linking of peptidoglycans (Hugonnet and Blanchard, 2007). However, BlaC (Rv2068c, β-lactamase) in M. tuberculosis is a broad-spectrum hydrolase that renders ineffective the vast majority of relevant β-lactam compounds currently in use (Lu et al., 2020). Therefore, the development of inhibitors that bind and inhibit BlaC but cannot be hydrolyzed by BlaC is urgently needed. Some studies showed that meropenem in combination with clavulanic acid (a BlaC inhibitor) enhance its activity to effectively kill non-replicating M. tuberculosis (Hugonnet and Blanchard, 2007; Tiberi et al., 2016; van Rijn et al., 2019). Tolerability of intravenous meropenem with amoxicillin-clavulanate was poor at all phase II clinical trial doses, which maybe an obstacle of meropenem in second-line regimens (De Jager et al., 2022).
The structure of M. tuberculosis peptidoglycan is atypical in that it is mainly a 3 → 3 cross-link formed by the L,D-transpeptidase (Ldts; Lavollay et al., 2008; Gupta et al., 2010; Dubee et al., 2012). Penicillin and cephalosporin classes of β-lactams cannot inhibit L,D- transpeptidase function; however, carbapenems (eg. meropenem and imipenem) inactivate its function (Soroka et al., 2015; Lopez Quezada et al., 2020). Avibactam is another β-lactamase inhibitor based on a diazabicyclooctane (DBO) scaffold containing a cyclic urea rather than a β-lactam ring (Edoo et al., 2018). Ceftazidime was first marketed almost 40 years ago and has no activity against M. tuberculosis. However, Ceftazidime-avibactam has remarkable sterilizing effect at clinically achievable concentrations (Deshpande et al., 2017). Edoo et al. (2018) optimized the diazabicyclooctane (DBO) scaffold of avibactam (a β-lactamase inhibitor) and found that DBO 15a (DBO azide derivative) inhibited the LdtMt2 effectively. It was shown that optimization of avibactam to inhibit Ldt is an attractive strategy to obtain drugs with selective activity against Mycobacterium.
Currently, several potential drugs are under discovery and in a pre-clinical phase, in addition to Delamanid and PA-824, which are in clinical phase III. Some of the drugs in pre-clinical studies targeting the synthesis of cell wall are JSF-3285, CPZEN-45, TAM-16, and Indolcarboxamides (NITD-304, NITD-349). These drugs target common components such as PKs-13, WecA, KasA, and MmpL3 (Table 1). The TB drug development pipeline is promising in researching new drugs targeting DprE1 such as BTZ-043, PBTZ-169, TBA7371, and SQ109 (Table 1). The safety, tolerability, and efficacy of these drugs on healthy individuals and TB patients is under evaluation.
TB remains to be one of the leading causes of morbidity and mortality illness throughout the world. With the increasing number of multidrug-resistant and extensively drug-resistant TB worldwide, more effective drugs need to be developed to shorten the treatment time. However, the discovery of new drugs requires a better understanding of the virulence factors of M. tuberculosis in the host. The cell wall of M. tuberculosis plays an important role in the long-term infection and virulence of this pathogen. The biosynthetic pathways of the cell wall components reveal valuable drug targets (essential for the growth of M. tuberculosis and lack of their homologs in mammalian systems) that are the basis of present drugs and/or have the potential for new anti-TB drugs discovery.
XX wrote the original draft. JZ reviewed and edited the manuscript with help from CG and LP. ZH and BD contributed the schematic diagram. JZ and CW provided the conceptualization and fundings. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (31400040), the Fundamental Research Funds for the Central Universities (YJ201985).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Agarwal, M., Soni, V., Kumar, S., Singha, B., and Nandicoori, V. K. (2021). Unique C-terminal extension and interactome of mycobacterium tuberculosis GlmU impacts it’s in vivo function and the survival of pathogen. Biochem. J. 478, 2081–2099. doi: 10.1042/BCJ20210170
Aggarwal, A., Parai, M. K., Shetty, N., Wallis, D., Woolhiser, L., Hastings, C., et al. (2017). Development of a novel Lead that targets M. tuberculosis polyketide synthase 13. Cells 170, 249–259.e25. doi: 10.1016/j.cell.2017.06.025
Alderwick, L. J., Birch, H. L., Mishra, A. K., Eggeling, L., and Besra, G. S. (2007). Structure, function and biosynthesis of the mycobacterium tuberculosis cell wall: arabinogalactan and lipoarabinomannan assembly with a view to discovering new drug targets. Biochem. Soc. Trans. 35, 1325–1328. doi: 10.1042/BST0351325
Alderwick, L. J., Harrison, J., Lloyd, G. S., and Birch, H. L. (2015). The mycobacterial Cell Wall--peptidoglycan and arabinogalactan. Cold Spring Harb. Perspect. Med. 5:a021113. doi: 10.1101/cshperspect.a021113
Alfhili, M. A., Hussein, H. A. M., Park, Y., Lee, M. H., and Akula, S. M. (2021). Triclosan induces apoptosis in Burkitt lymphoma-derived BJAB cells through caspase and JNK/MAPK pathways. Apoptosis 26, 96–110. doi: 10.1007/s10495-020-01650-0
Asselineau, J., and Lederer, E. (1950). Structure of the mycolic acids of mycobacteria. Nature 166, 782–783.
