 Kamyar Mogodiniyai Kasmaei1,2
Kamyar Mogodiniyai Kasmaei1,2 Dayanand C. Kalyani1
Dayanand C. Kalyani1 Tom Reichenbach1
Tom Reichenbach1 Amparo Jiménez-Quero3
Amparo Jiménez-Quero3 Francisco Vilaplana3
Francisco Vilaplana3 Christina Divne1*
Christina Divne1*- 1Department of Industrial Biotechnology, School of Engineering Sciences in Chemistry, Biotechnology, and Health (CBH), KTH Royal Institute of Technology, Stockholm, Sweden
- 2Department of Animal Nutrition and Management, Swedish University of Agricultural Sciences, Uppsala, Sweden
- 3Division of Glycoscience, Department of Chemistry, School of Engineering Sciences in Chemistry, Biotechnology, and Health (CBH), KTH Royal Institute of Technology, Stockholm, Sweden
Ferulic acid is a common constituent of the plant cell-wall matrix where it decorates and can crosslink mainly arabinoxylans to provide structural reinforcement. Microbial feruloyl esterases (FAEs) specialize in catalyzing hydrolysis of the ester bonds between phenolic acids and sugar residues in plant cell-wall polysaccharides such as arabinoxylan to release cinnamoyl compounds. Feruloyl esterases from lactic acid bacteria (LAB) have been highlighted as interesting enzymes for their potential applications in the food and pharmaceutical industries; however, there are few studies on the activity and structure of FAEs of LAB origin. Here, we report the crystal structure and biochemical characterization of a feruloyl esterase (LbFAE) from Lentilactobacillus buchneri, a LAB strain that has been used as a silage additive. The LbFAE structure was determined in the absence and presence of product (FA) and reveals a new type of homodimer association not previously observed for fungal or bacterial FAEs. The two subunits associate to restrict access to the active site such that only single FA chains attached to arabinoxylan can be accommodated, an arrangement that excludes access to FA cross-links between arabinoxylan chains. This narrow specificity is further corroborated by the observation that no FA dimers are produced, only FA, when feruloylated arabinoxylan is used as substrate. Docking of arabinofuranosyl-ferulate in the LbFAE structure highlights the restricted active site and lends further support to our hypothesis that LbFAE is specific for single FA side chains in arabinoxylan.
Introduction
Ferulic acid (FA; 4-hydroxy-3-methoxy-cinnamic acid) is widely distributed in nature as a structural reinforcement component of plant cell walls (Lambert et al., 1999), where it is incorporated into the cell wall matrix by esterifying hemicelluloses, mainly glucuronoarabinoxylans (Carpita, 2001), as well as forming ether bonds to lignin and proteins (Kondo et al., 1990). Additionally, ferulic acid can form cross-links between lignin and arabinoxylan (AX) chains to produce lignin-ferulate-polysaccharide complexes (Terrett and Dupree, 2019). Furthermore, diferulate dimers can form cross-links between individual AX chains (Ralph et al., 1994). Agricultural residues constitute an important source of renewable carbon, containing some 0.5 to 3.0% FA (w/w), and these residues are therefore promising sources for FA production (Zhao and Moghadasian, 2008). Several methods have been developed to produce FA, including chemical and enzymatic hydrolysis (Antonopoulou et al., 2022). Nevertheless, enzymatic extraction catalyzed by feruloyl esterase (FAE) is more efficient, safe, and environmentally friendly than chemical extraction methods that commonly use harsh alkali solvents (Liu et al., 2021).
Feruloyl esterases (FAEs; E.C. 3.1.1.73) or cinnamoyl esterases (CEs), have been classified in the CAZy database as belonging to the carbohydrate esterase (CE) family (Drula et al., 2022). They are hydrolases belonging to the subclasses of carboxylic-acid esterases that catalyze the hydrolysis of ester bonds between phenolic acids (ferulic acid, p-coumaric acid, etc.) and sugar residues in plant cell-wall polysaccharides (arabinoxylan, pectin, etc.). FAEs are attractive due to their broad range of potential applications in biofuel, food, pharmaceutical and paper and pulp industries (Koseki et al., 2009; Dilokpimol et al., 2016). In the presence of FAE, feruloylated AX fibers are de-esterified into FA, di-FA, and the AX polysaccharide core, which have antioxidant, anti-inflammatory, and antimicrobial properties (Oliveira et al., 2019). The configuration of plant cell walls can be disrupted by FAEs acting on the lignin-ferulate-polysaccharide complexes, and thereby enhancing bioavailability of structural carbohydrates (cellulose and hemicelluloses) for enzymatic hydrolysis, e.g., in the rumen, biogas reactors, etc. (Tabka et al., 2006). Microbial FAEs were originally classified into four subfamilies, A to D, mainly based on substrate preference and the ability to release 5,5’-diFA from plant cell walls (Crepin et al., 2004). More recently, several groups have further expanded the classification (Benoit et al., 2008; Dilokpimol et al., 2016, 2017, 2018; Udatha et al., 2011, 2012). Of these reports, only Udatha et al. (2011) have offered some information regarding the classification of FAEs of bacterial and plant origins.
FAEs of fungal origin are currently the best characterized, and of considerable industrial interest (Dilokpimol et al., 2016; Underlin et al., 2020). However, several bacterial FAEs have also been characterized structurally and biochemically, including the FAEs from Clostridium thermocellum (Tabouriech et al., 2005), Dickeya dadantii (Hassan and Hugouvieux-Cotte-Pattat, 2011), Lactobacilli and Bifidobacteria (Lai et al., 2011; Fritsch et al., 2017; Xu et al., 2017; Zhang et al., 2021), Butyrivibrio proteoclasticus (Goldstone et al., 2010), Bacteroides intenstinalis (Pereira et al., 2021), Streptomyces cinnamoneus (Uraji et al., 2018), and uncultured bacteria from metagenomic studies (Holck et al., 2019).
FAEs derived from Lactobacillus or lactic acid bacteria (LAB) constitute an intriguing group of enzymes with potential applications in the food and pharmaceutical industries (Xu et al., 2020). In addition to having beneficial effects on the human and animal intestinal tracts (Wang et al., 2004; Liu et al., 2021), these bacteria also play an important role in silage fermentation, which relies on the production of organic acids to inhibit the growth of unwanted microorganisms (Mogodiniyai Kasmaei et al., 2020). Several LABs demonstrate high FAE activity, including L. acidophilus, L. amylovorus, L. farciminis, L. fermentum, L. gasseri, L. helveticus, L. johnsonii, and L. reuteri, L. farciminis, L. reuteri, and L. crispatus (Wang et al., 2004; Lai et al., 2011; Esteban-Torres et al., 2013; Xu et al., 2020; Liu et al., 2021; Zhang et al., 2021), but few studies have investigated the structural basis of the substrate binding and catalytic mechanisms.
In the present study, we expressed and purified FAE from Lentilactobacillus buchneri LN 4017 (LbFAE) and characterized its enzymatic properties. The crystal structure of LbFAE was determined in complex with FA, which allowed identification of the key residues responsible for FA binding, as well as predicting the binding mode of arabinofuranosyl ferulate (Araf-FA) binding. The present work offers new insight regarding the structural determinants of the substrate specificity and activity for LbFAE and provides a basis for further protein engineering for biotechnological applications.
Materials and methods
Substrates
Methyl ferulate (MF), methyl sinapinate and p-nitrophenyl ferulate (pNPF) were procured from Carbosynth Ltd. (United Kingdom). Methyl caffeate and methyl trans-p-coumarate were purchased from Tokyo Chemical Industry (USA). Polymeric feruloylated arabinoxylan (F-AX) was extracted from corn bran as described below and used as natural substrate.
Cloning and site-directed mutagenesis
The gene coding for a FAE annotated as a type-A FAE (faeA) from L. buchneri silage additive strain LN4017 (ATCC PTA-6138; GenBank ADI72729; UNP D7RU28) was synthesized by Integrated DNA Technologies (IDT, Iowa, USA), and cloned into the vector pNIC28-Bsa4 using ligation-independent cloning (LIC) technique. The LbFAE gene was amplified using the following forward and reverse primers:
Forward primer: 5′- TACTTCCAATCC ATGATCAAATTTGTAACTAC-3′.
Reverse primer: 5′- TATCCACCTTTACTG TTATTTTGCTTCTAACAACG-3′.
The 50 μl PCR mixture contained 50 ng plasmid DNA, 2 U Phusion High-Fidelity DNA polymerase (Thermo Fisher), 0.5 μM of each primer, 200 μM dNTP, 1.5 μl DMSO and 1 x HF Phusion buffer (Thermo Fisher). Thermocycling program was an initial denaturation at 98°C for 30 s followed by 30 cycles of denaturation (98°C for 10 s), annealing (60°C for 30 s) and elongation (72°C for 90 s) before a final elongation at 72°C for 10 min. PCR products were treated with 10 U of DpnI (Thermo Fisher) to digest methylated copies of DNA before purification with GeneJET PCR Purification Kit (Thermo Fisher). After linearization of pNIC28-Bsa4 vector with Eco31I (Thermo Fisher) and treatment of vector and insert with T4 DNA Polymerase to create overhang, the insert was annealed into the vector. This was followed by transforming the vector into chemically competent E. coli DH5α cells (Invitrogen) and plating on Luria-Bertani (LB) agar fortified with 50 μg ml−1 kanamycin at 37°C for 16 h.
The catalytic serine (Ser114) was replaced by alanine using site-directed mutagenesis to generate the LbFAE variant S114A. Site-directed mutagenesis was performed with the QuikChange II site-directed mutagenesis kit (Agilent Technologies) as described by the manufacturer, and using the following forward and reverse primers:
S114A _fwd (5′- CATTGCTGGGCGAAGCACTGGGTAGTGTTGC-3′).
S114A _rev (5′- GCAACACTACCCAGTGCTTCGCCCAGCAATG-3′).
The wild-type gene sequence was used as template for generating the S114A mutant. The mutated gene sequence was confirmed by DNA sequencing before transformation into E. coli BL21 (DE3)-T1 for protein production.
