
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 30 September 2022
Sec. Microbial Physiology and Metabolism
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.1020932
This article is part of the Research Topic Role of Transcription Factors and Sigma Factors in Bacterial Stress Physiology View all 5 articles
 Eukene O. Bensig1
Eukene O. Bensig1 Cecilio Valadez-Cano2ZiYu Kuang2Isabela R. Freire2
Cecilio Valadez-Cano2ZiYu Kuang2Isabela R. Freire2 Adrian Reyes-Prieto2
Adrian Reyes-Prieto2 Shawn R. MacLellan2*
Shawn R. MacLellan2*Genes of unknown function constitute a considerable fraction of most bacterial genomes. In a Tn5-based search for stress response genes in the nitrogen-fixing facultative endosymbiont Sinorhizobium (Ensifer) meliloti, we identified a previously uncharacterized gene required for growth on solid media with increased NaCl concentrations. The encoded protein carries a predicted thioredoxin fold and deletion of the gene also results in increased sensitivity to hydrogen peroxide and cumene hydroperoxide. We have designated the gene srlA (stress resistance locus A) based on these phenotypes. A deletion mutant yields phenotypic revertants on high salt medium and genome sequencing revealed that all revertants carry a mutation in genes homologous to either cenK or cenR. srlA promoter activity is abolished in these revertant host backgrounds and in a strain carrying a deletion in cenK. We also observed that the srlA promoter is autoregulated, displaying low activity in a wildtype (wt) host background and high activity in the srl deletion mutant background. The srlA promoter includes a conserved inverted repeat directly upstream of the predicted −35 subsequence. A mutational analysis demonstrated that the site is required for the high promoter activity in the srlA deletion background. Electromobility shift assays using purified wildtype CenR response regulator and a D55E phosphomimetic derivative suggest this protein acts as a likely Class II activator by binding promoter DNA. These results document the first identified CenK–CenR regulon member in S. meliloti and demonstrate this two-component regulatory system and gene srlA influences cellular growth and persistence under certain stress-inducing conditions.
Identifying genetic loci required for bacterial growth and persistence in varying physicochemical environments is an important pursuit. The nitrogen-fixing facultative endosymbiont Sinorhizobium (Ensifer) meliloti (Kuzmanović et al., 2022) has a large (6.7 Mb) tripartite genome (Galibert et al., 2001). This bacterium can persist as a free-living soil bacterium or as an intracellular legume symbiont where it undergoes physiological and morphological changes and begins fixing atmospheric nitrogen that can stimulate the growth of wild or agriculturally important host plants. These cellular lifestyles occur in very different environments and the soil environment particularly can involve widely varying physicochemical conditions. Accordingly, S. meliloti has been the subject of attempts to identify genes that when disrupted conditionally impair growth under stressful conditions that include high salinity (Wei et al., 2004; Domínguez-Ferreras et al., 2006; Miller-Williams et al., 2006; Vriezen et al., 2013; Calatrava-Morales et al., 2017), low pH (Reeve et al., 1998; Lagares et al., 2016; Albicoro et al., 2021), oxidative stress (Davies and Walker, 2007; Flechard et al., 2009; Lehman and Long, 2018) and other physical or chemical conditions.
Stress responses require linked sensory perception/transcriptional regulatory systems and in bacteria this requirement is largely met by three major protein classes: the Extracytoplasmic Function (ECF) σ factors and their associated sensory anti-σ factors (Lonetto et al., 1994; Helmann, 2002), one-component transcription factors (Ulrich et al., 2005), and two-component regulatory systems (TCS) that usually include an integral membrane sensory histidine kinase and cognate DNA-binding response regulator (Zschiedrich et al., 2016). The genome of S. meliloti includes genes encoding 11 ECF σ factors, over 40 TCSs and, relative to its genome size, an unusually large number (approximately 390) of one-component transcription factors (Ulrich et al., 2005). Given its facultative lifestyle, relatively large genome, agricultural importance, and its potential for linking sensory perception to transcriptional response, S. meliloti is a good candidate for investigating the regulatory mechanisms of stress responses.
A previous initiative to investigate the global transcriptional impact of deleting all 11 S. meliloti ECF σ factors (Lang et al., 2018) demonstrated that over 380 genes are collectively regulated by the ECF complement. In this work we describe the generation of a strain in which 10 of the 11 ECF σ factors have been deleted, including the general stress response σ factor, RpoE2 (Sauviac et al., 2007; Bastiat et al., 2010) that itself regulates the expression of at least 320 genes (Lang et al., 2018). This provides a genetic background in which an important sensory and transcriptional regulatory complement has been largely eliminated. Our long-term goal is to identify potentially redundant stress response genes in S. meliloti, where their physiological import is only manifest in a strain lacking the ECF σ factor complement.
In this initial attempt to pursue that strategy using our ECF σ factor-deficient strain as host, we employed Tn5 mutagenesis to identify novel genes conditionally important for survival on a high salinity stress-inducing media formulation. Here we show that the insertional inactivation or deletion of a gene of unknown function (locus SMc03845) renders cells unable to grow on solid medium supplemented with 0.16 M NaCl. The predicted protein encoded by this gene includes a thioredoxin-like fold and cells also display increased sensitivity to hydrogen peroxide and cumene hydroperoxide. Our subsequent analysis demonstrated that in this instance, the growth impairment phenotype was not related to the absence of the ECF σ factor complement, but this is nevertheless the first report of a phenotype associated with SMc03845, currently annotated as a conserved hypothetical gene of unknown function. We characterized the SMc03845 promoter and show its activity is subject to both positive and negative regulation and is dependent upon CenK-CenR (Skerker et al., 2005; Lakey et al., 2022), a TCS that is emerging as an important regulator of cell envelope-related functions in the α-proteobacteria.
The bacterial strains and plasmids used in this study are listed in Supplementary Table S1. S. meliloti strains were cultured at 30°C and E. coli strains were grown at 37°C. Both species were grown on LB (Miller) broth or this medium solidified with 1.6% agar. For S. meliloti, LB broth was supplemented with 2.5 mM MgSO4 and 2.5 mM CaCl2. Other supplementations are as noted elsewhere.
Unmarked deletions in genes rpoE1–rpoE9 and ECF σ-like gene SMc01150 (Supplementary Table S2) were generated using the sacB suicide vector pJQ200 (Quandt and Hynes, 1993). Briefly, 500–600 bp regions upstream and downstream of each gene were amplified and cloned into pJQ200 using the enzyme-free AQUA method (Beyer et al., 2015). Individually each plasmid was conjugated into strain RmP110 (Yuan et al., 2006) and single cross-over events were selected on LB medium supplemented with streptomycin (500 μg/ml) and gentamicin (60 μg/ml). Cells from a single colony were grown overnight without selection and plated on medium supplemented with 5% sucrose. From these plates several colonies were screened for sucrose resistance, gentamicin sensitivity, and these isolates were subjected to PCR with primers flanking the gene to identify those containing the required gene deletion. Deletion of all 10 ECF σ factor genes in strain NB1836 was confirmed using PCR and by whole genome sequencing. This strain does not include deletion of the ECF σ factor FecI, which a transcriptomic analysis (Lang et al., 2018) suggested regulates the expression of only 3 of the approximately 380 genes collectively regulated by ECF σ factors in S. meliloti.
Unmarked deletions of genes srlA and cenK were generated and confirmed by PCR as described above using primers listed in Supplementary Table S1, and by genome sequencing.
To carry out mutagenesis of strain NB1836, 100 μl of an overnight culture was mixed with 100 μl of E. coli cells carrying pUT-miniTn5-Kn (Modal Inc.), spotted on an LB plate and incubated at 30°C overnight. After resuspension of the mating spot in 1 ml saline, 100 μl volumes were spread-plated on selective medium containing streptomycin (500 μg/ml) and neomycin (100 μg/ml). Typically, colonies arising from ten individual matings were resuspended in LB broth to generate a library of Tn5 insertion mutants. To screen for NaCl-sensitive insertion mutants, cells from the library stock were plated on 150 mm plates to a density of approximately 500 cfu/plate. After growth for 3 days, colonies were replica-plated onto plates containing growth medium supplemented with relevant antibiotics and this medium supplemented with 0.16 M NaCl. This concentration of NaCl was used based on preliminary trials in our laboratory using wt cells, where it delayed the rate of colony growth on solid medium (from 3 to 5 days) indicating stress, but ultimately did not prevent colony growth from single cells. Cells from colonies unable to grow on the salt-supplemented medium were selected for further analysis that included confirmation of the salt-sensitive phenotype and M12 phage transduction into the parental strain to confirm linkage of phenotype and insertion.
Arbitrary PCR was used to map the location of Tn5 insertions. Briefly, using purified genomic DNA as template, a combination of IS50 primers (Pr794 and Pr795) and degenerate primers (Pr791 and Pr796) were used in the first round of PCR. For the second round, nested primers (Pr793 and Pr797) were used. Amplified products extracted from agarose gels were subjected to Sanger sequencing using primers Pr793 and Pr797.
Sinorhizobium meliloti strains were tested for salt-sensitivity on LB plates supplemented with 0.16 M NaCl. Typically, 5 μl from an overnight culture was spotted onto a plate containing the required antibiotics and streaked for isolated colonies. For testing sensitivity to oxidizing agents, 20 μl of an overnight culture was mixed with 5 ml of LB top agar (0.4% agar) and poured over a 100 mm petri plate containing 20 ml of solidified LB agar. Filter discs (40 mm) were supplemented with 10 μl of the stock oxidizing agent and placed onto the solidified top agar. Inhibition zones were measured after 24 h incubation at 30°C.