Bahuguna, A., and Rawat, D. S. (2020). An overview of new antitubercular drugs, drug candidates, and their targets. Med. Res. Rev. 40, 263–292.
Belisle, J. T., Vissa, V. D., Sievert, T., Takayama, K., Brennan, P. J., and Besra, G. S. (1997). Role of the major antigen of mycobacterium tuberculosis in cell wall biogenesis. Science 276, 1420–1422. doi: 10.1126/science.276.5317.1420
Bell, L. C. K., and Noursadeghi, M. (2018). Pathogenesis of HIV-1 and mycobacterium tuberculosis co-infection. Nat. Rev. Microbiol. 16, 80–90. doi: 10.1038/nrmicro.2017.128
Bhat, Z. S., Rather, M. A., Maqbool, M., Lah, H. U. L., Yousuf, S. K., and Ahmad, Z. (2017). Cell wall: a versatile fountain of drug targets in mycobacterium tuberculosis. Biomed. Pharmacother. 95, 1520–1534. doi: 10.1016/j.biopha.2017.09.036
Bhatt, A., Kremer, L., Dai, A. Z., Sacchettini, J. C., and Jacobs, W. R. Jr. (2005). Conditional depletion of Kas A, a key enzyme of mycolic acid biosynthesis, leads to mycobacterial cell lysis. J Bacteriol. 187, 7596–7606. doi: 10.1128/JB.187.22.7596-7606.2005
Bhutani, I., Loharch, S., Gupta, P., Madathil, R., and Parkesh, R. (2015). Structure, dynamics, and interaction of mycobacterium tuberculosis (Mtb) Dpr E1 and Dpr E2 examined by molecular modeling, simulation, and electrostatic studies. PLoS One 10:e0119771. doi: 10.1371/journal.pone.0119771
Bukhdruker, S., Varaksa, T., Grabovec, I., Marin, E., Shabunya, P., Kadukova, M., et al. (2020). Hydroxylation of Antitubercular drug candidate, SQ109, by mycobacterial cytochrome P450. Int. J. Mol. Sci. 21. doi: 10.3390/ijms21207683
Burki, T. (2019). BPaL approved for multidrug-resistant tuberculosis. Lancet Infect Dis. 19, 1063–1064. doi: 10.1016/S1473-3099(19)30489-X
Caminero, J. A., Sotgiu, G., Zumla, A., and Migliori, G. B. (2010). Best drug treatment for multidrug-resistant and extensively drug-resistant tuberculosis. Lancet Infect Dis. 10, 621–629. doi: 10.1016/S1473-3099(10)70139-0
Chatterji, M., Shandil, R., Manjunatha, M. R., Solapure, S., Ramachandran, V., Kumar, N., et al. (2014). 1,4-azaindole, a potential drug candidate for treatment of tuberculosis. Antimicrob. Agents Chemother. 58, 5325–5331. doi: 10.1128/AAC.03233-14
Choi, K. H., Kremer, L., Besra, G. S., and Rock, C. O. (2000). Identification and substrate specificity of beta -ketoacyl (acyl carrier protein) synthase III (mtFabH) from mycobacterium tuberculosis. J. Biol. Chem. 275, 28201–28207. doi: 10.1074/jbc.M003241200
Conradie, F., Diacon, A. H., Ngubane, N., Howell, P., Everitt, D., Crook, A. M., et al. (2020). Treatment of highly drug-resistant pulmonary Tuberculosis. N. Engl. J. Med. 382, 893–902. doi: 10.1056/NEJMoa1901814
Daffe, M. (2015). The cell envelope of tubercle bacilli. Tuberculosis 95, S155–S158. doi: 10.1016/j.tube.2015.02.024
De Jager, V., Gupte, N., Nunes, S., Barnes, G. L., van Wijk, R. C., Mostert, J., et al. (2022). Early bactericidal activity of meropenem plus clavulanate (with or without rifampin) for tuberculosis: The COMRADE randomized, phase 2A clinical trial. Am J. Respir. Crit. Care 205, 1228–1235. doi: 10.1164/rccm.202108-1976OC
Delaine, T., Bernardes-Génisson, V., Quémard, A., Constant, P., Meunier, B., and Bernadou, J. (2010). Development of isoniazid-NAD truncated adducts embedding a lipophilic fragment as potential bi-substrate InhA inhibitors and antimycobacterial agents. Eur. J. Med. Chem. 45, 4554–4561. doi: 10.1016/j.ejmech.2010.07.016
Deshpande, D., Srivastava, S., Chapagain, M., Magombedze, G., Martin, K. R., Cirrincione, K. N., et al. (2017). Ceftazidime-avibactam has potent sterilizing activity against highly drug-resistant tuberculosis. Sci. Adv. 3:e1701102. doi: 10.1126/sciadv.1701102
Dhaked, D. K., Bala Divya, M., and Guruprasad, L. (2019). A structural and functional perspective on the enzymes of mycobacterium tuberculosis involved in the L-rhamnose biosynthesis pathway. Prog. Biophys. Mol. Biol. 145, 52–64. doi: 10.1016/j.pbiomolbio.2018.12.004
Domenech, P., Reed, M. B., and Barry, C. E. 3rd. (2005). Contribution of the Mycobacterium tuberculosis MmpL protein family to virulence and drug resistance. Infect Immun. 73, 3492–3501. doi: 10.1128/IAI.73.6.3492-3501.2005
