 Qi Liang Zhu1,2†Kun Yan1†Nian Zhao Wang1Shu Qi Ma1De Shan Lu1
Qi Liang Zhu1,2†Kun Yan1†Nian Zhao Wang1Shu Qi Ma1De Shan Lu1 Xiao Hua Su2,3
Xiao Hua Su2,3 Zheng Sai Yuan2,3Yu Feng Dong4
Zheng Sai Yuan2,3Yu Feng Dong4 Yan Ping Wang1*
Yan Ping Wang1* Chang Jun Ding2,3*
Chang Jun Ding2,3*- 1Taishan Forest Ecosystem Research Station of State Forestry Administration, College of Forestry, Shandong Agricultural University, Tai’an, China
- 2State Key Laboratory of Tree Genetics and Breeding, Research Institute of Forestry, Chinese Academy of Forestry, Beijing, China
- 3Key Laboratory of Tree Breeding and Cultivation of State Forestry Administration, Research Institute of Forestry, Chinese Academy of Forestry, Beijing, China
- 4Shandong Academy of Forest, Jinan, China
The interaction between plants and microbes dominates plant growth and fitness in specific environments. The study of the relationship between plant genotypes and rhizobacterial community structure would provide a deep insight into the recruitment strategies of plants toward soil bacteria. In this study, three genotypes of 18-year-old mature poplar (H1, H2, and H3) derived from four different parents were selected from a germplasm nursery of Populus deltoides. Rhizosphere soil carbon, nitrogen, and phosphorus properties as well as the 16S rDNA sequences of rhizobacterial communities were analyzed to determine the relationship between poplar genotypes and rhizobacterial communities assembly. The results showed there were significant differences in the diversity (Chao1, ACE index, and Shannon index) of rhizobacterial communities between H1 and H2, as well as between H2 and H3, but no difference between H1 and H3. Principal component analysis also revealed a similar structure of rhizobacterial communities between H1 and H3, whereas the rhizobacterial communities of H2 demonstrated significant differences from H1 and H3. Linear discriminant effect size analysis indicated that there were 11 and 14 different biomarkers in the H1 and H3 genotype, respectively, but 42 in the H2 genotype. Co-occurrence network analysis indicated that the rhizobacterial communities of H2 had a distinct network structure compared to those of the other two genotypes, whereas H1 and H3 had a similar pattern of co-occurrence network. Threshold indicator taxa analysis revealed that 63 genera responded significantly to NO3–-N content and 58 genera to NH4+-N/NO3–-N ratio. Moreover, the stochastic assembly process was found to be decreased with increasing NO3–-N content and fluctuated with increasing NH4+-N/NO3–-N ratio. All results indicated that the structure of poplar rhizobacterial communities were influenced by host genotypes, and available nitrogen might play a dominant role in the assembly of rhizobacterial communities. This study would promote the future selection and utilization of rhizobacteria in poplar breeding.
Introduction
Rhizosphere microorganisms play key roles in plant growth and fitness (Ren et al., 2020). Studies on the relationships between plants and microbes are showing profound impacts on modern agriculture. Researchers are trying to employ diverse microbes to improve plant growth and tolerance to stress. The relationship between soil habitat and microbial community has got a lot of attention (Williams and de Vries, 2020), while the interaction between soil microbiota and plant genotype is often ignored (Hemmerle et al., 2022; Qi et al., 2022). The genetic conservation makes the morphological and physiological traits of progenies biologically equivalent to their parents (Zanewich et al., 2018). However, hybridization between parents with different genotypes could create some new distinct phenotypic traits (Shen et al., 2021), so that hybrid progenies usually demonstrate more advantages of phenotype (Zanewich et al., 2018). The genetic distances between parent lines seem to be responsible for the progeny phenotype in out-crossing plants. Theoretically, rhizosphere microbial communities of plants are related to plant breeding practice. For example, a recent study showed that self-cross of rice (Oryza sp.) decreased the differences in rhizosphere bacterial and fungal communities between parental lines and progeny generations (Chang et al., 2021). So, it is necessary to combine traditional plant cross-breeding with soil microbiomes (Nerva et al., 2022). The genotypic differences of host plants may also play more significant roles in the recruitment and assembly of the rhizosphere microbiome (Cregger et al., 2018; Morella et al., 2019; Veach et al., 2019), thereby determining the plant growth and fitness (Brachi et al., 2022). Although the impact of host genotype on the assembly of rhizosphere microbial communities is still under debate (Lladó et al., 2017; Hartman and Tringe, 2019; Dove et al., 2021), a further step to investigate the relationship between host genotypes and rhizobacterial communities in woody plants is urgently required, which would provide a deeper insight into the potential utilization of soil microbes in trees breeding.
Poplar is very important for biofuel feedstock and timber production all over the world (Sannigrahi et al., 2010). However, the excellent poplar cultivar with high yield and quality wood is still in shortage in the practice of wood industry. Since the draft genome of black cottonwood (Populus trichocarpa) was published (Tuskan et al., 2006), it has been the model species in woody plants. And, genetic engineering based on molecular biology has markedly promoted the development of genetic-modified poplar breeding. However, as a dioecious tree species, hybridization between male and female parent lines still plays an important role in the practice of poplar selective breeding (Shen et al., 2021). For example, Populus deltoides, a rapid-growing poplar species with high timber production and tolerance to stress, is used as important parental lines in poplar cross-breeding. Because of the extensive distribution in mid-latitude countries and regions throughout the world, many natural variations of the species were observed and provided valuable genetic resources for modern poplar breeding (Fahrenkrog et al., 2017). Since some excellent germplasm resources of P. deltoids were introduced into China in the 1970s, the trees have formed many hybrid lineages with other numerous Populus species, and some of them displayed rapid growth and high yield even in some barren areas (e.g., arid and infertile sandy soils). To date, China has emerged as one of the countries with the largest plantations of P. deltoides in the world, contributing significantly to global wood production. The interaction between poplar and microbe has been paid more and more attention to in the sustainable development of poplar plantations. The examination of microbiomes related to poplar trees and the exploitation of core microbial communities have contributed much to poplar plantation silviculture. Currently, the profile of the poplar rhizosphere microbiome is becoming more and more clear (Schaefer et al., 2013; Wang et al., 2017). As dioecious tree species, their rhizobacterial communities were also shown to be significantly different between male and female poplar trees (Zhu et al., 2022), and their metabolic exudation of roots was also distinct (Xia et al., 2021). The temporal factors dominating the assembly of microorganisms associated with poplar have got extensive attention (Dove et al., 2021).
However, the limitations of previous studies are also obvious. For example, the observed trees were mostly young poplar, which limits the practical application of research achievements about beneficial microbes to mature poplar trees (Liu et al., 2021; Kristy et al., 2022). Obviously, the rhizosphere microbial communities are greatly affected by plant roots and appear to dramatically change with plant development and soil environmental fluctuations, which may finally disrupt the correct determination on the relationship between poplar and microbes. By contrast, mature poplar would provide a better model to investigate the interaction between trees and microbes (Shakya et al., 2013). Long-term interaction between trees and soils could develop a relatively stable ecosystem (Trivedi et al., 2020) and the impact of host genotype on soil microbiomes may emerge as tree ages (Dove et al., 2021). Additionally, the affinities among poplar genotypes in the previous studies were not clear, which may greatly influence the determination on the effect of poplar genotypes on microbial communities. Therefore, three mature poplar genotypes with well recorded parent lines were selected from a germplasm nursery of P. deltoides in this study. Their rhizobacterial communities were compared via 16S rDNA sequences, and the relationship between soil nutrients and rhizobacterial communities assembly was examined. Here, we tested two hypotheses, (1) the composition and structure of rhizobacterial communities would be different among poplar genotypes; and (2) the assembly of rhizobacterial communities may be determined by specific soil nutrients. The study would facilitate the integration of soil microbiomes with poplar breeding practice.
Materials and methods
Study site
The study site is located in the Dashahe national forestry farm of Shandong Province, 34°79’ (N) latitude and 116°08’ (E) longitude, with a warm temperate semi-humid continental monsoon climate and a mean annual temperature of 13.9°C. The frost-free period is approximately 206 d per year, and the annual average precipitation is approximately 737 mm. In terms of geographical location, the forestry farm is right on the Yellow River alluvial plain, and the soil texture mainly consists of sandy particles (more than 75%), which resulted in poor soil organic matter and nutrient conditions. Because the water- and fertilizer-holding capacity of the soil was extremely low, little irrigation and fertilization were applied in forest management. A germplasm nursery of Populus deltoides including 28 poplar genotypes was built in 2002, and the spacing between trees and rows was approximately 4 and 6 m, respectively. Twenty to thirty trees of each genotype were grown in a row, but all the genotypes regrettably had no replicates. Considering the influence of sex differences between male and female poplar trees on microbial communities (Xia et al., 2021; Zhu et al., 2022), the identical sex of poplar trees is required. In addition, to avoid soil heterogeneity, the poplar genotypes need to be neighboring as possible. Thus, only three male genotypes (H1, H2, and H3) were finally included in the study (Supplementary Figure 1A). According to the records of their parental lines, the three genotypic poplars are from four poplar parents (i.e., P. deltoides “Zhonghe 1,” “L23,” “T66,” and “I72”) and could be divided into three hybrid progeny groups (Supplementary Figure 1C): with the same female parent but a different male parent (SF, including H1 and H3); with the same male parent but a different female parent (SM, including H2 and H3); and without the same parent (NS, including H1 and H2). The understory vegetation of each genotype block mostly consisted of some common annual or perennial herbs, with no shrubs or other tree species.