Transcription activity from gene promoters was estimated by measuring GFPUV fluorescence in a Spectramax M3 fluorimeter (Molecular Devices) with an excitation wavelength of 340 nm and emission wavelength of 510 nm. For the srlA promoter, we measured GFP fluorescence in cultures grown overnight (24 h) and diluted, and cultures grown to an O.D of 0.5 from an overnight culture with both strategies yielding similar results. For convenience, we therefore used diluted overnight cultures for all promoter activity assays. Typically, S. meliloti cells carrying derivatives of the gfp reporter vector pOT1 were grown for 24 h at 30°C with shaking at 200 rpm. Cells from a 1 ml volume were washed once in saline (0.85% NaCl) and diluted (1,3) in saline. 200 μl volumes were measured for fluorescence and optical density at 600 nm. Specific activity was calculated from arbitrary GFP fluorescence units divided by optical density.
The transcription start site of srlA was mapped using the protocol and components supplied by the 5´ RACE System for Rapid Amplification of cDNA Ends kit (Invitrogen). Briefly, S. meliloti strain NB1854 cells carrying plasmid pEB57 were grown to an OD600 of 0.5. Total RNA was purified using the protocol and components of the TRIzol Max Bacterial RNA Isolation kit (Ambion). Primers used for nested PCR amplifications are listed in Supplementary Table S1.
Gene SMc03820 (cenR) DNA was amplified from RmP110 genomic DNA and ligated into pET17b generating pEB55. We used a Quikchange (Stratagene)-based site-directed mutagenesis protocol and primers Pr909 and Pr910 to change an aspartate codon at position 55 to a glutamate codon generating pEB56. Both the wt and D55E variant protein expression plasmids were transformed into E. coli BL21 (DE3; pLysS) cells. Expression of the proteins carrying a C-terminal hexahistidine tag was induced using 0.5 mM IPTG. A large fraction of protein formed inclusion bodies when expressed at 37°C. We therefore grew 1 l volumes at 37°C to an OD600 of 0.4 and transferred the volumes to 22°C before induction. After 18 h induction, cells were collected, subjected to 2 freeze–thaw cycles, resuspended in 20 ml lysis buffer (50 mM phosphate buffer [pH 7.5], 300 mM NaCl and 10 mM imidazole) and lysed using sonication. Insoluble material was pelleted by centrifugation for 20 min at 17,000 rpm and the clarified supernatant was supplemented with 0.5 ml (drained volume) of washed His-Bind Ni-NTA beads (Millipore). Protein binding occurred for 1 h at RT with slow rotation. After collection by low-speed centrifugation, beads were washed twice with 10 ml volumes of lysis buffer and transferred to a disposable chromatography column. To elute bound proteins, we used a step elution process (0.5 ml volumes, from 20 mM imidazole to 150 mM imidazole in 10 mM imidazole steps) and a final elution using 0.5 ml buffer containing 250 mM imidazole. Analysis by SDS-PAGE revealed essentially pure CenR protein in the 150 and 250 mM elution volumes. Buffer exchange to 50 mM phosphate (pH 7.5) lacking NaCl and imidazole was conducted using Amicon 10 kDa mw cutoff spin kits (Millipore). Protein concentration was determined using a Bradford-based protein assay (Bio-Rad) and bovine serum albumin (BSA) as standard.
Double-stranded probe (60 bp, biotin labeled at each 5′ end) including the SMc03845 inverted repeat was purchased from IDT. A typical 14 μl binding reaction included: 50 mM phosphate buffer (pH 7.5), 0.5 μg BSA, 0.05 μg poly (dI-dC), 20 fmol labeled probe, and CenR concentrations as specified. When applicable, 2 pmol of unlabeled wt and mutant competitor DNA was used per binding reaction. Each binding reaction was incubated at RT for 20 m prior to separation on a pre-run 6% acrylamide minigel in 0.5 × TBE at 10°C. After transferring DNA to positively charged nylon membrane (BrightStar, Ambion) and UV crosslinking, the labeled probe was detected using the LightShift Chemiluminscent EMSA kit (Pierce). A Chemidoc MP Imaging System (Bio-Rad) was used for data collection.
Genomic DNA from parental strains NB1854, NB1855 and six revertant derivatives of each parental strain was extracted using the GenCatch Genomic DNA Extraction Kit (Epoch Life Sciences). After measuring DNA concentration and quality (NanoVue Plus UV/Vis Spectrophotometer, GE), 150–300 ng/μl samples were submitted to The Center for Applied Genomics (TCAG; Toronto) for Nextera XT library preparation and whole genome sequencing (paired end, Illumina HiSeq2500). Quality checking of raw reads was performed using FastQC (Babraham Bioinformatics) analysis software followed by trimming using fastp (Chen et al., 2018). The trimmed reads were mapped to the S. meliloti Rm1021 reference genome (NCBI Accession number: ASM696v1) with Bowtie2 (Langmead and Salzberg, 2012) and the output (.sam file) was converted into.bam format using SAMtools.
As a comparator, an assembly for all sequence reads was performed via the SPAdes assembler tool using an in-house python script and the output files were aligned to the S. meliloti reference genome. MindTheGap and Ingap were used to detect single nucleotide polymorphisms (SNPs) or gene deletions. A visualization analysis was carried out using Geneious Prime 8.0.
Sanger sequencing services were provided by Genome Quebec (Montreal). Genome sequencing was conducted by The Centre for Applied Genomics at The Hospital for Sick Children (Toronto). Analysis was conducted using an in-house pipeline. E. coli transformation was conducted using cells rendered competent using the CaCl2 method. Transfer of plasmids into S. meliloti was accomplished using overnight triparental mating with E. coli strain MT616 carrying pRK600 as a helper plasmid. Site directed mutagenesis was accomplished using mutagenic oligonucleotides and a protocol based upon the Strategene QuikChange method (Agilent Technologies).
As part of a strategy to identify novel stress response genes we generated a strain of S. meliloti (NB1836; Supplementary Table S2) lacking most of its ECF sigma factors and used this as a host for mini-Tn5 transposon mutagenesis. After screening for growth defects on medium formulations that presented stressful growth conditions, we identified one insertion mutant that showed an inability to grow on LB (Miller) agar medium supplemented with 0.16 M NaCl. The transfer of neomycin resistance from the isolate to the parental strains NB1836 and RmP110 via transduction demonstrated that the salt-sensitive phenotype was linked to the Tn5 insertion. Using arbitrary PCR, we found that the transposon was inserted after nucleotide 89 in the open reading frame of gene SMc03845. Subsequently, the generation of an unmarked deletion of SMc03845 in NB1836 (generating NB1855), confirmed that deletion of the gene resulted in the same salt-sensitivity observed in the original transposon insertion mutant. Subsequently, the generation of unmarked SMc03845 deletions in wt strains Rm1021 (generating NB1903) and RmP110 (Yuan et al., 2006; generating NB1854) showed the same salt-sensitive phenotype (Figure 1; plate sectors b). We amplified the SMc03845 open reading frame (ORF) including 174 bp upstream of the ORF and ligated the product into plasmid pOT1 (Allaway et al., 2001). When this plasmid (pEB60) was conjugated into the two ∆SMc03845 strains (NB1903 and NB1854), the salt-sensitive phenotype was complemented in each case (Figure 1; plate sectors c). The fortuitous presence of two unique PvuI sites in the SMc03845 ORF enabled us to generate an in-frame 52 codon deletion in the gene carried by pEB60. When this plasmid (pEB66) was transferred into the ∆SMc03845 strains, the salt-sensitive phenotype was no longer complemented (Figure 1; plate sectors d). Using the wildtype strains Rm1021 and RmP110 in this analysis demonstrated that the mutant phenotype was not related to the absence of ten of the ECF σ factors in strain NB1836 and most of the remainder of our characterization of this gene therefore involved only the parental strains RmP110 or Rm1021 and ∆SMc03845 derivatives thereof. Based upon our phenotypic characterization of SMc03845, we have re-named SMc03845 srlA (stress resistance locus A).