Dubee, V., Triboulet, S., Mainardi, J. L., Etheve-Quelquejeu, M., Gutmann, L., Marie, A., et al. (2012). Inactivation of Mycobacterium tuberculosis l, d-transpeptidase LdtMt(1) by carbapenems and cephalosporins. Antimicrob Agents Chemother. 56, 4189–4195. doi: 10.1128/AAC.00665-12
Edoo, Z., Iannazzo, L., Compain, F., de la Sierra, L., Gallay, I., van Tilbeurgh, H., et al. (2018). Synthesis of avibactam derivatives and activity on beta-lactamases and peptidoglycan biosynthesis enzymes of Mycobacteria. Chemistry. 24, 8081–8086. doi: 10.1002/chem.201800923
Farjallah, A., Chiarelli, L. R., Forbak, M., Degiacomi, G., Danel, M., Goncalves, F., et al. (2021). A coumarin-based analogue of thiacetazone as dual covalent inhibitor and potential fluorescent label of HadA in Mycobacterium tuberculosis. ACS Infect Dis. 7, 552–565. doi: 10.1021/acsfecdis.0c00325
Freundlich, J. S., Wang, F., Vilcheze, C., Gulten, G., Langley, R., Schiehser, G. A., et al. (2009). Triclosan derivatives: towards potent inhibitors of drug-sensitive and drug-resistant Mycobacterium tuberculosis. Chem. MedChem. 4, 241–248. doi: 10.1002/cmdc.200800261
Gavalda, S., Léger, M., van der Rest, B., Stella, A., Bardou, F., Montrozier, H., et al. (2009). The Pks13/FadD32 crosstalk for the biosynthesis of mycolic acids in mycobacterium tuberculosis. J. Biol. Chem. 284, 19255–19264. doi: 10.1074/jbc.M109.006940
Giri, A., Safi, H., Cabibbe, A. M., Gupta, S., Narang, A., Tyagi, G., et al. (2019). Lack of association of novel mutation Asp397Gly in aftB gene with ethambutol resistance in clinical isolates of Mycobacterium tuberculosis. Tuberculosis (Edinb). 115, 49–55. doi: 10.1016/j.tube.2019.01.004
Grzegorzewicz, A. E., Pham, H., Gundi, V. A. K. B., Scherman, M. S., North, E. J., Hess, T., et al. (2012). Inhibition of mycolic acid transport across the mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 8, 334–341. doi: 10.1038/nchembio.794
Guardia, A., Gulten, G., Fernandez, R., Gómez, J., Wang, F., Convery, M., et al. (2016). N-Benzyl-4-((heteroaryl)methyl)benzamides: a new class of direct NADH-dependent 2-trans Enoyl-acyl carrier protein reductase (InhA) inhibitors with Antitubercular activity. ChemMedChem 11, 687–701. doi: 10.1002/cmdc.201600020
Gupta, R., Lavollay, M., Mainardi, J. L., Arthur, M., Bishai, W. R., and Lamichhane, G. (2010). The mycobacterium tuberculosis protein LdtMt2 is a nonclassical transpeptidase required for virulence and resistance to amoxicillin. Nat. Med. 16, 466–469. doi: 10.1038/nm.2120
Han, X., Chen, C., Yan, Q., Jia, L., Taj, A., and Ma, Y. (2019). Action of Dicumarol on Glucosamine-1-phosphate acetyltransferase of GlmU and mycobacterium tuberculosis. Front. Microbiol. 10:1799. doi: 10.3389/fmicb.2019.01799
Hariguchi, N., Chen, X., Hayashi, Y., Kawano, Y., Fujiwara, M., Matsuba, M., et al. (2020). OPC-167832, a novel Carbostyril derivative with potent Antituberculosis activity as a DprE1 inhibitor. Antimicrob. Agents Chemother. 64:e02020-19. doi: 10.1128/AAC.02020-19
He, X., Alian, A., and Ortiz de Montellano, P. R. (2007). Inhibition of the mycobacterium tuberculosis enoyl acyl carrier protein reductase InhA by arylamides. Bioorg. Med. Chem. 15, 6649–6658. doi: 10.1016/j.bmc.2007.08.013
Heinrich, N., Dawson, R., du Bois, J., Narunsky, K., Horwith, G., Phipps, A. J., et al. (2015). Early phase evaluation of SQ109 alone and in combination with rifampicin in pulmonary TB patients. J. Antimicrob. Chemother. 70, 1558–1566. doi: 10.1093/jac/dku553
Hugonnet, J. E., and Blanchard, J. S. (2007). Irreversible inhibition of the mycobacterium tuberculosis beta-lactamase by clavulanate. Biochemistry 46, 11998–12004. doi: 10.1021/bi701506h
Inoyama, D., Awasthi, D., Capodagli, G. C., Tsotetsi, K., Sukheja, P., Zimmerman, M., et al. (2020). A preclinical candidate targeting mycobacterium tuberculosis KasA. Cell. Chem. Biol. 27, 560–570.e10. doi: 10.1016/j.chembiol.2020.02.007
Ioerger, T. R., O’Malley, T., Liao, R., Guinn, K. M., Hickey, M. J., Mohaideen, N., et al. (2013). Identification of new drug targets and resistance mechanisms in mycobacterium tuberculosis. PLoS One 8:e75245. doi: 10.1371/journal.pone.0075245
Ishizaki, Y., Hayashi, C., Inoue, K., Igarashi, M., Takahashi, Y., Pujari, V., et al. (2013). Inhibition of the first step in synthesis of the mycobacterial cell wall core, catalyzed by the GlcNAc-1-phosphate transferase WecA, by the novel caprazamycin derivative CPZEN-45. J. Biol. Chem. 288, 30309–30319. doi: 10.1074/jbc.M113.492173