Production and purification of LbFAE
The recombinant plasmids (wild type and S114A variant) were transformed into E. coli BL21(DE3)-T1 competent cells and the bacteria was cultured in Terrific Broth supplemented with 50 μg ml−1 kanamycin at 37°C. After reaching an OD600 value of ~0.6, the temperature was reduced to 17°C and when the OD600 value reached ~1.0, β-D-1-thiogalactopyranoside (IPTG) was added at 0.2 mM to induce FAE gene expression. After ~18 h incubation, bacterial cells were harvested by centrifugation at 4500 × g (Beckman Coulter JA-10 fixed-angle rotor). The bacterial cells were resuspended in a buffer containing 25 mM K2HPO4 (pH 7.2), 150 mM NaCl and 5% (v/v) glycerol with one tablet of cOmplete™ Protease Inhibitor Cocktail (Roche) before homogenization with AVESTIN Emulsiflex-C3 (Avestin Europe, GmbH), and centrifugation (Beckman Coulter Avanti J-20 XP, California, USA) at 12,096 × g for 10 min at 4°C.
The Ni-NTA agarose resin was equilibrated with buffer containing 25 mM K2HPO4 (pH 7.2), 150 mM NaCl and 5% (v/v) glycerol. The supernatant was incubated with equilibrated Ni-NTA agarose resin for 1 h at 4°C and then loaded onto Bio-Rad Econo-Pac. The bound proteins to the resin was first washed with a buffer containing 25 mM K2HPO4 (pH 7.2), 150 mM NaCl, 30 mM imidazole and 5% (v/v) glycerol and thereafter with a buffer containing 25 mM K2HPO4 (pH 7.2), 300 mM NaCl, 50 mM imidazole and 5% (v/v) glycerol before eluting bound proteins with an elution buffer (25 mM K2HPO4 pH 7.2) 150 mM NaCl, 500 mM imidazole and 5% (v/v) glycerol). Washing and elution were carried out at 4°C.
The eluted proteins were concentrated using Vivaspin20 centrifugal concentrators (polyethersulfone filter, 10 kDa molecular weight cut-off) and were subjected to size exclusion chromatography using HiLoad 16/60 Superdex 200 prep grade column (GE Healthcare Life Sciences) and a mobile phase containing 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (Hepes) pH 7.5, 150 mM NaCl, and 5% (v/v) glycerol. The recovered fractions of FAE were analyzed using SDS-PAGE and pooled, and further concentrated with a Vivaspin20 centrifugal spin concentrator. Approximately 27 mg of recombinant LbFAE was obtained from 1 liter of bacterial culture.
Optimal pH, optimal temperature, substrate specificity and kinetic parameters
The optimal pH and temperature of LbFAE activity were determined using MF as substrate. A 200 μl mixture contained 102 μg ml−1 LbFAE and 100 μM MF and 1% DMSO. The optimal pH was evaluated in a buffer system comprising three buffers: 100 mM citrate–phosphate (pH 4.0, 4.5, 5.0, 5.5, 6.0); 100 mM sodium phosphate (pH 6.0, 6.5, 7.0, 7.5, 8.0); and 100 mM Tris–HCl (pH 8.0, 8.5, 9.0).
The reaction was carried out at 30°C for 10 min and terminated by placing the reaction in a boiling water bath for 5 min. Released FA was quantified using HPLC and a Photodiode Array Detector at 319 nm (Waters Assoc., USA). The column used was Luna Omega PS C18 (250 × 4.6 mm, particle size 5 μm; Phenomenex Inc., Torrance, USA). The column temperature was set at 30°C. Acetonitrile:acetic acid (10%) 20:80 (v:v) was the mobile phase. The optimal temperature of LbFAE was determined in sodium-phosphate buffer (100 mM, pH 6.5) for the temperature range 10–70°C with samples taken at 10°C intervals and analyzed as above.
The catalytic activities of LbFAE against methyl p-coumarate, methyl caffeate and methyl sinapinate and S114A LbFAE against MF were evaluated at the optimal pH and temperature determined for MF. The FAE activity using pNPF substrate was measured spectrophotometrically as described previously (Mastihuba et al., 2002). The kinetic constants (Vmax, Km) for MF and pNPF were calculated using a 200 μl−1 reaction mixture including 10 or 50 μg ml−1 LbFAE and 0.0125–2.0 mM of substrate at the derived optimal pH and temperature. Vmax and Km were determined by fitting the Michaelis–Menten equation to the data using nonlinear regression (R2 = 0.9724) in GraphPad Prism 5 program (GraphPad Software, San Diego California USA). All assays were performed in triplicates.
Degradation of feruloylated arabinoxylan
Feruloylated arabinoxylan (F-AX) was extracted from corn bran (provided by Cargill Deutschland GmbH, Krefeld, Germany from their wet steeping process of corn grain) and characterized as reported previously (Rudjito et al., 2020). The de-starching step and the subcritical water extraction (SWE) of F-AX were performed at the pilot plant by Celabor (Chaineux, Belgium). Phenolic acid profile of F-AX was determined after saponification and HPLC-UV as described previously (Rudjito et al., 2020).
The F-AX was solubilized in phosphate buffer (100 mM, pH 6.5) at 5 mg ml−1. The enzyme was diluted in phosphate buffer and added into 2 ml substrate medium (in triplicate) to reach a concentration of 1 mg enzyme per g of F-AX. The reaction was carried out at 40°C and aliquots (100 ul) were taken after 10 min, 2, 4, 8, and 24 h, directly inactivated at 100°C for 5 min and stored in dark at 4°C for the analysis of phenolic compounds.
Sinapic acid, p-coumaric acid, ferulic acid, and diferulic acids (5–5′ di-FA and 8–8′ di-FA) were quantified by HPLC as described previously (Rudjito et al., 2020). In short, aliquots were diluted 5-times in methanol:2% acetic acid mixture (1.1 v/v) and 10 μl were injected (1 ml min−1) onto the HPLC system (Waters 2,695 separation module, USA) equipped with a C18 guard column and an SB-C18 separation column (Zorbax SB-C18 5 μm particle size, 4.6 × 250 mm, Agilent, USA). The separation was performed at 25°C using as eluent A 2% acetic acid in H2O (v/v); and eluent B 100% methanol. The gradient used for the separation was: 100%–75% A (11 min), 71.25% A (4 min), 64% A (10 min), 55% A (10 min), 35% A (3 min), 100% A (3 min) and 100% A (4 min), and measurements were made on a photodiode array detector (Waters 2,996, USA) at 200–400 nm. Concentrations between 0.005 g l−1 and 0.1 g l−1 were used for standard calibration.
Analysis of oligomeric state using size-exclusion chromatography
The oligomeric state of LbFAE was investigated using size-exclusion chromatography (SEC) on a HiLoad 16/60 Superdex 200 prep grade column equilibrated with 20 mM Hepes (pH 7.5) containing 150 mM NaCl at a flow rate of 1 ml min−1 at 4°C. For estimation of the LbFAE molecular weight, the standard proteins used were ferritin (440 kDa), aldolase (158 kDa), conalbumin (75 kDa), and ribonuclease A (13.7 kDa).
Protein crystallization and structure determination
LbFAE could be crystallized under several conditions using vapor diffusion in hanging drops at room temperature. Crystals of wild-type and S114A LbFAE were obtained under two different reservoir conditions, either by mixing 1 μl 20 mg ml−1 protein in 50 mM Hepes pH 7.5, 150 mM NaCl, 5% (v/v) glycerol with 0.5 μl reservoir solution 1 (0.1 M 2,2-Bis(hydroxymethyl)-2,2′,2″-nitrilo-triethanol (Bis-Tris) buffer (pH 5.5), 0.2 M sodium chloride, 25% (w/v) polyethylene glycol (PEG) 3,350); or reservoir solution 2 (0.1 M sodium cacodylate (pH 6.5), 0.2 M calcium acetate and 18% (w/v) PEG 8000). The S114A crystals used for data collection were soaked with either methyl ferulate (MF) or methyl sinapinate (MS) prior to harvest and flash-freezing. All crystals were flash-frozen in liquid nitrogen prior to data collection.
X-ray diffraction data were collected using synchrotron radiation, and the XDS package was used for processing and scaling (Kabsch, 2010). As mentioned above, LbFAE crystallizes in different space groups, but without a clear correlation to the crystallization conditions used. A tetragonal wild-type dataset (1.9 Å resolution) was phased by molecular replacement with PHENIX Phaser (Adams et al., 2010) using the prepared homology model. A clear solution was obtained, and the phases were further improved by density modification using RESOLVE implemented in the PHENIX suite. A complete initial model could be built, and further improved by iterative manual model correction using COOT (Emsley et al., 2010) guided by σA-weighted 2Fo-Fc electron-density maps and crystallographic refinement in PHENIX. All model refinement was performed using phenix.refine and included reciprocal-space refinement of x, y, z coordinates, and individual B factor refinement.
The refined, initial model was used to phase three datasets using molecular replacement with PHENIX (Supplementary Table S1): a 1.90-Å resolution dataset from a wild-type crystal (reservoir 2) soaked with a grain of FA added to the mother liquor prior to crystal harvest; a 1.50-Å dataset of the wild type without ligand (reservoir 1); and a 1.45-Å dataset of the S114A variant soaked with MS (reservoir 2). The resulting molecular-replacement solutions generated models that were manually corrected and refined as described above. Although several LbFAE S114A crystals soaked with MF or MS were screened, no structure with bound MF or MS could be identified, and only the unliganded mutant structure was refined.
Electrostatic potential surfaces were calculated using APBS-PDB2PQR software suite at the PoaissonBolzmann server (Jurrus et al., 2018)1. PDB2PQR was run using PROPKA to assign protonation states at pH 6.5, and the PARSE forcefield. The output was used as input to APBS calculations using automatically configured finite difference Poisson-Boltzmann calculations (mg-auto). Electrostatic potential surfaces were visualized with PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC).
Sequence analysis of FAEs with known 3D structure
Sequences for the putative FAEs listed in Supplementary Table S2 with known three-dimensional (3D) structure were structurally aligned using the multiple structure 3D alignment algorithm in PDBeFold v.2.59 (Krissinel and Henrick, 2004)2, and the aligned multiple sequence alignment (MSA) used as input for generating a phylogenetic tree. The MSA was visualized using ESPript 3.0 (Robert and Gouet, 2014)3. A phylogenetic tree was computed using the Simple Phylogeny option at the EMBL-EBI server (Madeira et al., 2019)4 using the pre-aligned sequences as input. The tree was visualized using iTOL v.6.4.3 (Interactive Tree of Life5; Letunic and Bork, 2021). An alignment was also generated using PDBeFold v.2.59 including only the closest relatives (LJ0536 and Est1E), also visualized using ESPript 3.0.
