
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 24 November 2022
Sec. Terrestrial Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.1018940
This article is part of the Research Topic Geomicrobes: Life in Terrestrial Deep Subsurface, volume II View all 6 articles
 Sunanda Mandal1
Sunanda Mandal1 Himadri Bose2
Himadri Bose2 Kheerthana Ramesh1
Kheerthana Ramesh1 Rajendra Prasad Sahu2Anumeha Saha2
Rajendra Prasad Sahu2Anumeha Saha2 Pinaki Sar2
Pinaki Sar2 Sufia Khannam Kazy1*
Sufia Khannam Kazy1*Characterization of inorganic carbon (C) utilizing microorganisms from deep crystalline rocks is of major scientific interest owing to their crucial role in global carbon and other elemental cycles. In this study we investigate the microbial populations from the deep [up to 2,908 meters below surface (mbs)] granitic rocks within the Koyna seismogenic zone, reactivated (enriched) under anaerobic, high temperature (50°C), chemolithoautotrophic conditions. Subsurface rock samples from six different depths (1,679–2,908 mbs) are incubated (180 days) with CO2 (+H2) or HCO3− as the sole C source. Estimation of total protein, ATP, utilization of NO3- and SO42− and 16S rRNA gene qPCR suggests considerable microbial growth within the chemolithotrophic conditions. We note a better response of rock hosted community towards CO2 (+H2) over HCO3−. 16S rRNA gene amplicon sequencing shows a depth-wide distribution of diverse chemolithotrophic (and a few fermentative) Bacteria and Archaea. Comamonas, Burkholderia-Caballeronia-Paraburkholderia, Ralstonia, Klebsiella, unclassified Burkholderiaceae and Enterobacteriaceae are reactivated as dominant organisms from the enrichments of the deeper rocks (2335–2,908 mbs) with both CO2 and HCO3−. For the rock samples from shallower depths, organisms of varied taxa are enriched under CO2 (+H2) and HCO3−. Pseudomonas, Rhodanobacter, Methyloversatilis, and Thaumarchaeota are major CO2 (+H2) utilizers, while Nocardioides, Sphingomonas, Aeromonas, respond towards HCO3−. H2 oxidizing Cupriavidus, Hydrogenophilus, Hydrogenophaga, CO2 fixing Cyanobacteria Rhodobacter, Clostridium, Desulfovibrio and methanogenic archaea are also enriched. Enriched chemolithoautotrophic members show good correlation with CO2, CH4 and H2 concentrations of the native rock environments, while the organisms from upper horizons correlate more to NO3−, SO42−, Fe and TIC levels of the rocks. Co-occurrence networks suggest close interaction between chemolithoautotrophic and chemoorganotrophic/fermentative organisms. Carbon fixing 3-HP and DC/HB cycles, hydrogen, sulfur oxidation, CH4 and acetate metabolisms are predicted in the enriched communities. Our study elucidates the presence of live, C and H2 utilizing Bacteria and Archaea in deep subsurface granitic rocks, which are enriched successfully. Significant impact of depth and geochemical controls on relative distribution of various chemolithotrophic species enriched and their C and H2 metabolism are highlighted. These endolithic microorganisms show great potential for answering the fundamental questions of deep life and their exploitation in CO2 capture and conversion to useful products.
Recent explorations of the deep biosphere in the crystalline basement of continents and oceanic crust have produced a growing awareness on life extending deep interior of the igneous provinces (Nyyssönen et al., 2014; Lau et al., 2016; Dutta et al., 2018, 2019; Magnabosco et al., 2018; Bell et al., 2020; Purkamo et al., 2020; Suzuki et al., 2020; Sahu et al., 2022). The subsurface beneath the Earth’s continental crust is constrained with multiple physical and chemical extremities including high temperature and pressure, lack of sufficient pore space for fluid and organismic mobility, water and readily metabolizable nutrients (Kieft, 2016). Yet, evidence supports life’s presence at depths down to ~1 km beneath seafloor, and ~5 km in continental settings harbouring ~12–20% of the total biomass of microorganisms on Earth, mostly in the continental subsurface (Magnabosco et al., 2018; Purkamo et al., 2020). Previous evidence indicates that terrestrial subsurface holds diverse and novel microbes with potential for answering questions cardinal to the origin, adaptation and evolution of life, their relevance in elemental cycling, and in production of valuable resources (Escudero et al., 2018). However, despite these observations, the deep biosphere has been largely overlooked and the understanding of the metabolic functions, especially mechanisms of carbon and energy metabolism of the deep continental subsurface microbiome, has remained less explored.
The primary energy source in the deep biosphere is proposed to be geochemical (Gold, 1992; Amend and Teske, 2005; Colwell and D’Hondt, 2013). Terrestrial Deep subsurface, having limited photosynthetically derived energy substrates, are fueled primarily by microbial chemotrophy, where versatile chemolithotrophs, capable of inorganic C assimilation, oxidation of H2 and S compounds coupled with the reduction of diverse available electron acceptors are often found to be predominant. The existence of a specific hydrogen-driven microbial ecosystem (subsurface lithoautotrophic microbial ecosystems; SLiMEs), energetically powered by the geologically derived sources, is also proposed to drive the oligotrophic subsurface community (Nealson et al., 2005; Osburn et al., 2014; Bell et al., 2020). Rocks themselves can act as the source for different mineral nutrients and ions produced due to high temperature, water-rock interactions and other geochemical reactions. Geogenic supply of CO2 and CH4 (as C sources), H2, CH4, NH4+, Fe2+ and S2− (as electron donors) and NO3−, Fe3+, Mn4+, SO42− and CO2 (as electron acceptors) have potential roles in providing nutrient and energy supplies to the residing community (Colwell and D’Hondt, 2013; Konn et al., 2015; Kazy et al., 2021). Even though the dominance of lithoautotrophic metabolism has been reported from several terrestrial subsurface environments including Fennoscandian shield (Nyyssönen et al., 2014) and Witwatersrand Basin (Lau et al., 2016), the role of heterotrophic organisms, capable of utilizing the metabolic products of autotrophic reactions and driving the overall community function has also been highlighted (Purkamo et al., 2015; Bomberg et al., 2016; Momper et al., 2017; Sahu et al., 2022). Several of these studies have highlighted an intricate and interconnective association of autotrophic and heterotrophic processes in driving the overall communities.
Samples from different subsurface settings have been previously used to investigate the abundance, nature, and substrate specific response of deep dwelling organisms via several cultivation-based methods and enrichment studies (Kotelnikova and Pedersen, 1998; Hallbeck and Pedersen, 2008, 2012; Fichtel et al., 2015; Rajala et al., 2015; Russell et al., 2016; Purkamo et al., 2017, 2020; Rajala and Bomberg, 2017; Leandro et al., 2018; Imachi et al., 2019; Sanz et al., 2021; Nuppunen-Puputti et al., 2022). The outcome of these studies, resulted in the enrichment of microorganisms mostly belonging to bacterial phyla Proteobacteria, Firmicutes, Actinobacteria, Chloroflexi and Bacteroidetes (Rastogi et al., 2009, 2013; Fichtel et al., 2012; Fortunato and Huber, 2016; Rajala and Bomberg, 2017; Leandro et al., 2018) and archaeal members including Euryarchaeota, Thaumarchaeota and Crenarchaeota (Zeng et al., 2009; Nuppunen-Puputti et al., 2018; Imachi et al., 2020; Alain et al., 2021; Courtine et al., 2021; Li et al., 2021). “In the last few years, metagenomic investigations of terrestrial subsurface microbiome” (crustal/vent, faults or fractures fluids, aquifer samples have shown considerable species diversity with potential for assimilation of C and inorganic resources for energy metabolism (Nyyssönen et al., 2014; Purkamo et al., 2015; Hubalek et al., 2016; Lau et al., 2016; Magnabosco et al., 2016; Wu et al., 2016; Momper et al., 2017; Bell et al., 2020; Purkamo et al., 2020). In contrast to subsurface fluid-based studies, reactivation of inhabitant microbiota of Archean basement rocks has remained relatively less explored. Recently, deep subsurface studies on rock cores from Iberian Pyrite and a few other crystalline bedrocks and basalt samples from Deccan Traps have been carried out (Dutta et al., 2018, 2019; Leandro et al., 2018; Purkamo et al., 2020; Sanz et al., 2021; Sahu et al., 2022). However, reactivation and enrichment of deep endolithic microorganisms of the granitic bedrock are inadequately studied and their metabolic capabilities remain less known.
Scientific continental drilling of deep boreholes around the Koyna seismogenic zone (KSZ) in the southwestern part of the Deccan Volcanic Province (DVP) enabled a rare access to the basement beneath the Deccan Traps, the nature of which has been enigmatic for longtime (Roy et al., 2013; Roy, 2017). The Deccan Traps (~65 Ma) represents a massive continental flood basalt province comprising of erupted lavas and associated rocks that rests on 2.5 Ga crystalline basement (Schoene et al., 2015; Bhaskar Rao et al., 2017; Dutta et al., 2018). The Koyna–Warna region has gained substantial research interest owing to the intermittent reservoir-induced seismic activity that started after the impoundment of the Shivajisagar (Koyna) artificial water reservoir in 1962 (Gupta, 2017). A scientific borehole (3 km deep, named as Koyna pilot borehole, KFD1), was drilled in 2017 for setting up a deep borehole observatory in the region (Goswami et al., 2020). Rock cores obtained from various depth intervals (1,679–2,908 mbs) through this borehole were used to reactivate and enrich microorganisms under chemolithoautotrophic conditions. The specific goals for this study were to (i) understand the response of endolithic microbes (from different depths) towards the growth substrates CO2 (along with H2 as electron donor) or HCO3− as the sole C source (ii) identify the nature of reactivated microbial community members through amplicon based 16S rRNA gene sequencing and to assess their depth-wide distribution pattern, and (iii) elucidate the metabolic potential of the enriched microbial communities, through predicted metabolic profiling, with specific emphasis on C and energy metabolism processes.
Subsurface rock core samples were obtained from different depths (1,679–2,908 mbs) of the Koyna pilot borehole (17°17″57.27” N, 73°44″19.07″ E) of the Deccan Traps, India. Samples were collected following standard techniques and stored anaerobically in sterile containers at 0–4°C and aseptically transported to the laboratory. The detailed processes of handling and storage procedures of the samples were mentioned in the previous study from our group (Sahu et al., 2022). For microbiological investigations, rocks were powdered in N2-filled condition, using electrical coring machine, and the interior rock pieces/powder were obtained (Sahu et al., 2022). Details of the six rock samples (designated as C1, C2, C3, C4, C6 and C7), have been furnished in Supplementary Table 1, and were used for further enrichment studies.
Reactivation of indigenous microorganisms from the six rock core samples was performed by setting up microcosm-based enrichment setups. The rock powder was checked for the presence of contamination by estimating the sodium fluorescein concentration (used as a marker of potential contamination of drilling fluid) as per the protocol described earlier (Dutta et al., 2018). Rock powders devoid of any sodium fluorescein were used for further analysis. 0.05 g of rock powder samples were incubated anaerobically (in crimp sealed serum vials) at high temperature (50°C) using a basal medium [formulated based on the overall geochemistry of our rock samples as reported in (Sahu et al., 2022) and named as the Koyna medium] and other supplements. A Don Whitley anaerobic workstation (Model A-35) operating with N2/H2 + CO2 + N2 (10:10:80) gas environment was used to setup the enrichments. The detailed composition of the medium and incubation conditions, etc. have been provided in Supplementary Table 2. All medium components were prepared separately, sterilized either by autoclaving or filtration, (as appropriate) and used. The pH of the medium was set at pH 9.0. For the present study, each of the six samples (C1, C2, C3, C4, C6 and C7) were incubated with CO2 (along with H2 as electron donor) or 2 mM HCO3− (along with NH4 as electron donor) as sole C source. CO2 was added from a cylinder containing high purity gas mixture of CO2 and H2 in a premixed concentration ratio of 80:20. Ten ml of the gas mixture was added using the liquid displacement technique. A total of 12 experimental setups [marked as C1–C7 HC (for H2 + CO2 enriched sets) and C1–C7 BC (for HCO3− enriched sets)] and two controls (setups devoid of rock samples) were incubated for 180 days at 50°C and considered for further study post-incubation.
Total protein concentration of the enrichment cultures was measured to estimate the microbial biomass content. Protein concentration was measured through a fluorometric method (Qubit, Thermo-Fisher Scientific), using the Qubit Protein Assay Kit. In brief, enrichment samples were sonicated with 30% amplitude and pulse of 15 s on and 15 s off, for 10 min in order to obtain the total protein from the enrichment cultures. After sonication, the solution was centrifuged at 10,000 rpm for 5 min and the supernatant was collected and used as test samples for the estimation. The working solution as well as assay tubes were prepared and the estimation was done in triplicate using the manufacturers protocol (https://research.fredhutch.org/content/dam/stripe/hahn/methods/mol_biol/Qubit_Protein_Assay_QR.pdf). The fluorometer was calibrated against the standards and the test samples were measured in μg ml−1.
A Luminometer (Glomax 20/20 System, Promega with BacTiter-Glo™ Microbial Cell Viability Assay kit) was used for estimating the total ATP of the living cells present in the enrichment samples (Adhikari and Kallmeyer, 2010). The details of the protocol were same as provided by the manufacturer, i.e the BacTiter-Glo™ Microbial Cell Viability Assay Technical Bulletin #TB337. Light emission was measured (as relative light units per second, RLU s−1) for three, five seconds intervals with a five seconds delay before each interval, which was then quantified as mM with reference to standard ATP curve. Sterile tips were used in transferring all solutions and samples to exclude ATP contamination of pipettes and solutions. All procedures were performed in triplicates in the dark and all plastic material, solutions, and pipettes were stored in the dark as light could hamper the results causing delayed fluorescence.
The utilization/reduction of electron acceptors might act as a significant sign of growth of microorganisms. In this study, the initial and final concentrations of terminal electron acceptors like SO42− and NO3−, were measured to obtain information about the change in their concentration. Required amounts of enrichment cultures were obtained, centrifuged at 10,000 rpm for 30 min and the supernatant was used for further analysis. SO42− estimation was performed through the BaCl2 based turbidimetric spectroscopy method (Chesnin and Yien, 1951). For measurement of NO3− concentration, the samples were treated with salicylic acid-H2SO4 reagent, followed by NaOH, and estimated via colorimetric procedures (Cataldo et al., 1975). All analyses were performed in triplicates.
Formation of different metabolites as intermediates or end-products of various pathways could provide an indication of successful utilization of the provided enrichment substrates. Metabolites such as lactate, acetate, and formate in the enrichment setups were measured through Ion Chromatography (Eco IC, Metrohm) with PRP-X300 (Hamilton) column following the manufacturer’s protocol. The standard solutions were prepared using highly purified grade sodium salts. 15 ml culture was aseptically and anaerobically withdrawn from each of the serum vials, centrifuged (10,000 rpm for 30 min) and the supernatant was used for IC analysis. The samples were acidified using 0.1 N HCl, filtered using 0.22 μm filter (Merck Milipore, Millex GP) and transferred to the autosampler for further processing.
The entire culture volume of the serum vials (~100 ml) from each enrichment was centrifuged at 10,000 rpm for 30 min and the pellet (containing the rock powder and microorganisms) was used for extraction of total DNA in multiple replicates (n = 4) using QIAGEN power soil DNA isolation kit with certain modifications (30 min incubation after adding C2 and C3 solutions each). The total volume of eluted DNA from each sample was pooled, concentrated and quantified through a fluorometric method (Qubit, Thermo-Fisher Scientific). The V4 region of 16S rRNA gene was amplified from the isolated total DNA with V4-specific barcoded primers 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACVSGGGTATCTAAT-3′, Bates et al., 2011). The V4 amplicon libraries were purified, processed and sequenced through an in-house Ion S5 (Thermo-Fisher Scientific). Details of the sequencing procedures are described elsewhere (Dutta et al., 2018). Total DNA was extracted from sequencing reagent and sequenced using the same procedure, as control. The sequence reads were submitted to the Short Read Archive (NCBI) under BioProject ID PRJNA642293 (for H2 + CO2 set) and PRJNA643233 (for HCO3− set).
Bacterial abundance in the enrichment cultures was quantified by estimating the copy number of bacterial specific 16S rRNA genes (V3 region). All the amplification reactions were set up in triplicate. Quantitative PCR was performed in QuantStudio 5 using Power SYBR green PCR master mix (Invitrogen), with primer concentration of five pM and the following amplification conditions: 95°C for 10 min, 40 cycles of 95°C for 15 s, 55°C for 30 s and 72°C for 30 s. Melting curve analysis was run after each assay to check PCR specificity. The primers used were P1 (5’-CCTACGGGAGGCAGCAG-3′) and P2 (5’-ATTACCGCGGCTGCTGG-3′). Other details are same as described earlier (Dutta et al., 2018). Samples were run in at least three dilutions to check for PCR inhibitions. Bacterial 16S rRNA gene copy numbers were determined in each sample by comparing the amplification result to a standard dilution series ranging from 102 to 1010 of plasmid DNA containing the V3 region of the 16S rRNA gene of Achromobacter sp. MTCC 12117. We could not perform the quantification (as we did not obtain any amplification) of archaeal populations, because of lower DNA concentrations.
Raw sequence reads obtained after sequencing were quality filtered (removal of primers, homopolymers run of less than six bp, read length beyond the range of 250–300 bp, mean quality score below minimum of 25, with minimum six primer mismatches) and analysed in QIIME 1.9.1 pipeline (Caporaso et al., 2010). Denovo-based clustering (97% sequence similarity) of filtered reads to OTUs was performed using UCLUST under QIIME workflow. Taxonomic assignment of representative reads from each OTU was performed using SILVA 132 database.1 The taxonomic assignments could not be performed in newer versions of SILVA database due to unavailability of required computational facilities. OTUs detected in reagent control, singleton OTUs (the cumulative read for which is one amongst all sets, Reitmeier et al., 2021) and OTUs affiliated to potential contaminants of deep subsurface, i.e., members of taxa Staphylococcaceae, Streptococcaceae, Propionibacteriaceae, Lactobacillaceae, E. coli-Shigella, Corynebacterium (Hubalek et al., 2016; Sheik et al., 2018), were removed from the OTU table before further analysis. Samples were normalized to 14,800 reads by random sampling for alpha diversity analysis.