Jankute, M., Cox, J. A. G., Harrison, J., and Besra, G. S. (2015). Assembly of the mycobacterial Cell Wall. Annu. Rev. Microbiol. 69, 405–423. doi: 10.1146/annurev-micro-091014-104121
Jankute, M., Grover, S., Rana, A. K., and Besra, G. S. (2012). Arabinogalactan and lipoarabinomannan biosynthesis: structure, biogenesis and their potential as drug targets. Future Microbiol. 7, 129–147. doi: 10.2217/fmb.11.123
Kalscheuer, R., Palacios, A., Anso, I., Cifuente, J., Anguita, J., Jacobs, W. R. Jr., et al. (2019). The mycobacterium tuberculosis capsule: a cell structure with key implications in pathogenesis. Biochem. J. 476, 1995–2016. doi: 10.1042/BCJ20190324
Kamsri, P., Koohatammakun, N., Srisupan, A., Meewong, P., Punkvang, A., Saparpakorn, P., et al. (2014). Rational design of InhA inhibitors in the class of diphenyl ether derivatives as potential anti-tubercular agents using molecular dynamics simulations. SAR QSAR Environ. Res. 25, 473–488. doi: 10.1080/1062936X.2014.898690
Kaur, P., Potluri, V., Ahuja, V. K., Naveenkumar, C. N., Krishnamurthy, R. V., Gangadharaiah, S. T., et al. (2021). A multi-targeting pre-clinical candidate against drug-resistant tuberculosis. Tuberculosis (Edinb). 129:102104. doi: 10.1016/j.tube.2021.102104
Kolly, G. S., Mukherjee, R., Kilacsková, E., Abriata, L. A., Raccaud, M., Blaško, J., et al. (2015). GtrA protein Rv3789 is required for Arabinosylation of arabinogalactan in mycobacterium tuberculosis. J. Bacteriol. 197, 3686–3697. doi: 10.1128/JB.00628-15
Kouassi, A. F., Kone, M., Keita, M., Esmel, A., Megnassan, E., N’Guessan, Y. T., et al. (2015). Computer-aided design of orally bioavailable pyrrolidine carboxamide inhibitors of enoyl-acyl carrier protein reductase of Mycobacterium tuberculosis with favorable pharmacokinetic profiles. Int. J. Mol. Sci. 16, 29744–29771. doi: 10.3390/ijms161226196
Kremer, L., Douglas, J. D., Baulard, A. R., Morehouse, C., Guy, M. R., Alland, D., et al. (2000). Thiolactomycin and related analogues as novel anti-mycobacterial agents targeting KasA and KasB condensing enzymes in mycobacterium tuberculosis. J. Biol. Chem. 275, 16857–16864. doi: 10.1074/jbc.M000569200
Kremer, L., Nampoothiri, K. M., Lesjean, S., Dover, L. G., Graham, S., Betts, J., et al. (2001). Biochemical characterization of acyl carrier protein (AcpM) and malonyl-CoA:AcpM transacylase (mtFabD), two major components of Mycobacterium tuberculosis fatty acid synthase II. J. Biol. Chem. 276, 27967–27974. doi: 10.1074/jbc.M103687200
Kumar, P., Saumya, K. U., and Giri, R. (2020). Identification of peptidomimetic compounds as potential inhibitors against MurA enzyme of mycobacterium tuberculosis. J. Biomol. Struct. Dyn. 38, 4997–5013. doi: 10.1080/07391102.2019.1696231
Lai, C. Y., and Cronan, J. E. (2003). Beta-ketoacyl-acyl carrier protein synthase III (FabH) is essential for bacterial fatty acid synthesis. J. Biol. Chem. 278, 51494–51503. doi: 10.1074/jbc.M308638200
Lavollay, M., Arthur, M., Fourgeaud, M., Dubost, L., Marie, A., Veziris, N., et al. (2008). The peptidoglycan of stationary-phase mycobacterium tuberculosis predominantly contains cross-links generated by L D-transpeptidation. J. Bacteriol. 190, 4360–4366. doi: 10.1128/JB.00239-08
Lee, W., and Engels, B. (2013). Clarification on the decarboxylation mechanism in KasA based on the protonation state of key residues in the acyl-enzyme state. J. Phys. Chem. B 117, 8095–8104. doi: 10.1021/jp403067m
Lenaerts, A. J., Gruppo, V., Marietta, K. S., Johnson, C. M., Driscoll, D. K., Tompkins, N. M., et al. (2005). Preclinical testing of the nitroimidazopyran PA-824 for activity against mycobacterium tuberculosis in a series of in vitro and in vivo models. Antimicrob. Agents Chemother. 49, 2294–2301. doi: 10.1128/AAC.49.6.2294-2301.2005
Li, W., Upadhyay, A., Fontes, F. L., North, E. J., Wang, Y., Crans, D. C., et al. (2014). Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58, 6413–6423. doi: 10.1128/AAC.03229-14
Li, W., Xin, Y., McNeil, M. R., and Ma, Y. (2006). rmlB and rmlC genes are essential for growth of mycobacteria. Biochem. Biophys. Res. Commun. 342, 170–178. doi: 10.1016/j.bbrc.2006.01.130