Results
SEC analysis of the oligomeric state
The theoretical molecular weight of LbFAE including the His6 tag and TEV-cleavage site is 31.6 kDa, which was confirmed as a single band of approximately 30 kDa by SDS-PAGE analysis (Supplementary Figure S1), and a SEC elution profile that is consistent with a molecular mass of approximately 60 kDa (Supplementary Figure S1), which provides support for a homodimeric state.
Characterization of the catalytic activity of LbFAE
The optimal pH and temperature of the enzyme were 6.5 and 40°C, respectively (Figures 1A,B). LbFAE released (per mg of enzyme) 53 nmol min−1 FA, 29 nmol min−1 p-coumaric acid, and 12 nmol sinapic acid min−1 from the corresponding methyl esters under the experimental conditions used (Figure 1C). The lowest activity was observed for methyl caffeate where only 2 nmol min−1 (per mg enzyme) was released. Further, the S114A LbFAE showed no catalytic activity on MF.
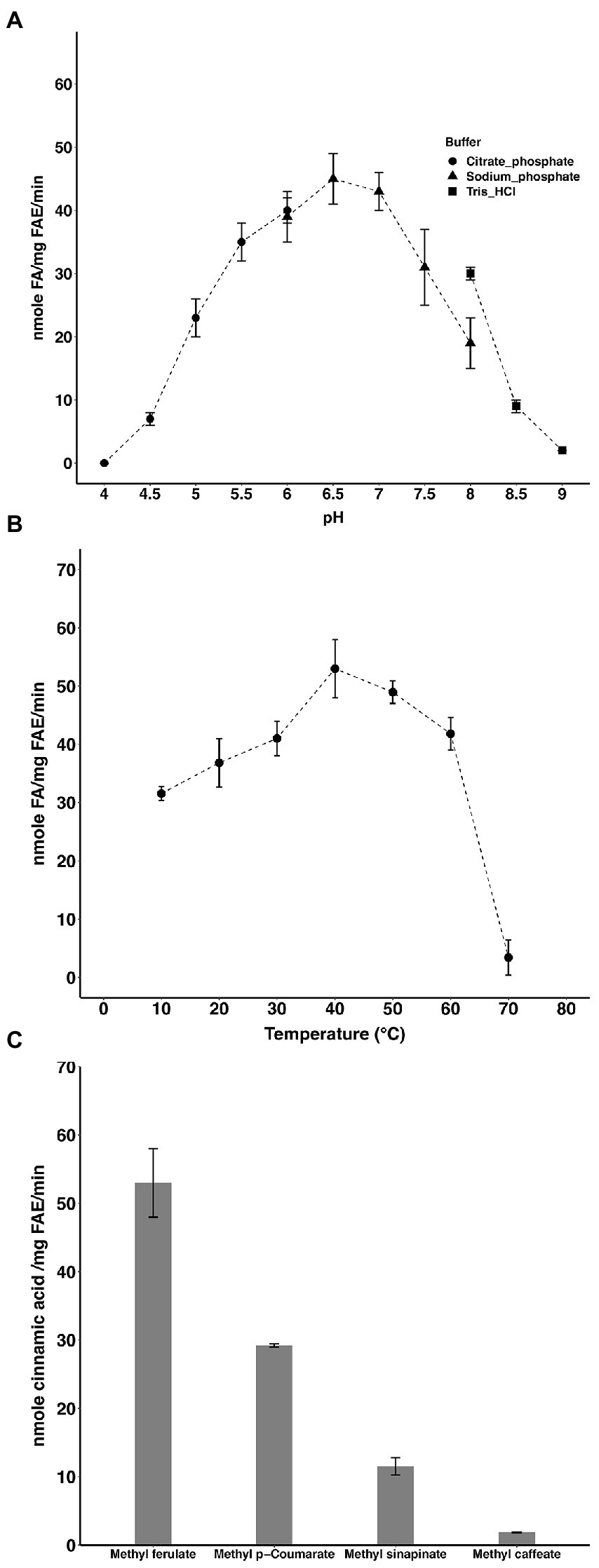
Figure 1. Activity of LbFAE. pH (A) and temperature (B) optimum of activity with MF as substrate. (C) Activity on different hydroxycinnamic acids. Measurements were performed in triplicates and the error bars indicate the mean ± SD.
Curves showing the enzyme velocity versus substrate concentration for LbFAE toward the MF and pNPF (Figure 2) demonstrate a significant difference in velocity of the enzyme reaction with the two substrates. The kinetic parameters (kcat and Km) were determined from the fit of the Michalis-Menten reaction to the data (Table 1). LbFAE shows an apparent higher affinity for MF than pNPF, as the estimated Km is 13-fold lower for MF than pNPF. The kcat/Km ratio shows that LbFAE hydrolyses MF 34 times more efficiently than pNPF. The high kcat/Km ratio for MF is attributed to the very fast reaction (high kcat) and the high substrate affinity (low Km) as compared to pNPF.
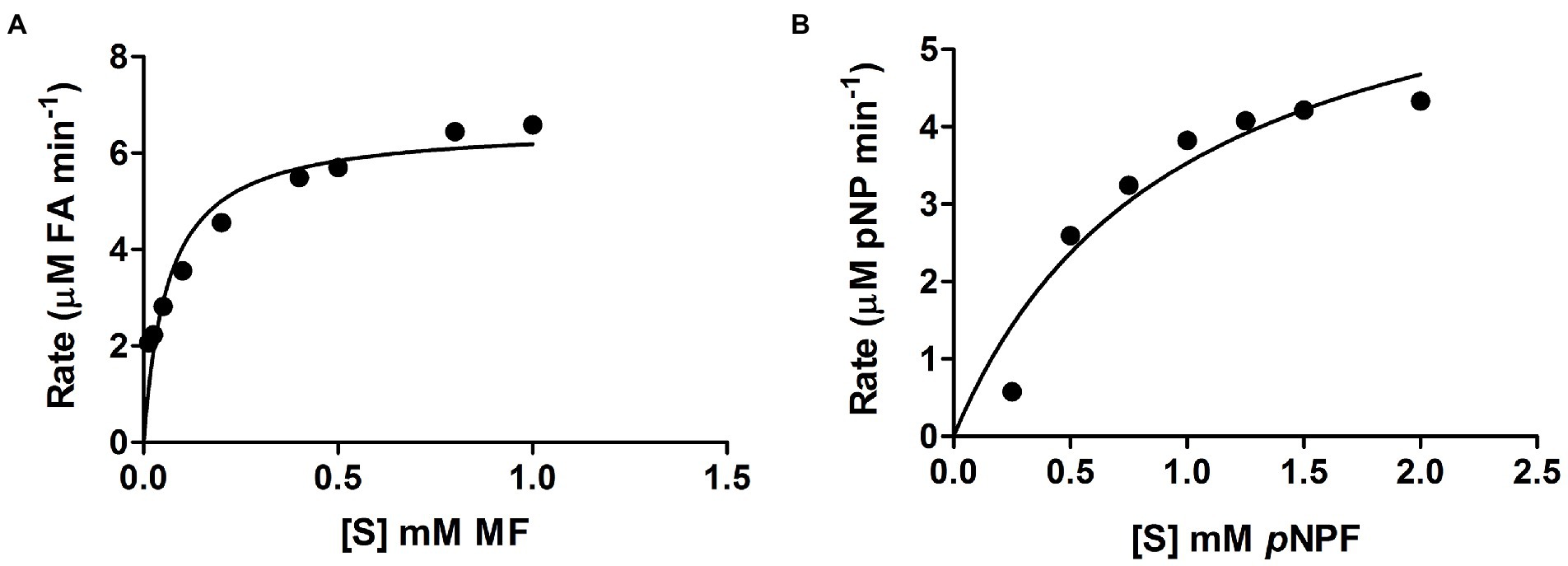
Figure 2. Velocity versus substrate concentration. (A) Methyl ferulate (MF), and (B) p-nitrophenyl ferulate (pNPF). Values are shown as the mean and SD for triplicate experiments.

Table 1. Kinetic parameters of LbFAE determined against the pNPF and MF.
Degradation of feruloylated arabinoxylan
The hydroxycinnamic-acid profile of F-AX after saponification was as follows: 26.6 mg FA; 1.4 mg p-coumaric acid; 0.6 mg sinapic acid; 1.1 mg 5–5′ diferulic acid; and 0.7 mg 8–8′ diferulic acid. Per g substrate. FA was the only phenolic component detected from F-AX after treatment with LbFAE (Figure 3). We did not detect p-coumaric acid, sinapic acid or FA dimers. After 2 h incubation, LbFAE released 1.9 mg FA per g substrate, increasing slightly to 2.4 mg after 24 h incubation, corresponding to 9% of the total amount of FA present in F-AX.
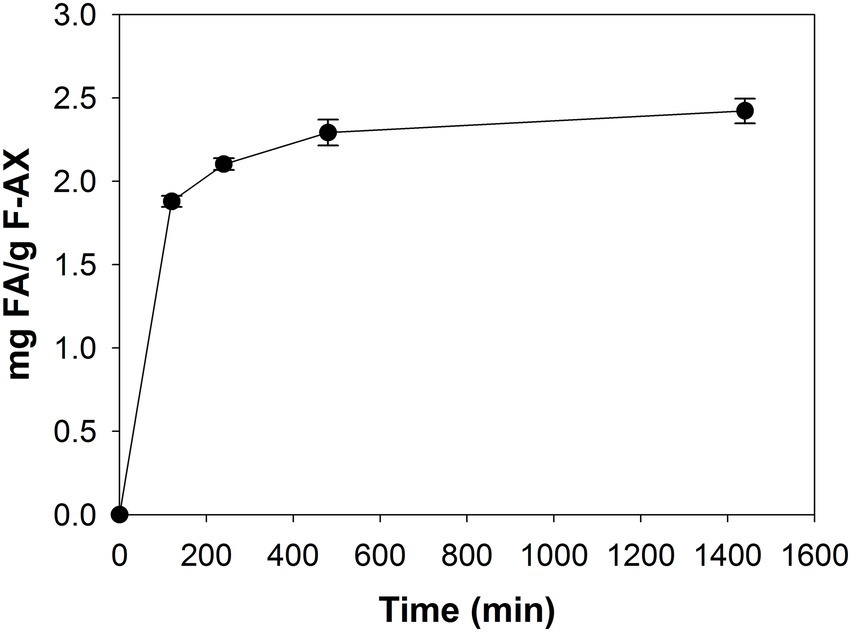
Figure 3. Degradation of feruloylated arabinoxylan. Release of FA from F-AX by incubation with LbFAE for 24 h. Error bars indicate the mean ± SD.