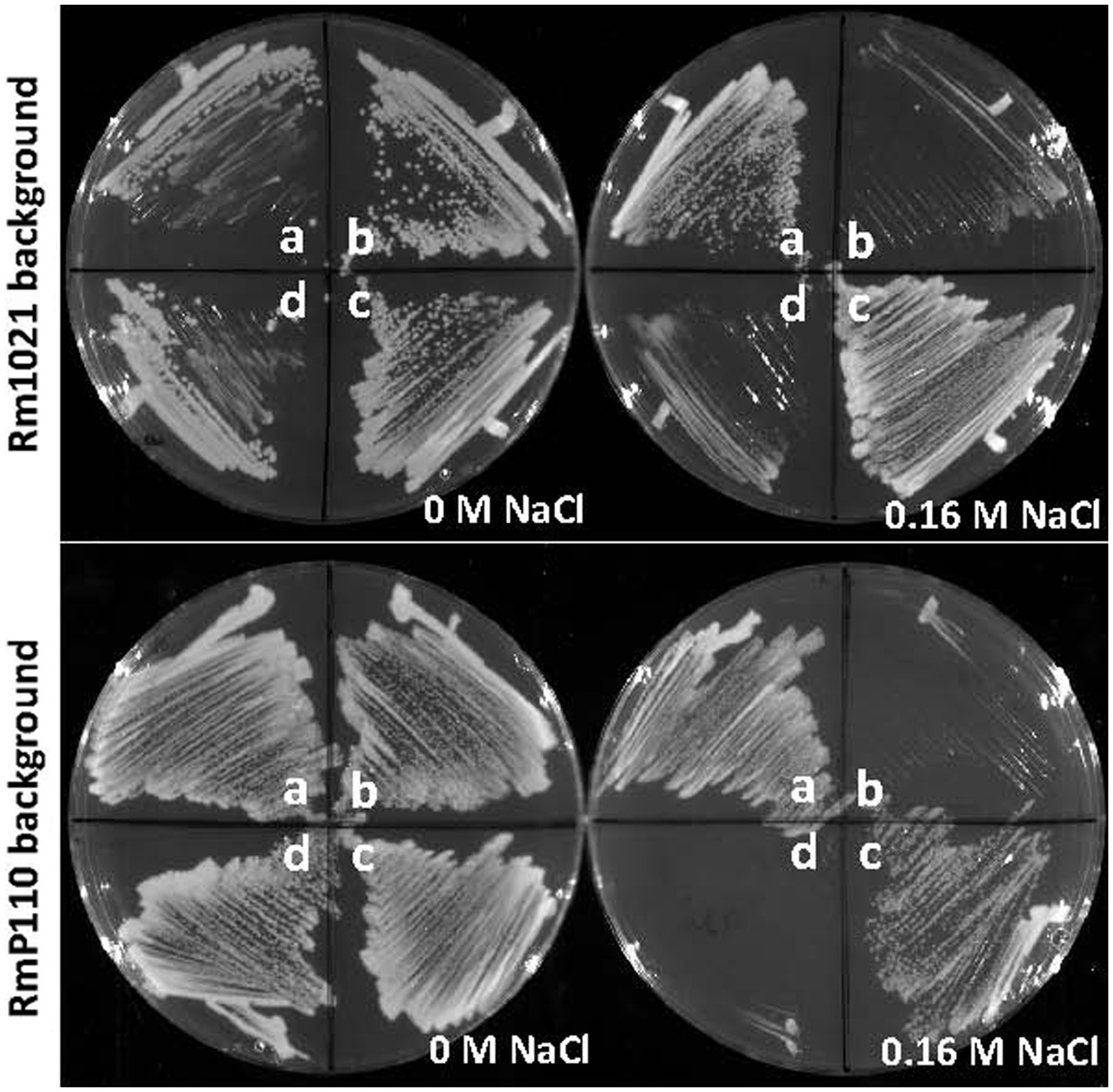
Figure 1. Salt-sensitive phenotype of srlA deletion strains. srlA was deleted in two wt host backgrounds: Rm1021 (top plates) and RmP110 (bottom plates) generating strains NB1903 and NB1854, respectively. Sectors a–d for all plates are as follows: (A), wt strain; (B), ∆srlA strain; (C), ∆srlA strain complemented with pEB60; (D), ∆srlA strain complemented with pEB66. Strains NB1903 and NB1854 fail to grow in LB medium supplemented with 0.16 M NaCl (sectors b). pEB60 carries the wt srlA ORF and complements the salt sensitive phenotype of a genomic srlA deletion (sectors c). pEB66 carries the srlA ORF with a 52 codon deletion and fails to complement the salt sensitive phenotype of a genomic srlA deletion (sectors d).
srlA is annotated as a conserved gene of unknown function and it is broadly distributed amongst the α-proteobacteria. A homolog of the protein from Agrobacterium fabrum strain C58 (protein Atu2684, 58% identity with SrlA) has been crystalized and structurally characterized (PDB 2AXO). We modeled the structure of SrlA from Atu2684 using the protein structure prediction program Phyre2 (Kelley et al., 2015) and Chimera (Pettersen et al., 2004). It carries a thioredoxin fold (Supplementary Figure S1) and is predicted to possess an N-terminal signal sequence. Although strain NB1854 (RmP110 [∆srlA]) is unable to grow on solid LB (Figure 1) supplemented with 0.16 M NaCl, we were unable to reproducibly detect a similar salt-sensitive growth defect during growth in liquid medium. Since SrlA appears to carry a redox-reactive thioredoxin fold, we tested sensitivity to a selection of inorganic and organic oxidizing agents. Relative to the parental strain RmP110, NB1854 demonstrated significantly (p < 0.0001) increased sensitivity to H2O2 and cumene hydroperoxide (Figure 2). Neither the parental strain nor the ∆srlA mutant showed sensitivity to paraquat, diquat, or ethyl viologen at the concentrations tested. Testing of benzyl viologen and tert-butyl hydroperoxide resulted in poorly demarcated inhibition zones that were difficult to accurately measure. No differential growth impairment was noted during growth at low pH (pH 5.6), elevated temperature (40°C), or on M9 minimal medium vs. LB medium for any of conditions tested (data not shown). Thus, the phenotypic consequences of srlA deletion appears rather restricted to elevated NaCl concentrations and certain oxidizing agents although our phenotypic characterization is continuing.
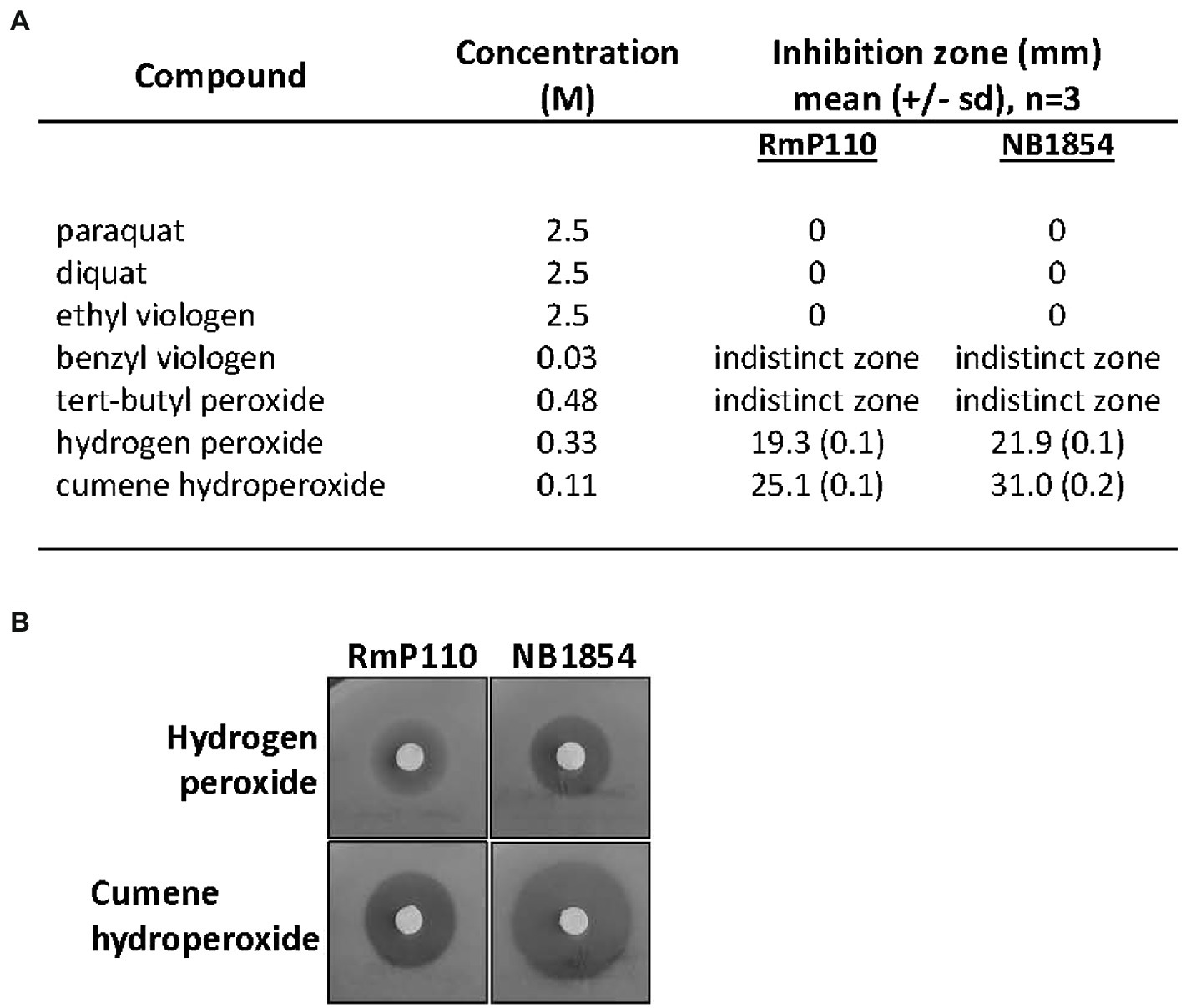
Figure 2. Effect of srlA deletion on resistance to oxidizing agents. (A) effects of compounds tested against wt strain RmP110 and ∆srlA strain NB1854. Benzyl peroxide and tert-butyl peroxide resulted in inhibition zones with indistinct margins that were difficult to measure. Mean differences between host strain zone diameters were significant for both hydrogen peroxide and cumene hydroperoxide (unpaired t-test, p < 0.0001). (B) representative disc experiments testing hydrogen peroxide and cumene hydroperoxide.
