 Peijie Zheng
Peijie Zheng Yue Qi
Yue Qi Xueyong Li1
Xueyong Li1 Yuewu Liu
Yuewu Liu Yuhua Yao
Yuhua Yao Guohua Huang
Guohua Huang- 1School of Electrical Engineering, Shaoyang University, Shaoyang, China
- 2College of Information and Intelligence, Hunan Agricultural University, Changsha, China
- 3School of Mathematics and Statistics, Hainan Normal University, Haikou, China
Transcription factors (TFs) are typical regulators for gene expression and play versatile roles in cellular processes. Since it is time-consuming, costly, and labor-intensive to detect it by using physical methods, it is desired to develop a computational method to detect TFs. Here, we presented a capsule network-based method for identifying TFs. This method is an end-to-end deep learning method, consisting mainly of an embedding layer, bidirectional long short-term memory (LSTM) layer, capsule network layer, and three fully connected layers. The presented method obtained an accuracy of 0.8820, being superior to the state-of-the-art methods. These empirical experiments showed that the inclusion of the capsule network promoted great performances and that the capsule network-based representation was superior to the property-based representation for distinguishing between TFs and non-TFs. We also implemented the presented method into a user-friendly web server, which is freely available at http://www.biolscience.cn/Capsule_TF/ for all scientific researchers.
Introduction
Transcription factors (TFs) are also sequence-specific DNA-binding factors, a family of proteins that control the expression of target genes (Karin, 1990; Latchman, 1997). The TFs are widely distributed, and their numbers vary with the size of the genome (Nimwegen, 2006). The larger genomes are likely to have a larger number of TFs on average. Approximately 10% of genes in the human genome are conservatively estimated to code for TFs. Consequently, the TFs are the potentially largest family of proteins in humans. The TFs exert regulating roles alone or together with other proteins in a complex by hindering or facilitating the recruitment of RNA polymerase (a type of enzyme) to specific DNA regions (Roeder, 1996; Nikolov and Burley, 1997). The regulation roles of the TFs are either positive or negative. The TFs promote the recruitment of RNA polymerase function as activators and contrarily ones to hold back recruitment as repressors. The TFs are involved in many important cellular processes including transcription regulation. Some TFs are responsible for cell differentiation (Wheaton et al., 1996), some respond to intercellular signals (Pawson, 1993), and some reply to environmental changes (Shamovsky and Nudler, 2008). Mutations in the TFs are discovered to be implied in many diseases (Bushweller, 2019). The TFs are a control switch to turn on or off to ensure when, where, and how many genes are accurately expressed. Thus, it is a fundamental problem but a therapeutic opportunity for drug discovery and development to accurately identify TFs. Physical or chemical methods (called wet experiments) are a prime alternative to identify TFs. The wet experiments include SELEX-based methods (Roulet et al., 2002), MITOMI (Rockel et al., 2012), and ChIP-based assays (Yashiro et al., 2016). Most known TFs were discovered by wet experiments and deposited in public databases (Wingender et al., 1996; Riaño-Pachón et al., 2007; Zhu et al., 2007; Zhang et al., 2020). The wet experiments accumulated a limited number of TFs at the expense of an enormous amount of time and money. It is only by the wet experiments that it is impossible and insufficient to discover all TFs in all the tissues or species all over the world. With advances in artificial intelligence, it is becoming possible to learn a computational model from these known TFs to recognize new unknown TFs which will be subsequently examined by the wet experiments. The computational methods shrank greatly the numbers of potential TFs that the wet experiments scanned, and thus, save a vast volume of time and money. The computational methods are becoming essentially complementary to the wet experiments, and both are jointly accelerating the exploration of the TFs.
To the best of our knowledge, Liu et al. (2020) pioneered the first computational method for discriminating TFs from non-TFs. Liu et al. extracted three types of sequence features: composition/transition/distribution (CTD) (Tan et al., 2019), split amino acid composition (SAAC), and dipeptide composition (DC) (Ding and Li, 2015). Comprehensively comparing the contribution of features and performances of five frequently used machine learning algorithms: logistic regression, random forest, k-nearest neighbor, XGBoost, and support vector machine (SVM). Liu et al. finally chose 201 optimal features and SVM for building the classifier. Liu et al. opened an avenue to identify TFs. Lately, Li et al. (2022) created a different idea from Liu et al. to distinguish TFs and non-TFs. Instead of designing sophisticated features. Li et al. directly took the sequence as input, split three amino acid residues as a basic unit, and employed long short-term memory (LSTM) for capturing semantic differences between TFs and non-TFs. Li et al. promoted the predictive accuracy to 86.83%. The LSTM is a special recurrent neural network (RNN) which suffered from the long-distance dependency. The capsule network proposed is a novel neural work architecture (Sabour et al., 2017), whose remarkable advantage is to capture relationship between local parts. This just made up for the deficiency of LSTM. Inspired by this, we proposed a capsule network-based method for TFs prediction.
Materials and methods
Data
The training and the testing data were downloaded from the website1 (Li et al., 2022), which was manually collected by Liu et al. (2020). The original dataset contained 601 human and 129 mouse TFs which preferred methylated DNA (Graves and Schmidhuber, 2005; Wang et al., 2018) and 286 TFs which preferred non-methylated DNA (Yin et al., 2017). Liu et al. (2020) conducted the following steps for improving the quality of the dataset. The sequences containing illegal characters such as “X”, “B”, and “Z” were first removed. Then, the CD-HIT, which is a clustering tool (Huang et al., 2010; Zou et al., 2020), was used to decrease redundancy between sequences. The cutoff threshold was set to 0.25, meaning that the sequence identity between any two sequences was no more than 0.25. Third, less than 50 amino acid sequences were excluded. A total of 522 TFs were finally preserved as positive samples after the above three processes. Liu et al. sampled the same number of non-TFs from the UniProt database (release 2019_11) which meets the following five requirements: (1) reviewed proteins, (2) proteins with evidence at protein level, (3) proteins in full length and of more than 50 amino acid residues, (4) proteins without DNA-binding TF activities, and (5) Homo sapiens proteins with less than 25% sequence identity in the CD-HIT. Liu et al. divided the data further into the training and the independent test dataset at the ratio of 8:2, with the former containing 406 positive and 406 negative samples, and the latter containing 106 positive and 106 negative samples.