Overall structure of LbFAE
Wild-type crystals grew as either triclinic (P1) or tetragonal (P43212) crystals under identical crystallization conditions, whereas crystals of the S114A variant were isomorphous with the P43212 crystal form despite being produced under different conditions. Several LbFAE crystals were screened, and one dataset of the wild type was used for initial phase determination. A homology model was produced manually based on aligning the LbFAE sequence with that of the crystal structure of Lactobacillus johnsonii cinnamoyl esterase LJ0536 (PDB 3PFB; Lai et al., 2011) and mutating the amino acids of the template in COOT (Emsley et al., 2010).
The LbFAE monomer structure includes 260 amino acids that forms the canonical α/β-hydrolase superfamily fold with a conserved Ser-His-Asp catalytic triad (Figure 4). The structure is characterized by a central open twisted β-sheet with eight β-strands starting with an N-terminal β-hairpin followed by six parallel β-strands, which includes three complete βαβ motifs. The active site is located at the C-terminal ends of the parallel β-strands of the α/β-hydrolase core where the crossover connection between βαβ motifs occur. An extensive domain (residues 140–186), inserted between β5 and α5, folds over the active site to form a large “lid.” The conserved catalytic triad (Figure 4) is situated at the end of β5 (Ser114), and in the loops following β8 (His233) and β7 (Asp206). The nucleophilic serine (Ser114 in LbFAE) is positioned at the center of a conserved lipase (EC 3.1.1.3)-esterase (EC 3.1.1.1) pentapeptide G-X-S-X-G motif (Brenner, 1988).
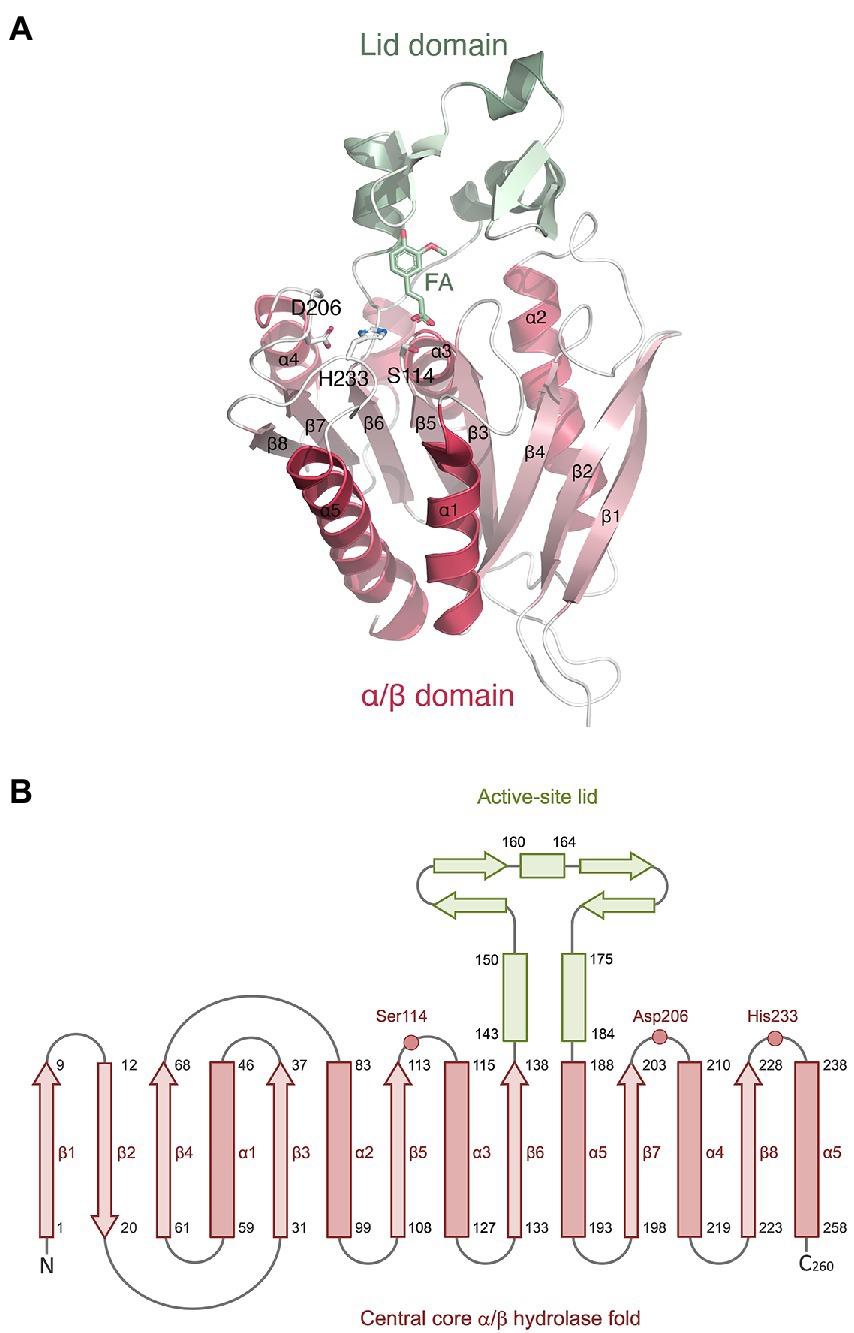
Figure 4. Monomer structure of LbFAE. (A) Ribbon drawing showing the eight-stranded α/β hydrolase fold (red) and the lid domain (green). Ferulic acid (FA) and the side chains of the catalytic triad (Ser114, His233, Asp206) are shown as ball-and-stick objects. (B) Topology diagram showing the same information as in (A).
The protein forms a homodimer with two-fold symmetry where the active sites from the two monomers are oriented facing each other (Figure 5A). The active sites can be accessed only through a groove formed between the two monomers at one end of the homodimer (the opening to the groove is seen in the bottom view in Figure 5B). The homodimeric structure observed in the crystal structure agrees well with the results from SEC analysis, as well as analysis of the dimer interface using PDBePISA v.1.52 (Krissinel and Henrick, 2007)6. PISA calculations returned a CSS (complexation significance score) of 1.0, which confirms a stable homodimeric association. Homodimer formation is associated with a solvation free energy gain (ΔGint) of −6.0 kcal mol−1 and the free energy of assembly dissociation (ΔGdiss) of 2.3 kcal mol−1. The stability of the dimer is supported by 19 hydrogen bonds and eight salt bridges across the interface, and an interface area of 1366.6 Å2. A total of 2,730 Å2 of the solvent-accessible surface area of the LbFAE monomers are buried upon dimer assembly. Possible interface salt bridges involve Arg42 Nη1, Nη2, Nε with the Glu73 oxygen atoms Oε1 and Oε2; Arg65 Nη1 and Nη2 with Glu73 Oε2. Electrostatic potential surfaces calculated at the pH optimum of activity (pH 6.5) shows that the buried active site has a net positive charge, whereas the area around the entrance is negatively charged (Figure 5B).
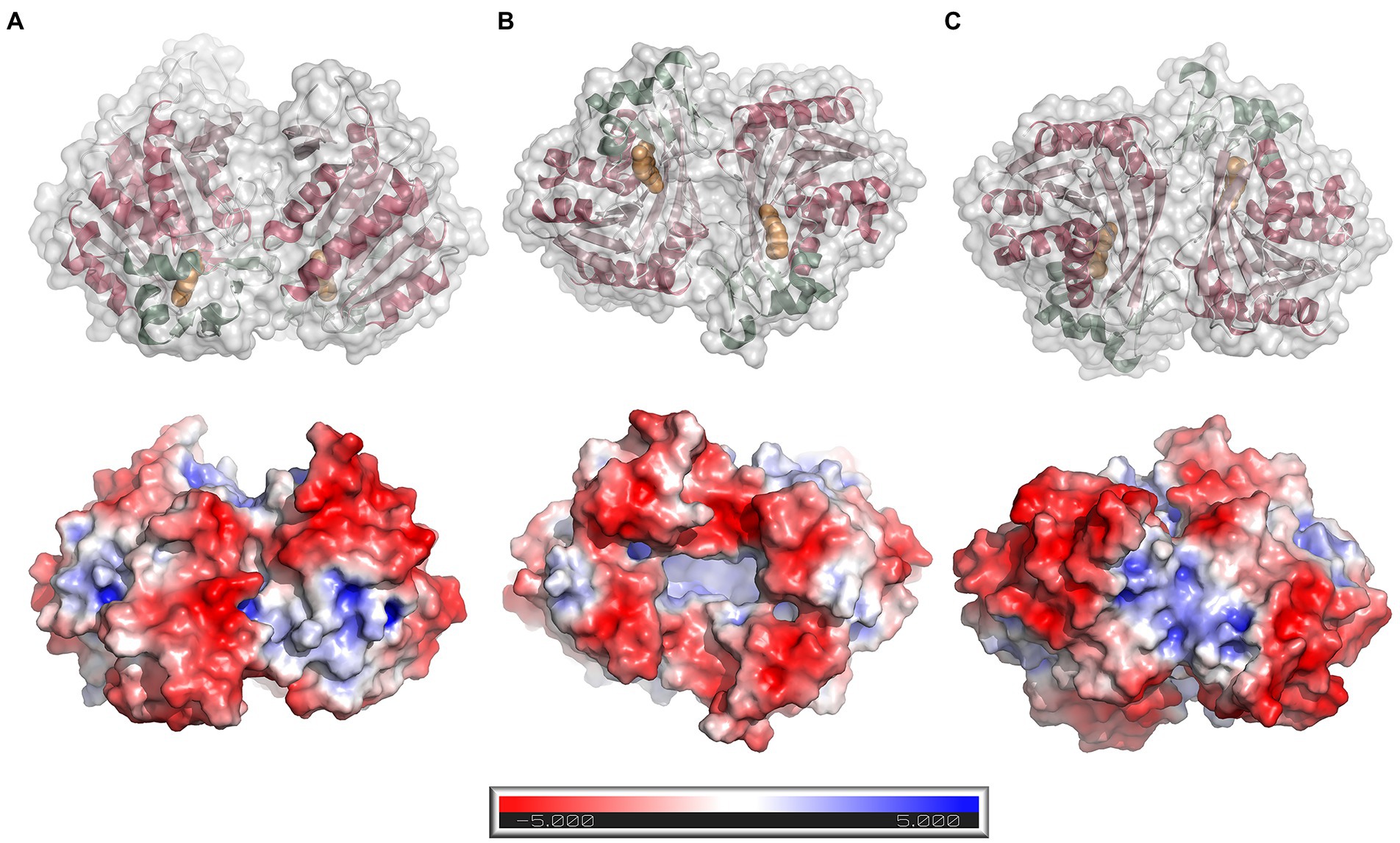
Figure 5. Dimer structure and surface electrostatics. Top row: ribbon drawings with an overlaid semitransparent gray molecular surface showing the LbFAE homodimer in three orientations (A) side view, (B) bottom view, and (C) top view. The α/β domain is colored red, and the lid domain is green. Ferulic acid is shown in orange. Bottom row: electrostatic potential surfaces calculated at pH 6.5 and colored in the range − 5.0 to 5.0 kT/e. The entrance to the active site is located at the bottom of the dimer, which is emphasized in orientation B.