We amplified a 127 bp region upstream of the srlA ORF and ligated the product into the GFP reporter vector pOT1 in both orientations generating plasmids pEB57 and pEB58. Only the construct with the srlA fragment in the same orientation as the gfp gene (pEB57) displayed significant (p < 0.01) promoter activity relative to the empty vector (Figure 3). This construct displayed low promoter activity in the parental RmP110 host but much higher activity in the ∆srlA host NB1854 suggesting an autorepression effect. To examine this more closely, we measured transcriptional activity from pEB60 (carrying the srlA promoter contiguous with its entire ORF). In the ∆srlA host this construct showed low promoter activity statistically indistinguishable from activity in the wt host (Figure 4). In contrast, the transcriptional activity from pEB66 (the pEB60 derivative with an in-frame 52 codon PvuI deletion in the srlA ORF) was high and statistically indistinguishable (p < 0.01) from promoter activity observed in the host cells carrying a genomic srlA deletion and just the promoter in trans. Thus, activity of the srlA promoter is low when cells harbor an intact srlA gene (either on the chromosome or on a plasmid), but high in the absence of srlA suggesting that protein SrlA negatively regulates (either directly or indirectly) its own transcription.
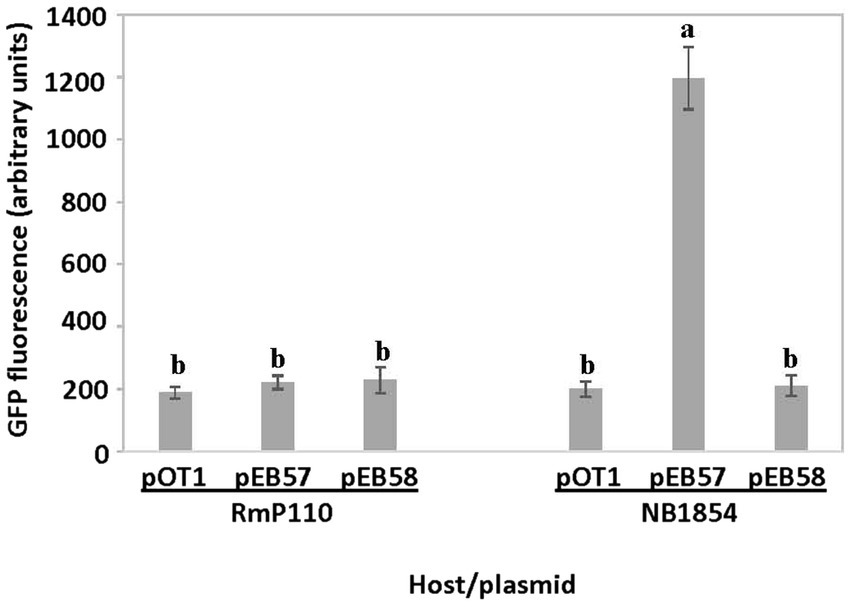
Figure 3. srlA promoter activity in wt and ∆srlA host backgrounds. Transcription activity detected from reporter vector pOT1, pEB57 (127 bp region upstream of srlA in same orientation as gfp) and pEB58 (127 bp region upstream of srlA in opposite orientation as gfp) in RmP110 (wt) and NB1854 (∆srlA) host strains. GFP activity reported as mean ± sd, n = 3. Letters indicate whether means are significantly different (p < 0.01, ANOVA and Tukey HSD).
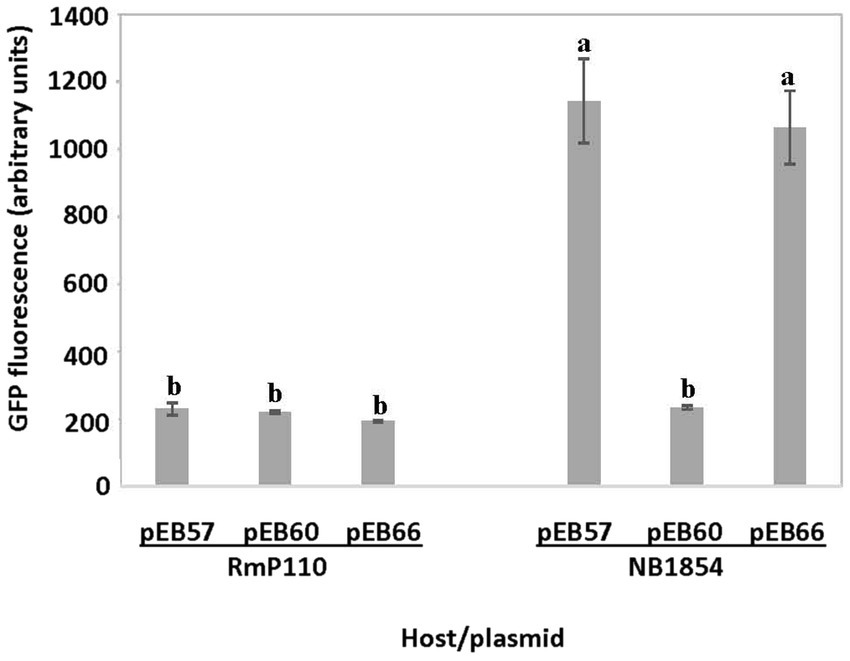
Figure 4. in cis repression of srlA promoter activity. Transcription activity detected from pEB57 (srlA promoter-gfp fusion) and pEB60 (srlA promoter-srlA ORF-gfp fusion) in RmP110 (wt) and NB1854 (∆srlA) host strains. pEB66 is a pEB60 derivative carrying an in-frame deletion in the srlA ORF. GFP activity reported as mean ± sd, n = 3. Letters indicate whether means are significantly different (p < 0.01, ANOVA and Tukey HSD).
SrlA carries a predicted thioredoxin-like fold (see Supplementary Figure S1). We found that a derivative carrying a C61S substitution mutation in its CXXC motif (pEB67) was still able to complement the salt-sensitive phenotype of a ∆srlA strain (Figure 5A) indicating that the derivative protein is expressed and functional. Interestingly however, the Cys substitution did diminish the autorepression phenotype (Figure 5B) yielding promoter activity that was statistically indistinguishable (p < 0.05) from the activity when repression was abolished by an in-frame deletion in plasmid-borne srlA.
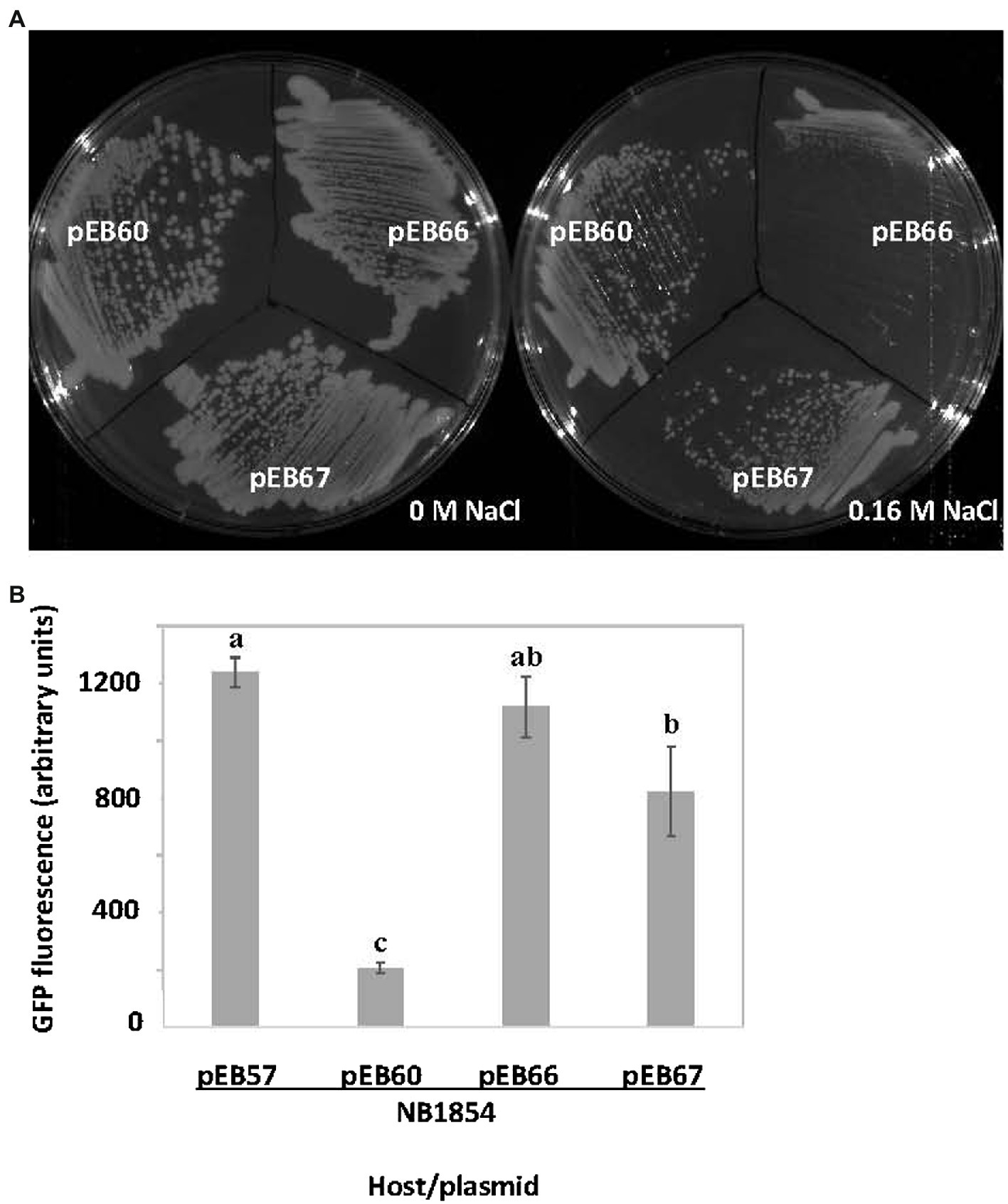
Figure 5. Effects of cysteine substitution mutation in SrlA thioredoxin fold. (A) the pEB60 (wt srlA ORF) derivative pEB67 carrying a C61S substitution mutation complements the salt-sensitive phenotype of ∆srlA strain NB1854. (B) the C61S SrlA substitution mutant expressed from pEB67 does not fully repress the srlA promoter as compared with expression of wt SrlA from pEB60. pEB66 is a pEB60 derivative carrying an in-frame deletion in the srlA ORF and can neither complement nor repress promoter. GFP activity reported as mean ± sd, n = 3. Letters indicate whether means are significantly different (p < 0.05, ANOVA and Tukey HSD).