Poplar rhizosphere soil collection
Six sample trees were selected in each poplar genotype for rhizosphere soil collection. Briefly, five soil blocks (50 cm in length, 50 cm in width, and 20 cm in depth) were excavated at 0.5 m around the trunk based on the distribution of poplar fine roots (Supplementary Figure 1B; Zhu et al., 2018). After removing the surface herbaceous vegetation, poplar roots were collected from the soil block. Due to the absence of other woody plants in the plot, poplar roots can be easily distinguished from herbaceous roots based on their color and flexibility. The coarse roots of poplar in the soil block are helpful to assure that the fine roots were exactly from sample tree. However, only fine roots with a diameter of less than 2 mm were collected from coarse roots for this study. The fine roots were immediately placed in sterile bags and stored at 4°C for rhizosphere soil collection. Three quadrates (5 m × 5 m) were selected at random in each poplar genotype to obtain five bulk soil samples (Supplementary Figure 1B). Similarly, the bulk soils of three quadrates were mixed into a single sample. In the laboratory, a subset of fine roots from each sample tree was placed in a 50 mL centrifuge tube to which 30 mL PBS (phosphate buffer solution) was added and vortexed for 3 min. Following the removal of fine roots, rhizosphere soil was collected from tubes by centrifugation at 7,000 × g for 10 min (Li et al., 2014) and stored at −80°C for microbial communities analysis. Another subset of fine roots was collected for soil C, N and P analysis by brushing the fine root surface with a small brush. Thus, a total of 18 rhizosphere soil samples (6 tree samples × 3 genotypes) and 3 bulk soil samples were collected for this study.
Soil carbon, nitrogen, and phosphorus analysis
The total carbon (TC) and total nitrogen (TN) contents of the soil samples were measured using an elemental analyzer (Vario MACRO cube, Elementar, Germany). The total phosphorus (TP) and available phosphorus (AP) contents in the soil samples were measured using an automatic chemical analyzer (SmartChem® 200, AMS/Westco Scientific Instruments, Italy). The soil nitrate (NO3–) and ammonium (NH4+) contents were measured using an automatic flow analyzer (SEAL AutoAnalyzer AA3 HR, Germany). Finally, the soil stoichiometric characteristics were calculated.
DNA extraction and amplification
Approximately 100 mg soil of each sample was taken from each sample for total soil microbial DNA extraction using the MAG-BIND Soil DNA Extraction Kit (Omega Bio-Tek, USA, Cat. # M5635-02). DNA quality was checked using an UV spectrophotometer (RS232G, Eppendorf, Germany). The V3-V4 region of bacterial 16S rDNA was amplified using the specific primers 338F (5′-ACTCCTACGGGAGGCAGCA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′). The 250 bp paired-ends amplicons were sequenced using the Illumina platform (Illumina Inc., San Diego, California, USA).
Bioinformatics
Sequence data were analyzed with QIIME 2 2019.4 (Bolyen et al., 2018). Briefly, raw data were demultiplexed using the demux plugin, followed by primers removal with the cutadapt plugin (Martin, 2011). Sequences were delineated into amplicon sequence variants (ASVs) after quality filtration, mergence, and chimera sequence removal using the DADA2 algorithm (Callahan et al., 2016). According to the Silva database, the representative sequences were assigned a taxonomic classification using the classify-sklearn na1̈ve Bayes taxonomy classifier, and then the host contaminants (e.g., chloroplast or mitochondrial sequence) were removed. Finally, a total of 11,733 ASVs were obtained. Using the QIIME feature-table rarefy function, these ASVs were randomly selected according to 95% of the minimum sample sequence size to obtain the rarefied ASV table (Kemp and Aller, 2004). And then, the relative abundance of ASV, which is the amount of ASV in a sample as a percentage of the total abundance of that sample, was calculated for subsequent analysis. The raw data were submitted to the NCBI Sequence Read Archive (SRA) database (PRJNA881800).
Statistical analysis
All statistical analyses and plots in this study were generated in R 4.0.5 (R Core Team, 2020). One-way analysis of variance (ANOVA) and least significant difference (LSD) multiple comparisons were used to test the differences in soil nutrition content, α diversity (Chao1, ACE, Shannon, and PD index), NTI of bacterial communities among groups (α = 0.05). Linear discriminant analysis1 was used to identify biomarkers at different taxonomic levels. Correlation analysis was performed using the ‘‘psych’’ package (V.2.0.9)2 and visualized using the ‘‘corrplot’’ package (V.0.84).3 The ‘‘TITAN24 “package was used for threshold indicator taxa analysis to determine the response of the bacterial communities to environmental factors. This method determines the response threshold of the soil bacterial community based on the change points of each taxon along the environmental gradient and the synchronization of the change points (Gao et al., 2020). Gephi software (V.0.9.2)5 was used to visualize and analyze the microbial co-occurrence network (Bastian et al., 2009). Taxa with 0 abundance were excluded from the data for each group, and only significant correlations were included in the network (r > 0.8, P < 0.01). Nodes with higher within-module connectivity (Zi) and among-module connectivity (Pi) were defined as hub nodes (Shi et al., 2016). Phylogenetic trees were constructed using FastTree2 software (Price et al., 2010), and the nearest taxon index (NTI) was calculated using the “picante” package (V.1.8.2) (Kembel et al., 2010). The NTI was used to assess phylogenetic processes; NTI > 0 indicates phylogenetic clustering, and NTI < 0 indicates phylogenetic overdispersion (Kembel, 2009). The assembly processes of the microbial community were evaluated using a null model (Stegen et al., 2012). The standard deviation of the observed β-mean-nearest taxon distance (βMNTD) from the mean of the null distribution is defined as the β-nearest taxon index (βNTI). | βNTI| > 2 indicates the dominance of deterministic processes, including homogeneous selection (βNTI < −2) and variable selection (βNTI > 2), and | βNTI| < 2 indicates the dominance of stochastic processes. The source code is available at https://github.com/yankun212/Frontiers_PGR.git.
Results
Comparisons of carbon, nitrogen, and phosphorus contents in rhizosphere soils of poplar genotypes
The NO3–-N content in the rhizosphere soil of H2 was more than those of other soils, while the NH4+-N/NO3–-N ratio was less (Table 1). Specifically, the NO3–-N content of H1 and H2 rhizosphere soils differed significantly (P < 0.05). Similarly, the content of NO3–-N and NH4+-N/NO3–-N ratio of H2 and H3 showed significant differences (P < 0.05). However, the nutrient content and stoichiometric characteristics of rhizosphere soils did not differ significantly between H1 and H3. Therefore, the soil available nitrogen contents appeared closely relative to poplar genotypes.
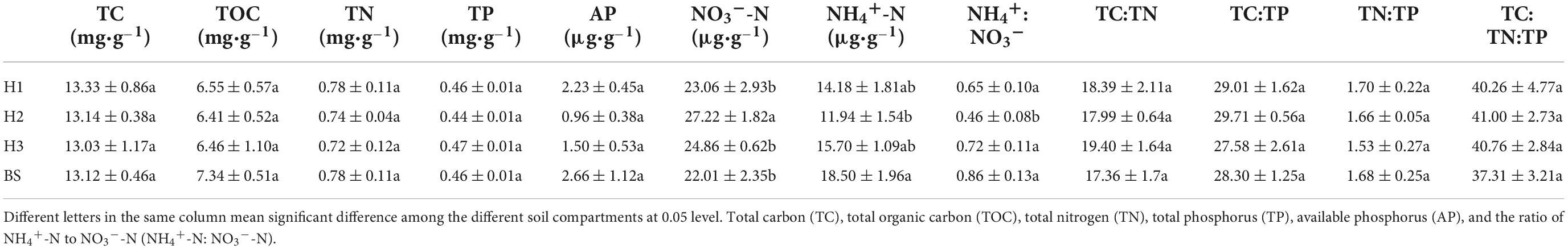
Table 1. The soil carbon, nitrogen, phosphorus content, and stoichiometric characteristic in rhizosphere and bulk soils of poplar genotypes (mean ± SE).
Comparisons of rhizobacterial community diversity among poplar genotypes
Analysis of rhizobacterial community diversity revealed that the richness index (Chao1, ACE) and phylogenetic index (PD) of H1 and H2 differed significantly (P < 0.05), as did those of H2 and H3. However, there were no significant differences of diversity indices between H1 and H3 (Figures 1A–D). These findings indicated that the structures of rhizobacterial communities differed among poplar genotypes. Furthermore, principal component analysis (PCA) showed significant differences in the rhizobacterial community between H1 and H2 (R2 = 0.23, P = 0.017), as well as between H2 and H3 (R2 = 0.25, P = 0.002), but not between H1 and H3 (R2 = 0.19, P = 0.1) (Figures 1E–H). The three tests of Adonis, ANOSIM, and MRPP revealed identical differences among the three poplar genotypes (Supplementary Table 1). The results indicated that the structure of rhizobacterial communities was significantly related to poplar genotypes.
