 Xue Yang
Xue Yang Tiejun Zhang
Tiejun Zhang Chang-Wei Lei
Chang-Wei Lei Qin Wang
Qin Wang Zheren Huang
Zheren Huang Hong-Ning Wang
Hong-Ning Wang- Animal Disease Prevention and Food Safety Key Laboratory of Sichuan Province, College of Life Sciences, Sichuan University, Chengdu, China
Antibiotic resistance genes (ARGs) as a novel type of environmental pollutant pose a health risk to humans. Oxazolidinones are one of the most important antibiotics for the treatment of Gram-positive bacterial infections in humans. Although oxazolidinones are not utilized in the livestock industry, florfenicol is commonly used on farms to treat bacterial infections, which may contribute to the spread of the cfr, optrA, and poxtA genes on farms. Using metagenomics sequencing, we looked into the antibiotic resistome context of florfenicol and oxazolidinone in 10 large-scale commercial farms in China. We identified 490 different resistance genes and 1,515 bacterial genera in the fecal samples obtained from 10 farms. Florfenicol-resistant Kurthia, Escherichia, and Proteus were widely present in these samples. The situation of florfenicol and oxazolidone resistance in pig farms is even more severe. The total number of genes and the abundance of drug resistance genes were higher in pigs than in chickens, including optrA and poxtA. All the samples we collected had a high abundance of fexA and floR. Through nanopore metagenomic analysis of the genetic environment, we found that plasmids, integrative and conjugative element (ICE), and transposons (Tn7-like and Tn558) may play an important role in the spread of floR, cfr, and optrA. Our findings suggest that florfenicol and oxazolidinone resistance genes have diverse genetic environments and are at risk of co-transmission with, for example, tetracycline and aminoglycoside resistance genes. The spread of florfenicol- and oxazolidinone–resistant bacteria on animal farms should be continuously monitored.
Introduction
Antibiotics have been used extensively for more than half a century as effective medicines for the treatment and prevention of human and animal diseases, as well as to accelerate animal growth. However, with the extensive use of antibiotics, the global situation of drug resistance became progressively worse (Zhang et al., 2015; He et al., 2019). It is estimated that antimicrobial resistance will kill 10 million people per year by 2050, according to O’Neill (2014).
Florfenicol is a synthetic animal-specific drug that has been approved by the Chinese government in 1999. However, due to improper use, Escherichia coli resistant to florfenicol started to emerge, and the prevalence of drug-resistant strains increased year after year, garnering a lot of attention. Several florfenicol resistance genes have been found, including floR, fexA, fexB, cfr, and others (Arcangioli et al., 1999; Liu et al., 2012; Shen et al., 2013). Linezolid is a first-generation oxazolidinone antibiotic with a potent antibacterial action against Gram-positive infections, commonly known as the last line of defense against infections by multidrug-resistant Gram-positive pathogens (Bozdogan and Appelbaum, 2004). In 2006, the cfr gene was found to mediate linezolid resistance (Long et al., 2006). New resistance genes (optrA and poxtA) can be resistant to oxazolidinones and amide alcohols (Wang et al., 2015; Antonelli et al., 2018). Animal usage of linezolid has never been authorized. Interestingly, three linezolid resistance genes (cfr, optrA, and poxtA) were detected in bacteria of animal origin (Shen et al., 2013; Wang et al., 2015; Lei et al., 2019). It is unclear whether the use of florfenicol has led to the emergence of linezolid-resistant genes. Serious public health concerns have been raised as a result of the widespread distribution of linezolid resistance genes in bacteria.
Continuous monitoring of drug resistance requires not only identifying the microbial resistance group and the host of the resistant group, but also the evaluation of mobile genetic elements (MGEs) (Frost et al., 2005). Pure culture isolation in combination with whole-genome sequencing has been and continues to be a major tool for determining phenotypic and genotypic correlations in the pathogenic bacteria and identifying MGEs with which they are related (Zhao S. et al., 2016; Guo et al., 2017; Xia et al., 2017). However, only a small percentage of bacteria in feces can be cultured and isolated, and this proportion does not reflect the general state of the sample, severely limiting the use of pure culture isolation to investigate the fecal resistome (Luo et al., 2017).
The diversity and abundance of ARGs in samples have considerably risen, thanks to metagenome sequencing (Ma et al., 2016). Although Illumina sequencing has a high level of precision, the read length is short, making it challenging to determine the species and genetic background of ARG. In comparison to short-read sequencing, Oxford Nanopore Method’s (ONT) long-read sequencing technology has a significantly high sequencing error rate, but can generate long reads that can span most repetitive sequences and link the ARGs and their flanking regions, and thus, the knowledge and technology gap mentioned above can be bridged (Che et al., 2019; Li et al., 2022). This study utilized Nanopore and Illumina sequencing technology to evaluate the genomic location of florfenicol ARGs more efficiently and correctly.
Materials and methods
Sample collection and pretreatment
Five swine fecal samples and five chicken fecal samples were collected separately from 10 industrialized farms. Chicken samples were collected from four provinces, including Sichuan, Chongqing, Anhui, and Beijing. All the swine farms were located in Sichuan province (Supplementary Table 1). We selected five houses on each farm and used the five-point sampling method to collect the samples in each house and mix them into one. Samples were moved from the field to the lab in a low-temperature box with dry ice. Five samples from each farm were combined into one. Five grams of mixed samples from each farm was weighed, mixed thoroughly by adding 5 ml of phosphate-buffered saline (PBS), and filtered through gauze.
Cultivation of resistant bacteria, DNA extraction, and detection
Approximately, 100 μ L of the filtrate was plated onto an Luria-Bertani (LB) agar plate supplemented with 16 μ g/mL florfenicol. After incubating overnight (12 h) in aerobic conditions at 37°C, each sample was collected by washing all colonies/isolates grown on the incubated LB plate three times using PBS. DNA was extracted from the 10 post-culture sample colonies collected above using the QIAampPower fecal DNA kit (QIAGEN, Netherlands)1 according to the manufacturer’s instructions. These DNA samples were extracted for subsequent metagenomic sequencing. For library building and sequencing, sufficient supplies of high-quality nucleic acids must be obtained. We used agarose gel electrophoresis (AGE) to assess the DNA purity and integrity, and Qubit to determine the DNA concentration.
Library preparation, sequencing, and assembly
Illumina library preparation and sequencing
For the DNA sample preparation, a total of 1 μg of DNA per sample was used as the input material. Sequencing libraries were created using the New England Biolabs (NEBNext)® UltraTM DNA Library Prep Kit for Illumina (NEB, USA). After passing the library check, Illumina sequencing using the PE150 strategy was performed at Novogene Corporation (Beijing, China).
Illumina data pretreatment and metagenome assembly
To obtain the clean data for further analysis, Readfq (V82) was used to pre-process the raw data. Clean data must be blasted to the host database, which uses the Bowtie 2.2.4 software3 with default parameters to filter reads from the host and remove the reads with high alignment similarity. MEGAHIT4 was used to assemble clean data created from each sample from scratch.
Nanopore library preparation and sequencing
According to the Illumina sequencing results, two samples (C.2 and S.4) with a high abundance of drug resistance genes were chosen for nanopore sequencing.