Binding of ferulic acid
A variant with the catalytic serine replaced by an alanine (S114A) was generated to enable binding of different alkyl hydroxycinnamate compounds. Several S114A crystals were soaked with substrates, including MF and MS, but no binding was observed in the crystal structures. Thus, the refined crystal structure of the MS-soaked S114A variant instead contains an acetate molecule in the active site that originates from the buffer. Wild-type crystals soaked with substrates were also screened, but only FA (i.e., the product) resulted in a complex with LbFAE.
As mentioned above, the two active sites are only accessible from one face of the homodimer, the other end being closed off by close-packed side-chain atoms. When viewed down the entrance, aromatic side chains are observed to line the entrance opening such that Phe207-Phe38-Phe169-Tyr169-Phe46 from each monomer together form an oval, almost closed collar (Figure 6A). Both carboxylic oxygen atoms O1 and O2 of FA are within hydrogen-bonding distance of Ser114 Oγ (Figure 6B). Moreover, FA O1 can hydrogen bond to the backbone amide group of Phe38, and FA O2 can form an ionic bond with His233 Nε2.
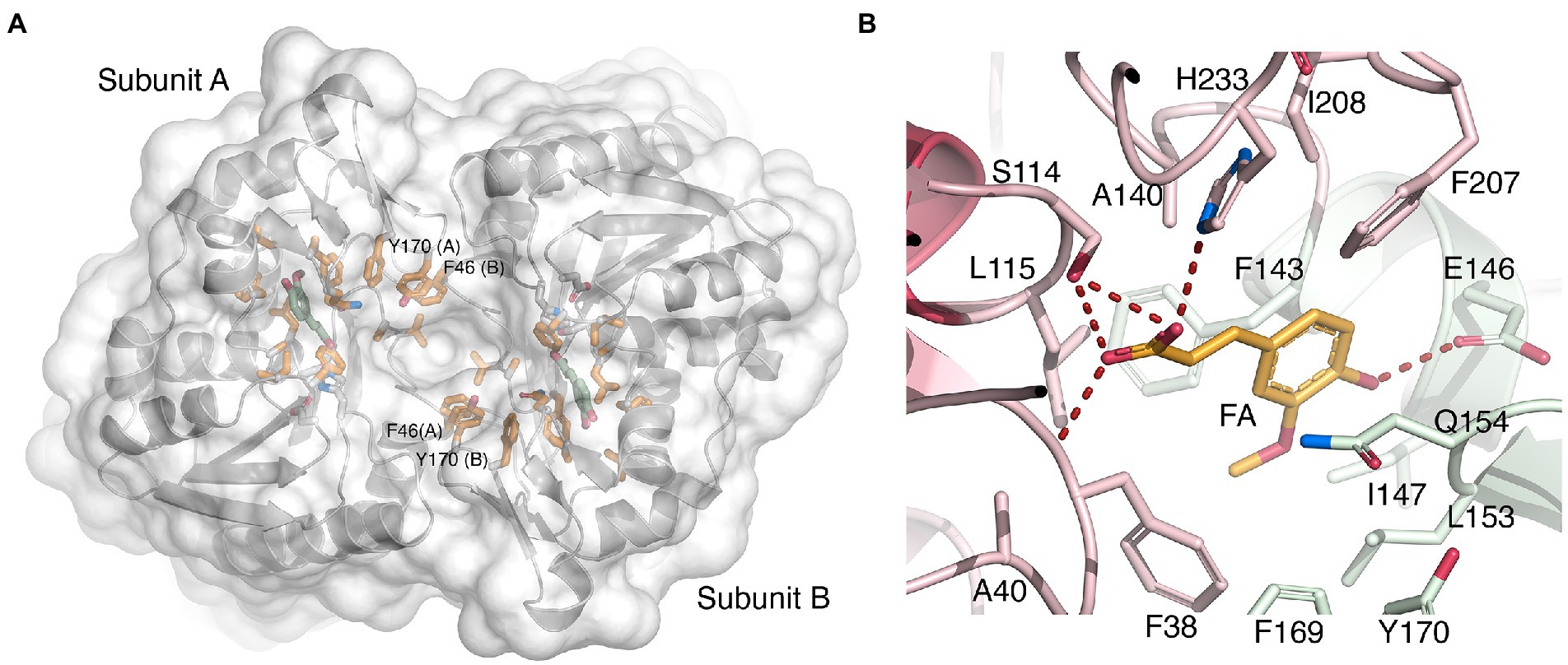
Figure 6. Binding of ferulate to LbFAE. (A) Ribbon drawing of the LbFAE homodimer with an overlaid water-accessible surface viewed from the entrance to the active-site groove (view from bottom as in Figure 4B). The ferulate molecule is colored green, and hydrophobic residues lining the groove are colored orange. An arc of aromatic residues from each monomer (Phe207-Phe38-Phe169-Tyr169-Phe46) form an oval collar around the active-site entrance. The narrow opening to the active sites is seen as a deep indentation at the center of the dimer. (B) Details of the key enzyme-ligand interactions. The ferulate molecule is colored orange, and residues belonging to the core and lid domains are colored red and green, respectively. See text for additional details.
Additionally, the O4 hydroxyl group of the cinnamoyl ring offers a hydrogen bond to Glu146 Oε1 (Figure 6B). Hydrophobic interactions with the cinnamoyl ring of FA involves Phe143 Cα and Cβ, and Gln154 Cγ, and the C10 atom of the methoxy substituent which engages in a CH/π interaction with the nearby Phe38 ring. The unliganded wild-type and S114A structures do not reveal significant conformational changes compared with the FA-bound wild-type model, which suggests that binding of substrate, and release of product, may not require major structural rearrangements.
Structural comparison with related enzymes
Sequence searches using BLASTP7 to find similar FAEs with known experimental 3D structure returned the cinnamoyl esterase LJ0536 from L. johnsonii as the closest match (Lai et al., 2011), which was therefore used as search model for molecular replacement calculations. The refined LbFAE model in complex with FA was submitted to the DALI server (Holm, 2020)8 to identify similar 3D structures. The search returned only two models with root-mean-square deviation (r.m.s.d.) values less than 2.0 Å, namely the expected L. johnsonii feruloyl esterase LJ0536 (PDB 3PFB, Lai et al., 2011; r.m.s.d. 1.5 Å; 30% sequence identity), and feruloyl esterase Est1E from Butyrivibrio proteoclasticus (PDB 2WTN, Goldstone et al., 2010; r.m.s.d. 1.9 Å; 26% sequence identity).
LJ0536 and Est1E form homodimers, and interestingly, these homodimers are fundamentally different from that of LbFAE, with their shallow and open active sites exposed toward the solvent rather than facing each other (Figure 7A). Our PDBePISA calculations on 2WTN shows that the Est1E dimer in the crystal is predicted to be stable (CSS 1.0), but with a modest ΔGint of −0.2 kcal mol−1. The authors report that results from dynamic light scattering were consistent with a dimeric Est1E assembly (Goldstone et al., 2010). According to PDBePISA, the LJ0536 dimer (3PFB) is not likely to represent a stable assembly (CSS 0.0), and no experiments were reported that confirm that LJ0536 is a dimer in solution. Several other crystal structures of FAEs have been shown to form stable, homodimeric assemblies (Supplementary Table S2), but these have monomer structures that are more distantly related to LbFAE, LJ0536 and Est1E.