Li, W., Sanchez-Hidalgo, A., Jones, V., de Moura, V. C., North, E. J., and Jackson, M. (2017). Synergistic interactions of MmpL3 inhibitors with antitubercular compounds in vitro. Antimicrob Agents chemother. 61, e02399–e02316. doi: 10.1128/AAC.02399-16
Lopez, Q. L., Smith, R., Lupoli, T. J., Edoo, Z., Li, X., Gold, B., et al. (2020). Activity-based protein profiling reveals that cephalosporins selectively active on non-replicating Mycobacterium tuberculosis bind multiple protein families and spare peptidoglycan transpeptidases. Front Microbiol. 11:1248. doi: 10.3389/fmicb.2020.01248
Lu, Z., Wang, H., Zhang, A., Liu, X., Zhou, W., Yang, C., et al. (2020). Structures of Mycobacterium tuberculosis penicillin-binding protein 3 in complex with five beta-lactam antibiotics reveal mechanism of inactivation. Mol. Pharmacol. 97, 287–294. doi: 10.1124/mol.119.118042
Makarov, V., Lechartier, B., Zhang, M., Neres, J., Sar, A. M., Raadsen, S. A., et al. (2014). Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 6, 372–383. doi: 10.1002/emmm.201303575
Makarov, V., Manina, G., Mikusova, K., Möllmann, U., Ryabova, O., Saint-Joanis, B., et al. (2009). Benzothiazinones kill mycobacterium tuberculosis by blocking arabinan synthesis. Science 324, 801–804. doi: 10.1126/science.1171583
Manjunatha, U., Boshoff, H. I. M., and Barry, C. E. (2009). The mechanism of action of PA-824: novel insights from transcriptional profiling. Commun. Integr. Biol. 2, 215–218. doi: 10.4161/cib.2.3.7926
Manjunatha, U. H., SP, S. R., Kondreddi, R. R., Noble, C. G., Camacho, L. R., Tan, B. H., et al. (2015). Direct inhibitors of InhA are active against Mycobacterium tuberculosis. Sci. Transl. Med. 7:269ra263. doi: 10.1126/scitranslmed.3010597
Matsumoto, M., Hashizume, H., Tomishige, T., Kawasaki, M., Tsubouchi, H., Sasaki, H., et al. (2006). OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 3:e466. doi: 10.1371/journal.pmed.0030466
McNeil, M. B., O’Malley, T., Dennison, D., Shelton, C. D., Sunde, B., and Parish, T. (2020). Multiple mutations in Mycobacterium tuberculosis MmpL3 increase resistance to MmpL3 inhibitors. mSphere. 5, e00985–e00920. doi: 10.1128/mSphere.00985-20
Miotto, P., Zhang, Y., Cirillo, D. M., and Yam, W. C. (2018). Drug resistance mechanisms and drug susceptibility testing for tuberculosis. Respirology. 23, 1098–1113. doi: 10.1111/resp.13393
Nguyen, T. V. A., Anthony, R. M., Cao, T. T. H., Bañuls, A. L., Nguyen, V. A. T., Vu, D. H., et al. (2020). Delamanid resistance: update and clinical management. Clin. Infect. Dis. 71, 3252–3259. doi: 10.1093/cid/ciaa755
Oh, T. J., Daniel, J., Kim, H. J., Sirakova, T. D., and Kolattukudy, P. E. (2006). Identification and characterization of Rv3281 as a novel subunit of a biotin-dependent acyl-CoA carboxylase in mycobacterium tuberculosis H37Rv. J. Biol. Chem. 281, 3899–3908. doi: 10.1074/jbc.M511761200
Ouyang, F., Tang, N., Zhang, H. J., Wang, X., Zhao, S., Wang, W., et al. (2018). Maternal urinary triclosan level, gestational diabetes mellitus and birth weight in Chinese women. Sci. Total Environ. 626, 451–457. doi: 10.1016/j.scitotenv.2018.01.102
Parish, T., Roberts, G., Laval, F., Schaeffer, M., Daffé, M., and Duncan, K. (2007). Functional complementation of the essential gene fabG1 of mycobacterium tuberculosis by mycobacterium smegmatis fabG but not Escherichia coli fabG. J. Bacteriol. 189, 3721–3728. doi: 10.1128/JB.01740-06
Portevin, D., de Sousa-D’Auria, C., Houssin, C., Grimaldi, C., Chami, M., Daffé, M., et al. (2004). A polyketide synthase catalyzes the last condensation step of mycolic acid biosynthesis in mycobacteria and related organisms. Proc. Natl. Acad. Sci. U. S. A. 101, 314–319. doi: 10.1073/pnas.0305439101
Portevin, D., de Sousa-D’Auria, C., Montrozier, H., Houssin, C., Stella, A., Lanéelle, M. A., et al. (2005). The acyl-AMP ligase FadD32 and AccD4-containing acyl-CoA carboxylase are required for the synthesis of mycolic acids and essential for mycobacterial growth: identification of the carboxylation product and determination of the acyl-CoA carboxylase components. J. Biol. Chem. 280, 8862–8874. doi: 10.1074/jbc.M408578200