For the MinION platform (Oxford Nanopore Technologies, Oxford, UK), using a total of 8 μg of DNA per sample as input material, sequencing libraries were created using the Ligation Sequencing Kit (SQK-LSK109). The DNA was disrupted with a Megaruptor (Diagenode, Denville, NJ, USA), and the fragments larger than 10 kb were screened with Bluepippin. The barcodes were applied after the end repair and addition of the A tail, and then the fragment length was detected. The linker and template were then purified, and samples with different barcodes were combined in equimolar quantities. The clustering of the bar-coded samples was performed with PromethION Flow Cell Priming Kit (EXPFLP001.PRO.6) according to the manufacturer’s instructions.
MinION data pretreatment and metagenome assembly
The original format of nanopore sequencing down data is fast5 format, and fast5 was converted to fastq format using Guppy (Oxford Nanopore built-in software) basecalling. We used the software NanoPlot (Version: NanoPlot 1.18.2) selection threshold Q > 7 for quality control of the data. The blasr (version 5.3.55) software was used to remove host contamination by default.
Flye (Version: 2.4.2-release6) was used to assemble and analyze the clean data. Following data preprocessing, the assembled scaftigs were compared to the clean data of each sample by Bowtie 2.2.4 (see text footnote 2) to obtain the paired-end reads (PE) reads that were not used. Filter scaftig fragments that are less than 500 bp in length for statistical analysis resulting from a single assembly.
Bioinformatics analysis
Gene prediction and abundance analysis
The open-reading frame (ORF) predictions for all assembled scaftigs (>500 bp) were performed using MetaGeneMark (V2.107) and filtered for length information less than 100 nt. The predicted ORFs were de-redundant using the cd-hit program in the CD-HIT software (V4.5.88). These procedures are used to produce a non-redundant gene catalog (Unigenes).
Species and resistance gene annotation
Species annotation: DIAMOND (V0.9.99) compares Unigenes with bacteria, fungi, and archaea extracted from National Center for Biotechnology Information (NCBI’s) NCBI RefSeq (NR) (Version: 2018.01) database. Species annotation information was determined to use the systematic classification function of the MEGAN6.10
Resistance gene annotation: Unigenes were aligned with the Comprehensive Antibiotic Resistance Database (CARD) using resistance gene identifier (RGI) (v4.2.2) to identify antibiotic resistance genes (ARGs). The relative abundance of each ARO was computed using the RGI alignment findings and the abundance data from Unigenes. All analyses make use of the default settings.
Other analysis
We perform principal component analyze (PCoA) analysis based on Bray--Curtis distance. The distribution patterns of the bacterial genera of samples were also clustered and characterized using a principal component analyses (PCoAs) approach by R package and QIIME.11 The Pearson correlation test was performed using the Hmisc software package of R language to analyze the correlation between ARGs. The correlation was screened using the threshold of P < 0.01, r ≥ 0.9, and then visualized using Gephi 0.9.2.12 The histogram is drawn using the ggplot2 package in R software, and the circle chart is drawn using the circos.13 MinION-assembled contigs were searched for MGEs against CARD database, and the location of the contig on the chromosome or plasmid was ascertained using plasflow.14 The genetic environments were analyzed using rast website,15 Easyfig v2.2.2,16 and Adobe Illustrator 2020 software.
Results
Illumina and MinION sequencing read statistics
Illumina sequencing on average generated 12.28 GB of clean data with an average of 74,815 scaftig numbers (Supplementary Table 2). For the two samples (C.2 and S.4) for which the nanopore data assembly was performed, the length of the nanopore reads (avg. 52 kb; N50 198 kb) was much longer than the contigs (avg. 1.3 kb; N50 1.8 kb) assembled using Illumina short reads (Supplementary Table 3). A total of 11 long scaftigs over 2 GB were generated, and these sequences may represent the complete genome of the strain. With the use of these longer nanopore readings, we can examine the genomic localization of ARGs in more completely assembled sequences.
Composition of fecal communities
The PCoA intuitively displayed the differences in microbial communities between different samples and two groups (G1 indicates the chicken farms and G2 indicates the swine farms). The microbial communities from G1 and G2 have a big difference (Figure 1A), and the number of genes in swine farms is higher than that in chicken farms.
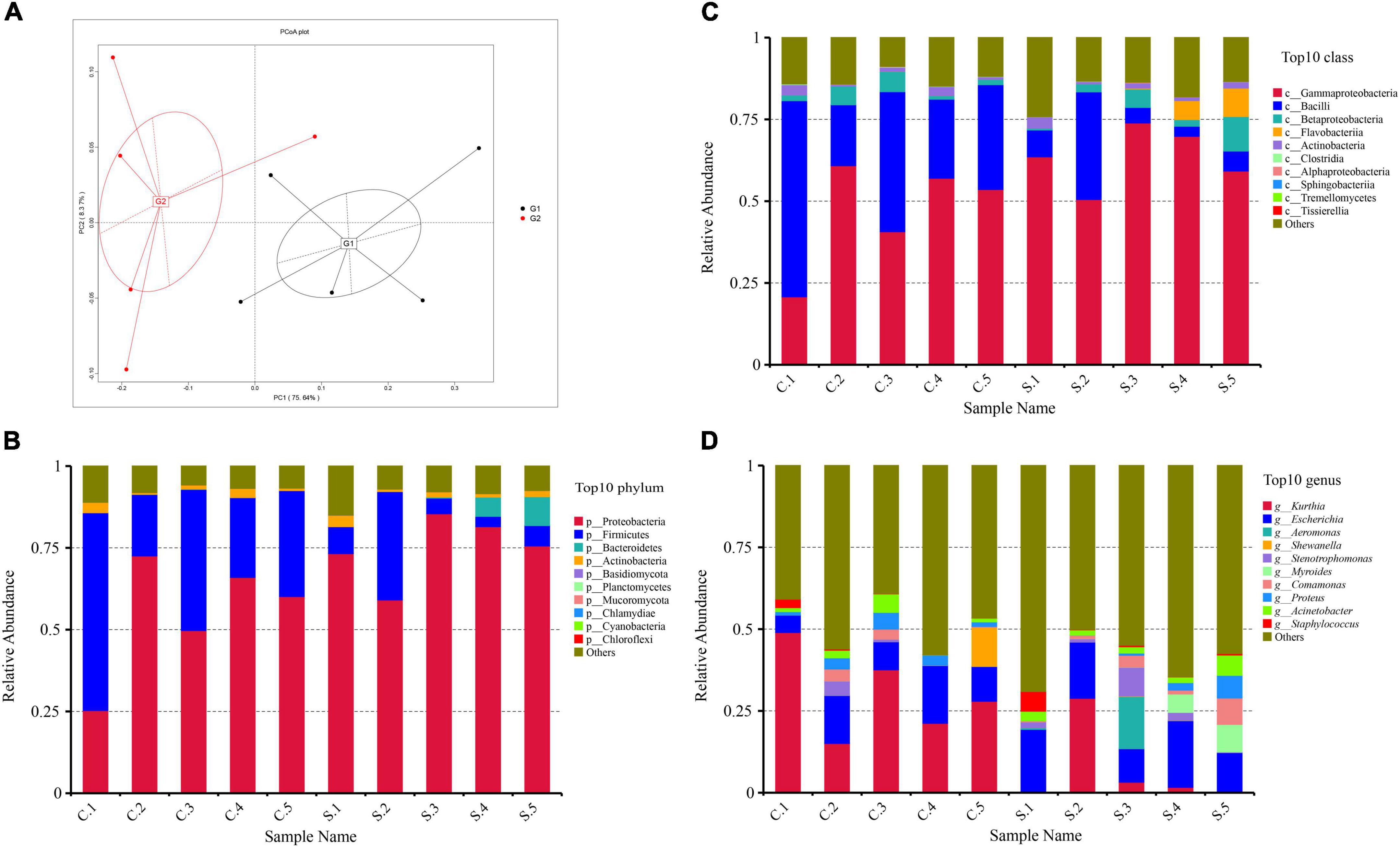
Figure 1. (A) Principal component analyze (PCoA) of the 10 samples based on Bray–Curtis distance. (B) Relative abundance of the top 20 phyla; (C) Relative abundance of the top 20 classes; (D) Relative abundance of the top 20 genera.
