 Zhenda Shang1†Yaping Wang2*†Miao An2†
Zhenda Shang1†Yaping Wang2*†Miao An2† Xiushuang Chen3,4
Xiushuang Chen3,4 Muhammad Fakhar-e-Alam Kulyar2Zhankun Tan1Suozhu Liu1*
Muhammad Fakhar-e-Alam Kulyar2Zhankun Tan1Suozhu Liu1* Kun Li3,4*
Kun Li3,4*- 1College of Animal Science, Tibet Agricultural & Animal Husbandry University, Nyingchi, China
- 2College of Veterinary Medicine, Huazhong Agricultural University, Wuhan, China
- 3Institute of Traditional Chinese Veterinary Medicine, College of Veterinary Medicine, Nanjing Agricultural University, Nanjing, China
- 4MOE Joint International Research Laboratory of Animal Health and Food Safety, College of Veterinary Medicine, Nanjing Agricultural University, Nanjing, China
The soil microbiome is crucial in determining contemporary realistic conditions for future terrestrial ecological and evolutionary development. However, the precise mechanism between the fecal deposition in livestock grazing and changes in the soil microbiome remains unknown. This is the first in-depth study of bacterial and fungal taxonomic changes of excrement contaminated soils in the plateau (>3,500 m). This suggests the functional shifts towards a harmful-dominated soil microbiome. According to our findings, excrement contamination significantly reduced the soil bacterial and fungal diversity and richness. Furthermore, a continuous decrease in the relative abundance of microorganisms was associated with nutrient cycling, soil pollution purification, and root-soil stability with the increasing degree of excrement contamination. In comparison, soil pathogens were found to have the opposite trend in the scenario, further deteriorating normal soil function and system resilience. Such colonization and succession of the microbiome might provide an important potential theoretical instruction for microbiome-based soil health protection measures in the plateau of China.
Introduction
Soil harbors are home to various microorganisms, including bacteria and fungi, referred to as the soil microbiome (Geisen et al., 2015). Soil microbiome is a dynamic and complex ecosystem in the rhizosphere and/or in the bulk soil, which regulates primary production, organic-matter degradation and carbon sequestration in soil, etc., (Bardgett and van der Putten, 2014; Ruiz et al., 2020). The interaction relationships between below-ground and aboveground microorganisms influence the composition, production, succession formation, and dynamic changes in soil communities (Kardol and Wardle, 2010). Both deterministic and stochastic processes may influence community assembly processes in soil ecosystems. The former involves niche-based mechanisms, including natural selection (e.g., competition, inhibition, predation, and mutualisms), while the latter reflect random shifts in the relative abundances of microorganisms (Chase, 2010; Vellend, 2010; Rosindell et al., 2011). In turn, the activities and interactions of soil microorganisms have a major impact on life-sustaining processes such as soil productivity and nutrient cycling (Cardinale, 2012). The soil microbiome composition, e.g., bacteria and fungi, can be influenced by the competitive relationships arising from resource sharing or coexisting interactions (de Boer et al., 2005; Mille-Lindblom et al., 2006). However, our knowledge about this hidden biodiversity is less established than aboveground community dynamics (Bardgett and van der Putten, 2014). This inadequate understanding hampers us from anticipating the subsequent outcomes of livestock grazing, which induces a soil microbial diversity shift (Wardle, 2002).
Livestock production is indispensable to agriculture throughout the globe, especially in highland areas of China. The Qinghai-Tibet Plateau (>3,000 m) has extremely harsh living conditions, such as low temperature, hypoxia, and intense ultraviolet rays (Li et al., 2019). The local nomads carried on the ancient civilization, whose primary source of income was grazing yaks, a distinctive plateau species (Wang et al., 2021). Previous studies reported that the microbiome composition is influenced by environmental conditions (Li and Zhao, 2015). Another long-standing issue in micro-ecology is the intensity of grazing that poses a significant challenge and pressure on land. For example, animal movement and continuous grazing disrupted soil aggregates, resulting in a negative soil stability index associated with the variation in soil structure and/or soil porosity (Imhoff et al., 2000; Di et al., 2001). Aside from soil compaction, excremental contamination has reduced soil microbial diversity (Bardgett and van der Putten, 2014; Schutzius et al., 2019). Pathogenic bacteria (even anthropozoonosis) are carried in animal excrement that enters surface water or soil after surface runoff and percolation/infiltration after precipitation events, causing significant harm to human health (Harwood et al., 2014; Guo et al., 2019). Several methods have been developed to accomplish soil protection, one of which is to enhance soil immunity, i.e., the ability of soil to defend against invading pathogens (Raaijmakers and Mazzola, 2016). It has been proved that soil functional microorganisms’ activities determine soil immunity (Wang and Li, 2019). Here, characterizing the soil microbiome colonization and succession based on grazing-dependent impacts may be important if adopted a microbiome-based strategy to improve soil resilience and immunity.