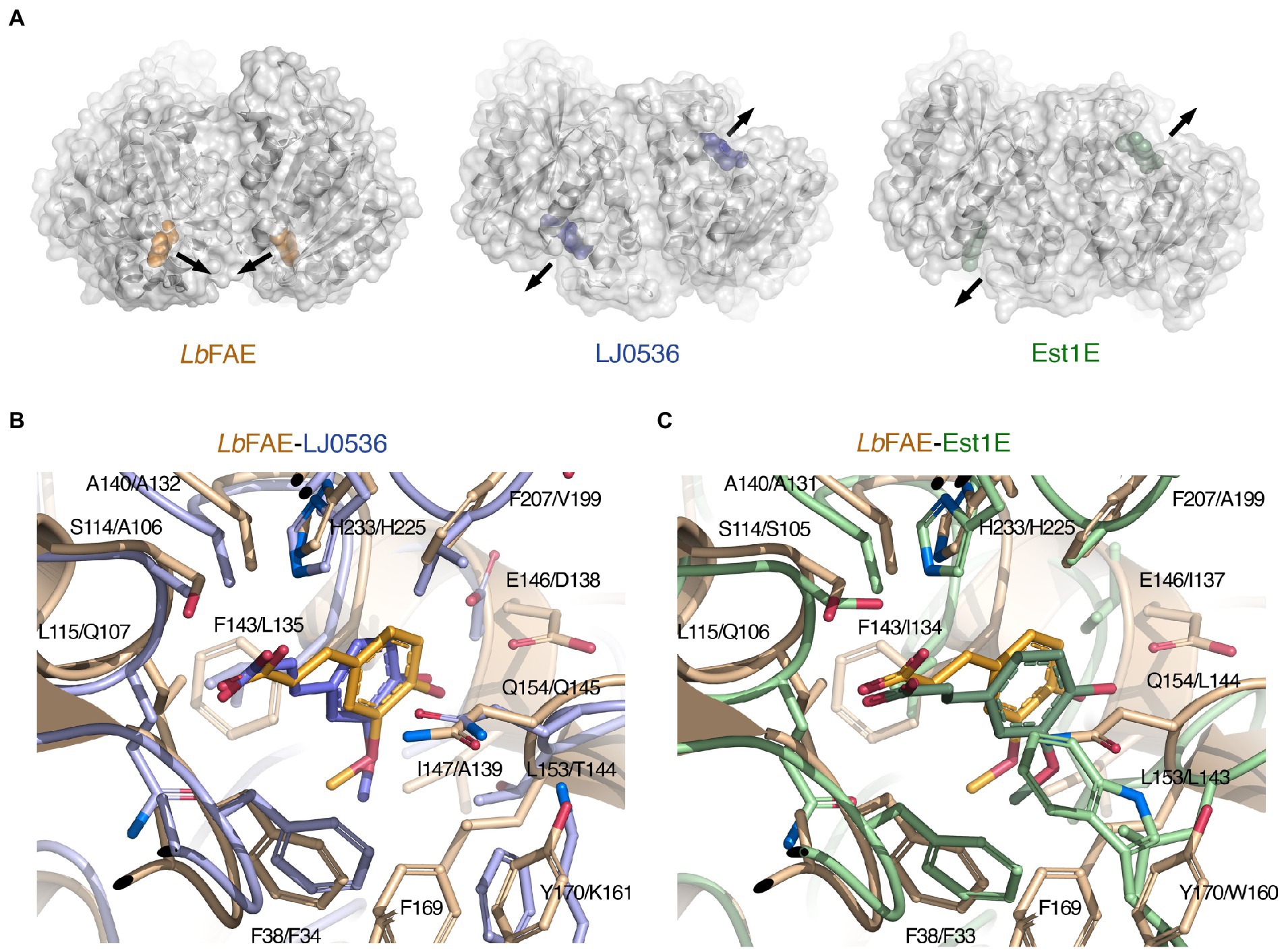
Figure 7. Comparison of LbFAE with the closest known FAE structures. (A) Ribbon drawings with overlaid molecular surfaces for LbFAE in complex with ferulate in orange, L. johnsonii cinnamoyl esterase LJ0536 in complex with ethylferulate in blue (PDB 3PFB) and B. proteoclasticus Est1E in complex with ferulate in green (PDB 2WTN). For the LJ0536 and Est1E dimers, the active sites are situated on opposite faces of the homodimer and are surface-exposed. In LbFAE, the active sites are facing each other and are buried below the protein surface. The arrows indicate the active-site entrances. Overlaid active site in the wild-type LbFAE ferulate complex (orange) with the (B) S106A-LJ0536 ferulate complex (blue; PDB 3PFC), and with the (C) wild-type Est1E ferulate complex (green; PDB 2WTN). The first residue in each label is for LbFAE. The ferulate molecule assumes identical orientations but slightly different conformation in LbFAE and LJ0536, whereas the ferulate molecule in Est1E has a different orientation in relation to the active site.
There are 14 unique experimental 3D structures available for bacterial enzymes with predicted FAE activity (one additional structure has not been released), and five unique structures corresponding to fungal FAEs (Supplementary Table S2). To visualize the phylogenetic relationship between these enzymes the amino-acid sequences were aligned based on the best matching of their corresponding atomic 3D coordinates to generate a structure-based sequence alignment with PDBeFold (Supplementary Figure S2). This type of alignment provides correct matching of non-identical residues, gaps, and insertions, which is particularly important when the sequence identity is low, as in the case of FAEs. Indeed, the low sequence similarity is evident from the alignment with only five residues strictly conserved across the 19 FAE members (Supplementary Figure S2). The resulting phylogenetic tree confirms LJ0536 and Est1E as the closest neighbors to LbFAE among FAEs with known 3D structure (Supplementary Figure S3).
A structural alignment using only the three closest structures LbFAE (FA complex, chain A, tag removed), LJ0536 (PDB 3PFC, FA complex, chain A, tag removed) and Est1E (PDB 2WTN, FA complex, chain A, tag removed) reveals 38 shared identical residues with an overall r.m.s.d. of 1.42 Å (Supplementary Figure S4). Pairwise structural comparisons give the following r.m.s.d. and sequence identity: LbFAE-LJ0536 of 1.45 Å and 31.2%; LbFAE-Est1E 1.66 Å and 26.9%; and LJ0536-Est1E 1.10 Å and 31.2%. If instead chain B of Est1E entry 2WTN is used, r.m.s.d. and sequence-identity values for LbFAE-Est1E are 1.69 Å and 27.0%, and for the LJ0536-Est1E pair 1.05 Å and 31.3%.
Although the active sites in LbFAE, LJ0536 and Est1E bear obvious similarities, including the catalytic triad and the G-X-S-X-G motif, there are also differences that indicate differences in substrate specificity (Supplementary Figure S4). As already noted above, the active sites in LJ0536 and Est1E are shallow and surface-exposed (Figure 7A), but in LbFAE is accessible only from a central opening of the homodimer. Beyond the difference in active-site accessibility, the precise conformations of the lid loops that covers the active sites differ, as do the identities and conformation of several key side chains. Of the two homologs, the active site in LJ0536 is clearly more similar to LbFAE than Est1E is. The most pronounced conformational differences that directly affect ligand binding occur in the β7-α4 loop (residues 204–209) where the catalytic histidine is situated, and in the first half (residues 139–169) of the lid domain. Another important difference relates to the different dimerization mode in LbFAE, where Phe46 from subunit B reaches into the active site of subunit A to stack with Tyr170 in subunit A (Figure 6A) and spatially restrict the active site compared with the more exposed active sites in the homologs.
There are more obvious differences at the detailed structural level. In both LbFAE and LJ0536, the FA molecule is bound in the same orientation and conformation in both subunits of the homodimers, whereas in Est1E, the FA is bound in two distinctly different orientations in the active site of subunits A and B, where the binding mode observed for Est1E subunit B agrees with that in LJ0536 and LbFAE. Thus, the FA molecule in LbFAE, LJ0536 (PDB 3PFC; Figure 7B) and Est1E subunit B (PDB 2WTN chain B; Figure 7C) have the same orientation. The only difference is the precise conformation with the C2-C1-C6-C8 dihedral angle being in cis configuration (0°) in LbFAE, and trans configuration (180°) in LJ0536 and Est1E (Figures 7B,C).
While several residues are identical (Figures 6B,C; Supplementary Table S3), the precise side-chain conformation typically differ somewhat, and in Est1E particularly, the different orientation of the FA molecule in chain A engages interactions not observed in LbFAE and LJ0536. Leu144 in Est1E chain B is probably the most obvious example, where the leucine side chain in Est1E moves to assume the position of the cinnamoyl ring in LbFAE, LJ0536 and Est1E chain B.
Despite about 40% of the active-site residues being identical between the three enzymes (Supplementary Table S3), the emerging picture suggests a more hydrophobic active-site cavity in LbFAE, which is mainly due to the amino-acid replacements at positions 115, 143, 147, 153, 178, and 207 (indicated by an asterisk in Supplementary Table S3). As discussed above, the hydrophobic character of the LbFAE active site is further emphasized by an oval collar of Phe residues around the central entrance between the two subunits of the homodimer (Figure 6A).
Structures of complexes with different ligands have been determined for LJ0536, namely FA (PDB 3PFC), ethyl ferulate (PDB 3PFB, 3QM1) and caffeate (PDB 3S2Z; Lai et al., 2011). In the LJ0536-FA complex, Asp138 Oδ2 can form a hydrogen bond to the C4 hydroxyl group of the hydroxycinnamoyl ring (Glu146 Oε1 in LbFAE). This appears to be a suitable candidate for hydroxycinnamate versus cinnamate selectivity. Lai and co-workers confirmed by site-directed mutagenesis that both Asp138 and Gln145 are critical for substrate recognition (Glu146 and Gln154 in LbFAE). As for Gln154 in LbFAE, Gln145 in LJ0536 interacts with its Cγ hydrogen atoms with the hydroxycinnamoyl ring through CH/π interaction. In Est1E, Asp138 in LJ0536 (Glu146 in LbFAE) is occupied by Ile137, which is unable to interact with the hydroxycinnamoyl ring. Moreover, Gln145 in LJ0536 (Gln154 in LbFAE) is replaced by Leu144 in Est1E which can only offer van der Waals interactions with the O3-C10 group of FA. These differences may suggest a different substrate-ring selectivity mechanism in Est1E.
All three enzymes can provide suitably similar, but not identical, hydrophobic interactions with the ethyl moiety of the hydroxycinnamoyl ethoxy group. However, this region in Est1E differs substantially between the two subunits. In subunit A, two lid loops (residues 142–148 and 158–164) rearrange to flip FA toward the entrance of the active site. This type of flexibility is not observed in our structures of unbound and FA-bound LbFAE, and neither in the structures of LJ0536 (Lai et al., 2011). In LbFAE, the lid loop 168–173, which corresponds to the flexible lid loop 158–164 in Est1E, is part of the dimer interface in LbFAE.
This is not the case for Est1E, which forms a different type of dimer. Therefore, we consider a similar conformational change of the lid loop and reorganization of the FA product unlikely in LbFAE since it would disrupt the LbFAE homodimer. A conformational change corresponding to that in Est1E has also been deemed unlikely for LJ0536 (Lai et al., 2011). Taken together, the absence of the hydroxycinnamoyl-selectivity determinants in Est1E present in both LbFAE and LJ0536, and the highly flexible and surface-exposed lid loops further suggest that the substrate specificity and selectivity of Est1E is fundamentally different.
Discussion
In this work, we have shown that the structure of LbFAE reveals a new type of dimerization compared with previously characterized bacterial homodimeric FAEs. Correlating the structural details to differences in substrate specificity and enzyme function is challenging since the substrates used for biochemical characterization of the three enzymes LbFAE, LJ0536 and Est1E differ, and all enzymes have slightly different structural determinants for substrate binding. Est1E is produced by the rumen bacterium B. proteoclatsicus and displays broad activity on plant-derived substrates, including ethyl ferulate, ethyl cinnamate, ethyl-3-coumarin carboxylate, ryegrass hemicellulose and birchwood xylan with the highest specific activity observed for ethyl-3-coumarin carboxylate (Goldstone et al., 2010).
In-silico docking of arabinofuranose to Est1E suggested that an arabinofuranosyl group attached to a hydroxycinnamoyl compound can be accommodated in the active site, and that the apparent flexibility of loops in the lid domain would allow large substrates. In the case of LbFAE however, the restricted access to the active sites and seemingly low flexibility in the lid domain suggest that the natural substrate is limited to accessible arabinofuranosyl-ferulate side chains on arabinoxylan chains. This agrees with our observations that LbFAE did not produce diferulic acid, and that relatively low amounts of FA were released from F-AX (Figure 3). The limited accessibility of the active site of LbFAE would probably make this enzyme inefficient on lignin-ferulate-polysaccharide complexes.