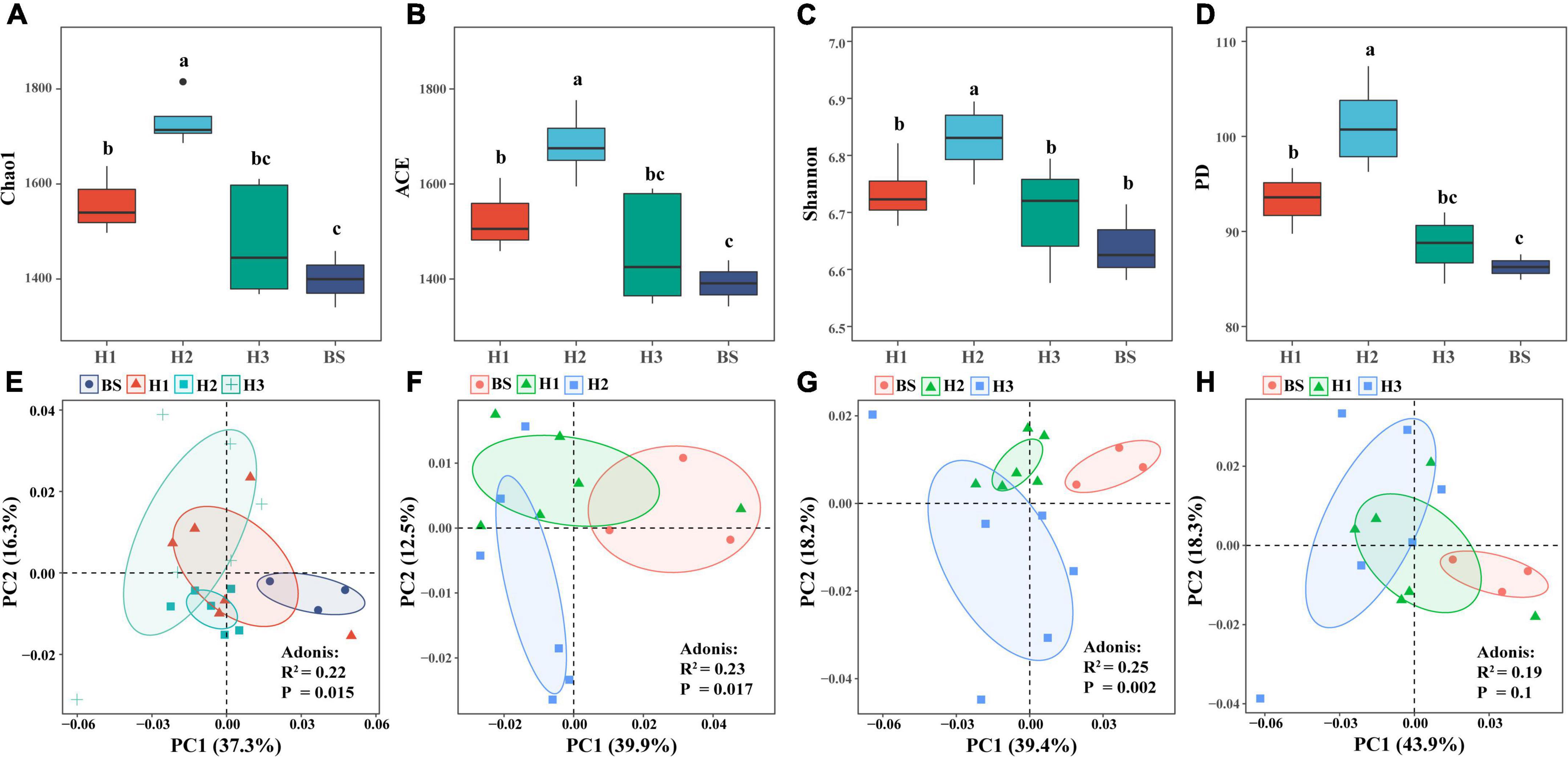
Figure 1. α-diversity (A–D) and β-diversity (E–H) of bacterial community in rhizosphere and bulk soils of poplar genotypes.
Differences in the composition of rhizobacterial communities among poplar genotypes
A total of 36 phyla and 666 genera of bacteria were annotated in all the sample soils, with 10 phyla showing a relative abundance of over 1% (Figure 2). Proteobacteria and Planctomycetes had significantly different relative abundances between H1 and H2, as well as between H2 and H3 (P < 0.05). However, the relative abundance of the above two bacterial phyla did not differ between H1 and H3 (P > 0.05). The F-values of nine abundant phyla in the SF group (H1 and H3 genotype) were smaller than those of the other two groups (SM and NS) (Supplementary Table 2). The VENN diagram showed that the number of rhizobacteria ASVs shared by H1 and H3 (741 + 1,705) was greater (Supplementary Figure 2). Linear discriminant effect size analysis indicated that there were 11 and 14 specific biomarkers in the H1 and H3 genotypes, respectively, while it was up to 42 biomarkers in the H2 genotype (Figure 3). So, H1 and H3 had a similar composition and structure of rhizobacterial communities and were different from the H2 genotype. Additionally, co-occurrence network analysis showed that H1 and H3 had similar average network connectivity and modularity (Figure 4). However, H2 had more network nodes, edges, and higher average degrees (Supplementary Table 3). The above results suggested that the composition and structure of rhizobacterial communities in poplar genotypes were different.
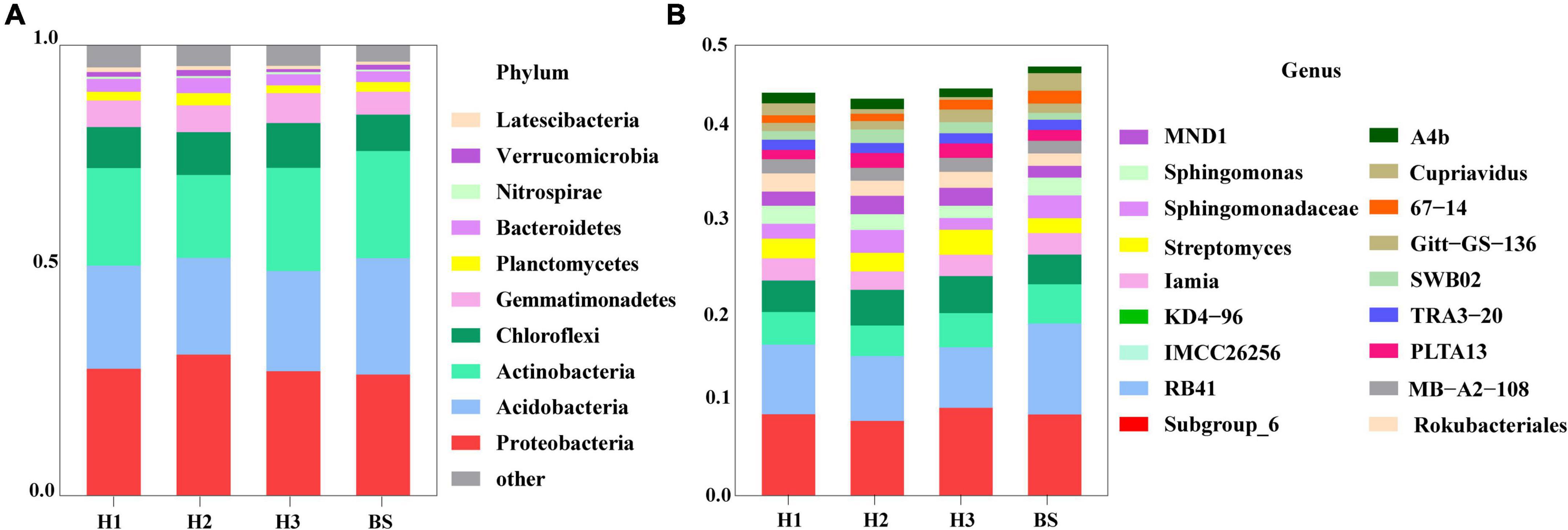
Figure 2. The composition of bacterial community in rhizosphere and bulk soils at the level of phylum (A) and genus (B).
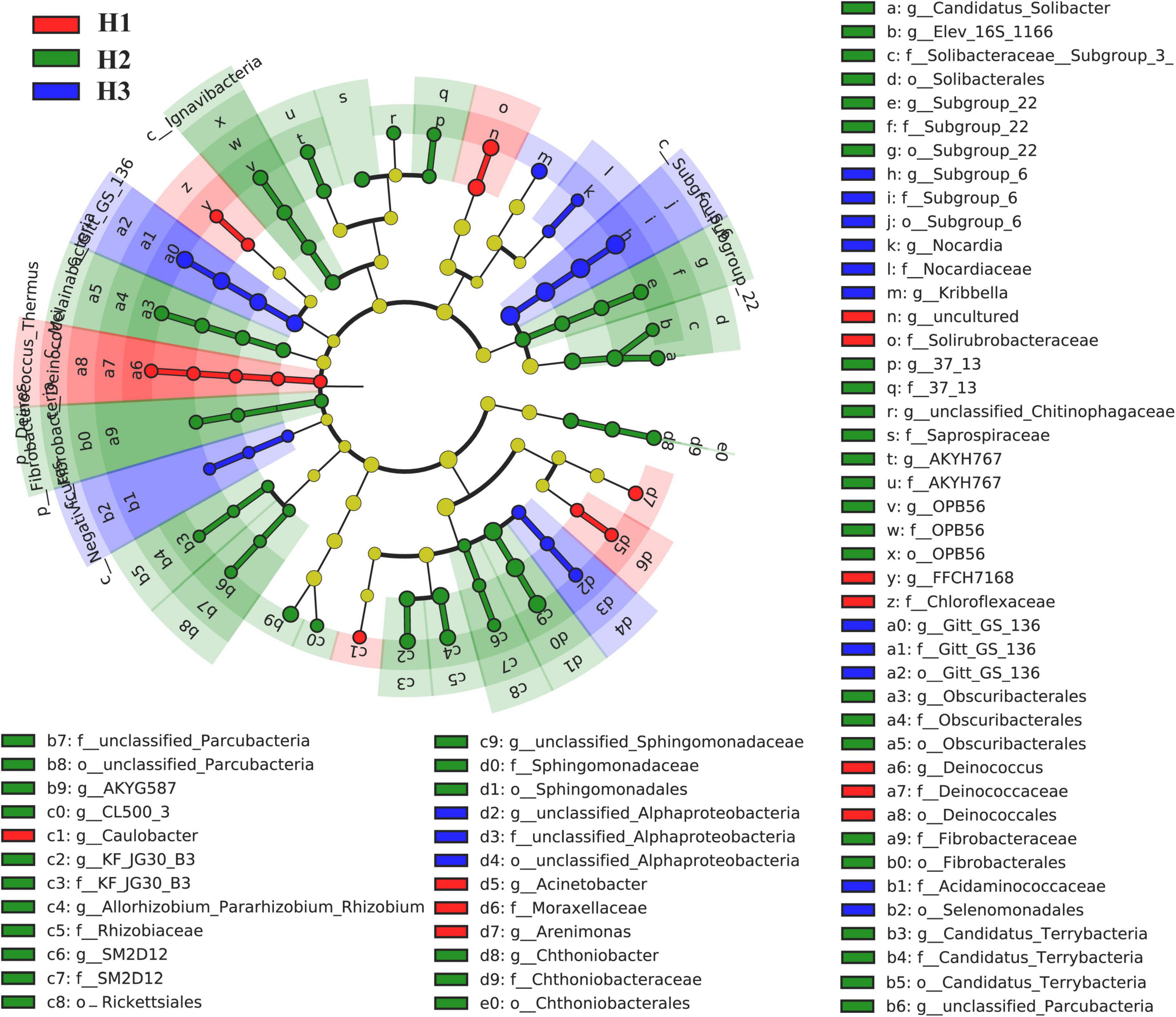
Figure 3. Linear discriminant effect size analysis (LEfSe) on the rhizobacteria biomarkers of poplar genotypes.