A likely transcription start site (TSS, + 1) for srlA was previously deduced in a global transcriptomic analysis (Schlüter et al., 2013). Since that study also indicated other possible (probably artifactual) TSSs for the locus, we used 5-RACE to experimentally determine the srlA TSS and confirmed its location 99 bp upstream of the start codon (Supplementary Figure S2). From previous data (Schlüter et al., 2013), this analysis, and a comparative sequence alignment using analogous regions from other rhizobial genomes, we were able to deduce the approximate location of the promoter −10 and − 35 subsequences (Figure 6). The comparative sequence alignment also revealed the presence of an inverted repeat (AGTCAC–NNCNNN–GTGACT) directly upstream and adjacent to the predicted srlA-35 subsequence (Figure 6). To assess the importance of the repeat to srlA promoter activity, we generated two mutations: C(−47)G in the left arm of repeat and G(−38)T in the right arm of repeat and compared activities with the wt repeat promoter. Both the C(−47)G mutation and, to a lesser extent, the G(−38)T mutation significantly (p < 0.01) reduced promoter activity relative to the wt promoter. The promoter carrying both mutations showed the least activity but was not statistically distinguishable from the construct carrying only the C(−47)T mutation. (Figure 7). These data, and the configuration and location of the repeat, suggest it is likely a recognition sequence for a Class II activator protein, a class of transcription factor that binds at or adjacent to the promoter −35 subsequence and activates transcription via protein–protein interaction with RNA polymerase holoenzyme (specifically, domain 4 of the σ factor subunit; Browning and Busby, 2016)
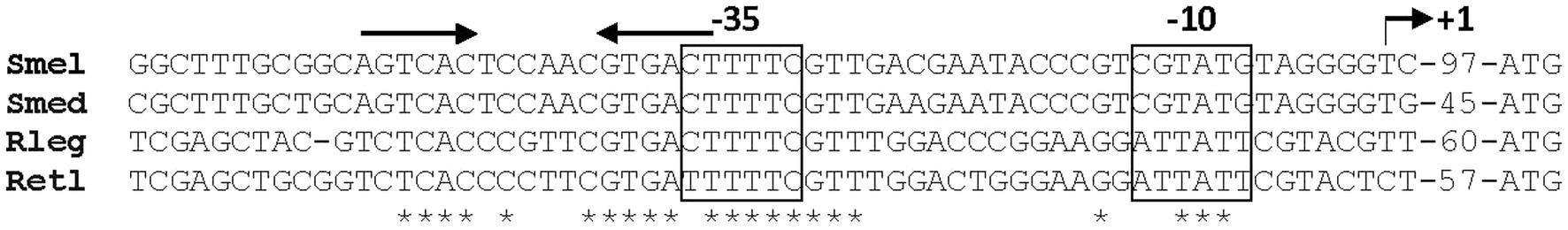
Figure 6. Alignment of srlA promoter region and analogous regions from other rhizobial genomes. srlA TSS (+1) from Schlüter et al. (2013) and experimentally verified (angled arrow) and distance to ATG start codon indicated. –10 and −35 subsequences (boxed) from Schlüter et al. (2013). Conserved inverted repeat indicated by arrows. Nucleotide conservation indicated by asterisks. Smel, S. meliloti 1,021 (locus SMc03845, srlA); Smed, Sinorhizobium medicae WSM419 (locus Sme_3082); Rleg, Rhizobium leguminosarum 3,841 (locus RL4535); Retl, Rhizobium etli CFN42 (locus RHE_CH03943).
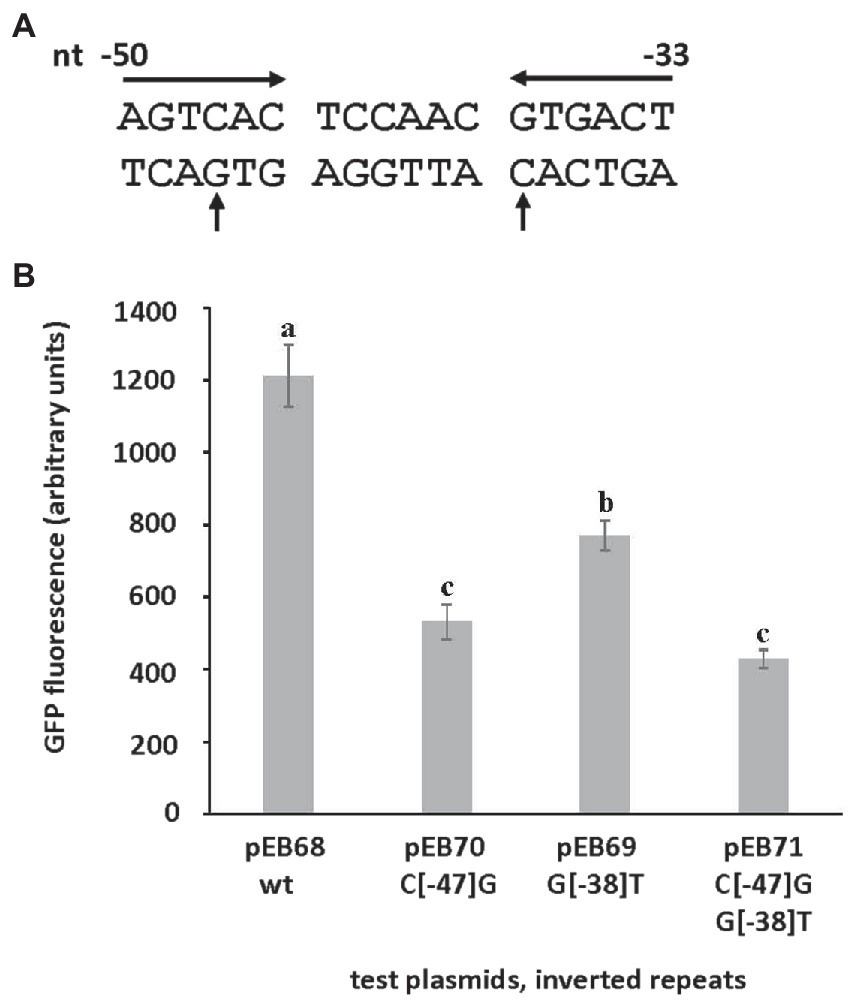
Figure 7. The effect of mutations in inverted repeat on srlA promoter activity. (A) srlA promoter repeat sequence (inverted arrows). Nucleotide numbering from + 1 TSS. Mutated nucleotides (C−47 and G−38) indicated (vertical arrows). (B) wt and mutant repeat srlA promoter activities in NB1854 host cells. GFP activity reported as mean ± sd, n = 3. Letters indicate whether means are significantly different (p < 0.01, ANOVA and Tukey HSD).
Using the perfect repeat found in the srlA promoter and allowing one mismatch, a computational search for similar inverted repeat sequences in the S. meliloti Rm1021 genome revealed three additional imperfect repeats in the predicted promoter regions of genes SMc01226, SMc00517 and chvI (Table 1). In addition to strong conservation in the left and right arms of the repeats, all four share a conserved C nucleotide in the spacer region between the repeats. Interestingly, the inverted repeat in SMc01226 is in the same position as the repeat in srlA—directly upstream and adjacent to the −35 subsequence. In contrast, in both SMc00517 and chvI, the repeats are almost identically located close to their respective −10 subsequences and TSSs.

Table 1. Related inverted repeats in four promoter sequences.
As described above, deletion of srlA results in the inability to form colonies on solid medium supplemented with 0.16 M NaCl, even after prolonged incubation. However, we frequently observed the emergence of a very few colonies on plates inoculated with high cell densities and these colonies formed with a growth rate similar to that of wt strain colony formation on the same high salt medium. We isolated several of these phenotypic revertants arising in two different ∆srlA host backgrounds: five from the NB1854 background and six from the ECF σ factor deletion strain (NB1855) in which we first detected the Tn5 insertional inactivation of srlA. Genome sequencing of these independently acquired revertants revealed the striking finding that every revertant possessed a mutation in either gene SMc01716 (encoding a predicted histidine kinase) or in gene SMc03820, encoding a likely DNA-binding response regulator (Table 2). These proteins are homologs of the Caulobacter crescentus histidine kinase CenK and response regulator CenR and were previously predicted (Skerker et al., 2005) to comprise a likely two-component regulatory system in S. meliloti. The concordance of mutations in either SM01716 or SMc03820 in every revertant isolate (Table 2) suggests these proteins do indeed form a cognate pair of regulators in S. meliloti. Here we will refer to this S. meliloti TCS as CenK–CenR after the nomenclature used in C. crescentus. Discounting identical mutations within backgrounds (likely sibling effects) we documented four unique mutations (two in cenK and two in cenR) amongst the NB1854 reversion isolates and five unique mutations (three in cenK and two in cenR) in the NB1855 isolates (Table 2). Taken together, seven of the nine unique isolates possessed point mutations leading to amino acid substitutions in their respective proteins. Of the two without amino acid substitution mutations, revertant 2A had a single nucleotide substitution ten nucleotides upstream the cenR ATG start codon. This changes a likely ribosome-binding site sequence (GGAA) to GTAA and possibly reduces accumulation of CenR in the cell. Revertant 1B had a single nucleotide insertion causing a frameshift mutation early (after codon 85) in the 506 codon cenK gene (Table 2). The amino acid substitution mutations and their structural and functional consequences will be the subject of a future communication.