At the phylum rank, the compositions of the florfenicol-resistant microbial communities in each sample were relatively similar. Proteobacteria (65%), Firmicutes (23%), and Bacteroidetes (15%) were the most abundant bacterial phyla in all 10 samples (Figure 1B). The predominant classes were Gammaproteobacteria (55%), Bacilli (23%), and Betaproteobacteria (4%) (Figure 1C). The predominant genera were Kurthia (19%), Escherichia (14%), and Proteus (2.4%) (Figure 1D). At the genus level, the drug-resistant bacteria in the feces of pigs and chickens are quite different. The main resistant bacteria in the chicken farms is Kurthia, while in the pig farms it is Escherichia. Florfenicol-resistant Staphylococcus is more abundant in pig farms than that in chicken farms. Some samples also displayed unique features, such as a high abundance of Staphylococcus in S1 and an unusually high abundance of Aeromonas and Stenotrophomonas in S3 (Supplementary File 1).
Composition of the resistome
A total of 490 different resistance genes were detected in 10 samples; tet(A), qnrD, floR, sul2, erm(C), and tetM were the predominant resistance types in all the samples (Supplementary File 2). The abundance of total drug resistance genes and the abundance of floR in pig farms were significantly higher than in chicken farms. Chicken farms have a higher abundance of fexA (Figure 2A).
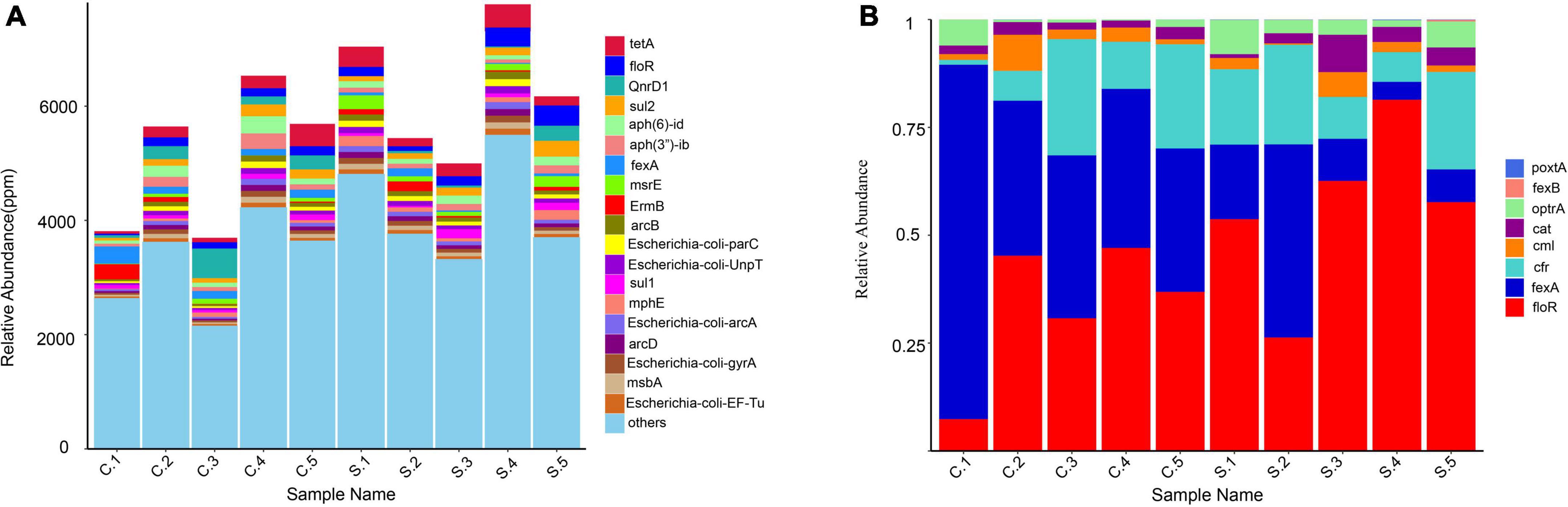
Figure 2. (A) Relative abundance of the top 20 antibiotic resistance genes (ARGs); (B) Relative abundance of resistance genes associated with florfenicol.
We estimated the relative abundance of associated genes for the clear analysis of bacterial resistance to florfenicol and oxazolidinone in pig and chicken farms. FloR, fexA, and oxazolidinone resistance genes cfr and optrA are distributed on all farms. PoxtA was distributed in nine farms except for C3, although its abundance was low. The abundance of optrA and poxtA in swine farms was higher than that in chicken farms. The prevalence of chloramphenicol resistance genes cml and cat remained relatively high (Figure 2B).
In this study, the resistance mechanism results were based on CARD databases (Figure 3). In our study, resistance mechanisms were classified into five major categories, namely, efflux pump, antibiotic inactivation, target alteration, antibiotic target protection, and antibiotic target replacement. Additional combinatorial mechanisms also exist. According to our results, antibiotic efflux pumps were detected as the main resistance mechanism in all samples, followed by antibiotic inactivation and target alteration. Proteobacteria have the most resistance genes, and the resistance mechanism is mainly by efflux pumps. It is noteworthy that many chromosomal genes are included in the CARD database. It is problematic as to whether chromosomal genes should be counted as ARGs, since they are resistant only when a mutation occurs, for example, quinolone resistance genes (gyrA and parC).