Prasad, M. S., Bhole, R. P., Khedekar, P. B., and Chikhale, R. V. (2021). Mycobacterium enoyl acyl carrier protein reductase (InhA): a key target for antitubercular drug discovery. Bioorg. Chem. 115:105242. doi: 10.1016/j.bioorg.2021.105242
Priyanka, T. A., Maske, P., Mote, C., and Dighe, V. (2020). Gestational and lactational exposure to triclosan causes impaired fertility of F1 male offspring and developmental defects in F2 generation. Environ. Pollut. 257:113617. doi: 10.1016/j.envpol.2019.113617
Prosser, G. A., and de Carvalho, L. P. (2013). Kinetic mechanism and inhibition of mycobacterium tuberculosis D-alanine:D-alanine ligase by the antibiotic D-cycloserine. FEBS J. 280, 1150–1166. doi: 10.1111/febs.12108
Qu, D., Zhao, X., Sun, Y., Wu, F. L., and Tao, S. C. (2021). Mycobacterium tuberculosis thymidylyltransferase RmlA is negatively regulated by Ser/Thr protein kinase PknB. Front Microbiol. 12:643951. doi: 10.3389/fmicb.2021.643951
Raghavendra, T., Patil, S., and Mukherjee, R. (2018). Peptidoglycan in mycobacteria: chemistry, biology and intervention. Glycoconj. J. 35, 421–432. doi: 10.1007/s10719-018-9842-7
Rani, C., and Khan, I. A. (2016). UDP-GlcNAc pathway: potential target for inhibitor discovery against M. tuberculosis. Eur. J. Pharm. Sci. 83, 62–70. doi: 10.1016/j.ejps.2015.12.013
Rani, J., Silla, Y., Borah, K., Ramachandran, S., and Bajpai, U. (2020). Repurposing of FDA-approved drugs to target MurB and MurE enzymes in Mycobacterium tuberculosis. J. Biomol. Struct Dyn. 38, 2521–2532. doi: 10.1080/07391102.2019.1637280
Rao, S. P., Lakshminarayana, S. B., Kondreddi, R. R., Herve, M., Camacho, L. R., Bifani, P., et al. (2013). Indolcarboxamide is a preclinical candidate for treating multidrug-resistant tuberculosis. Sci. Transl. Med. 5:214ra168. doi: 10.1126/scitranslmed.3007355
Ravichandran, R., Ridzwan, N. F. W., and Mohamad, S. B. (2020). Ensemble-based high-throughput virtual screening of natural ligands using the Super Natural-II database against cell-wall protein dTDP-4-dehydrorhamnose reductase (RmlD) in Mycobacterium tuberculosis. J. Biomol. Struct Dyn. 40, 5069–5078. doi: 10.1080/07391102.2020.1867641
Robertson, G. T., Ramey, M. E., Massoudi, L. M., Carter, C. L., Zimmerman, M., Kaya, F., et al. (2021). Comparative analysis of pharmacodynamics in the C3HeB/FeJ mouse tuberculosis model for DprE1 inhibitors TBA-7371, PBTZ169, and OPC-167832. Antimicrob. Agents Chemother. 65:e0058321. doi: 10.1128/AAC.00583-21
Sacco, E., Covarrubias, A. S., O’Hare, H. M., Carroll, P., Eynard, N., Jones, T. A., et al. (2007). The missing piece of the type II fatty acid synthase system from mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 104, 14628–14633. doi: 10.1073/pnas.0704132104
Sacksteder, K. A., Protopopova, M., Barry, C. E., Andries, K., and Nacy, C. A. (2012). Discovery and development of SQ109: a new antitubercular drug with a novel mechanism of action. Future Microbiol. 7, 823–837. doi: 10.2217/fmb.12.56
Salomon, J. J., Galeron, P., Schulte, N., Morow, P. R., Severynse-Stevens, D., Huwer, H., et al. (2013). Biopharmaceutical in vitro characterization of CPZEN-45, a drug candidate for inhalation therapy of tuberculosis. Ther Deliv. 4, 915–923. doi: 10.4155/tde.13.62
Sangshetti, J. N., Joshi, S. S., Patil, R. H., Moloney, M. G., and Shinde, D. B. (2017). Mur ligase inhibitors as anti-bacterials: a comprehensive review. Curr. Pharm Des. 23, 3164–3196. doi: 10.2174/1381612823666170214115048
Shi, J., Lu, J., Wen, S., Zong, Z., Huo, F., Luo, J., et al. (2018). In vitro activity of PBTZ169 against multiple mycobacterium species, Antimicrob agents Chemother, vol. 62. doi: 10.1128/AAC.01314-18
Shirude, P. S., Shandil, R., Sadler, C., Naik, M., Hosagrahara, V., Hameed, S., et al. (2013). Azaindoles: noncovalent DprE1 inhibitors from scaffold morphing efforts, kill mycobacterium tuberculosis and are efficacious in vivo. J. Med. Chem. 56, 9701–9708. doi: 10.1021/jm401382v
Singh, R., Manjunatha, U., Boshoff, H. I. M., Ha, Y. H., Niyomrattanakit, P., Ledwidge, R., et al. (2008). PA-824 kills non-replicating mycobacterium tuberculosis by intracellular NO release. Science 322, 1392–1395. doi: 10.1126/science.1164571
Singh, A., Somvanshi, P., and Grover, A. (2019). Drug repurposing against arabinosyl transferase (EmbC) of mycobacterium tuberculosis: essential dynamics and free energy minima based binding mechanics analysis. Gene 693, 114–126. doi: 10.1016/j.gene.2019.01.029
