 Siwei Liu1†
Siwei Liu1† Shan Yu
Shan Yu Hailin Yang
Hailin Yang Yuanyuan Li
Yuanyuan Li Hailong Lu
Hailong Lu- 1School of Earth and Space Sciences, Peking University, Beijing, China
- 2Beijing International Center for Gas Hydrate, School of Earth and Space Sciences, Peking University, Beijing, China
- 3National Research Center for Geoanalysis, Beijing, China
- 4Guangzhou Marine Geological Survey, Guangzhou, China
Biogenic and thermogenic gas are two major contributors to gas hydrate formation. Methane hydrates from both origins may have critical impacts on the ecological properties of marine sediments. However, research on microbial diversity in thermogenic hydrate-containing sediments is limited. This study examined the prokaryotic diversity and distributions along a sediment core with a vertical distribution of thermogenic gas hydrates with different occurrences obtained from the Qiongdongnan Basin by Illumina sequencing of 16S rRNA genes as well as molecular and geochemical techniques. Here, we show that gas hydrate occurrence has substantial impacts on both microbial diversity and community composition. Compared to the hydrate-free zone, distinct microbiomes with significantly higher abundance and lower diversity were observed within the gas hydrate-containing layers. Gammaproteobacteria and Actinobacterota dominated the bacterial taxa in all collected samples, while archaeal communities shifted sharply along the vertical profile of sediment layers. A notable stratified distribution of anaerobic methanotrophs shaped by both geophysical and geochemical parameters was also determined. In addition, the hydrate-free zone hosted a large number of rare taxa that might perform a fermentative breakdown of proteins in the deep biosphere and probably respond to the hydrate formation.
Introduction
Natural gas hydrates, mainly occurring in continental slope sediments under relatively high pressure and low temperature conditions, are crystalline solids containing low molecular gases in cage like structures constructed by water molecules, such as methane, ethane, CO2, etc. (Macdonald et al., 1994). Natural gas hydrates constitute an abundant form of carbon on Earth (Kvenvolden, 1993) and the most significant intermediate sink for the greenhouse gas methane, which appears to be dynamically stored in or released from hydrate reservoirs (Dickens, 2001b; Dickens, 2001a; Kvenvolden, 2002; Boetius and Wenzhöfer, 2013).
In nature, methane gas can be generated by two dominant processes, either “biogenic” or “thermogenic” (Whiticar, 1999). Biogenic methane is produced by microbial communities as a part of anaerobic respiration, whereas thermogenic gases are generated from thermocatalytic degradation of organic compounds at elevated temperatures (Schoell, 1988; Wuebbles and Hayhoe, 2002). The biogenic formation of methane is conducted by methanogenic archaea via three primary pathways: hydrogenotrophic, acetoclastic, and methylotrophic methanogenesis (Liu and Whitman, 2008; Thauer et al., 2008; Mayumi et al., 2016). In marine sediment, the majority of methane reservoir is consumed by microorganisms via the anaerobic oxidation of methane (AOM) mainly coupled with the reduction of sulfate, though iron, manganese, nitrate, and nitrite can also be used as electron acceptors in AOM processes (Barnes and Goldberg, 1976; Raghoebarsing et al., 2006; Beal et al., 2009; Ettwig et al., 2010; Yang et al., 2021). Typically, AOM happens in sulfate–methane transition zone (SMTZ) and is performed by microbial consortia where archaeal anaerobic methanotrophs (ANMEs) conduct reverse methanogenesis in association with bacterial partners (Barnes and Goldberg, 1976; Boetius et al., 2000; Reeburgh, 2007).
Relationships between the microbial structure and distribution and hydrated methane in deep marine sediments have received worldwide attention. Extensive studies have been conducted to characterize microbial diversity, activity and importance in methane hydrate-bearing subseafloor sediments, such as those in the Cascadia Margin (Cragg et al., 1995; Bidle et al., 1999; Lanoil et al., 2001, 2005; Marchesi et al., 2001; Mills et al., 2003, 2005; Knittel et al., 2005; Inagaki et al., 2006; Nunoura et al., 2008), Nankai Trough (Reed et al., 2002; Kormas et al., 2003; Newberry et al., 2004; Mills et al., 2012; Katayama et al., 2016, 2022), East Japan Sea (Yanagawa et al., 2014), Andaman Sea (Briggs et al., 2012), Ulleung Basin (Jeong et al., 2010; Lee et al., 2013; Ryu et al., 2013; Cho et al., 2017), Arctic (Carrier et al., 2020) and Shenhu area of the South China Sea (Liao et al., 2009; Lin et al., 2014; Jiao et al., 2015; Cui et al., 2019, 2020). A few of these studies were performed to compare the microbial communities in marine sediments with or without gas hydrate (Inagaki et al., 2006; Yanagawa et al., 2014; Jiao et al., 2015; Cui et al., 2020). Nevertheless, due to the wide distribution of biogenic hydrates, and the important role taken by microbes in methane metabolization, nearly all previous research had focused on the marine sediments containing biogenic methane, trying to figure out the microbiological processes that cycle the carbon in sediments rich in hydrates (Inagaki et al., 2006; Yanagawa et al., 2014; Jiao et al., 2015). It should not be neglected that the formation or dissociation of gas hydrate, even if they were not biogenic produced, could alter the geophysical and geochemical properties of the subseafloor sediments and further impact the microbial communities therein (Tomaru et al., 2004; Lee et al., 2008). In addition, heavier hydrocarbons (C2+) prevalent in the thermogenic hydrate-related gases might also influence the microbial distribution and metabolic potentials (Dong et al., 2019, 2020).
So far, it is still unclear the compositions and distributed characteristics of microbial communities inhabiting thermogenic gas hydrate-associated marine sediments, and whether they could also be distinguished from those found in the typical marine sediments without gas hydrates, as reported in the marine sedimentary environment with biogenic hydrates.
The Qiongdongnan Basin (QDNB) is one of the marine gas hydrate exploration targets in the South China Sea, where highly saturated gas hydrates were detected (Cui et al., 2019; Wei et al., 2019; Ye et al., 2019). Based on the carbon isotopic and field analysis of the composition of the hydrate-related gas, previous research has suggested that thermogenic gas might be the primary source of the hydrates in QDNB (Liang et al., 2019; Ye et al., 2019; Geng et al., 2021). This study aimed at characterizing the abundance and diversity of microorganisms in the marine sediments associated with thermogenic gas hydrates and determining spatial variations of the community structure along with hydrates distribution. We thus surveyed the microbial communities in a sediment core with the presence of multi-layer hydrates collected from the stable gas hydrate zone of QDNB, using molecular, geochemical techniques and statistical community analyses. We report that the microbial communities in thermogenic hydrate-bearing marine sediments of a QDNB core can be statistically distinguished from typical sediments with a significantly higher abundance and lower diversity, a remarkable stratified distribution of archaea along with various hydrate occurrences is also discovered. And it is speculated that a large population of rare taxa performing fermentative protein degradation metabolism was reduced by the gas hydrates formation.
Materials and methods
Site description and sample collection
The study region is in the Qiongdongnan Basin, which is one of the potential gas hydrate-bearing basins on the northwestern continental slope of the South China Sea (Figure 1). The main components of the sediments are hemipelagic clayey silt and silty clay (Meng et al., 2021). Geophysical and geological characteristics related to fluid migration and gas hydrate deposits including mud diapirs, bottom simulating reflectors and gas chimneys are widespread in the area (Zhang et al., 2019). Previous studies had suggested that thermogenic gas might be the predominant source of hydrates at some sites in QDNB according to the carbon isotopic composition analyses (Huang et al., 2016; Liang et al., 2019; Lai et al., 2022; Ren et al., 2022), low C1/C2+ (3–68) values of the hydrate-bound gases also indicated that thermogenic gas is the primary driver for the gas hydrate formation in QDNB (Ye et al., 2019).
Figure 1. Location map of the Qiongdongnan Basin (QDNB), South China Sea. The red point refers to the coring site W01.
Sediment core samples were obtained from the Qiongdongnan area at Site W01 which was drilled and cored to a depth of 176.7 m below seafloor (mbsf) in a water depth of 1513.89 m by pressure coring tools with Ball valve (PCTB) during the GMGS6 drilling expedition conducted in 2019 (Meng et al., 2021; Figure 1). Site W01 was positioned above deep subsurface gas chimneys, suggesting upstream gas fluxes within sediments (Meng et al., 2021). According to previous reports, the majority of natural gas in W01 is methane, supplemented by C2-C5 gases (Huang et al., 2016).
Detailed geochemical profiles which will be published elsewhere show that Core W01 exhibited significant gas hydrate indicators, and the pore-water sulfate was depleted throughout the core, indicating the sulfate–methane transition depth is located at 0 mbsf. Within the Core W01, subsamples used in this study were collected from three depth intervals: two gas hydrates bearing layers of 4.5–27.5 mbsf and 41–69 mbsf, respectively, and the 70–176 mbsf depth interval which was identified as a gas hydrate-free layer according to the drilling results. Based on the gas hydrate morphology classification method created by Holland et al., type II gas hydrates (fracture-filling gas hydrates exhibit vein, massive and nodular morphologies) were discovered in the 4.5–27.5 mbsf interval, while type I gas hydrates (disseminated pore-filling gas hydrate) were developed in the 41–69 mbsf (Holland et al., 2008; Meng et al., 2021). The occurrences of gas hydrates also seem to be affected by compositional changes in the hosting sediment, thus the hydrates of the studied layers could be further differentiated into three subtypes (II-b, I-b and I-a) according to lithological properties (Bahk et al., 2013).
In this study, a total of nine subsamples were retrieved from Core W01 using 50-mL cut-end syringes, including six samples with three hydrate types (I-a, I-b, and II-b) and three samples within the deep gas hydrate-free zone. The detailed sample information was summarized in Table 1. Subsamples were stored onboard at −20°C, transported on dry ice to the laboratory at the end of the expedition, and stored at −80°C until further processing.
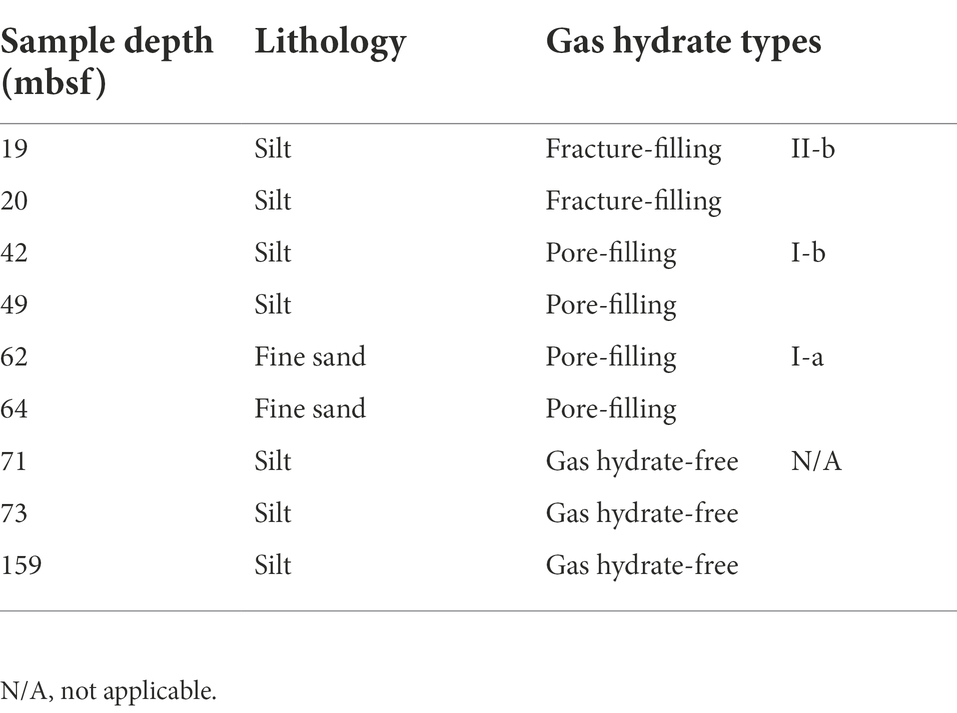
Table 1. Information of the nine retrieved samples from Core W01 of QDNB.