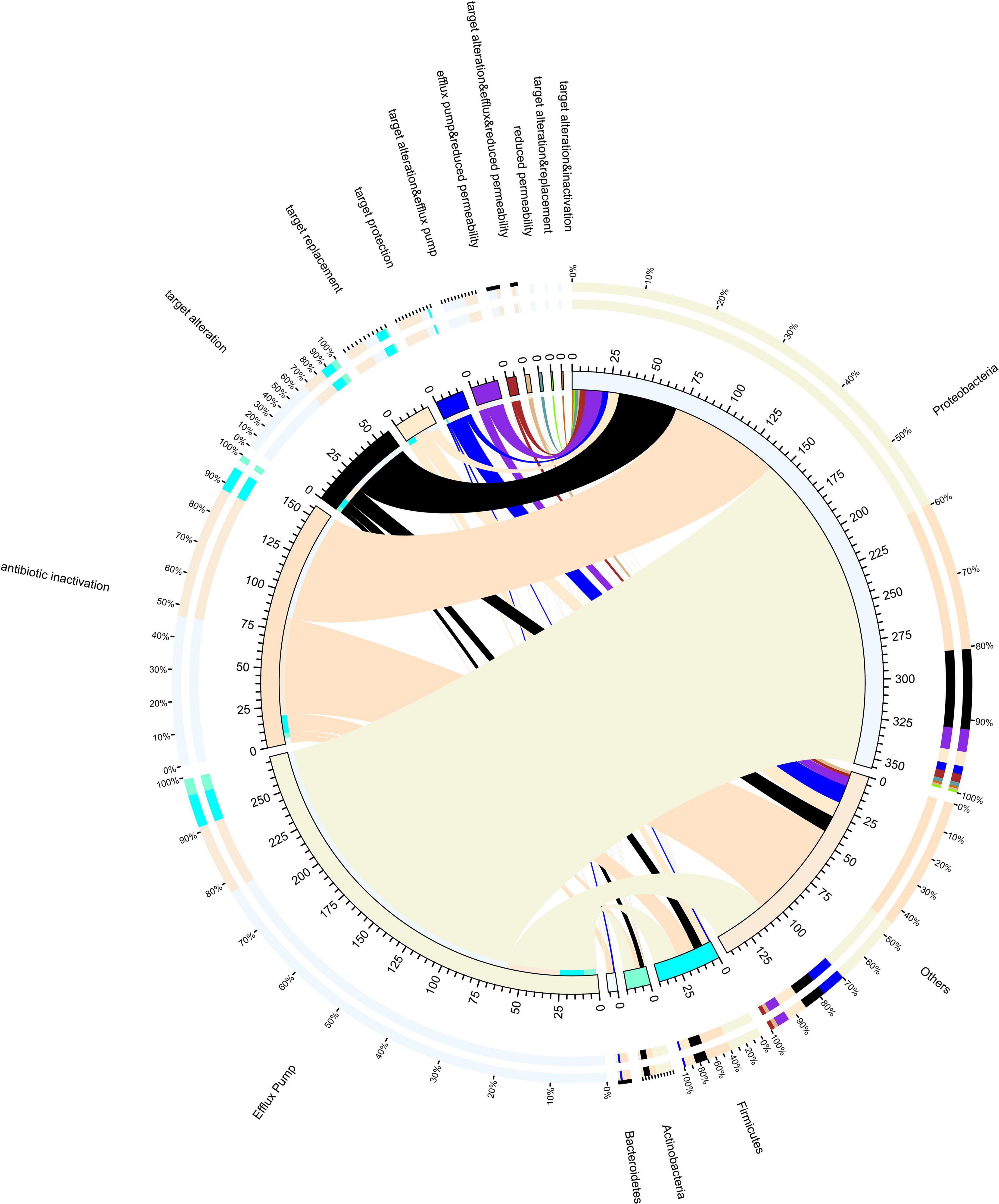
Figure 3. Resistance mechanisms and species circle map. On the right is the phylum-level species information, and on the left is the resistance mechanism information. Different colors of the inner circle indicate different species and resistance mechanisms of resistance, and the scale is the number of genes.
We selected high abundance resistance genes and clinically important resistance genes to construct the network map (Figure 4). We found that a co-occurrence relationship existed among many drug resistance genes. It is particularly noteworthy that the florfenicol resistance gene floR has a strong correlation with the tetracycline resistance gene tet(X), and fexA is also positively correlated with tet(M) and aminoglycoside resistance gene aph3-llla. PoxtA, a crucial oxazolidinone resistance gene that has undergone extensive research in recent years, has a favorable correlation with both tet(X) and qacH.
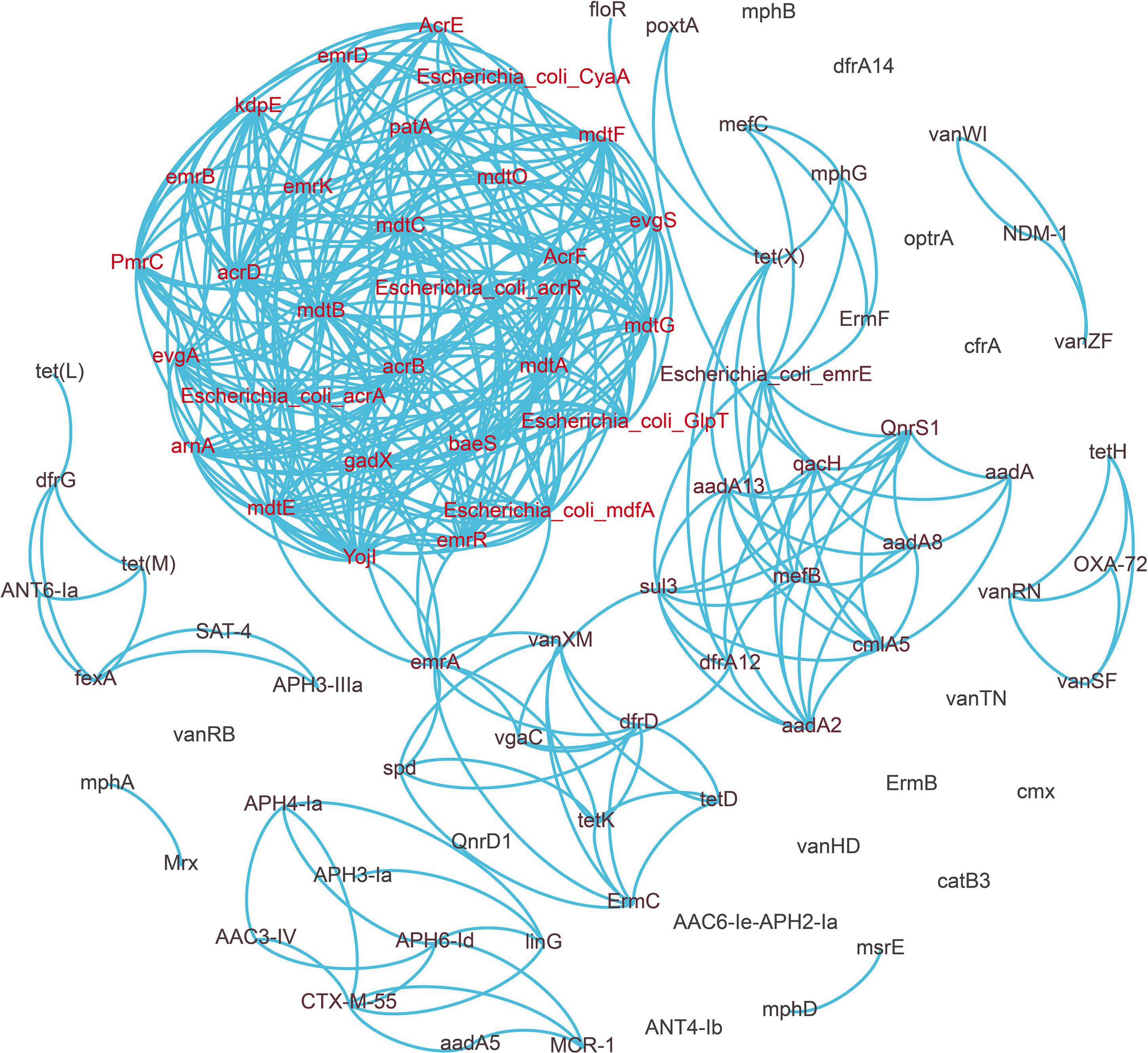
Figure 4. Network analysis revealing the co-occurrence patterns between antibiotic resistance genes (ARGs). A connection represents a strong relationship between ARGs (Pearson’s correlation coefficient r ≥ 0.9, p < 0.01).