Principal Component Analysis (PCA) and Canonical Correspondence Analysis (CCA) were performed using PAST software version 4.06 (Hammer et al., 2001) on the basis of (i) growth parameters and (ii) concentration of selected geochemical parameters along with abundant taxa (top 50% relative abundance) in both enrichment sets, respectively. Non-metric multidimensional scaling analysis (NMDS, Dixon, 2003; Doi and Okamura, 2011) was performed for comparing the similarity/dissimilarity of the microbial communities at taxonomic level by using metaMDS function of ‘vegan’ package in R software 4.2.1 (Kuhnert et al., 2005; Van der Loo, 2012) which helped to depict the effect of different substrates and depth on the microbial communities. Similarity percentage (SIMPER) analysis was done by using Simper function in ecological vegan package of R software 4.2.1 (R Development Core Team, 2017) to identify the microbial taxa responsible for the dissimilarity between different depths in both the substrate types (HC and BC). OTU overlap among the enrichments from different substrates and among different rocks in the same enrichment were elucidated using InteractiVenn (Heberle et al., 2015). Phyla belonging to individual enrichments were selected for correlation analysis and the correlation heatmap was constructed on the basis of Spearman correlation using METAGENassist (Arndt et al., 2012).
16S rRNA sequence reads of various OTUs were used to construct a phylogenetic tree using a distance matrix-based Neighbor-joining method in MEGA 11 software with 1,000 bootstrap re-iterations (Tamura et al., 2021). Initially, top 15 abundant OTUs from each enrichment set and the OTUs affiliated to Cyanobacteria from the enrichments were analysed by homology search for the closest sequence relatives (matches) using the National Centre for Biotechnology Information (NCBI) BLASTn nucleotide database. Multiple sequence alignments were performed with CLUSTALW package of MEGA 11. The sequences were represented with their accession number followed by the organism’s identity and its location. Finally, phylogenetic tree reconstruction and validation were performed using Neighbor-Joining (NJ) algorithm (Saitou and Nei, 1987) with 1,000 bootstrap re-iterations using Jukes-Cantor distance model (Jukes and Cantor, 1969).
Co-occurrence network analysis was performed at two different levels, considering (i) OTUs enriched in at least four samples out of total six at the lowest taxa level and (ii) association of taxa strongly correlated with depth of the rock samples and with relevant geochemical parameters of the rocks. Correlation among different taxa was calculated using otu.association command in mothur (Schloss et al., 2009). Correlation among different taxa and geochemical parameters was calculated using the PAST software version 4.06 (Hammer et al., 2001). The Spearman correlation values r ≥ ±0.9 and r ≥ ±0.3 were used for construction of co-occurrence network amongst taxa and amongst taxa and parameters, respectively. Visualization of the interaction network was done using Cytoscape 3.7.1 (Shannon et al., 2003).
Microbial metabolic pathways were estimated based on the 16S rRNA gene data from the closed OTU picking method using the PICRUSt v2 software (Douglas et al., 2020). PICRUSt v2 compares 16S rRNA marker gene data with the nearest match of the known genome sequence to allocate the functional attributes. A weighted nearest sequenced taxon index (NSTI) was calculated to evaluate the accuracy of prediction. The predicted KO numbers were plotted on KEGG pathway maps2 separately. Abundances of genes related to general metabolic pathways, different C fixation pathways, lithotrophy related metabolisms, secondary metabolite production and xenobiotic degradation were selected for detailed analysis.
Microbial enrichment and response in anaerobic high temperature enrichment microcosms was assessed by estimating the cell biomass in terms of total protein, ATP, as a metabolic marker, utilization of SO42− and NO3− (provided as terminal electron acceptor) and bacterial 16S rRNA gene copy numbers (Figures 1A–E). Substantial amounts of protein and ATP were obtained in both setups indicating a positive response of the rock communities to the substrate provided. Consumption of ~50% of the initial SO42− concentration or ~ 94% of the initial NO3− concentration could be observed. The total amount of bacterial gene copy number in each enrichment culture as estimated by 16S rRNA gene targeted qPCR analysis indicated 2.9 × 1010 ml−1 to 6.5 × 1010 ml−1 copies in HC enrichment while in BC enrichments it ranged from 3.7 × 1010 ml−1 to 5.8 × 1010 ml−1. However, archaeal 16S rRNA genes could not be amplified during qPCR analysis. A PCA biplot on the basis of observed growth parameters reflected the distribution of the rock core enrichment samples in two halves of the plot (Figures 2A,B).
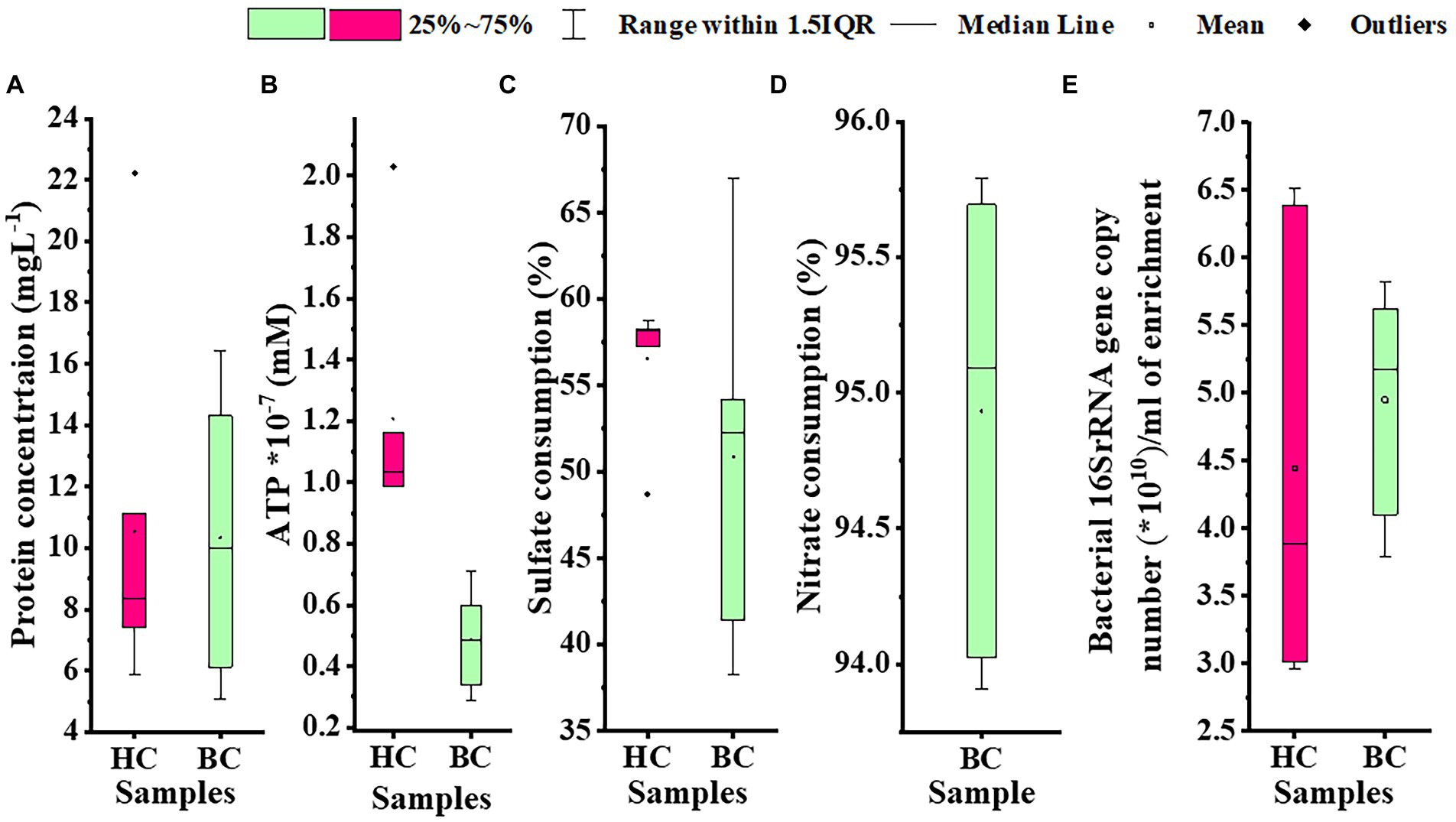
Figure 1. Assessment of (A) cell biomass in terms of total protein, (B) ATP, (C) utilization of sulfate; provided as TEA, (D) utilization of nitrate; as TEA, (E) Bacterial 16S rRNA gene copy number per mL of enrichment in HC and BC sets. The black center line denotes the median value (50th percentile), the empty square denotes the mean value and the pink (HC) and green (BC) boxes contain the 25–75th percentiles of the data sets. The values beyond these upper and lower boundaries are considered as outliers, marked with black filled diamond shapes.
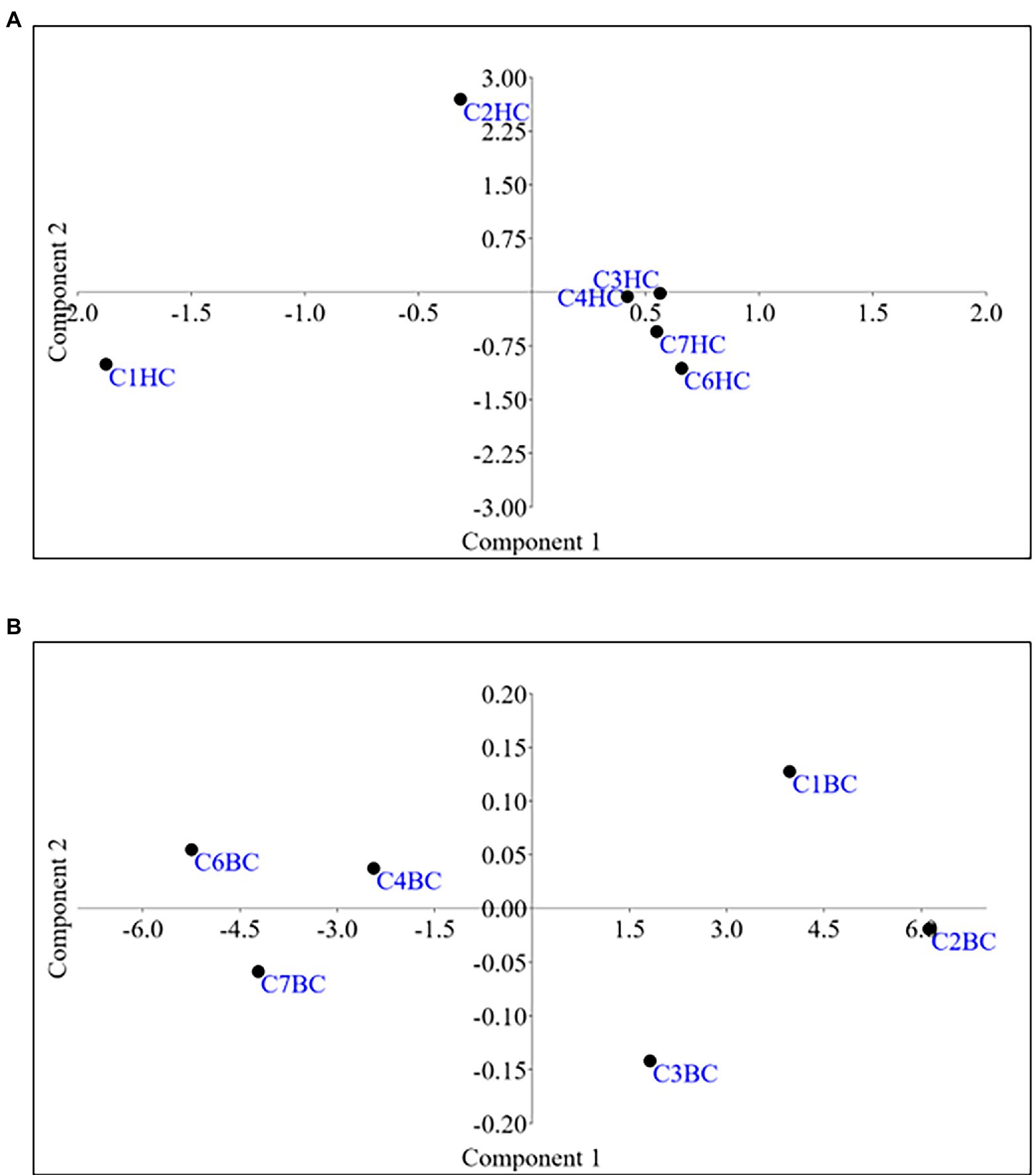
Figure 2. Principal Component Analysis for (A)HC/(B)BC enrichments based on the growth parameters.
Identification of different metabolites (small organic acids), via ion chromatography was done. Approximately 77 mgl−1 and 1.0 mgl−1 formate as well as 191.1 mgl−1 and 6.6 mgl−1 lactate in HC and BC microcosms, respectively were estimated. However, ~9.5 mgl−1 acetate was detected only in the BC sets (Supplementary Table 3).
Details of the sequence information have been summarized in Table 1. A total of 389,051 (HC) or 86,100 (BC) 16S rRNA reads was obtained. In HC setup, a maximum of 141,124 and a minimum of 18,136 quality filtered reads were obtained from C1HC and C4HC sets, respectively. Whereas in case of BC enrichments, a maximum of 23,686 and a minimum of 4,400 quality filtered reads were obtained from C7BC and C3BC sets, respectively. On an average more than 99% of the quality filtered reads could be classified as bacteria (368,807 in HC and 85,124 in BC sets) and archaea (16,989 in HC and 286 in BC sets). Following the removal of control and potential erroneous reads, a total of 18,258 OTUs (HC) and 10,482 OTUs (BC) were obtained (Supplementary Figure 1). The Venn diagram illustrated in Supplementary Figures 2A,B depicted the sharing of OTUs amongst the six samples in each enrichment. It was observed that the percentage of unique OTUs and their cumulative abundance decreased as the depth of the rock (used as sample for enrichment) increased. Only 34 and 9 OTUs were common amongst all six samples in HC and BC enrichments, respectively.
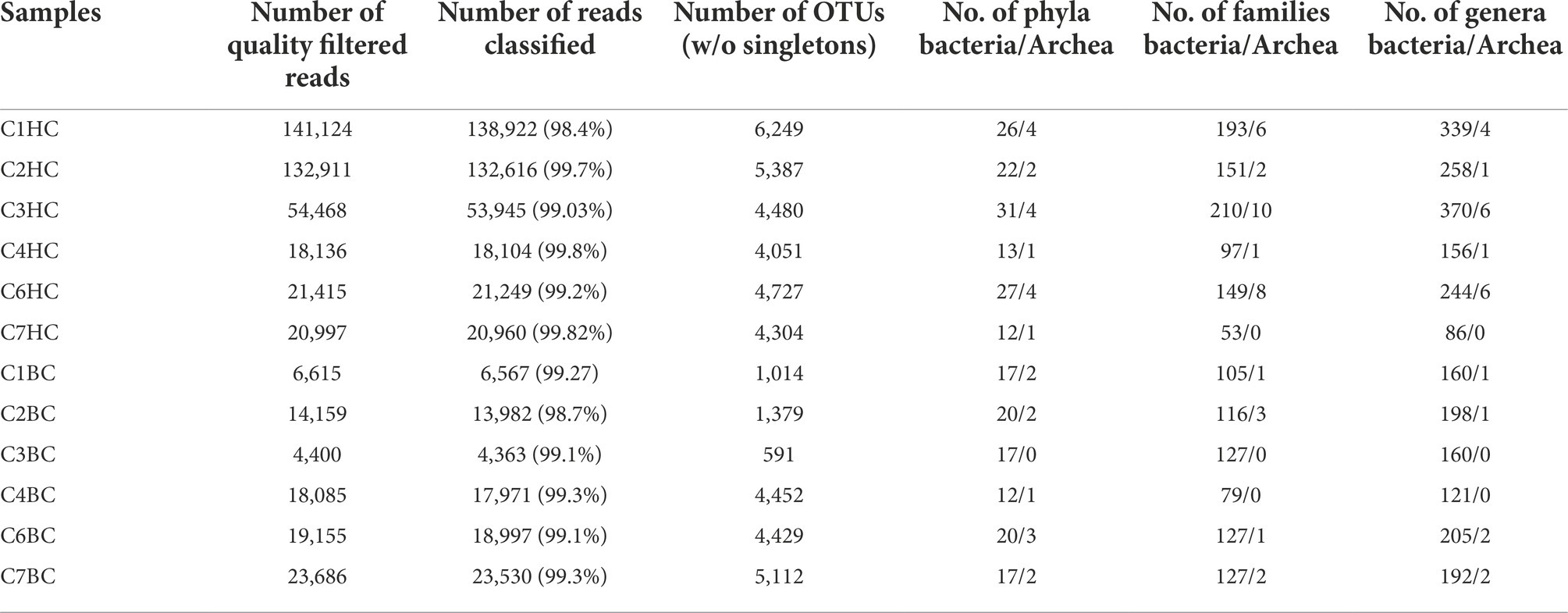
Table 1. Sequencing information of samples in H2 + CO2 (designated as C1–C7 HC) and HCO3− (are designated as C1–C7 BC) enrichment samples.
Details of the alpha diversity indices were summarized in Table 2. Chao1 estimator, which estimated the minimum total number of OTUs, ranged from ~4176 (C2HC) to ~7629 (C6HC) in HC sets and 770 (C3BC) to 7236 (C7BC) in the BC microcosms. The Shannon diversity index, which was calculated to evaluate species abundance and evenness in each sample, showed values between 8.4 (C2HC) to 10.1 (C4HC) in HC enrichments, whereas in case of BC enrichments, the values ranged between 7.8 (C3BC) to 10.3 (C7BC). The Shannon index values were slightly higher in the deeper horizon samples (C4–C7) under both enrichment conditions. Simpson index, an indicator of species dominance in a community, ranged between 0.98–0.99 across all enrichment samples.