Methods
As shown in Figure 1, the proposed method called Capsule_TF is a deep learning-based method. It mainly contains five layers, namely, embedding layer, bidirectional LSTM layer, capsule network layer, and three fully connected layers. The protein sequence as input goes through the embedding layer and is then embedded into low-dimensional vectors. The bidirectional LSTM layer and the capsule network layer are used to extract high-level representations of protein sequences. Three fully connected layers are finally used to discriminate TFs from non-TFs. The Capsule_TF is an end-to-end deep learning model without designing any features.
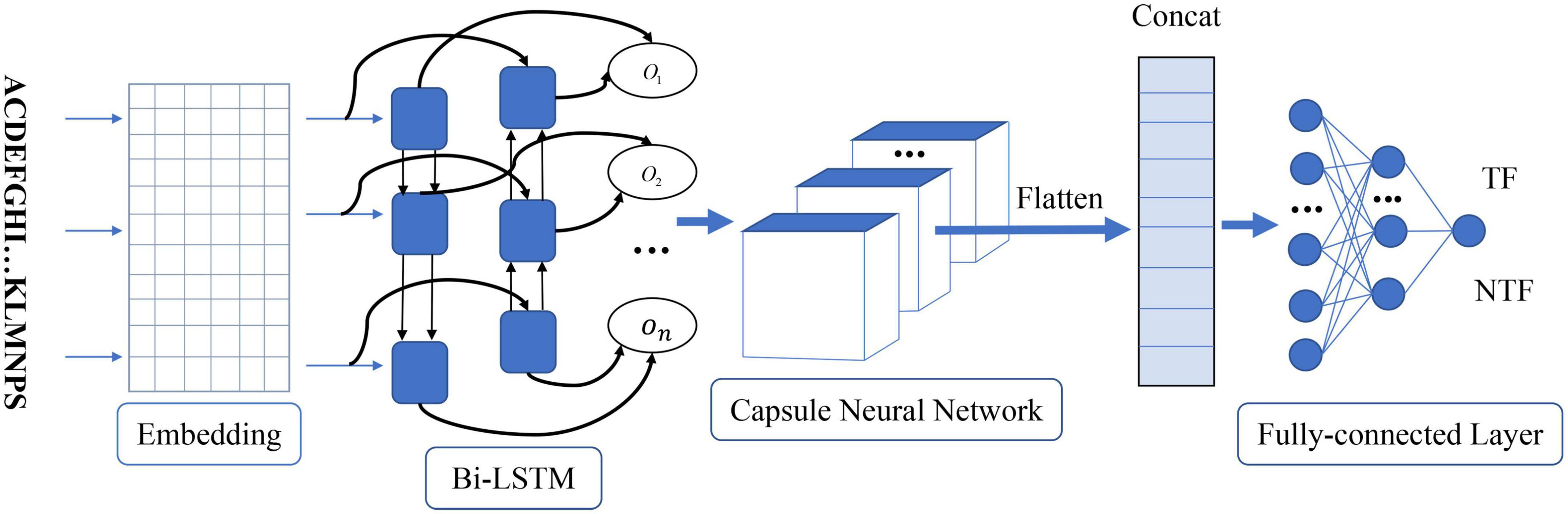
Figure 1. The architecture of the proposed capsule network-based method. On represents the word vector after the bidirectional LSTM layer. TF and NTF represent the final predicted outcome as a transcription factor or not.
Embedding
It is mandatory for text sequence input to be converted into digital sequences which are suitable to be processed by the subsequent machine learning algorithms. There are many ways of converting text sequences into digital sequences, such as a one-hot encoding scheme (Buckman et al., 2018) and Word2vec (Rong, 2014). The one-hot encoding scheme fails to capture relationships between words and is opt to yield sparse representation when the vocabulary is large. It is a common practice to use embedding to translate text sequences into dense digital vectors. In the field of text analysis by the deep neural network, the embedding is generally the first layer generally defined by
where denotes the embedding of the word, xi represents input, and We ∈ Rn×k denotes a lookup table that stores the embedding of words. We is the learnable parameter.
