
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 09 December 2022
Sec. Evolutionary and Genomic Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.1055055
This article is part of the Research Topic Epitranscriptomics in Microbiology View all 5 articles
 Chu-Yu Tang1
Chu-Yu Tang1 Jie Wang1Xin Liu1Jian-Bo Chen1Jing Liang1Tao Wang1
Jie Wang1Xin Liu1Jian-Bo Chen1Jing Liang1Tao Wang1 Wayne Roydon Simpson2Yu-Ling Li1*
Wayne Roydon Simpson2Yu-Ling Li1* Xiu-Zhang Li1*
Xiu-Zhang Li1*Ophiocordyceps sinensis is widely utilized due to its pharmaceutical value. Mycelial protein forms a key active component of O. sinensis and determines the medicinal potential of fungus. Here, we describe the development of an optimized fermentation medium to obtain more mycelial soluble protein from O. sinensis using response surface methodology (RSM) and investigate the increased mycelial protein content using transcriptomics. The maximum mycelial protein content of 2.11% was obtained using a medium consisting of 20% beef broth, 0.10% peptone, 2% glucose, 0.15% yeast extract, 0.20% KH2PO4, and 0.02% MgSO4. Transcriptome analysis identified 790 differentially expressed genes (DEGs), including 592 up-regulated genes and 198 down-regulated genes, optimisation resulted in more up-regulated genes. The main DEGs were enriched in metabolic pathways, ABC transporters, starch and sucrose metabolism, tyrosine metabolism, and glutathione metabolism. In addition, some DEGs associated with mycelial protein enhancement such as tyrosinase (TYR), glutathione S-transferase (GST), glutamine synthetase (glnA), and β-glucosidase may contribute to increased mycelial protein content. Real-time quantitative PCR (RT-qPCR) was used to confirm gene expression and the results support the accuracy of RNA-Seq and DEG analysis. This study provides an optimized fermentation method for enhancing the mycelial protein content of O. sinensis and a reference for the effective development of O. sinensis protein.
Chinese cordyceps or ‘Dong Chong Xia Cao’ is rare in nature. It is a complex of the larvae of endemic ghost moths (Hepialidae) parasitized by Ophiocordyceps sinensis and distributed on hillsides of the Tibetan Plateau at altitudes of 3,000–5,000 meters (Qiu et al., 2020; Li et al., 2021). The growth niche presents a hostile environment that leads to unstable Chinese cordyceps production and a variable metabolism that affects the production of active metabolites. This contingent production of Chinese cordyceps contributes to the fungus being extremely expensive and overexploited (Wei et al., 2021). The scarcity and high commercial value of Chinese cordyceps have stimulated the demand for Cordyceps-like products (He, 2018). Contemporary pharmacological studies indicated that the mycelial products of Ophiocordyceps sinensis, produced using fermentation technologies, have similar pharmaceutical values to Chinese cordyceps demonstrating influence over the respiratory, cardiovascular, immune systems and the treatment of chronic diseases (Dong et al., 2009; Zhang et al., 2012, 2020; Li et al., 2021; Yuan et al., 2022; Wu et al., 2022). This method of production has gradually been accepted as an alternative to the harvesting of naturally occurring fungus (Hsieh et al., 2005; Li et al., 2011; Wang et al., 2014). In terms of pharmacological activity, studies have shown that the substances with immune and antioxidant effects are Cordyceps proteins (Kong et al., 2021). With progressive research on Cordyceps proteins, the basic structures of the peptides have been elucidated (Jia et al., 2005) and most soluble proteins have been shown to be mostly functional proteins in O. sinensis, whose diversity is generally associated with the versatility of the medicinal fungal effects (Ren et al., 2013). Many of the proteins are extracellular or intracellular proteases, additionally a protein has been identified that in the form of a novel protein-binding polysaccharide HS002-II demonstrates significant immunomodulatory activity (He et al., 2013; Rakhee et al., 2021). A previous study has identified at least two fibrinolytic enzymes from O. sinensis mycelium with research implications for the development of drugs for thrombotic diseases (Zang et al., 2020). With the exception of adenosine, signature proteins differ from other Cordyceps genera and can used as markers in identification and quality determination assays (Chan et al., 2018; Qian et al., 2019). The elevation of O. sinensis mycelial protein content and examination of the factors influencing efficacy are basic requirements for the exploitation of Cordyceps mycoproteins.
The Plackett-Bruman design (PBD) is a preferred way to screen for significant biological factors and is often used in conjunction with the response surface methodology (RSM) containing the Central Composite Design (CCD) and the Box–Behnken design (BBD), where the BBD includes all global designs and only requires three layers of factors to be operational (Addelman, 1979; Dong et al., 2009; Zhang et al., 2022). In fermentation process research, RSM technology has been used to optimize all influencing parameters collectively and improve product content (Guo et al., 2018). It can be used to evaluate the relative importance of several influencing factors. At the same time, the variability in the fermentation process can be reduced, the output response can be monitored, and the response of the output to the nominal and target requirements can be closely confirmed, which has the advantages of reducing development time and overall cost (Elibol, 2002). Most of the designs have been successfully applied to optimize the extraction method and medium. Hsieh et al. optimized the medium for obtaining high-content Cordyceps sinensis polysaccharide and found that the optimal polysaccharide production medium was composed of 6.17% sucrose, 0.53% corn steep powder, 0.5% (NH4)2 HPO4, and 0.15% KH2PO4 at pH 4.44 (Hsieh et al., 2005). However, there is no report concerning the optimization of mycelial protein in media for O. sinensis.
The transcriptome is the basis for the investigation of gene function and structure. It has become increasingly popular in medicine, animals, plants, and fungi since it can rapidly reveal the molecular mechanisms of physiological changes in organisms and can inform subsequent proteomic and metabolomic studies (Hao et al., 2017; Wan et al., 2022; Wei et al., 2022). Previous studies have utilized transcriptomic analysis of O. sinensis to determine genes involved in key pathways of fruiting body development (Xiang et al., 2014; Tong et al., 2020). It has been demonstrated that genes such as nucleoside diphosphate kinase gene (ndk) and β subunit of the fatty acid synthase gene (cel-2) play a crucial role in the development of both growth and fruiting body development (Tong et al., 2020). Studies examining DEGs have also shown that some key genes related to fatty acid metabolism are upregulated in Chinese cordyceps with unsaturated fatty acid production being high in Chinese Cordyceps compared to cultivated fungus by transcriptome analysis (Zhang et al., 2021). Current information regarding the transcriptome of O. sinensis following optimization of the culture medium is scant. The results of this study provide a valuable reference for the cultivation and utilization of high mycelial protein of O. sinensis.
The mycelia of O. sinensis (TOS-1) was originally isolated from Chinese cordyceps collected from Zaduo County (E 95°8′23″, N 32°47′38″, H: 4601 m), Qinghai Province, China and deposited at the State Key Laboratory of Plateau Ecology and Agriculture, Qinghai Academy of Animal and Veterinary Sciences, Qinghai University. The sequence of TOS-1 was submitted to the National Center for Biotechnology Information with the corresponding serial number: OP379526. Phylogenetic analysis confirmed that the characteristic strain of this species was O. sinensis. The strain was activated and inoculated in a new 300 ml conical flask containing 150 ml of fermentation medium at an inoculation rate of 10% (v/v). It was cultured on a rotary shaker at 18°C for 70 d at 135 rpm using a fermentation medium containing (1:2) 50% beef broth, 0.50% peptone, 0.15% yeast powder, 3% glucose, 0.02% MgSO4 and 0.20% KH₂PO4. At the conclusion of liquid culture, the mycelium balls were harvested by centrifugation for 20 min at 6,000 ×g, washed with deionized water 3–4 times, the clean mycelium was placed in an incubator at 24 ~ 26°C to keep it dry and its weight constant and stored at −80°C for further analysis.
The mycelial protein of O. sinensis was extracted using Minute™ Total Protein Extraction Kit for Microbes with Thick Cell Walls (Invent Biotechnologies Inc., United States) and store at −20°C for further analysis. Bradford’s assay was performed to determine the mycelial soluble protein content of the O. sinensis (Bradford, 1976). Bovine serum albumin (BSA) was used as the standard. The protein concentration was calculated from the absorbance and the final protein content was determined according to the following equation:
where C (mg/mL) is the mycelial protein concentration, V (mL) is the volume of the extract, and M (mg) is the mass of the mycelium.
Screening and evaluation of important media components affecting mycelial protein content of O. sinensis was performed using a Plackett-Burman design. In this study, all treatments based on the principal components of six variables (beef broth, peptone, yeast extract, glucose, KH₂PO4, MgSO4) of the medium were performed in triplicate according to the designed matrix, 10% (v/v) strain suspension was inoculated in each 150 ml Erlenmeyer flask containing sterilized substrate, flasks were incubated at 18°C for 24 days. This designed matrix using the equation includes two levels, low (−1) and high (+1), shown in (Supplementary Table S1).
where Y represents the response, β0 represents the constant, and βi is the regression coefficient, X is the independent variable and k is the number of variables.
The three variables affecting mycelial protein content were selected based on the results of the PBD design, and the optimal levels of the variables were selected using the path of the steepest ascent method. The optimal region with the highest content was approached based on the positive and negative effects of the key variables and reasonable step size was designed. The central point of RMS can be obtained by the path of the steepest ascent method. The direction change and the step size range were designed as shown in Supplementary Table S2.
Three important factors, beef broth, peptone, and glucose, were determined by PBD to affect the mycelial protein of O. sinensis. Each variable was studied independently at three levels (−1, 0, 1). 17 treatments with details of variables and levels are shown in Supplementary Table S3. Independent variables were calculated using quadratic polynomial equations:
where Y represents the response, β0 represents the constant, and βi, βii, and βij are the regression coefficients of the three main effects, respectively.
All data were analyzed by Design-Expert 11.0 and GraphPad Prism 8.0.2 was used to plot the data. All experiments are expressed as mean ± standard error (SEM).
The mycelia of O. sinensis optimized medium (BBD) and unoptimized medium (CK) samples were obtained under the culture conditions of 24 days at 18°C. Then, the clean mycelium was placed in an incubator at 24 ~ 26°C to keep it dry and its weight constant. Three replicate samples were prepared for each treatment. After collection, all samples were immediately frozen in liquid nitrogen at −80°C prior to RNA isolation.
Total RNA was isolated using Trizol (Invitrogen, United States) according to the manufacturer’s instructions. RNA degradation and contamination was monitored on 1% agarose gels, and the concentration and integrity of RNA were measured using Qubit® RNA Assay Kit in Qubit® 2.0 Flurometer (Life Technologies, CA, United States) and the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, United States). Sequencing libraries were generated using NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, United States) following the manufacturer’s recommendations. The quality of the library was assessed using the Agilent Bioanalyzer 2100 system. The cDNA libraries were sequenced on an Illumina NovaSeq 6000 platform (Metware Biological Science and Technology Co., Ltd., Wuhan, China) and 150 bp paired-end reads were generated. The original data were processed to remove adapter sequences, ploy-N sequences, low-quality reads, and very short-length reads. All subsequent analyses are based on clean reads.