Geochemical analyses
The temperature was monitored by the inner T/P sensors during coring and processing. On board, pH measurements were made on pore water with a pH meter immediately after collection. Barium sulfate nephelometry and refractometry were used to quantify the pore water sulfate and salinity, respectively. The contents of anions (Cl−and Br−) were quantified using the standard method of ion chromatography (Metrohm 790 IC), with an analytical precision of 2%. The grain sizes of sediment samples were determined using a laser diffraction particle size analyzer (Mastersizer 3000, Malvern, United Kingdom) as described previously (Luo et al., 2012). The total organic carbon (TOC) of the sediments was measured by using a carbon/sulfur determinator (Series CS 744, Leco) in National Research Center for Geoanalysis, Beijing China. In detail, 200–300 mg samples were combusted at ~1,100°C inside the oven of the CS744 analyzer and the amount of carbon dioxide (CO2) generated was measured by an infrared cell. Samples were analyzed in duplicate to QA/QC for the homogeneity of the aliquots taken and analytical precision (Baux et al., 2019). Sediment samples were analyzed for major element compositions using X-Ray Fluorescence (XRF, GeniusIF, Xenemetrix Ltd.) with a precision of better than 8%. The δ13C and δ18O values of the bulk carbonate phase of subsamples were measured using a stable isotope ratio mass spectrometer (IRMS, Thermo Delta V) with an automated carbonate preparation device (GasBench II) as previously described (Liu et al., 2022).
DNA extraction, PCR amplification, and Illumina MiSeq sequencing
For amplicon sequencing, microbial genomic DNA was extracted from 0.35 g of sediment using the FastDNA Spin Kit for Soil (MP Biomedicals) for polymerase chain reaction (PCR) amplification following the manufacturer’s instructions. For quantitative PCR (qPCR), microbial DNA was extracted and purified using the Natarajan et al. modified SDS-based method (Natarajan et al., 2016). The DNA concentrations were determined using a NanoDrop™ 2000 spectrophotometer (Thermo Fisher Scientific, United States).
The hypervariable V4 region of bacterial and archaeal 16S rRNA genes was amplified by PCR using a barcoded universal primer set Univ519F/Univ802R (Supplementary Table 2). PCR reaction mixtures contained 1 to 10 ng diluted DNA extracts, 1 × FastPfu buffer, 0.4 μmol/l of each primer, 250 μmol/l deoxynucleotides (dNTPs), 1 μl FastPfu polymerase (TransGen, China), and Mill-Q water added to a final volume of 50 μl. Thermal cycling was performed under the following conditions: initial denaturation at 95°C for 5 min, followed by 29 cycles of 94°C for 20 s, 50°C for 20 s, and 72°C for 25 s, and a final extension at 72°C for 10 min. A negative (no-template) control was used to exclude contamination. For each sample, amplicons from at least six independent PCR products were pooled and purified using EZNA Gel Extraction Kit (Omega Bio-Tek, United States) according to the manufacturer’s instructions and subjected to high-throughput sequencing using the Illumina MiSeq platform (Illumina Inc., United States) at Majorbio BioPharm Technology Co., Ltd., Shanghai China.
Quantification of the bacterial, archaeal 16S rRNA genes and mcrA gene
To quantify the abundance of bacteria and archaea domain as well as anaerobic methanotrophs in the sediment samples, SYBR-Green-based qPCR was conducted with extracted DNA, using published specific primer sets (Supplementary Table 2). Each reaction (20 μl) contained 1× KOD SYBR qPCR Mix (TOYOBO, Co. Ltd), 5–8 ng template DNA, and 0.8 μM of each forward and reverse primer. All qPCR reactions were carried out using a CFX Connect Real-Time System (Bio-Rad, CA, United States).
Standard curves were generated using 10-fold dilutions of the known amount of purified PCR products of the target gene fragments. Each reaction was performed in triplicate. Melting curves were analyzed to detect the presence of primer dimers. The assay conditions are listed in Supplementary Table 2.
Sequence processing
The 16S rRNA gene sequences were processed using Git for windows 2.28.0, R 4.2.0, Rstudio 1.4.1106, VSEARVH v2.15.2 (Edgar, 2010), and USEARCH v10.0.240 (Rognes et al., 2016). Specifically, raw sequence data were firstly checked by FastQC1 and processed as follows: joined paired-end reads and renamed by sample with the “-fastq_mergepairs” command, filtered low-quality reads (−fastx_filter command) after removing barcodes and primers by “-fastx_truncate,” removed redundant reads and singletons with the “-derep_fulllength” command. Thereafter, operational classification units (OTUs) were constructed using the “-cluster_otus” command at 97% sequence identity. OTUs were then mapped against the Silva 123 database (Quast et al., 2012) to remove sequences from chimera with the UCHIME algorithm in VSEARCH (−uchime_ref command). The taxonomy of the OTUs was classified with the Silva 123 database based on the sintax algorithm in VSEARCH (−sintax command) and the OTUs assigned to the chloroplasts and mitochondria were removed. Subsequently, the OTU table was obtained by USEARCH using the “fastx_getseqs” command.
Bioinformatic and statistical analysis
For alpha and beta diversity, samples were rarefied at 37,320 sequences using R package vegan (Quast et al., 2012) corresponding to the lowest number of sequences in one sample. α-diversity indices, such as the specie richness, Chao 1, ACE, Shannon, Simpson, and Goods coverage, were calculated using the “-alpha_div” command of USEARCH, and the rarefaction curve was depicted by the package ggplot2. Distance matrices of Unifrac, Bray_Curtis, Jaccard, Manhatten and Euclidean were calculated with the “-beta_div” command, after constructing evolutionary trees based on OTUs. Heat maps of dissimilarity indices of the prokaryotic communities were depicted by GraphPad Prism 8.0.2, and the microbial communities’ dissimilarities among sediment groups were tested by ANOSIM.
Principal component analysis (PCoA) plot based on weighted Unifrac distances and Unweighted Pair Group Method with Arithmetic Means (UPGMAs) Clustering were displayed by vegan package, stats package and ggplot2 package in R software. Venn plots were constructed using R package ggvenn. Significantly different OTUs were visualized with a Volcano plot using the ggplot2 package. The heatmap of differentially abundant microbial taxa between hydrate-bearing and-free sediment groups was constructed by R package pheatmap based on the t-test (BH-adjusted p < 0.05) at the order level after scale normalization. The analysis of the process and scripts mainly referred to the study of Yongxin Liu (Liu et al., 2021).
The microbial function profiles of Core W01 microbiota were predicted using PICRUSt (phylogenetic investigation of communities by reconstruction of unobserved states, conducted on the ehbio online platform)2 to normalize the OTU table for 16S rRNA gene copy number variations, and then impute the functional microbial content of each sample based on KEGG orthologs (KO) and pathways. The heat map was then constructed using the R package devtools.
The effects of environmental variables on sediment microbial orders were judged using distance-based redundancy analysis (dbRDA) with the R package vegan. The Variance Inflation Factor Analysis (VIF) was performed prior to the RDA analysis in order to eliminate collinearity between environmental components and enhance efficiency. ANOVA-like permutation testing of constrained ordinations was performed using the R package statis. The plot was then visualized using the Canoco program for Windows 5.0.
The co-occurrence network was constructed to explore the associations between methanotrophs and other microbial taxa by calculating in R using the WGCNA package (Langfelder and Horvath, 2008). After the identification of the potential positive correlations between microbial families with Pearson’s R > 0.8 and BH-FDR-corrected p < 0.001, only families of methanotrophs and their associated taxa were retained for visualization using the interactive platform Gephi 0.9.5 with undirected network and the Fruchterman-Reingold layout (Bastian et al., 2009).
Results
Sediment geochemical conditions
To understand the geochemical conditions where microbial communities occur, sediment samples from Core W01 were analyzed for total organic carbon (TOC), grain size, major elemental contents, stable carbon and oxygen isotopes composition of carbonate (δ13Ccarb and δ18Ocarb). TOC values greater than 1% were detected in the top three sediment samples collected at 19, 20, 42 mbsf, while other deeper samples were with a lower TOC below 0.5% (Figure 2A). One sand layer was observed in Core W01 according to the results of particle size distribution: two sediment samples at 62 and 64 mbsf were fine sands with average grain sizes of 77.6 and 80.9 μm, respectively, while other sediment samples were composed of silt with smaller average particle sizes ranging from 7.43 to 13.18 μm (Table 1; Figure 2B; Supplementary Table 1). The lowest TOC content, chlorinity, Br concentration, salinity, and relative higher alkalinity were observed in these two samples (Supplementary Table 1), In addition, concentrations of Fe, Mn, Al, and K in the sandy samples at 62 and 64 mbsf were much lower while concentrations of Si and Ti exhibited higher values (Figure 2E), suggesting the geochemical anomaly of these two samples. Further, to confirm the origin of methane flux in the Site W01, we detected the carbon and oxygen isotope composition of carbonate in sediments which are affected by the percentage of AOM-driven carbonate. The results showed that the δ13C and δ18O values of carbonate in sediment (δ13Ccarb, δ18Ocarb) from Core W01 ranged from −2.50 to 1.42‰ and −6.64 to −1.05‰, respectively (Figure 2F; Supplementary Table 1), indicating a thermogenic origin of methane in the deep subsurface (Whiticar, 1999).
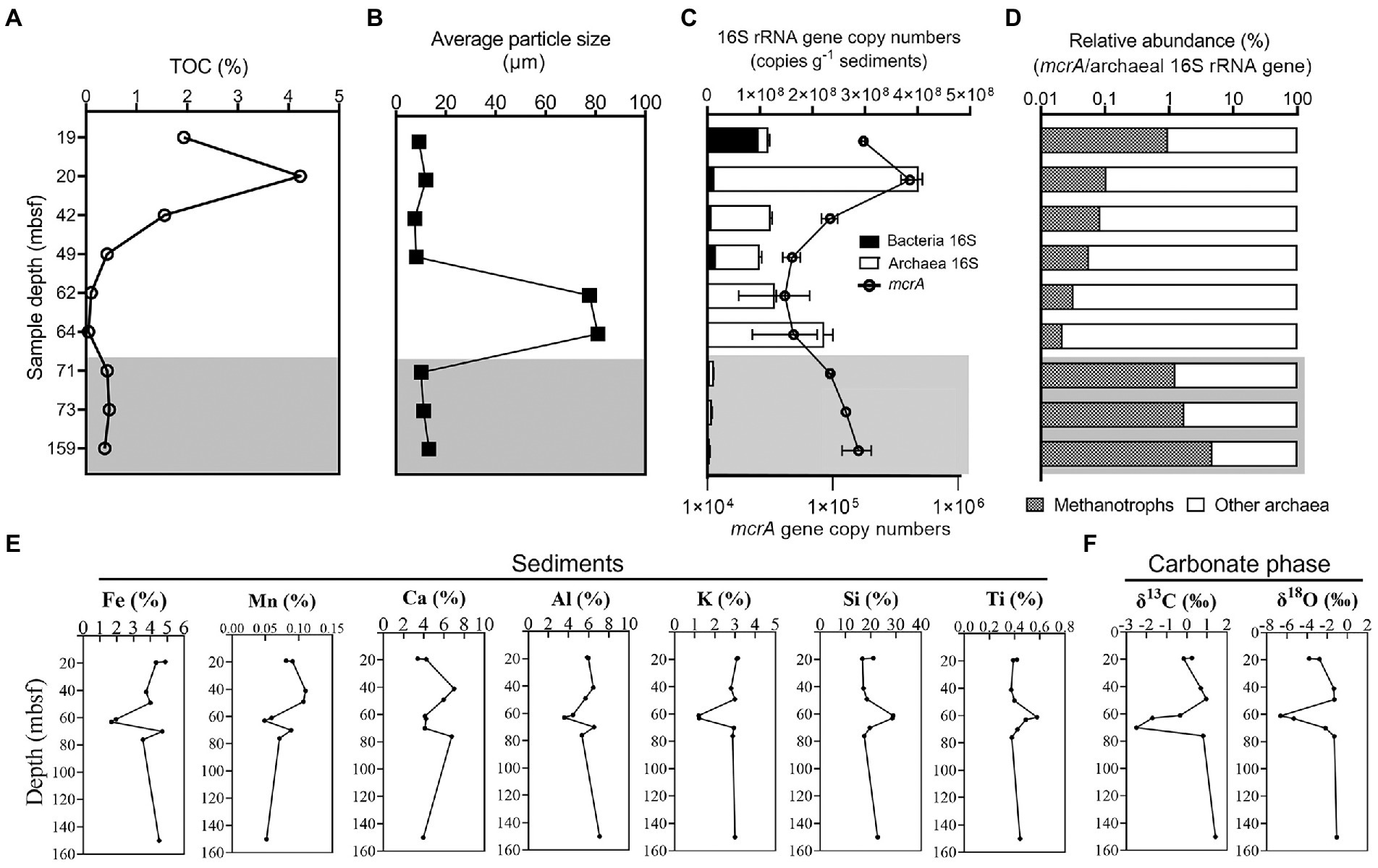
Figure 2. Depth profiles of TOC (A), average particle size (B), gene abundance of bacterial and archaeal 16S rRNA genes and mcrA (C), the ratio of mcrA gene copies in archaeal 16S rRNA gene copies (D), major elemental contents concentration (E) and stable carbon and oxygen isotopes composition of carbonate (F) of the sediment samples from Core W01. The gas hydrate-free zone is indicated by shaded area.