Currently, studies related to soil microbial biodiversity are entering a new era. More and more scientists are becoming aware of the critical role of soil biodiversity in providing important ecological services to human society. They are using the latest advanced tools to explore the micro-biodiversity of soil (Larigauderie et al., 2012). Here we explore the variation in soil microbial community, including bacteria and fungi, caused by animal excrement contamination in plateau using 16S ribosomal DNA (16S rDNA) and Internal Transcribed Spacer (ITS) high-throughput sequencing techniques. Exploring interactions between excrement contamination and the soil microbial community may lead to novel ways to manage soil-borne diseases in the plateau.
Materials and methods
Soil sampling and processing
During May 2021, soil samples were collected at a place in Aba Autonomous Prefecture (31°51′-33°33’ N, 101°51′-103°22′ E, > 3,500 m). Furthermore, yaks in the sampling sites were allowed to graze freely without any treatment for their excrement. According to the natural habit of the yaks, all animals were grazed freely in the highland pasture (defined as NM, 12,000 m2 per yak) from 06:00 h and 18:00 h, and the rest of the day was in the housed area (defined as NJ, 3 m2 per yak). Total 18 soil cores (0–15 cm depths and 2.5 cm diameter) were collected randomly from the three plots by using sterile aluminum core tubes, no-grazing soil (NF), grazing effect soil (NM) and yaks’ housed area soil (NJ), with intact cores being delivered to the laboratory, homogenized and sieved (2.0 mm mesh) to remove plant debris and flash-subsequently frozen in −80°C for further analysis.
DNA extraction
Through high-throughput sequencing, 16S rDNA and ITS genes characterized bacterial and fungal diversity. Following the instruction procedure, the genomic DNA (gDNA) was extracted per soil sample using FastDNA Spin Kit (MP Biochemicals, United States). Afterward, the purity of extracted DNA was done using a Universal DNA Purification Kit (TIANGEN Biotech Co., Ltd., Beijing, China). The quality and integrity of gDNA were evaluated by 1% agarose gel electrophoresis and UV–Vis spectrophotometer (Thermo Fisher Scientific, Nanodrop™, Waltham, United States).
16S rDNA and its genes amplification and sequencing
The specific primers based on the V3-V4 region for bacteria of the 16S rDNA gene and ITS region for fungi of the 18S rRNA gene were synthesized for PCR amplification (F: 5’-ACTCCTACGGGAGGCAGCA-3′ and R: 5’-GGACTACHVGGGTWTCTAAT-3′) and (F: 5’-NNNNNNNNGCATCGATGAAGAACGCAGC-3′ and 5’-TCCTCCGCTTATTGATATGC-3′), respectively (Hu et al., 2016). The melting temperature of bacterial was 55°C and PCR cycles were 35; whereas the melting temperature of fungal was 58°C for 50 s, and PCR cycles were 35. After cleaning and normalizing, subsequent quality control of PCR amplicons was executed using a High Sensitive DNA Kit (Agilent Technologies, United States). The sequencing library was prepared according to the standard process provided by Illumina (Illumina, Inc., SanDiego, California, United States). The qualified library was validated in the following rules: library with one peak and no linker was selected; using Library Quantification Kit (KAPA Biosystems, United Kingdom) to quantify the libraries and the concentrations were kept above 2 nM. Finally, the qualified library was sequenced paired ends on the Illumina Novaseq platform (Illumina, Inc., SanDiego, California, United States) to define the phylogenetic types.