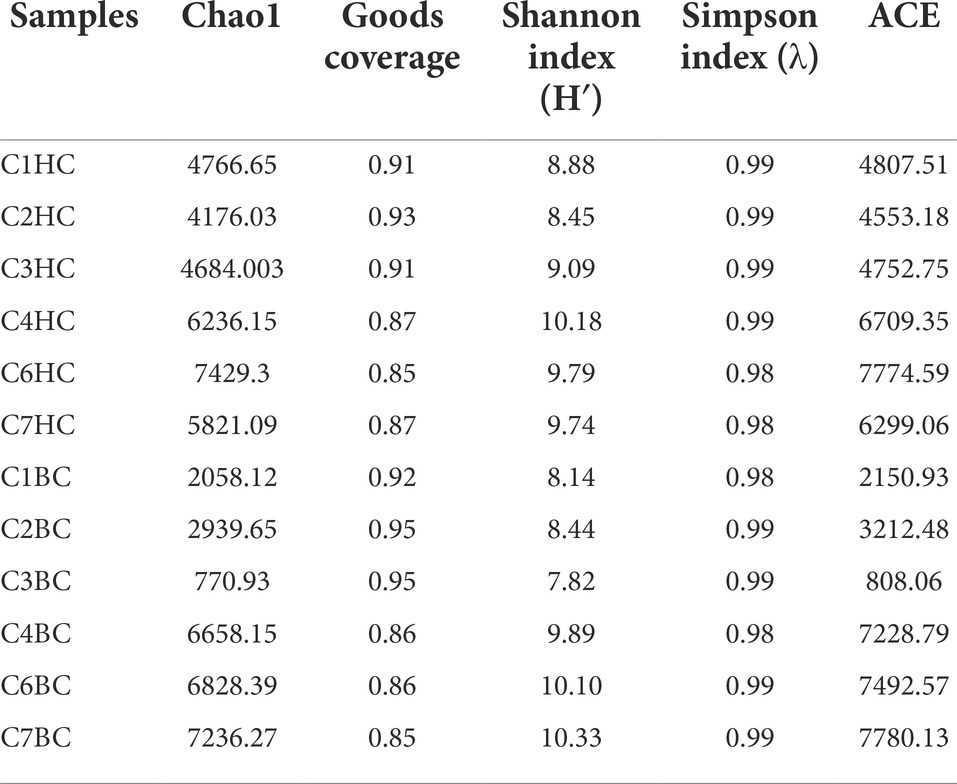
Table 2. Alpha diversity indices for H2 + CO2 (designated as C1–C7 HC) and HCO3− (are designated as C1–C7 BC) enrichment samples.
The details of sequence affiliations in each taxonomic level were presented in Table 1. Assigned reads from HC enrichments (total number: 385796) were assigned to 576 genera belonging to 41 phyla (37 bacteria and 4 archaea). Compared to HC, number of taxa detected in BC setups were less. Assigned reads from the BC enrichments (85410) were affiliated to 435 genera belonging to 32 phyla (29 bacteria and 3 archaea).
Members of the phylum Proteobacteria constituted the most dominant group (upto ~63%) across the enrichment conditions (Supplementary Figure 3). The other abundant phyla were Actinobacteria (11%,), Firmicutes (8%), Bacteriodetes (8%), Thaumarchaeota (1.3%), Verrucomicrobia (1.13%), Chloroflexi (2%), Acidobacteria (1.2%) and Cyanobacteria (0.7%). Interestingly, it was observed that for both the enrichment conditions relative abundance of Gammaproteobacteria* (excluding Betaproteobacteriales) and Betaproteobacteriales members were higher in the deeper rock (C4–C7) based enrichments (28 and 53%) than in relatively shallower rock (C1–C3) enrichments (13.5 and 10%). However, the distribution of Actinobacteria, Firmicutes, Bacteroidetes, Cyanobacteria were found to be more in the upper horizon samples (C1–C3HC; C1–C3BC). Members of the phylum Chloroflexi were found to be one of the dominant groups with ~13% abundance in C3BC, whereas Acidobacteria members were found to be relatively abundant in sample C3, in both the HC and BC enrichments.
At the lowest taxonomic level, the most abundant microbial groups in HC enrichments were found to constitute >45% of the total community in each setup (Figure 3A). Comamonas, was found to be the major taxon in HC enrichments. Other major taxa included the members of Burkholderia-Caballeronia-Praburkholderia, Klebsiella, Ralstonia, Enterobacter, unclassified Enterobacteriaceae and Burkholderiaceae. Interestingly, these taxa were more abundant in the rock enrichments from the deeper horizons (C4–C7). On the other hand, mixotrophic Pseudomonas, Rhodanobacter, Methyloversatilis, members of Group 1.1c Thaumarchaeota and Conexibacter members were more abundant in enrichments of shallower rocks with HC (C1–C3). Noticeably, presence of metabolically versatile Oxyphotobacteria (belonging to Cyanobacteria) was observed in these enrichments with an average relative abundance of 0.7%, with higher abundance in the shallower samples (1.25%). In BC enrichments the top taxa contributed around ~45% in average (Figure 3B). The major groups belonged to similar taxa as in HC enrichment of the deeper horizon rocks. Comamonas, Burkholderia-Caballeronia-Paraburkholderia, Klebsiella, unclassified Enterobacteriaceae, Burkholderiaceae, Enterobacter and Ralstonia were the most dominant taxa with higher distribution in enrichments from deeper rocks. Nocardiodes, Sphingomonas, Aeromonas dominated the BC enrichments of shallower rock samples. Other less abundant taxa (relative abundance <0.1%) in HC enrichment microcosms showed the presence of important hydrogen oxidizing and/or CO2 utilizing taxa like Chloroflexi, Curvibacter, Hydrogenophaga, Hydrogenispora, Methanobacterium, Methanolinea, Acidithiobacillus and Thiobacillus as well as desiccation and radiation resistant, endolithic, extremophilic Chroococcidiopsis. Syntrophic, chemolithoautotrophic, sulfur oxidizing groups belonging to Syntrophobacter, facultatively chemolithoautotrophic Thermincola, Pelobacter and autotrophic Curvibacter, aromatic compound degrader Azoarcus were observed as minor groups in BC enrichment setups. Supplementary Figures 4A,B illustrates the relative abundances of 50 representative less abundant taxa in HC and BC enrichments, respectively.
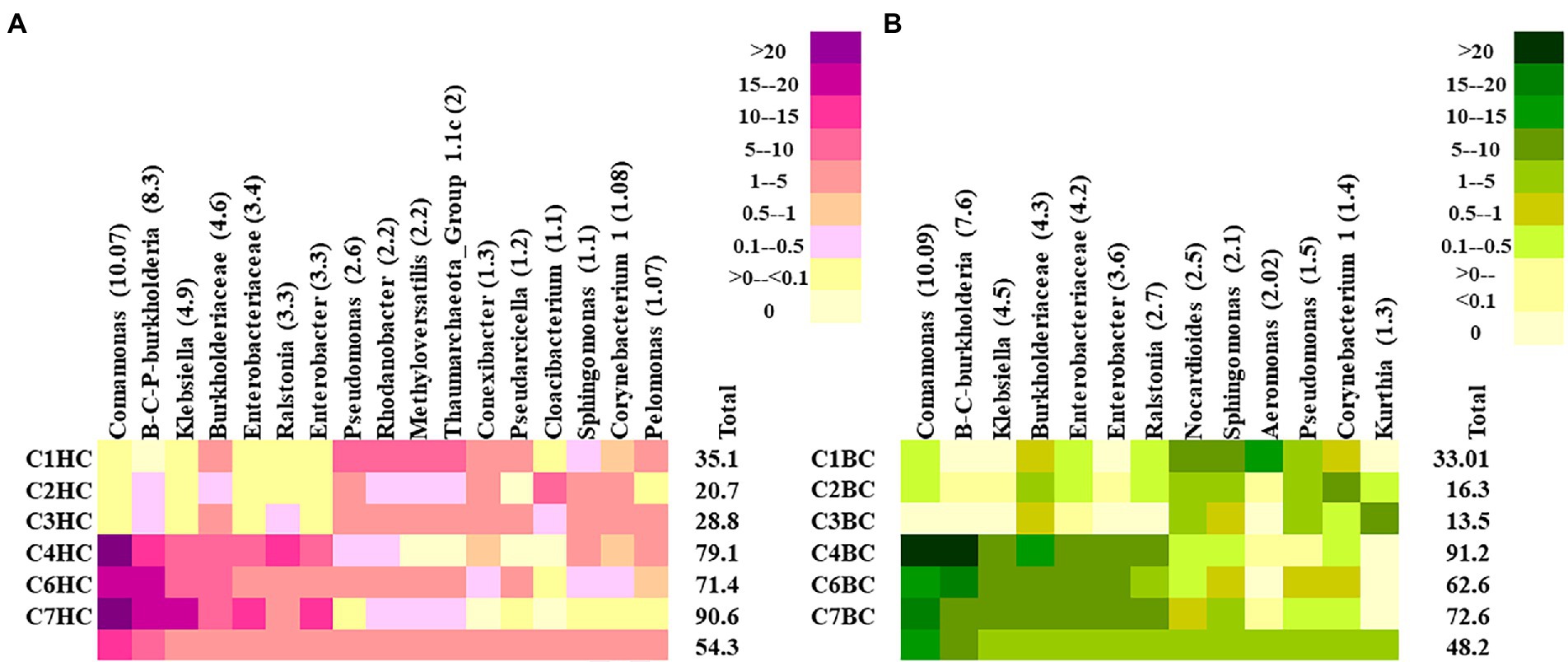
Figure 3. Heatmap displaying the relative abundance of major taxa (abundance ≥1%) detected in (A) HC and (B) BC enrichments. Burkholderia-Caballeronia-Paraburkholderia is designated as B-C-P-burkholderia.
Rank abundance of observed OTUs was analysed to get an idea of the dominant reactivated community members under different enrichment conditions. In HC enrichments the top 50 OTUs cumulatively contributed to 30% abundance of the total. Rank abundance histograms and taxonomic distribution showed that an OTU, belonging to Burkholderia-Caballeronia-Paraburkholderia was the most abundant one with an average relative abundance of 4.8% (Figure 4A). It was followed by the OTUs related to denitrifying Rhodanobacter (2.1%), Thaumarchaeota (1.8%), Methyloversatilis (1.7%), Pseudarcicella (1.2%), Cloacibacterium (1.1%), taxa capable of hydrogen oxidation like Conexibacter (1%) and Pelomonas (1%). Interestingly, the presence of nine of these top 10 OTUs mentioned (except Conexibacter) were observed to have been enriched with bicarbonate, as enrichment substrate, as well. The other less abundant group contained OTUs belonging to potential H2 and CO2 utilizing autotrophic organisms like Ralstonia, Chloroflexi_KD4-96, Acidobacteraceae (Subgroup 1), Comamonas, Luteolibacter, Methylobacterium, Enterobacteraceae and facultative anaerobic Sporichthyaceae.
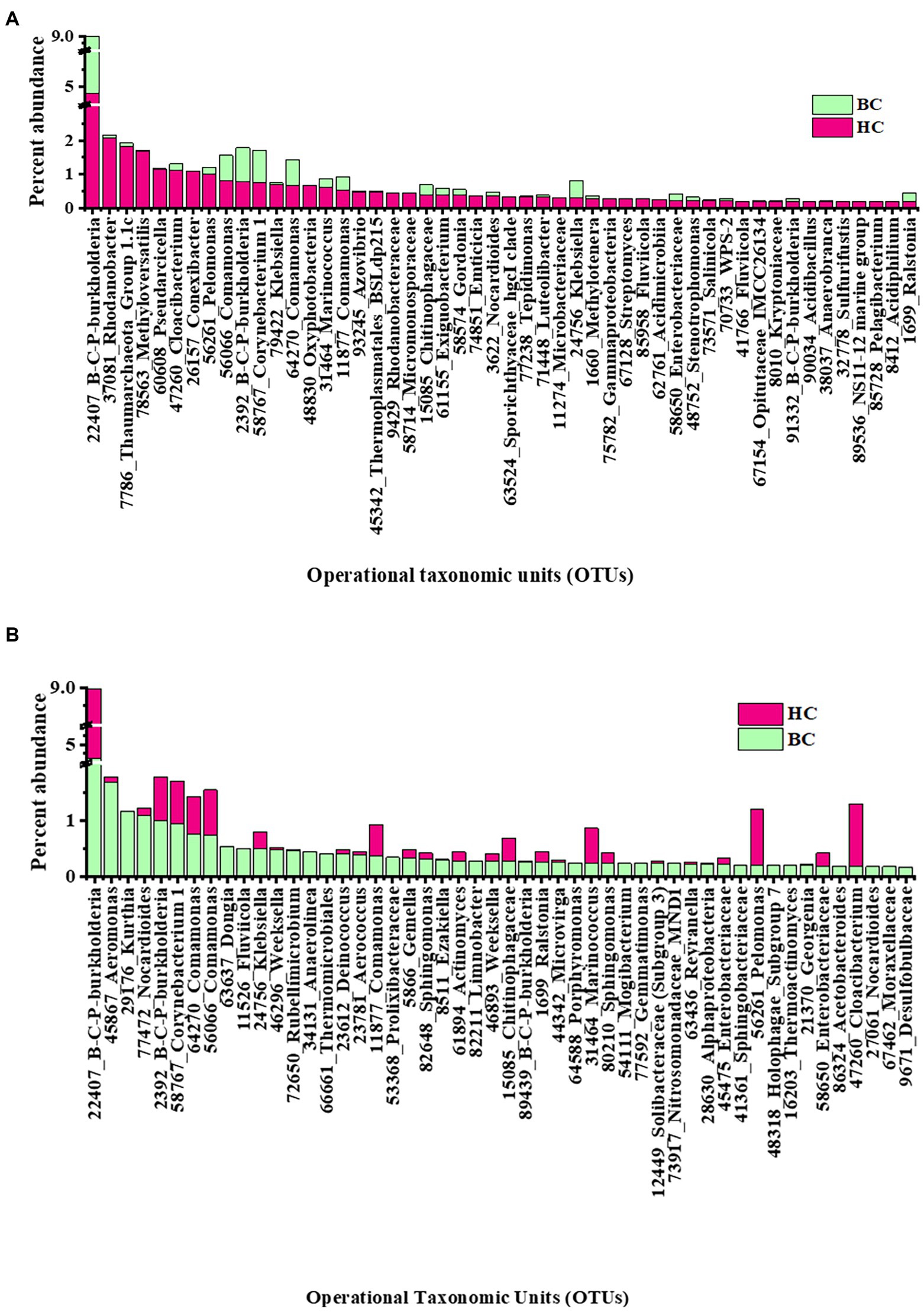
Figure 4. Rank abundance (based on relative percentage abundance) of top 50 OTUs detected in (A) HC and (B) BC enrichments. Burkholderia-Caballeronia-Paraburkholderia is designated as B-C-P-burkholderia.
In BC enrichment microcosms, rank abundance analysis (Figure 4B) of top 50 OTUs, cumulative abundance 24%, revealed that the most abundant OTU to be the same (affiliated to facultative chemolithotrophic Burkholderia-Caballeronia-Paraburkholderia) as in the HC enrichment. OTUs related to Aeromonas (1.6%), Kurthia (1.1%), Nocardiodes (1.1%) and Burkholderia-Caballeronia-Paraburkholderia (1.01%) followed the dominant one. OTUs related to Aeromonas, Pseudomonas, Burkholderiaceae, Aquabacterium, Rhodanobacteraceae Enterobacter, Klebsiella, Nakamurella, Lautropia, constituted the less abundant OTUs. Supplementary Figures 5A,B illustrates the rank abundances of 50 representative less abundant (relative abundance <0.1%) OTUs in HC and BC enrichments, respectively.
The Non-metric Multidimensional scaling (NMDS) ordination was performed to understand the collective role of enrichment substrates and rock sample’s depths on the distribution of microbial taxa. The output reflected that deeper (C4–C7) rock hosted enriched communities occupied a distinct ordination space separated from shallower (C1–C3) rock enriched communities for each substrate (Figure 5). It was observed that microbial communities were clustered based on depth rather than the enrichment substrates (HC or BC). Stress was found to be ≤0.06 which indicated good fit of ordination (Dexter et al., 2018; Mullin et al., 2020). The partitioning was observed clearly in between the two halves of the plot. A canonical correspondence analysis (CCA) based on selected geochemical parameters along with enriched microbial taxa was drawn (Figures 6A,B), which revealed strong significant correlation of enriched microbial taxa with depth and parameters. Separate guilds were formed which were found to be guided by depth and geochemical parameters. Investigation of the individual enrichment revealed positive correlation of Mn with CCA axes 1 and 2 in HC enrichment sets (Supplementary Table 4A). Parameters like CO2, H2, and CH4 correlated positively with CCA axis 1 and negatively with CCA axis 2. Positive correlation of axis 2 was observed with TIC, Fe, NO3 - and SO42−, which on the other hand correlated negatively with axis 1. For BC enriched samples, CO2, H2, and CH4 correlated positively with both the axes (Supplementary Table 4B). Mn correlated positively with CCA axis 1 and negatively with CCA axis 2. Negative correlation effect of TIC and SO42− was observed for both the axes, whereas Fe and NO3 - correlated positively with CCA axis 2. The distribution of the microorganisms in the deeper rock (C4–C7) enrichments were strongly constrained by Mn, CO2, H2, and CH4 whereas the composition of the organisms in the shallower depth enrichment samples (C1–C3) could be guided by TIC, Fe, NO3 - and SO42− in both the enrichments. SIMPER analysis was done to determine the microbial taxa responsible for the differences between different depths in both enrichment conditions (Tables 3, 4). The topmost discriminating taxa were Comamonas, Burkholderia-Caballeronia-Paraburkholderia, Klebsiella, Enterobacter, Burkholderiaceae, Enterobacteriaceae and Ralstonia.