Long short-term memory
The LSTM (Hochreiter and Schmidhuber, 1997) belongs to the family of recurrent neural networks (RNNs) (Sherstinsky, 2020), which is typically a neural network sharing parameters at all time steps. The LSTM was pioneered by Hochreiter and Schmidhuber (Hochreiter and Schmidhuber, 1997) and later was continuously improved. The structure of the current LSTM was mainly made up of the cell state, the hidden state, the input, and the output. Figure 2 demonstrates the structure of the LSTM at the time step t which is identical at all the time steps. The cell state preserved memories for preceding words but was regulated by the gates to determine how much information was conveyed to the next time step. There are three gates in the LSTM: forget gate, input gate, and output gate. The forget gate is defined as
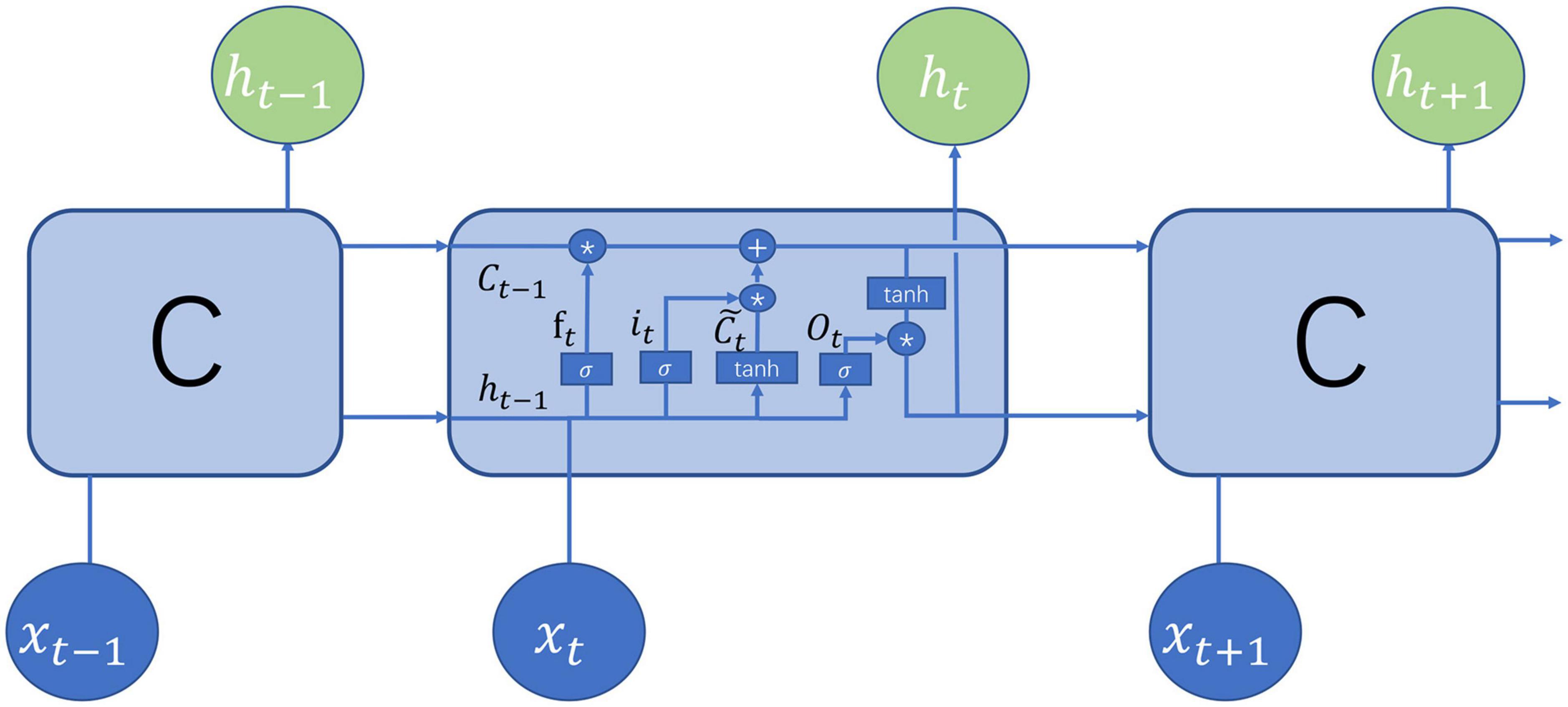
Figure 2. Illustration of long short-term memory (LSTM) structure (Olah, 2015).
where ht–1 denotes the hidden state at time step t-1, xt is the input at time step t, Wf and bf are learnable parameters, and σ is the sigmoid function. Obviously, the output of the forget gate falls between 0 and 1. The input gate and the candidate cell are defined, respectively, as
and
where Wi, Wc, bi, and bc are learnable parameters. The cell state is updated by
The preceding information is all forgotten if the forget gate is 0, namely, ft = 0, all the information is born in mind if ft = 1, and part are born if ft is more than 0 but less than 1. Obviously, the forget gate determines how much memories for preceding words are preserved. The input gate and the candidate cell determine how much new information about the time step is added to the cell state. The contribution of the time step t to the cell state is nearly nothing if the second item in Equation (5) is equal to 0. The hidden states are updated jointly by the cell state and the output gate
where Ot denotes the output gate which is computed by
Compared with the traditional RNN, the LSTM solved well long-term dependency issues by the cell state conveying memory. To capture both directional dependencies between words, the bidirectional LSTM was used here. Due to its efficiency and effectiveness in sequence analysis, the LSTM has been widely applied to the N6-methyladenosine prediction (Chen et al., 2022), speech recognition (Sak et al., 2015), continuous B-cell epitope prediction (Saha and Raghava, 2006), N4-Acetylcytidine prediction (Zhang et al., 2022), lysine succinylation identification (Huang et al., 2021), sentiment analysis (Arras et al., 2017), and action recognition (Du et al., 2015).
Capsule network
The capsule network is a newly developed neural network in 2017 (Sabour et al., 2017). The capsule network is different from the conventional neural network. The basic unit of the capsule network is capsules which are defined as a set of neurons, while the latter consists of neurons. The neuron is generally a scalar value that represents a single pattern, while the capsules are a multi-dimensional vector, being able to represent multi-patterns. In addition, the capsule network is capable of capturing links between different local properties (Jia and Meng, 2016; Xi et al., 2017), which the convolution neural network (Shin et al., 2016) fail to discover. At the heart of the capsule network lies the dynamic routing as illustrated in Figure 3. vi was assumed to be the capsules in the layer L, whose prediction vectors are defined by