The reference genome information of O. sinensis was downloaded from1 (Shu et al., 2020). Clean reads of each sample with three replicates were aligned to the reference genome using HISAT2 (v2.1.0). Gene alignment was calculated using feature Counts (version v1.6.2) along with Fragments Per Kilobase of exon model per Million mapped fragments (FPKM) of each gene based on the gene length. DESeq2 (version v1.22.1) was used to analyze the differential expression between the two groups, and the p value was corrected using the Benjamini & Hochberg method. Differentially expressed genes (DEGs) were identified using |log2FC (log2fold-change)| ≥1 and false discovered rate (FDR) <0.05 as statistically significant. Enrichment tests were performed based on Gene Ontology (GO) functions and Kyoto Encyclopedia of Genes and Genomes (KEGG2) pathways. The raw Illumina sequencing results of O. sinensis were deposited in the NCBI Sequence Read Archive (SRA) with the accession number: (PRJNA874172).
Thirteen DEGs were arbitrarily selected for transcriptome sequencing analysis and validated by RT-qPCR experiments in triplicate. The ACT1 gene was used to normalize the expression level. The specific primer sequences are listed in Supplementary Table S4. The reaction system contains 10 μl of Hieff® RT-qPCR SYBR Green Master Mix (Low Rox Plus) (Yeasen Biotechnology Co., Shanghai, China), 0.4 μl of each primer (0.2 μM), 1 μl of cDNA, and 8.2 μl of RNase-free H2O. The cycling conditions were as follows: 95°C for 5 min, 40 cycles of 95°C for 10 s and 60°C for 30 s. Analysis of relative gene expression levels by the 2−ΔΔCt method. The GraphPad Prism 8.0.2 (version 8.0.2, GraphPad Software, Inc.) was used for statistical analysis.
PBD was used to screen for medium variables affecting the mycelium, 6 variables were investigated for their effect. The 12 treatments and their correspondences are shown in Table 1. Design Expert 11.0 was used to fit the treatment data in Table 1, the factor contribution rate and variance analysis results are shown in Table 2. p < 0.05 suggests significant model terms. The corrected coefficient of determination R2adj was 0.8853, indicating that 88.53% of the variability of the treatment data could be explained by the model. The effect of beef broth (X1), peptone (X2), and glucose (X4) on mycelial protein content was highly significant (p < 0.01). Therefore, X1, X2, and X4 can be considered the main factors affecting mycelia protein content. The following model equation has been obtained for mycelia protein content (Y). Among the significant factors, the regression coefficients of factors X1 and X4 are negative, indicating that their effect on mycelial protein content is negative, and X2 is positive, suggesting that the addition of X1 and X4 may be appropriately reduced and the content of X2 may be increased in subsequent studies. According to the size analysis of the contribution rate and correlation coefficient in Table 2, the secondary factors X3, X5 and X6 were no longer added in the subsequent treatments, and it was determined that the addition amount of the original medium was 0.15, 0.2, 0.02%, respectively.
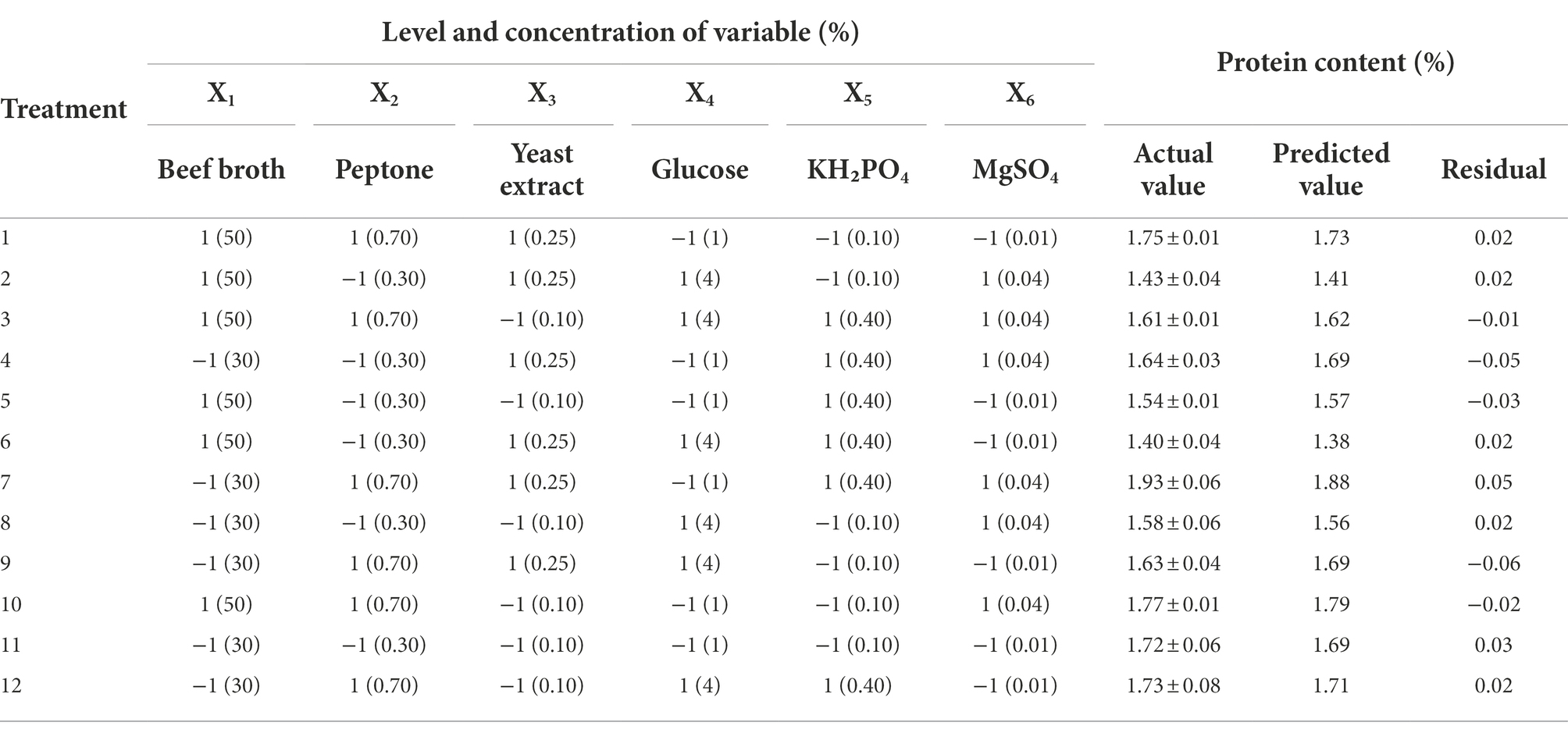
Table 1. Plackett-Burman experimental design matrix for screening composition of the growth medium.
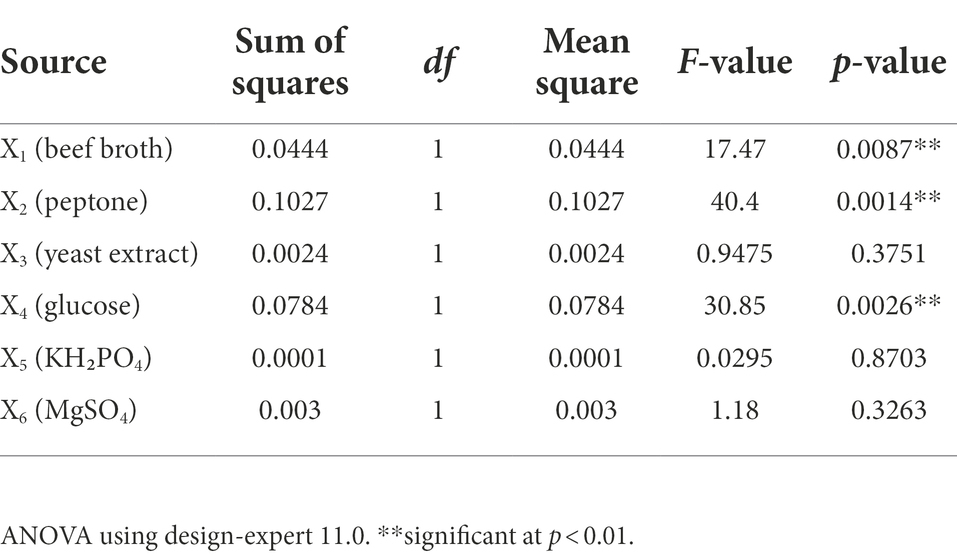
Table 2. Analysis of the significance of PB design.
According to Table 3, it was found that the highest mycelial protein content was reached in the second group of treatments, so the corresponding beef broth content (30%), peptone (0.3%), and glucose (2%) in this group were used as the central points for the subsequent response surface experiments.
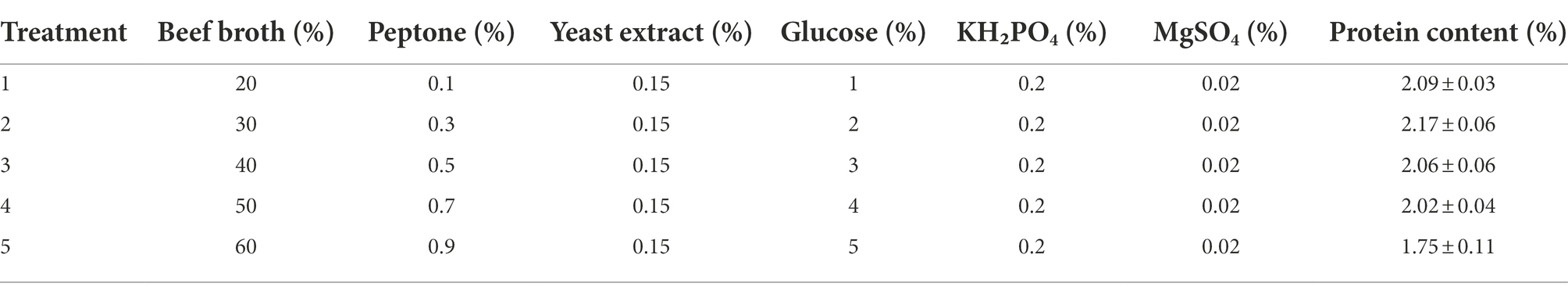
Table 3. Design and results of steepest ascent method.
Multiple regressions were fitted to the data in Table 4 using Design Expert 11.0 software to construct regression models for the extraction process parameters. With O. sinensis mycelial protein content as the response variable, beef broth, peptone, and glucose as the test variables, the response and test variables can be developed into a quadratic multinomial regression prediction model equation.