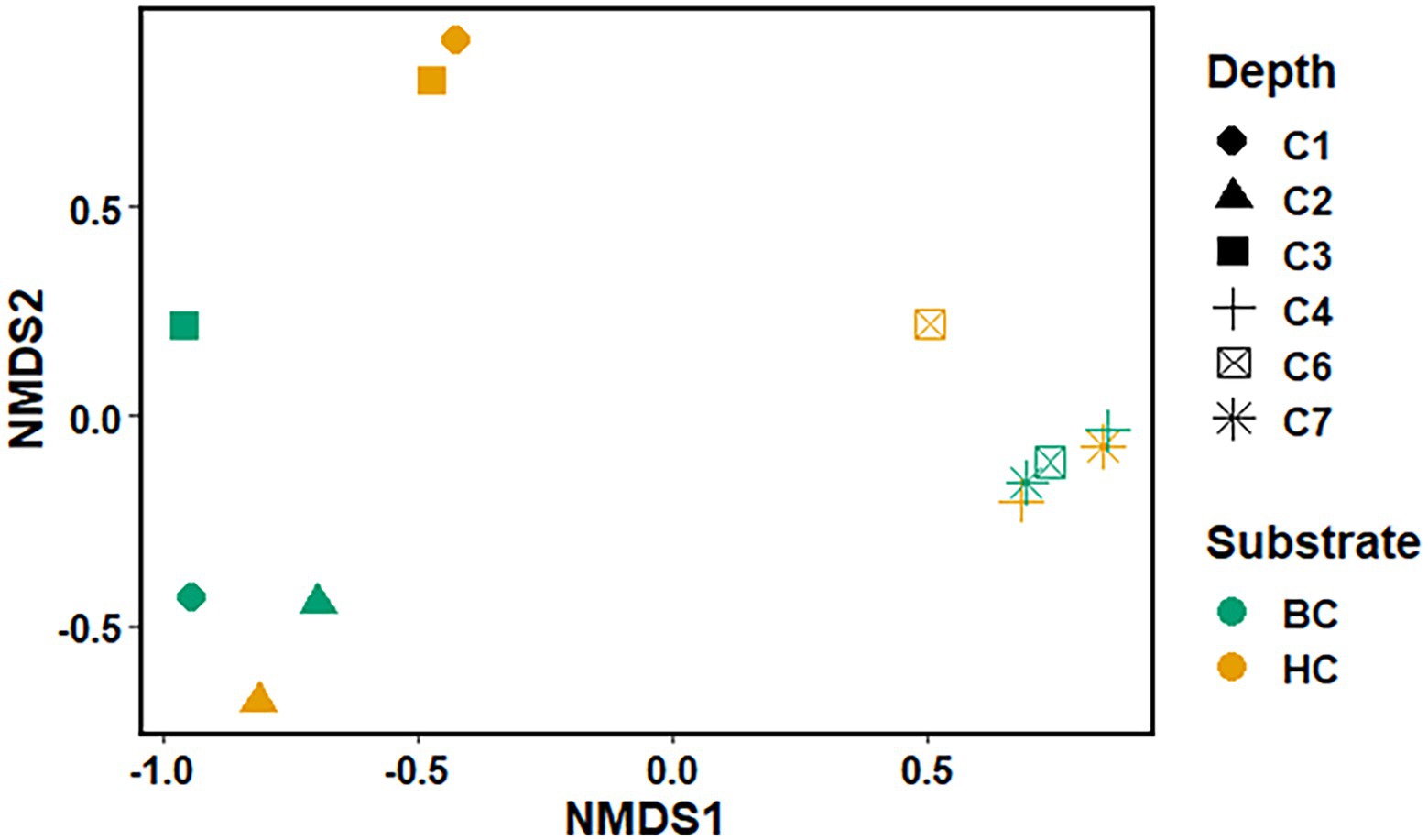
Figure 5. Non-metric Multidimensional scaling of microbial community (at the taxa level) for microbial taxa belonging HC and BC enrichment based on their association with depth.
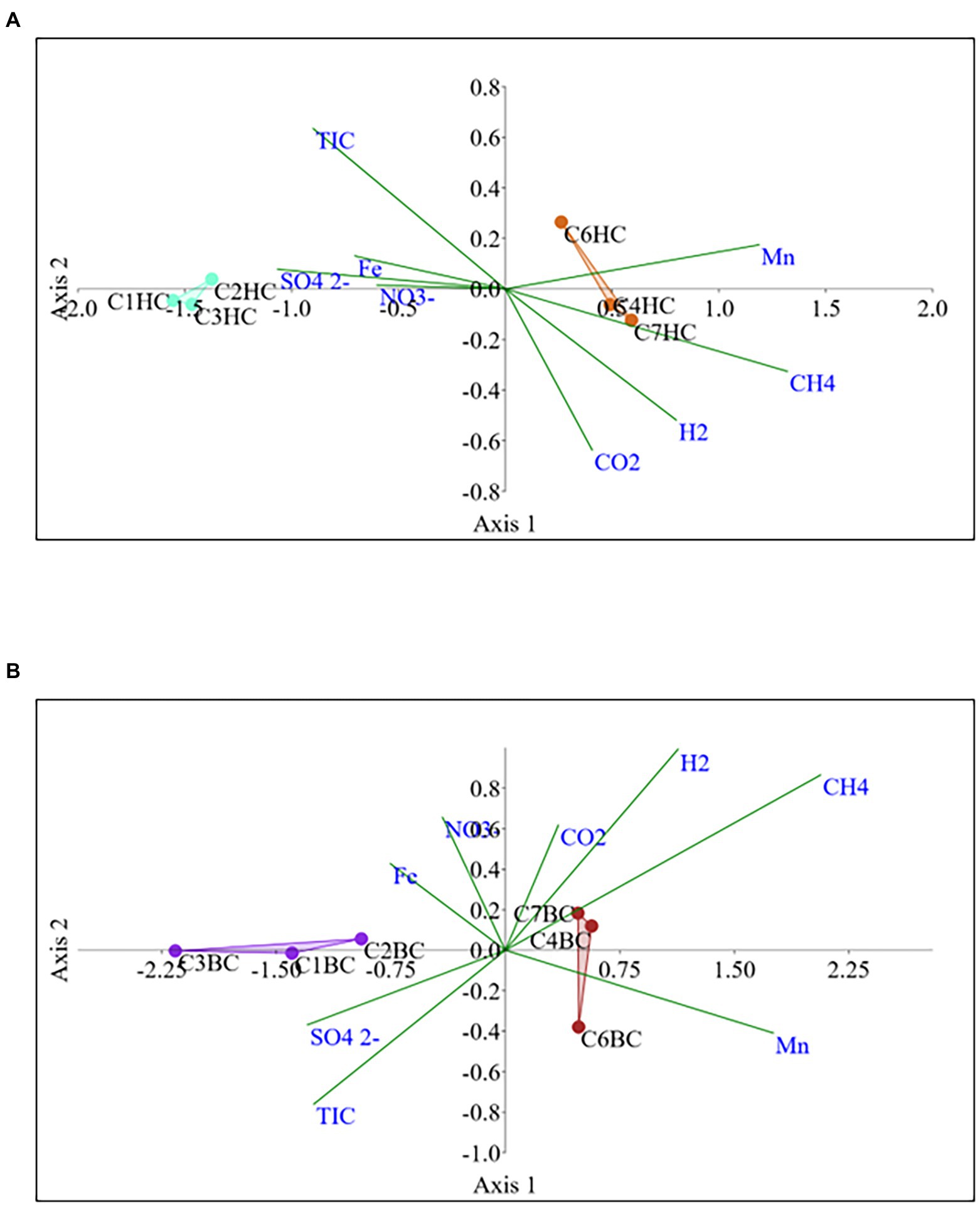
Figure 6. Canonical correspondence analysis for microbial taxa belonging (A) HC and (B) BC enrichment based on their association with depth and relevant geochemical parameters.
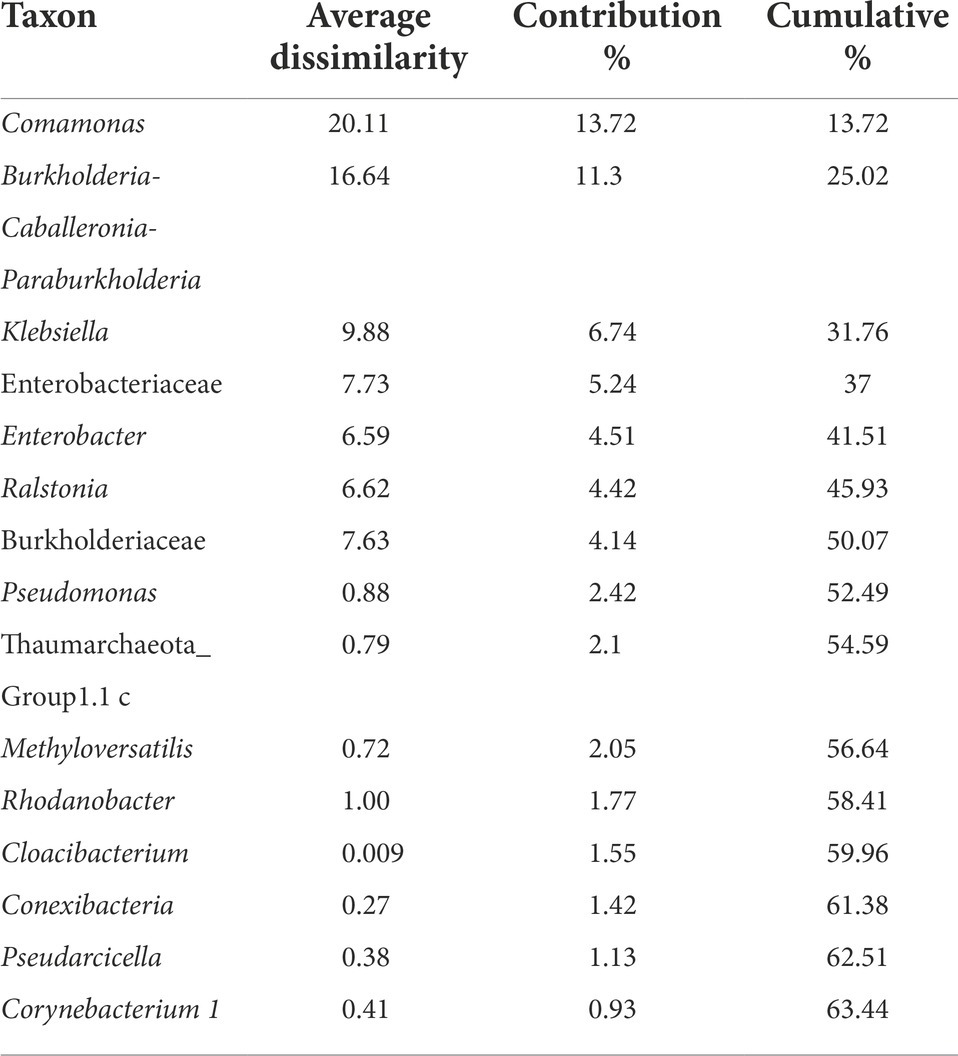
Table 3. SIMPER analysis of microbial communities from H2 + CO2 enrichments using rock samples from different depth.
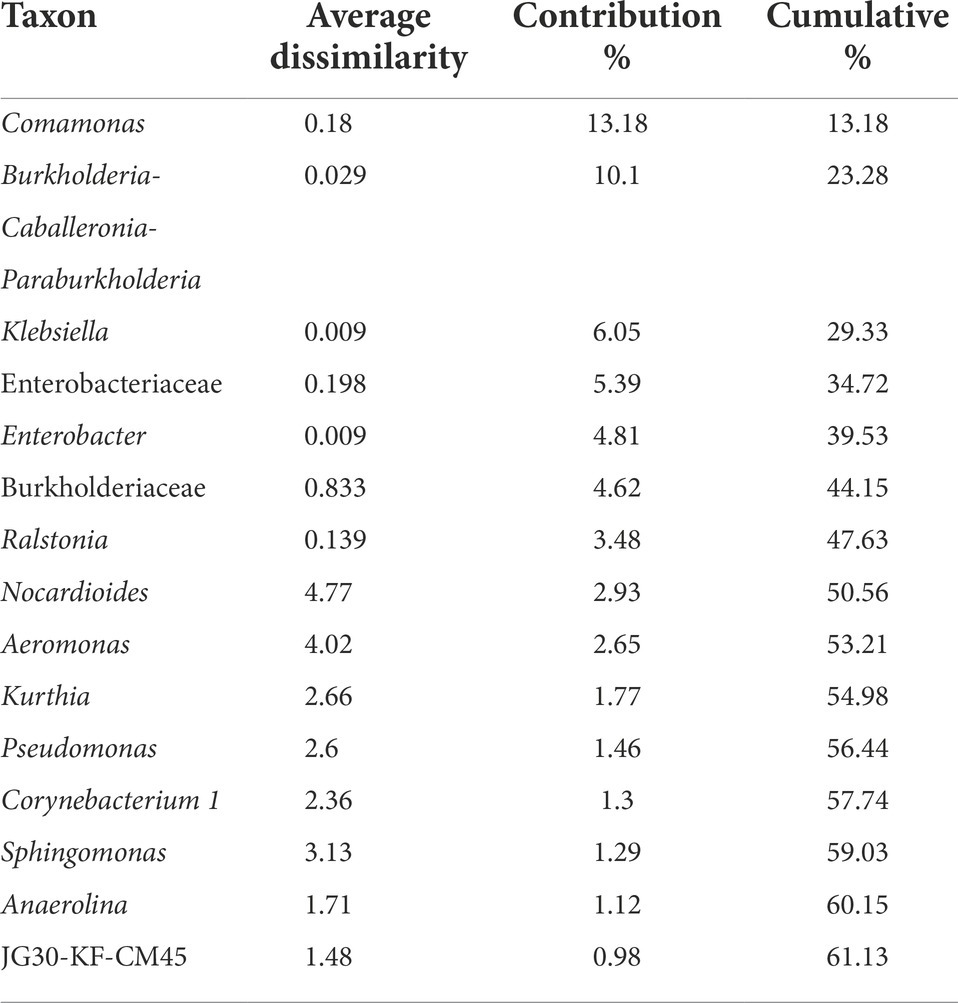
Table 4. SIMPER analysis of microbial communities from HCO3− enrichments using rock samples from different depth.
The Spearman correlation was used to identify the population at the phylum level that shared strong correlation amongst each other. In HC amended sets (Supplementary Figure 6), enriched members of the bacterial phyla Bacteroidetes, Spirochaetes, Cyanobacteria, Chloroflexi, WPS-2, and Patescibacteria as well as archaeal Euryarchaeota and Thaumarchaeota members constituted a strongly correlated clade. Armatimonadetes, Chlamydiae, Actinobacteria, Fusobacteria, Planctomycetes and Firmicutes populations formed the second guild. Coprothermobactereota, Altiarchaeota, Crenarchaeota, Caldiserica, Thermotogae, Verrucomicrobia constituted the third group. Lentisphaerae, Omnitraphechaeota, Synergistis and members belonging to FCPU426, Latescibacteria and Zixibacteria formed smaller mutually correlated clade. Proteobacteria, in spite of being the most abundant phylum, showed negative correlation with other enriched phyla. In the case of BC enrichments (Supplementary Figure 7) Chloroflexi, Gemmatimonadetes, Verrucomicrobia, Firmicutes, Actinobacteria and Bacteroidetes formed a strongly correlated group. The other two groups with strong correlation were formed by Cyanobacteria, Euryarchaeota, Deinicoccus Thermus and Crenarchaeota, Fibrobacteres, Tenericutes, respectively. Broadly, chemo/litho/autotrophic members of Cyanobacteria, Choloroflexi, Verrucomicrobia, Altiarchaeota were observed to correlate with members of Coprothermobactereota, Tenericutes and Gemmatimonadetes that are known to follow hetero/organotrophic lifestyle mostly with fermentative type of metabolism.
The co-occurrence pattern of the H2 + CO2 or HCO3− enriched microorganisms was analysed using network inferences based on strong and significant correlations, at the lowest taxonomic levels for OTUs present in at least four samples (Figures 7A,B). In HC enriched sets, the resulting microbial network consisted of 51 nodes and 187 edges (Figure 7A). The network consisted of two major hotspots (regions with highly connected taxa), with Bradyrhizobium, Brachybacterium, Streptomyces, Acinetobacter, Corynebacterium 1, being the taxa with the highest number of connections (16 each), followed by Sphingomonadaceae (15 connections). These taxa were found to be connected to Conexibacter, Nocardiodes, Pseudonocardia, Methylobacterium, Marinococcus, Stenotrophomonas, Actinobacillus, Curvibacter, Comamonas and Thermoplasmatales members. The second hotspot was formed by members belonging to Thaumarchaeota_Group 1.1c, Methyloversatilis, Rhodanobacter, Pseudomonas, Microtrichales, Smithella, Ralstonia and Cupriavidus. The most dominant taxa of the HC enrichments like Comamonas, Burkholderia-Caballeronia-Paraburkholderia, Enterobacteriaceae members (maximally dominant in C4–C7 enrichments) showed very few connections (mostly negative) with the other groups, hinting upon the fact that depth of the samples also played a role in determining the correlation amongst the enriched members. Members belonging to Burkholderiaceae, Chloroflexi, Alicycliphylus, Brachymonas, Citrobacter, Burkholderia-Caballeronia-Paraburkholderia, Comamonas, Cupriavidus, Ralstonia and Hydrogenophaga showed strong positive correlation with depth, and other geochemical parameters like CH4, Mn, H2, CO2 as depicted in Supplementary Figure 8A. Bacterial taxa like Anoxybacillus, Conexibacter, Sideroxydans, Anaerococcus, Anaerobranca, Chloroflexi_KD4-96, Pseudomonas as well as archaeal taxa belonging to Methanomassillicoccalles and Methanosaeta were observed to correlate positively with Fe, SO42− and NO3−, and negatively with depth and CH4, Mn, H2 (Supplementary Figure 8B).