Microbial abundance
Bacterial, archaeal 16S rRNA gene, and mcrA copy numbers were estimated by qPCR to represent the abundances of bacteria, archaea, and anaerobic methanotrophs, respectively. The results showed that bacterial and archaeal 16S rRNA gene copy numbers ranged from 1.14 × 106 to 9.97 × 107 and from 3.36 × 106 to 3.88 × 108 copies g−1 wet weight sediment, respectively (Figure 2C). The abundance of the archaeal 16S rRNA gene was generally one order of magnitude greater than that of bacteria but in one instance, the uppermost sample at 19 mbsf, where bacteria comprised 84% of the total prokaryotic community. Pearson’s analysis showed the total abundance of bacterial and archaeal 16S rRNA genes, which reached its maximum in the sample retrieved at 20 mbsf and decreased sharply in the three deepest hydrate-free samples at 71, 73, and 159 mbsf, was positively correlated to the TOC (r = 0.74, p = 0.024; Supplementary Figure 1) of the sediments.
The number of gene copies of mcrA, a methanotrophic archaea-specific gene, was detected in all collected sediment samples, varied between 4.16 × 104 to 4.12 × 105 copies g−1 wet weight sediment (Figure 2C). The proportion of methane metabolizing group in total archaeal cells of each sediment sample, estimated by the ratio of copy number of mcrA to archaeal 16S rRNA genes, decreased with depth in the upper hydrate-bearing sediment samples, from 0.96 and 0.02%, while raised in the three deep non-gas hydrate sediment samples retrieved below the gas hydrate zone, from 1.27 to 4.79% (Figure 2D).
Alpha-diversity analysis
Nine sediment samples covering the shallower hydrate-bearing layers to the deep hydrate-free zone of the Core W01 (for details, Table 1) were used for DNA isolation and prokaryotic diversity analysis. A total of 496,887 high-quality sequences were generated by MiSeq sequencing for all sediment samples (median = 59,094 sequences, ranging from 37,320 to 67,775 sequences). At a threshold of 97% sequence identity, 5,505 OTUs were identified (median = 674 OTUs, ranging from 182 to 3,088 OTUs, which comprises 5,388 bacterial OTUs and 117 archaeal OTUs, respectively (Supplementary Table 3). An overall dominance of bacterial sequences and high absolute numbers of bacteria OTUs were found throughout the core, accounting for 92.2–99.9% of the total sequence obtained from each sample. Species rarefaction curves all plateaued under the current sequencing depth (Figure 3A), suggesting that saturation in sequencing was achieved and those sequences covered all microbial species in the investigated W01 samples.
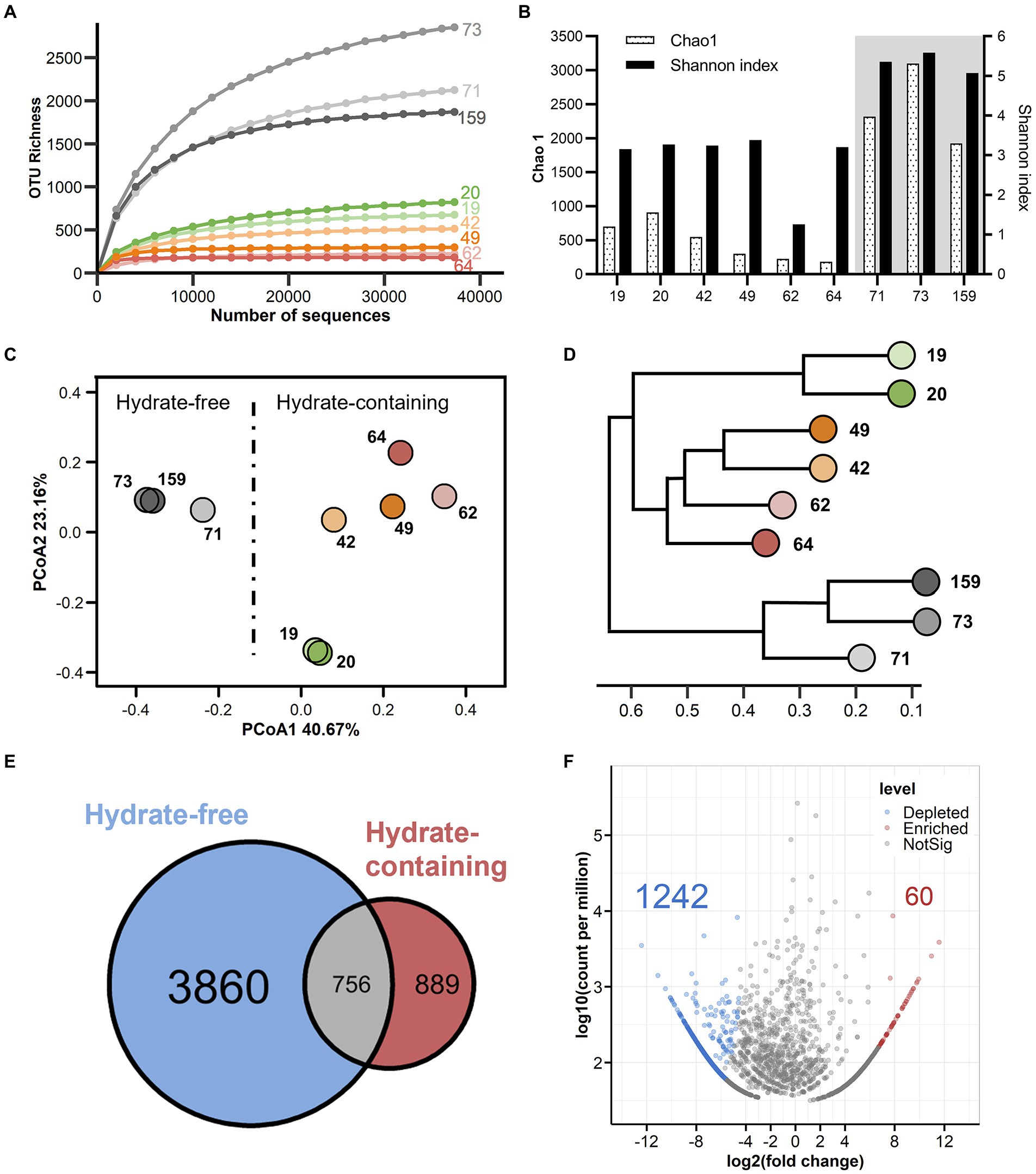
Figure 3. Alpha and beta diversity across W01 sediment column. (A) OTU-level rarefaction (observed species) curves. (B) Chao 1 estimated richness and Shannon index. The hydrate-free zone is indicated by shaded area. (C) Shift of microbial community structure in W01 sediment samples as visualized by Principal Co-ordinate Analysis (PCoA) based on the weighted Unifrac distance matrix. Each point represents the entire microbiological assemblage recovered from one sample; samples plotting closer to each other are more similar in microbial composition. (D) Dendrogram showing hierarchical clustering of the samples based on OTUs. (E) A Venn diagram displaying the degree of overlap of microbial OTUs between hydrate-free and hydrate-containing samples. (F) A Volcano plot illustrating OTUs that were significantly enriched (red) and depleted (blue) by hydrate-containing compared with hydrate-free samples as determined by differential abundance analysis. Enriched and depleted OTUs represent OTUs with two-fold higher or lower relative abundance (p < 0.05) in the hydrate-containing sediments, respectively.
A vertical variation in microbial diversity was observed in W01. Gas hydrate-bearing sediments were characterized by relatively low Chao1 diversity reducing along with the depth from 19 to 64 mbsf, while surprisingly, Chao1 indexes steepened in the three samples collected from the deep hydrate-free zone, from 71 to 159 mbsf. The lowest values were calculated in the two sandy samples retrieved at 62 and 64 mbsf. Shannon indices revealed the same trends as the Chao1 estimator (Figure 3B; Supplementary Table 4). In addition, Chao 1, Shannon index, OTU Richness, Inverse Simpson index, and Ace index were all significantly increased in the three deepest samples, suggesting an apparent higher prokaryotic diversity in the gas hydrate-free zone of the Core W01 (Supplementary Table 5).
Microbial composition and distribution
Bacterial community
The microbial community structure was obtained based on the 16S rRNA genes V4 hypervariable regions with universal 16S primers targeting both bacteria and archaea. We revealed highly diverse bacterial communities with up to 76 phyla in W01 sediments. In general, the recovered sequences were dominated by Proteobacteria (with α-and γ-subdivisions), Actinobacterota, JS1 (Atribacteria), Firmicutes, and Bacteroidota at all depths, which comprised 85.12% of the total sequences. Compared to the hydrate-free samples, JS1 lineage and Aerophobota were generally detected with greater abundance in the silty gas hydrate-containing sediments, but not in sandy ones. Nevertheless, accounting for a few percent of the total, many taxa such as Acidobacteriota, Chloroflexi, Planctomycetota, Cyanobacteria, Gemmatimonadota and Myxococcota were more abundant in the hydrate-free zone (Figure 4A left panel). Additionally, sandy hydrate-associated samples from 62 and 64 mbsf were characterized by the lowest phylum varieties and dominated by fewer phyla, which is consistent with the results of the alpha-diversity analysis (Figure 3A left panel, Supplementary Table 4).
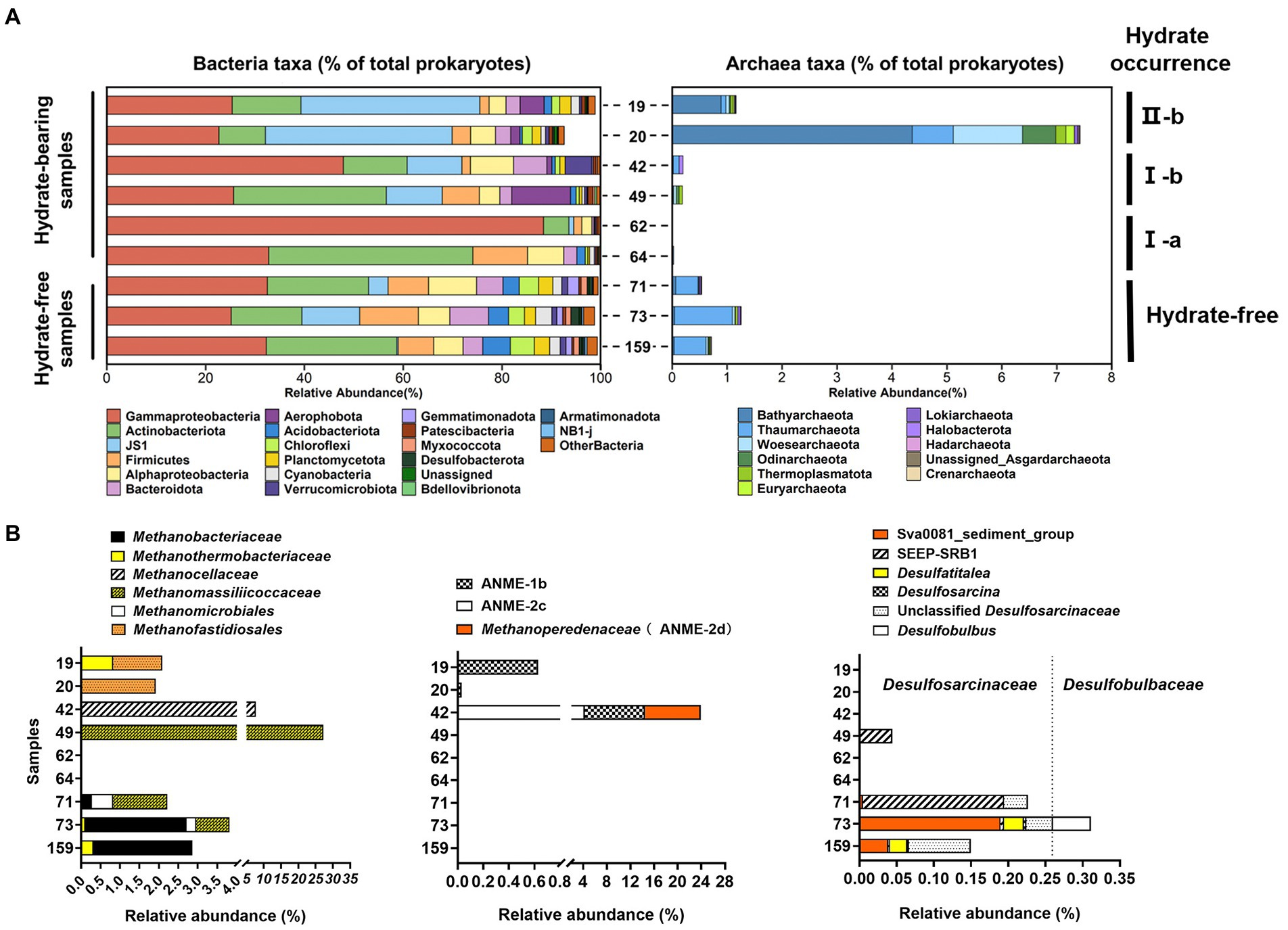
Figure 4. Microbial communities of nine sediment samples of Core W01 from QDNB based on Illumina MiSeq sequencing of 16S rRNA gene tagged amplicons. The percentage of the designated taxa within each sample is shown by colored bars. The distribution of (A) bacterial and archaeal communities are shown at the phylum level. Top 20 most abundant bacterial phylum and all archaeal phylum are listed. Relative abundance of sequences associated to (B) methanogen, ANME and deltaproteobacterial members of AOM consortia in sediments of Core W01. Percentages refer to the abundance of sequences of methanotrophs and sulfate reducers in total archaeal and bacterial sequences of each sample, respectively.