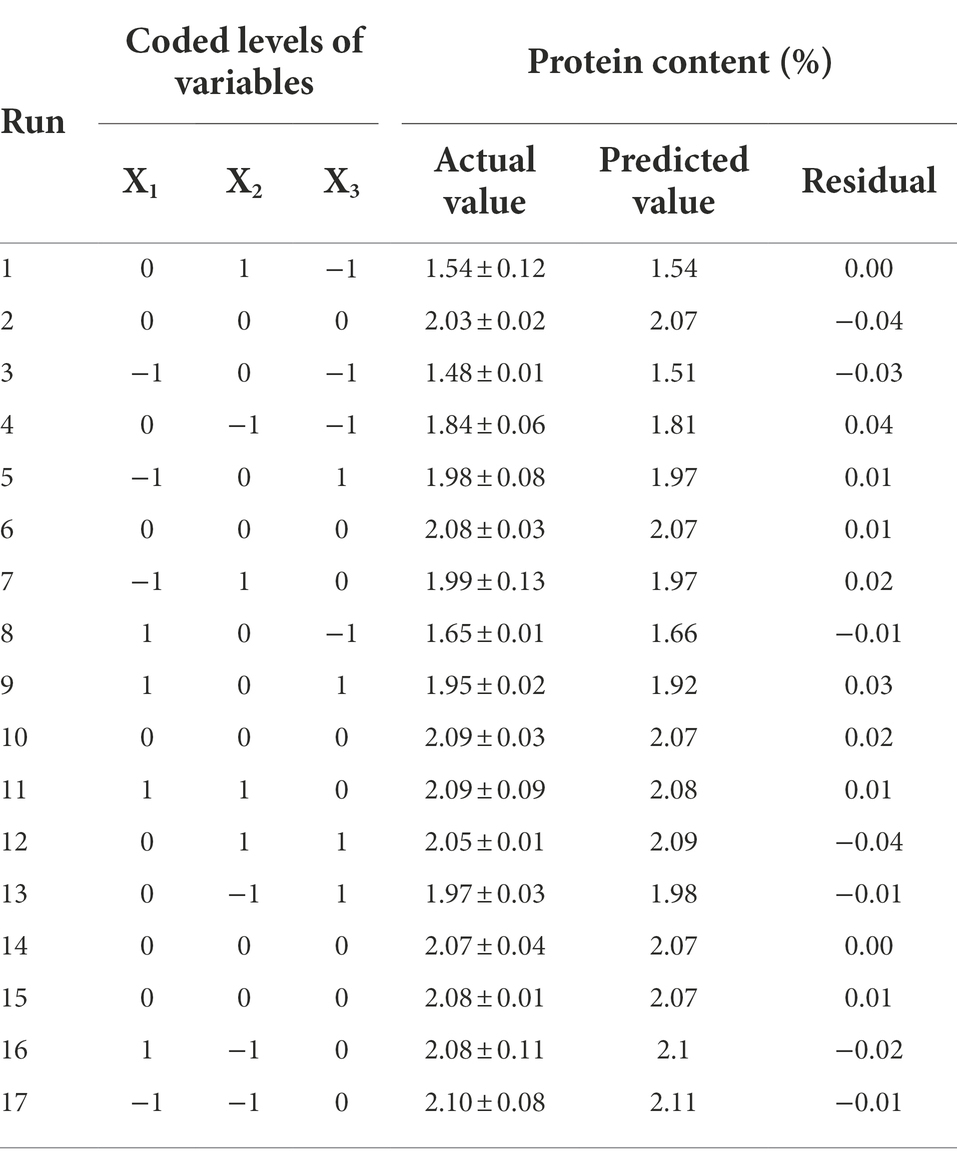
Table 4. Box–Behnken design with experimental and predicted values of mycelial protein content.
The results are shown in Table 5 that the model p < 0.0001 and lacks a fit term p = 0.1393, which indicates a significant regression and an insignificant misfit model. The correlation coefficient of the model R2 = 0.988 indicates a good fit for the model. The correction coefficient of determination R2adj = 0.9725 indicates that approximately 97.25% of the variation in response values (Y) can be explained by the model. The model coefficient of variation (CV) value of 1.7% < 10% indicates that the Box–Behnken test has good confidence and accuracy and the model is appropriate. The linear coefficients (X2 and X3), cross-product coefficients (X1X3 and X2X3), and quadratic term coefficients (X12, X22, and X32) were significant (p < 0.05), whereas the other terms coefficients were not significant (p > 0.05). The F values confirmed the order of factors affecting mycelial protein content as X3 > X2 > X1 and the order effect of interactions as X2X3 > X1X3 > X1X2. Design Expert 11.0 software was applied to optimize the parameters of the proposed model, and the maximum mycelial protein content was 2.11% when the concentration of beef broth was 20%, peptone was 0.10%, and glucose was 2%. The optimized medium formulation was finally determined as 20% beef broth, 0.10% peptone, 2% glucose, 0.15% yeast extract, 0.20% KH₂PO4, 0.02% MgSO4.
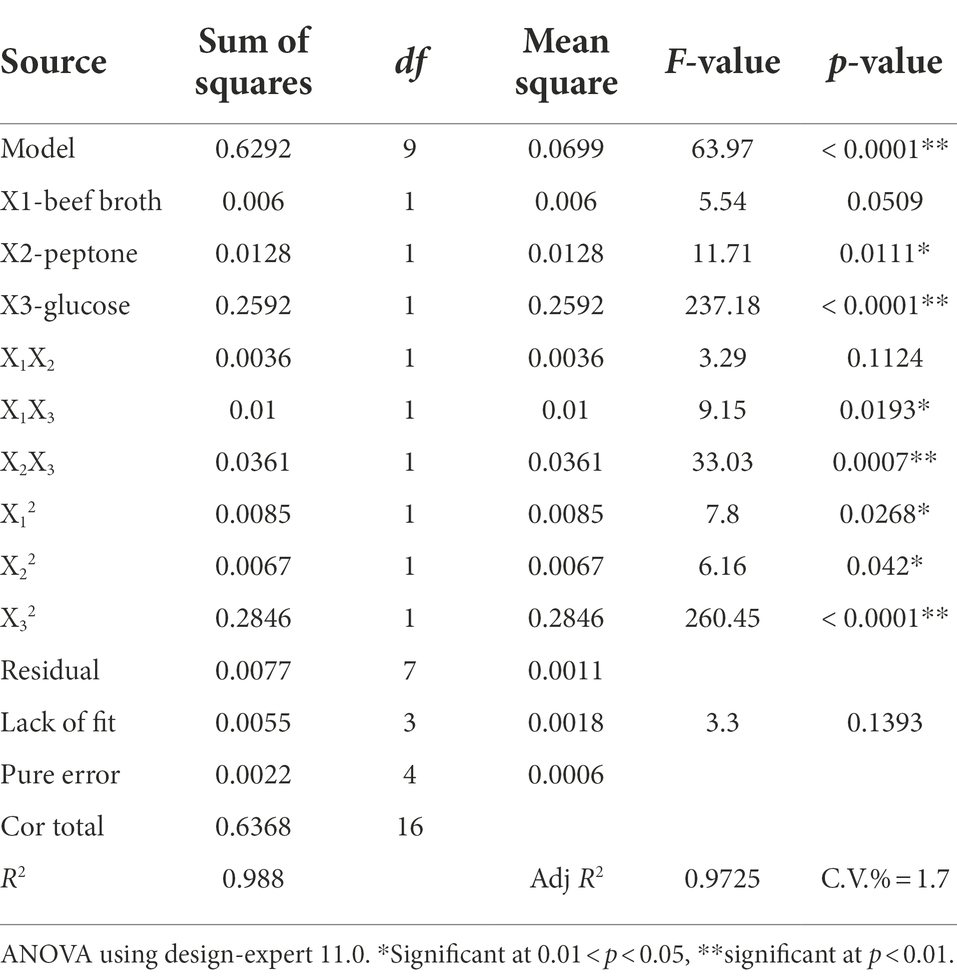
Table 5. ANOVA of the fitted polynomial quadratic model.
To visually analyze the magnitude of the effect of the other two factors and their interactions on the response values, one beef broth (X1), peptone (X2), and glucose (X3) was fixed at the centroid level (0 levels), respectively. The contours of the regression model and their response surfaces are shown in Figure 1. It can be seen from the figure that the slope of B and C is steeper than that of A, indicating that the significant interaction between B (X2 and X3) and C (X1 and X3). The average value of three replicate experiments was obtained and the mycelial protein content of O. sinensis mycelia was (2.03 ± 0.01) %, which was extremely close to the predicted value of 2.11% and fitted well with the predicted value, indicating that the above model can better predict the mycelial protein content of mycelia production.
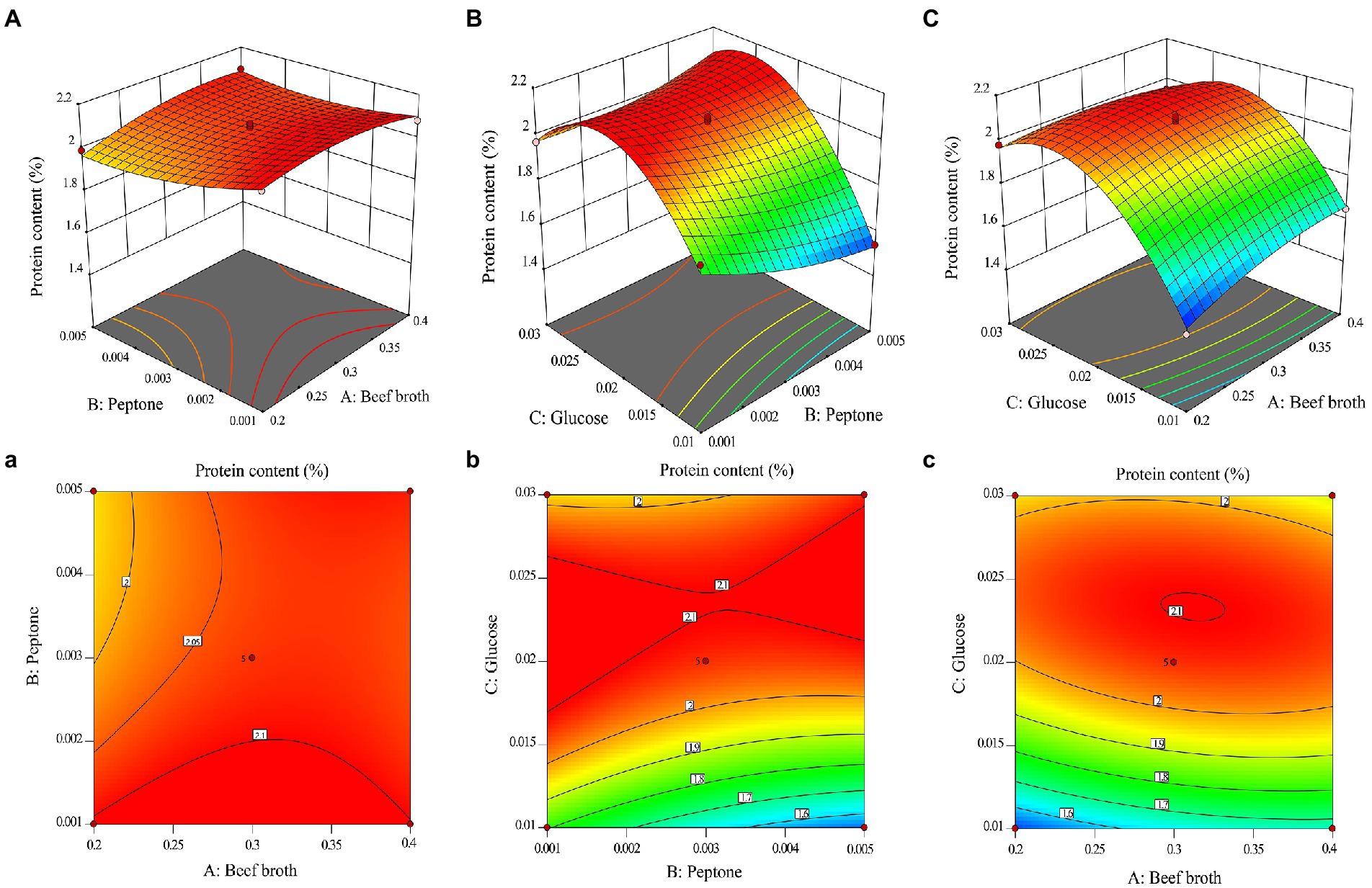
Figure 1. 3D response surface and corresponding contour line of mycelial protein content: (A) Effect of beef broth and peptone addition on mycelial protein content; (B) Effect of peptone and glucose addition on mycelial protein content; (C) Effect of beef broth and glucose addition on mycelial protein content.