In order to acquire more accurate and reliable sequencing results (effective reads), the raw data from high-throughput sequencing were filtered to eliminate the low quality by the following pre-procedures: (1) The raw reads were filtered using Trimmomatic software (v0.33, Germany), subsequent the primer sequences were identified and removed by using Cutadapt software (v1.9.1, Germany) to acquire clean reads, (2) Clean paired-end sequences were overlapped and merged to tags using Usearch software (v1.0, United States), (3) those reads contaminated by chimeric sequences were identified and removed with the UCHIME (v4.2, United States) tool to acquire the final effective reads. Subsequently, Quality control and taxonomically classified operational taxonomic unit (OTU, with 97% sequence similarity) mapping to the SILVA (for bacteria), and UNITE (for fungi) databases were performed using the QIIME platform (v1.8.0, United States).
Bioinformatics and statistical analysis
Venn diagram, drawn using R software (v3.0.3, Austria), visually showed the counts of unique and common OTUs among groups. The Rarefaction curve, Shannon index, Rank abundance curve, and Coverage index were set for assessing high-throughput sequencing data volume, depth, richness, and uniformity of the species contained in the sample. The Quantitative Insights into Microbial Ecology (v1.8.0, QIIME, United States) was used for constructing a genus-level phylogenetic tree and subsequently drawn by Python software. ACE index, Chao1 index, Shannon index, and Simpson index, which reflect alpha diversity, were evaluated by QIIME software (v1.8.0, United States). The similarities and clustering between individuals or groups were visually exhibited using Principal Coordinates Analysis (PCoA) and Unweighted Pair-group Method with Arithmetic Mean (UPGMA). Metastats software was used to perform T-test for finding the differentially abundant taxa (phylum, class, and genus) among groups. Student’s t-test was used to compare the alpha diversities among groups and perform calculations in SPSS software (v17.0, United States). The values were expressed as the mean ± SD, and the value of p (corrected) less than 0.05 indicated statistical significance.