Genetic environment of floR, cfr, and optrA
A total of 506 drug resistance genes were detected in the two samples (C.2 and S.4) revealed by nanopore sequencing. We discovered that ARGs on plasmids made up 32% of all ARGs using the software plasflow, indicating that they have a high potential for transfer into recipient settings. To systematically observe the resistance genes and genetic environment of florfenicol and oxazolidinone, we use nanopore sequencing reads to analyze relevant resistance genes located on MGEs. Specific information on the positioning of floR, cfr, optrA, and fexA is shown in Supplementary File 3. The genetic analysis of the selected 12 reads showed that these genes were associated with plasmids, integrative and conjugative element (ICE), and transposons.
Four different chromosomal and plasmid-harbored floR clusters were found. First, we found that floR is located in Tn7-like transposons from Proteus mirabilis in Reads1. Reads2–Reads5 represent three plasmids carrying floR (Figure 5A). They harbored the replication initiator genes repQ1, repFIC, repFIA, and repFIB, respectively. On the majority of plasmids, we could locate more than one floR gene. Other resistance genes, including blaTEM–1, aac(3)-IId, tet(A), and tet(R), were also found in these genomes. We discovered that IS26 and ISVsa3 were frequently present in the surroundings, indicating that IS26 and ISVsa3 might promote the transfer of floR among different plasmids or other MGEs.
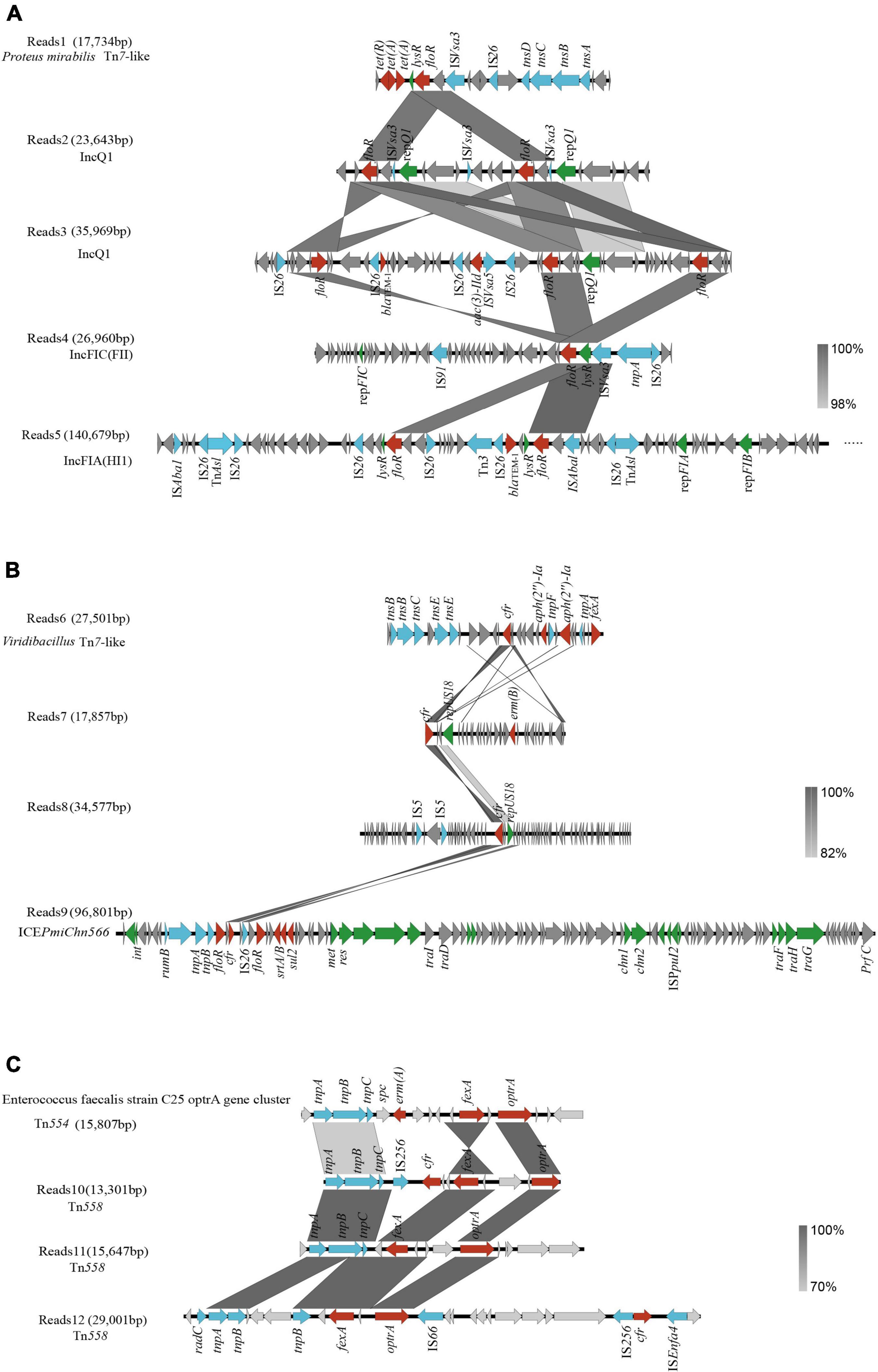
Figure 5. Genetic context of floR, cfr, and optrA genes. (A) floR genetic context; (B) cfr genetic context; (C) optrA genetic context. Genes and open-reading frames (ORFs) are shown as arrows, and their orientations of transcription are indicated by the arrowheads. Antimicrobial resistance genes are in red, transposase elements are in blue, replication genes and other protein genes are in green, and hypothetical protein genes are in gray.
Reads6 harbored a cfr-carrying gene fragment (Figure 5B). The BLAST analysis showed that it was 99.81% identical to the corresponding region of Macrococcus caseolyticus strain 5459_5_49 (GenBank accession no. BK012114) with 48% coverage. The transposase gene tnsABCDE is the intrinsic structure of the transposon Tn7 (Luo et al., 2021). The chromosomal location and the presence of tnsABCDE suggest that cfr-aph(2″)-Ia-fexA is carried in Viridibacillus by a novel transposon distantly related to Tn7.
Two cfr-carrying plasmids with sizes ranging from 17.8 to 34.5 kb were obtained after assembly (Reads7 and Reads8). They carried the replicon repUS18 (Figure 5B). A previous report showed that the repUS18 gene has been linked to the antimicrobial resistance gene transfer in Enterococci, and is frequently found on broad host-range Inc18 plasmids (Luo et al., 2021). We also found a novel complete SXT/R391 ICE in the genome of P. mirabilis named ICEPmiChn566 (Figure 5B; He et al., 2021a). ICEPmiChn566 is 96,801 bp in size, with a G+C content of 47.5%. BLAST analysis showed that ICEPmiChn566 had 99.94% nucleotide identity to ICEPmiChn1 with 96% coverage (Lei et al., 2016). ICEPmiChn566 carries cfr and lacks tet(G)-tetR region compared with ICEPmiChn1.