Archaeal community
In this study, a total of eleven archaeal phyla were found in the Core W01 samples, with the dominance of Bathyarchaeota, Thaumarchaeota, Woesearchaeota, Odinarchaeota, Thermoplasmatota, and Euryarchaeota accounting for 93.18% of total archaeal sequences. Remarkably, a sharp shift in archaeal composition along the vertical profile of the Core W01 was observed. The high relative abundance of archaea was found in the organic-rich sediment layers (19 and 20 mbsf) associated with II-b type gas hydrates and was dominated by Bathyarchaeota, accounting for 72.4 and 55.8% of the archaeal sequences in each sample, respectively. By contrast, archaeal groups were scarce within type I gas hydrated-containing sediments (from 42 to 64 mbsf). The lowest archaeal abundances (less than 0.3%) were detected in the sandy samples associated with I-a type gas hydrates from 62 to 64 mbsf, while archaeal communities in the deep hydrate-free zone (at 71, 73, and 159 mbsf) with moderate sequence abundance around 1% were dominated by Thaumarchaeota, accounting for an average relative abundance of 78.98% in the archaeal community of each sample (Figure 4A right panel).
Anaerobic methanotrophs
A remarkable niche partitioning of the methanotrophic community along with the Core W01 was found. Sequences affiliated with methanogens and ANMEs represented 3.08 and 0.60% of the total archaeal sequences for W01 sediments, respectively. Except for the two sandy samples at 62 and 64 mbsf where methanotroph-related sequences were not recovered, methanogen-affiliated sequences were detected throughout the sampling depth with relative abundances from 2 to 30% in archaea. Methanogens in samples collected at 19 and 20 mbsf were dominated by Methanofastidiosales, accounting for 1.83% of archaeal reads in these two samples. Samples at 42 and 49 mbsf where the relative abundances of methanogens reached their peak, hosted Methanocellaceae and Methanomassiliicoccaceae respectively, accounting for 10 and 30% of the total archaeal sequences in each sample, respectively. Methanobacteriaceae and Methanomicrobiales were only found in the deep hydrate-free zone as dominant clades, while Methanothermobacteriaceae and Methanomassiliicoccaceae were found in both hydrate-containing and-free layers (Figure 4B left panel).
ANME group within W01 sediments was only detected in the top three sediment samples containing hydrates, samples at 19 and 20 mbsf were composed solely of ANME-1b, accounting for 0.66 and 0.03% of archaeal reads of each sample, respectively. Sample 42 contained the highest abundance of the ANME group with 24.35% of archaeal reads, comprised of ANME-1b, ANME-2c, and Methanoperedenaceae (ANME-2d; Figure 4B middle panel). However, δ-proteobacterial sulfate-reducing bacteria were only found in the hydrate-free zone with low abundance (Figure 4B right panel), indicating no niche overlap between ANMEs and their potential sulfate-reducing partner, we speculated that some sulfate reduction processes in these sediments were coupled to the oxidative breakdown of organic matter.
Beta-diversity analysis
The variation in the community structure of the W01 sediment samples was analyzed by Principal Co-ordinates Analysis (PCoA) and hierarchical clustering analysis. The first two PCoA axes explained a total of 63.8% of the variation. Sediment samples retrieved from the hydrate-bearing and hydrate-free zones separated along axis 1 on the PCoA plot based on the weighted Unifrac distance (Figure 3C). Also, hydrate-bearing and-free sediment samples clustered separately on the UPGMA tree based on the Bray-Curtis analysis. These results indicated that hydrate-containing and-free sediments contained distinct microbial communities (Figure 3D). Notably, sediment-associated microbial assemblages also demonstrated clear differentiation among the four studied layers (II-b, I-b, I-a and hydrate-free layers) of Core W01 with various hydrate occurrences according to ANOSIM tests with limited samples (Supplementary Figure 2E).
In the set of taxa of this study, the prokaryotic communities were composed of very few abundant taxa and a great many rare ones, with the top three most abundant OTUs comprising 45.61% of all sequences (data not shown). Venn diagrams were used to display the common and unique OTU numbers to intuitively describe the similarity and overlap between sample groups. Our results showed that a high number of unique OTUs were detected in the hydrate-free sediment samples, accounting for 69.70% of the total OTUs and 10.03% of the total sequences (Figure 3E; Supplementary Figure 2D). Furthermore, differentially OTUs between these two groups were further visualized in a Volcano plot. In detail, compared to the hydrate-free sediments, the hydrate-bearing samples showed 60 enhanced OTUs (1.09%) and 1,242 depleted OTUs (22.56%). Among the depleted OTUs, 1,237 OTUs (86.67%) were rare taxa (abundances below 0.1%), indicating that the rare community in the hydrate-free samples made major contributions to the significant difference between the two groups (Figure 3F). This conclusion was further confirmed by the analysis of the microbial order with significant differences (p < 0.05), except for uncultured Thermoleophilia, 71 of the total 72 differentially abundant orders in hydrate-bearing sediments were depleted (Supplementary Figure 3). These 72 orders accounted for 9.85% of the total sequences, with an average abundance of each order of only 0.14%. Together, these differences indicated that the formation of gas hydrates in Core W01 might modulate the taxonomic microbial groups by reducing the number and abundance of deep marine rare taxa.
Further, the sequence data were employed in PICRUSt analysis to predict the functional diversity of the microorganisms in each sample. A heatmap was made from the top 20 KEGG orthologue (KO) groups according to functional annotation and abundances, and all groups were clustered based on functional relative abundance. As seen from the heatmap, the functional potential of microbial communities from Core W01 differed along the depth, in particular, gas hydrate-free samples had a higher KO abundance in protein metabolic pathways such as “Arginine and proline metabolism” and “Peptidases” (Figure 5).
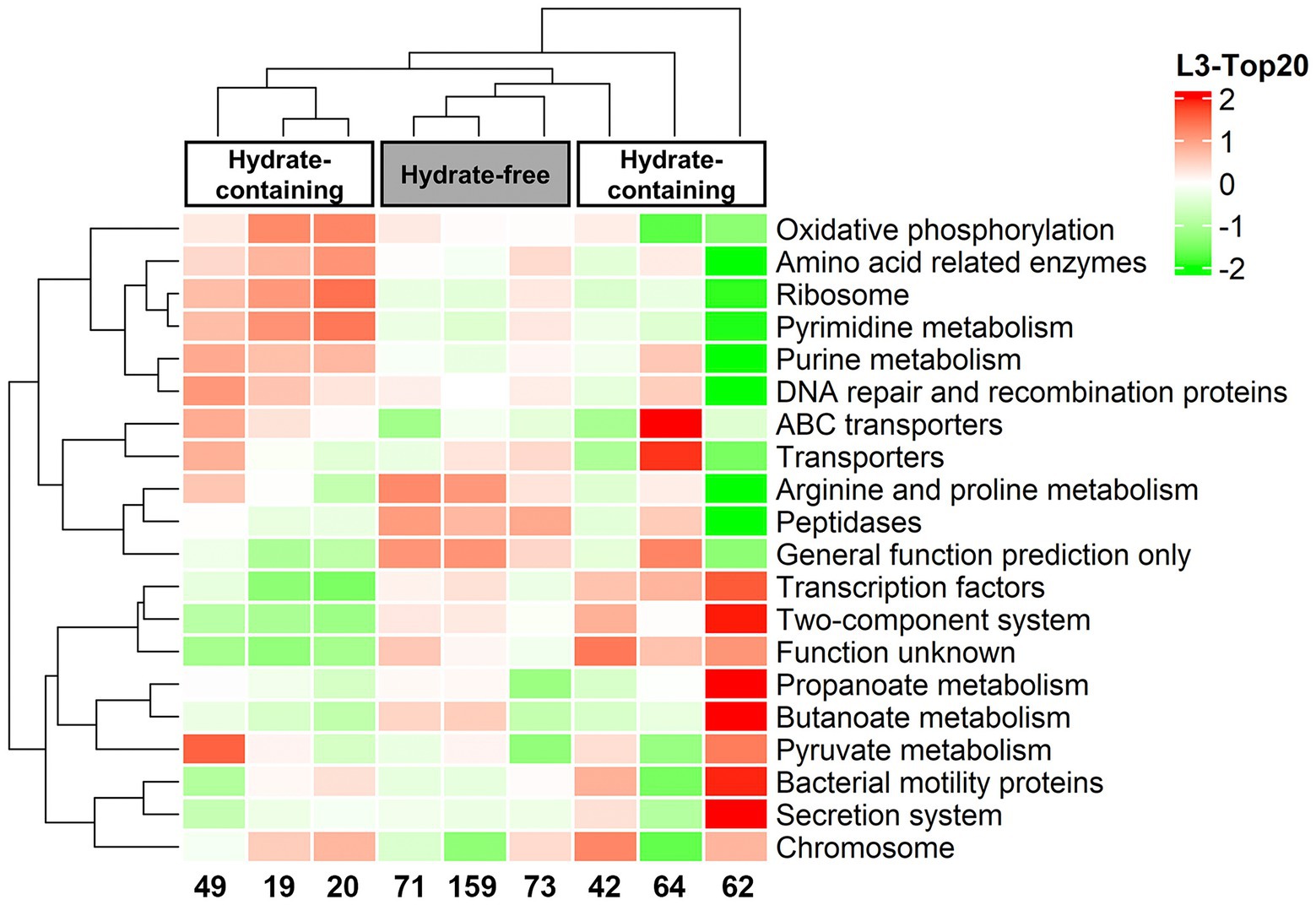
Figure 5. Heatmap profile of the top 20 most abundant KEGG (level-3) metabolic pathways created by metabolic function prediction associated to the microbiome profiles.
Environmental drivers shaping the distribution of microbial taxa
To determine the effect of sediment properties on microbial community composition, the property variables were analyzed using Distance-based Redundancy Analysis (dbRDA), where depth, TOC and concentrations of Ti were the most contributing factors as environmental input (Figure 6A). The first dbRDA axis correlating with all variables of the sediment explained 41.14% of the total variation and separated the top from bottom sediment samples, and the second axis of dbRDA explained 19.81% of the fitted variation. dbRDA analysis revealed that the abundance of JS1 lineage, Oceanospirillales and Bathyarchaeia were related to TOC and concentrations of Mn, and the abundance of Christensenellales, Thermomicrobiales, and Bacteroidales had positive correlations with depth and Ti concentrations (Figure 6A).