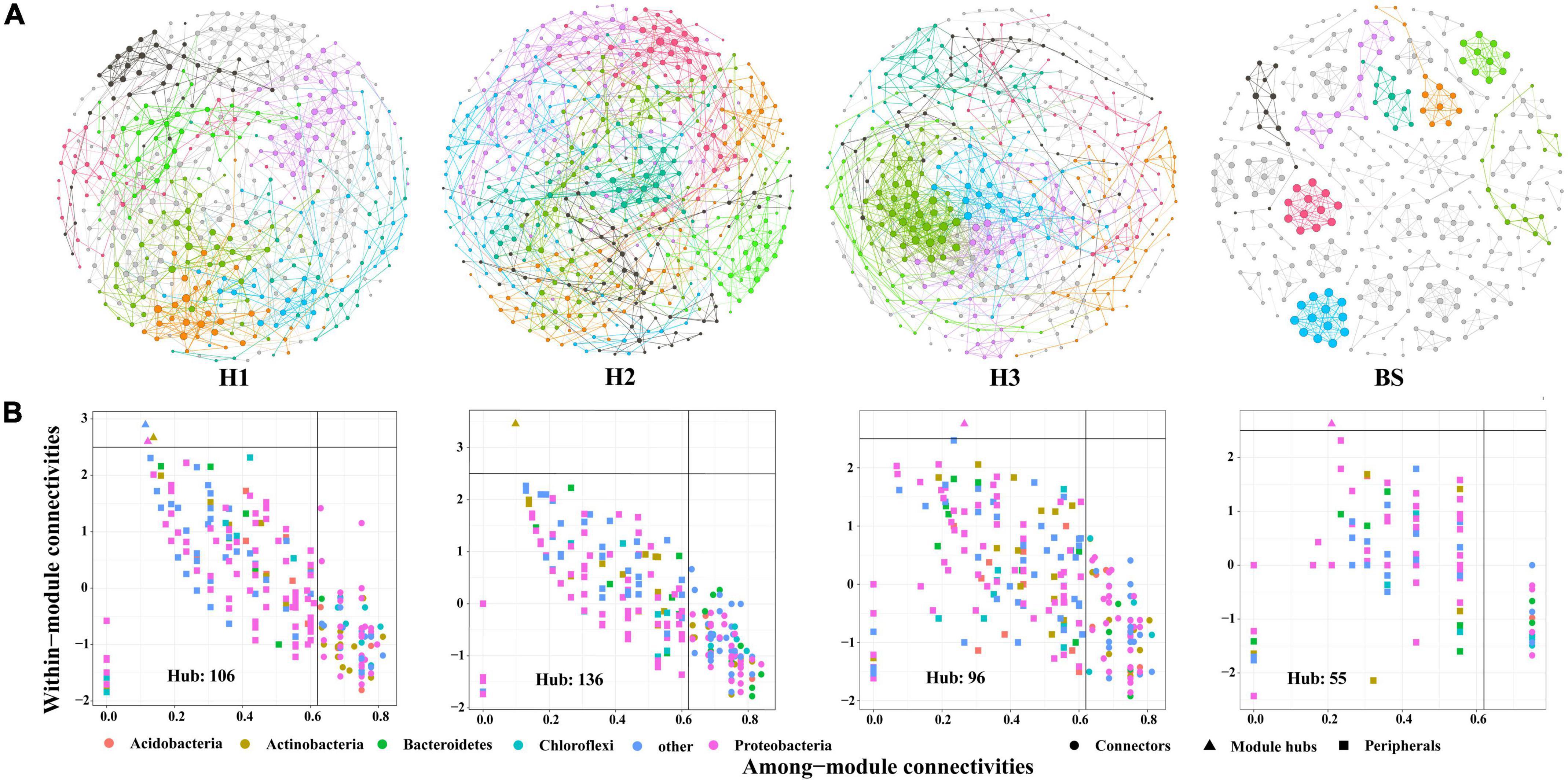
Figure 4. The co-occurrence networks (A) and hub nodes (B) of bacteria in rhizosphere and bulk soils. Different colors in the network represent different modules.
Correlation between rhizobacterial community and soil nutrients
Among the rhizobacteria genera with a relative abundance over 1%, 16 genera demonstrated a significant correlation with soil nutrients, and the majority of genera (up to nine genera) demonstrated a significant correlation with soil NO3–-N content (Figure 5). Meanwhile, threshold indicator taxa analysis revealed that 63 genera were responsive to NO3–-N changes and 58 genera were responsive to NH4+-N/NO3–-N changes (Figure 6). These findings indicate that the content of NO3–-N and the ratio of NH4+-N/NO3–-N have a significant impact on the composition of the poplar rhizobacterial community.
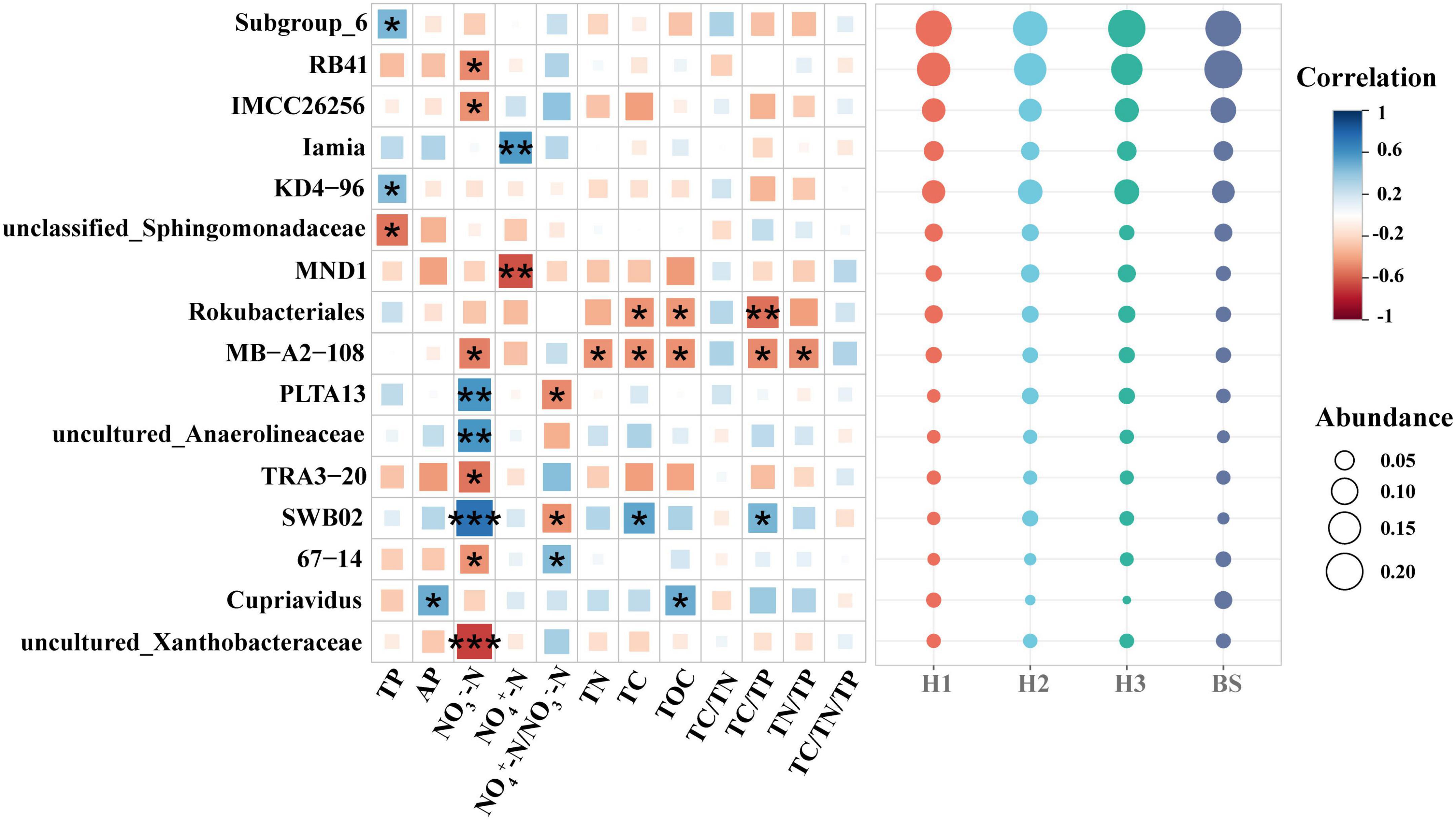
Figure 5. The correlation matrix between soil nutrient properties and abundant bacteria genera (the relative abundance over 1%) in rhizosphere and bulk soils of poplar. ***, ** and * show significant correlation at the 0.001, 0.01 and 0.05 level, respectively.