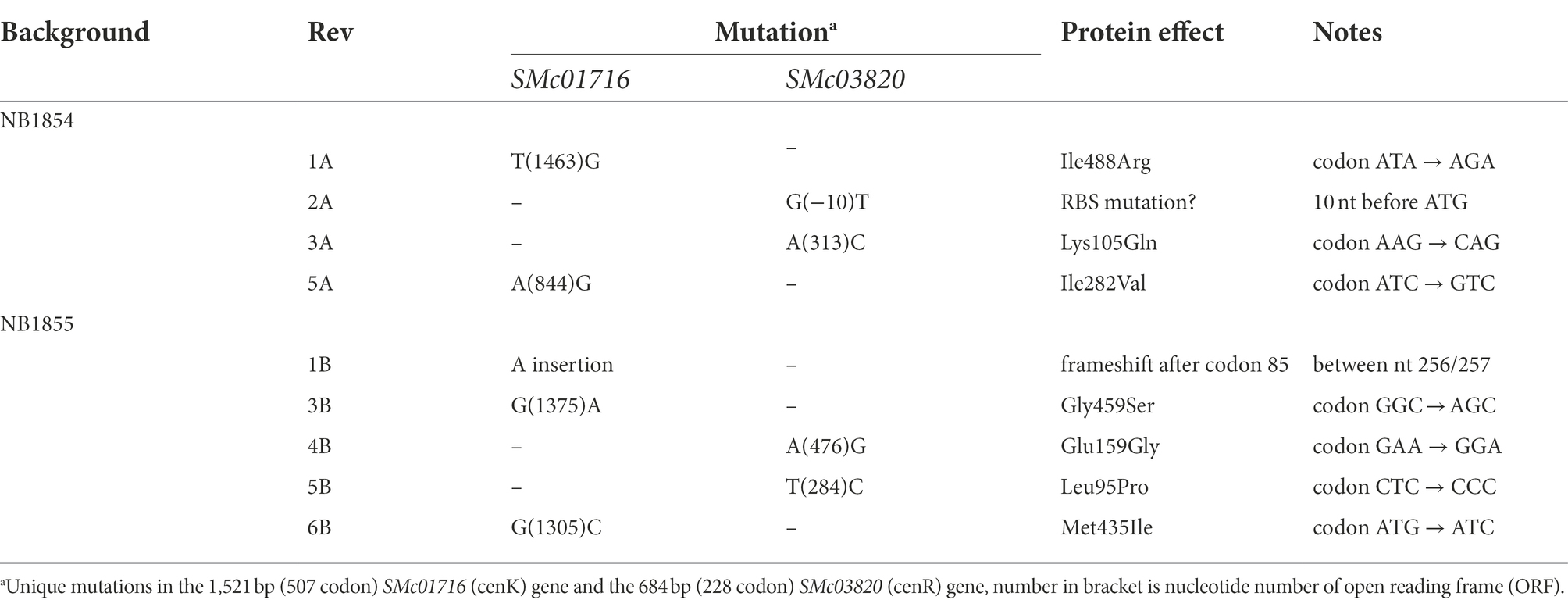
Table 2. Unique mutations detected in salt-resistant ∆srlA phenotypic revertant strains.
Two lines of genetic evidence suggests that S. meliloti CenK-CenR regulates the expression of SrlA. First, while the srlA promoter-gfp transcriptional fusion in pEB57 displays high promoter activity in a ∆srlA background, the promoter activity is very low in a ∆srlA ∆cenK double mutant (Figure 8A) and is statistically indistinguishable (p < 0.01) from the low (repressed) promoter activity in the wt host background. Second, when we transferred the promoter-gfp reporter vector into the revertant strains 1A, 2A, 3A, and 5A (carrying the various point mutations in either cenK or cenR, Table 2) srlA promoter activity was also abolished in every instance (Figure 8A). These data demonstrate that CenK–CenR, either directly or indirectly, regulates transcription from the srlA promoter.
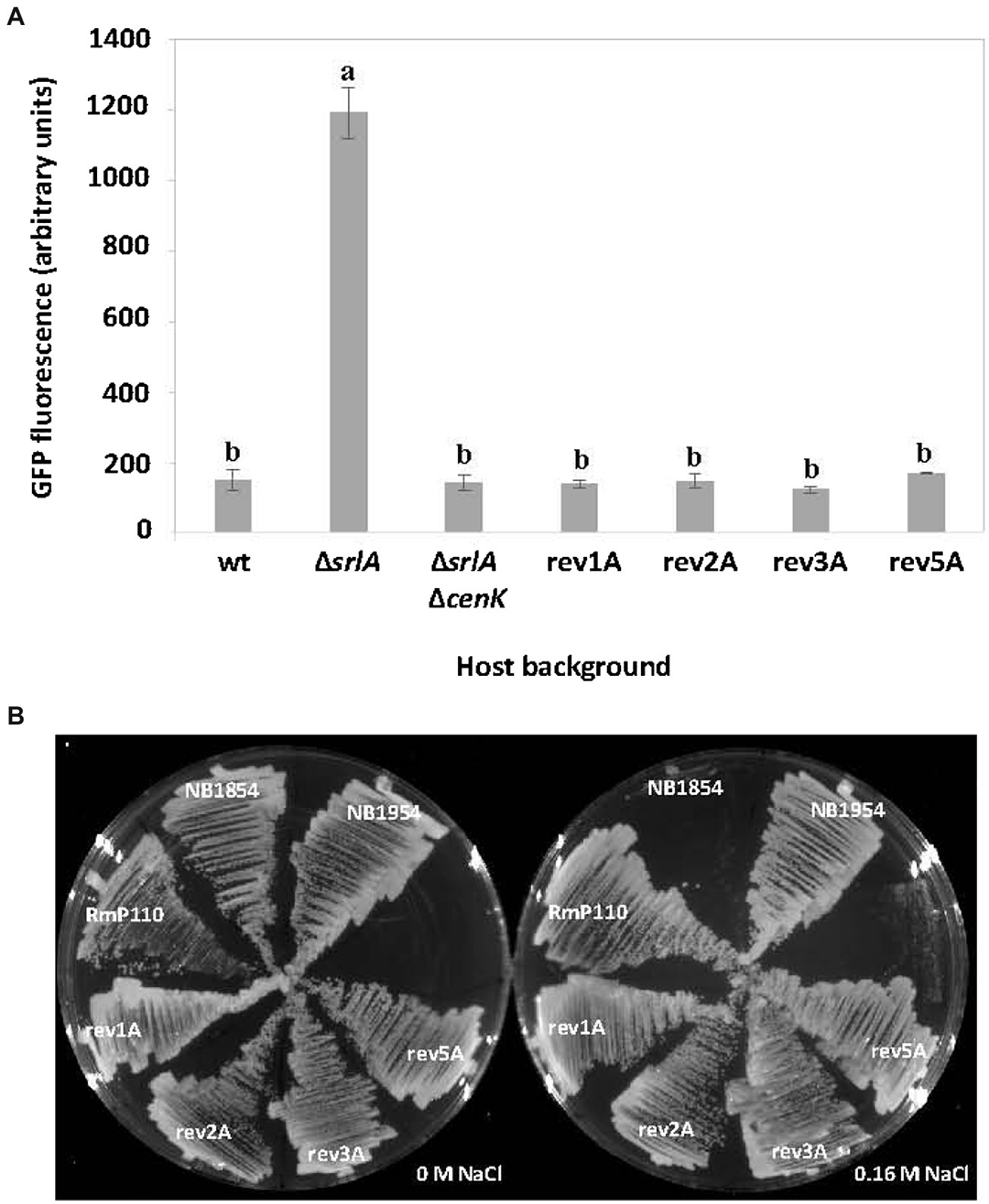
Figure 8. The effect of host cenK and cenR mutations on srlA promoter activity and salt-sensitivity. (A) transcription activity detected from srlA-gfp transcriptional fusion in plasmid pEB57 in genetic hosts: RmP110 (wt), NB1854 (∆srlA), NB1954 (∆srlA ∆cenK), and revertant strains 1A, 2A, 3A, and 5A (see Table 2 for revertant strain genotypes). GFP activity reported as mean ± sd, n = 3. Letters indicate whether means are significantly different (p < 0.01, ANOVA and Tukey HSD). (B) growth of strains on LB medium supplemented with 0 and 0.16 M NaCl.
Finally, since mutations in either cenK or cenR cause reversion of the salt-sensitive phenotype of strain NB1854 we reasoned that an unmarked deletion of cenK in strain NB1854 should have the same effect. Indeed, the ∆srlA ∆cenK double mutant (NB1954) grows as well as the revertants and the wildtype strain on medium supplemented with 0.16 M NaCl (Figure 8B).
To ascertain whether the response regulator CenR directly regulates activity from the srlA promoter we expressed and purified (Supplementary Figure S3) hexahistidine-tagged wt CenR protein and a phosphomimetic mutant derivative of CenR, CenRD55E, and conducted electromobility shift assays (EMSAs) using srlA promoter DNA as a probe. The previously described inverted repeat centered at position −42 of the srlA promoter was centrally located in the 60 bp double-stranded DNA we used as probe (Supplementary Figure S4).