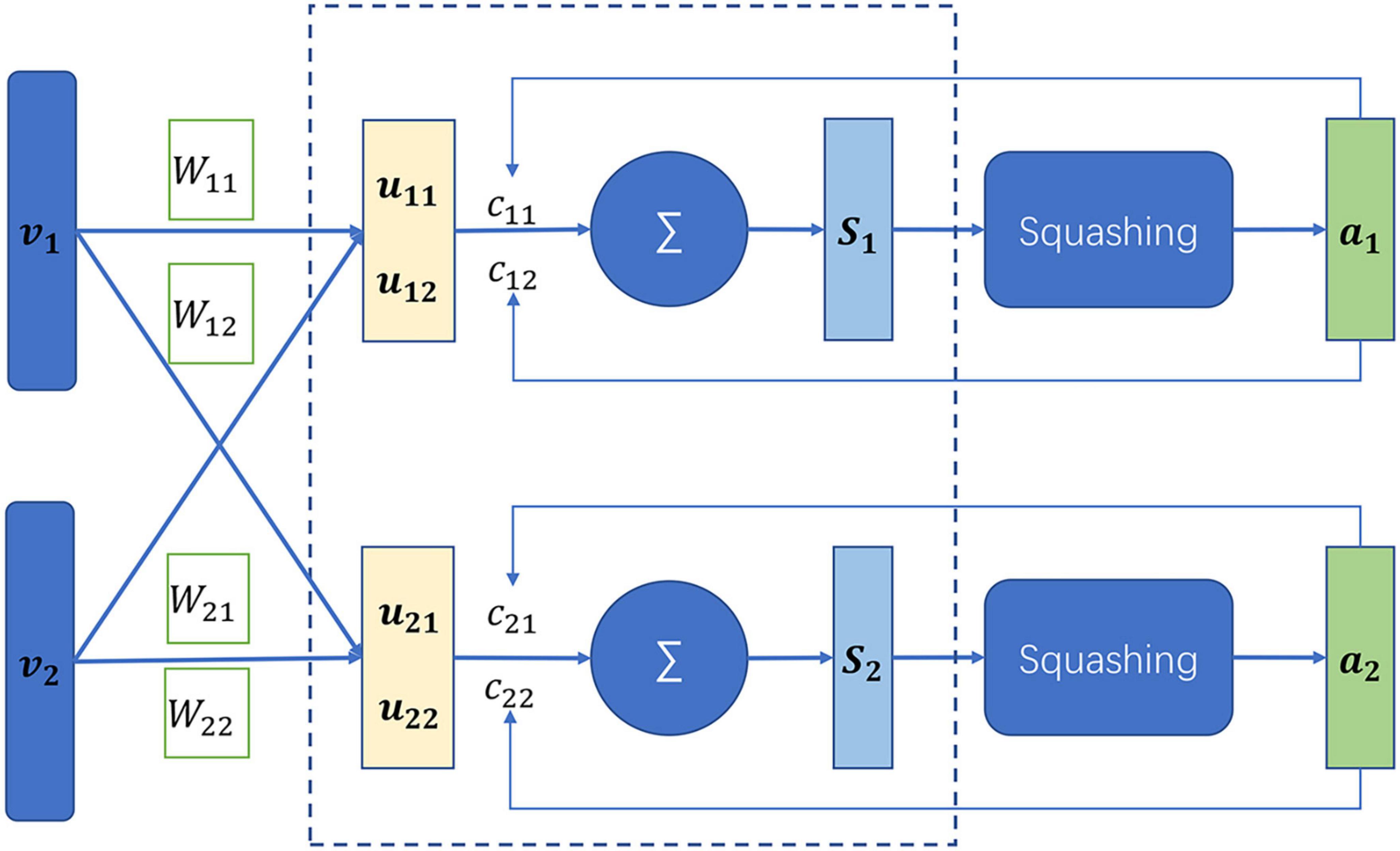
Figure 3. Illustration of dynamic routing in the capsule network (Lee, 2018).
where Wij is a learnable matrix. The capsule sj in the layer L+1 denotes a weighted sum over the prediction vectors, which is computed by
where cij is the coupling coefficient. The output of the capsule sj is further activated by a non-linear “squashing” function so that short vectors get shrunk to almost zero length and long vectors get shrunk to a length slightly below 1.
The coupling coefficient represents the probability of two capsules to the couple. The more consistent the two capsules, the large the coupling coefficient. The coupling coefficient is initialized as the log prior probabilities that the capsule j was coupled to the capsule i.
The prior probabilities are updated by the dynamic routing algorithm
The dynamic routing algorithm is to iterate the Equations (9) to (12).
Metrics
For binary classification, there are four common metrics: sensitivity (Sn), specificity (Sp), accuracy (Acc), and Matthews correlation coefficient (MCC), which are defined by
where TP and TN are the numbers of correctly predicted positive and negative samples, respectively, as well as FP and FN are the numbers of wrongly predicted positive and negative samples, respectively. In addition, we also employed the receiver operating characteristic (ROC) to evaluate performances. The area under the ROC curve (AUC) lies between 0 and 1. The more the AUC, the better the performance.
Results
There are two state-of-the-art methods for predicting TFs. One is the deep learning-based method by Li et al. (2022), which is called Li’s method, and another is the sequence feature-based method by Liu et al. (2020), which is called Liu’s method. To examine the Capsule_TF for efficiency and effectiveness in identifying TFs, we compared it with these two methods by the independent test. As shown in Table 1, the Capsule_TF is completely superior to the two methods. The Capsule_TF increased the Sn by 0.0283 over Li’s and even 0.1132 over Liu’s. The Capsule_TF increased MCC by 0.0386 over Li’s and even 0.1044 over Liu’s.
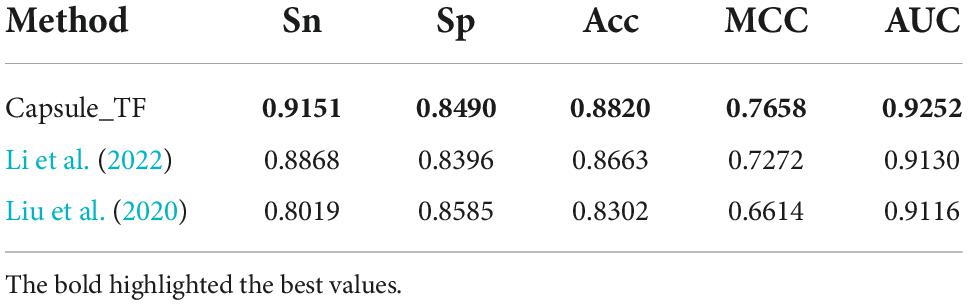
Table 1. Comparison with two states of the art methods in the independent test.
Discussion
Effect of position
The length of amino acid sequences varies with TFs. The longest reached 4,834 amino acid residues, the shortest is only 51 residues, and each TFs have an average of 536 residues. It is compulsory that the input is of the unified length in the machine learning algorithm. We investigated the effects of the number of amino acid residues at different positions on discriminating TFs from non-TFs. We chose 500 amino acid residues at the start, at the middle, and the end, respectively. As shown in Table 2, their predictive performances are approximately equivalent, meaning that positions have little effect. A potential reason is that 500 amino acid residues might contain sufficient information about TFs.