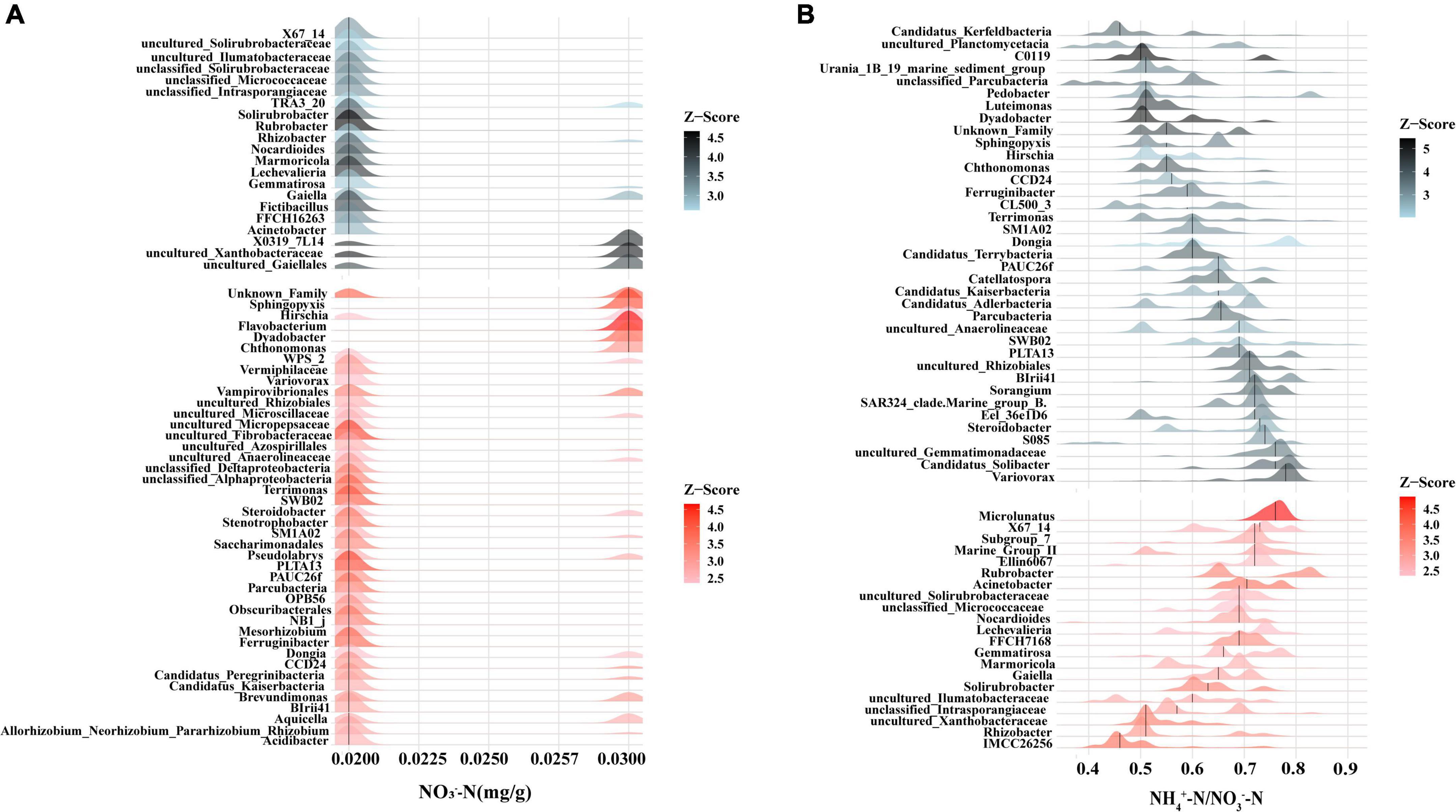
Figure 6. Threshold indicator taxa analysis (TITAN) on the response of bacteria genus to NO3–-N content (A) and the ratio of NH4+-N/NO3–-N (B). Red color represents positive response (i.e., abundance increases), black color represents negative response (i.e., abundance decrease).
NTI of rhizobacterial communities did not show significant differences among poplar genotypes (Figure 7A), indicating phylogenetic clustering of rhizobacterial taxa in the poplar genotype (NTI > 0). Furthermore, the rhizobacterial assembly was dominated by deterministic processes, however, the bacterial assembly of bulk soil was dominated by stochastic processes (Figure 7B). Pairwise comparisons of βNTI had a positive relationship with the difference of soil NO3–-N content (R2 = 0.229; P < 0.001) and NH4+-N/NO3–-N ratio (R2 = 0.052; P < 0.001) (i.e., the Euclidean dissimilarity) (Figures 7C,D), indicating that changes in soil nutrient content drive the transformation of bacterial assembly patterns. Furthermore, it is found that stochastic processes decreased with increasing NO3–-N content and fluctuated with increasing NH4+-N/NO3–-N ratio (Figures 7E,F). NO3–-N may be one of the dominant edaphic factors influencing bacterial community assembly in poplar rhizosphere soils.
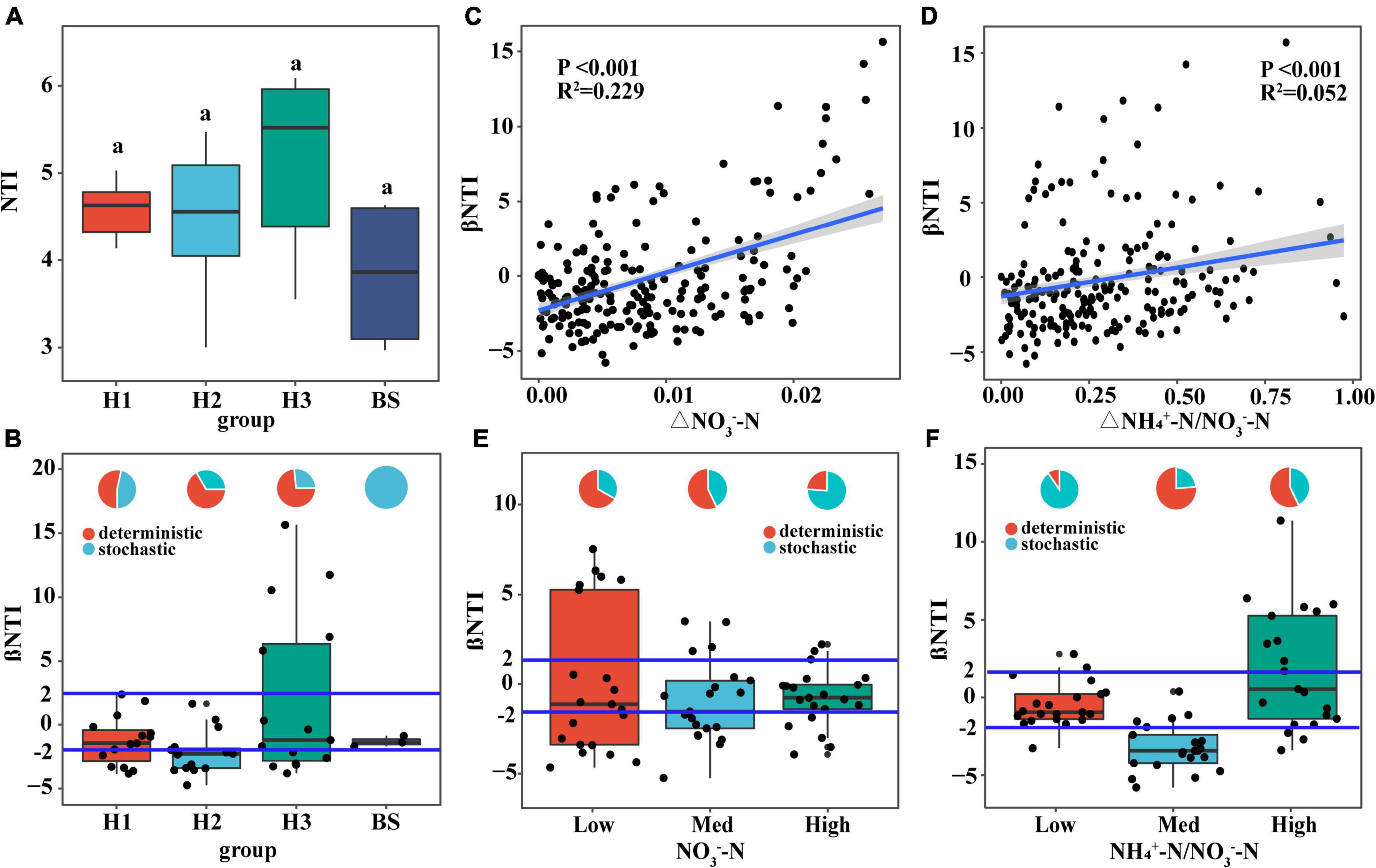
Figure 7. The relationship between the available nitrogen and the assembly of soil bacterial communities. Distribution of NTI value (A) and βNTI value (B,E,F) of soil bacteria community, and the relationship of βNTI value (C,D) with the difference of NO3–-N content and NH4+-N/NO3–-N. The small graphs in panels (B,E,F) represent the proportions of stochastic and deterministic processes in different groups of bacterial communities.