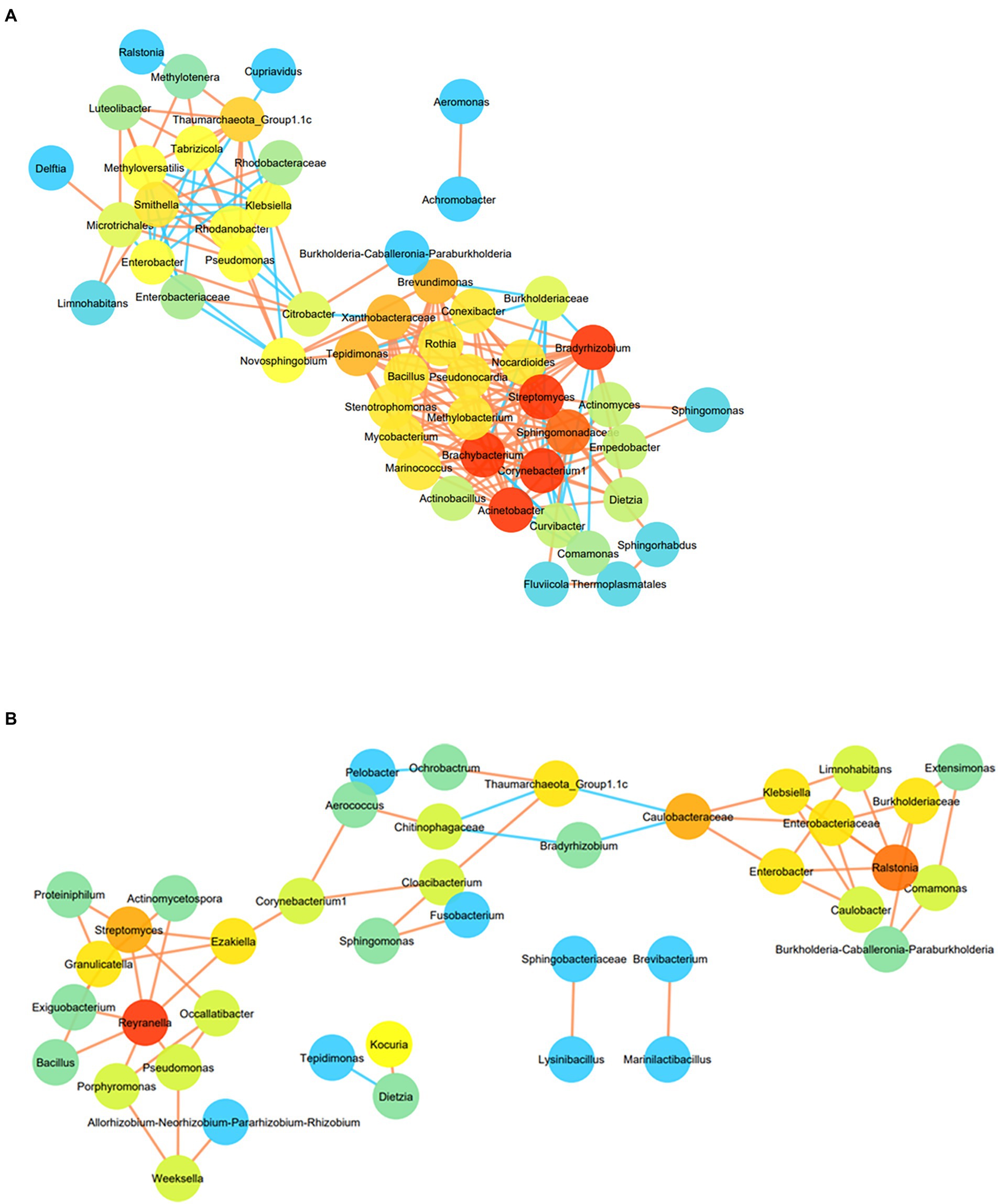
Figure 7. (A) Co-occurrence network of OTUs enriched from at least 4 samples (at the lowest taxa level) in HC enrichment. Node colour are proportional to the number of degrees. Nodes with higher degree are orange in colour than those with lower connections (blue in colour). Colour of the edges represents the strength of the Spearman’s correlations between the nodes. Edges with stronger correlation are orange in colour. For the Spearman correlation values |r| > 0.9 are only considered. (B) Co-occurrence network of OTUs enriched from at least 4 samples (at the lowest taxa level) in BC enrichment. Node colour are proportional to the number of degrees. Nodes with higher degree are orange in colour than those with lower connections (blue in colour). Colour of the edges represents the strength of the Spearman’s correlations between the nodes. Edges with stronger correlation are orange in colour. For the Spearman correlation values |r| > 0.9 are only considered.
Co-occurrence network for BC enriched samples were formed of three interconnected hotspots comprising of 41 nodes and 56 edges (Figure 7B). Reyranella was found to be the taxa with highest degree connected to seven other taxa nodes including Exiguobacterium, Bacillus, Streptomyces, Actinomycetospora and Pseudomonas. Ralstonia was found to act as the nucleus node of the second hotspot connecting to six other taxa namely, Enterobacteriaceae, Burkholderiaceae, Klebsiella, Comamonas, Enterobacter and Limnohabitans. Members belonging to Thaumarchaeota, Aerococcus, Sphingomonas and Chitinophagaceae formed the third hotspot. Methanolinea, Chroococcidiopsis, Xanthobacter, Ralstonia, Cupriavidus, Curvibacter, Methylophilus, Syntrophaceae and Burkholderiaceae members were found to be correlated positively with depth and parameters including CO2, H2, CH4 and Mn (Supplementary Figure 9A), whereas members of Thaumarchaeota_Group1.1c, Thermomicrobiales, Methylophilaceae, Thermoactinomyces, Thiomonas, Sporosarcina had strong positive correlation with SO42−, Fe and NO3 - but was found to correlate negatively with depth, Mn, CH4 and H2 (Supplementary Figure 9B).
To determine the evolutionary history and gain insight about the relationships of the reactivated organisms, Neighbour Joining (NJ) phylogenetic analysis of 15 most abundant OTUs and cyanobacterial OTUs from both inorganic enrichments (HC and BC, making a total of 36 query OTUs) were performed by constructing a bootstrap tree (Supplementary Figure 10). The presence and relative abundance of these OTUs were depicted in the form of heat map beside each OTU. OTUs observed under both conditions were designated in green, the ones enriched only in BC sets were designated in blue and the OTU that belongs only to HC enrichment set was marked in red. OTUs enriched under both conditions, belonging to bacterial members like Burkholderia-Caballeronia-Paraburkholderia, Klebsiella, Rhodanobacter, Methyloversatilis, Pelomonas, Comamonas, Aeromonas, Kurthia, Nocardiodes, Corynebacterium 1, Dongia, Gemella, Deinococcus, etc. and archaeal group Thaumarchaeota were observed to be phylogenetically connected to sequences retrieved from deep terrestrial subsurface sediments from USA, subsurface clay rock, sediment from Eyreville drill corehole in Chesapeake bay, deep sea, deep hydrothermal vent in Nankai Trough, Hadal Deep Biosphere Mariana Trench and hydrothermal aquifer in Naica Mines. OTU affiliated to Conexibacter (unique to HC enrichment) was found to be phylogenetically similar (with 1,000 bootstrap) to an endolithic bacterial sequence from serpentinite-hosted ophiolite massif of the Alpine-Apennine chain in Italy. Sequences belonging to Fluviicola, Anaerolinea and Prolixibacteraceae were enriched only under BC enrichment condition and were found to be similar to sequences from deep subsurfaces of sedimentary rock Japan, Paris basin aquifer and Mariana Trench. Cyanobacterial sequences enriched from both sets showed strong matches with other cyanobacterial sequences retrieved from granitic subsurface environment in Tokyo, Clay rock borehole water of Switzerland, deep subsurface of South Africa and Mariana Trench.
The bacterial community analysis performed in this study suggested the presence of diverse bacterial populations with varied predicted metabolic functions in deep terrestrial subsurface. PICRUSt v2 based predictive metabolic profiling was done to gain insights into the metabolic potential of such microorganisms that help them to sustain in a subsurface environment. The relative abundances of genes related to different metabolic traits, that were predicted from our enrichments have been presented in Figure 8. Analysis revealed that the genes known to be involved in carbon cycling pathways, mainly in carbon fixation viz. acetyl-CoA decarbonylase (acsABCD), formylmethanofuran dehydrogenase (fmdABCDE; genes for CO2 fixing WL pathway in acetogens and methanogens respectively), acetyl-CoA carboxylase, (accABCD; for HCO3− fixing 3-HP bicycle), phosphoenolpyruvate carboxylase (ppc), 4-hydroxybutyryl-CoA dehydratase, (abfD; for DC/4HB cycle, CO2 and HCO3− fixing), ribulose-bisphosphate carboxylase (rbcS/L), phosphoribulokinase (prk; for CO2 fixing CBB cycle) and Incomplete reductive TCA cycle, were predicted in all enrichment samples.
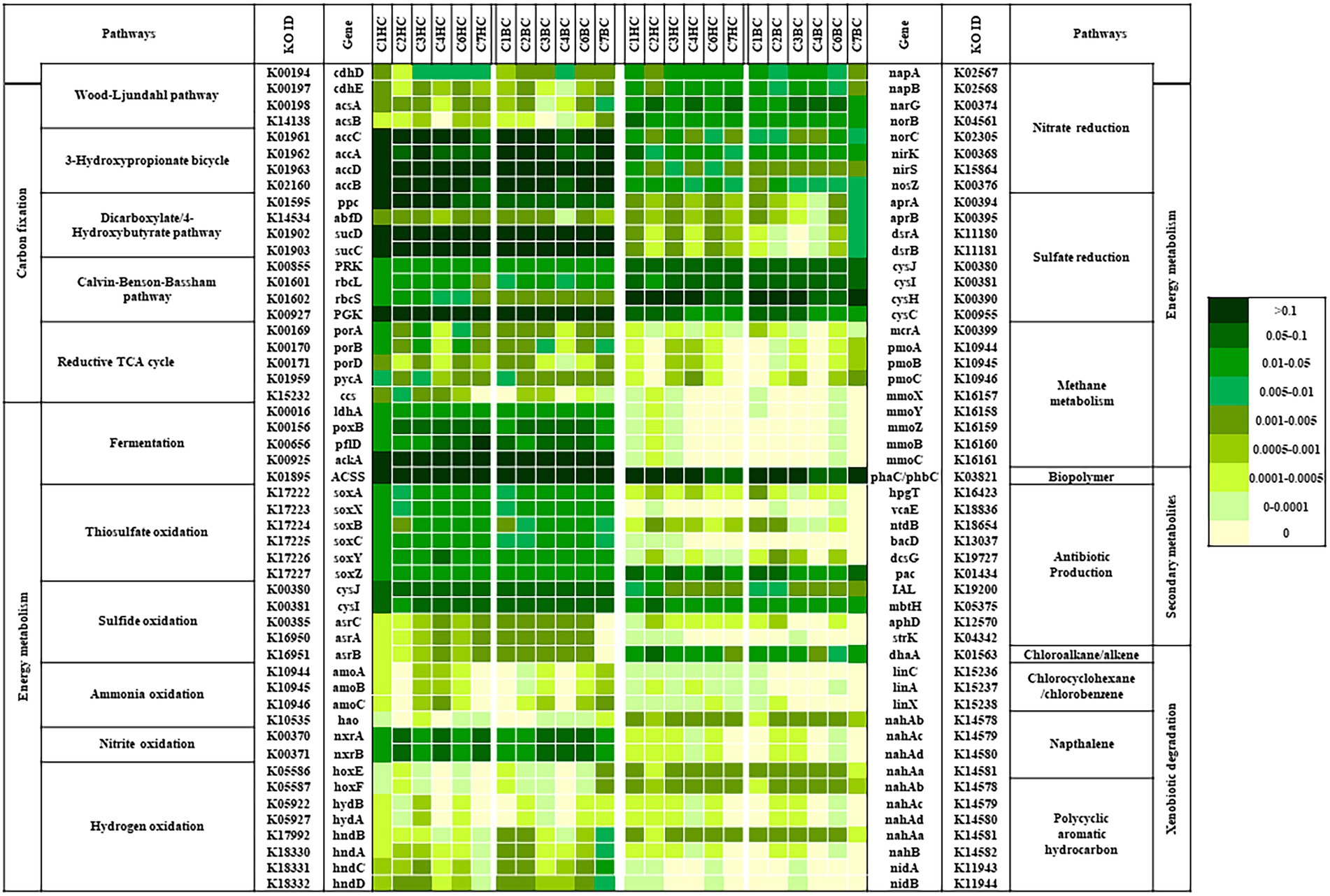
Figure 8. Relative abundance of predicted genes related to CO2 fixation pathway, lithotrophy related/energy metabolism and genes related to other important metabolism (amongst metabolic genes) as predicted by PICRUSt v2.
Lithotrophic metabolisms other than carbon dioxide and bicarbonate fixation play an important role in driving the energy cycle in the deep (Lovley and Chapelle, 1995; Osburn et al., 2014). The relative abundance of genes related to various lithotrophy related metabolism (as predicted) were presented in Figure 8, Energy metabolism.
Methane metabolism: Genes related to methane production (methyl-coenzyme M reductase; mcrA) and oxidation (methane monooxygenase; pmo, mmo) were predicted in most of the enrichment samples except in C7HC and C4BC. Overall relative abundance of genes mcr, pmo or mmo was found to be considerably lower than the abundance of other metabolic genes related to lithotrophy.
Nitrogen metabolism: The predicted presence and abundance of nitrogen metabolism genes in all enrichment sets hinted upon the significance of such metabolism. Genes related to nitrite oxidation (nitrate reductase / nitrite oxidoreductase; nxr), nitrate reduction (nitrate reductase; nar/nap, nitrite reductase; nir, nitric oxide reductase; nor and nitrous-oxide reductase; nos) and ammonia oxidation (ammonia monooxygenase; amo, hydroxylamine dehydrogenase; hao) were predicted in all enrichment sets.
Sulfur metabolism: Genes known to be involved in sulfide (anaerobic sulfite reductase; asr, cys) and thiosulfate (soxABCXYZ) oxidation, sulfate reduction (adenylylsulfate reductase; apr, dissimilatory sulfite reductase; dsr, sulfite reductase; cys), were also predicted from the enriched samples. However, the predicted relative abundance of dissimilatory sulfate reduction genes was lower than genes involved in assimilatory sulfate reduction.
Hydrogen oxidizing genes (hydrogenases; hnd, hyd, hox) were predicted to be more abundant in the deeper rock sample enrichments. Genes for production of acetate, lactate and pyruvate (ldh, pox, pfl, ack, ACSS) were predicted in all the enrichment samples.
Presence of genes related to synthesis of secondary metabolites like polyhydroxyalkanoate, polyhydroxybutyrate (pha/phb), antibiotic (vca, ntd, bac, dcs, pac, mbt, str), as well as degradation of chloroalkane (dha), chlorocyclohexane (lin), naphthalene (nah) and polycyclic aromatic hydrocarbon (nah, nid) could also be predicted from both the enrichment sets (Figure 8; Secondary metabolites and Xenobiotic degradation).
In spite of extreme high temperatures, lithostatic pressure, increasing dryness and aridity, decreasing porosity, scarcity of survival space and nutrient deprivation, deep terrestrial subsurface environments have been found to be inhabited by diverse microbial communities that have been shaped by the prevailing physical and geochemical factors (Nyyssonen et al., 2014; Kieft, 2016; Dutta et al., 2018, 2019; Sahu et al., 2022). The life in deep biosphere is considered to be driven by geologically produced CO2, H2 and/or reduced S and N driven chemoautotrophic production of small organic molecules which can be utilized further as nutrient resources. (Purkamo et al., 2015; Adhikari et al., 2016). In the present study, we have provided chemolithoautotrophic conditions, viz., two different inorganic carbon sources (CO2/HCO3-), H2 / NH4+ as electron donors and NO3− and/or SO42− as electron acceptors, to the deep subsurface rock hosted communities to enrich chemolithotrophic microorganisms. It has resulted in successful enrichment of diverse groups of microorganisms (Bacteria and Archaea) as evident from the outcome of the growth measurement experiments (Figure 1) and formation of different metabolites like lactate, formate and acetate. In taxon-based approaches, accurate assessment of species richness is considered as it is suggested to be necessary for effective analysis of the bacterial community (Kim et al., 2017). The diversity indices have suggested variable levels of diversity and richness in both HC and BC amended incubations. The outputs of Chao1 and ACE (richness estimators) indices mark the enrichment sets as microbially rich which is also supported by the Shannon diversity index values and corroborated well with the previous observations, on granitic samples belonging to KSZ (Dutta et al., 2018). The contrast between increase of gene copy numbers (on average) and the decrease in the number of OTUs (in deeper rock enrichments) has suggested that specific microbial taxa members dominated the enrichments from deeper rocks that also supported our taxonomic data where fewer groups made up the enriched communities of deeper rocks. On the other hand, enrichment cultures of the relatively shallower rocks exhibited more diverse organisms. The high percentage of unique OTUs in each enrichment set (Supplementary Figures 2A,B) possibly indicated that the community composition of the rock enrichment cultures were significantly shaped by the intrinsic nature of the rock samples obtained from different depths. 16S rRNA gene sequencing indicated the ubiquitous predominance of phylum Proteobacteria, (Gamma-, Alpha-, Delta-, Betaproteobacteriales), with increasing abundance in the deeper rock enrichment samples (Supplementary Figure 3). Several members of Proteobacteria are reported to follow autotrophic mode of nutrition along with H2/Fe2+/S oxidizing potential (Nakagawa and Takai, 2008; Brazelton et al., 2012; Purkamo et al., 2015; Kumar et al., 2018). Some of the members were reported to be capable of dark bicarbonate assimilation (Alonso-Saez et al., 2010). However, plenty of proteobacterial taxa follow heterotrophic mode of nutrition and it might be a possibility that these heterotrophic members survive on the organic metabolic byproducts of other autotrophic organisms. Actinobacteria, Firmicutes, Bacteroidetes, Acidobacteria are among the other dominant phyla found to be enriched in this study. Presence of Actinobacteria in the enrichment samples has corroborated well with extreme stress-tolerant ability (including high temperature and salinity) of its members as well as previous reports on their abundance within the igneous crust underneath the Deccan Traps (Idris et al., 2017; Dutta et al., 2018; Bull and Goodfellow, 2019; Hui et al., 2021; Sahu et al., 2022). Members of Firmicutes are found to be capable of extracting energy and other nutrients from diverse inorganic resources and also assimilate CO2 (Salehizadeh et al., 2020). Acidobacterial members are reported to harbour a versatile repertoire of genes necessary to use ammonia and/or nitrate/nitrite as their N sources and H2 at atmospheric concentrations (Eichorst et al., 2018). Thermophilic, anaerobic, chemolithoautotrophic Chloroflexi members are described to be environmentally widespread and reported to harbour genes involved in adaptation against various environmental stress conditions (Wasmund et al., 2014; Kadnikov et al., 2019; Vuillemin et al., 2020). They are reported to contain potential for fermentation, CO2 fixation and acetogenesis via the Wood–Ljungdahl (WL) pathway (Vuillemin et al., 2020). Presence of Thaumarchaeota and Euryarchaeota (known to harbor thermophilic ammonia oxidizers and methanogens, respectively) in the enrichments probably hinted about their chemolithoautotrophic potential that helped them to thrive within this extremely energy-limited terrestrial subsurface environments while playing a prominent biogeochemical role (Seán et al., 2003; Ragon et al., 2013; Cardarelli et al., 2020). The enriched taxa that are found capable of adaptation to diverse environmental stresses, CO2 fixation, and lithotrophic metabolisms could possibly be attributed and linked to the existing environmental and geochemical conditions of the KSZ deep subsurface (Dutta et al., 2018, 2019; Sahu et al., 2022). The rocks in this dark biosphere have not seen sunlight since the geologic past (Dutta et al., 2018), yet interestingly, enrichment of Cyanobacteria (thought to require sunlight to survive) were observed in HC and BC (minute fraction) enrichment sets. Previous studies have shown presence of Cyanobacteria in igneous crust (basalt and granitic rocks) from deep subsurface of the Deccan traps (Dutta et al., 2018). Members of Cyanobacteria have been reported to be ecologically versatile and to be associated with a potential for H2-based lithoautotrophic metabolism in deep terrestrial subsurface rocks (Momper et al., 2017; Kadnikov et al., 2018; Puente-Sánchez et al., 2018). Considering the recent report on presence of H2 in the deep subsurface of Koyna region (Podugu et al., 2019), the presence of Cyanobacterial members (mainly in HC enrichments) could probably be linked to their H2-based lithoautotrophic metabolic ability. Previous investigations suggested that Cyanobacterial members can also switch to fermentative lifestyle when required (Dutta et al., 2018 and references therein).