Results
16S rDNA and its high-throughput sequence data analysis
A total of 1,318,076 and 1,412,260 high-quality bacterial and fungal reads were obtained after quality control, which was grouped into 2,065 and 1,139 OTUs. The average number of effective reads generated per sample was 73,226 and 78,459 from V4 and ITS regions, respectively (Supplementary Tables S1, S2). Multi-indicators, Rarefaction curve, Shannon index, and Species accumulation curve per sample tended to be flat and extended to the right end of the x-axis, indicating that almost all species were annotated in bacteria and fungi populations (Supplementary Figure S1). Furthermore, the estimated coverage rate (Coverage) values were greater than 99%, indicating that the sequencing depth sufficiently represented the diversity of bacterial and fungal communities (Figures 1A,B).
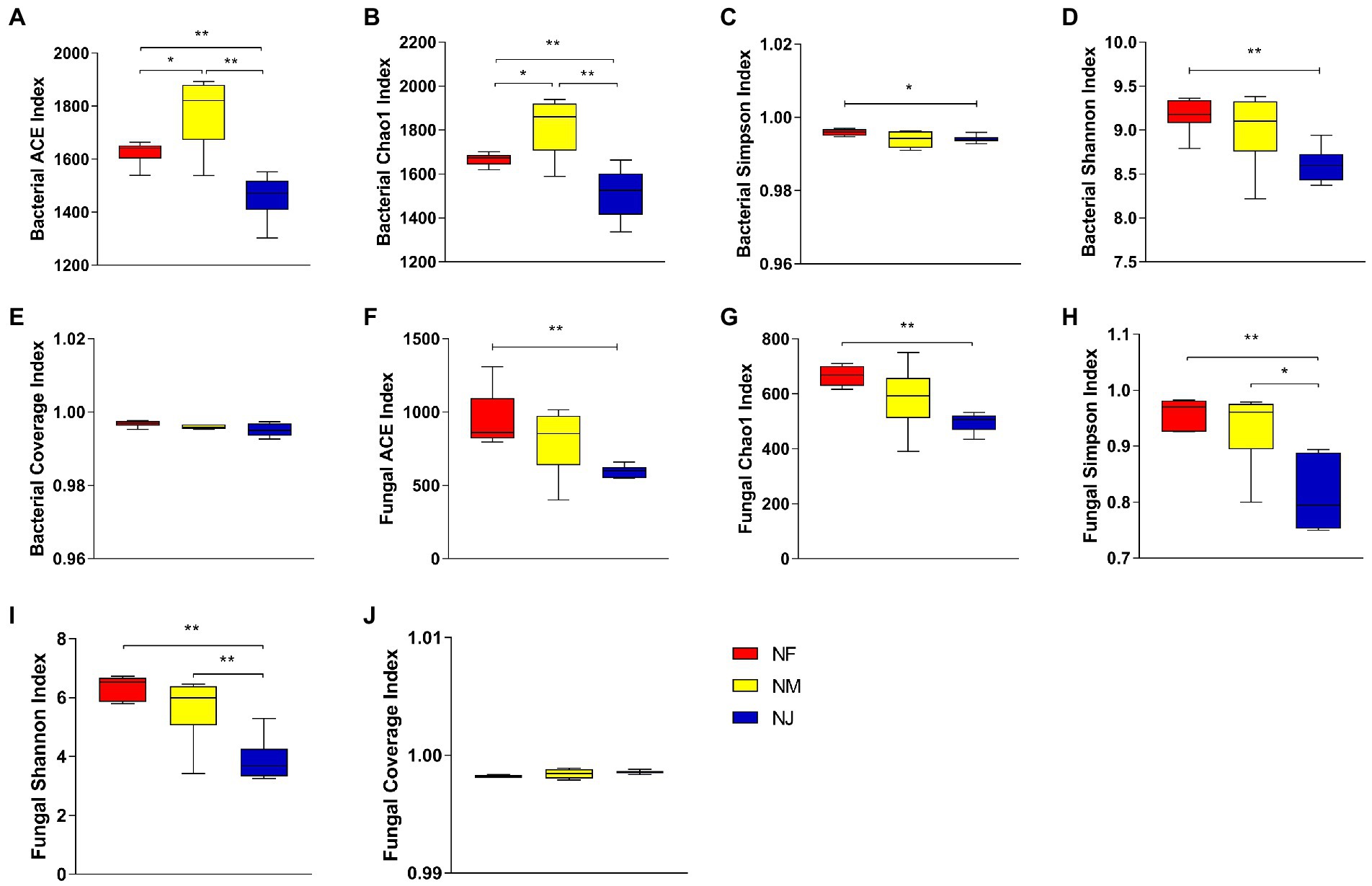
Figure 1. Evaluation of alpha diversity indices. (A, C–F) Represented soil bacterial, Coverage indices, ACE, Chao1, and Simpson, Shannon, respectively. (B, G–H) Represented fungal, Coverage indices, ACE, Chao1, Simpson, and Shannon, respectively. “*” represented that “p < 0.05”; “**” represented that “p < 0.01.”
Among detected 3,204 OTUs clustered at 97% sequence similarity, 1,556 bacterial and 517 fungal core OTUs were identified in all soil samples, accounting for approximately 48.6 and 16.1% of the total OTUs (Figures 2A,B). As expected, we detected a far higher number of core OTUs than unique OTUs per group, indicating that the population differences between groups were higher than those within groups (Figures 2C–E, 2F–H). Using the Python program, the representative sequences of the genus classification level were subjected to numerous sequence alignments to build a phylogenetic tree and image based on the OTU classification (Supplementary Figures 2A,B). The results of Anosim (analysis of similarities) also support these speculations (Figures 3C,F).