Discussion
The differences of rhizobacterial community among progenies
Evidence suggests that differences of plant rhizosphere microbial communities are associated with the rhizosphere environment (Naylor et al., 2017; Wang et al., 2018). NO3–-N is an important form of available nitrogen which is directly utilized by the majority of plant species (Von Wirìn et al., 1997). The present study showed that the NO3–-N content in the rhizosphere soil was significantly different among the poplar genotypes (Table 1), implying the utilization or transformation of NO3–-N in the rhizosphere soil is related to poplar genotypes. Further, 9 of 16 bacterial genera with a relative abundance of over 1% had a significant correlation with NO3–-N (Figure 5), implying that poplar trees recruited a large number of microorganisms in the rhizosphere for nitrogen cycling. Studies have shown that plants usually provide various substrates for rhizosphere microorganisms via root exudation (Bakker et al., 2018; Williams and de Vries, 2020) to recruit some beneficial microorganisms (e.g., Bacillus subtilis GB03, Bacillus subtilis FB17) in the rhizosphere soil (Bulgarelli et al., 2012; Hashem et al., 2019; Matthews et al., 2019), which was helpful to improve soil nutrient availability and the adaptation of plants to specific environments (Berendsen et al., 2012; Trivedi et al., 2020). For example, the profiles of root metabolism revealed that the influences of poplar root phenolic metabolomes on rhizobacterial communities were dependent on poplar sex and soil properties (Xia et al., 2021, 2022). Meanwhile, soil nutrient availability also affect rhizosphere microbial communities (Delgado-Baquerizo et al., 2017). According to the growth rate hypothesis, the carbon and nitrogen substrates from roots simultaneously affect the soil ecological process (i.e., rhizosphere priming effect) (Lu et al., 2019) and demonstrate significant regulation of soil microorganisms (Nyawade et al., 2019). Additionally, other soil elements (e.g., phosphorus) also play an important role in microbial colonization and plant growth (Graham, 2002; Peñuelas and Sardans, 2009). So, the rhizobacterial communities are dependent on soil nutrient conditions.
Meanwhile, rhizosphere microbial communities are closely related to plant genotypes (Bokulich et al., 2014; Rochefort et al., 2019). For example, the diversity and co-occurrence networks of rhizosphere microbial communities were shown differently among genotypes of Arabidopsis (Arabidopsis thaliana) and blueberry (Vaccinium ashei), respectively (Lundberg et al., 2012; Jiang et al., 2017). Mycorrhizal infections were significantly increased by secreted enzyme activities (e.g., β-xylosidase, cellobiohydrolase, β-glucosidase, acid phosphatase, and laccase) in mycorrhizal roots of different poplar genotypes (Courty et al., 2010). Based on the available literature, the differences in rhizosphere bacterial communities among plant cultivars are often observed (Zhu et al., 2012; Bokulich et al., 2014; Li et al., 2014). In the study, we also found that the rhizobacterial compositions were different among the three genotypes of P. deltoides (Figure 2E). This difference may be attributable to the various root exudates of different genotypic plants (Matthews et al., 2019). However, there are still few studies that could provide enough evidence about the genetic effects of parental lines on rhizosphere microbiomes of plant genotypes, except that ectomycorrhizal fungal communities of hybrid progenies were observed to be different from parents in Populus (Lamit et al., 2021). In this study, three poplar genotypes can be classified into three hybrid progeny groups based on their parental lines (Supplementary Figure 1). Thus, we attempted to clarify the potential effect of hybridization on the assembly of poplar rhizobacterial communities. We observed significant differences in rhizobacterial community diversity both in NS and SM groups (Figures 1A,B,D,F). However, the similar composition and diversity of the poplar rhizobacterial community were shown in the SF group (Figure 1H). Further analysis revealed that the number of co-shared ASVs in the SF group was greater than those in the SM and NS groups, whereas it was comparable between the NS and SM groups (Supplementary Figure 2). Meanwhile, the least biomarkers and the highest degree of similarity were observed in the H1 and H3 (SF group) (Figure 3), as well as a similar co-occurrence network of rhizobacterial communities (Figure 4). All these findings suggest that the rhizobacterial community composition of poplar genotypes may be related to the genetic effects of their parental lines. Regrettably, the results of this study are only observational and not from a formally designed field experiment. However, we believe that common garden experiments are helpful to examine the differences in rhizosphere microbial communities between progenies and parents in the future, which would provide more accurate and reliable information about the genetic effects of poplar on rhizobacterial communities.
Assembly process of rhizobacterial communities and its dominant factors
Quantifying the assembly process of microbial communities is important in microbial ecology research (Zhou and Ning, 2017). Community assembly of environmental microorganisms can be divided into stochastic and deterministic process. The stochastic process is random diffusion, while the deterministic process is environment selection, which explains the different responses of microbes to specific niches. Meanwhile, stochastic and deterministic process may accompany the entire life history of a host, but the balance between them is often influenced by environmental factors (Jiao et al., 2020). In harsh environments (such as limited nutrient resource), natural selection pressure limits the diversity and abundance of bacterial communities (Zhou and Ning, 2017), and the assembly of rhizosphere bacteria was dominated by a deterministic process (Dove et al., 2021). Our results indicated that the assembly of bacterial communities were influenced by a stochastic process in bulk soil but a deterministic process in rhizosphere soil (Figure 7). The difference may be attributable to the distinct soil properties. Compared to bulk soils, plant rhizosphere is a very complex micro-environment affected by many factors (e.g., root morphology, nutrient utilization preference of plant, and root exudation) (Gottel et al., 2011; Dove et al., 2021; Herms et al., 2022). During plant growth and development, plant roots can selectively recruit microorganisms colonizing the rhizosphere or rhizoplane via dead residues and root exudation (Hartmann et al., 2009), resulting in significant differences in microbial communities between bulk soil and rhizosphere soil (Gregory, 2006; Uroz et al., 2010). So, the richness of the bacterial communities in rhizosphere soil was significantly greater than that in bulk soil, and the assembly process of bacterial communities may be different between the two soil environments.
In tandem with the root-soil interaction process, the assembly of the rhizobacterial community is constantly in a state of flux. Study have also shown that the functional requirements (e.g., soil nutrients) of the host plant play an important role in the assembly of rhizobacterial communities (Ren et al., 2020). Soil available nutrients and their stoichiometry can drive the changes in soil microbial communities (Delgado-Baquerizo et al., 2017). Nitrogen is very important for rapid-growth poplar trees. In this study, the NO3–-N contents in the rhizosphere soils of different poplar genotypes were found to be significantly different (Table 1), which may greatly affect the assembly of rhizobacteria communities. Although there was no significant difference in rhizobacterial NTI among poplar genotypes (Figure 7A), βNTI was significantly correlated positively with the differences of soil NO3–-N content and the NH4+-N/NO3–-N ratio (Figures 7B,C). Thus, when available nitrogen in the rhizosphere is deficient, microbial growth is limited so that both positive and negative priming effects occur in the rhizosphere (Dijkstra et al., 2013). At this time, some rhizobacteria are recruited through deterministic heterogeneous selection, such as Nitrosospira (Zhang et al., 2019), Noviherbaspirillum (Ishii et al., 2017), and Mesorhizobium (Siddiqi et al., 2019), which accelerates soil mineralization to meet the plant’s demand for available nutrients (Blagodatskaya et al., 2007). When the available nitrogen in the rhizosphere is abundant, plants secrete large amounts of carbohydrates into rhizosphere soils. Rhizosphere microbes would use these root exudates as nitrogen and carbon substrates (Kuzyakov and Cheng, 2004; Dijkstra et al., 2013). Consequently, the rhizobacterial community tended to be randomly assembled across nitrogen content gradients (Figures 7E,F). All the preceding findings suggest that available nitrogen may be a dominant edaphic factor influencing the assembly of rhizobacterial communities.
Conclusion
This study revealed that the available nitrogen content in rhizosphere soil varied among the poplar genotypes. Moreover, the composition and structure of the rhizobacterial community were closely related to the poplar genotypes. Soil available nitrogen may play a significant role in determining the assembly of the rhizosphere bacterial community. The study could aid in the development of plant-microbe symbiotic breeding in the future.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA881800.
Author contributions
QZ, KY, NW, SM, DL, and ZY conducted the field work, data collection, and statistical analysis. QZ and KY conducted visualization and writing. XS, YW, YD, and CD conducted methodology design, data curation, and manuscript reviewing and writing. YW and CD executed the figure drawing and manuscript writing. All authors read and approved the final manuscript.
Funding
This work was funded by the National Key Research and Development Program of China (2021YFD2201205), the National Natural Science Foundation of China (32071751 and 31870662), and the Key Research and Development Program of Shandong Province (2021SFGC020503).
Acknowledgments
Many thanks to reviewers for the helpful comments on the manuscript. Thanks to Editage company for the language-editing service.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1052567/full#supplementary-material
Footnotes
- ^ http://huttenhower.sph.harvard.edu/galaxy/
- ^ https://CRAN.R-project.org/package=psych
- ^ https://github.com/taiyun/corrplot
- ^ https://CRAN.R-project.org/package=TITAN2
- ^ https://gephi.org
References
Bakker, P. A. H. M., Pieterse, C. M. J., de Jonge, R., and Berendsen, R. L. (2018). The soil-borne legacy. Cell 172, 1178–1180. doi: 10.1016/j.cell.2018.02.024
Bastian, M., Heymann, S., and Jacomy, M. (2009). Gephi: An open-source software for exploring and manipulating networks. Proc. Int. AAAI Conf. Web Soc. Media 3, 361–362.