NMDS analyses (Figure 5) has illustrated the community assemblages and the role of subsurface geochemistry (including depth and different enrichment substrates) in shaping the microbial community. The role of geochemical factors on microbial community structure and function in various crystalline deep biospheres have previously been described (Brazelton et al., 2013; Magnabosco et al., 2014). In the present study, depth is found to be the influential environmental variable which significantly affected the microbial community structure. Even though substrate has an influence on microbial composition in enrichment sets, it is subtle compared to the stark effect of depth of the different rocks used for enrichments. Depth wise distribution of the enrichment samples irrespective of the enrichment substrates is also observed in the CCA analysis (Figures 6A,B) along with strong constraint of Mn, CO2, H2, and CH4 in deeper rock (C4–C7) enrichments, and TIC, Fe, NO3− and SO42− in the shallower rock (C1–C3) enrichments. The results of SIMPER analysis has indicated that facultatively lithotrophic CO or H2-oxidizing, NO3−reducing and the most abundant Comamonas (Willems and De Vos, 2006) to be the maximum contributor in depth dependent average dissimilarity.
Correlation among the enriched microbial phyla based on their abundance highlighted the possible interplay among the enriched groups and a marked effect of depth. The groups constituting each of the strongly correlated clades has shown variety in their abundances with the change in the sample depth, either following a pattern or predominating at some specific depths. This difference in abundances could be attributed to the existing environmental conditions of that sample which may have favoured the growth of specific groups. Syntropy has been reported to play an important role in deep subsurface environments (Matsushita et al., 2020). Autotrophic taxa proficient of C fixation (e.g., Cyanobacteria), also capable H2-based metabolism (Puente-Sánchez et al., 2018), Chloroflexi, and NH4 oxidizing/ methanogenic archaeal groups are observed to associate with organotrophic acetate assimilating Bacteroidetes (Kadnikov et al., 2020), fermentative Firmicutes and Fusobacteria members (Rogers et al., 1998; Gupta et al., 2018).
Microbial co-occurrence analysis has indicated the presence of distinct and well-defined networks across the enriched samples (Figures 7A,B), which corroborated well with the correlation-based observation of association between autotrophic and heterotrophic communities. Taxa affiliated to the first hotspot have previously been reported to be involved in diverse, yet deep biosphere relevant metabolic processes. Organisms belonging to Bradyrhizobium, Brachybacterium, Streptomyces, Acinetobacter, Conexibacter, Pseudonocardia, Curvibacter, and Comamonas have been known to tolerate enhanced CO2 level or utilize H2 and/or CO/CO2 as sole energy and carbon source (Willems and De Vos, 2006; Greening et al., 2018; Takors et al., 2018; Gülay et al., 2018; Islam et al., 2020). These organisms are observed to correlate strongly amongst each other and with organotrophic Nocardiodes, methylotrophic Methylobacterium and mixotrophic Thermoplasmatales members (Huber and Stetter, 2006; Yoon and Park, 2006). The second hotspot depicted strong connections between lithotrophic organisms like ammonia oxidizing, dark carbon fixing Thaumarchaeota_ Group1.1c, SO42−, CO and H2 utilizing Microtrichales, lithoautotrophic Rhodobacteraceae members, methylotrophic Methyloversatilis and metabolically versatile Pseudomonas members (Ghosh and Dam, 2009; Lapidus et al., 2011; Pohlner et al., 2019; Cardarelli et al., 2020). The network of HCO3- enrichments depicted the presence of smaller interconnected subnetworks wherein dominant taxa of deeper rock enrichments (Ralstonia, Comamonas, Burkholderia-Caballeronia-Paraburkholderia, Burkholderiaceae etc.), and the dominant taxa of shallower rock enrichments (Sphingomonas, Nocardiodes, Corynebacterium 1, Aerococcus etc.) formed two hotspots. The interconnected taxa in these hotspots were reported to follow chemolithoautotrophy or organotrophy. The third hotspot mainly consisted of highly connected organotrophic and metabolically versatile taxa members (Yoon and Park, 2006; Cardarelli et al., 2020).
Overall, these correlation studies and co-occurrence networks distinctly highlighted the strong mixotrophic interconnection and association between enriched lithoautotrophic and organotrophic organisms, where, based on their preferred modes of nutrition they help in the successful utilization of the available substrates and drive the energy requirement of the enriched community. Similar associations amongst autotrophic and heterotrophic populations have been reported in previous studies on the deep subsurface settings and igneous rock environments (Hallbeck and Pedersen, 2012; Pedersen, 2013; Purkamo et al., 2015; Adhikari et al., 2016; Wu et al., 2016; Momper et al., 2017; Dutta et al., 2018; Sahu et al., 2022). A possible reason for this association can be the utilization of organic by products (exudated by chemolithoautotrops) by the interacting heterotrophic community (Wu et al., 2016).
The observation on the correlation networks of the enriched taxa with depth and other geochemical parameters corroborated well with similar reports from other deep subsurface environments with dominant chemolithotrophic mode of nutrition (Sahl et al., 2008; Yoon et al., 2008; Cramm, 2009; Panda et al., 2009; Purkamo et al., 2015; Carr et al., 2018; Gülay et al., 2018; Hädrich et al., 2019; Cardarelli et al., 2020; Nangle et al., 2020; Jakus et al., 2021). Members of Burkholderia-Caballeronia-Paraburkholderia (one of the major taxa enriched from our study and having positive correlation with sample’ depth) have also shown highest correlation with depth and connections with geochemical parameters like H2 and CH4 in an earlier study related to the rock metagenome (Sahu et al., 2022).
A similarity search of the most abundant OTUs revealed a significant relationship with already reported species from similar dark, aphotic and extremophilic deep subsurface habitats with chemoauto/organotrophic metabolic characteristics (Brown and Balkwill, 2009; Ragon et al., 2013; Ino et al., 2018; Supplementary Figure 10). These significant matches revealed that the groups enriched from our study might follow similar chemolithoautotrophic nutritional modes as reported previously. Considering anaerobic environmental conditions, SO42−, NO3−, Mn4+, Fe3+ and CO2, could be used as the most “auspicious” (Purkamo et al., 2015) terminal electron acceptors, and organisms metabolising the same, could thrive and play a dominant role in these enrichment samples.
Understanding the dynamics of processes carried out by ubiquitous microorganisms of these deep terrestrial systems has been important as it holds broad significance for the global biogeochemical cycles (Hernsdorf et al., 2017). Investigation of the 16S rRNA gene amplicon from the total community DNA of the enrichment cultures for function prediction elucidated the metabolic capacities of the cultured communities, especially in terms of their carbon and energy metabolism processes. The prediction of major metabolic pathways gave us an insight of the metabolic repertoire present in these enrichment cultures (Figure 8).
CO2 assimilation: Chemically stable and unreactive CO2 must be reduced to enable its incorporation into biological molecules. In the recent past, CO2 fixation pathways used by autotrophic microorganisms have received interest for biotechnological applications, since it could provide biological routes for de novo generation of fuels and small organic molecules (Hawkins et al., 2013). Analysis of the metabolic repertoire of the samples predicted the prevalence of mixotrophic (both autotrophic and organotrophic) modes of C-assimilation. 3HP bicycle, DC/4HB cycle, CBB and Wood-Ljungdahl pathways are predicted to be the most significant autotrophic C assimilation processes. The WL pathway have been described as the most energy inexpensive pathway with net synthesis of ATP along with carbon fixation (Berg, 2011; Liu et al., 2020). Prediction of the WL pathway key genes in the enriched microbiome could be justified owing to its suitability as a favoured C fixation processes in these extreme energy deprived habitats (Momper et al., 2017). Bioavailability of surface-derived organic C has been found to be limited in the terrestrial granitic subsurface and hence many resident heterotrophs rely on in-situ production of fixed carbon by chemoautotrophs. Key genes for 3-HP pathway and DC/4HB pathway are predicted from the cultures. These pathways have been known to assimilate bicarbonate via acetyl-CoA/propionyl-CoA carboxylase/ PEP carboxylase respectively, and produce pyruvate and acetyl CoA (Liu et al., 2020). Both cycles have very high energy requirements, and the reason these cycles have survived through evolution might be due to their oxygen tolerance and bicarbonate assimilation instead of CO2 (Liu et al., 2020). The latter is advantageous, as the intracellular bicarbonate concentration can be much higher than the intracellular CO2 concentration. Putative presence of the key gene Acetyl-CoA carboxylase (acc) also suggested the possibility of dark C assimilation in the deep subsurface by the enriched community (Purkamo et al., 2015). This enzyme converts Acetyl-CoA to Malonyl-CoA, which can be used in diverse biosynthetic pathways (e.g., fatty acid biosynthesis and secondary metabolism, Momper et al., 2017). The DC/4HB cycle has been known to fix one mole of CO2 via pyruvate synthase and one mole of bicarbonate via phosphoenolpyruvate (PEP) carboxylase, in anaerobes, as well as in facultative aerobes (Berg et al., 2010; Liu et al., 2020; Saini et al., 2011). The putative presence of genes related to this pathway might provide an added advantage of fixing both CO2 and bicarbonate via the same thereby managing the energy expenditure in these enriched communities. The energy requirement in this pathway is found to be lower than the other two bicarbonate fixing pathways (Berg et al., 2010; Liu et al., 2020). Organisms enriched in our samples, belonging to taxa like Chloroflexi-KD4-96, Microtrichales, Altiarchaeia, Methanolinea, Ralstonia, Rhodobacteria, Acidithiobacillus, Conexibacter, Hydrogenophaga, Methanobacterium etc., have been reported earlier to be capable of autotrophic fixation of CO2 (Beulig et al., 2015; Liu et al., 2020; Salehizadeh et al., 2020).
Lithotrophy related energy metabolism: Enriched endolithic microbial communities inhabiting rocks might carry out in situ primary metabolite production (Lovley and Chapelle, 1995; Stevens, 1997). Available evidence suggested the use of different chemolithoautotrophic mechanisms for exploiting this energy in the deep subsurface, such as S, N, Fe, Mn metabolism, H2 oxidation, CH4 metabolism and acetogenesis (Stevens, 1997; Osburn et al., 2014). The analysis of 16S rRNA gene amplicon inventory predicted the presence of genes for S (aps, dsr, cys, asr, sox), N (nar, nir, nif, nap, nos,), CH4 (pmo, mmo, mcr) metabolism, NH4 oxidation (amo, hao) and acetogenesis (WL pathway) in our enrichment cultures. The predictive abundance of SO42− reducing genes in the H2- induced enrichments hinted upon the fact that sulfate reduction dependent H2 oxidation, might be an ongoing process in the deeper rock enrichments (Pedersen, 2013). Predictive presence of H2 oxidizing genes (hnd, hyd, hox), mostly in the deeper sample enrichments, and the enrichment of members belonging to Hydrogenophaga, Methanolinea, Ralstonia, Methanobacterium, Cupriavidus and Dehalococcidia (all being previously reported as hydrogenotrophic organisms and positively correlated to depth in our study), indicated the value of H2 as an energy source and suggested that the enriched microbes could be involved in its utilization (Adhikari et al., 2016; Hernsdorf et al., 2017; Nangle et al., 2020; Yang et al., 2020). The enriched organisms (Thaumarchaeota_ Group1.1c, Thermodesulfovibrio, and Burkholderia-Caballeronia-Paraburkholderia, Thiomonas) are predicted to be capable of utilizing inorganic compounds to harbour energy for their sustenance where reduced S and N, could act as potent electron donors and SO42−, NO3−, Mn4+ as electron acceptors to fuel the metabolism of the enriched community (Panda et al., 2009; Cardarelli et al., 2020). The predicted ability of the enriched taxa to carry out lithotrophic metabolisms could possibly be attributed and linked to the presence of the mentioned electron donors and acceptors in the KSZ deep subsurface environments (Dutta et al., 2018, 2019; Sahu et al., 2022). Putative presence of fermentative pathways for acetate (pox, ACSS, ack), lactate (ldh), pyruvate (pfl), and formate production could be an indication of organotrophy (Dutta et al., 2018), thereby corroborating with earlier reports of interplay between autotrophic and heterotrophic enriched community.
Other associated metabolisms (secondary metabolite, antibiotic production and xenobiotic degradation): Secondary metabolites resulting from biosynthetic reactions have tremendous potential to be novel agents performing as chemotherapeutics, nutraceuticals or other value-added products. The deep biosphere has been investigated for bioactive natural products, unique polysaccharides, lipids and enzymes (Pettit, 2011). Genes related to synthesis of different antimicrobial and medicinal compounds, biodegradable polymers and xenobiotic degradation were predicted from the 16S rRNA gene amplicon inventory of our study. This prediction could be linked to the enrichment of taxa like Actinobacteria, Pseudomonas, Aeromonas, Alcaligenes, Ralstonia and Marinobacter, few of which were reported earlier to be capable of production natural biodegradable polyesters (PHA, PHBs), antimicrobial compounds and degradation of xenobiotic compounds (Mullis et al., 2019; Moradali and Rehm, 2020; Miglani et al., 2022).
In conclusion, this study provided a detailed overview of indigenous microorganisms enriched from granitic rock core samples, drilled out from the reservoir triggered seismogenic zone in Koyna, Maharashtra, India. Our enrichment-based work showed that the deep, hot, crystalline granitic rocks host live microorganisms (Bacteria and Archaea) and these can be brought to culture using chemolithotrophic, anaerobic conditions, with inorganic carbon substrates like CO2 (along with H2 as electron donor) or HCO3− (along with NH4 as electron donor) as the sole C source. Furthermore, the taxonomic compositional analyses indicated a statistically significant, distinct depth-wide partitioning of diverse chemolithotrophic Bacteria and Archaea enriched from these rocks. 16S rRNA gene-based community composition showed Proteobacterial members to be the dominantly enriched bacterial group with their abundance being higher in the deeper rock sample enrichments. Based on the results of predictive metabolic profiling, it can be assumed that assimilation of carbon with utilization of sulfur, hydrogen and nitrogen compounds coupled with fermentation and anaerobic respiration could act as the main energy drivers.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
SK and PS conceived the study, designed the experiments, arranged all the resources, prepared the manuscript and mentored the study. SM carried out the microcosm setups, microbiological analysis, statistical analysis, data analysis, and manuscript preparation. HB assisted in sampling, sub-coring of rocks and carrying out some experiments. KR carried out the statistical analysis. RS assisted in qPCR and sequence analysis. AS performed the sequencing. All authors contributed to the article and approved the submitted version.
This study was supported by the Ministry of Earth Sciences (MoES), Government of India (project ID: MoES/P.O. (Seismo)/1(288)/2016 dated 16 March 2017).
The authors are thankful to Dr. Sukanta Roy, Project Director, MoES-BGRL, Karad, and all other members of the Borehole Geophysics Research Laboratory, Karad, India, engaged in the deep drilling of the Koyna pilot borehole for their help and support in sampling. SM gratefully acknowledges the fellowship provided by National Institute of Technology Durgapur. KR gratefully acknowledges the MHRD fellowship provided by National Institute of Technology Durgapur. RS gratefully acknowledges the fellowship provided by Council of Scientific and Industrial Research, Government of India. AS gratefully acknowledges the fellowship provided by Indian Institute of Technology Kharagpur.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at:
https://www.frontiersin.org/articles/10.3389/fmicb.2022.1018940/full#supplementary-material
Supplementary Figure 1 | OTU overlap within HC and BC enrichments to determine the unique and shared OTUs.
Supplementary Figure 2 | OTU overlap within all (A)HC and (B)BC enrichments.
Supplementary Figure 3 | Taxonomic distribution of the microbial communities in HC and BC enrichments at the phylum/class/order level.
Supplementary Figure 4 | Heatmap displaying the relative abundance of less abundant taxa (abundance <0. 1%) detected in (A) HC and (B) BC enrichments.
Supplementary Figure 5 | Rank abundance (based on relative percentage abundance) of less abundant OTUs (abundance <0. 1%) detected in (A) HC and (B) BC enrichments.
Supplementary Figure 6 | Heatmap of the Spearman correlation based on abundance of all microbial phyla detected in HC enrichment (abundance values are mentioned in the first bracket).
Supplementary Figure 7 | Heatmap of the Spearman correlation based on abundance of all microbial phyla detected in BC enrichment (abundance values are mentioned in the first bracket).
Supplementary Figure 8A | Co-occurrence network of microbial communities (taxa level) in HC enrichment positively correlated with sample’s depth. Node colour are proportional to the number of degrees. Nodes with higher degree are orange in colour than those with lower connections. Colour of the edges represents the strength of the Spearman’s correlations between the nodes. Edges with stronger correlation are orange in colour. Burkholderia-Caballeronia-Paraburkholderia is designated as Bur-Cab-Para.
Supplementary Figure 8B | Co-occurrence network of microbial community (taxa level) in HC enrichment negatively correlated with sample’s depth. Node colour are proportional to the number of degrees. Nodes with higher degree are green in colour than those with lower connections. Colour of the edges represents the strength of the Spearman’s correlations between the nodes. Edges with stronger correlation are green in colour.
Supplementary Figure 9A | Co-occurrence network of microbial community (taxa level) in BC enrichment positively correlated with sample’s depth. Node colour are proportional to the number of degrees. Nodes with higher degree are orange in colour than those with lower connections. Colour of the edges represents the strength of the Spearman’s correlations between the nodes. Edges with stronger correlation are orange in colour. Burkholderia-Caballeronia-Paraburkholderia is designated as Bur-Cab-Para.
Supplementary Figure 9B | Co-occurrence network of microbial community (taxa level) in BC enrichment negatively correlated with sample’s depth. Node colour are proportional to the number of degrees. Nodes with higher degree are green in colour than those with lower connections. Colour of the edges represents the strength of the Spearman’s correlations between the nodes. Edges with stronger correlation are green in colour.
Supplementary Figure 10 | Neighbour joining tree based on top 30 OTUs and cyanobacterial OTU sequences and their related organisms showing phylogenetic relationship. Numbers at the nodes represent the bootstrap value (1,000 replicates). The colour codes depict the presence and abundance of OTUs in sample zones. S-Shallower rock enrichments (average of C1–C3), D- Deeper rock enrichments (average of C4–C7), HC- sets amended with H2 + CO2, BC- sets amended with Bicarbonate.