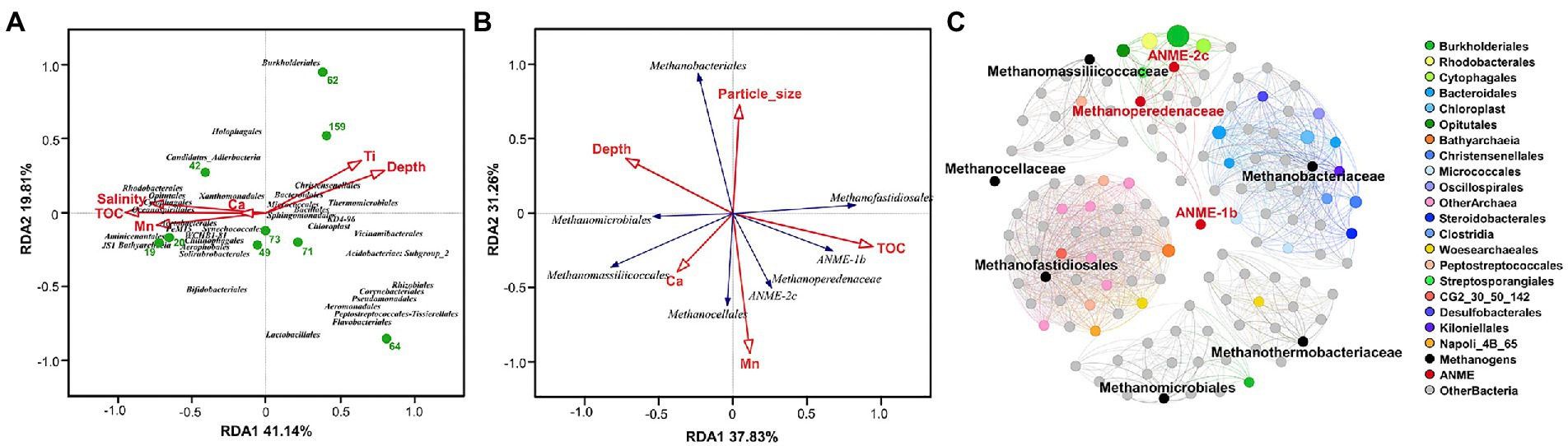
Figure 6. The impact of different environmental parameters on microbial community and the co-occurrence network analysis. Distance-based redundancy analysis (dbRDA) plot based on environmental variables fitted to the taxonomic composition of the microbial taxa (A) and anaerobic methanotrophs (B) at the order level. The green dots represent 9 detected sediment samples in the QDNB sediments; environmental parameters and microbial taxa are represented as red arrows and blue arrows, respectively. (C) Co-occurrence patterns of the microbial families regarding the relationship of the methanotrophic archaea with other taxa in Core W01. Edges joining the nodes signify significantly strong positive correlations (p < 0.001, r > 0.8). Nodes are colored by corresponding orders assigned to different lineages, and the node size reflects the relative abundance of each microbial family.
We further identified which set of variables better explains variations in the relative abundance of ANME and methanogen groups by using dbRDA. In this case, 70.37% of the correlation between methane-metabolizing archaeal composition and environmental data was explained by two axes, demonstrating the reliability of the results. A strong niche preference was shown with TOC and Mn concentrations being the main predictor variables (Figure 6B). ANME groups are all located in the bottom right quadrant of the dbRDA plot, showing strong correlations with Mn concentration and TOC. Methanobacteriales showed the highest positive correlation with the average particle size, Methanofastidiosales were associated with high levels of TOC, Methanocellales showed strong positive correlations with Mn concentrations, while Methanomicrobiales and Methanomassiliicoccales correlated strongly with depth (Figure 6B).
The co-occurrence network analysis showed that methanogenic and ANME families formed individual clusters with other corresponding taxa and there was no shared node (Figure 6C), further confirming the niche separation of the methane-metabolizing archaea in the Core W01. In particular, ANME families were significantly and positively correlated with highly abundant bacterial orders Burkholderiales, Rhodobacterales and Cytophagales; Methanofastidiosales were significantly and positively correlated mainly with Bathyarchaeia and low abundant families in both bacterial and archaeal domain; Methanobacteriaceae were significantly and positively correlated with Bacteroidales, Chloroplast, Kiloniellales and Oscillopirales (Figure 6C). The ecological interactions between methanogens/ANMEs and other microbial taxa are in line with the fact that the methanotrophs and some heterotrophic microbial species in deep-sea sediments could interact metabolically via an anaerobic food chain.
Discussion
In marine sediments where hydrates are present, a considerable proportion of the methane is thermogenic. In addition to QDNB, gas hydrates containing thermogenic hydrocarbons have been discovered in a variety of locations, including the Gulf of Mexico (Sassen and MacDonald, 1994; Sassen et al., 2004), the Caspian Sea (Diaconescu et al., 2001), the Black Sea (Woodside et al., 2003; Mazzini et al., 2004), and the Japan Sea (Matsumoto et al., 2005). Thermogenic gas hydrates are more likely to be found in deep sediments about 1,000–2,000 mbsf (or even deeper) in most regions and much below the methane hydrate stability zone (Lu et al., 2007). It is anticipated that more thermogenic complex gas hydrates will be discovered when deeper wells are drilled. The relationships between microbial distributions, cell abundance, and sediment geochemistry as well as physical characteristics in thermogenic hydrate systems have not yet been fully investigated. Our study characterized microbial biogeography with respect to this environmental system using sediment core samples from the QDNB area.
High relative abundance of archaea in Core W01
In this study, qPCR results showed that the abundance of 16S rRNA gene of archaea is generally ten times more than that of bacteria except for in the shallowest sample at 19 mbsf, however, our sequencing results with a universal primer set showed an overwhelming dominance of bacterial sequences. It has been methodologically challenging to accurately quantify the cell numbers of a certain microbial group in marine environments (Morono et al., 2014), and there is no universally acknowledged method for quantifying bacteria and archaea in marine sediments, and different techniques have yielded highly conflicting results with identical samples (Lloyd et al., 2013a). According to Lipp et al., which is consistent with our qPCR results, archaea are more abundant than bacteria in marine systems (Lipp et al., 2008), previous studies on marine hydrate-rich environments might underestimate archaeal biomass due to the limitation of detection techniques (Lipp et al., 2008; Nunoura et al., 2008; Ye et al., 2009). We speculate that the inconsistency between the two methods was caused by the PCR primers discrepancy or the incomplete archaeal cell lysis using the DNA extraction kit. Although the primer pair we adopted for the amplicon sequencing was reported to have superior predicted coverage of the taxonomic diversity of archaea and bacteria (Klindworth et al., 2012; Starnawski et al., 2017), it appears to be possible that our primer set is a poor match to the 16S rRNA gene sequences of most archaea in the Core W01. In future studies, sequence amplification with Archaea-specific primers might be helpful to produce the most informative profiles of archaea and to further confirm our observation in the QDNB area. Commercial DNA extraction kits are thought to produce better comparable results, especially among different individuals due to the process standardization (Natarajan et al., 2016), but archaeal cells from the W01 sediment samples might not be completely lysed because certain archaeal cells are resistant to lytic protocols of the Kit that are effective for bacterial cells, due to unusual cell wall structures (Roopnarain et al., 2017). Our results indicated that the handcrafted SDS-Based DNA extraction method we used for the qPCR template preparation might recover a larger amount of the genomic DNA of archaea with better performance of cell lysis efficiency. Archaea are predicted to be better adapted in extreme low-energy settings like those present in the deep biosphere, while bacteria thrive in more dynamic environments (Valentine, 2007). However, due to the longer life cycles and community turnover times in the deep subsurface, archaea with higher abundance do not necessarily govern the biogeochemical processes (Lipp et al., 2008).
Potential microbial indicators for marine hydrate systems
The association between the microbial community composition and the occurrence of gas hydrate and depth has been established earlier in hydrate-containing marine sediments (Lanoil et al., 2001; Inagaki et al., 2006). In the Core W01, similarly, of the present study, microbial community structure between the thermogenic hydrate-containing and-free sediments was significantly different. For gas hydrate exploration, microbial taxa that can serve as complementary indicators have long been looked for. In the Core W01, notably, the JS1 lineage and Aerophobetes (Aerophobota phylum) were found to be more abundant in silty hydrate-containing sediment compared to hydrate-free samples, but not in sandy ones where they presented at very low abundance. JS1 is a prevalent bacterial group in subseafloor sediments and has been frequently found to be dominant in some strictly anoxic organic-rich sediments with gas hydrates or significantly correlated to the hydrate presence, indicating a hydrate-containing habitat preference (Reed et al., 2002; Inagaki et al., 2006; Nunoura et al., 2008; Jeong et al., 2010; Lee et al., 2013; Ryu et al., 2013; Yanagawa et al., 2014; Katayama et al., 2016; Cho et al., 2017). A recent genomic study suggested that JS1 has the potential for fermentation using a variety of substrates and for syntrophic acetate oxidation coupled with hydrogen or formate scavenging methanogens (Lee et al., 2018). Aerophobota is a newly defined bacterial phylum that is widely distributed in deep-sea sediments (Chaumeil et al., 2019). Although some previous research has shown evidence of saccharolytic and fermentative metabolism in at least some members of Aerophobetes, the diversity and metabolic capacities of this taxon are still poorly understood (Wang et al., 2007; Rinke et al., 2013; Wang et al., 2016). Aerophobetes have been detected in the gas hydrate-bearing zone of sediment cores in relative higher abundance compared to hydrate-free sediments in the South China Sea earlier (Cui et al., 2019, 2020) and in some cold seep environments (Wang et al., 2016; Huang et al., 2019). It has been discovered that gas hydrate dynamics could be affected by properties of gas hydrate-bearing sediments, such as particle size, sediment components and porosity (Lu et al., 2011; Kumari et al., 2021). At depths of 62 and 64 mbsf, elements Fe, Mn, Ca, Al, and K in sediments showed sharp decreases, while Si and Ti increased. The anomaly of these major element content suggests a significant difference in paleoclimate and mineralogical composition in these two sandy samples, which might mainly comprise quartz or silicate (Wei et al., 2003; von Eynatten et al., 2016). It is possible that microbial activities may also associate with the change of these elements. The dbRDA showed a significant impact of Mn, Ca and Ti content on certain microbial groups, though the reason for this is not clear, we speculated elements with electrovalence change such as manganese content in the sediments might reflect a redox change in the local environment (Ingram et al., 2016).
It should be noted that the correlation between hydrate presence and these two bacterial lineages with hydrate-containing habitat preference was only detected in the silty samples, suggesting the limitations of the microbial index while applied to sediments of different lithology.
Further, our results showed that in the Core W01 the archaeal communities, including anaerobic methanotrophic groups shifted sharply along with discrete layers, while the bacterial changes along these layers were rather moderate. Remarkably, sandy hydrate-bearing samples at 62 and 64 mbsf showed the lowest microbial diversity, and only four archaeal OTUs were found in this layer with low abundances (data not shown). This vertically stratified distribution of microbial communities in the subseafloor has been reported in several studies, emphasizing the significance of environmental characteristics, including the physical and chemical factors, on microbial populations (Reed et al., 2006; Wilms et al., 2006; Li et al., 2008; Pachiadaki et al., 2011). Particularly, Mills et al. showed a significant difference of archaea communities between the sediment-entrained hydrate and interior hydrate in Gulf of Mexico hydrate ecosystems whereas bacteria were not distinguishable, indicating that archaea might be less tolerant to environmental fluctuations and more specialized for particular environmental circumstances (Mills et al., 2005). The observed congruence of archaeal community and vertical changes with gas hydrate-bearing dynamics in our study suggested that archaeal communities might respond to the occurrence of gas hydrates and are more sensitive to lithology and hydrate morphologies. Microorganisms, especially anaerobic methanotrophs exhibited distinct distributions according to geochemical variables previously unidentified. Thus, the copresence of archaeal members related to hydrate-bearing marine sediments could be further investigated to look for the microbiological indicators for the potential locales, accumulation, and morphologies of the thermogenic hydrate in deep marine sediments.
Methanotrophs in Core W01
In sediment samples of Core W01, methanogens only exist in a small fraction of marine sediments where a considerable amount of methane is present, indicating the trivial contribution of the methanogenesis process. A total of five orders of methanogen were found, accounting for approximately 0.03% of prokaryotes. Among them, Methanobacteriales, Methanomicrobiales, and Methanocellales are hydrogenotrophs, which are able to reduce CO2 using H2 as an electron donor to produce methane (Liu and Whitman, 2008). Methanofastidiosa and Methanomassiliicoccales, two newly described novel methanogens, were reported to perform methanogenesis by an H2–dependent methylotrophic pathway, according to recent genomic and transcriptomic evidence (Zhang et al., 2020). Thus, our results indicated that hydrogenotrophic and methylotrophic methanogenesis is the dominant pathways for biogenic methane production in Core W01. In addition, the ANMEs group, including ANME-1b, ANME-2c, and Methanoperedenaceae (ANME-2d) were detected in the uppermost three samples where no 16S rRNA gene fragments of sulfate-reducing deltaproteobacterial partners were found. ANME-1b which was the only subgroup found in all these three samples constituted a major component of the ANME cluster, accounting for 50% of the ANME sequences. As mentioned in previous studies, ANME-1 could be independent of bacterial association, our findings support the above observation. Based primarily on the observations that ANME-1 was more prevalent in the sulfate-depleted methanogenic zone of sediments, several studies have suggested that ANMEs, and ANME-1 in particular, may be capable of producing methane, primarily (Lloyd et al., 2011; Yanagawa et al., 2011; Jagersma et al., 2012; Bertram et al., 2013). However, Yoshinaga et al. (2014) pointed out that due to the carbon back flux from AOM, SMTZ may extend into the methanogenic zone where ANME may actually carry out AOM under sulfate-depleted conditions (Yoshinaga et al., 2014). We failed to amplify mcrA genes to use as markers for anaerobic methanotrophs despite many attempts with different primer sets reported by previous studies. We speculated the reason could be the measly percentage of genomic DNA from methanotrophs in the extracted PCR templates, it is also possible that the organization of the mcrA in methanotrophs from the W01 site is slightly different than in other counterparts.