To investigate the expression changes before (CK) and after (BBD) optimization of O. sinensis, this study performed a transcriptomic study with three biological replicates for each sample, respectively. A total of 40.81 Gb of clean data was obtained, the average GC content was 61.04%. The mapping results are shown in Supplementary Table S5. Gene expression levels between replicates for each sample exhibited high Pearson correlation coefficient values (more than 0.97), indicating good reproducibility between replicates.
Currently, FPKM is the most used commonly method to estimate gene expression levels. Based on the gene FPKM values, the expression level of genes with FPKM>60 in the reference transcriptome indicates high expression, while genes with FPKM of 0 ~ 1 were expressed at low levels or not expressed. The results showed that there were some differences in gene expression levels of pre-optimized BBD and CK 8947 genes (FPKM≥1) were expressed in BBD, accounting for 93.48% of the total number of genes, among which 3,383 genes were highly expressed (FPKM>60), accounting for 35.35% of the total number of genes; while 8,892 genes (FPKM≥1) were expressed in CK, accounting for 92.91% of the total number of genes. Among them, 3,173 genes were highly expressed (FPKM>60), accounting for 33.15% of the total number of genes. Further analysis of the control group CK and the experimental group BBD showed that 26 genes in the CK group were extremely highly expressed (FPKM>3,000) and 40 genes in the BBD group were extremely highly expressed (FPKM>2,000).
Sequences from 6 libraries were searched and annotated with the KEGG, GO, Swiss-Prot, Pfam, KOG, and NR public databases by Blast software. The percentages of annotated sequences based on the KEGG, GO, Swiss-Prot, Pfam, KOG and NR databases were 60.42, 69.72, 56.58, 70.32, 60.01, and 92.44%, respectively. KEEG classification results showed that 5,783 unigenes were enriched in 133 pathways. The main typical pathways are amino acid metabolism (360 genes), carbohydrate metabolism (548 genes) and lipid metabolism genes (120 genes). The amino acid metabolism subclass involves 13 pathways, namely (ko00220) arginine biosynthesis, (ko00250) alanine, aspartate and glutamate metabolism, (ko00260) glycine, serine and threonine metabolism, (ko00270) cysteine and methionine metabolism, (ko00280) valine, leucine and isoleucine degradation, (ko00290) valine, leucine and isoleucine biosynthesis, (ko00300) lysine biosynthesis, (ko00310) lysine degradation, (ko00340) histidine metabolism, (ko00350) tyrosine metabolism, (ko00360) phenylalanine metabolism, (ko00380) tryptophan metabolism, (ko00400) phenylalanine, tyrosine and tryptophan biosynthesis. The Carbohydrate metabolism subclass involves 15 pathways, especially including (ko00010) glycolysis/gluconeogenesis, (ko00020) citrate cycle (TCA cycle), (ko00500) starch and sucrose metabolism etc.
Based on the threshold of |log2FC| ≥ 1 and FDR < 0.05, 790 significantly differentially expressed genes of O. sinensis mycelium were obtained when comparing BBD and CK, among which 592 genes were up-regulated and 198 genes were down-regulated. Among the differential genes within the threshold of|log2FC| ≥ 5, 12 genes with higher difference folds were up-regulated and one gene was down-regulated. Volcano plots that demonstrate significant DEGs between each comparison are shown in Figure 2A. The hierarchical clustering analysis for DEGs showed that the expression patterns of the two treatments were different (Figure 2B).
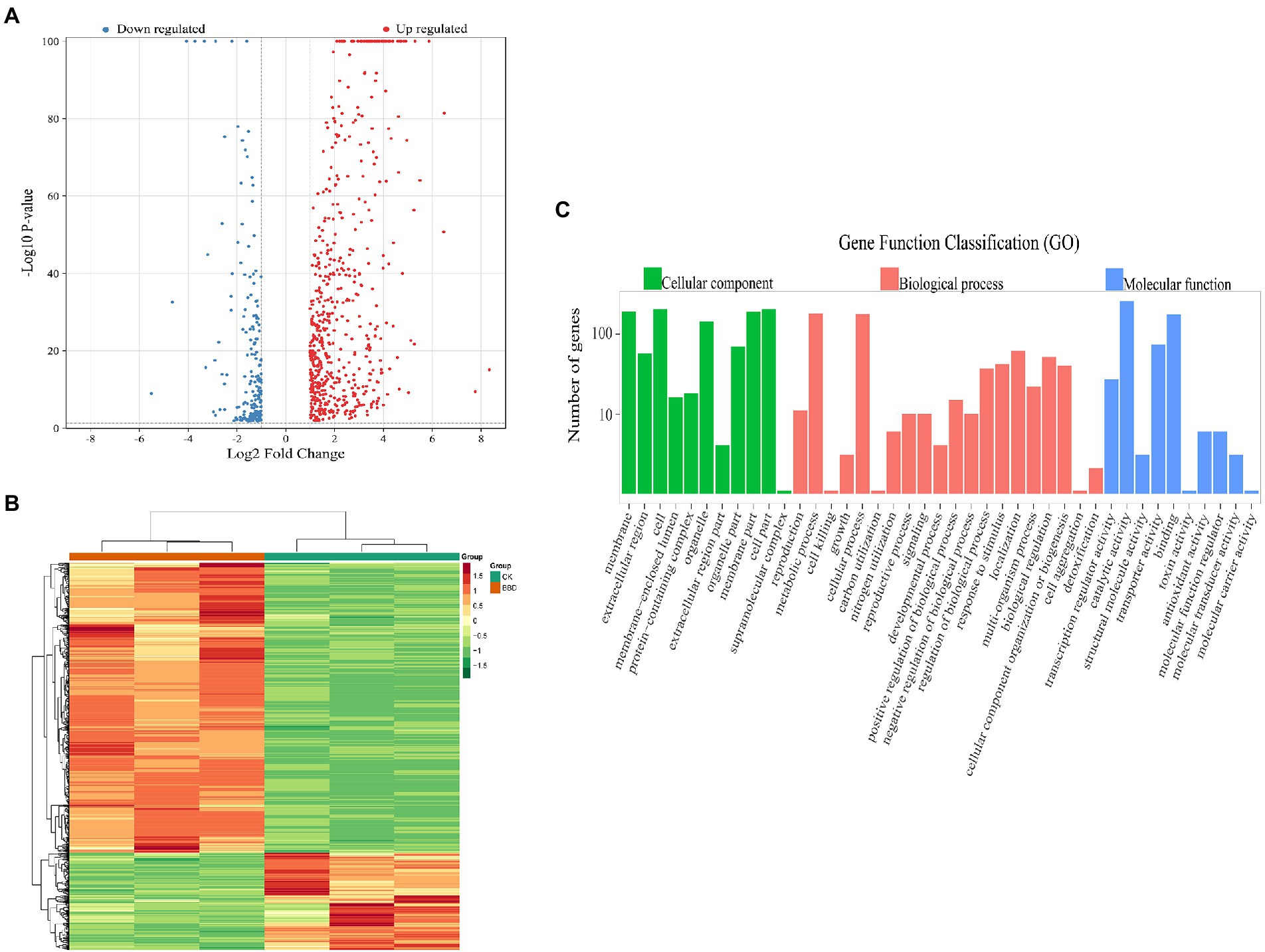
Figure 2. (A) Volcano plot of differentially expressed genes (DEGs); (B) Heatmap analysis of six samples; (C) Histogram of GO classification of DEGs. The abscissa represents the secondary GO entry and the ordinate represent the data of the differential genes of the GO entry.
Based on the GO database, the functional classification of differentially expressed O. sinensis mycelium genes in BBD and CK sets can provide a macroscopic understanding of the functional distribution of differentially expressed genes (Figure 2C). The results show that a total of 790 DEGs were annotated to GO functional classifications, including biological processes, cellular component, and molecular functions. Among biological processes, there were 20 subtypes annotated, with most genes involved in metabolic processes (GO:0008152), cellular processes (GO:0009987), and localization (GO:0051179), with 181 (22.91%), 178 (22.53%), and 62 (7.84%), respectively. There are 11 subtypes in the category of cellular components, most of the genes were assigned to genes involved in cell (GO:0005623), a cellular fraction (GO:0044464), and membrane (GO:0016020), 207 (26.20%), 207 (26.20%) and 193 (24.43%), respectively. 10 subtypes were annotated in terms of molecular function, with most genes involved in catalytic activity (GO:0003824), binding (GO:0005488), and transporter protein activity (GO:0005215), with percentages of 261 (33.04%), 177 (22.41%), and 74 (9.37%), respectively.
DEG functions were annotated based on the KOG (Clusters of orthologous groups for eukaryotic complete genomes) database. In this study, 271 (34.3%) DEGs were classified into 26 functional categories (Figure 3A). The main categories are general cluster general function prediction only (54, 19.93%), secondary metabolite biosynthesis, transport, and catabolism (44, 16.24%), amino acid transport and metabolism (25, 9.23%), posttranslational modification, protein turnover, chaperones (24, 8.86%), followed by carbohydrate transport and metabolism (22, 8.12%), lipid transport and metabolism (20, 7.38%), inorganic ion transport and metabolism (17, 6.27%), and signal transduction mechanisms (12, 4.43%).

Figure 3. (A) A cluster of orthologous groups annotations of assembled DEGs. (B) KEGG enrichment of DEGs. Rich ratio = term candidate genes num/term genes num. P-value ≤0.05 indicates significant enrichment.