Siricilla, S., Mitachi, K., Skorupinska-Tudek, K., Swiezewska, E., and Kurosu, M. (2014). Biosynthesis of a water-soluble lipid I analogue and a convenient assay for translocase I. Anal. Biochem. 461, 36–45. doi: 10.1016/j.ab.2014.05.018
Siricilla, S., Mitachi, K., Wan, B., Franzblau, S. G., and Kurosu, M. (2015). Discovery of a capuramycin analog that kills nonreplicating mycobacterium tuberculosis and its synergistic effects with translocase I inhibitors. J. Antibiot. (Tokyo) 68, 271–278. doi: 10.1038/ja.2014.133
Slama, N., Jamet, S., Frigui, W., Pawlik, A., Bottai, D., Laval, F., et al. (2016). The changes in mycolic acid structures caused by hadC mutation have a dramatic effect on the virulence of Mycobacterium tuberculosis. Mol Microbiol. 99, 794–807. doi: 10.1111/mmi.13266
Soroka, D., Li de la Sierra-Gallay, I., Dubée, V., Triboulet, S., van Tilbeurgh, H., Compain, F., et al. (2015). Hydrolysis of clavulanate by mycobacterium tuberculosis beta-lactamase BlaC harboring a canonical SDN motif. Antimicrob. Agents Chemother. 59, 5714–5720. doi: 10.1128/AAC.00598-15
Squeglia, F., Ruggiero, A., and Berisio, R. (2018). Chemistry of peptidoglycan in mycobacterium tuberculosis life cycle: an off-the-wall balance of synthesis and degradation. Chemistry 24, 2533–2546. doi: 10.1002/chem.201702973
Tadolini, M., Garcia-Garcia, J. M., Blanc, F. X., Borisov, S., Goletti, D., Motta, I., et al. (2020). On tuberculosis and COVID-19 co-infection. Eur. Respir J. 56:2002328. doi: 10.1183/13993003.02328-2020
Tahlan, K., Wilson, R., Kastrinsky, D. B., Arora, K., Nair, V., Fischer, E., et al. (2012). SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of mycobacterium tuberculosis. Antimicrob. Agents Chemother. 56, 1797–1809. doi: 10.1128/AAC.05708-11
Tan, Y. Z., Rodrigues, J., Keener, J. E., Zheng, R. B., Brunton, R., Kloss, B., et al. (2020a). Cryo-EM structure of arabinosyltransferase EmbB from mycobacterium smegmatis. Nat. Commun. 11:3396. doi: 10.1038/s41467-020-17202-8
Tan, Y. Z., Zhang, L., Rodrigues, J., Zheng, R. B., Giacometti, S. I., Rosário, A. L., et al. (2020b). Cryo-EM structures and regulation of Arabinofuranosyltransferase AftD from mycobacteria. Mol. Cell 78, 683–699.e11. doi: 10.1016/j.molcel.2020.04.014
Tiberi, S., Sotgiu, G., D’Ambrosio, L., Centis, R., Abdo Arbex, M., Alarcon Arrascue, E., et al. (2016). Comparison of effectiveness and safety of imipenem/clavulanate- versus meropenem/clavulanate-containing regimens in the treatment of MDR- and XDR-TB. Eur. Respir. J. 47, 1758–1766. doi: 10.1183/13993003.00214-2016
Torrelles, J. B., DesJardin, L. E., MacNeil, J., Kaufman, T. M., Kutzbach, B., Knaup, R., et al. (2009). Inactivation of mycobacterium tuberculosis mannosyltransferase pimB reduces the cell wall lipoarabinomannan and lipomannan content and increases the rate of bacterial-induced human macrophage cell death. Glycobiology 19, 743–755. doi: 10.1093/glycob/cwp042
Trefzer, C., Rengifo-Gonzalez, M., Hinner, M. J., Schneider, P., Makarov, V., Cole, S. T., et al. (2010). Benzothiazinones: prodrugs that covalently modify the decaprenylphosphoryl-beta-D-ribose 2′-epimerase DprE1 of mycobacterium tuberculosis. J. Am. Chem. Soc. 132, 13663–13665. doi: 10.1021/ja106357w
Tyagi, S., Nuermberger, E., Yoshimatsu, T., Williams, K., Rosenthal, I., Lounis, N., et al. (2005). Bactericidal activity of the nitroimidazopyran PA-824 in a murine model of tuberculosis. Antimicrob. Agents Chemother. 49, 2289–2293. doi: 10.1128/AAC.49.6.2289-2293.2005
van der Beek, S. L., Zorzoli, A., Çanak, E., Chapman, R. N., Lucas, K., Meyer, B. H., et al. (2019). Streptococcal dTDP-L-rhamnose biosynthesis enzymes: functional characterization and lead compound identification. Mol. Microbiol. 111, 951–964. doi: 10.1111/mmi.14197
van Rijn, S. P., Zuur, M. A., Anthony, R., Wilffert, B., van Altena, R., Akkerman, O. W., et al. (2019). Evaluation of carbapenems for treatment of multi- and extensively drug-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother. 63, e01489–e01418. doi: 10.1128/AAC.01489-18