Adhikari, R. R., Glombitza, C., Nickel, J. C., Anderson, C. H., Dunlea, A. G., Spivack, A. J., et al. (2016). Hydrogen utilization potential in subsurface sediments. Front. Microbiol. 7:8. doi: 10.3389/fmicb.2016.00008
Adhikari, R. R., and Kallmeyer, J. (2010). Detection and quantification of microbial activity in the subsurface. Geochemistry 70, 135–143. doi: 10.1016/j.chemer.2010.05.003
Alain, K., Vince, E., Courtine, D., Maignien, L., Zeng, X., Shao, Z., et al. (2021). Thermococcus henrietii sp. nov., a novel extreme thermophilic and piezophilic sulfur-reducing archaeon isolated from a deep-sea hydrothermal chimney. Int. J. Syst. Evol. Microbiol. 71:004895. doi: 10.1099/ijsem.0.004895
Alonso-Saez, L., Galand, P. E., Casamayor, E. O., Pedros-Alio, C., and Bertilsson, S. (2010). High bicarbonate assimilation in the dark by Arctic bacteria. ISME J. 4, 1581–1590. doi: 10.1038/ismej.2010.69
Amend, J. P., and Teske, A. (2005). Expanding frontiers in deep subsurface microbiology. Palaeogeogr. Palaeoclimatol. Palaeoecol. 219, 131–155. doi: 10.1016/j.palaeo.2004.10.018
Arndt, D., Xia, J., Liu, Y., Zhou, Y., Guo, A. C., Cruz, J. A., et al. (2012). METAGENassist: a comprehensive web server for comparative metagenomics. Nucleic Acids Res. (Web Server issue): W88-95 40, W88–W95. doi: 10.1093/nar/gks497
Bates, S. T., Berg-Lyons, D., Caporaso, J. G., Walters, W. A., Knight, R., and Fierer, N. (2011). Examining the global distribution of dominant archaeal populations in soil. ISME J. 5, 908–917. doi: 10.1038/ismej.2010.171
Bell, E., Lamminmäki, T., Alneberg, J., Andersson, A. F., Qian, C., Xiong, W., et al. (2020). Active sulfur cycling in the terrestrial deep subsurface. ISME J. 14, 1260–1272. doi: 10.1038/s41396-020-0602-x
Berg, I. A. (2011). Ecological aspects of the distribution of different autotrophic CO2 fixation pathways. Appl. Environ. Microbiol. 77, 1925–1936. doi: 10.1128/AEM.02473-10
Berg, I. A., Ramos-Vera, W. H., Petri, A., Huber, H., and Fuchs, G. (2010). Study of the distribution of autotrophic CO2 fixation cycles in Crenarchaeota. Microbiology 156, 256–269. doi: 10.1099/mic.0.034298-0
Beulig, F., Heuer, V. B., Akob, D. M., Viehweger, B., Elvert, M., Herrmann, M., et al. (2015). Carbon flow from volcanic CO2 into soil microbial communities of a wetland mofette. ISME J. 9, 746–759. doi: 10.1038/ismej.2014.148
Bhaskar Rao, Y. J., Sreenivas, B., Vijaya Kumar, T., Khadke, N., Kesava Krishna, A., and Babu, E. V. S. S. K. (2017). Evidence for Neoarchean basement for the Deccan volcanic flows around Koyna-Warna region, western India: zircon U-Pb age and Hf-isotopic results. J. Geol. Soc. India 90, 752–760. doi: 10.1007/s12594-017-0787-4
Bomberg, M., Lamminmäki, T., and Itävaara, M. (2016). Microbial communities and their predicted metabolic characteristics in deep fracture groundwaters of the crystalline bedrock at Olkiluoto, Finland. Biogeosciences 13, 6031–6047. doi: 10.5194/bg-13-6031-2016
Brazelton, W. J., Nelson, B., and Schrenk, M. O. (2012). Metagenomic evidence for H2 oxidation and H2 production by serpentinite-hosted subsurface microbial communities. Front. Microbio. 2:268. doi: 10.3389/fmicb.2011.00268
Brazelton, W. J., Morrill, P. L., Szponar, N., and Schrenk, M. O. (2013). Bacterial communities associated with subsurface geochemical processes in continental serpentinite springs. Appl. Environ. Microbiol. 79, 3906–3916. doi: 10.1128/AEM.00330-13
Brown, M. G., and Balkwill, D. L. (2009). Antibiotic resistance in bacteria isolated from the deep terrestrial subsurface. Microbiol. Ecol. 57, 484–493. doi: 10.1007/s00248-008-9431-6
Bull, A. T., and Goodfellow, M. (2019). Dark, rare and inspirational microbial matter in the extremobiosphere: 16000 m of bioprospecting campaigns. Microbiology 165, 1252–1264.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data intensity normalization improves colour calling in SOLiD sequencing. Nat. Met. 7, 335–336. doi: 10.1038/nmeth.f.303
Cardarelli, E. L., Bargar, J. R., and Francis, C. A. (2020). Diverse Thaumarchaeota dominate subsurface ammonia-oxidizing communities in semi-arid floodplains in the western United States. Microbiol. Ecol. 80, 778–792. doi: 10.1007/s00248-020-01534-5
Carr, S. A., Schubotz, F., Dunbar, R. B., Mills, C. T., Dias, R., Summons, R. E., et al. (2018). Acetoclastic Methanosaeta are dominant methanogens in organic-rich Antarctic marine sediments. ISME J. 12, 330–342. doi: 10.1038/ismej.2017.150
Cataldo, D. A., Maroon, M., Schrader, L. E., and Youngs, V. L. (1975). Rapid colorimetric determination of nitrate in plant tissue by nitration of salicylic acid. Commun. Soil Sci. Plant Anal. 6, 71–80. doi: 10.1080/00103627509366547
Chesnin, L., and Yien, C. H. (1951). Turbidimetric determination of available sulfates. Soil Sci. Soc. Am. J. 15:149. doi: 10.2136/sssaj1951.036159950015000C0032x
Colwell, F. S., and D’Hondt, S. (2013). Nature and extent of the deep biosphere. Rev. Mineral. Geochem. 75, 547–574.
Courtine, D., Vince, E., Maignien, L., Philippon, X., Gayet, N., Shao, Z., et al. (2021). Thermococcus camini sp. nov., a hyperthermophilic and piezophilic archaeon isolated from a deep-sea hydrothermal vent at the Mid-Atlantic Ridge. Int. J. Syst. Evol. Microbiol. 71:004853. doi: 10.1099/ijsem.0.004853
Cramm, R. (2009). Genomic view of energy metabolism in Ralstonia eutropha H16. Microb. Physiol. 16, 38–52. doi: 10.1159/000142893
Dexter, E., Rollwagen-Bollens, G., and Bollens, S. M. (2018). The trouble with stress: a flexible method for the evaluation of nonmetric multidimensional scaling. Limnol. Oceanogr. Met. 16, 434–443. doi: 10.1002/lom3.10257
Dixon, P. (2003). VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x
Doi, H., and Okamura, H. (2011). Similarity indices, ordination, and community analysis tests using the software R. Jpn. J. Ecol. 61, 3–20.
Douglas, G. M., Maffei, V. J., Zaneveld, J., Yurgel, S. N., Brown, J. R., Taylor, C. M., et al. (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688. doi: 10.1038/s41587-020-0548-6
Dutta, A., Dutta Gupta, S., Gupta, A., Sarkar, J., Roy, S., Mukherjee, A., et al. (2018). Exploration of deep terrestrial subsurface microbiome in late cretaceous Deccan traps and underlying Archean basement, India. Sci. Rep. 8, 1–16. doi: 10.1038/s41598-018-35940-0
Dutta, A., Sar, P., Sarkar, J., Dutta Gupta, S., Gupta, A., Bose, H., et al. (2019). Archaeal communities in deep terrestrial subsurface underneath the Deccan traps, India. Front. Microbiol. 10:1362. doi: 10.3389/fmicb.2019.01362
Eichorst, S. A., Trojan, D., Roux, S., Herbold, C., Rattei, T., and Woebken, D. (2018). Genomic insights into the Acidobacteria reveal strategies for their success in terrestrial environments. Environ. Microbiol. 20, 1041–1063. doi: 10.1111/1462-2920.14043
Escudero, C., Oggerin, M., and Amils, R. (2018). The deep continental subsurface: the dark biosphere. Int. Microbiol. 21, 3–14. doi: 10.1007/s10123-018-0009-y
Fichtel, K., Logemann, J., Fichtel, J., Rullkötter, J., Cypionka, H., and Engelen, B. (2015). Temperature and pressure adaptation of a sulfate reducer from the deep subsurface. Front. Microbiol. 6:1078. doi: 10.3389/fmicb.2015.01078
Fichtel, K., Mathes, F., Könneke, M., Cypionka, H., and Engelen, B. (2012). Isolation of sulfate-reducing bacteria from sediments above the deep-subseafloor aquifer. Front. Microbiol. 3:65. doi: 10.3389/fmicb.2012.00065
Fortunato, C., and Huber, J. (2016). Coupled RNA-SIP and metatranscriptomics of active chemolithoautotrophic communities at a deep-sea hydrothermal vent. ISME J. 10, 1925–1938. doi: 10.1038/ismej.2015.258
Ghosh, W., and Dam, B. (2009). Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol. Rev. 33, 999–1043. doi: 10.1111/j.1574-6976.2009.00187.x
Gold, T. (1992). The deep, hot biosphere. Proc. Natl. Acad. Sci. U. S. A. 89, 6045–6049. doi: 10.1073/pnas.89.13.6045
Goswami, D., Hazarika, P., and Roy, S. (2020). In situ stress orientation from 3 km borehole image logs in the Koyna Seismogenic zone, Western India: implications for transitional faulting environment. Tectonics 39:e2019TC005647. doi: 10.1029/2019TC005647
Greening, C., Berney, M., Hards, K., Gregory, M., Ralf, C., Conrad Gülay, A., et al. (2018). Diversity of iron oxidizers in groundwater-fed rapid sand filters: evidence of Fe (II)-dependent growth by Curvibacter and Undibacterium spp. Front. Microbiol. 9:2808. doi: 10.3389/fmicb.2018.02808
Gülay, A., Çekiç, Y., Musovic, S., Albrechtsen, H. J., and Smets, B. F. (2018). Diversity of Iron Oxidizers in Groundwater-Fed Rapid Sand Filters: Evidence of Fe(II)-Dependent Growth by Curvibacter and Undibacterium spp. Front. Microbiol. 9:2808. doi: 10.3389/fmicb.2018.02808
Gupta, H. K. (2017). Koyna, India, an ideal site for near field earthquake observations. J. Geol. Soc. India 90, 645–652. doi: 10.1007/s12594-017-0771-z
Gupta, A., Dutta, A., Sarkar, J., Panigrahi, M. K., and Sar, P. (2018). Low-abundance members of the Firmicutes facilitate bioremediation of soil impacted by highly acidic mine drainage from the Malanjkhand copper project, India. Front. Microbiol. 9:2882. doi: 10.3389/fmicb.2018.02882
Hädrich, A., Taillefert, M., Akob, D. M., Cooper, R. E., Litzba, U., Wagner, F. E., et al. (2019). Microbial Fe(II) oxidation by Sideroxydans lithotrophicus ES-1 in the presence of Schlöppnerbrunnen fen-derived humic acids. FEMS Microbiol. Ecol. 95, 1–19. doi: 10.1093/femsec/fiz034
Hallbeck, L., and Pedersen, K. (2008). Characterization of microbial processes in deep aquifers of the Fennoscandian shield. Appl. Geochem. 23, 1796–1819. doi: 10.1016/j.apgeochem.2008.02.012
Hallbeck, L., and Pedersen, K. (2012). Culture-dependent comparison of microbial diversity in deep granitic groundwater from two sites considered for a Swedish final repository of spent nuclear fuel. FEMS Microbiol. Ecol. 81, 66–77. doi: 10.1111/j.1574-6941.2011.01281.x
Hammer, Ø., Harper, D. A. T., and Ryan, P. D. (2001). PAST: paleontological statistics Sofware package for education and data analysis. Palaeontol. Electron. 4, 1–9.
Hawkins, A. S., McTernan, P. M., Lian, H., Kelly, R. M., and Adams, M. W. (2013). Biological conversion of carbon dioxide and hydrogen into liquid fuels and industrial chemicals. Curr. Opin. Biotechnol. 24, 376–384. doi: 10.1016/j.copbio.2013.02.017
Heberle, H., Meirelles, G. V., da Silva, F. R., Telles, G. P., and Minghim, R. (2015). InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformat. 16:169. doi: 10.1186/s12859-015-0611-3
Hernsdorf, A., Amano, Y., Miyakawa, K., Ise, K., Suzuki, Y., Anantharaman, K., et al. (2017). Potential for microbial H2 and metal transformations associated with novel bacteria and archaea in deep terrestrial subsurface sediments. ISME J. 11, 1915–1929. doi: 10.1038/ismej.2017.39
Hubalek, V., Wu, X., Eiler, A., Buck, M., Heim, C., Dopson, M., et al. (2016). Connectivity to the surface determines diversity patterns in subsurface aquifers of the Fennoscandian shield. ISME J. 10, 2447–2458. doi: 10.1038/ismej.2016.36
Huber, H., and Stetter, K.O. (2006). Thermoplasmatales M. Dworkin, S. Falkow, E. Rosenberg, KH. Schleifer, and E Stackebrandt. (eds) The Prokaryotes. Springer, New York, NY.
Hui, M. L., Tan, L. T., Letchumanan, V., He, Y. W., Fang, C. M., Chan, K. G., et al. (2021). The extremophilic actinobacteria: from microbes to medicine. Antibiotics 10, 1–15. doi: 10.3390/antibiotics10060682
Idris, H., Goodfellow, M., Sanderson, R., Asenjo, J. A., and Bull, A. T. (2017). Actinobacterial rare biospheres and dark matter revealed in habitats of the Chilean Atacama Desert. Sci. Rep. 7, 1–11.
Imachi, H., Nobu, M. K., Nakahara, N., Morono, Y., Ogawara, M., Takaki, Y., et al. (2020). Isolation of an archaeon at the prokaryote–eukaryote interface. Nature 577, 519–525. doi: 10.1038/s41586-019-1916-6
Imachi, H., Tasumi, E., Takaki, Y., Hoshino, T., Schubotz, F., Gan, S., et al. (2019). Cultivable microbial community in 2-km-deep, 20-million-year-old subseafloor coalbeds through~ 1000 days anaerobic bioreactor cultivation. Sci. Rep. 9, 1–16. doi: 10.1038/s41598-019-38754-w
Ino, K., Hernsdorf, A. W., Konno, U., Kouduka, M., Yanagawa, K., Kato, S., et al. (2018). 1198 ecological and genomic profiling of anaerobic methane-oxidizing archaea in a deep granitic 1199 environment. ISME J. 12, 31–47. doi: 10.1038/ismej.2017.140
Islam, Z. F., Welsh, C., Bayly, K., Grinter, R., Southam, G., Gagen, E. J., et al. (2020). A widely distributed hydrogenase oxidises atmospheric H2 during bacterial growth. ISME J. 14, 2649–2658. doi: 10.1038/s41396-020-0713-4
Jakus, N., Blackwell, N., Osenbrück, K., Straub, D., Byrne, J. M., Wang, Z., et al. (2021). Nitrate removal by a novel lithoautotrophic nitrate-reducing, iron (II)-oxidizing culture enriched from a pyrite-rich limestone aquifer. Appl. Environ. Microbiol. 87:e0046021. doi: 10.1128/AEM.00460-21
Jukes, T. H., and Cantor, C. R. (1969). “Evolution of protein molecules” in Mammalian Protein Metabolism. ed. H. N. Munro (New York: Academic Press), 21–132.
Kadnikov, V. V., Mardanov, A. V., Beletsky, A. V., Banks, D., Pimenov, N. V., Frank, Y. A., et al. (2018). A metagenomic window into the 2-km-deep terrestrial subsurface aquifer revealed multiple pathways of organic matter decomposition. FEMS Microbiol. Ecol. 94, 1–16.
Kadnikov, V. V., Mardanov, A. V., Beletsky, A. V., Karnachuk, O. V., and Ravin, N. V. (2019). Genome of the candidate phylum Aminicenantes bacterium from a deep subsurface thermal aquifer revealed its fermentative saccharolytic lifestyle. Extremophiles 23, 189–200. doi: 10.1007/s00792-018-01073-5
Kadnikov, V. V., Mardanov, A. V., Beletsky, A. V., Karnachuk, O. V., and Ravin, N. V. (2020). Microbial life in the deep subsurface aquifer illuminated by metagenomics. Front. Microbiol. 11, 1–15. doi: 10.3389/fmicb.2020.572252
Kazy, S. K., Sahu, R. P., Mukhopadhyay, S., Bose, H., Mandal, S., and Sar, P. (2021). “Microbial life in deep terrestrial continental crust” in Extreme Environments. eds. A. Pandey and A. Sharma (Boca Raton: CRC Press), 263–291.
Kieft, T. L. (2016). “Microbiology of the deep continental biosphere” in Their world: A diversity of microbial environments. Advances in environmental microbiology, Vol. 1. ed. C. Hurst (Cham: Springer), 225–249.