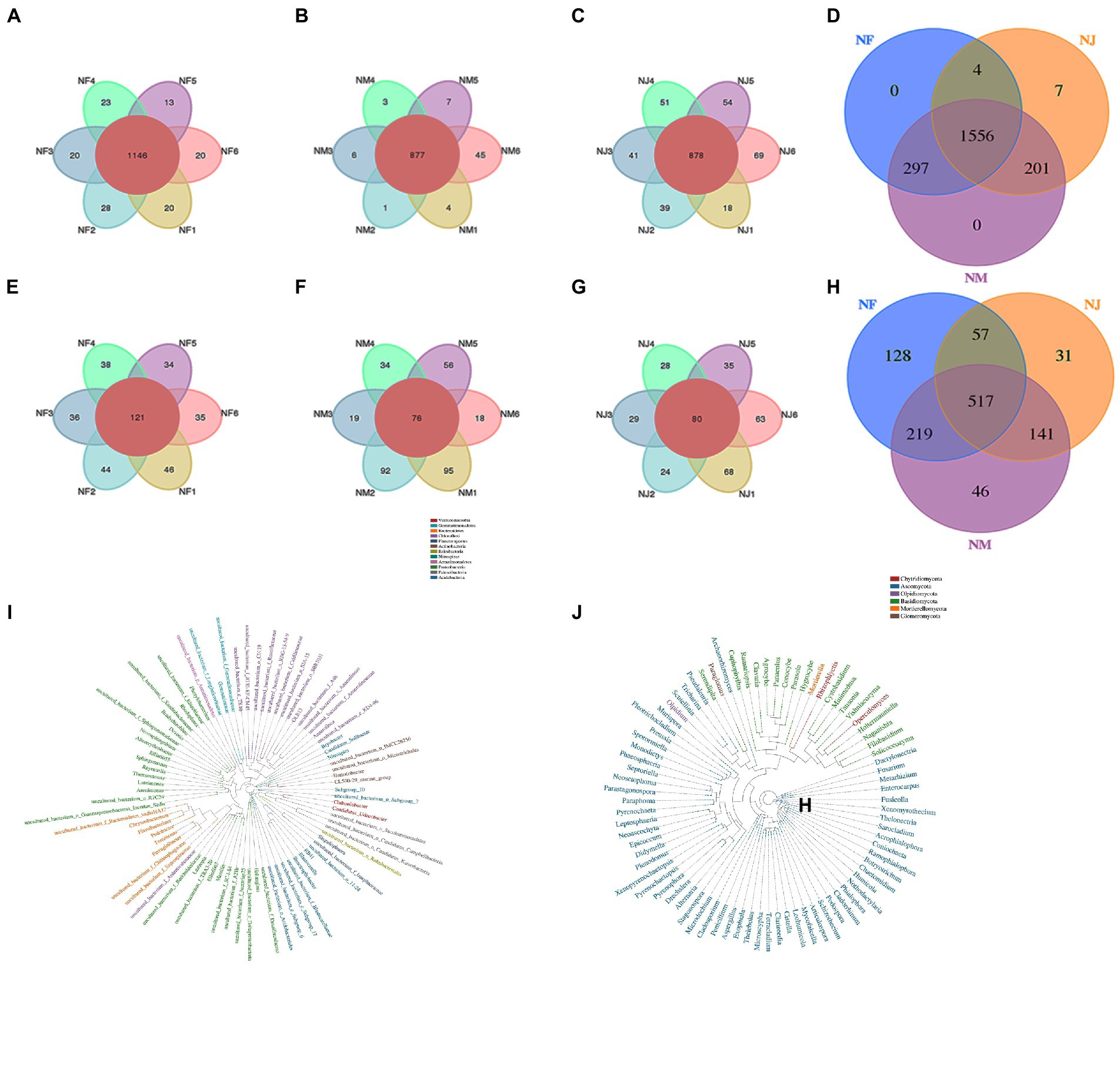
Figure 2. Analysis of Venn diagrams. (A, C–E) Venn diagrams for bacterial OTUs compositions in different groups. (B, F–H) Venn diagrams for fungal OTUs compositions in different groups.