Berendsen, R. L., Pieterse, C. M., and Bakker, P. A. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Blagodatskaya, E. V., Blagodatsky, S. A., Anderson, T. H., and Kuzyakov, Y. (2007). Priming effects in Chernozem induced by glucose and N in relation to microbial growth strategies. Appl. Soil Ecol. 37, 95–105. doi: 10.1016/j.apsoil.2007.05.002
Bokulich, N. A., Thorngate, J. H., Richardson, P. M., and Mills, D. A. (2014). Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc. Natl. Acad. Sci. U.S.A. 111, 139–148. doi: 10.1073/pnas.1317377110
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C., Al-Ghalith, G. A., et al. (2018). QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. Peer J. Prepr. 6:e27295v2. doi: 10.7287/peerj.preprints.27295v2
Brachi, B., Filiault, D., Whitehurst, H., Darme, P., Gars, P. L., and Mentec, M. L. (2022). Plant genetic effects on microbial hubs impact host fitness in repeated field trials. Proc. Natl. Acad. Sci. U.S.A. 119:e2201285119. doi: 10.1073/pnas.2201285119
Bulgarelli, D., Schlaeppi, K., Spaepen, S., van Themaat, E. V. L., and Schulze-Lefert, P. (2012). Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 64, 807–838. doi: 10.1146/annurev-arplant-050312-120106
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Chang, J., Shi, S., Tian, L., Leite, M. F., Chang, C., and Ji, L. (2021). Self-crossing leads to weak co-variation of the bacterial and fungal communities in the rice rhizosphere. Microorganisms 9:175. doi: 10.3390/microorganisms9010175
Courty, P. E., Labbé, J., Kohler, A., Marçais, B., Bastien, C., Churin, J. L., et al. (2010). Effect of poplar genotypes on mycorrhizal infection and secreted enzyme activities in mycorrhizal and non-mycorrhizal roots. J. Exp. Bot. 62, 249–260. doi: 10.1093/jxb/erq274
Cregger, M. A., Veach, A. M., Yang, Z. K., Crouch, M. J., Vilgalys, R., Tuskan, G. A., et al. (2018). The Populus holobiont: Dissecting the effects of plant niches and genotype on the microbiome. Microbiome 6:31. doi: 10.1186/s40168-018-0413-8
Delgado-Baquerizo, M., Reich, P. B., Khachane, A. N., Campbell, C. D., Thomas, N., and Freitag, T. E. (2017). It is elemental: Soil nutrient stoichiometry drives bacterial diversity. Environ. Microbiol. 19, 1176–1188. doi: 10.1111/1462-2920.13642
Dijkstra, F. A., Carrillo, Y., Pendall, E., and Morgan, J. A. (2013). Rhizosphere priming: A nutrient perspective. Front. Microbiol. 4:216. doi: 10.3389/fmicb.2013.00216
Dove, N. C., Veach, A. M., Muchero, W., Wahl, T., Stegen, J. C., Schadt, C. W., et al. (2021). Assembly of the Populus microbiome is temporally dynamic and determined by selective and stochastic factors. mSphere 6, e1316–e1320. doi: 10.1128/msphere.01316-20
Fahrenkrog, A. M., Neves, L. G., ResendJr, M. F., Dervinis, C., Davenport, R., and Barbazuk, W. B. (2017). Population genomics of the eastern cottonwood (Populus deltoides). Ecol. Evol. 7, 9426–9440. doi: 10.1002/ece3.3466
Gao, C., Montoya, L., Xu, L., Madera, M., Hollingsworth, J., Purdom, E., et al. (2020). Fungal community assembly in drought-stressed sorghum shows stochasticity, selection, and universal ecological dynamics. Nat. Commun. 11:34. doi: 10.1038/s41467-019-13913-9
Gottel, N. R., Castro, H. F., Kerley, M., Yang, Z., Pelletier, D. A., Podar, M., et al. (2011). Distinct microbial communities within the endosphere and rhizosphere of Populus deltoides roots across contrasting soil types. Appl. Environ. Microb. 77, 5934–5944. doi: 10.1128/aem.05255-11
Graham, H. (2002). Ecological stoichiometry: Biology of elements from molecules to the biosphere. Sterner, R.W. and Elser, J.J. Princeton university press, princeton, NJ, USA. ISSN 0-691-07491-7. J. Plankton Res. 25:1183. doi: 10.1093/plankt/25.9.1183
Gregory, P. J. (2006). Roots, rhizosphere and soil: The route to a better understanding of soil science? Eur. J. Soil Sci. 57, 2–12. doi: 10.1111/j.1365-2389.2005.00778.x
Hartman, K., and Tringe, S. G. (2019). Interactions between plants and soil shaping the root microbiome under abiotic stress. Biochem. J. 476, 2705–2724. doi: 10.1042/BCJ20180615
Hartmann, A., Schmid, M., Tuinen, D. V., and Berg, G. (2009). Plant-driven selection of microbes. Plant Soil 321, 235–257. doi: 10.1007/s11104-008-9814-y
Hashem, A., Tabassum, B., and Abd-Allah, E. F. (2019). Bacillus subtilis: A plant-growth promoting rhizobacterium that also impacts biotic stress. Saudi J. Biol. Sci. 26, 1291–1297. doi: 10.1016/j.sjbs.2019.05.004
Hemmerle, L., Maier, B. A., Bortfeld-Miller, M., Ryback, B., Gabelein, C. G., Ackermann, M., et al. (2022). Dynamic charater displacement among a pair of bacterial phyllosphere commensals in situ. Nat. Commun. 13:2836. doi: 10.1038/s41467-022-30469-3
Herms, C. H., Hennessy, R. C., Bak, F., Dresbøll, D. B., and Nicolaisen, M. H. (2022). Back to our roots: Exploring the role of root morphology as a mediator of beneficial plant–microbe interactions. Environ. Microbiol. 24, 3264–3272. doi: 10.1111/1462-2920.15926
Ishii, S., Ashida, N., Ohno, H., Segawa, T., Yabe, S., and Otsuka, S. (2017). Noviherbaspirillum denitrificans sp. nov., a denitrifying bacterium isolated from rice paddy soil and Noviherbaspirillum autotrophicum sp. nov., a denitrifying, facultatively autotrophic bacterium isolated from rice paddy soil and proposal to reclassify Herbaspirillum massiliense as Noviherbaspirillum massiliense comb. nov. Int. J. Syst. Evol. Microbiol. 67, 1841–1848. doi: 10.1099/ijsem.0.001875
Jiang, Y., Li, S., Li, R., Zhang, J., Liu, Y., Lv, L., et al. (2017). Plant cultivars imprint the rhizosphere bacterial community composition and association networks. Soil Biol. Biochem. 109, 145–155. doi: 10.1016/j.soilbio.2017.02.010
Jiao, S., Yang, Y. F., Xu, Y. Q., Zhang, J., and Lu, Y. H. (2020). Balance between community assembly processes mediates species coexistence in agricultural soil microbiomes across eastern China. ISME J. 14, 202–216. doi: 10.1038/s41396-019-0522-9
Kembel, S. W. (2009). Disentangling niche and neutral influences on community assembly: Assessing the performance of community phylogenetic structure tests. Ecol. Lett. 12, 949–960. doi: 10.1111/j.1461-0248.2009.01354.x
Kembel, S. W., Cowan, P. D., Helmus, M. R., Cornwell, W. K., Morlon, H., Ackerly, D. D., et al. (2010). Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26, 1463–1464. doi: 10.1093/bioinformatics/btq166
Kemp, P. F., and Aller, J. Y. (2004). Bacterial diversity in aquatic and other environments: What 16S rDNA libraries can tell us. FEMS Microbiol. Ecol. 47, 161–177. doi: 10.1016/s0168-6496(03)00257-5
Kristy, B. D., Carrell, A. A., Johnston, E. R., Klingeman, D. M., Gwinn, K., Syring, K. C., et al. (2022). Chronic drought differentially alters the belowground microbiome of drought tolerant and drought susceptible genotypes of Populus trichocarpa. Phytobiomes J. doi: 10.1094/PBIOMES-12-21-0076-R [Epub ahead of print].
Kuzyakov, Y., and Cheng, W. (2004). Photosynthesis controls of CO2 efflux from maize rhizosphere. Plant Soil 263, 85–99. doi: 10.1023/B:PLSO.0000047728.61591.fd
Lamit, L. J., Meinhardt, K. A., Flores-Rentería, L., Kovacs, Z. I., Zinkgraf, M., Whitham, T. G., et al. (2021). Ectomycorrhizal fungal communities differ among parental and hybrid Populus cross types within a natural riparian habitat. Fungal Ecol. 52:101059. doi: 10.1016/j.funeco.2021.101059
Li, X. Z., Rui, J. P., Mao, Y. J., Yannarell, A., and Mackie, R. (2014). Dynamics of the bacterial community structure in the rhizosphere of a maize cultivar. Soil Biol. Biochem. 68, 392–401. doi: 10.1016/j.soilbio.2013.10.017
Liu, M., Wang, Y., Liu, X., Korpelainen, H., and Li, C. (2021). Intra- and intersexual interactions shape microbial community dynamics in the rhizosphere of Populus cathayana females and males exposed to excess Zn. J. Hazard. Mater. 402:123783. doi: 10.1016/j.jhazmat.2020.123783
Lladó, S., López-Mondéjar, R., and Baldrian, P. (2017). Forest soil bacteria: Diversity, involvement in ecosystem processes, and response to global change. Microbiol. Mol. Biol. R. 81:e00063. doi: 10.1128/MMBR.00063-16
Lu, J. Y., Dijkstra, F. A., Wang, P., and Cheng, W. X. (2019). Roots of non-woody perennials accelerated long-term soil organic matter decomposition through biological and physical mechanisms. Soil Biol. Biochem. 134, 42–53. doi: 10.1016/j.soilbio.2019.03.015
Lundberg, D. S., Lebeis, S. L., Paredes, S. H., Yourstone, S., Gehring, J., Malfatti, S., et al. (2012). Defining the core Arabidopsis thaliana root microbiome. Nature 488, 86–90. doi: 10.1038/nature11237
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet 17, 10–12. doi: 10.14806/ej.17.1.200
Matthews, A., Pierce, S., Hipperson, H., and Raymond, B. (2019). Rhizobacterial community assembly patterns vary between crop species. Front. Microbiol. 10:581. doi: 10.3389/fmicb.2019.00581
Morella, N. M., Weng, F. C.-H., Joubert, P. M., Metcalf, C. J. E., Lindow, S., and Koskella, B. (2019). Successive passaging of a plant-associated microbiome reveals robust habitat and host genotype-dependent selection. Proc. Natl. Acad. Sci. U.S.A. 117, 1148–1159. doi: 10.1073/pnas.1908600116
Naylor, D., DeGraaf, S., Purdom, E., and Coleman-Derr, D. (2017). Drought and host selection influence bacterial community dynamics in the grass root microbiome. ISME J. 11, 2691–2704. doi: 10.1038/ismej.2017.118
Nerva, L., Sandrini, M., Moffa, L., Velasco, R., Balestrini, R., and Chitarra, W. (2022). Breeding toward improved ecological plant–microbiome interactions. Trends Plant Sci. 27, 1134–1143. doi: 10.1016/j.tplants.2022.06.004
Nyawade, S. O., Karanja, N. N., Gachene, C. K. K., Gitari, H. I., Schulte-Geldermann, E., and Parker, M. L. (2019). Short-term dynamics of soil organic matter fractions and microbial activity in smallholder potato-legume intercropping systems. Appl. Soil Ecol. 142, 123–135. doi: 10.1016/j.apsoil.2019.04.015
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Qi, M., Berry, J. C., Veley, K. W., O’Connor, L., Finkel, O. M., Salas-González, I., et al. (2022). Identification of beneficial and detrimental bacteria impacting sorghum responses to drought using multi-scale and multi-system microbiome comparisons. ISME J. 16, 1957–1969. doi: 10.1038/s41396-022-01245-4
R Core Team (2020). R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing.