Kim, B. R., Shin, J., Guevarra, R. B., Lee, J. H., Kim, D. W., Seol, K. H., et al. (2017). Deciphering diversity indices for a better understanding of microbial communities. J. Microbiol. Biotechnol. 27, 2089–2093. doi: 10.4014/jmb.1709.09027
Konn, C., Charlou, J. L., Holm, N. G., and Mousis, O. (2015). The production of methane, hydrogen, and organic compounds in ultramafic-hosted hydrothermal vents of the Mid-Atlantic Ridge. Astrobiology 15, 381–399. doi: 10.1089/ast.2014.1198
Kotelnikova, S., and Pedersen, K. (1998). Distribution and activity of methanogens and homoacetogens in deep granitic aquifers at Äspö Hard Rock Laboratory, Sweden. FEMS Microbiol. Ecol. 26, 121–134. doi: 10.1111/j.1574-6941.1998.tb00498.x
Kuhnert, P., Venables, B., and Zocchi, S.S. (2005). An introduction to R: Software for statistical modelling & computing (29–35). USP/ESALQ/LCE
Kumar, S., Herrmann, M., Blohm, A., Hilke, I., Frosch, T., Trumbore, S. E., et al. (2018). Thiosulfate-and hydrogen-driven autotrophic denitrification by a microbial consortium enriched from groundwater of an oligotrophic limestone aquifer. FEMS Microbiol. Ecol. 94:fiy141. doi: 10.1093/femsec/fiy141
Lapidus, A., Clum, A., Labutti, K., Kaluzhnaya, M. G., Lim, S., Beck, D. A., et al. (2011). Genomes of three methylotrophs from a single niche reveal the genetic and metabolic divergence of the methylophilaceae. J. Bacteriol. 193, 3757–3764. doi: 10.1128/JB.00404-11
Lau, M. C., Kieft, T. L., Kuloyo, O., Linage-Alvarez, B., Van Heerden, E., Lindsay, M. R., et al. (2016). An oligotrophic deep-subsurface community dependent on syntrophy is dominated by sulfur-driven autotrophic denitrifiers. Proc. Natl. Acad. Sci. U. S. A. 113, E7927–E7936. doi: 10.1073/pnas.1612244113
Leandro, T., Rodríguez, N., Sanz, J. L., da Costa, M. S., and Amils, R. (2018). Study of methanogenic cultures of rocks from the deep subsurface of the Iberian Pyrite belt. Heliyon 4:e00605. doi: 10.1016/j.heliyon.2018.e00605
Li, X. G., Tang, H. Z., Zhang, W. J., Qi, X. Q., Qu, Z. G., Xu, J., et al. (2021). Thermococcus aciditolerans sp. nov., a piezotolerant, hyperthermophilic archaeon isolated from a deep-sea hydrothermal vent chimney in the Southwest Indian Ridge. Int. J. Syst. Evol. Microbiol. 71:004934. doi: 10.1099/ijsem.0.004934
Liu, Z., Wang, K., Chen, Y., Tan, T., and Nielsen, J. (2020). Third-generation biorefineries as the means to produce fuels and chemicals from CO2. Nat. Cat. 3, 274–288. doi: 10.1038/s41929-019-0421-5
Lovley, D. R., and Chapelle, F. H. (1995). Deep subsurface microbial processes. Rev. Geophys. 33, 365–381. doi: 10.1029/95RG01305
Magnabosco, C., Lin, L. H., Dong, H., Bomberg, M., Ghiorse, W., Stan-Lotter, H., et al. (2018). The biomass and biodiversity of the continental subsurface. Nat. Geosci. 11, 707–717. doi: 10.1038/s41561-018-0221-6
Magnabosco, C., Ryan, K., Lau, M. C., Kuloyo, O., Sherwood Lollar, B., Kieft, T. L., et al. (2016). A metagenomic window into carbon metabolism at 3 km depth in Precambrian continental crust. ISME J 10, 730–741. doi: 10.1038/ismej.2015.150
Magnabosco, C., Tekere, M., Lau, M. C. Y., Linage, B., Kuloyo, O., Erasmus, M., et al. (2014). Comparisons of the composition and biogeographic distribution of the bacterial communities occupying south African thermal springs with those inhabiting deep subsurface fracture water. Front. Microbiol. 5:679. doi: 10.3389/fmicb.2014.00679
Matsushita, M., Ishikawa, S., Magara, K., Sato, Y., and Kimura, H. (2020). The potential for CH4 production by syntrophic microbial communities in diverse deep aquifers associated with an accretionary prism and its overlying sedimentary layers. Microbes Environ. 35:ME19103. doi: 10.1264/jsme2.ME19103
Miglani, R., Parveen, N., Kumar, A., Ansari, M. A., Khanna, S., Rawat, G., et al. (2022). Degradation of xenobiotic pollutants: an environmentally sustainable approach. Meta 12:818. doi: 10.3390/metabo12090818
Momper, L., Jungbluth, S. P., Lee, M. D., and Amend, J. P. (2017). Energy and carbon metabolisms in a deep terrestrial subsurface fluid microbial community. ISME J. 11, 2319–2333. doi: 10.1038/ismej.2017.94
Moradali, M. F., and Rehm, B. H. A. (2020). Bacterial biopolymers: from pathogenesis to advanced materials. Nat. Rev. Microbiol. 18, 195–210. doi: 10.1038/s41579-019-0313-3
Mullin, S. W., Wanger, G., Kruger, B. R., Sackett, J. D., Hamilton-Brehm, S. D., and Bhartia, R. (2020). Patterns of in situ mineral colonization by microorganisms in a ~60°C deep continental subsurface aquifer. Front. Microbiol. 11, 1–23. doi: 10.3389/fmicb.2020.536535
Mullis, M. M., Rambo, I. M., Baker, B. J., and Reese, B. K. (2019). Diversity, ecology, and prevalence of antimicrobials in nature. Front. Microbiol. 10:2518. doi: 10.3389/fmicb.2019.02518
Nakagawa, S., and Takai, K. (2008). Deep-sea vent chemoautotrophs: diversity, biochemistry and ecological significance. FEMS Microbiol. Ecol. 65, 1–14. doi: 10.1111/j.1574-6941.2008.00502.x
Nangle, S. N., Ziesack, M., Buckley, S., Trivedi, D., Loh, D. M., Nocera, D. G., et al. (2020). Valorization of CO2 through lithoautotrophic production of sustainable chemicals in Cupriavidus necator. Metab. Eng. 62, 207–220. doi: 10.1016/j.ymben.2020.09.002
Nealson, K. H., Inagaki, F., and Takai, K. (2005). Hydrogen-driven subsurface lithoautotrophic microbial ecosystems (SLiMEs): do they exist and why should we care? Trends Microbiol. 13, 405–410. doi: 10.1016/j.tim.2005.07.010
Nuppunen-Puputti, M., Kietäväinen, R., Raulio, M., Soro, A., Purkamo, L., Kukkonen, I., et al. (2022). Epilithic microbial community functionality in deep oligotrophic continental bedrock. Front. Microbiol. 13:826048. doi: 10.3389/fmicb.2022.826048
Nuppunen-Puputti, M., Purkamo, L., Kietäväinen, R., Nyyssönen, M., Itävaara, M., Ahonen, L., et al. (2018). Rare biosphere archaea assimilate acetate in Precambrian terrestrial subsurface at 2.2 km depth. Geosciences 8:418. doi: 10.3390/geosciences8110418
Nyyssönen, M., Hultman, J., Ahonen, L., Kukkonen, I., Paulin, L., Laine, P., et al. (2014). Taxonomically and functionally diverse microbial communities in deep crystalline rocks of the Fennoscandian shield. ISME J. 8, 126–138. doi: 10.1038/ismej.2013.125
Osburn, M. R., LaRowe, D. E., Momper, L. M., and Amend, J. P. (2014). Chemolithotrophy in the continental deep subsurface: Sanford underground research facility (SURF), USA. Front. Microbiol. 5:610. doi: 10.3389/fmicb.2014.00610
Panda, S. K., Jyoti, V., Bhadra, B., Nayak, K. C., Shivaji, S., Rainey, F. A., et al. (2009). Thiomonas bhubaneswarensis sp. nov., an obligately mixotrophic, moderately thermophilic, thiosulfate-oxidizing bacterium. Int. J. Syst. Evol. Microbiol. 59, 2171–2175. doi: 10.1099/ijs.0.007120-0
Pedersen, K. (2013). Metabolic activity of subterranean microbial communities in deep granitic groundwater supplemented with methane and H2. ISME J. 7, 839–849. doi: 10.1038/ismej.2012.144
Pettit, R. K. (2011). Small-molecule elicitation of microbial secondary metabolites. Microbiol. Biotechnol. 4, 471–478. doi: 10.1111/j.1751-7915.2010.00196.x
Podugu, N., Mishra, S., Wiersberg, T., and Roy, S. (2019). Chemical and noble gas isotope compositions of formation gases from a 3 km deep scientific borehole in the Koyna Seismogenic zone, Western India. Geofluids 2019, 1–16. doi: 10.1155/2019/1078942
Pohlner, M., Dlugosch, L., Wemheuer, B., Mills, H., Engelen, B., and Reese, B. K. (2019). The majority of active rhodobacteraceae in marine sediments belong to uncultured genera: a molecular approach to link their distribution to environmental conditions. Front. Microbiol. 10:659. doi: 10.3389/fmicb.2019.00659
Puente-Sánchez, F., Arce-Rodríguez, A., Oggerin, M., García-Villadangos, M., Moreno-Paz, M., Blanco, Y., et al. (2018). Viable cyanobacteria in the deep continental subsurface. Proc. Natl. Acad. Sci. U. S. A. 115, 10702–10707. doi: 10.1073/pnas.1808176115
Purkamo, L., Bomberg, M., Nyyssönen, M., Ahonen, L., Kukkonen, I., and Itävaara, M. (2017). Response of deep subsurface microbial community to different carbon sources and electron acceptors during∼ 2 months incubation in microcosms. Front. Microbiol. 8:232. doi: 10.3389/fmicb.2017.00232
Purkamo, L., Bomberg, M., Nyyssönen, M., Kukkonen, I., Ahonen, L., and Itävaara, M. (2015). Heterotrophic communities supplied by ancient organic carbon predominate in deep Fennoscandian bedrock fluids. Microbiol. Ecol. 69, 319–332. doi: 10.1007/s00248-014-0490-6
Purkamo, L., Kietäväinen, R., Nuppunen-Puputti, M., Bomberg, M., and Cousins, C. (2020). Ultradeep microbial communities at 4.4 km within crystalline bedrock: implications for habitability in a planetary context. Life 10:2. doi: 10.3390/life10010002
R Development Core Team. (2017). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing.
Ragon, M., Van Driessche, A. E. S., García-Ruíz, J. M., Moreira, D., and López-García, P. (2013). Microbial diversity in the deep-subsurface hydrothermal aquifer feeding the giant gypsum crystal-bearing Naica Mine, Mexico. Front. Microbiol. 4:37. doi: 10.3389/fmicb.2013.00037
Rajala, P., and Bomberg, M. (2017). Reactivation of deep subsurface microbial community in response to methane or methanol amendment. Front. Microbiol. 8:431. doi: 10.3389/fmicb.2017.00431
Rajala, P., Bomberg, M., Kietäväinen, R., Kukkonen, I., Ahonen, L., Nyyssönen, M., et al. (2015). Rapid reactivation of deep subsurface microbes in the presence of C-1 compounds. Microorganisms 3, 17–33. doi: 10.3390/microorganisms3010017
Rastogi, G., Gurram, R. N., Bhalla, A., Gonzalez, R., Bischoff, K. M., Hughes, S. R., et al. (2013). Presence of glucose, xylose, and glycerol fermenting bacteria in the deep biosphere of the former Homestake gold mine, South Dakota. Front. Microbiol. 4:18. doi: 10.3389/fmicb.2013.00018
Rastogi, G., Muppidi, G. L., Gurram, R. N., Adhikari, A., Bischoff, K. M., Hughes, S. R., et al. (2009). Isolation and characterization of cellulose-degrading bacteria from the deep subsurface of the Homestake gold mine, Lead, South Dakota, USA. J. Ind. Microbiol. Biotechnol. 36, 585–598. doi: 10.1007/s10295-009-0528-9
Reitmeier, S., Hitch, T. C., Treichel, N., Fikas, N., Hausmann, B., Ramer-Tait, A. E., et al. (2021). Handling of spurious sequences affects the outcome of high-throughput 16S rRNA gene amplicon profiling. ISME Commun. 1, 1–12. doi: 10.1038/s43705-021-00033-z
Rogers, A. H., Chen, J., Zilm, P. S., and Gully, N. J. (1998). The behaviour of Fusobacterium nucleatum chemostat-grown in glucose- and amino acid-based chemically defined media. Anaerobe 4, 111–116. doi: 10.1006/anae.1997.0140
Roy, S., Rao, N. P., Akkiraju, V. V., Goswami, D., Sen, M., Bansal, B. K., et al. (2013). Granitic basement below Deccan traps unearthed by drilling in the Koyna Seismic zone, western India. J. Geol. Soc. India 81:289. doi: 10.1007/s12594-013-0034-6
Russell, J. A., León-Zayas, R., Wrighton, K., and Biddle, J. F. (2016). Deep subsurface life from north pond: enrichment, isolation, characterization and genomes of heterotrophic bacteria. Front. Microbiol. 7:678. doi: 10.3389/fmicb.2016.00678
Sahl, J. W., Schmidt, R., Swanner, E. D., Mandernack, K. W., Templeton, A. S., Kieft, T. L., et al. (2008). Subsurface microbial diversity in deep-granitic-fracture water in Colorado. Appl. Environ. Microbiol. 74, 143–152. doi: 10.1128/AEM.01133-07
Sahu, R. P., Kazy, S. K., Bose, H., Mandal, S., Dutta, A., Saha, A., et al. (2022). Microbial diversity and function in crystalline basement beneath the Deccan traps explored in a 3 km borehole at Koyna, western India. Environ. Microbiol. 24, 2837–2853. doi: 10.1111/1462-2920.15867
Saini, R., Kapoor, R., Kumar, R., Siddiqi, T. O., and Kumar, A. (2011). CO2 utilizing microbes—a comprehensive review. Biotechnol. Adv. 29, 949–960. doi: 10.1016/j.biotechadv.2011.08.009
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Salehizadeh, H., Yan, N., and Farnood, R. (2020). Recent advances in microbial CO2 fixation and conversion to value-added products. Chem. Eng. J. 390:124584. doi: 10.1016/j.cej.2020.124584
Sanz, J. L., Rodriguez, N., Escudero, C., Carrizo, D., and Amils, R. (2021). Biological production of H2, CH4 and CO2 in the deep subsurface of the Iberian Pyrite Belt. Environ. Microbiol. 23, 3913–3922. doi: 10.1111/1462-2920.15561
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schoene, B., Samperton, K. M., Eddy, M. P., Keller, G., Adatte, T., Bowring, S. A., et al. (2015). U-Pb geochronology of the Deccan Traps and relation to the end-cretaceous mass extinction. Science 347, 182–184. doi: 10.1126/science.aaa0118
Seán, P., O'Connell, R., Lehman, M., Snoeyenbos-West, O., Winston, V. D., and Cummings, D. E. (2003). Detection of Euryarchaeota and Crenarchaeota in an oxic basalt aquifer. FEMS Microbiol. Ecol. 44, 165–173. doi: 10.1016/S0168-6496(02)00465-8
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Sheik, C. S., Reese, B. K., Twing, K. I., Sylvan, J. B., Grim, S. L., Schrenk, M. O., et al. (2018). Identification and removal of contaminant sequences from ribosomal gene databases: lessons from the census of deep life. Front. Microbiol. 9:840. doi: 10.3389/fmicb.2018.00840
Stevens, T. (1997). Lithoautotrophy in the subsurface. FEMS Microbiol. Rev. 20, 327–337. doi: 10.1111/j.1574-6976.1997.tb00318.x
Suzuki, Y., Yamashita, S., Kouduka, M., Ao, Y., Mukai, H., Mitsunobu, S., et al. (2020). Deep microbial proliferation at the basalt interface in 33.5–104-million-year-old oceanic crust. Commun. Biol. 3:136. doi: 10.1038/s42003-020-0860-1
Takors, R., Kopf, M., Mampel, J., Bluemke, W., Blombach, B., Eikmanns, B., et al. (2018). Using gas mixtures of CO, CO2 and H2 as microbial substrates: the do's and don'ts of successful technology transfer from laboratory to production scale. Microb. Biotechnol. 11, 606–625. doi: 10.1111/1751-7915.13270
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Van der Loo, M. P. (2012). Learning RStudio for R Statistical Computing. Packt Publishing Ltd., Birmingham.
Vuillemin, A., Kerrigan, Z., D’Hondt, S., and Orsi, W. D. (2020). Exploring the abundance, metabolic potential and gene expression of subseafloor Chloroflexi in million-year-old oxic and anoxic abyssal clay. FEMS Microbiol. Ecol. 96, 1–14. doi: 10.1093/femsec/fiaa223
Wasmund, K., Schreiber, L., Lloyd, K., Petersen, D. G., Schramm, A., Stepanauskas, R., et al. (2014). Genome sequencing of a single cell of the widely distributed marine subsurface Dehalococcoidia, phylum Chloroflexi. ISME J. 8, 383–397. doi: 10.1038/ismej.2013.143
Willems, A., and De Vos, P. (2006). “Comamonas,” in The Prokaryotes. eds. M. Dworkin, S. Falkow, E. Rosenberg, K. H. Schleifer, and E. Stackebrandt (New York, NY: Springer)
Wu, X., Holmfeldt, K., Hubalek, V., Lundin, D., Åström, M., Bertilsson, S., et al. (2016). Microbial metagenomes from three aquifers in the Fennoscandian shield terrestrial deep biosphere reveal metabolic partitioning among populations. ISME J. 10, 1192–1203. doi: 10.1038/ismej.2015.185
Yang, Y., Sanford, R., Yan, J., Chen, G., Cápiro, N. L., Li, X., et al. (2020). Roles of organohalide-respiring Dehalococcoidia in carbon cycling. mSystems 5, 1–12. doi: 10.1128/mSystems.00757-19
Yoon, J. H., and Park, Y. H. (2006). “The genus Nocardioides,” in The Prokaryotes. eds. M. Dworkin, S. Falkow, E. Rosenberg, K. H. Schleifer, and E. Stackebrandt (New York, NY: Springer).
Yoon, K. S., Tsukada, N., Sakai, Y., Ishii, M., Igarashi, Y., and Nishihara, H. (2008). Isolation and characterization of a new facultatively autotrophic hydrogen-oxidizing Betaproteobacterium, Hydrogenophaga sp. AH-24. FEMS Microbiol. Lett. 278, 94–100. doi: 10.1111/j.1574-6968.2007.00983.x
Keywords: Koyna seismogenic zone, terrestrial subsurface granitic rock, deep biosphere, inorganic carbon metabolism, chemolithotrophic microorganisms
Citation: Mandal S, Bose H, Ramesh K, Sahu RP, Saha A, Sar P and Kazy SK (2022) Depth wide distribution and metabolic potential of chemolithoautotrophic microorganisms reactivated from deep continental granitic crust underneath the Deccan Traps at Koyna, India. Front. Microbiol. 13:1018940. doi: 10.3389/fmicb.2022.1018940
Edited by:
Malin Bomberg, VTT Technical Research Centre of Finland Ltd, FinlandReviewed by:
D’Arcy Renee Meyer-Dombard, University of Illinois at Chicago, United StatesCopyright © 2022 Mandal, Bose, Ramesh, Sahu, Saha, Sar and Kazy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sufia Khannam Kazy, c3VmaWEua2F6eUBidC5uaXRkZ3AuYWMuaW4=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.