Important role played by the rare taxa
Despite the fact that microbial communities commonly contain a large number of rare taxa that constitute the majority of the observed membership, it is unknown what role this microbial “rare biosphere” plays in the dynamics of the community. Previous studies suggested that most environmental metabarcoding studies are not sequencing deep enough and OTUs/taxa represented by low-abundance sequences such as singletons, doubletons, and tripletons may be valuable and informative in highlighting rare lineages in communities (Zhan et al., 2014). Rare microbial taxa may contribute to nutrient cycling in great diversity, serving as a reservoir that can rapidly respond to environmental changes and foster community stability in a variety of settings (Shade et al., 2014). In this study, a unique structural perspective of the subseafloor microbial populations was produced by the highly resolved sequence dataset obtained at an average sequencing depth of ten million reads per sample. Our results showed numerous microbial OTUs (5,436 out of 5,505 total OTUs) that individually represent less than 0.1% of the sequence highlighting the community’s overall diversity. Surprisingly, a prominent high microbial diversity comprised of a large number of rare taxa was observed in the deep hydrate-free sediments with TOC of less than 1%, indicating the presence of gas hydrate in marine sediments could have a selection effect on the microbial communities.
Below the methanogenesis zone, the food chain begins with fermentation and is rather brief (Parkes et al., 2000). According to Lomstein et al. (2012), most of the available organic materials in deep sediments are microbial necromass. Because cells are primarily composed of proteins by weight, most of the fermentable necromass is in the form of proteins and amino acids (Lomstein et al., 2012). It has been suggested that protein degradation seems to be a crucial metabolism for some subsurface microorganisms (Lloyd et al., 2013b) and peptidases were more active than other enzymes in similar settings (Coolen and Overmann, 2000; Jacobson Meyers et al., 2014). Our results of predictive functional profiles of microbial communities in W01 sediment samples showed that peptidases synthesis, as well as arginine and proline metabolism, were significantly more abundant in the deep hydrate-free sediment samples in contrast to hydrate-containing sediments, indicating fermentative breakdown of proteins is a crucial strategy to support deep biospheres and could be inhibited by the gas hydrate formation.
Conclusion
In this study, high-throughput sequencing of the prokaryotic 16S rRNA genes combined with molecular and geochemical analyses was applied to investigate the microbial characteristics in the Core W01 of QDNB with multi-layer thermogenic hydrates. Based on our results, the following conclusions have been reached:
1. In the Core W01, the abundance of archaea cells was ten times higher than that of bacteria.
2. Distinct microbial abundance and diversity related to the thermogenic gas hydrate presence were identified.
3. The bacterial taxa of all samples are dominated by Gammaproteobacteria and Actinobacteria. The phyla ‘Atribacteria’ lineage JS1 and Aerophobota were significantly abundant in the silty hydrate-containing sediments, other than in sandy ones; while the archaeal communities changed dramatically throughout the vertical profile of sediment layers and a spatial heterogenicity of anaerobic methanotrophs was also determined.
4. The deep hydrate-free sediments harbored a large number of distinct rare microbial groups presumed to perform protein degradation and might respond to the hydrate formation.
5. The distribution of microbial taxa is related to the hydrate existence, occurrences and environmental parameters including sedimentary facies.
Our research offers a novel perspective on the distributions of microbial species associated with thermogenic gas hydrates and adds new insights to the ecological role of the methane reservoir on Earth.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: http://gsa.big.ac.cn/index.jsp, PRJCA011124.
Author contributions
SY designed the study, conducted the experiment, helped with analysis, and wrote the manuscript in consultation with all other authors. SL performed the phylogenetic and statistical analyses and revised this manuscript. XL performed the experiments for nucleic acid extractions and quantitative PCR and revised this manuscript. HY, YL, and XX did the geochemical experiments. YF collected and shipped the samples and provided porewater parameters. HL revised this manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by China Geological Survey (grant number DD20221703).
Acknowledgments
We are grateful to the crew and scientists of the GMGS6 drilling expedition for their support in sediment sample collection and analysis. We also thank the scientists of the Guangzhou Marine Geological Survey for measuring porewater parameters and providing these unpublished data.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1032851/full#supplementary-material
Footnotes
1. ^http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
2. ^http://www.ehbio.com/ImageGP/index.php/Home/Index/PiCrust.html
References
Bahk, J. J., Kim, D. H., Chun, J. H., Son, B. K., Kim, J. H., Ryu, B. J., et al. (2013). Gas hydrate occurrences and their relation to host sediment properties: results from second Ulleung Basin gas hydrate drilling expedition, East Sea. Mar. Pet. Geol. 47, 21–29. doi: 10.1016/j.marpetgeo.2013.05.006
Barnes, R., and Goldberg, E. (1976). Methane production and consumption in anoxic marine sediments. Geology 4, 297–300. doi: 10.1130/0091-7613(1976)4<297:MPACIA>2.0.CO;2
Bastian, M., Heymann, S., and Jacomy, M. (2009). "Gephi: an Open Source Software for Exploring and Manipulating Networks", in: Proceedings of the International AAAI Conference on web and social media), 361–362.
Baux, N., Murat, A., Faivre, Q., Lesourd, S., Poizot, E., Méar, Y., et al. (2019). Sediment dynamic equilibrium, a key for assessing a coastal anthropogenic disturbance using geochemical tracers: application to the eastern part of the bay of seine. Cont. Shelf Res. 175, 87–98. doi: 10.1016/j.csr.2019.02.002
Beal, E. J., House, C. H., and Orphan, V. J. (2009). Manganese-and iron-dependent marine methane oxidation. Science 325, 184–187. doi: 10.1126/science.1169984
Bertram, S., Blumenberg, M., Michaelis, W., Siegert, M., Kruger, M., and Seifert, R. (2013). Methanogenic capabilities of ANME-archaea deduced from C-13-labelling approaches. Environ. Microbiol. 15, 2384–2393. doi: 10.1111/1462-2920.12112
Bidle, K. A., Kastner, M., and Bartlett, D. H. (1999). A phylogenetic analysis of microbial communities associated with methane hydrate containing marine fluids and sediments in the Cascadia margin (ODP site 892B). FEMS Microbiol. Lett. 177, 101–108. doi: 10.1111/j.1574-6968.1999.tb13719.x
Boetius, A., Ravenschlag, K., Schubert, C. J., Rickert, D., Widdel, F., Gieseke, A., et al. (2000). A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 407, 623–626. doi: 10.1038/35036572
Boetius, A., and Wenzhöfer, F. (2013). Seafloor oxygen consumption fuelled by methane from cold seeps. Nat. Geosci. 6, 725–734. doi: 10.1038/ngeo1926
Briggs, B. R., Inagaki, F., Morono, Y., Futagami, T., Huguet, C., Rosell-Mele, A., et al. (2012). Bacterial dominance in subseafloor sediments characterized by methane hydrates. FEMS Microbiol. Ecol. 81, 88–98. doi: 10.1111/j.1574-6941.2012.01311.x
Carrier, V., Svenning, M. M., Grundger, F., Niemann, H., Dessandier, P.-A., Panieri, G., et al. (2020). The impact of methane on microbial communities at marine Arctic gas hydrate bearing sediment. Front. Microbiol. 11:1932. doi: 10.3389/fmicb.2020.01932
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2019). GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36, 1925–1927. doi: 10.1093/bioinformatics/btz848
Cho, H., Hyun, J.-H., You, O.-R., Kim, M., Kim, S.-H., Choi, D.-L., et al. (2017). Microbial community structure associated with biogeochemical processes in the sulfate–methane transition zone (SMTZ) of gas-hydrate-bearing sediment of the Ulleung Basin, East Sea. Geomicrobiol. J. 34, 207–219. doi: 10.1080/01490451.2016.1159767
Coolen, M. J., and Overmann, J. R. (2000). Functional exoenzymes as indicators of metabolically active bacteria in 124, 000-year-old sapropel layers of the eastern Mediterranean Sea. Appl. Environ. Microbiol. 66, 2589–2598. doi: 10.1128/AEM.66.6.2589-2598.2000
Cragg, B., Parkes, R., Fry, J., Weightman, A., Rochelle, P., Maxwell, J., et al. (1995). "27. The Impact of Fluid and Gas Venting on Bacterial Populations and Processes in Sediments from the Cascadia Margin Accretionary System (Sites 888-892) and the Geochemical CONSEQUENCES1", in: Proceedings of the Ocean Drilling Program, Scientific Results 146, 399–144.
Cui, H., Su, X., Chen, F., Holland, M., Yang, S., Liang, J., et al. (2019). Microbial diversity of two cold seep systems in gas hydrate-bearing sediments in the South China Sea. Mar. Environ. Res. 144, 230–239. doi: 10.1016/j.marenvres.2019.01.009
Cui, H., Su, X., Liang, J., Chen, F., Holland, M., Yang, S., et al. (2020). Microbial diversity in fracture and pore filling gas hydrate-bearing sediments at site GMGS2-16 in the Pearl River Mouth Basin, the South China Sea. Mar. Geol. 427:106264. doi: 10.1016/j.margeo.2020.106264
Diaconescu, C. C., Kieckhefer, R. M., and Knapp, J. H. (2001). Geophysical evidence for gas hydrates in the deep water of the South Caspian Basin, Azerbaijan. Mar. Pet. Geol. 18, 209–221. doi: 10.1016/S0264-8172(00)00061-1
Dickens, G.R. (2001a). Modeling the Global Carbon Cycle with a Gas Hydrate Capacitor: Significance for the Latest Paleocene Thermal Maximum. Washington, DC American Geophysical Union Geophysical Monograph Series 124, 19–38.
Dickens, G. R. (2001b). The potential volume of oceanic methane hydrates with variable external conditions. Org. Geochem. 32, 1179–1193. doi: 10.1016/S0146-6380(01)00086-9
Dong, X., Greening, C., Rattray, J. E., Chakraborty, A., Chuvochina, M., Mayumi, D., et al. (2019). Metabolic potential of uncultured bacteria and archaea associated with petroleum seepage in deep-sea sediments. Nat. Commun. 10:1816. doi: 10.1038/s41467-019-09747-0
Dong, X., Rattray, J. E., Campbell, D. C., Webb, J., Chakraborty, A., Adebayo, O., et al. (2020). Thermogenic hydrocarbon biodegradation by diverse depth-stratified microbial populations at a Scotian Basin cold seep. Nat. Commun. 11:5825. doi: 10.1038/s41467-020-19648-2
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Ettwig, K. F., Butler, M. K., Le Paslier, D., Pelletier, E., Mangenot, S., Kuypers, M. M. M., et al. (2010). Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464, 543–548. doi: 10.1038/nature08883
Geng, M., Zhang, R., Yang, S., Guo, J., and Chen, Z. (2021). Focused fluid flow, shallow gas hydrate, and cold seep in the Qiongdongnan Basin, Northwestern South China Sea. Geofluids 2021, 1–11. doi: 10.1155/2021/5594980
Holland, M., Schultheiss, P., Roberts, J., and Druce, M. (2008). “Observed gas hydrate morphologies in marine sediments” in 6th International Conference on Gas Hydrates (Vancouver, BC, Canada: Chevron), 6–10.