Ren, Y., Xun, W., Yan, H., Ma, A., Xiong, W., and Shen, Q. (2020). Functional compensation dominates the assembly of plant rhizospheric bacterial community. Soil Biol. Biochem. 150:107968. doi: 10.1016/j.soilbio.2020.107968
Rochefort, A., Briand, M., Marais, C., Wagner, M.-H., Laperche, A., Vallée, P., et al. (2019). Influence of environment and host plant genotype on the structure and diversity of the Brassica napus Seed microbiota. Phytobiomes J. 3, 326–336. doi: 10.1094/PBIOMES-06-19-0031-R
Sannigrahi, P., Ragauskas, A. J., and Tuskan, G. A. (2010). Poplar as a feedstock for biofuels: A review of compositional characteristics. Biofuel. Bioprod Bior. 4, 209–226. doi: 10.1002/bbb.206
Schaefer, A. L., Lappala, C. R., Morlen, R. P., Pelletier, D. A., Lu, T. S., Lankford, P. K., et al. (2013). LuxR- and LuxI-Type quorum-sensing circuits are prevalent in members of the Populus deltoides microbiome. Appl. Environ. Microb. 79, 5745–5752. doi: 10.1128/aem.01417-13
Shakya, M., Gottel, N., Castro, H., Yang, Z. K., Gunter, L., Labbé, J., et al. (2013). A multifactor analysis of fungal and bacterial community structure in the root microbiome of mature Populus deltoides trees. PLoS One 8:e76382. doi: 10.1371/journal.pone.0076382
Shen, H., Xing, X., Guan, Y., Zhou, L., Liu, S., and Gao, H. (2021). Radial variation studies on wood properties of Populus deltoides parents and their hybrids. BioResources 16, 4905–4923. doi: 10.15376/biores.16.3.4905-4923
Shi, S., Nuccio, E. E., Shi, Z. J., He, Z., Zhou, J., and Firestone, M. K. (2016). The interconnected rhizosphere: High network complexity dominates rhizosphere assemblages. Ecol. Lett. 19, 926–936. doi: 10.1111/ele.12630
Siddiqi, M. Z., Thao, N. T. P., Choi, G., Kim, D. C., Lee, Y. W., Kim, S. Y., et al. (2019). Mesorhizobium denitrificans sp. nov., a novel denitrifying bacterium isolated from sludge. J. Microbiol. 57, 238–242. doi: 10.1007/s12275-019-8590-0
Stegen, J. C., Lin, X. J., Konopka, A. E., and Fredrickson, J. K. (2012). Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 6, 1653–1664. doi: 10.1038/ismej.2012.22
Trivedi, P., Leach, J. E., Tringe, S. G., Sa, T., and Singh, B. K. (2020). Plant–microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 18, 607–621. doi: 10.1038/s41579-020-0412-1
Tuskan, G. A., DiFazio, S., Jansson, S., Bohlmann, J., Grigoriev, I., Hellsten, U., et al. (2006). The genome of black cottonwood. Populus Trichocarpa Sci. 313, 1596–1604. doi: 10.1126/science.1128691
Uroz, S., Buée, M., Murat, C., Frey, K. P., and Martin, F. (2010). Pyrosequencing reveals a contrasted bacterial diversity between oak rhizosphere and surrounding soil. Environ. Microbiol. Rep. 2, 281–288. doi: 10.1111/j.1758-2229.2009.00117.x
Veach, A. M., Morris, R., Yip, D. Z., Yang, Z. K., Engle, N. L., Cregger, M. A., et al. (2019). Rhizosphere microbiomes diverge among Populus trichocarpa plant-host genotypes and chemotypes, but it depends on soil origin. Microbiome 7:76. doi: 10.1186/s40168-019-0668-8
Von Wirìn, N., Gazzarrini, S., and Frommer, W. B. (1997). Regulation of mineral nitrogen uptake in plants. Plant Soil 196, 191–199. doi: 10.1023/A:1004241722172
Wang, Q. T., Wang, N., Wang, Y. P., Wang, Q. K., and Duan, B. L. (2017). Differences in root-associated bacterial communities among fine root branching orders of poplar (Populus × euramericana (Dode) Guinier.). Plant Soil. 421, 123–135. doi: 10.1007/s11104-017-3449-9
Wang, Y., Mao, Z., Bakker, M. R., Kim, J. H., Brancheriau, L., Buatois, B., et al. (2018). Linking conifer root growth and production to soil temperature and carbon supply in temperate forests. Plant Soil 426, 33–50. doi: 10.1007/s11104-018-3596-7
Williams, A., and de Vries, F. T. (2020). Plant root exudation under drought: Implications for ecosystem functioning. N. Phytol. 225, 1899–1905. doi: 10.1111/nph.16223
Xia, Z. C., He, Y., Yu, L., Li, Z. J., Korpelainen, H., and Li, C. Y. (2021). Revealing interactions between root phenolic metabolomes and rhizosphere bacterial communities in Populus euphratica plantations. Biol. Fert. Soils 57, 421–434. doi: 10.1007/s00374-020-01527-z
Xia, Z., He, Y., Korpelainen, H., Niinemets, Ü, and Li, C. Y. (2022). Sex-specific interactions shape root phenolics and rhizosphere microbial communities in Populus cathayana. Forest Ecol. Manag. 504:119857. doi: 10.1016/j.foreco.2021.119857
Zanewich, K. P., Pearce, D. W., and Rood, S. B. (2018). Heterosis in poplar involves phenotypic stability: Cottonwood hybrids outperform their parental species at suboptimal temperatures. Tree Physiol. 38, 789–800. doi: 10.1093/treephys/tpy019
Zhang, Q., Li, Y., He, Y., Liu, H., Dumont, M. G., Brookes, P. C., et al. (2019). Nitrosospira cluster 3-like bacterial ammonia oxidizers and Nitrospira-like nitrite oxidizers dominate nitrification activity in acidic terrace paddy soils. Soil Biol. Biochem. 131, 229–237. doi: 10.1016/j.soilbio.2019.01.006
Zhou, J. Z., and Ning, D. L. (2017). Stochastic community assembly: Does it matter in microbial ecology? Microbiol. Mol. Biol. Rev. 81:e00002-e17. doi: 10.1128/MMBR.00002-17
Zhu, Q. L., Yan, K., Dong, Y. F., and Wang, Y. P. (2022). Rhizosphere bacterial communities and soil nutrient conditions reveal sexual dimorphism of Populus deltoides. J. For. Res. 1–11. doi: 10.1007/s11676-022-01517-x [Epub ahead of print].
Zhu, W. R., Sang, Y. L., Zhu, Q. L., Duan, B. L., and Wang, Y. P. (2018). Morphology and longevity of different-order fine roots in poplar (Populus × euramericana) plantations with contrasting forest productivities. Can. J. For. Res. 48, 611–620. doi: 10.1139/cjfr-2017-0296
Keywords: poplar genotype, rhizobacterial community, community assembly, co-occurrence network, plant-microbe relationship
Citation: Zhu QL, Yan K, Wang NZ, Ma SQ, Lu DS, Su XH, Yuan ZS, Dong YF, Wang YP and Ding CJ (2022) The structure and assembly of rhizobacterial communities are influenced by poplar genotype. Front. Microbiol. 13:1052567. doi: 10.3389/fmicb.2022.1052567
Received: 24 September 2022; Accepted: 14 November 2022;
Published: 29 November 2022.
Edited by:
Md. Motaher Hossain, Bangabandhu Sheikh Mujibur Rahman Agricultural University, BangladeshReviewed by:
Xia Zhichao, Hangzhou Normal University, ChinaNicholas LeBlanc, Crop Improvement and Protection Research, Agricultural Research Service (USDA), United States
Joshua R. Herr, University of Nebraska-Lincoln, United States
Copyright © 2022 Zhu, Yan, Wang, Ma, Lu, Su, Yuan, Dong, Wang and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Ping Wang, c2RhdXd5cEBob3RtYWlsLmNvbQ==; Chang Jun Ding, Y2hhbmdqdW5kaW5nQGNhZi5hYy5jbg==
†These authors have contributed equally to this work and share first authorship