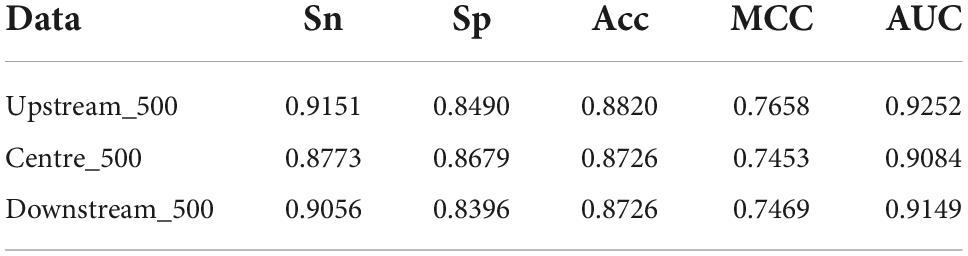
Table 2. Predictive performance of amino acid residues from different positions.
Contribution of capsule network
In comparison with Li’s method, the remarkable characteristic of the Capsule_TF is to utilize the capsule network. In order to investigate the contribution of the capsule network to classifying TFs, we removed it. The predictive performance after excluding the capsule network is listed in Table 3. Obviously, all metrics except Sp. decreased precipitously. Sn decreased from 0.9151 to 0.6320, Acc from 0.8820 to 0.7594, MCC from 0.7658 to 0.5365, and AUC from 0.9252 to 0.8120. The results indicated that the capsule network contributed much to identifying TFs.
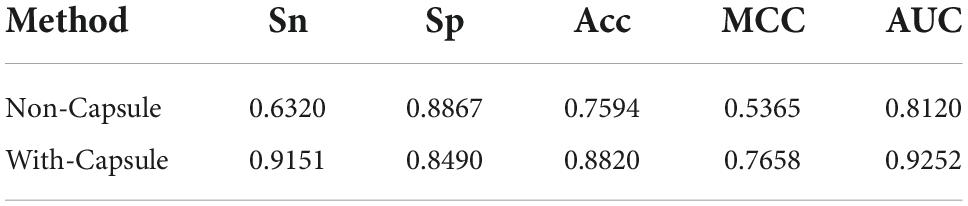
Table 3. Predictive performance of the method without capsule network.
Comparison with feature-based methods
The discriminative features provide a potential explanation to distinguish between both classes of samples. We compared three frequently used property-based features with the capsule network-based features. Three property-based features are PKx, relative amino acid propensity (RAA), and physicochemical characteristics (Li et al., 2008, 2021; Zhang et al., 2019). The output of the capsule layer was considered as the capsule network-based feature. Figure 4 visualizes the first two components of four types of features. The first two components were computed by PCA (Yang et al., 2004). Obviously, the first two components of the capsule network-based features are more discriminative than those of the other three types of features. We used the SVM (Noble, 2006) to compare the discriminative abilities of these features. As shown in Table 4, the capsule network-based feature is superior to the three property-based features. We also compared the logistic regression and LDA with the Capsule_TF. As listed in Tables 5, 6, the Capsule_TF is superior to the logistic regression and the LDA, and the capsule network-based features are superior to the conventional representations.
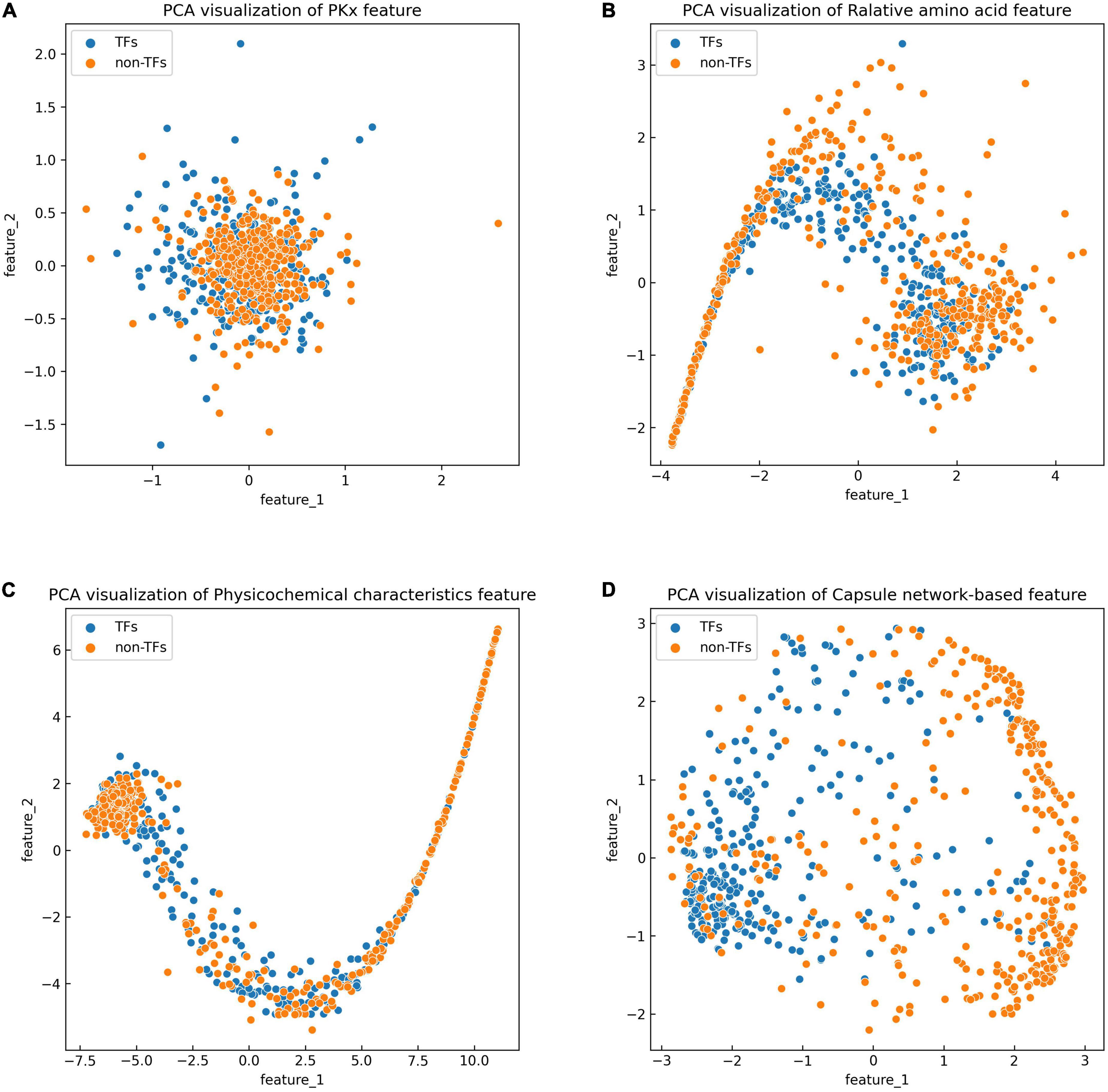
Figure 4. Principal component analysis (PCA) visualization about the different features: (A) PKx, (B) relative amino acid propensity, (C) physicochemical characteristics, and (D) capsule network-based features. As seen in this image, orange represents non-TFs and blue represents TFs.