Vilcheze, C., and Jacobs, W. R. Jr. (2014). Resistance to isoniazid and Ethionamide in mycobacterium tuberculosis: genes, mutations, and causalities. Microbiol. Spectr. 2:MGM2-0014-2013. doi: 10.1128/microbiolspec.MGM2-0014-2013
Vilcheze, C., and Jacobs, W. R. Jr. (2019). The isoniazid paradigm of killing, resistance, and persistence in mycobacterium tuberculosis. J. Mol. Biol. 431, 3450–3461. doi: 10.1016/j.jmb.2019.02.016
Vilcheze, C., Molle, V., Carrere-Kremer, S., Leiba, J., Mourey, L., Shenai, S., et al. (2014). Phosphorylation of KasB regulates virulence and acid-fastness in Mycobacterium tuberculosis. PLoS Pathog. 10:e1004115. doi: 10.1371/journal.ppat.1004115
Vosatka, R., Kratky, M., and Vinsova, J. (2018). Triclosan and its derivatives as antimycobacterial active agents. Eur. J. Pharm Sci. 114, 318–331. doi: 10.1016/j.ejps.2017.12.013
Wall, M. D., Oshin, M., Chung, G. A. C., Parkhouse, T., Gore, A., Herreros, E., et al. (2007). Evaluation of N-(phenylmethyl)-4-[5-(phenylmethyl)-4,5,6,7-tetrahydro-1H-imidazo[4,5-c]pyridin- 4-yl]benzamide inhibitors of mycobacterium tuberculosis growth. Bioorg. Med. Chem. Lett. 17, 2740–2744. doi: 10.1016/j.bmcl.2007.02.078
Wilson, R., Kumar, P., Parashar, V., Vilchèze, C., Veyron-Churlet, R., Freundlich, J. S., et al. (2013). Antituberculosis thiophenes define a requirement for Pks13 in mycolic acid biosynthesis. Nat. Chem. Biol. 9, 499–506. doi: 10.1038/nchembio.1277
World Health, O. (2021) Global tuberculosis report 2021. Geneva: World Health Organization. Licence: CC BY-NY-SA 3.0 IGO
Xavier, A. S., and Lakshmanan, M. (2014). Delamanid: a new armor in combating drug-resistant tuberculosis. J. Pharmacol. Pharmacother. 5, 222–224. doi: 10.4103/0976-500X.136121
Xu, J., Li, S. Y., Almeida, D. V., Tasneen, R., Barnes-Boyle, K., Converse, P. J., et al. (2019). Contribution of pretomanid to novel regimens containing bedaquiline with either linezolid or moxifloxacin and pyrazinamide in murine models of tuberculosis. Antimicrob Agents Chemother. 63, e00021–e00019. doi: 10.1128/AAC.00021-19
Yu, M., Dou, C., Gu, Y., and Cheng, W. (2018). Crystallization and structure analysis of the core motif of the Pks13 acyltransferase domain from Mycobacterium tuberculosis. PeerJ. 6:e4728. doi: 10.7717/peerj.4728
Yu, X., Zeng, X., Shi, W., Hu, Y., Nie, W., Chu, N., et al. (2018). Validation of cycloserine efficacy in treatment of multidrug-resistant and extensively drug-resistant tuberculosis in Beijing. China. Antimicrob Agents Chemother. 62, e01824–e01817. doi: 10.1128/AAC.01824-17
Zhang, J., Angala, S. K., Pramanik, P. K., Li, K., Crick, D. C., Liav, A., et al. (2011). Reconstitution of functional mycobacterial arabinosyltransferase AftC proteoliposome and assessment of decaprenylphosphorylarabinose analogues as arabinofuranosyl donors. ACS Chem. Biol. 6, 819–828. doi: 10.1021/cb200091m
Zhang, W., Liu, L. L., Lun, S., Wang, S. S., Xiao, S., Gunosewoyo, H., et al. (2021). Design and synthesis of mycobacterial pks13 inhibitors: Conformationally rigid tetracyclic molecules. Eur. J. Med. Chem. 213:113202. doi: 10.1016/j.ejmech.2021.113202
Zhang, L., Zhao, Y., Gao, Y., Wu, L., Gao, R., Zhang, Q., et al. (2020). Structures of cell wall arabinosyltransferases with the anti-tuberculosis drug ethambutol. Science 368, 1211–1219. doi: 10.1126/science.aba9102
Zhang, G., Guo, S., Cui, H., and Qi, J. (2018). Virtual screening of small molecular inhibitors against DprE1. Molecules. 23:524. doi: 10.3390/molecules23030524
Zhang, B., Li, J., Yang, X., Wu, L., Zhang, J., Yang, Y., et al. (2019). Crystal structures of membrane transporter MmpL3, an anti-TB drug target. Cell. 176:e613. doi: 10.1016/j.cell.2019.01.003
Keywords: Mycobacterium tuberculosis, anti-tuberculosis drug, cell envelope, drug target, lead compounds
Citation: Xu X, Dong B, Peng L, Gao C, He Z, Wang C and Zeng J (2022) Anti-tuberculosis drug development via targeting the cell envelope of Mycobacterium tuberculosis. Front. Microbiol. 13:1056608. doi: 10.3389/fmicb.2022.1056608
Edited by:
Sukhendu Mandal, University of Calcutta, IndiaReviewed by:
Vikram Saini, All India Institute of Medical Sciences, IndiaCopyright © 2022 Xu, Dong, Peng, Gao, He, Wang and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jumei Zeng, WmVuZ0p1bWVpQHNjdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.