Based on the KEGG database, the DEGs were mapped into the KEGG database to perform enrichment analysis of metabolic pathways and further study their functions. The results showed that 200 DEGs were successfully annotated in 98 metabolic pathways. This study selected the top 6 metabolic pathways with significantly enriched differential genes (Figure 3B), including metabolic pathways (140, 70%), ABC transporters (13, 6.5%), starch and sucrose metabolism (11, 5.5%), tyrosine metabolism (10, 5%), non-homologous end-joining (6, 3%) and glutathione metabolism (8, 4%). To get a more comprehensive understanding of the expression status of differential genes before and after optimization of the medium, genes associated with increased mycelial protein content were screened based on transcriptomic data, the study concluded that the up-regulated genes were mainly present in metabolic pathways, ABC transporters, starch and sucrose metabolism, and tyrosine metabolism. However, it is noteworthy that 13 genes were enriched on ABC transporter proteins, which may be related to the increase in nutrient uptake by mycelium.
Based on the transcriptome data of O. sinensis, it was found that most DEGs were enriched in amino acid metabolic pathways, such as tyrosine metabolism, glutathione metabolism, cysteine and methionine metabolism, alanine, aspartate and glutamate metabolism. This means that the optimization of medium stimulates mycelium growth and development and substance synthesis. The tyrosine metabolic pathway was significantly enriched in the amino acid metabolic pathway (Figure 4A). Notably, tyrosinase and catechol-O-methyltransferase played important roles as significantly upregulated enzymes in the tyrosine metabolic pathway. Some DEGs annotated as tyrosinase (G6O67_008272/G6O67_003412/G6O67_005660/G6O67_008019) were considerably more expressed in BBD than in CK. Previous study has indicated that tyrosinase catalyzes the oxidation of tyrosine to two catecholamines (dopamine and dopamine), which are key enzymes in melanin synthesis and control the activity of melanocytes in living organisms (Sánchez Ferrer et al., 1995; Masuoka and Maekawa, 2016). Besides the effect from mycelial protein content, the difference in mycelial pigmentation of BBD and CK might be indirectly related to tyrosinase (Seo et al., 2003). Furthermore, some DEGs were annotated as homogentisate 1,2-dioxygenase (HGD), alcohol dehydrogenase 1/7 alcohol dehydrogenase 1/7 (ADH1_7) were both upregulated. It is noteworthy that the primary-amine oxidase (AOC3) was significantly down-regulated.
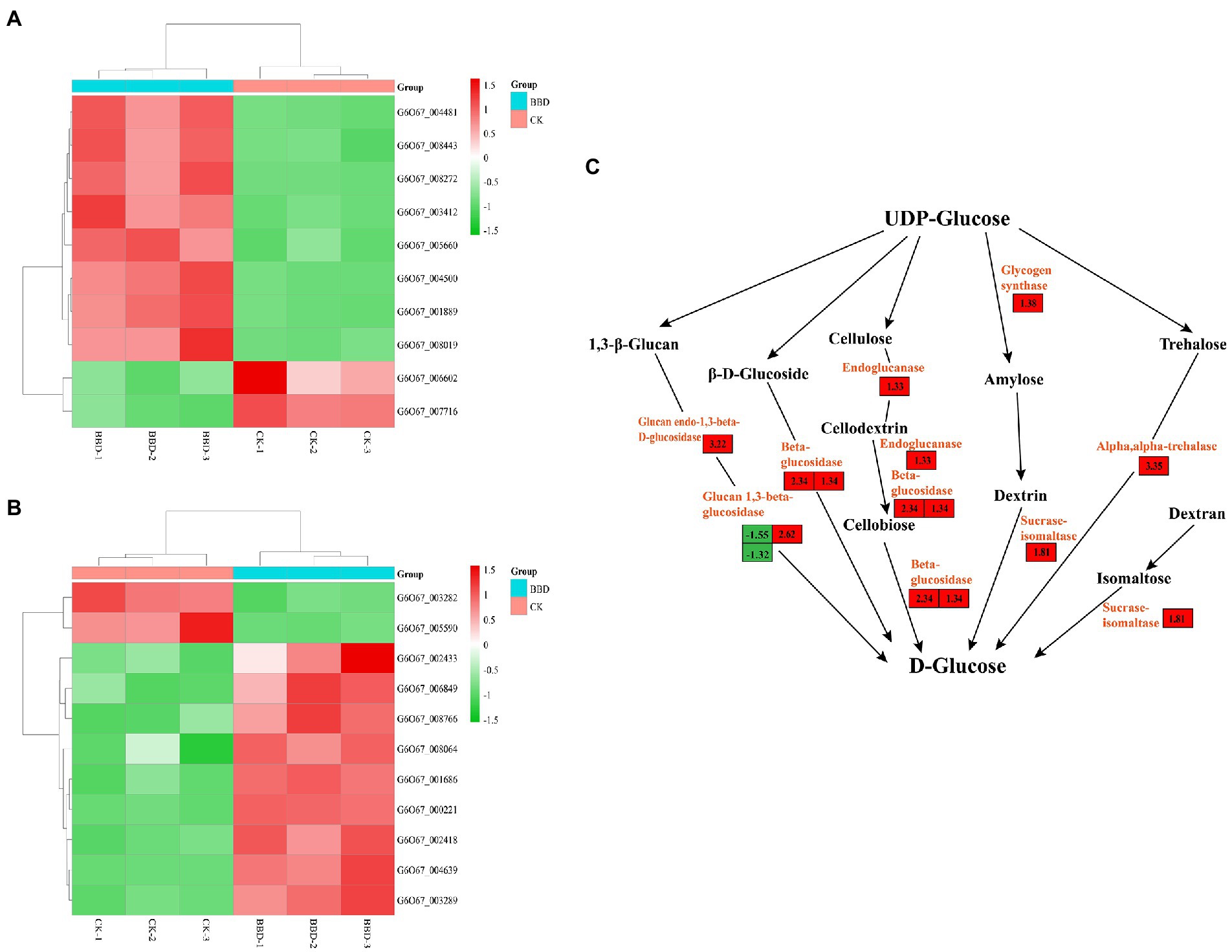
Figure 4. Based on KEGG analysis of differential genes in O. sinensis mycelium BBD and CK. Heatmaps illustrate the relative expression levels of these genes. (A) Tyrosine metabolism; (B) Starch and sucrose metabolism; (C) DEGs in starch and sucrose metabolism.
DEGs related to glutathione metabolism γ-glutamylcyclo-transferase (G6O67_001865), γ-glutamyl transpeptidase/glutathione hydrolase (G6O67_004614), isocitrate dehydrogenase (G6O67_006510) and glutathione S-transferase (G6O67_008888, G6O67_000596, G6O67_001875, G6O67_005094) were significantly up-regulated. Their final products are 5-oxoproline, L-cysteine, glutathione etc. Research has shown that fungi are high in bioactive peptides or amino acids such as glutathione and L-cysteine, which are useful for immunity, antioxidant, detoxification, and cancer treatment (Lv et al., 2019; Minich and Brown, 2019). Additionally, L-cysteine is an essential sulfur-containing amino acid for the synthesis of reduced glutathione. Glutathione S-transferase (G6O67_008888, G6O67_000596, G6O67_001875, G6O67_005094) was found to be the most significantly upregulated gene expressed in BBD. Previous studies have reported that GSTs are a major detoxifying enzyme in organisms, catalyzing the binding of glutathione to electrophilic substrates, increasing the water solubility of toxic substances, and eliminating reactive oxygen species produced in organisms subjected to oxidative stress (Morel et al., 2009). Therefore, glutathione s-transferase may be an important factor leading to the increase of mycelial active protein content, improving the antioxidant function of mycelium.
In alanine, aspartate and glutamate metabolism, most of enzymes containing adenylosuccinate synthase (purA), glutamate dehydrogenase (GDH), glutamine synthetase (glnA), glutamine-fructose-6-phosphate transaminase (glmS) and carbamoyl-phosphate synthase (CAD) were significantly upregulated. It was found that the upregulation of glutamate dehydrogenase and glutamine synthetase accelerates the synthesis of glutamate and glutamine during the synthesis of amino acids. As a major metabolic center in many organisms, glutamate, in addition to its role in protein synthesis, is involved in a variety of processes such as amino acid synthesis and the formation of secondary natural products and nitrogen assimilation (Walker and van der Donk, 2016). Glutamate is usually produced through two pathways, both of which lead to the overall conversion of 2-oxoglutarate (an intermediate of the citric acid cycle) to glutamate. One of these pathways is the reduction of ammonia as a nitrogen donor through glutamate dehydrogenase (GDH) to aminate 2-oxoglutarate (Brosnan and Brosnan, 2013). GDH belongs to the family of amino acid dehydrogenases, with other members acting on leucine, phenylalanine or valine, and tends physiologically in the direction of deamination (Tang and Hutchinson, 1993). Additionally, glutamine is produced by the action of glutamine synthetase, which uses ammonium as a nitrogen source to catalyze the ATP-dependent amination of glutamate. This finding implies that, because of the upregulation of glutamate dehydrogenase in relation to glutamate and glutamine synthesis, the variety of amino acids synthesized after medium optimization may be more in the non-optimized medium.
To dig deeper into the relevant genes affecting mycelial protein content, the DEGs annotated in KEGG based on the starch and sucrose metabolic pathway were first targeted and a total of 11 differential genes were found, nine of which were up-regulated and two down-regulated (Figure 4B). Six pathways for the synthesis of D-glucose were found to exist in this pathway (Figure 4C). The largest number of genes annotated was β-glucosidase, which is an important component of the cellulolytic enzyme family. It mainly catalyzes the generation of cellobiose from cellulose dextrin and then is catalyzed by β-glucosidase to glucose. Meanwhile, β-glucosidase can also catalyze the generation of glucose from β-D-glucoside. During optimization, β-glucosidase was higher than the unoptimized level. Other differential genes were annotated as endoglucanases, which often work together with β-glucosidase to completely hydrolyze cellulose molecules into glucose. Therefore, we speculate that the differential genes may provide an additional carbon source for the improved protein content of the optimized mycelium.
After transcriptome data analysis, this study found that DEGs were mainly enriched in four major categories involving metabolism, genetic information processing, environmental information processing and cellular processes. Among the subclass of the carbohydrate metabolism pathway, this research particularly focused on the Citrate cycle (TCA cycle) (Eprintsev et al., 2018). Notably, pyruvate carboxylase (pyc), isocitrate dehydrogenase (IDH) and citrate synthase (gltA) were significantly upregulated in the TCA cycle. Citrate synthase and isocitrate dehydrogenase are the rate-limiting enzymes in the TCA cycle (Akram, 2014). They are essential for the intermediate products of the cycle (e.g., oxaloacetate, α-ketoglutarate), which are the raw materials for the synthesis of sugars, amino acids and fats (Fernie et al., 2004; Xie et al., 2022). Isocitrate dehydrogenase has important oxidoreductase and decarboxylation functions as a component of the TCA cycle. It catalyzes the decarboxylation of isocitrate to produce α-ketoglutarate, releasing CO2 and NADPH (Spaans et al., 2015). Previous studies have also demonstrated that isocitrate dehydrogenase is a major source of NADPH and has an active role in the synthesis of metabolites such as amino acids (Becker et al., 2007; Papagianni, 2012). In addition, IDH plays an important role in cellular defense against oxidative damage and ROS detoxification (Mailloux et al., 2007; Paul et al., 2007). The presence of these enzymes suggests that the gradual increase in protein content after medium optimization probably influences the promotion of the tricarboxylic acid cycle in mycelium.