Nevertheless, docking of arabinofuranosyl ferulate (Araf-FA) to the LbFAE active site is straightforward. Despite the restricted active site and narrow opening between the two subunits, the Araf moiety is easily accommodated and is positioned in open cavity leading to the active sites (Figure 8). Several favorable interactions are possible with Glu146 coordinating the hydroxycinnamoyl hydroxyl group, Ser114 and His233 can provide hydrogen bonds to the ester oxygen of the bond to be cleaved, and Glu44 can form two hydrogen bonds to the O2’ and O3 hydroxyl groups of the arabinofuranosyl moiety of the leaving group. In addition, His233 is within hydrogen-bonding distance to the nucleophilic Ser114 and to the endocyclic O4’ oxygen of the arabinofuranosyl ring. Thus, the substrate is anchored in three critical positions, the aromatic ring, the ester oxygen and the sugar ring.
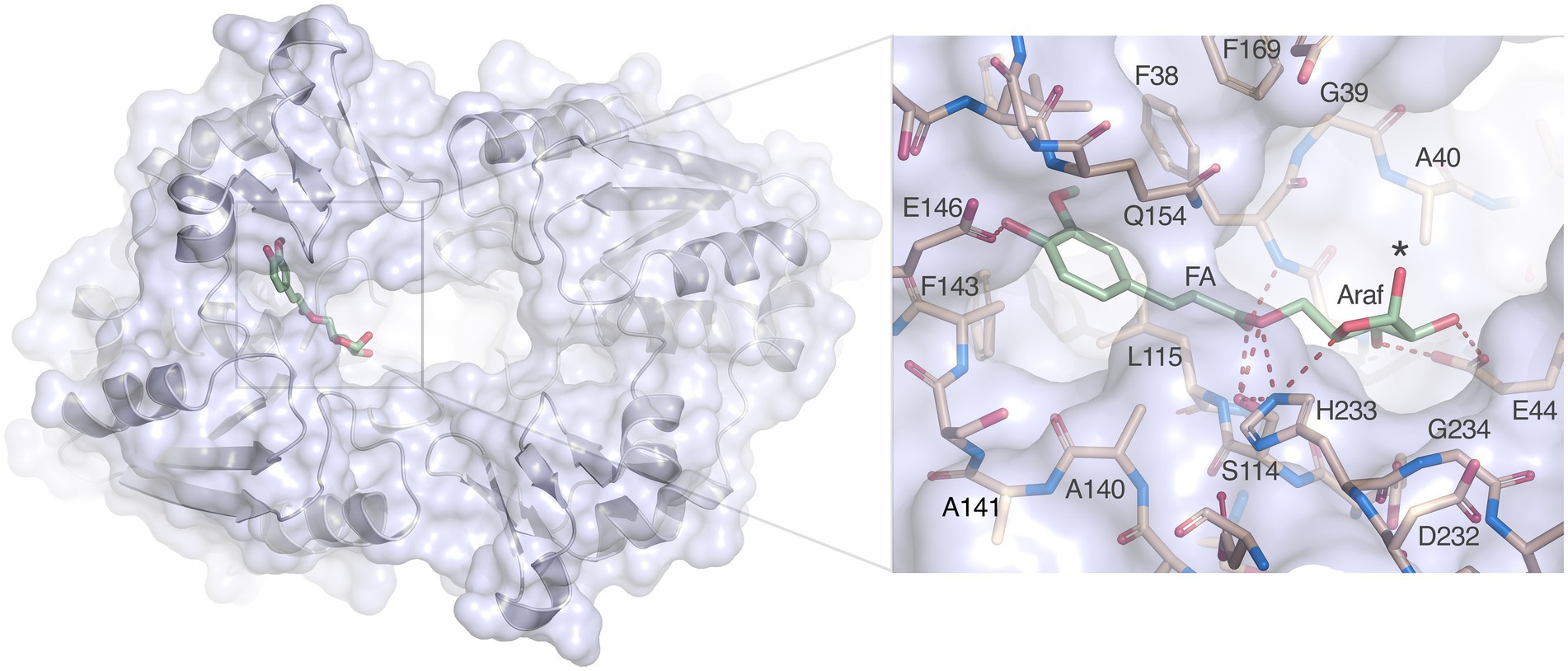
Figure 8. Docking of arabinofuranosyl ferulate. Arabinofuranosyl ferulate (Araf-FA; green) docked in the active site of LbFAE. The view is from the bottom of the homodimer where the entrance to the active site is located. The arabinofuranosyl moiety is exposed to the opening at the dimer interface. The inset shows possible hydrogen bonds between the ferulate and arabinofuranosyl moieties as dashed red lines.
While arabinofuranosyl ferulate is well accommodates, how LbFAE accesses this substrate also needs to be considered. We therefore modeled a xylotetraose with arabinofuranosyl ferulate linked to the reducing-end xylose residue (Supplementary Figure S5). The architecture of the homodimer makes it likely that LbFAE approaches xylan chain ends decorated with arabinofuranosyl ferulate with an exo-type approach, rather than cleaving arabinofuranosyl ferulate groups attached internally in the xylan backbone. In this binding mode, the hydroxyl group of Tyr170 could form a hydrogen bond with O3 of the second xylose residue, and the third xylose residue would stack favorably with the ring of Phe207 (Supplementary Figure S5). It is possible that conformational changes in LbFAE upon encountering the polymeric substrate would allow also internal arabinofuranosyl ferulate groups to be cleaved.
A histidine-ring flip mechanism has been proposed based on NMR studies for the protonation of the leaving group (Ash et al., 2000). Recently, results from quantum mechanics/molecular mechanics (QM/MM)-based transition-path sampling (TPS) on A. niger FaeA has provided strong support for an alternative mechanism whereby the histidine shuttles a proton from the nucleophile to the leaving group as a concerted event without ring flip (Silveira et al., 2021). In our proposed interaction network, His233 would indeed be suitably oriented to shuttle a proton from Ser114 to the leaving group in a concerted mechanism without the need for conformational changes.
Data availability statement
The data presented in the study are deposited in the Protein Data Bank repository (http://www.rcsb.org), accession numbers 7Z2X, 7Z2U, and 7Z2V.
Author contributions
CD and KK conceived the research. KK, DK, TR, AJ-Q, FV, and CD designed and performed experiments and analyzed the data. All authors contributed to the article and approved the submitted version.
Funding
This study was partially supported by the Swedish Research Council Formas (Grant number 2016–01449) to KK and additionally enabled by faculty funding to CD. FV acknowledges the Swedish Research Council Formas (Grant number 942–2016-119) for financial support.
Acknowledgments
We are grateful for support with data collection at beamlines I24 and I03, Diamond Light Source (UK), and at the BESSY II beamline 14.2 (Germany). Part of this work was facilitated by the Protein Science Facility at Karolinska Institutet/SciLifeLab (http://psf.ki.se). We thank Markus Keskitalo for fruitful discussions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1050160/full#supplementary-material
Footnotes
1. ^https://server.PoissonBolzmann.org
2. ^https://www.ebi.ac.uk/msd-srv/ssm/
3. ^https://espript.ibcp.fr/ESPript/ESPript/
4. ^https://www.ebi.ac.uk/Tools/phylogeny/simple_phylogeny
6. ^https://www.ebi.ac.uk/pdbe/pisa
References
Adams, P. D., Afonine, P. V., Bunkóczi, G., Chen, V. B., Davis, I. W., Echols, N., et al. (2010). PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221. doi: 10.1107/S0907444909052925
Antonopoulou, I., Sapountzaki, E., Rova, U., and Christakopoulos, P. (2022). Ferulic acid from plant biomass: a phytochemical with promising antiviral properties. Front. Nutr. 8:777576. doi: 10.3389/fnut.2021.777576
Ash, E. L., Sudmeier, J. L., Day, R. M., Vincent, M., Torchilin, E. V., Haddad, K. C., et al. (2000). Unusual 1H NMR chemical shifts support (his) Cε1–Ḥ··O==C H-bond: proposal for reaction-driven ring flip mechanism in serine protease catalysis. Proc. Natl. Acad. Sci. U. S. A. 97, 10371–10376. doi: 10.1073/pnas.97.19.10371
Benoit, I., Danchin, E. G. J., Bleichrodt, R. J., and de Vries, R. P. (2008). Biotechnological applications and potential of fungal feruloyl esterases based on prevalence, classification and biochemical diversity. Biotechnol. Lett. 30, 387–396. doi: 10.1007/s10529-007-9564-6
Brenner, S. (1988). The molecular evolution of genes and proteins: a tale of two serines. Nature 334, 528–530. doi: 10.1038/334528a0
Carpita, N. C. (2001). Cell wall architecture of the elongating maize coleoptile. Plant Physiol. 127, 551–565. doi: 10.1104/pp.010146
Crepin, V. F., Faulds, C. B., and Connerton, I. F. (2004). Functional classification of the microbial feruloyl esterases. Appl. Microbiol. Biotechnol. 63, 647–652. doi: 10.1007/s00253-003-1476-3
Dilokpimol, A., Mäkelä, M. R., Aguilar-Pontes, M. V., Benoit-Gelber, I., Hildén, K. S., and de Vries, R. P. (2016). Diversity of fungal feruloyl esterases: updated phylogeneti classification, properties, and industrial applications. Biotechnol. Biofuels 9:231. doi: 10.1186/s13068-016-0651-6
Dilokpimol, A., Mäkelä, M. R., Mansouri, S., Belova, O., Waterstraat, M., Bunzel, M., et al. (2017). Expanding the feruloyl esterase gene family of Aspergillus Niger by characterization of a feruloyl esterase, FaeC. New Biotechnol. 37, 200–209. doi: 10.1016/j.nbt.2017.02.007
Dilokpimol, A., Mäkelä, M. R., Varriale, S., Zhou, M., Cerullo, G., Gidijala, L., et al. (2018). Fungal feruloyl esterases: functional validation of genome mining based enzyme discovery including uncharacterized subfamilies. New Biotechnol. 41, 9–14. doi: 10.1016/j.nbt.2017.11.004
Drula, E., Garron, M.-L., Dogan, S., Lombard, V., Henrissat, B., and Terrapon, N. (2022). The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res. 50, D571–D577. doi: 10.1093/nar/gkab1045
Emsley, P., Lohkamp, B., Scott, W. G., and Cowtan, K. (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501. doi: 10.1107/S0907444910007493
Esteban-Torres, M., Reverón, I., Miguel Mancheño, J., De Las Rivas, B., and Muñoz, R. (2013). Characterization of a feruloyl esterase from lactobacillus plantarum. Appl. Environ. Microbiol. 79, 5130–5136. doi: 10.1128/AEM.01523-13
Fritsch, C., Jänsch, A., Ehrmann, M. A., Toelstede, S., and Vogel, R. F. (2017). Characterization of cinnamoyl esterases from different lactobacilli and Bifidobacteria. Curr. Microbiol. 74, 247–256. doi: 10.1007/s00284-016-1182-x
Goldstone, D. C., Villas-Boas, S. G., Till, M., Kelly, W. J., Attwood, G. T., and Arcus, V. L. (2010). Structural and functional characterization of a promiscuous feruloyl esterase (Est1E) from the rumen bacterium Butyrivibrio proteoclasticus. Proteins 78, 1457–1469. doi: 10.1002/prot.22662
Hassan, S., and Hugouvieux-Cotte-Pattat, N. (2011). Identification of two feruloyl esterases in Dickeya dadantii 3937 and induction of the major feruloyl esterase and of pectate lyases by ferulic acid. J. Bacteriol. 193, 963–970. doi: 10.1128/JB.01239-10