The horizontal transfer of the optrA is mostly influenced by the genetic environment. In addition to IS1216, which has a significant impact on the transmission of optrA, the genetic environment surrounding optrA might include Tn558 or Tn554 which consist of three transposase-encoding genes (tnpA, tnpB, and tnpC) (He et al., 2016). Our research also confirms this fact, that is, Reads11 and 12 (Figure 5C). Interestingly, we found (Reads10) a Tn558 connected downstream to cfr-fexA-optrA (Figure 5C). As far as we know, the localization of cfr-fexA-optrA on a single Tn558 was first reported in China. BLAST analysis showed that Reads10 was 99.82% identical to the corresponding region of Enterococcus faecium strain C25 optrA gene clusterl sequence (GenBank accession no. MK251150) with 32% coverage (Kang et al., 2019).
Discussion
Antimicrobial resistance varies dramatically across countries, regions, and even individual herds (Zhou et al., 2022). Researchers in Hong Kong made significant progress in their investigation of the antibiotic resistome in four wastewater treatment plants using Nanopore and Illumina sequencing techniques (Che et al., 2019). We adopted their methods to investigate the composition, and abundance of fecal microbial and resistome composition in chickens and pigs from large-scale breeding farms in China. The findings of this study add to our understanding of the breadth and extent of florfenicol and oxazolidinone resistance in China’s large-scale industrial swine and chicken farms.
We were able to identify and quantify a total of 60 phyla, 72 classes, and 1,515 bacterial genera that can exist under the selection pressure of florfenicol in all of the samples using a metagenomics sequencing technique. The population of Bacteroidia was the most numerous in uncultured stool samples (Munk et al., 2018). But Proteobacteria has risen to the top of the food chain in our samples. It should not be overlooked that our experiments were carried out under aerobic conditions, which are highly selective for species abundance. The low abundance of Bacteroidetes in the samples should be attributed to the aerobic culture conditions used in this study, as Bacteroidetes are anaerobic. In contrast, the widespread presence of Kurthia, Escherichia, and Proteus in these samples is not only due to their high resistance to florfenicol but is also related to aerobic culture. The oxazolidinone antibiotic linezolid has effective antibacterial activity against staphylococci. The presence of a high abundance of florfenicol-resistant staphylococci in animal feces is a phenomenon that deserves our continued attention.
In our samples, compared to chicken farms, swine farms have a higher total number of genes and gene abundance for resistance to florfenicol and oxazolidinone, and there are significant variances between farms. On the one hand, it is related to the differences in the structure and composition of the intestine between pigs and chickens. On the other hand, this may be attributed to the huge use of florfenicol in pig farms and the prohibition of the use of antibiotics in laying hens during the laying period (Zhao et al., 2010; Zhao Q. et al., 2016; Munk et al., 2018). Previous research has shown although the use of chloramphenicol in food-producing animals was banned in China in 2002,17 florfenicol is still in use, which may cause a still relatively high prevalence of the chloramphenicol resistance genes (Yang et al., 2022). The cml and cat genes were also found to be highly abundant in our study. In this study, the main resistance mechanism of these resistant bacteria was found to be the efflux pump. Through correlation analysis, we discovered extensive colinearity of florfenicol and oxazolidinone resistance genes with other resistance genes, such as tigecycline and aminoglycoside.
Importantly, long sequences make it possible to analyze the genetic context of the multiple drug resistance genes in binned genomes. At the same time, we recognize that ARGs localized on MGEs may be at risk of transmission. Our study demonstrates that plasmids, ISs, transposons, and ICE all contribute significantly to facilitating the dissemination of floR, cfr, and optrA genes. The genetic structures of the drug-resistant plasmids were different, indicating the diversity of plasmids carrying the floR and cfr genes. IncF and IncQ plasmids are among the most prevalent incompatibility types involved in the transfer of resistance determinants in Enterobacteriaceae and have been reported worldwide (Muthuirulandi Sethuvel et al., 2019). LncQ and lncF carriage of floR has also been widely identified and reported, which is also compatible with our findings (Loftie-Eaton and Rawlings, 2012; Mohsin et al., 2021).
By comparison, it was found that the tetracycline resistance genes [tet(A) and tet(R)] and the beta-lactam antibiotic resistance gene blaTEM–1 frequently existed on the plasmid containing the floR gene, indicating that there may be co-transmission between these resistance genes. At the same time, it was also found that multiple insertion sequences IS26 and ISVsa3 appeared on the plasmid carrying the floR gene, which may spread through plasmid recombination and mediate bacterial multidrug resistance. One study found the existence of ISVsa3 on the reported ICE carrying the floR gene and verified that ISVsa3 mediates the transfer of floR (He et al., 2021b). The mobile element IS26, a member of the IS6 family, is essential for the recombination, clustering, and spread of resistance genes (Liu et al., 2021). ICEPmiChn566, which had 99.94% nucleotide identity to ICEPmiChn1, has one more cfr gene added to VRn (He et al., 2015; Liu et al., 2021). Most likely, the IS26 insertion sequence mediated the transfer of this cfr region within the chromosome. FloR and cfr were carried in the Tn7-like structure, which indicated that Tn7-like elements might play a role in mediating floR and cfr transmission. Tn558 has been an important dissemination media of optrA and has been reported all over the world (Tsilipounidaki et al., 2019; Almeida et al., 2020a,b). Tn558 normally surrounds optrA and mediates their transfer (He et al., 2016). In our study, we identified a genetic environment downstream of Tn558 as cfr-fexA-optrA. We should take appropriate measures to control the further spread of cfr and optrA and the emergence of mutant linezolid resistance.
These findings reveal that florfenicol and oxazolidone resistance is a serious issue in pig and chicken farms. Diverse resistant bacteria and resistant genes exist in farms, and these drug-resistant genes have a high risk of transmission. Antibiotic resistance in farms could pose a significant hazard to the environment, food safety, and human health. We should conduct continuous monitoring of drug-resistant bacteria, drug-resistant genes, and MGEs on farms. In addition, adequate management of animal waste in farms should be implemented, and florfenicol usage should be limited to prevent the spread of antibiotic resistance.
Conclusion
In this study, we applied Illumina and Nanopore metagenomic sequencing techniques to investigate the ARGs and genetic contexts of ARGs related to florfenicol and oxazolidinone across 10 farms in China. The findings revealed a wide spread of florfenicol-resistant bacteria and ARGs in feces, including floR, fexA, and the oxazolidinone resistance determinants cfr, optrA, and poxtA. More substantial drug resistance issues exist in pig farms than in chicken farms. MGEs, including plasmids, ICE, transposons, and insertion sequences, all play a role in the dissemination of these genes, according to genome binning and genetic context analysis. Our results demonstrate that the florfenicol and oxazolidone resistance genes are closely associated with other important resistance genes, such as tigecyclines and aminoglycoside antibiotics. Pig and chicken farms are an important repository of oxazolidinone resistance genes.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/search/all/?term=PRJNA843911 and https://www.ncbi.nlm.nih.gov/search/all/?term=PRJNA843901.