To confirm the reliability of the RNA-Seq analysis, RT-qPCR analysis of the expression levels of randomly selected genes in the major pathways enriched by DEGs was performed in this study. RT-qPCR was performed on 13 genes to verify differential gene expression obtained by RNA-Seq (Figure 5). This result found that the direction of fold change detected by RT-qPCR of gene expression changes was similar to Illumina sequencing results, with four genes down-regulated and nine genes up-regulated. These outcomes demonstrate the dependability of our transcriptome data.
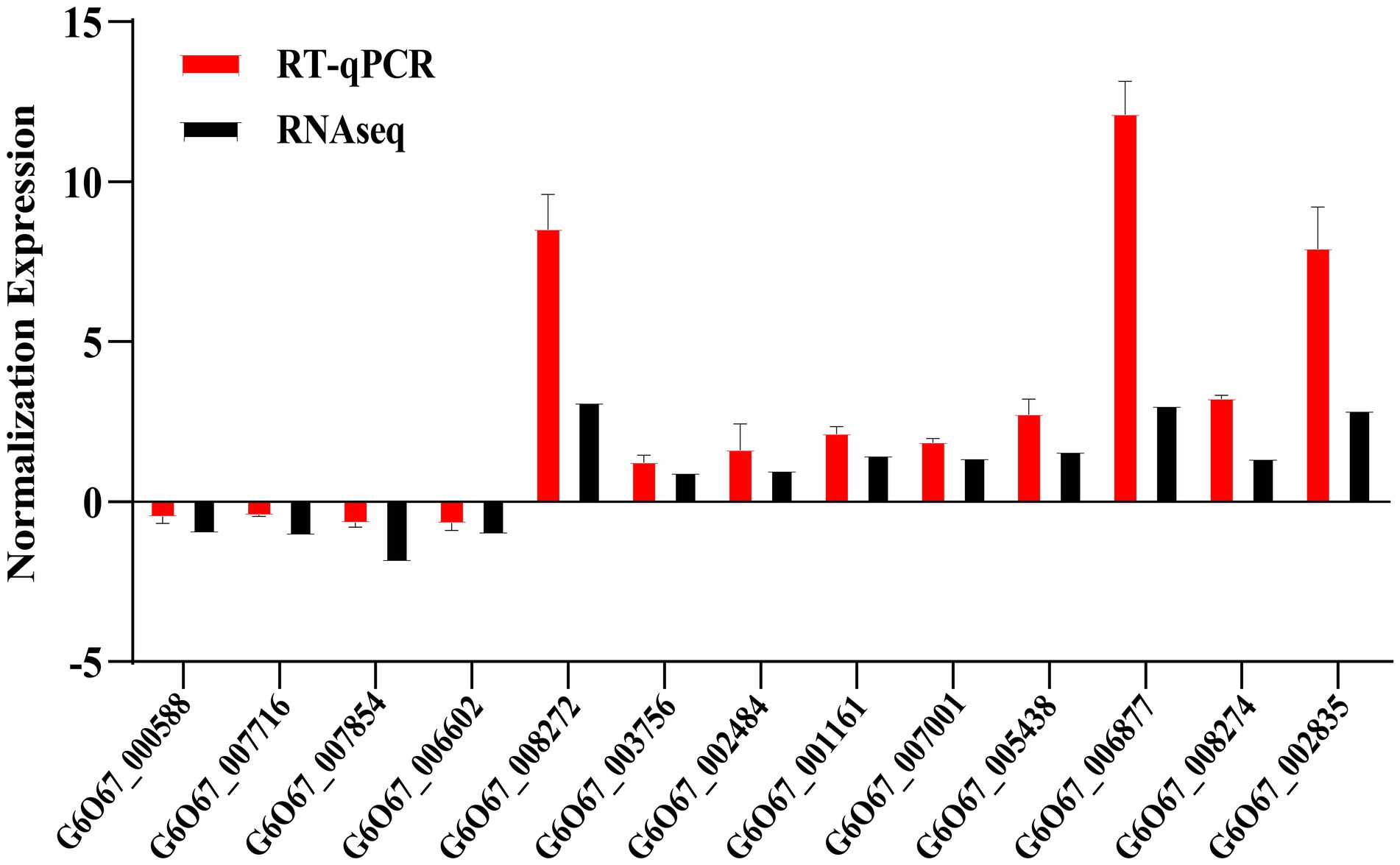
Figure 5. Quantitative identification of differentially expressed genes. Red bars represent RT-qPCR results (2−ΔΔCt). Black bars indicate RNA-seq results (log2 fold change). Error bars represent standard errors. ACT1 gene was the internal reference.
Fungal products from O. sinensis are an important alternative to Chinese Cordyceps. The main ingredient found to have a good development prospect is Cordyceps protein, and its level can also be used as an important index for quality evaluation (Wang et al., 2018). Therefore, it is crucial to gain a reliable culture optimization method to improve the enzymatic activity and total protein of mycelium. Traditional methods for optimization of culture media often use an orthogonal test method and response surface method, which can be used to consider several factors simultaneously and find the best combination of factor levels but cannot obtain a clear regression equation of factors and response values over the whole region (Kim and Na, 1997; Zhang et al., 2010). However, PBD and RSM can be used to determine the influence of the test factors and the interaction between the factors on the response values during the treatment (Elibol, 2002). A previous study using PBD and CCD design has shown the optimal concentrations of NaH2PO4, Na2HPO4, NaCl, yeast extract and corn pulp, which resulted in the highest productivity of succinic acid (Stylianou et al., 2021). In addition, it has been found in previous studies that the use of PBD and RSM for media optimization are very effective methods and produce satisfactory results (Guo et al., 2018).
In this research, the medium of O. sinensis was optimized using RSM, which included medium components containing a carbon source, nitrogen source and trace elements. It was found that a carbon source contributed the most to the increase of mycelial protein content of O. sinensis. Carbohydrates have a great influence on the fermentation process when the microorganisms undergo mass multiplication by fermentation culture. Sufficient carbon must be available for biosynthesis and energy production to allow for the survival of the cells (Manirafasha and Lu, 2015). It was found that in the culture of O. sinensis, carbon and nitrogen sources are the main building block sources for protein synthesis, and appropriate changes in the ratio have an effect on mycelium production and active proteins or enzymes (Dong et al., 2009). In the three-dimensional surface plot and contour plot of the interaction effects, indicating two factors that are close to elliptical and have significant interaction are beef broth and glucose. These provide a carbon source, and their ratios play an important role in the growth of the fungi. When the addition of peptone was 0.1%, the mycelial protein content increased and then decreased with the addition of both, with the same trend, but the contour density along the direction of beef broth was greater than that along the direction of glucose, indicating that increasing the amount of beef broth was more effective than increasing glucose within a certain range.
It is noteworthy that the addition of glucose at the highest mycelial protein content during medium optimization gives an intermediate value, indicating that too much or too little may hinder the growth of mycelium. It was found that the combination of medium components had a profound effect on the metabolic pathways of microorganisms regulating the production of many metabolites. This implies that the metabolic pathways of mycelia cultured under the optimized medium are active when the glucose concentration reaches 2%. This is consistent with the results of the transcriptome analysis.
The statistical results support the view of this study in the RSM hypothesis that carbohydrates play an important role in the transcriptome. Transcriptome analysis revealed that DEGs are associated with GO terms ‘metabolic processes’, ‘cellular processes’ and ‘catalytic activity’. This implies that the expression of metabolic processes in altered medium composition is associated with mycelial dynamics and that proteins or enzymes expressed by DEGs contribute to medium utilization by the mycelium (Xie et al., 2022). Increased mycelial protein content implies an increase in protein or protein mixtures produced by the fungus and excreted to the culture medium. Increased mycelial protein content usually manifests as, for example, an increased level of enzyme activity, such as cellulase, hemicellulase, protease, or production of more total extracellular protein (Pakula et al., 2013). Previous studies have shown that cellulase, hemicellulase, ligninase, and pectinase production is susceptible to regulation primarily at the transcriptional level in filamentous fungi (Aro et al., 2005).
KEGG enrichment of DEGs showed that they are mainly involved in ATP binding cassette (ABC) transport proteins, ‘starch and sucrose metabolism’, ‘amino acid metabolism’ (e.g., alanine, aspartate and glutamate metabolism, tyrosine metabolism), fatty acid degradation, and fatty acid biosynthesis. These pathways are associated with basic metabolic types and are involved in the synthesis of primary and secondary metabolites. Previous studies have shown that starch and sucrose metabolism has a key role in fungal growth (Chen et al., 2021). Carbohydrates are important components of fungal cell walls and fungal growth makes higher metabolic demands (Coronado et al., 2008). Nine genes involved in the “starch metabolism process” were significantly up-regulated and two were significantly down-regulated. Annotation showed DEGs encoding a glucan endo−1,3-beta-D-glucosidase (G6O67_004639), an endoglucanase (G6O67_001686), and DEGs encoding alpha-trehalase (G6O67_008766) in this transcriptome. Endoglucanase is one of the components of cellulase, which hydrolyzes the β − 1,4-glycosidic bond in the internal non-crystalline region of the cellulose molecule, ultimately hydrolyzing cellulose to glucose, providing the fungus with more carbon and access to more exogenous proteins (Pakula et al., 2013). Not only did the sucrose and starch pathways contribute to the increase in mycelial protein content during medium optimization, but starch and sucrose metabolism are associated with the production of energy and active compounds for fruiting body maturation (Tong et al., 2020).
Notably, the 11 upregulated genes were enriched in five ATP-binding cassette (ABC) transporter protein subfamilies, namely ABCA (ABCA3), ABCB (ABCB1), ABCC (ABCC1), ABCD (PXA1/2), ABCG (PDR5, SNQ2), which were found in previous studies to be possibly related to nutrient uptake by the fungus (Adour et al., 2010; Wu et al., 2022). GO analysis showed that the transporter proteins are involved in carbohydrate derivative binding, ATP binding and have active transmembrane transporter protein activity. The increased expression of ATP-binding cassette (ABC) transport proteins could facilitate the transport of glucose, which requires entry into the cytoplasmic membrane (Frelet and Klein, 2006). In the mycelium culture, the optimized medium composition may have activated the ABC transporter proteins to using the energy generated by ATP hydrolysis to transport the substrate through a reverse concentration gradient. In addition, ABC transport proteins are also involved in cellular processes such as DNA repair, transcription and regulation of gene expression (Braz et al., 2004). Three transport proteins are particularly highly expressed in sclerotia and show the positive effects on transport proteins and the diffusion of specific molecules such as peptides and proteins (Zhao et al., 2020). The highly expressed transporter proteins provide the conditions for substrate transport and contribute to the growth and development of mycelium and protoplasts.