Holck, J., Fredslund, F., Møller, M. S., Brask, J., Krogh, K. B. R. M., Lange, L., et al. (2019). A carbohydrate-binding family 48 module enables feruloyl esterase action on polymeric arabinoxylan. J. Biol. Chem. 294, 17339–17353. doi: 10.1074/jbc.RA119.009523
Holm, L. (2020). Using Dali for protein structure comparison. Methods Mol. Biol. 2112, 29–42. doi: 10.1007/978-1-0716-0270-6_3
Jurrus, E., Engel, D., Star, K., Monson, K., Brandi, J., Felberg, L. E., et al. (2018). Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128. doi: 10.1002/pro.3280
Kondo, T., Mizuno, K., and Kato, T. (1990). Cell wall-bound p-coumaric acids in Italian ryegrass. Can. J. Plant Sci. 70, 495–499. doi: 10.4141/cjps90-058
Koseki, T., Fushinobu, S., Ardiansyah Shirakawa, H., and Komai, M. (2009). Occurrence, properties, and applications of feruloyl esterases. Appl. Microbiol. Biotechnol. 84, 803–810. doi: 10.1007/s00253-009-2148-8
Krissinel, E., and Henrick, K. (2004). Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D 60, 2256–2268. doi: 10.1107/S0907444904026460
Krissinel, E., and Henrick, K. (2007). Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797. doi: 10.1016/j.jmb.2007.05.022
Lai, K. K., Stogios, P. J., Vu, C., Xu, X., Xui, H., Molloy, S., et al. (2011). An inserted α/β subdomain shapes the catalytic pocket of lactobacillus johnsonii cinnamoyl esterase. PLoS One 6:e23269. doi: 10.1371/journal.pone.0023269
Lambert, N., Kroon, P. A., Faulds, C. B., Plumb, G. W., McLauchlan, W. R., Day, A. J., et al. (1999). Purification of cytosolic beta-glucosidase from pig liver and its reactivity towards flavonoid glycosides. Biochim. Biophys. Acta 1435, 110–116. doi: 10.1016/s0167-4838(99)00213-7
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Liu, S., Soomro, L., Wei, X., Yuan, X., Gu, T., Li, Z., et al. (2021). Directed evolution of feruloyl esterase from lactobacillus acidophilus and its application for ferulic acid production. Bioresour. Technol. 332:124967. doi: 10.1016/j.biortech.2021.124967
Madeira, F., Park, Y., Lee, J., Buso, N., Gur, T., Madhusoodanan, N., et al. (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641. doi: 10.1093/nar/gkz268
Mastihuba, V., Kremnický, L., Mastihubová, M., Willett, J. L., and Côté, G. L. (2002). A spectrophotometric assay for feruloyl esterases. Anal. Biochem. 309, 96–101. doi: 10.1016/S0003-2697(02)00241-5
Mogodiniyai Kasmaei, K., Schlosser, D., Sträuber, H., and Kleinsteuber, S. (2020). Does glucose affect the de-esterification of methyl ferulate by lactobacillus buchneri? Microbiol. Open 9:e971. doi: 10.1002/mbo3.971
Oliveira, D. M., Mota, T. R., Oliva, B., Segato, F., Marchiosi, R., Ferrarese-Filho, O., et al. (2019). Feruloyl esterases: biocatalysts to overcome biomass recalcitrance and for the production of bioactive compounds. Bioresour. Technol. 278, 408–423. doi: 10.1016/j.biortech.2019.01.064
Pereira, G. V., Abdel-Hamid, A. M., Dutta, S., D’Alessandro-Gabazza, C. N., Wefers, D., Farris, J. A., et al. (2021). Degradation of complex arabinoxylans by human colonic Bacteroidetes. Nat. Commun. 12:459. doi: 10.1038/s41467-020-20737-5
Ralph, J., Quideau, S., Grabber, J. H., and Hatfield, R. D. (1994). Identification and synthesis of new ferulic acid dehydrodimers present in grass cell walls. J. Chem. Soc. Perkin Trans. 1, 3485–3498. doi: 10.1039/P19940003485
Robert, X., and Gouet, P. (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324. doi: 10.1093/nar/gku316
Rudjito, R. C., Jiménez-Quero, A., Hamzaoui, M., Kohnenb, S., and Vilaplana, F. (2020). Tuning the molar mass and substitution pattern of complex xylans from corn fibre using subcritical water extraction. Green Chem. 22, 8337–8352. doi: 10.1039/D0GC02897E
Silveira, R. L., Knott, B. C., Pereira, C. S., Crowley, M. F., Skaf, M. S., and Beckham, G. T. (2021). Transition path sampling study of the feruloyl esterase mechanism. J. Phys. Chem. B 125, 2018–2030. doi: 10.1021/acs.jpcb.0c09725
Tabka, M. G., Herpoël-Gimbert, I., Monod, F., Asther, M., and Sigoillot, J. C. (2006). Enzymatic saccharification of wheat straw for bioethanol production by a combined cellulase xylanase and feruloyl esterase treatment. Enzyme Microb. Technol. 39, 897–902. doi: 10.1016/j.enzmictec.2006.01.021
Tabouriech, N., Prates, J. A. M., Fontes, C. M. G. A., and Davies, G. J. (2005). Molecular determinants of substrate specificity in the feruloyl esterase module of xylanase 10B from clostridium thermocellum. Acta Crystallogr. D 61, 194–197. doi: 10.1107/S0907444904029695
Terrett, O. M., and Dupree, P. (2019). Covalent interactions between lignin and hemicelluloses in plant secondary cell walls. Curr. Opin. Biotechnol. 56, 97–104. doi: 10.1016/j.copbio.2018.10.010
Udatha, D. B. R. K., Kouskoumvekaki, I., Olsson, L., and Panagiotou, G. (2011). The interplay of descriptor-based computational analysis with pharmacophore modeling builds the basis for a novel classification scheme for feruloyl esterases. Biotechnol. Adv. 29, 94–110. doi: 10.1016/j.biotechadv.2010.09.003
Udatha, D. B. R. K., Mapelli, V., Panagiotou, G., and Olsson, L. (2012). Common and distant structural characteristics of feruloyl esterase families from Aspergillus oryzae. PLoS One 7:e39473. doi: 10.1371/journal.pone.0039473
Underlin, E. N., Frommhagen, M., Dilokpimol, A., van Erven, G., de Vries, R. P., and Kabel, M. A. (2020). Feruloyl esterases for biorefineries: subfamily classified specificity for natural substrates. Front. Bioeng. Biotechnol. 8:332. doi: 10.3389/fbioe.2020.00332
Uraji, M., Tamura, H., Mizohata, E., Arima, J., Wan, K., Ogawa, K., et al. (2018). Loop of Streptomyces feruloyl esterase plays an important role in the enzyme’s catalyzing the release of ferulic acid from biomass. Appl. Environ. Microbiol. 84, e02300–e02317. doi: 10.1128/AEM.02300-17
Wang, X., Geng, X., Egashira, Y., and Sanada, H. (2004). Purification and characterization of a feruloyl esterase from the intestinal bacterium lactobacillus acidophilus. Appl. Environ. Microbiol. 70, 2367–2372. doi: 10.1128/AEM.70.4.2367-2372.2004
Xu, Z., He, H., Zhang, S., Guo, T., and Kong, J. (2017). Characterization of feruloyl esterases produced by the four lactobacillus species: L. amylovorus, L. acidophilus, L. farciminis and L. fermentum, isolated from ensiled corn Stover. Front. Microbiol. 8, 8:941. doi: 10.3389/fmicb.2017.00941
Xu, Z., Kong, J., Zhang, S., Wang, T., and Liu, X. (2020). Comparison of enzyme secretion and ferulic acid production by Escherichia coli expressing different lactobacillus feruloyl esterases. Front. Microbiol. 11:568716. doi: 10.3389/fmicb.2020.568716
Zhang, H., Wen, B., Liu, Y., Du, G., Wei, X., Uddin Imam, K. M. S., et al. (2021). A reverse catalytic triad asp containing loop shaping a wide substrate binding pocket of a feruloyl esterase from lactobacillus plantarum. Int. J. Biol. Macromol. 184, 92–100. doi: 10.1016/j.ijbiomac.2021.06.033
Keywords: feruloyl esterase, ferulic acid, Lentilactobacillus buchneri, crystal structure, arabinoxylan
Citation: Kasmaei KM, Kalyani DC, Reichenbach T, Jiménez-Quero A, Vilaplana F and Divne C (2022) Crystal structure of the feruloyl esterase from Lentilactobacillus buchneri reveals a novel homodimeric state. Front. Microbiol. 13:1050160. doi: 10.3389/fmicb.2022.1050160
Edited by:
Kang Lan Tee, The University of Sheffield, United KingdomReviewed by:
Mohamed Habib, Cairo University, EgyptFranz Josef St John, Forest Products Laboratory, Forest Service (USDA), United States
Copyright © 2022 Kasmaei, Kalyani, Reichenbach, Jiménez-Quero, Vilaplana and Divne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christina Divne, ZGl2bmVAa3RoLnNl