Author contributions
XY and C-WL conceived the study, designed the experiments, and analyzed the data. TZ analyzed the data. QW, ZH, and XC performed the experiments. XY wrote the manuscript. H-NW guided the experiments and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (U21A20257 and 31830098), National System for Layer Production Technology (CARS-40-K14), and Sichuan Science and Technology Program (2021ZDZX0010, 2020YFN0147, 2020NZZJ001, and 2021YFYZ0030).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1018901/full#supplementary-material
Supplementary Table 1 | Sample information.
Supplementary Table 2 | Illumina sequencing supplementary data.
Supplementary Table 3 | Nanopore sequencing supplementary data.
Supplementary File 1 | Species information (Illumina sequencing).
Supplementary File 2 | ARGs relative abundance (Illumina sequencing).
Supplementary File 3 | MGE information of florfenicol resistance gene mapping.
Footnotes
- ^ https://www.qiagen.com/au/products/new-products/qiaamp-powerfecal-pro-dna-kit/
- ^ https://github.com/cjfields/readfq
- ^ http://bowtie-bio.sourceforge.net/bowtie2/index.shtml
- ^ https://github.com/voutcn/megahit
- ^ https://anaconda.org/bioconda/blasr
- ^ https://github.com/fenderglass/Flye/
- ^ http://topaz.gatech.edu/GeneMark/
- ^ http://www.bioinformatics.org/cd-hit
- ^ https://github.com/bbuchfink/diamond/
- ^ https://software-ab.informatik.uni-tuebingen.de/download/megan6/welcome.html
- ^ https://qiime2.org/
- ^ https://gephi.org/
- ^ http://circos.ca/software/download/
- ^ https://plasflow.co.uk/
- ^ https://rast.nmpdr.org/
- ^ http://mjsull.github.io/Easyfig/
- ^ http://www.moa.gov.cn/ztzl/ncpzxzz/flfg/200709/t20070919_893091.htm
References
Almeida, L. M., Gaca, A., Bispo, P. M., Lebreton, F., Saavedra, J. T., Silva, R. A., et al. (2020a). Coexistence of the oxazolidinone resistance–associated genes cfr and optrA in Enterococcus faecalis from a healthy piglet in Brazil. Front. Public Health 8:518. doi: 10.3389/fpubh.2020.00518
Almeida, L. M., Lebreton, F., Gaca, A., Bispo, P. M., Saavedra, J. T., Calumby, R. N., et al. (2020b). Transferable resistance gene optrA in Enterococcus faecalis from Swine in Brazil. Antimicrob. Agents Chemother. 64:e00142-20. doi: 10.1128/AAC.00142-20
Antonelli, A., D’Andrea, M. M., Brenciani, A., Galeotti, C. L., Morroni, G., Pollini, S., et al. (2018). Characterization of poxtA, a novel phenicol–oxazolidinone–tetracycline resistance gene from an MRSA of clinical origin. J. Antimicrob. Chemother. 73, 1763–1769. doi: 10.1093/jac/dky088
Arcangioli, M. A., Leroy-Sétrin, S., Martel, J. L., and Chaslus-Dancla, E. (1999). A new chloramphenicol and florfenicol resistance gene Łanked by two integron structures in Salmonella typhimurium DT104. FEMS Microbiol. Lett. 174, 327–332.
Bozdogan, B., and Appelbaum, P. C. (2004). Oxazolidinones: activity, mode of action, and mechanism of resistance. Int. J. Antimicrob. Agent 23, 113–119. doi: 10.1016/j.ijantimicag.2003.11.003
Che, Y., Xia, Y., Liu, L., Li, A., Yang, Y., and Zhang, T. (2019). Mobile antibiotic resistome in wastewater treatment plants revealed by Nanopore metagenomic sequencing. Microbiome 7:44. doi: 10.1186/s40168-019-0663-0
Frost, L. S., Leplae, R., Summers, A. O., and Toussaint, A. (2005). Mobile genetic elements: the agents of open source evolution. Nat. Rev. Microbiol. 9, 722–732.
Guo, J., Li, J., Chen, H., Bond, P. L., and Yuan, Z. (2017). Metagenomic analysis reveals wastewater treatment plants as hotspots of antibiotic resistance genes and mobile genetic elements. Water Res. 123, 468–478. doi: 10.1016/j.watres.2017.07.002
He, J., Lei, C., Li, C., Wang, X., Cui, P., and Wang, H. (2021a). Identification of a novel genomic resistance island PmGRI1-STP3 and an SXT/R391 integrative conjugative element in Proteus mirabilis of swine origin in China. J. Glob. Antimicrob. Res. 25, 77–81. doi: 10.1016/j.jgar.2021.02.018
He, J., Sun, L., Zhang, L., Leptihn, S., Yu, Y., Hua, X., et al. (2021b). A novel SXT/R391 integrative and conjugative element carries two copies of the blaNDM-1 Gene in Proteus mirabilis. mSphere 6:e58821. doi: 10.1128/mSphere.00588-21
He, L., He, L., Liu, Y., Zhang, M., Zhao, J., Zhang, Q., et al. (2019). Microbial diversity and antibiotic resistome in swine farm environments. Sci. Total Environ. 685, 197–207. doi: 10.1016/j.scitotenv.2019.05.369
He, S., Hickman, A. B., Varani, A. M., Siguier, P., Chandler, M., Dekker, J. P., et al. (2015). Insertion sequence IS26 reorganizes plasmids in clinically isolated multidrug-resistant bacteria by replicative transposition. mBio 6:e00762-15. doi: 10.1128/mBio.00762-15
He, T., Shen, Y., Schwarz, S., Cai, J., Lv, Y., Li, J., et al. (2016). Genetic environment of the transferable oxazolidinone/phenicol resistance gene optrA in Enterococcus faecalis isolates of human and animal origin. J. Antimicrob. Chemother. 71, 1466–1473. doi: 10.1093/jac/dkw016
Kang, Z., Lei, C., Kong, L., Wang, Y., Ye, X., Ma, B., et al. (2019). Detection of transferable oxazolidinone resistance determinants in Enterococcus faecalis and Enterococcus faecium of swine origin in Sichuan Province, China. J. Glob. Antimicrob. Res. 19, 333–337. doi: 10.1016/j.jgar.2019.05.021
Lei, C., Kang, Z., Wu, S., Chen, Y., Kong, L., and Wang, H. (2019). Detection of the phenicol–oxazolidinone–tetracycline resistance gene poxtA in Enterococcus faecium and Enterococcus faecalis of food-producing animal origin in China. J. Antimicrob. Chemother. 74, 2459–2461. doi: 10.1093/jac/dkz198
Lei, C., Zhang, A., Wang, H., Liu, B., Yang, L., and Yang, Y. (2016). Characterization of SXT/R391 integrative and conjugative elements in Proteus mirabilis isolates from food-producing animals in China. Antimicrob. Agents Chemother. 60, 1935–1938. doi: 10.1128/AAC.02852-15