Optimization of culture substrates or genetic modification of fungal strains are commonly used as the main ways of improving the protein production of filamentous fungi (Punt et al., 2002). Overexpression or deletion of genes is used to improve protein and enzyme yields. Yoon et al. disrupted decuple protease gene in the filamentous fungus Aspergillus oryzae to greatly improve heterologous protein production (Yoon et al., 2010). In addition, Genetic modifications can also be used to improve heterologous protein production when promoters or regulatory elements of genes encoding secreted proteins are used for heterologous expression (Pakula et al., 2013). Madhavan et al. demonstrated that after disrupting their natural β-galactosidase (lac4) promoter the fungal promoter was shown to have the potential to drive heterologous protein production in Kluyveromyces lactis (Madhavan and Sukumaran, 2014). The expression level of recombinant Pro-transglutaminase (pro-MTG) from Streptomyces mobaraensis achieved 87.6 U/ml and 70.7 U/ml under the control of two constitutive promoter PsodA and PydzA, respectively, demonstrating that optimized strong promoters can express extracellular proteins at high levels in Bacillus subtilis (Liu et al., 2018). However, this study identified DEGs and major enrichment pathways, but the use of promoters and modification of the genes has not been explored. Further attention and efforts are needed to explore the effects of genetic modifications of the fungus and the conditions of protein production in O. sinensis modified under suitable media.
In summary, this work used Plackett-Burman design, path of steepest ascent method, and Box–Behnken design to screen the culture medium of O. sinensis and obtained the best medium with the highest mycelial protein content, while transcriptomics was used to analyze the medium before and after optimization at the gene level. It found that changing the ratio of the medium components could increase the mycelial protein content of O. sinensis. However, excessive addition would inhibit protein synthesis. The micronutrients KH2PO4 and MgSO4 in the medium had no significant effect on the total mycelium protein content (p > 0.05). The highest mycelial protein content of 2.11% was obtained based on the optimization results, at which point the optimized medium formulation was finalized as 20% beef broth, 0.10% peptone, 2% glucose, 0.15% yeast extract, 0.20% KH2PO4, and 0.02% MgSO4. Most of the up-regulated differential genes affecting mycelium protein content were analyzed from a transcriptomic perspective. The identification of DEGs revealed 790 genes, of which the number of up-regulated genes was 592 and the number of down-regulated genes was 198 genes. The optimized expression pattern had more up-regulated genes than before optimization. The main enriched pathways of DEGs affecting protein content were ABC transporters, starch and sucrose metabolism, tyrosine metabolism and glutathione metabolism. In addition, it is noteworthy that individual enzymes such as tyrosinase (TYR), glutathione S-transferase (gst), glutamine synthetase (glnA), β-glucosidase, pyruvate carboxylase (pyc), isocitrate dehydrogenase (IDH1) and citrate synthase (gltA) potentially contribute more to the increase in mycelial protein content, while the difference in mycelium color before and after medium optimization was likely due to tyrosinase. In conclusion, this study not only lays a foundation for further research targeting the proteins of O. sinensis, but also benefits industrialization and development applications of O. sinensis.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
C-YT designed and performed the experiments and wrote the manuscript. X-ZL, WS, XL, and JW analyzed the interpretation of the data-designed experiments and revised them critically for intellectual content. J-BC, JL, and TW performed the experiments. Y-LL provided reagents and experimental equipment. All authors contributed to the article and approved the submitted version.
This research was funded by a grant of the major science and technology projects of Qinghai Province (2021-SF-A4).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1055055/full#supplementary-material
Addelman, S. (1979). Statistics for experimenters. Technometrics 21, 387–388. doi: 10.1080/00401706.1979.10489788
Adour, L., Bude, F., and Amrane, A. (2010). Sequential use of ammonium and leucine as nitrogen sources during growth of Geotrichum candidum on a glucose based medium. Electron. J. Biotechnol. 13, 13–14. doi: 10.4067/S0717-34582010000100013
Akram, M. (2014). Citric acid cycle and role of its intermediates in metabolism. Cell Biochem. Biophys. 68, 475–478. doi: 10.1007/s12013-013-9750-1
Aro, N., Pakula, T., and Penttilä, M. (2005). Transcriptional regulation of plant cell wall degradation by filamentous fungi. FEMS Microbiol. Rev. 29, 719–739. doi: 10.1016/j.femsre.2004.11.006
Becker, J., Klopprogge, C., Herold, A., Zelder, O., Bolten, C. J., and Wittmann, C. (2007). Metabolic flux engineering of L-lysine production in Corynebacterium glutamicum--over expression and modification of G6P dehydrogenase. J. Biotechnol. 1322, 99–109. doi: 10.1016/j.jbiotec.2007.05.026
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. doi: 10.1016/0003-2697(76)90527-3
Braz, A. S. K., Finnegan, J., Waterhouse, P., and Margis, R. (2004). A plant orthologue of RNase L inhibitor (RLI) is induced in plants showing RNA interference. J. Mol. Evol. 59, 20–30. doi: 10.1007/s00239-004-2600-4
Brosnan, J. T., and Brosnan, M. E. (2013). Glutamate: a truly functional amino acid. Amino Acids 45, 413–418. doi: 10.1007/s00726-012-1280-4
Chan, C. H., Lam, S., Fong, F., Chan, D., Lee, F., and Sze, E. (2018). Optimization of protein extraction and two-dimensional gel electrophoresis profiles for the identification of Cordyceps sinensis and other similar species. PLoS One 13:e0202779. doi: 10.1371/journal.pone.0202779
Chen, Z., Yu, J., Feng, Y., Ma, M., Wansong, Y., and Guo, X. (2021). Transcriptome different analysis of Tremella aurantialba at mycelium and fruiting body stages. Biotechnol. Bull. 376, 73–84. doi: 10.13560/j.cnki.biotech.bull.1985.2020-1038
Coronado, J., Mneimneh, S., Epstein, S., Qiu, W. G., and Lipke, P. (2008). Conserved processes and lineage-specific proteins in fungal Cell Wall evolution. Eukaryot. Cell 6, 2269–2277. doi: 10.1128/EC.00044-07
Dong, C. H., Xie, X. Q., Wang, X. L., Zhan, Y., and Yao, Y. J. (2009). Application of box-Behnken design in optimisation for polysaccharides extraction from cultured mycelium of Cordyceps sinensis. Food Bioprod. Process. 87, 139–144. doi: 10.1016/j.fbp.2008.06.004
Elibol, M. (2002). Response surface methodological approach for inclusion of perfluorocarbon in actinorhodin fermentation medium. Process Biochem. 38, 667–673. doi: 10.1016/S0032-9592(02)00171-1
Eprintsev, A. T., Fedorin, D. N., Dobychina, M. A., and Igamberdiev, A. U. (2018). Regulation of expression of the mitochondrial and peroxisomal forms of citrate synthase in maize during germination and in response to light. Plant Sci. 272, 157–163. doi: 10.1016/j.plantsci.2018.04.017
Fernie, A. R., Carrari, F., and Sweetlove, L. J. (2004). Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 7, 254–261. doi: 10.1016/j.pbi.2004.03.007
Frelet, A., and Klein, M. (2006). Insight in eukaryotic ABC transporter function by mutation analysis. FEBS Lett. 580, 1064–1084. doi: 10.1016/j.febslet.2006.01.024
Guo, F., Zheng, H., Cheng, Y., Song, S., Zheng, Z., and Jia, S. (2018). Medium optimization for ε-poly-L-lysine production by Streptomyces diastatochromogenes using response surface methodology. Lett. Appl. Microbiol. 66, 124–131. doi: 10.1111/lam.12812
Hao, K., Wang, F., Nong, X., McNeill, M., Liu, S., Wang, G., et al. (2017). Response of peanut Arachis hypogaea roots to the presence of beneficial and pathogenic fungi by transcriptome analysis. Sci. Rep. 7:964. doi: 10.1038/s41598-017-01029-3
He, J. (2018). Harvest and trade of caterpillar mushroom (Ophiocordyceps sinensis) and the implications for sustainable use in the Tibet region of Southwest China. J. Ethnopharmacol. 221, 86–90. doi: 10.1016/j.jep.2018.04.022
He, L., Ji, P., Cheng, J., Wang, Y., Qian, H., Li, W., et al. (2013). Structural characterization and immunostimulatory activity of a novel protein-bound polysaccharide produced by Hirsutella sinensis Liu, Guo. Yu Zeng. Food Chem. 141, 946–953. doi: 10.1016/j.foodchem.2013.04.053
Hsieh, C., Tsai, M., Hsu, T., Chang, D., and Lo, C. (2005). Medium optimization for polysaccharide production of Cordyceps sinensis. Appl Biochem Biotech. 120, 145–157. doi: 10.1385/ABAB:120:2:145
Jia, J. M., Ma, X. C., Wu, C. F., Wu, L. J., and Hu, G. S. (2005). Cordycedipeptide a, a new cyclodipeptide from the culture liquid of Cordyceps sinensis (Berk.) Sacc. Chem Pharm Bull (Tokyo). 53, 582–583. doi: 10.1248/cpb.53.582
Kim, S., and Na, S. (1997). Response surface method using vector projected sampling points. Struct. Saf. 19, 3–19. doi: 10.1016/S0167-4730(96)00037-9
Kong, B. H., AlvinRazif, C. S., Muhammad Fazril MohamadNg, S. T. T., SengFung, C., and Yee, S. (2021). Antioxidant and cytotoxic effects and identification of Ophiocordyceps sinensis bioactive proteins using shotgun proteomic analysis. J Food Technol Biotech. 59, 201–208. doi: 10.17113/ftb.59.02.21.7151
Li, Y., Wang, X., Jiao, L., Jiang, Y., Li, H., Jiang, S., et al. (2011). A survey of the geographic distribution of Ophiocordyceps sinensis. J. Microbiol. 49, 913–919. doi: 10.1007/s12275-011-1193-z
Li, Y., Yan, Y., Tang, Z., Wang, K., He, J. S., and Yao, Y. J. (2021). Conserving the Chinese caterpillar fungus under climate change. Biodivers. Conserv. 30, 547–550. doi: 10.1007/s10531-020-02109-z
Liu, X., Wang, H., Wang, B., and Pan, L. (2018). High-level extracellular protein expression in Bacillus subtilis by optimizing strong promoters based on the transcriptome of Bacillus subtilis and bacillus megaterium. Protein Expres Purif. 151, 72–77. doi: 10.1016/j.pep.2018.06.006
Lv, H., Zhen, C., Liu, J., Yang, P., Hu, L., and Shang, P. (2019). Unraveling the potential role of glutathione in multiple forms of cell death in cancer therapy. Oxidative Med. Cell. Longev. 2019, 3150145–3150116. doi: 10.1155/2019/3150145
Madhavan, A., and Sukumaran, R. (2014). Promoter and signal sequence from filamentous fungus can drive recombinant protein production in the yeast Kluyveromyces lactis. Bioresour. Technol. 165, 302–308. doi: 10.1016/j.biortech.2014.03.002
Mailloux, R. J., Bériault, R., Lemire, J., Singh, R., Chénier, D. R., Hamel, R. D., et al. (2007). The tricarboxylic acid cycle, an ancient metabolic network with a novel twist. PLoS One 2:e690. doi: 10.1371/journal.pone.0000690
Manirafasha, E., and Lu, Y. (2015). Optimization of Medium Components for Natamycin Production by Streptomyces Gilvosporeus. Xiamen: Xiamen University
Masuoka, Y., and Maekawa, K. (2016). Gene expression changes in the tyrosine metabolic pathway regulate caste-specific cuticular pigmentation of termites. Insect Biochem Molec. 74, 21–31. doi: 10.1016/j.ibmb.2016.04.006
Minich, D., and Brown, B. (2019). A review of dietary (Phyto) nutrients for glutathione support. Nutrients 11:2073. doi: 10.3390/nu11092073
Morel, M., Ngadin, A., Droux, M., Jacquot, J. P., and Gelhaye, E. (2009). The fungal glutathione S-transferase system. Evidence of new classes in the wood-degrading basidiomycete Phanerochaete chrysosporium. Cell. Mol. Life Sci. 66, 3711–3725. doi: 10.1007/s00018-009-0104-5
Pakula, T., Saloheimo, M., Hakkinen, M., Westerholm-Parvinen, A., Penttila, M., and Vitikainen, M. (2013). Method for Protein Production in Filamentous fungi. U.S. Patent No 13/701894. Washington, DC: U.S. Patent.