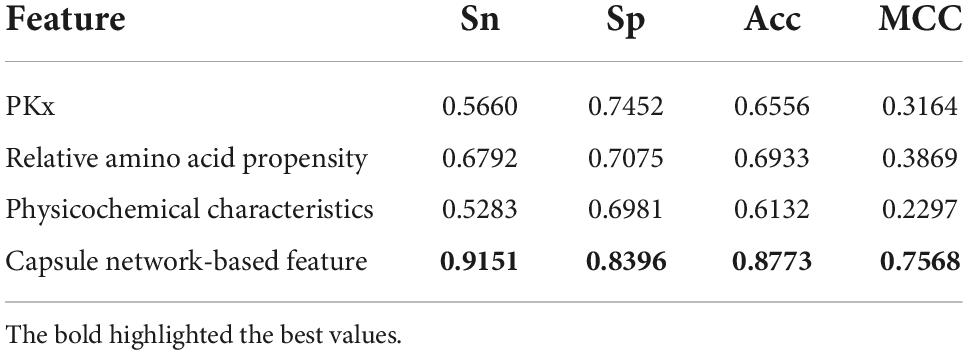
Table 4. Performance comparison across different features by SVM.
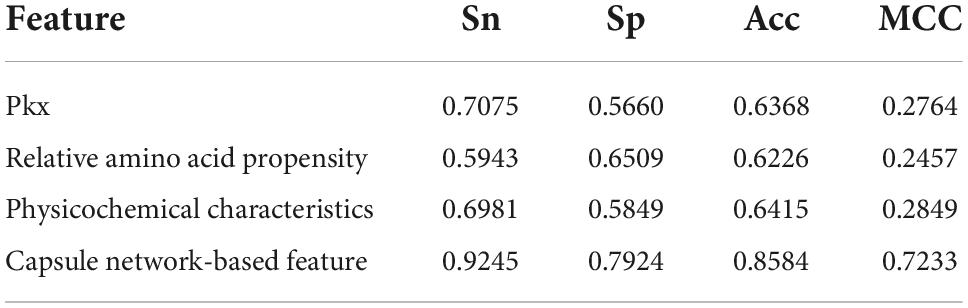
Table 5. Performance comparison across different features by logistic regression.
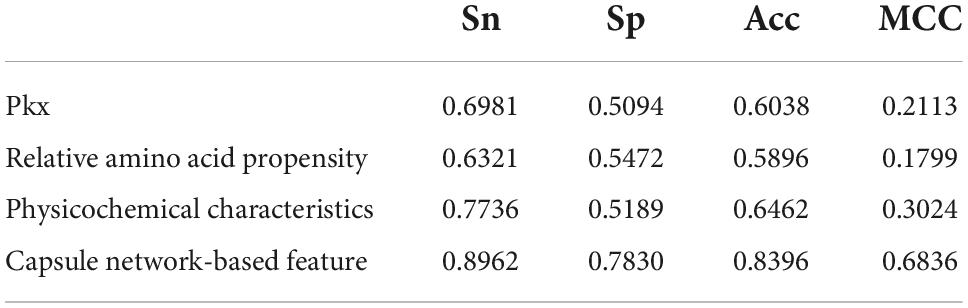
Table 6. Performance comparison across different features by linear discriminant analysis (LDA).
The previous results indicated that the Capsule_TF outperformed two state-of-the-art methods: Li’s method (Li et al., 2022) and Liu’s method (Liu et al., 2020). Li’s method (Li et al., 2022) is a Bi-LSTM-based method, while Capsule_TF not only employed Bi-LSTM but also utilized a capsule network. The inclusion of a capsule network effectively promoted the representation of protein sequences of TFs. The ablation experiments validated the contribution of the capsule network to the identification of TFs (Table 3). Liu’s method (Liu et al., 2020) is feature-based. We compared features extracted by Capsule_TF with traditional sequence property-based features. As shown in Figure 4 and Table 4, the capsule network-based feature is more discriminative than the traditional sequence property-based feature. Despite the Capsule_TF obtaining superior performances over the state-of-the-art methods, there were some limitations that need to be improved in the feature. First, the consumption time in dynamic routing is very large. Therefore, Capsule_TF is not suitable to deal with large-scale datasets. Second, the interpretability of Capsule_TF needs to be improved.
Web application
We realized the presented method into a web application which is freely available.2 The web application is based on the Django framework and utilized python and Tensorflow. The web application is very easy for users to use. The first thing is for the user to upload the predicted protein sequences in the FASTA format to the textbox or the file to the web. Clicking the “submit” button, users will obtain the results. The consuming time is directly proportional to the number of protein sequences. In addition, users could download the training and testing dataset in the experiments.