Li, L., Xiao, Y., Olsen, R. H., Wang, C., Meng, H., and Shi, L. (2022). Short- and long-read metagenomics insight into the genetic contexts and hosts of mobile antibiotic resistome in Chinese swine farms. Sci. Total Environ. 827:154352. doi: 10.1016/j.scitotenv.2022.154352
Liu, H., Wang, Y., Wu, C., Schwarz, S., Shen, Z., Jeon, B., et al. (2012). A novel phenicol exporter gene, fexB, found in enterococci of animal origin. J. Antimicrob. Chemother. 67, 322–325. doi: 10.1093/jac/dkr481
Liu, Y., He, D., Zhang, M., Pan, Y., Wu, H., Yuan, L., et al. (2021). The formation of two hybrid plasmids mediated by IS26 and Tn6952 in Salmonella enterica serotype enteritidis. Front. Microbiol. 12:676574. doi: 10.3389/fmicb.2021.676574
Loftie-Eaton, W., and Rawlings, D. E. (2012). Diversity, biology and evolution of IncQ-family plasmids. Plasmid 67, 15–34. doi: 10.1016/j.plasmid.2011.10.001
Long, K. S., Poehlsgaard, J., Kehrenberg, C., Schwarz, S., and Vester, B. (2006). The Cfr rRNA methyltransferase confers resistance to phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A antibiotics. Antimicrob. Agents Chemother. 50, 2500–2505. doi: 10.1128/AAC.00131-06
Luo, G., Li, B., Li, L., Zhang, T., and Angelidaki, I. (2017). Antibiotic resistance genes and correlations with microbial community and metal resistance genes in full-scale biogas reactors as revealed by metagenomic analysis. Environ. Sci. Technol. 51, 4069–4080. doi: 10.1021/acs.est.6b05100
Luo, X., Yin, Z., Zeng, L., Hu, L., Jiang, X., Jing, Y., et al. (2021). Chromosomal integration of huge and complex blaNDM-Carrying genetic elements in Enterobacteriaceae. Front. Cell Infect. Microbiol. 11:690799. doi: 10.3389/fcimb.2021.690799
Ma, L., Xia, Y., Li, B., Yang, Y., Li, L., Tiedje, J. M., et al. (2016). Metagenomic assembly reveals hosts of antibiotic resistance genes and the shared resistome in pig, chicken, and human feces. Environ. Sci. Technol. 50, 420–427. doi: 10.1021/acs.est.5b03522
Mohsin, M., Hassan, B., Martins, W. M. B. S., Li, R., Abdullah, S., Sands, K., et al. (2021). Emergence of plasmid-mediated tigecycline resistance tet(X4) gene in Escherichia coli isolated from poultry, food and the environment in South Asia. Sci. Total Environ. 787:147613. doi: 10.1016/j.scitotenv.2021.147613
Munk, P., Knudsen, B. E., Lukjancenko, O., Duarte, A. S. R., Van Gompel, L., Luiken, R. E. C., et al. (2018). Abundance and diversity of the faecal resistome in slaughter pigs and broilers in nine European countries. Nat. Microbiol. 3, 898–908. doi: 10.1038/s41564-018-0192-9
Muthuirulandi Sethuvel, D. P., Anandan, S., Devanga Ragupathi, N. K., Gajendiran, R., Kuroda, M., Shibayama, K., et al. (2019). IncFII plasmid carrying antimicrobial resistance genes in Shigella flexneri: vehicle for dissemination. J. Glob. Antimicrob. Res. 16, 215–219. doi: 10.1016/j.jgar.2018.10.014
O’Neill, J. (2014). Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. London: Review on Antimicrobial Resistance.
Shen, J., Wang, Y., and Schwarz, S. (2013). Presence and dissemination of the multiresistance gene cfr in Gram-positive and Gram-negative bacteria. J. Antimicrob. Chemother. 68, 1697–1706. doi: 10.1093/jac/dkt092
Tsilipounidaki, K., Gerontopoulos, A., Papagiannitsis, C., and Petinaki, E. (2019). First detection of an optrA-positive, linezolid-resistant ST16 Enterococcus faecalis from human in Greece. New Microbes New Infect. 29:100515. doi: 10.1016/j.nmni.2019.01.010
Wang, Y., Lv, Y., Cai, J., Schwarz, S., Cui, L., Hu, Z., et al. (2015). A novel gene, optrA, that confers transferable resistance to oxazolidinones and phenicols and its presence in Enterococcus faecalis and Enterococcus faecium of human and animal origin. J. Antimicrob. Chemother. 70, 2182–2190. doi: 10.1093/jac/dkv116
Xia, Y., Li, A., Deng, Y., Jiang, X., Li, L., and Zhang, T. (2017). MinION nanopore sequencing enables correlation between resistome phenotype and genotype of coliform bacteria in municipal sewage. Front. Microbiol. 8:2105. doi: 10.3389/fmicb.2017.02105
Yang, L., Shen, Y., Jiang, J., Wang, X., Shao, D., Lam, M. M. C., et al. (2022). Distinct increase in antimicrobial resistance genes among Escherichia coli during 50 years of antimicrobial use in livestock production in China. Nat. Food 3, 197–205. doi: 10.1038/s43016-022-00470-6
Zhang, Q., Ying, G., Pan, C., Liu, Y., and Zhao, J. (2015). Comprehensive evaluation of antibiotics emission and fate in the River Basins of China: source analysis, multimedia modeling, and linkage to bacterial resistance. Environ. Sci. Technol. 49, 6772–6782. doi: 10.1021/acs.est.5b00729
Zhao, L., Dong, Y. H., and Wang, H. (2010). Residues of veterinary antibiotics in manures from feedlot livestock in eight provinces of China. Sci. Total Environ. 408, 1069–1075. doi: 10.1016/j.scitotenv.2009.11.014
Zhao, Q., Wang, Y., Wang, S., Wang, Z., Du, X., Jiang, H., et al. (2016). Prevalence and abundance of florfenicol and linezolid resistance genes in soils adjacent to swine feedlots. Sci. Rep. 6:32192. doi: 10.1038/srep32192
Zhao, S., Tyson, G. H., Chen, Y., Li, C., Mukherjee, S., Young, S., et al. (2016). Whole-genome sequencing analysis accurately predicts antimicrobial resistance phenotypes in Campylobacter spp. Appl. Environ. Microb. 82, 459–466. doi: 10.1128/AEM.02873-15
Keywords: nanopore sequencing, Illumina sequencing, florfenicol, oxazolidinone, genetic context
Citation: Yang X, Zhang T, Lei C-W, Wang Q, Huang Z, Chen X and Wang H-N (2022) Florfenicol and oxazolidone resistance status in livestock farms revealed by short- and long-read metagenomic sequencing. Front. Microbiol. 13:1018901. doi: 10.3389/fmicb.2022.1018901
Received: 14 August 2022; Accepted: 26 September 2022;
Published: 20 October 2022.
Edited by:
Jian-Hua Liu, South China Agricultural University, ChinaReviewed by:
Ruichao Li, Yangzhou University, ChinaTao He, Jiangsu Academy of Agricultural Sciences (JAAS), China
Copyright © 2022 Yang, Zhang, Lei, Wang, Huang, Chen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong-Ning Wang, d2hvbmduaW5nQDE2My5jb20=