Both CenRwt and CenRD55E bound and shifted probe DNA (Figure 9A) and at least qualitatively it appears as if CenRD55E has a greater affinity for the probe than does CenRwt. The binding of CenRD55E to srlA promoter DNA appears to be specific and dependent upon the inverted repeat sequence since challenging the binding with unlabeled DNA carrying the inverted repeat diminishes the observed shift (Figure 9B) but challenging with unlabeled DNA carrying a mutated inverted repeat does not.

Figure 9. Binding of CenR protein to 60 bp srlA promoter region (see “Materials and methods”; Supplementary Figure S4). (A) Biotinylated probe (20 fmol) associated with no protein (lane 1) and increasing concentrations of purified (see Supplementary Figure S3) CenRD55E (lanes 2–3) or CenRwt (lanes 4–10) protein. (B) Probe binding challenged with unlabeled wt (lane 3) or unlabeled mutated DNA (lane 4).
Previous attempts to isolate salt-sensitive Tn5 mutants of S. meliloti identified a diverse group of genes that when disrupted led to an inability to tolerate elevated salt concentrations in growth media. In strain Rm1021, these genes included SMb2012 (a putative DNA ligase), fabG, pgm, exoF1, tig, and ftsE (Miller-Williams et al., 2006) and in strain 042BM included genes omp10 (a probable outer membrane lipoprotein), greA, relA, and nuoL (Wei et al., 2004).
Our attempt to detect salt-sensitive mutants resulted in the identification of an insertion in gene SMc03845, which led to increased sensitivity to elevated NaCl concentrations in growth medium, and sensitivity to certain oxidizing agents. This is the first attribution of a phenotype to gene SMc03845 (srlA), a conserved gene of unknown function. To our knowledge, previous attempts to identify salt or oxidizing agent-sensitive mutants did not identify this locus. At least in the case of salt sensitivity, earlier attempts to identify mutants may have failed to detect srlA for two reasons. First, we used strain RmP110, a derivative of Rm1021 in which a frame-shift mutation in the phosphate transport gene pstC was repaired (Yuan et al., 2006). We note that while Rm1021 (∆srlA) is also salt-sensitive and unable to grow on medium supplemented with 0.16 M NaCl, its sensitivity is somewhat more muted than a RmP110 (∆srlA) derivative and most earlier screens for similar mutant phenotypes have used Rm1021 as host. Second, the salt-sensitive phenotype is only displayed during growth on solid medium, not in liquid culture, and this might also have contributed to not detecting srlA mutants in earlier screens. We are continuing our examination of SrlA to characterize its subcellular location, its role in the cell and the function of its thioredoxin-like domain, the phenotypic consequences of its deletion, and how its synthesis contributes to its transcriptional autorepression.
In addition to the autorepression phenotype we detected, our analysis of srlA promoter regulation also revealed that the CenK–CenR TCS is required for srlA promoter activity. The effect of deletion of a cenR homolog has been studied in Brucella melitensis (Zhang et al., 2009) and homologous CenK–CenR systems have been characterized in C. crescentus (Skerker et al., 2005) and Rhodobacter sphaeroides (Burger et al., 2017; Lakey et al., 2022) but not in S. meliloti. Lakey et al. (2022) predicted a recognition sequence for CenR binding in R. sphaeroides and other selected α-proteobacteria. This sequence (TGA–N8–TGA) aligns reasonably well with the inverted repeat sequence (AGTCAC–N6–GTGACT) we identified in S. meliloti.
Several of the results in this initial characterization seem enigmatic but hint at a more complex regulatory scheme that awaits description. First, it is surprising that impairment of a TCS system required for srlA transcription would also revert the salt-sensitive phenotype of a ∆srlA mutant. We are currently using genomic approaches to determine the CenK–CenR regulon but surmise that the loss of expression of one or more other regulon members in a CenK mutant leads to a compensatory phenotype that nulls the salt-sensitive phenotype of a srlA deletion mutant. Secondly, the response regulator CenR is essential in C. crescentus (Skerker et al., 2005) and R. sphaeroides (Burger et al., 2017; Lakey et al., 2022) and based on a Tn-Seq analysis (diCenzo et al., 2018) was predicted to be essential in S. meliloti. In accord with this prediction, despite repeated attempts we were unable to delete cenR although cenK could be readily deleted. This parallels the recent finding in Rhodobacter sphaeroides (Lakey et al., 2022) that cenR, but not cenK, is essential in that bacterium. Thus, although CenK and CenR are cognate, CenR must be able to retain its essential function in the cell in the absence of the kinase. Possibly, CenR can be phosphorylated by an alternative kinase or its essential function in the cell is mediated by the unphosphorylated form of the protein. Notwithstanding these possibilities, the deletion of CenK does abolish srlA promoter activity in S. meliloti, suggesting that phosphorylated CenR is required at this promoter in the cell. From in vitro EMSA experiments, it does qualitatively appear as if CenRD55E binds srlA promoter DNA with greater affinity than CenRwt, but confirmation of this awaits a more quantitative analysis. Third, although CenR is essential in S. meliloti, we detected point mutations in cenR that leads to loss of srlA promoter activity and reversion of the salt-sensitive phenotype of a ∆srlA strain. Evidently, while these mutations in CenR debilitate its action at the srlA promoter, they are not lethally inactivating. Our current results suggest that CenR binds srlA promoter DNA with apparent specificity for the inverted repeat and this is consistent with our genetic examination of promoter activation. To strengthen the conviction that CenR directly acts upon the srlA promoter, we are currently continuing our DNA binding experiments and extending the analysis to the related repeats found in genes SMc01226, SMc00517, and chvI and will include purified CenR carrying the point mutations detected in our reversion analysis.
With respect to the similar promoter inverted repeats found in the S. meliloti genome, the differential location of these repeats, either adjacent to the −35 subsequences or near the TSS, suggests CenR can act either as an activator or repressor of transcription. It is worthwhile to note that while the cellular roles of proteins SrlA, SMc01226, and SMc00517 remain to be elucidated, ChvI is a particularly well-studied and important regulator of cell envelope-related functions in S. meliloti (Cheng and Walker, 1998; Bélanger et al., 2009; Chen et al., 2009; Bélanger and Charles, 2013; Ratib et al., 2018). The possibility this present work has identified an additional regulator of ChvI expression is intriguing.
Finally, the phenomenon of autorepression of the srlA promoter by SrlA expression also hints at a more complex regulatory network that awaits discovery. Although it is possible that SrlA directly binds srlA promoter DNA, it does not possess a recognizable DNA-binding motif and is predicted to possess an N-terminal signal sequence peptide. We are therefore exploring the possibility that secretion of SrlA stimulates a secondary signaling pathway that results in expression of a repressor protein that either acts directly upon the srlA promoter by occluding RNA polymerase binding, or by preventing the action of CenR at the srlA promoter. As documented in this work, a substitution mutation in one of the two cysteine residues in the thioredoxin fold of SrlA does not impair its ability to complement the salt-sensitive phenotype of a ∆srlA mutant but does diminish the autorepression effect. Whatever the primary role of SrlA in the cell and the importance of its thioredoxin-like domain to its function, this raises the possibility that at least the mechanistic path to autorepression involves a disulfide-dependent switch.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
EB and SM conducted the experimental research and wrote the manuscript. ZK and IF contributed to experimental work. CV-C and AR-P contributed to genome sequence analysis and other computational analyses. All authors contributed to the article and approved the submitted version.