Conclusion
The TFs are very influential in transcription regulation. It is a challenging task to accurately recognize TFs at present. We presented a capsule network-based method for identifying TFs, which outperformed the state-of-the-art methods in the experiments. The presented method benefits from the inclusion of a capsule network, which captures a more informative representation than the property-based method. We also developed a web application that facilitated the detection of TFs. The method and the web application are helpful to identify TFs and to further explore their roles. The TFs play typically regulating roles in gene expression by binding to short DNA sequences. The roles of TFs depend on their binding to DNA sequences. In the future, we hope to create an effective and efficient method to recognize such binding and interpret its mechanism from the semantics of both protein and DNA sequences.
Data availability statement
The datasets presented in this study can be found in online repositories at http://www.biolscience.cn/Capsule_TF/.
Author contributions
PZ: data curation, methodology, software, investigation, and writing. YQ: validation. XL, YL, and YY: conceptualization and writing. GH: conceptualization, funding acquisition, supervision, and writing – reviewing and editing. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (62272310 and 62162025), the Hunan Province Natural Science Foundation of China (2022JJ50177 and 2020JJ4034), the Scientific Research Fund of Hunan Provincial Education Department (21A0466 and 19A215), and the Shaoyang University Innovation Foundation for Postgraduate (CX2021SY052).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
Arras, L., Montavon, G., Müller, K.-R., and Samek, W. (2017). Explaining recurrent neural network predictions in sentiment analysis. arXiv [preprint]. doi: 10.48550/arXiv.1706.07206
Buckman, J., Roy, A., Raffel, C., and Goodfellow, I. (2018). “Thermometer encoding: one hot way to resist adversarial examples,” in Proceeding of the international conference on learning representations.
Bushweller, J. H. (2019). Targeting transcription factors in cancer—from undruggable to reality. Nat. Rev. Cancer 19, 611–624. doi: 10.1038/s41568-019-0196-7
Chen, J., Zou, Q., and Li, J. (2022). DeepM6ASeq-EL: prediction of human N6-methyladenosine (m6A) sites with LSTM and ensemble learning. Front. Comput. Sci. 16:1–7. doi: 10.1007/s11704-020-0180-0
Ding, H., and Li, D. (2015). Identification of mitochondrial proteins of malaria parasite using analysis of variance. Amino Acids 47, 329–333. doi: 10.1007/s00726-014-1862-4
Du, Y., Wang, W., and Wang, L. (2015). “Hierarchical recurrent neural network for skeleton based action recognition,” in Proceedings of the IEEE conference on computer vision and pattern recognition, 1110–1118.
Graves, A., and Schmidhuber, J. (2005). Framewise phoneme classification with bidirectional LSTM and other neural network architectures. Neural Networks 18, 602–610. doi: 10.1016/j.neunet.2005.06.042
Hochreiter, S., and Schmidhuber, J. (1997). Long short-term memory. Neural Comput. 9, 1735–1780. doi: 10.1162/neco.1997.9.8.1735
Huang, G., Shen, Q., Zhang, G., Wang, P., and Yu, Z. G. (2021). LSTMCNNsucc: a bidirectional LSTM and CNN-based deep learning method for predicting lysine succinylation sites. BioMed Res. Int. 2021, 112. doi: 10.1155/2021/9923112
Huang, Y., Niu, B., Gao, Y., Fu, L., and Li, W. (2010). CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682. doi: 10.1093/bioinformatics/btq003
Jia, X., and Meng, M. Q.-H. (2016). “A deep convolutional neural network for bleeding detection in wireless capsule endoscopy images,” in Proceeding of the 2016 38th annual international conference of the IEEE engineering in medicine and biology society (EMBC), (IEEE), 639–642.
Karin, M. (1990). Too many transcription factors: positive and negative interactions. New Biol. 2, 126–131.
Latchman, D. S. (1997). Transcription factors: an overview. Int. J. Biochem. Cell Biol. 29, 1305–1312. doi: 10.1016/S1357-2725(97)00085-X
Lee, H. Y. (2018). Capsule. Available online at: https://www.bilibili.com/video/av16583439/?from=search&seid=4942786181857065642
Li, H., Gong, Y., Liu, Y., Lin, H., and Wang, G. (2022). Detection of transcription factors binding to methylated DNA by deep recurrent neural network. Briefings Bioinform. 23:bbab533. doi: 10.1093/bib/bbab533
Li, N., Sun, Z., and Jiang, F. (2008). Prediction of protein-protein binding site by using core interface residue and support vector machine. BMC Bioinform. 9:1–13. doi: 10.1186/1471-2105-9-553
Li, Y., Golding, G. B., and Ilie, L. (2021). DELPHI: accurate deep ensemble model for protein interaction sites prediction. Bioinformatics 37, 896–904. doi: 10.1093/bioinformatics/btaa750
Liu, M.-L., Su, W., Wang, J.-S., Yang, Y.-H., Yang, H., and Lin, H. (2020). Predicting preference of transcription factors for methylated DNA using sequence information. Mol. Ther. Nucleic Acids 22, 1043–1050. doi: 10.1016/j.omtn.2020.07.035
Nikolov, D., and Burley, S. (1997). RNA polymerase II transcription initiation: a structural view. Proc. Natl. Acad. Sci.U.S.A. 94, 15–22. doi: 10.1073/pnas.94.1.15
Nimwegen, E. (2006). Scaling laws in the functional content of genomes. Trends Genet 19, 236–253. doi: 10.1007/0-387-33916-7_14
Noble, W. S. (2006). What is a support vector machine? Nat. Biotechnol. 24, 1565–1567. doi: 10.1038/nbt1206-1565
Olah, C. (2015). Understanding LSTM networks. Available online at: https://colah.github.io/posts/2015-08-Understanding-LSTMs/ (accessed October 11, 2022).