Huang, J. M., Baker, B. J., Li, J. T., and Wang, Y. (2019). New microbial lineages capable of carbon fixation and nutrient cycling in Deep-Sea sediments of the northern South China Sea. Appl. Environ. Microbiol. 85:e00523-19. doi: 10.1128/aem.00523-19
Huang, B., Tian, H., Li, X., Wang, Z., and Xiao, X. (2016). Geochemistry, origin and accumulation of natural gases in the Deepwater area of the Qiongdongnan Basin, South China Sea. Mar. Pet. Geol. 72, 254–267. doi: 10.1016/j.marpetgeo.2016.02.007
Inagaki, F., Nunoura, T., Nakagawa, S., Teske, A., Lever, M., Lauer, A., et al. (2006). Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments, on the Pacific Ocean margin. Proc. Natl. Acad. Sci. U. S. A. 103, 2815–2820. doi: 10.1073/pnas.0511033103
Ingram, W. C., Meyers, S. R., Shen, Z., Xu, H., and Martens, C. S. (2016). Manganese enrichments near a large gas-hydrate and cold-seep field: a record of past redox and sedimentation events. Depos. Rec. 2, 142–153. doi: 10.1002/dep2.18
Jacobson Meyers, M. E., Sylvan, J. B., and Edwards, K. J. (2014). Extracellular enzyme activity and microbial diversity measured on seafloor exposed basalts from Loihi seamount indicate the importance of basalts to global biogeochemical cycling. Appl. Environ. Microbiol. 80, 4854–4864. doi: 10.1128/aem.01038-14
Jagersma, C. G., Meulepas, R. J. W., Timmers, P. H. A., Szperl, A., Lens, P. N. L., and Stams, A. J. M. (2012). Enrichment of ANME-1 from eckernforde bay sediment on thiosulfate, methane and short-chain fatty acids. J. Biotechnol. 157, 482–489. doi: 10.1016/j.jbiotec.2011.10.012
Jeong, I. S., Cho, J. C., Bahk, J. J., Hyun, S. M., Kwon, K. K., Lee, J. H., et al. (2010). “Vertical profile of bacterial community in the sediment of Ulleung Basin: implication of the presence of methane-driven community,” in 3rd International Conference on Environmental, Industrial, and Applied Microbiology, 219–226.
Jiao, L., Su, X., Wang, Y., Jiang, H., Zhang, Y., and Chen, F. (2015). Microbial diversity in the hydrate-containing and-free surface sediments in the Shenhu area, South China. Geosci. Front. 6, 627–633. doi: 10.1016/j.gsf.2014.04.007
Katayama, T., Yoshioka, H., Kaneko, M., Amo, M., Fujii, T., Takahashi, H. A., et al. (2022). Cultivation and biogeochemical analyses reveal insights into methanogenesis in deep subseafloor sediment at a biogenic gas hydrate site. ISME J. 16, 1464–1472. doi: 10.1038/s41396-021-01175-7
Katayama, T., Yoshioka, H., Takahashi, H. A., Amo, M., Fujii, T., and Sakata, S. (2016). Changes in microbial communities associated with gas hydrates in subseafloor sediments from the Nankai trough. FEMS Microbiol. Ecol. 92:fiw093. doi: 10.1093/femsec/fiw093
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2012). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Knittel, K., Losekann, T., Boetius, A., Kort, R., and Amann, R. (2005). Diversity and distribution of methanotrophic archaea at cold seeps. Appl. Environ. Microbiol. 71, 467–479. doi: 10.1128/aem.71.1.467-479.2005
Kormas, K. A., Smith, D. C., Edgcomb, V., and Teske, A. (2003). Molecular analysis of deep subsurface microbial communities in Nankai trough sediments (ODP leg 190, site 1176). FEMS Microbiol. Ecol. 45, 115–125. doi: 10.1016/s0168-6496(03)00128-4
Kumari, A., Balomajumder, C., Arora, A., Dixit, G., and Gomari, S. R. (2021). Physio-chemical and mineralogical characteristics of gas hydrate-bearing sediments of the Kerala-Konkan, Krishna-Godavari, and Mahanadi basins. J. Marine Sci. Eng. 9:808. doi: 10.3390/jmse9080808
Kvenvolden, K. A. (1993). Gas hydrates—geological perspective and global change. Rev. Geophys. 31, 173–187. doi: 10.1029/93RG00268
Kvenvolden, K. A. (2002). Methane hydrate in the global organic carbon cycle. Terra Nova 14, 302–306. doi: 10.1046/j.1365-3121.2002.00414.x
Lai, H., Qiu, H., Liang, J., Kuang, Z., Fang, Y., Ren, J., et al. (2022). Geochemical characteristics and gas-to-gas correlation of two leakage-type gas hydrate accumulations in the Western Qiongdongnan Basin, South China Sea. Acta Geol. Sin. Engl. Ed. 96, 680–690. doi: 10.1111/1755-6724.14931
Langfelder, P., and Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 1–13. doi: 10.1186/1471-2105-9-559
Lanoil, B. D., La Duc, M. T., Wright, M., Kastner, M., Nealson, K. H., and Bartlett, D. (2005). Archaeal diversity in ODP legacy borehole 892b and associated seawater and sediments of the Cascadia margin. FEMS Microbiol. Ecol. 54, 167–177. doi: 10.1016/j.femsec.2005.03.015
Lanoil, B. D., Sassen, R., La Duc, M. T., Sweet, S. T., and Nealson, K. H. (2001). Bacteria and archaea physically associated with Gulf of Mexico gas hydrates. Appl. Environ. Microbiol. 67, 5143–5153. doi: 10.1128/AEM.67.11.5143-5153.2001
Lee, Y. M., Hwang, K., Lee, J. I., Kim, M., Hwang, C. Y., Noh, H.-J., et al. (2018). Genomic insight into the predominance of candidate phylum Atribacteria JS1 lineage in marine sediments. Front. Microbiol. 9:2909. doi: 10.3389/fmicb.2018.02909
Lee, J.-W., Kwon, K. K., Azizi, A., Oh, H.-M., Kim, W., Bahk, J.-J., et al. (2013). Microbial community structures of methane hydrate-bearing sediments in the Ulleung Basin, East Sea of Korea. Mar. Pet. Geol. 47, 136–146. doi: 10.1016/j.marpetgeo.2013.06.002
Lee, J. Y., Santamarina, J. C., and Ruppel, C. (2008). Mechanical and electromagnetic properties of northern Gulf of Mexico sediments with and without THF hydrates. Mar. Pet. Geol. 25, 884–895. doi: 10.1016/j.marpetgeo.2008.01.019
Li, Y., Li, F., Zhang, X., Qin, S., Zeng, Z., Dang, H., et al. (2008). Vertical distribution of bacterial and archaeal communities along discrete layers of a deep-sea cold sediment sample at the East Pacific rise (∼13°N). Extremophiles 12, 573–585. doi: 10.1007/s00792-008-0159-5
Liang, J., Zhang, W., Lu, J., Wei, J., Kuang, Z., He, Y., et al. (2019). Geological occurrence and accumulation mechanism of natural gas hydrates in the eastern Qiongdongnan Basin of the South China Sea: insights from site GMGS5-W9-2018. Mar. Geol. 418:106042. doi: 10.1016/j.margeo.2019.106042
Liao, L., Xu, X. W., Wang, C. S., Zhang, D. S., and Wu, M. (2009). Bacterial and archaeal communities in the surface sediment from the northern slope of the South China Sea. J. Zhejiang Univ. Sci. B 10, 890–901. doi: 10.1631/jzus.B0920181
Lin, L.-H., Wu, L.-W., Cheng, T.-W., Tu, W.-X., Lin, J.-R., Yang, T. F., et al. (2014). Distributions and assemblages of microbial communities along a sediment core retrieved from a potential hydrate-bearing region offshore southwestern Taiwan. J. Asian Earth Sci. 92, 276–292. doi: 10.1016/j.jseaes.2014.02.014
Lipp, J. S., Morono, Y., Inagaki, F., and Hinrichs, K.-U. (2008). Significant contribution of archaea to extant biomass in marine subsurface sediments. Nature 454, 991–994. doi: 10.1038/nature07174
Liu, Y., Qin, Y., Chen, T., Lu, M., Qian, X., Guo, X., et al. (2021). A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell 12, 315–330. doi: 10.1007/s13238-020-00724-8
Liu, Y., Wei, J., Li, Y., Chang, J., Miao, X., and Lu, H. (2022). Seep dynamics as revealed by authigenic carbonates from the eastern Qiongdongnan Basin, South China Sea. Mar. Pet. Geol. 142:105736. doi: 10.1016/j.marpetgeo.2022.105736
Liu, Y., and Whitman, W. B. (2008). Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann. N. Y. Acad. Sci. 1125, 171–189. doi: 10.1196/annals.1419.019
Lloyd, K. G., Alperin, M. J., and Teske, A. (2011). Environmental evidence for net methane production and oxidation in putative ANaerobic MEthanotrophic (ANME) archaea. Environ. Microbiol. 13, 2548–2564. doi: 10.1111/j.1462-2920.2011.02526.x
Lloyd, K. G., May, M. K., Kevorkian, R. T., and Steen, A. D. (2013a). Meta-analysis of quantification methods shows that archaea and bacteria have similar abundances in the subseafloor. Appl. Environ. Microbiol. 79, 7790–7799. doi: 10.1128/AEM.02090-13
Lloyd, K. G., Schreiber, L., Petersen, D. G., Kjeldsen, K. U., Lever, M. A., Steen, A. D., et al. (2013b). Predominant archaea in marine sediments degrade detrital proteins. Nature 496, 215–218. doi: 10.1038/nature12033
Lomstein, B. A., Langerhuus, A. T., D’Hondt, S., Jørgensen, B. B., and Spivack, A. J. (2012). Endospore abundance, microbial growth and necromass turnover in deep sub-seafloor sediment. Nature 484, 101–104. doi: 10.1038/nature10905
Lu, H., Kawasaki, T., Ukita, T., Moudrakovski, I., Fujii, T., Noguchi, S., et al. (2011). Particle size effect on the saturation of methane hydrate in sediments – constrained from experimental results. Mar. Pet. Geol. 28, 1801–1805. doi: 10.1016/j.marpetgeo.2010.11.007
Lu, H., Seo, Y. T., Lee, J. W., Moudrakovski, I., Ripmeester, J. A., Chapman, N. R., et al. (2007). Complex gas hydrate from the Cascadia margin. Nature 445, 303–306. doi: 10.1038/nature05463
Luo, X. X., Yang, S. L., and Zhang, J. (2012). The impact of the three gorges dam on the downstream distribution and texture of sediments along the middle and lower Yangtze River (Changjiang) and its estuary, and subsequent sediment dispersal in the East China Sea. Geomorphology 179, 126–140. doi: 10.1016/j.geomorph.2012.05.034
Macdonald, I. R., Guinasso, N. L., Sassen, R., Brooks, J. M., Lee, L., and Scott, K. T. (1994). Gas hydrate that breaches the sea-floor on the continental-slope of The Gulf-of-Mexico. Geology 22, 699–702. doi: 10.1130/0091-7613(1994)022<0699:Ghtbts>2.3.Co;2
Marchesi, J. R., Weightman, A. J., Cragg, B. A., Parkes, R. J., and Fry, J. C. (2001). Methanogen and bacterial diversity and distribution in deep gas hydrate sediments from the Cascadia margin as revealed by 16S rRNA molecular analysis. FEMS Microbiol. Ecol. 34, 221–228. doi: 10.1111/j.1574-6941.2001.tb00773.x
Matsumoto, R., Tomaru, H., Hiruta, A., Ishida, Y., Takeuchi, R., Snyder, G., et al. (2005). Gas Hydrate Layer and Prominent Flares of Gas Plumes in Naoetsu Basin, Eastern Margin of Japan Sea. AGU Fall Meeting Abstracts, 2005, OS41C-04.