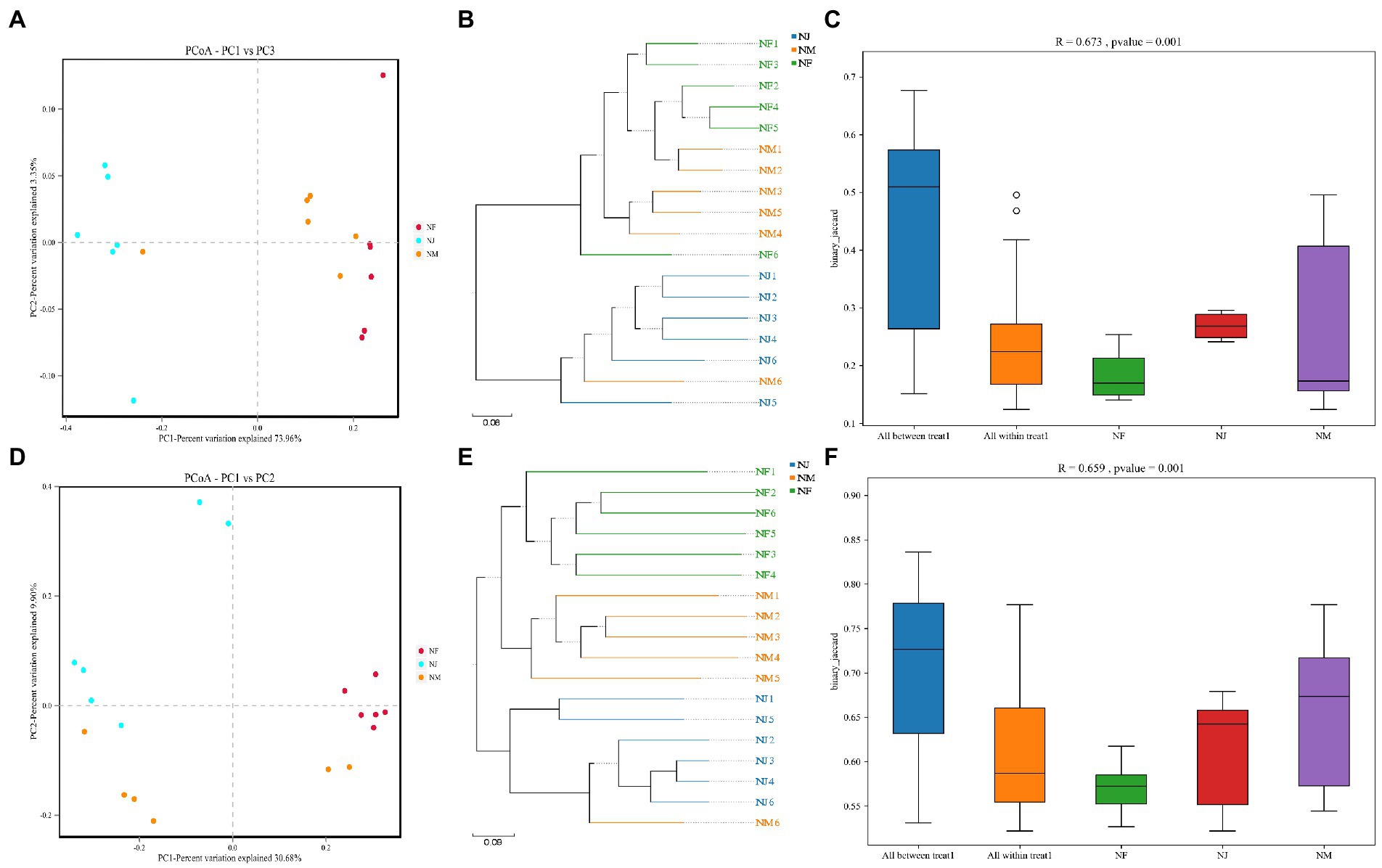
Figure 3. Similarity analysis of soil microbial structures in all groups. (A–C) represented the bacterial similarity between groups or individuals using PCoA, UPGMA, and Anosim box Figures. (D–F) represented the fungal similarity between groups or individuals using PCoA, UPGMA, and Anosim box Figures.
Changes in the diversity of bacterial and fungal communities under the influence of yaks’ excrement contamination
The relationship between soil bacterial and fungal alpha diversity and the yaks’ excrement contamination was explored in the three habitats’ soil layers. The ACE indices and Chao1 indices (reflected species richness), Shannon indices and Simpson indices (species diversity) of these three different soil communities were significantly different. Our results showed that the bacterial and fungal alpha diversity in NJ was lower than other soil properties. Specifically, the average bacterial ACE, Chao1, Shannon, and Simpson indices were 1626.326, 1666.811, 9.170, 0.996 in NF, 1778.276, 1818.304, 9.007, 0.994 in NM, whereas 1458.801, 1512.533, 8.602, 0.994 in NJ (Figures 1C–F). Results indicated that the soil bacterial microbiome in NF had the highest species diversity (p < 0.05 or p < 0.01). Unlike bacteria, NF soil had the highest fungal species richness and diversity among the three groups, followed by NM, while soil microbial alpha diversity in NJ comes last (Figures 1G–H).