Papagianni, M. (2012). Recent advances in engineering the central carbon metabolism of industrially important bacteria. Microb. Cell Factories 11:50. doi: 10.1186/1475-2859-11-50
Paul, L., Mishra, P. K., Blumenthal, R. M., and Matthews, R. G. (2007). Integration of regulatory signals through involvement of multiple global regulators: control of the Escherichia coli gltBDF operon by Lrp, IHF, Crp, and ArgR. BMC Microbiol. 7:2. doi: 10.1186/1471-2180-7-2
Punt, P. J., van Biezen, N., Conesa, A., Albers, A., Mangnus, J., and van den Hondel, C. (2002). Filamentous fungi as cell factories for heterologous protein production. Trends Biotechnol. 20, 200–206. doi: 10.1016/S0167-7799(02)01933-9
Qian, Z., Fan, J., Li, C., Yang, F., and Li, W. (2019). Effects of different drying conditions on protein in Cordyceps. China J Chin. Mater Med. 44, 1983–1988. doi: 10.19540/j.cnki.cjcmm.20181101.002
Qiu, X., Cao, L., and Han, R. (2020). Analysis of volatile components in different Ophiocordyceps sinensis and insect host products. Molecules 25:1603. doi: 10.3390/molecules25071603
Rakhee,, Mishra, J., Yadav, R. B., Meena, D. K., Arora, R., Sharma, R. K., et al. (2021). Novel formulation development from Ophiocordyceps sinensis (Berk.) for management of high-altitude maladies. 3 Biotech 11:9. doi: 10.1007/s13205-020-02536-3
Ren, Y., Qiu, Y., Wan, D., Lv, X., and Guo, J. (2013). Preliminary study on correlation between diversity of soluble proteins and producing area of Cordyceps sinensis. China J Chin. Mater Med. 3809, 1375–1377. doi: 10.4268/cjcmm20130923
Sánchez Ferrer, Á., Neptuno Rodríguez-López, J., García-Cánovas, F., and García-Carmona, F. (1995). Tyrosinase: a comprehensive review of its mechanism. Biochim. Biophys. Acta 1247, 1–11. doi: 10.1016/0167-4838(94)00204-T
Seo, S., Sharma, V. K., and Sharma, N. (2003). Mushroom tyrosinase: recent prospects. J Agrc Food Chem. 51, 2837–2853. doi: 10.1021/jf020826f
Shu, R., Zhang, J., Meng, Q., Zhang, H., Zhou, G., Li, M., et al. (2020). A new high-quality draft genome assembly of the Chinese cordyceps Ophiocordyceps sinensis. Genome Biol. Evol. 12, 1074–1079. doi: 10.1093/gbe/evaa112
Spaans, S. K., Weusthuis, R. A., van der Oost, J., and Kengen, S. W. (2015). NADPH-generating systems in bacteria and archaea. Front. Microbiol. 6:742. doi: 10.3389/fmicb.2015.00742
Stylianou, E., Pateraki, C., Ladakis, D., Vlysidis, A., and Koutinas, A. (2021). Optimization of fermentation medium for succinic acid production using Basfia succiniciproducens. Environ Technol Innov. 24:101914. doi: 10.1016/j.eti.2021.101914
Tang, L., and Hutchinson, C. R. (1993). Sequence, transcriptional, and functional analyses of the valine (branched-chain amino acid) dehydrogenase gene of Streptomyces coelicolor. J. Bacteriol. 175, 4176–4185. doi: 10.1128/jb.175.13.4176-4185.1993
Tong, X., Zhang, H., Wang, F., Xue, Z., Cao, J., Peng, C., et al. (2020). Comparative transcriptome analysis revealed genes involved in the fruiting body development of Ophiocordyceps sinensis. Peer J. 8:e8379. doi: 10.7717/peerj.8379
Walker, M. C., and van der Donk, W. A. (2016). The many roles of glutamate in metabolism. J. Ind. Microbiol. Biotechnol. 43, 419–430. doi: 10.1007/s10295-015-1665-y
Wan, S., Li, Q., Yu, H., Liu, S., and Kong, L. (2022). Transcriptome analysis based on dietary beta-carotene supplement reveals genes potentially involved in carotenoid metabolism in Crassostrea gigas. Gene 818:146226. doi: 10.1016/j.gene.2022.146226
Wang, P., Hung, Y., Li, W., Chiu, C., and Pan, T. (2014). Systematic revelation of the protective effect and mechanism of Cordyceps sinensis on diethylnitrosamine-induced rat hepatocellular carcinoma with proteomics. Oncotarget 7, 60270–60289. doi: 10.18632/oncotarget.11201
Wang, Y., Wang, Y., Chen, R., Wan, D., Peng, C., Shen, C., et al. (2018). Effect of protein extracts of Cordyceps on growth of A549 cells and immune activity of mouse peritoneal macrophages in vitro. Chinese J Exp Tradit Med Formulae. 241, 79–84. doi: 10.13422/j.cnki.syfjx.2018010079
Wei, Y., Zhang, L., Wang, J., Wang, W., Niyati, N., Guo, Y., et al. (2021). Chinese caterpillar fungus (Ophiocordyceps sinensis) in China: current distribution, trading, and futures under climate change and overexploitation. Sci. Total Environ. 755:142548. doi: 10.1016/j.scitotenv.2020.142548
Wei, J., Zheng, G., Dong, X., Li, H., Liu, S., Wang, Y., et al. (2022). Integration of transcriptome and proteome analysis reveals the mechanism of freezing tolerance in winter rapeseed. Plant Growth Regul. 96, 103–118. doi: 10.1007/s10725-021-00763-z
Wu, R. M., Jia, Q. Q., Li, X. Z., Ma, Y. F., Zhang, J., Li, Y. L., et al. (2022). Preparation of the sphingolipid fraction from mycelia of Cordyceps sinensis and its immunosuppressive activity. J. Ethnopharmacol. 291:115126. doi: 10.1016/j.jep.2022.115126
Wu, Z. Y., Li, Y. R., Xu, Y. B., Zhang, Y. P., Tao, G. J., Zhang, L., et al. (2022). Transcriptome analysis of bacillus licheniformis for improving bacitracin production. ACS Synth. Biol. 11, 1325–1335. doi: 10.1021/acssynbio.1c00593
Xiang, L., Li, Y., Zhu, Y., Luo, H., Li, C., Xu, X., et al. (2014). Transcriptome analysis of the Ophiocordyceps sinensis fruiting body reveals putative genes involved in fruiting body development and cordycepin biosynthesis. Genomics 103, 154–159. doi: 10.1016/j.ygeno.2014.01.002
Xie, L., Xie, J., Chen, X., Tao, X., Xie, J., Shi, X., et al. (2022). Comparative transcriptome analysis of Monascus purpureus at different fermentation times revealed candidate genes involved in exopolysaccharide biosynthesis. Food Res. Int. 160:111700. doi: 10.1016/j.foodres.2022.111700
Yoon, J., Maruyama, J. I., and Kitamoto, K. (2010). Disruption of ten protease genes in the filamentous fungus aspergillus oryzae highly improves production of heterologous proteins. Appl Microbiol Biot. 89, 747–759. doi: 10.1007/s00253-010-2937-0
Yuan, Q. H., Xie, F., Tan, J., Yuan, Y., Mei, H., Zheng, Y., et al. (2022). Extraction, structure and pharmacological effects of the polysaccharides from Cordyceps sinensis: a review. J. Funct. Foods 89:104909. doi: 10.1016/j.jff.2021.104909
Zang, W., Mei, Y., and Liang, Y. (2020). Purification and characterization of Ophiocordyceps sinensis plasmin from solid culture mycelia. Microbiol China 472, 562–570. doi: 10.13344/j.microbiol.china.190321
Zhang, G., Lei, H., He, X., and Shan, Y. (2010). Modern mathematical statistical methods in optimization of fermentation medium. Food Fermentation Ind. 365, 110–113. doi: 10.13995/j.cnki.11-1802/ts.2010.05.007
Zhang, Y., Li, E., Wang, C., Li, Y., and Liu, X. (2012). Ophiocordyceps sinensis, the flagship fungus of China: terminology, life strategy and ecology. Mycol. 3, 2–10. doi: 10.1080/21501203.2011.654354
Zhang, J., Yu, H., Li, S., Zhong, X., Wang, H., and Liu, X. (2020). Comparative metabolic profiling of Ophiocordyceps sinensis and its cultured mycelia using GC–MS. Food Res. Int. 134:109241. doi: 10.1016/j.foodres.2020.109241
Zhang, H., Yue, P., Tong, X., Gao, T., Peng, T., and Guo, J. (2021). Comparative analysis of fatty acid metabolism based on transcriptome sequencing of wild and cultivated Ophiocordyceps sinensis. Peer J. 9:e11681. doi: 10.7717/peerj.11681
Zhang, X., Zhu, X. Z., Shi, X. J., Yang, H., and Yi, Y. T. (2022). Extraction and purification of inulin from Jerusalem artichoke with response surface method and ion exchange resins. ACS Omega. 7, 12048–12055. doi: 10.1021/acsomega.2c00302
Keywords: Ophiocordyceps sinensis, mycelium, response surface methodology, transcriptome, soluble protein content
Citation: Tang C-Y, Wang J, Liu X, Chen J-B, Liang J, Wang T, Simpson WR, Li Y-L and Li X-Z (2022) Medium optimization for high mycelial soluble protein content of Ophiocordyceps sinensis using response surface methodology. Front. Microbiol. 13:1055055. doi: 10.3389/fmicb.2022.1055055
Edited by:
Yingnan Hou, Shanghai Jiao Tong University, ChinaReviewed by:
Chan Zhang, Beijing Technology and Business University, ChinaCopyright © 2022 Tang, Wang, Liu, Chen, Liang, Wang, Simpson, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu-Ling Li, eXVsaW5nbGkyMDAwQDE2My5jb20=; Xiu-Zhang Li, eGl1emhhbmcxMUAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.