Mayumi, D., Mochimaru, H., Tamaki, H., Yamamoto, K., Yoshioka, H., Suzuki, Y., et al. (2016). Methane production from coal by a single methanogen. Science 354, 222–225. doi: 10.1126/science.aaf8821
Mazzini, A., Ivanov, M. K., Parnell, J., Stadnitskaia, A., Cronin, B. T., Poludetkina, E., et al. (2004). Methane-related authigenic carbonates from the Black Sea: geochemical characterisation and relation to seeping fluids. Mar. Geol. 212, 153–181. doi: 10.1016/j.margeo.2004.08.001
Meng, M., Liang, J., Lu, J., Zhang, W., Kuang, Z., Fang, Y., et al. (2021). Quaternary deep-water sedimentary characteristics and their relationship with the gas hydrate accumulations in the Qiongdongnan Basin, northwest South China Sea. Deep Sea Res. Part I Oceanogr. Res. Pap. 177:103628. doi: 10.1016/j.dsr.2021.103628
Mills, H. J., Hodges, C., Wilson, K., MacDonald, I. R., and Sobecky, P. A. (2003). Microbial diversity in sediments associated with surface-breaching gas hydrate mounds in the Gulf of Mexico. FEMS Microbiol. Ecol. 46, 39–52. doi: 10.1016/s0168-6496(03)00191-0
Mills, H. J., Kiel Reese, B., Shepard, A., Dowd, S. E., Riedinger, N., Morono, Y., et al. (2012). Characterization of metabolically active bacterial populations in subseafloor Nankai trough sediments above, within, and below the sulfate–methane transition zone. Front. Microbiol. 3:113. doi: 10.3389/fmicb.2012.00113
Mills, H. J., Martinez, R. J., Story, S., and Sobecky, P. A. (2005). Characterization of microbial community structure in Gulf of Mexico gas hydrates: comparative analysis of DNA-and RNA-derived clone libraries. Appl. Environ. Microbiol. 71, 3235–3247. doi: 10.1128/AEM.71.6.3235-3247.2005
Morono, Y., Terada, T., Hoshino, T., and Inagaki, F. (2014). Hot-alkaline DNA extraction method for deep-subseafloor archaeal communities. Appl. Environ. Microbiol. 80, 1985–1994. doi: 10.1128/aem.04150-13
Natarajan, V. P., Zhang, X., Morono, Y., Inagaki, F., and Wang, F. (2016). A modified SDS-based DNA extraction method for high quality environmental DNA from seafloor environments. Front. Microbiol. 7:986. doi: 10.3389/fmicb.2016.00986
Newberry, C. J., Webster, G., Cragg, B. A., Parkes, R. J., Weightman, A. J., and Fry, J. C. (2004). Diversity of prokaryotes and methanogenesis in deep subsurface sediments from the Nankai trough, ocean drilling program leg 190. Environ. Microbiol. 6, 274–287. doi: 10.1111/j.1462-2920.2004.00568.x
Nunoura, T., Inagaki, F., Delwiche, M. E., Colwell, F. S., and Takai, K. (2008). Subseafloor microbial communities in methane hydrate-bearing sediment at two distinct locations (ODP Leg204) in the Cascadia margin. Microbes Environ. 23, 317–325. doi: 10.1264/jsme2.ME08514
Pachiadaki, M. G., Kallionaki, A., Dählmann, A., De Lange, G. J., and Kormas, K. A. (2011). Diversity and spatial distribution of prokaryotic communities along a sediment vertical profile of a Deep-Sea mud volcano. Microb. Ecol. 62, 655–668. doi: 10.1007/s00248-011-9855-2
Parkes, R. J., Cragg, B. A., and Wellsbury, P. (2000). Recent studies on bacterial populations and processes in subseafloor sediments: a review. Hydrogeol. J. 8, 11–28. doi: 10.1007/PL00010971
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Raghoebarsing, A. A., Pol, A., van de Pas-Schoonen, K. T., Smolders, A. J. P., Ettwig, K. F., Rijpstra, W. I. C., et al. (2006). A microbial consortium couples anaerobic methane oxidation to denitrification. Nature 440, 918–921. doi: 10.1038/nature04617
Reeburgh, W. S. (2007). Oceanic methane biogeochemistry. Chem. Rev. 107, 486–513. doi: 10.1021/cr050362v
Reed, D. W., Fujita, Y., Delwiche, M. E., Blackwelder, D. B., Sheridan, P. P., Uchida, T., et al. (2002). Microbial communities from methane hydrate-bearing deep marine sediments in a forearc basin. Appl. Environ. Microbiol. 68, 3759–3770. doi: 10.1128/aem.68.8.3759-3770.2002
Reed, A. J., Lutz, R. A., and Vetriani, C. (2006). Vertical distribution and diversity of bacteria and archaea in sulfide and methane-rich cold seep sediments located at the base of the Florida escarpment. Extremophiles 10, 199–211. doi: 10.1007/s00792-005-0488-6
Ren, J., Cheng, C., Xiong, P., Kuang, Z., Liang, J., Lai, H., et al. (2022). Sand-rich gas hydrate and shallow gas systems in the Qiongdongnan Basin, northern South China Sea. J. Pet. Sci. Eng. 215:110630. doi: 10.1016/j.petrol.2022.110630
Rinke, C., Schwientek, P., Sczyrba, A., Ivanova, N. N., Anderson, I. J., Cheng, J.-F., et al. (2013). Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437. doi: 10.1038/nature12352
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016:e2584. doi: 10.7717/peerj.2584
Roopnarain, A., Mukhuba, M., Adeleke, R., and Moeletsi, M. (2017). Biases during DNA extraction affect bacterial and archaeal community profile of anaerobic digestion samples. 3 Biotech 7:375. doi: 10.1007/s13205-017-1009-x
Ryu, B.-J., Collett, T. S., Riedel, M., Kim, G. Y., Chun, J.-H., Bahk, J.-J., et al. (2013). Scientific results of the second gas hydrate drilling expedition in the Ulleung Basin (UBGH2). Mar. Pet. Geol. 47, 1–20. doi: 10.1016/j.marpetgeo.2013.07.007
Sassen, R., and MacDonald, I. R. (1994). Evidence of structure H hydrate, Gulf of Mexico continental slope. Org. Geochem. 22, 1029–1032. doi: 10.1016/0146-6380(94)90036-1
Sassen, R., Roberts, H. H., Carney, R., Milkov, A. V., DeFreitas, D. A., Lanoil, B., et al. (2004). Free hydrocarbon gas, gas hydrate, and authigenic minerals in chemosynthetic communities of the northern Gulf of Mexico continental slope: relation to microbial processes. Chem. Geol. 205, 195–217. doi: 10.1016/j.chemgeo.2003.12.032
Schoell, M. (1988). Multiple origins of methane in the earth. Chem. Geol. 71, 1–10. doi: 10.1016/0009-2541(88)90101-5
Shade, A., Jones, S. E., Caporaso, J. G., Handelsman, J., Knight, R., Fierer, N., et al. (2014). Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. mBio 5, e01371–e01314. doi: 10.1128/mBio.01371-14
Starnawski, P., Bataillon, T., Ettema, T. J. G., Jochum, L. M., Schreiber, L., Chen, X., et al. (2017). Microbial community assembly and evolution in subseafloor sediment. Proc. Natl. Acad. Sci. 114, 2940–2945. doi: 10.1073/pnas.1614190114
Thauer, R. K., Kaster, A. K., Seedorf, H., Buckel, W., and Hedderich, R. (2008). Methanogenic archaea: ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6, 579–591. doi: 10.1038/nrmicro1931
Tomaru, H., Matsumoto, R., Lu, H., and Uchida, T. (2004). Geochemical process of gas hydrate formation in the Nankai trough based on chloride and isotopic anomalies in interstitial water. Resour. Geol. 54, 45–51. doi: 10.1111/j.1751-3928.2004.tb00186.x
Valentine, D. L. (2007). Adaptations to energy stress dictate the ecology and evolution of the archaea. Nat. Rev. Microbiol. 5, 316–323. doi: 10.1038/nrmicro1619
von Eynatten, H., Tolosana-Delgado, R., Karius, V., Bachmann, K., and Caracciolo, L. (2016). Sediment generation in humid Mediterranean setting: grain-size and source-rock control on sediment geochemistry and mineralogy (Sila massif, Calabria). Sediment. Geol. 336, 68–80. doi: 10.1016/j.sedgeo.2015.10.008
Wang, Y., Gao, Z.-M., Li, J.-T., Bougouffa, S., Tian, R. M., Bajic, V. B., et al. (2016). Draft genome of an Aerophobetes bacterium reveals a facultative lifestyle in deep-sea anaerobic sediments. Sci. Bull. 61, 1176–1186. doi: 10.1007/s11434-016-1135-6
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Wei, J., Liang, J., Lu, J., Zhang, W., and He, Y. (2019). Characteristics and dynamics of gas hydrate systems in the northwestern South China Sea - results of the fifth gas hydrate drilling expedition. Mar. Pet. Geol. 110, 287–298. doi: 10.1016/j.marpetgeo.2019.07.028
Wei, G., Liu, Y., Li, X., Shao, L., and Liang, X. (2003). Climatic impact on Al, K, Sc and Ti in marine sediments: Evidence from ODP site 1144, South China Sea. Geochem. J. 37, 593–602. doi: 10.2343/geochemj.37.593
Whiticar, M. J. (1999). Carbon and hydrogen isotope systematics of bacterial formation and oxidation of methane. Chem. Geol. 161, 291–314. doi: 10.1016/S0009-2541(99)00092-3
Wilms, R., Sass, H., Köpke, B., Jr, K., Cypionka, H., and Engelen, B. (2006). Specific bacterial, archaeal, and eukaryotic communities in tidal-flat sediments along a vertical profile of several meters. Appl. Environ. Microbiol. 72, 2756–2764. doi: 10.1128/AEM.72.4.2756-2764.2006
Woodside, J. M., Modin, D. I., and Ivanov, M. K. (2003). An enigmatic strong reflector on subbottom profiler records from the Black Sea—the top of shallow gas hydrate deposits. Geo-Mar. Lett. 23, 269–277. doi: 10.1007/s00367-003-0149-7
Wuebbles, D. J., and Hayhoe, K. (2002). Atmospheric methane and global change. Earth Sci. Rev. 57, 177–210. doi: 10.1016/S0012-8252(01)00062-9
Yanagawa, K., Kouduka, M., Nakamura, Y., Hachikubo, A., Tomaru, H., and Suzuki, Y. (2014). Distinct microbial communities thriving in gas hydrate-associated sediments from the eastern Japan Sea. J. Asian Earth Sci. 90, 243–249. doi: 10.1016/j.jseaes.2013.10.019
Yanagawa, K., Sunamura, M., Lever, M. A., Morono, Y., Hiruta, A., Ishizaki, O., et al. (2011). Niche separation of Methanotrophic archaea (ANME-1 and-2) in methane-seep sediments of the eastern Japan Sea offshore Joetsu. Geomicrobiol J. 28, 118–129. doi: 10.1080/01490451003709334
Yang, H., Yu, S., and Lu, H. (2021). Iron-coupled anaerobic oxidation of methane in marine sediments: a review. J. Marine Sci. Eng. 9:875. doi: 10.3390/jmse9080875
Ye, G., Wang, S., Jiang, L., Xiao, X., Wang, F., Noakes, J., et al. (2009). Distribution and diversity of bacteria and archaea in marine sediments affected by gas hydrates at Mississippi canyon in the Gulf of Mexico. Geomicrobiol J. 26, 370–381. doi: 10.1080/01490450902929308
Ye, J., Wei, J., Liang, J., Lu, J., Lu, H., Zhang, W., et al. (2019). Complex gas hydrate system in a gas chimney, South China Sea. Mar. Pet. Geol. 104, 29–39. doi: 10.1016/j.marpetgeo.2019.03.023
Yoshinaga, M. Y., Holler, T., Goldhammer, T., Wegener, G., Pohlman, J. W., Brunner, B., et al. (2014). Carbon isotope equilibration during sulphate-limited anaerobic oxidation of methane. Nat. Geosci. 7, 190–194. doi: 10.1038/ngeo2069
Zhan, A., Xiong, W., He, S., and MacIsaac, H. J. (2014). Influence of artifact removal on rare species recovery in natural complex communities using high-throughput sequencing. PLoS One 9:e96928. doi: 10.1371/journal.pone.0096928
Zhang, W., Liang, J., Su, P., Wei, J., Gong, Y., Lin, L., et al. (2019). Distribution and characteristics of mud diapirs, gas chimneys, and bottom simulating reflectors associated with hydrocarbon migration and gas hydrate accumulation in the Qiongdongnan Basin, northern slope of the South China Sea. Geol. J. 54, 3556–3573. doi: 10.1002/gj.3351
Keywords: microbial diversity, thermogenic gas, natural gas hydrate, Illumina sequencing, marine sediments, South China Sea
Citation: Liu S, Yu S, Lu X, Yang H, Li Y, Xu X, Lu H and Fang Y (2022) Microbial communities associated with thermogenic gas hydrate-bearing marine sediments in Qiongdongnan Basin, South China Sea. Front. Microbiol. 13:1032851. doi: 10.3389/fmicb.2022.1032851
Edited by:
Weishu Zhao, Shanghai Jiao Tong University, ChinaReviewed by:
Hiroyuki Imachi, Japan Agency for Marine-Earth Science and Technology (JAMSTEC), JapanJiwen Liu, Ocean University of China, China
Copyright © 2022 Liu, Yu, Lu, Yang, Li, Xu, Lu and Fang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shan Yu, c2hhbnl1QHBrdS5lZHUuY24=; Yunxin Fang, ZmFuZ3l1bnhpbjA4MjVAc2luYS5jbg==
†These authors share first authorship