The PCoA plots and UPGMA tree revealed the patterns of bacterial and fungal microbiota β diversity. Unsupervised clustering using PCoA based on UniFrac distance matrices suggested the variations in our dataset due to excrement contamination, with the bacterial and the fungal microbiota clustering independently from different groups (Figures 3A,D). Soil microbiome was sequestered into three branches based on shifts in evolutionary information per sample, indicating a difference in soil microbiome induced by excrement contamination (Figures 3B,E).
Significant alterations bacterial-microbiota composition and dominant species under the influence of yaks’ excrement contamination
Mapping the top ten most abundant bacterial taxa onto the species distribution histogram at different taxonomical levels (phylum, class, and genus) revealed some clustering of community structure by excrement contamination (Figures 4A–C). Regardless of excrement contamination, Proteobacteria was the dominating phylum, accounting for more than 29.81 percent of all sequences. In addition, Acidobacteria was the most abundant phylum in NF (29.41%) and NM (25.29%), whereas NJ had the dominant phylum of Bacteroidetes (20.12%). The top 3 most abundant bacterial classes in NF were Alphaproteobacteria (12.96%), Gammaproteobacteria (13.22%), and Subgroup_6 (10.74%), while Alphaproteobacteria (15.83, 14.23%), Gammaproteobacteria (14.84, 14.25%), and Bacteroidia (10.26, 19.31%) dominated the NM and NJ. At the genus level, The top 5 most abundant bacterial genera identified in NF were uncultured_bacterium_c_Subgroup_6 (10.16%), Candidatus_Udaeobacter (7.02%), uncultured_bacterium_f_Gemmatimonadaceae (3.06%), RB41 (3.92%), and uncultured_bacterium_f_SC-I-84 (2.32%); uncultured_bacterium_c_Subgroup_6 (8.67%), uncultured_bacterium_f_Blastocatellaceae (5.18%), Sphingomonas (5.77%), Candidatus_Udaeobacter (3.01%), and uncultured_bacterium_f_Burkholderiaceae (2.92%) in NM, while uncultured_bacterium_c_Subgroup_6 (6.25%), uncultured_bacterium_f_Blastocatellaceae (7.71%), uncultured_bacterium_f_Burkholderiaceae (3.11%), Chryseobacterium (4.35%), and uncultured_bacterium_f_A4b (2.66%) in NJ.