Pawson, T. (1993). Signal transduction—a conserved pathway from the membrane to the nucleus. Dev. Genet. 14, 333–338. doi: 10.1002/dvg.1020140502
Riaño-Pachón, D. M., Ruzicic, S., Dreyer, I., and Mueller-Roeber, B. (2007). PlnTFDB: an integrative plant transcription factor database. BMC Bioinform. 8:1–10. doi: 10.1186/1471-2105-8-42
Rockel, S., Geertz, M., and Maerkl, S. J. (2012). MITOMI: a microfluidic platform for in vitro characterization of transcription factor–DNA interaction. Methods Mol. Biol. 786, 97–114. doi: 10.1007/978-1-61779-292-2_6
Roeder, R. G. (1996). The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem. Sci. 21, 327–335. doi: 10.1016/S0968-0004(96)10050-5
Rong, X. (2014). word2vec parameter learning explained. arXiv [preprint]. doi: 10.48550/arXiv.1411.2738
Roulet, E., Busso, S., Camargo, A. A., Simpson, A. J., Mermod, N., and Bucher, P. (2002). High-throughput SELEX–SAGE method for quantitative modeling of transcription-factor binding sites. Nat. Biotechnol. 20, 831–835. doi: 10.1038/nbt718
Sabour, S., Frosst, N., and Hinton, G. E. (2017). Dynamic routing between capsules. Adv. Neural Inform. Proc. Syst. 2017:30.
Saha, S., and Raghava, G. P. S. (2006). Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct Function Bioinform. 65, 40–48. doi: 10.1002/prot.21078
Sak, H., Senior, A., Rao, K., and Beaufays, F. (2015). Fast and accurate recurrent neural network acoustic models for speech recognition. arXiv [preprint]. doi: 10.48550/arXiv.1507.06947
Shamovsky, I., and Nudler, E. (2008). New insights into the mechanism of heat shock response activation. Cell. Mol. Life Sci. 65, 855–861. doi: 10.1007/s00018-008-7458-y
Sherstinsky, A. (2020). Fundamentals of recurrent neural network (RNN) and long short-term memory (LSTM) network. Phys. D Nonlinear Phenomena 404:132306. doi: 10.1016/j.physd.2019.132306
Shin, H. C., Roth, H. R., Gao, M., Lu, L., Xu, Z., Nogues, I., et al. (2016). Deep convolutional neural networks for computer-aided detection: CNN architectures, dataset characteristics and transfer learning. IEEE Trans. Med. Imaging 35, 1285–1298. doi: 10.1109/TMI.2016.2528162
Tan, J. X., Li, S. H., Zhang, Z. M., Chen, C. X., Chen, W., Tang, H., et al. (2019). Identification of hormone binding proteins based on machine learning methods. Math Biosci. Eng. 16, 2466–2480. doi: 10.3934/mbe.2019123
Wang, G., Luo, X., Wang, J., Wan, J., Xia, S., Zhu, H., et al. (2018). MeDReaders: a database for transcription factors that bind to methylated DNA. Nucleic Acids Res. 46, D146–D151. doi: 10.1093/nar/gkx1096
Wheaton, K., Atadja, P., and Riabowol, K. (1996). Regulation of transcription factor activity during cellular aging. Biochem. Cell Biol. 74, 523–534. doi: 10.1139/o96-056
Wingender, E., Dietze, P., Karas, H., and Knüppel, R. (1996). TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res. 24, 238–241. doi: 10.1093/nar/24.1.238
Xi, E., Bing, S., and Jin, Y. (2017). Capsule network performance on complex data. arXiv [preprint]. doi: 10.48550/arXiv.1712.03480
Yang, J., Zhang, D., Frangi, A. F., and Yang, J. Y. (2004). Two-dimensional PCA: a new approach to appearance-based face representation and recognition. IEEE Trans. Pattern Analy. Mach. Intelli. 26, 131–137. doi: 10.1109/tpami.2004.1261097
Yashiro, T., Hara, M., Ogawa, H., Okumura, K., and Nishiyama, C. (2016). Critical role of transcription factor PU.1 in the function of the OX40L/TNFSF4 promoter in dendritic cells. Sci. Rep. 6, 1–11. doi: 10.1038/srep34825
Yin, Y., Morgunova, E., Jolma, A., Kaasinen, E., Sahu, B., Khund-Sayeed, S., et al. (2017). Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356:eaaj2239. doi: 10.1126/science.aaj2239
Zhang, G., Luo, W., Lyu, J., Yu, Z. G., and Huang, G. (2022). CNNLSTMac4CPred: a hybrid model for N4-acetylcytidine prediction. Int. Sci. Comput. Life Sci. 14, 439–451. doi: 10.1007/s12539-021-00500-0
Zhang, J., Ma, Z., and Kurgan, L. (2019). Comprehensive review and empirical analysis of hallmarks of DNA-, RNA- and protein-binding residues in protein chains. Briefings Bioinform. 20, 1250–1268. doi: 10.1093/bib/bbx168
Zhang, Q., Liu, W., Zhang, H.-M., Xie, G.-Y., Miao, Y.-R., Xia, M., et al. (2020). hTFtarget: a comprehensive database for regulations of human transcription factors and their targets. Geno. Proteomics Bioinform. 18, 120–128. doi: 10.1016/j.gpb.2019.09.006
Zhu, Q. H., Guo, A. Y., Gao, G., Zhong, Y. F., Xu, M., Huang, M., et al. (2007). DPTF: a database of poplar transcription factors. Bioinformatics 23, 1307–1308. doi: 10.1093/bioinformatics/btm113
Keywords: transcription factors, capsule network, deep learning, LSTM, semantics
Citation: Zheng P, Qi Y, Li X, Liu Y, Yao Y and Huang G (2022) A capsule network-based method for identifying transcription factors. Front. Microbiol. 13:1048478. doi: 10.3389/fmicb.2022.1048478
Received: 19 September 2022; Accepted: 26 October 2022;
Published: 06 December 2022.
Edited by:
Hao Lin, University of Electronic Science and Technology of China, ChinaReviewed by:
Wen Zhang, Huazhong Agricultural University, ChinaJiangning Song, Monash University, Australia
Copyright © 2022 Zheng, Qi, Li, Liu, Yao and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guohua Huang, Z3VvaHVhaGhuQDE2My5jb20=