This study was supported by a grant to SM from the Natural Sciences and Engineering Research Council of Canada (grant no. 386710). Molecular graphics were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
We thank Denise Clark, Madeleine Crawford, Rakshit Galwa, Jinny MacLellan, Eric Meng, and Cheryl Patten for technical assistance and helpful discussions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1020932/full#supplementary-material
Albicoro, F. J., Draghi, W. O., Martini, M. C., Salas, M. E., Torres Tejerizo, G. A., Lozano, M. J., et al. (2021). The two-component system ActJK is involved in acid stress tolerance and symbiosis in Sinorhizobium meliloti. J. Biotechnol. 329, 80–91. doi: 10.1016/J.JBIOTEC.2021.01.006
Allaway, D., Schofield, N. A., Leonard, M. E., Gilardoni, L., Finan, T. M., and Poole, P. S. (2001). Use of differential fluorescence induction and optical trapping to isolate environmentally induced genes. Environ. Microbiol. 3, 397–406. doi: 10.1046/j.1462-2920.2001.00205.x
Bastiat, B., Sauviac, L., and Bruand, C. (2010). Dual control of Sinorhizobium meliloti RpoE2 sigma factor activity by two PhyR-type two-component response regulators. J. Bacteriol. 192, 2255–2265. doi: 10.1128/JB.01666-09
Bélanger, L., and Charles, T. C. (2013). Members of the Sinorhizobium meliloti ChvI regulon identified by a DNA binding screen. BMC Microbiol. 13:132. doi: 10.1186/1471-2180-13-132
Bélanger, L., Dimmick, K. A., Fleming, J. S., and Charles, T. C. (2009). Null mutations in Sinorhizobium meliloti exoS and chvI demonstrate the importance of this two-component regulatory system for symbiosis. Mol. Microbiol. 74, 1223–1237. doi: 10.1111/j.1365-2958.2009.06931.x
Beyer, H. M., Gonschorek, P., Samodelov, S. L., Meier, M., Weber, W., and Zurbriggen, M. D. (2015). AQUA cloning: a versatile and simple enzyme-free cloning approach. PLoS One 10:e0137652. doi: 10.1371/journal.pone.0137652
Browning, D. F., and Busby, S. J. W. (2016). Local and global regulation of transcription initiation in bacteria. Nat. Rev. Microbiol. 14, 638–650. doi: 10.1038/nrmicro.2016.103
Burger, B. T., Imam, S., Scarborough, M. J., Noguera, D. R., and Donohue, T. J. (2017). Combining genome-scale experimental and computational methods to identify essential genes in Rhodobacter sphaeroides. mSystems 2, e00015–e00017. doi: 10.1128/MSYSTEMS.00015-17/SUPPL_FILE/SYS003172109SD9.PDF
Calatrava-Morales, N., Nogales, J., Ameztoy, K., van Steenbergen, B., and Soto, M. J. (2017). The NtrY/NtrX system of Sinorhizobium meliloti GR4 regulates motility, EPS i production, and nitrogen metabolism but is dispensable for symbiotic nitrogen fixation. Mol. Plant-Microbe Interact. 30, 566–577. doi: 10.1094/MPMI-01-17-0021-R
Chen, E. J., Fisher, R. F., Perovich, V. M., Sabio, E. A., and Long, S. R. (2009). Identification of direct transcriptional target genes of ExoS/ChvI two-component signaling in Sinorhizobium meliloti. J. Bacteriol. 191, 6833–6842. doi: 10.1128/JB.00734-09
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Cheng, H. P., and Walker, G. C. (1998). Succinoglycan production by rhizobium meliloti is regulated through the ExoS-ChvI two-component regulatory system. J. Bacteriol. 180, 20–26. doi: 10.1128/jb.180.1.20-26.1998
Davies, B. W., and Walker, G. C. (2007). Identification of novel Sinorhizobium meliloti mutants compromised for oxidative stress protection and symbiosis. J. Bacteriol. 189, 2110–2113. doi: 10.1128/JB.01802-06
diCenzo, G. C., Benedict, A. B., Fondi, M., Walker, G. C., Finan, T. M., Mengoni, A., et al. (2018). Robustness encoded across essential and accessory replicons of the ecologically versatile bacterium Sinorhizobium meliloti. PLoS Genet. 14:e1007357. doi: 10.1371/JOURNAL.PGEN.1007357
Domínguez-Ferreras, A., Pérez-Arnedo, R., Becker, A., Olivares, J., Soto, M. J., and Sanjuán, J. (2006). Transcriptome profiling reveals the importance of plasmid pSymB for osmoadaptation of Sinorhizobium meliloti. J. Bacteriol. 188, 7617–7625. doi: 10.1128/JB.00719-06
Flechard, M., Fontenelle, C., Trautwetter, A., Ermel, G., and Blanco, C. (2009). Sinorhizobium meliloti rpoE2 is necessary for H2O2 stress resistance during the stationary growth phase. FEMS Microbiol. Lett. 290, 25–31. doi: 10.1111/j.1574-6968.2008.01401.x
Galibert, F., Finan, T. M., Long, S. R., Pühler, A., Abola, P., Ampe, F., et al. (2001). The composite genome of the legume symbiont Sinorhizobium meliloti. Science 293, 668–672. doi: 10.1126/science.1060966
Helmann, J. D. (2002). The extracytoplasmic function (ECF) sigma factors. Adv. Microb. Physiol. 46, 47–110. doi: 10.1016/S0065-2911(02)46002-X
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. E. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Kuzmanović, N., Fagorzi, C., Mengoni, A., Lassalle, F., and Dicenzo, G. C. (2022). Taxonomy of Rhizobiaceae revisited: proposal of a new framework for genus delimitation. Int. J. Syst. Evol. Microbiol. 72. doi: 10.1099/ijsem.0.005243
Lagares, A., Draghi, W. O., del Papa, M. F., Hellweg, C., Watt, S. A., Watt, T. F., et al. (2016). A consolidated analysis of the physiologic and molecular responses induced under acid stress in the legume-symbiont model-soil bacterium Sinorhizobium meliloti. Sci. Rep. 6. doi: 10.1038/SREP29278
Lakey, B. D., Myers, K. S., Alberge, F., Mettert, E. L., Kiley, P. J., Noguera, D. R., et al. (2022). The essential Rhodobacter sphaeroides CenKR two-component system regulates cell division and envelope biosynthesis. PLoS Genet. 18:e1010270. doi: 10.1371/journal.pgen.1010270
Lang, C., Barnett, M. J., Fisher, R. F., Smith, L. S., Diodati, M. E., and Long, S. R. (2018). Most Sinorhizobium meliloti Extracytoplasmic function sigma factors control accessory functions. mSphere 3:3. doi: 10.1128/mspheredirect.00454-18
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lehman, A. P., and Long, S. R. (2018). OxyR-dependent transcription response of Sinorhizobium meliloti to oxidative stress. J. Bacteriol. 200:e00622–17. doi: 10.1128/JB.00622-17
Lonetto, M. A., Brown, K. L., Rudd, K. E., and Buttner, M. J. (1994). Analysis of the Streptomyces coelicolor sigE gene reveals the existence of a subfamily of eubacterial RNA polymerase σ factors involved in the regulation of extracytoplasmic functions. Proc. Natl. Acad. Sci. U. S. A. 91, 7573–7577. doi: 10.1073/pnas.91.16.7573
Miller-Williams, M., Loewen, P. C., and Oresnik, I. J. (2006). Isolation of salt-sensitive mutants of Sinorhizobium meliloti strain Rm1021. Microbiology (Reading) 152, 2049–2059. doi: 10.1099/MIC.0.28937-0
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Quandt, J., and Hynes, M. F. (1993). Versatile suicide vectors which allow direct selection for gene replacement in gram-negative bacteria. Gene 127, 15–21. doi: 10.1016/0378-1119(93)90611-6
Ratib, N. R., Sabio, E. Y., Mendoza, C., Barnett, M. J., Clover, S. B., Ortega, J. A., et al. (2018). Genome-wide identification of genes directly regulated by ChvI and a consensus sequence for ChvI binding in Sinorhizobium meliloti. Mol. Microbiol. 110, 596–615. doi: 10.1111/mmi.14119
Reeve, W. G., Tiwari, R. P., Wong, C. M., Dilworth, M. J., and Glenn, A. R. (1998). The transcriptional regulator gene phrR in Sinorhizobium meliloti WSM419 is regulated by low pH and other stresses. Microbiology (N Y) 144, 3335–3342. doi: 10.1099/00221287-144-12-3335
Sauviac, L., Philippe, H., Phok, K., and Bruand, C. (2007). An extracytoplasmic function sigma factor acts as a general stress response regulator in Sinorhizobium meliloti. J. Bacteriol. 189, 4204–4216. doi: 10.1128/JB.00175-07
Schlüter, J. P., Reinkensmeier, J., Barnett, M. J., Lang, C., Krol, E., Giegerich, R., et al. (2013). Global mapping of transcription start sites and promoter motifs in the symbiotic α-proteobacterium Sinorhizobium meliloti 1021. BMC Genom. 14:156. doi: 10.1186/1471-2164-14-156
Skerker, J. M., Prasol, M. S., Perchuk, B. S., Biondi, E. G., and Laub, M. T. (2005). Two-component signal transduction pathways regulating growth and cell cycle progression in a bacterium: a system-level analysis. PLoS Biol. 3:e334. doi: 10.1371/journal.pbio.0030334
Ulrich, L. E., Koonin, E. v., and Zhulin, I. B. (2005). One-component systems dominate signal transduction in prokaryotes. Trends Microbiol. 13, 52–56. doi: 10.1016/J.TIM.2004.12.006
Vriezen, J. A. C., de Bruijn, F. J., and Nüsslein, K. (2013). Identification and characterization of a NaCl-responsive genetic locus involved in survival during desiccation in Sinorhizobium meliloti. Appl. Environ. Microbiol. 79, 5693–5700. doi: 10.1128/AEM.01037-13
Wei, W., Jiang, J., Li, X., Wang, L., and Yang, S. S. (2004). Isolation of salt-sensitive mutants from Sinorhizobium meliloti and characterization of genes involved in salt tolerance. Lett. Appl. Microbiol. 39, 278–283. doi: 10.1111/j.1472-765X.2004.01577.x
Yuan, Z. C., Zaheer, R., and Finan, T. M. (2006). Regulation and properties of PstSCAB, a high-affinity, high-velocity phosphate transport system of Sinorhizobium meliloti. J. Bacteriol. 188, 1089–1102. doi: 10.1128/JB.188.3.1089-1102.2006
Zhang, X., Ren, J., Li, N., Liu, W., and Qing, W. (2009). Disruption of the BMEI0066 gene attenuates the virulence of Brucella melitensis and decreases its stress tolerance. Int. J. Biol. Sci. 5, 570–577. doi: 10.7150/ijbs.5.570
Keywords: transcription, two-component regulatory, cenK, chvI, alpha-proteobacteria, salt stress, oxidative stress, thioredoxin
Citation: Bensig EO, Valadez-Cano C, Kuang Z, Freire IR, Reyes-Prieto A and MacLellan SR (2022) The two-component regulatory system CenK–CenR regulates expression of a previously uncharacterized protein required for salinity and oxidative stress tolerance in Sinorhizobium meliloti. Front. Microbiol. 13:1020932. doi: 10.3389/fmicb.2022.1020932
Edited by:
Herb Schellhorn, McMaster University, CanadaReviewed by:
Chang Fu Tian, China Agricultural University, ChinaCopyright © 2022 Bensig, Valadez-Cano, Kuang, Freire, Reyes-Prieto and MacLellan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shawn R. MacLellan, c3JtQHVuYi5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.