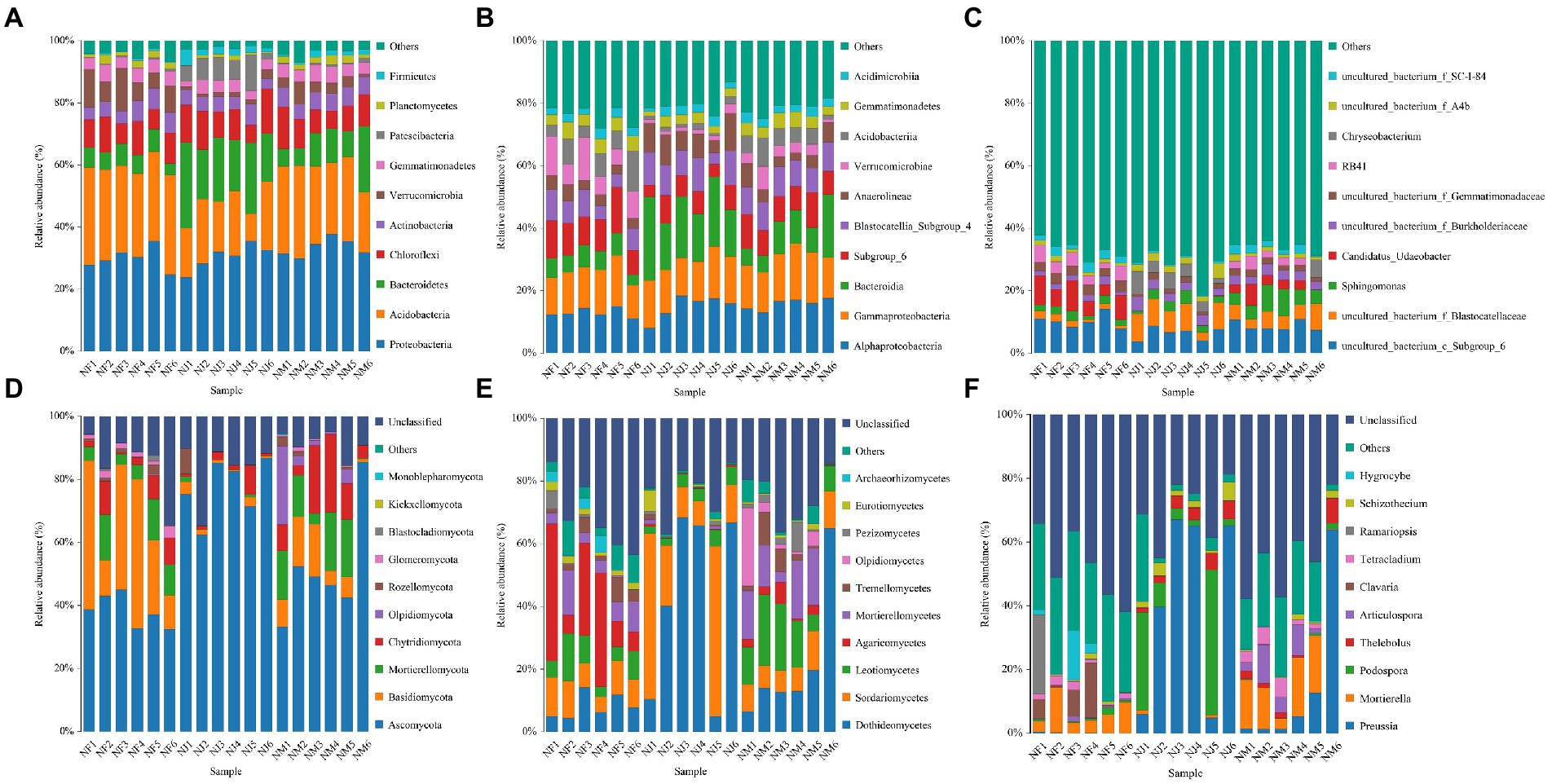
Figure 4. Histogram of the relative abundance distribution of species at different taxonomic levels. (A–C) Represented bacterial abundance at phylum, class, and genus levels, respectively. (D–F) Represented fungal abundance at phylum, class, and genus levels, respectively.
We used Metastst analysis to quantify the association of excrement contamination induced by intensive grazing with bacterial-microbiota composition. According to mapping results, the distribution of significantly altered clusters on the heatmap supported our hypothesis. At the phylum level, several taxonomic classifications in soil, Acidobacteria, Latescibacteria, Verrucomicrobia, Rokubacteria, Entotheonellaeota, and Gemmatimonadetes, significantly decreased with the increasing degree of excrement contamination (Figure 5A). In contrast, Bacteroidetes, Deinococcus-Thermus, Patescibacteria, Fibrobacteres, Actinobacteria, and Chloroflexi had the opposite trend (p < 0.05 or p < 0.01). Among the 14 classes affected by excretion contamination, the average relative abundance of 8 taxa (Figure 5B), Latescibacteria, Holophagae, Verrucomicrobiae, Dehalococcoidia, Phycisphaerae, Acidobacteriia, Gemmatimonadetes, and Thermoleophilia were lower in NM and NJ than in NF. At the same time, Chloroflexia, Coriobacteriia, Erysipelotrichia, Clostridia, Actinobacteria, and Gammaproteobacteria increased with the increasing degree of contamination (p < 0.05 or p < 0.01). Metastatic analysis confirmed the dominance of genera Ochrobactrum, Nordella, Rhizobacter, Candidatus_Xiphinematobacter, Mesorhizobium, Rhodoplanes, Gaiella, Chthoniobacter, Dysgonomonas, and Chryseolinea in the NF microbiota (Figure 5C). Romboutsia, Nannocystis, Terrimicrobium, Caenimonas, Propionivibrio, Barnesiella, Faecalibacterium, Alistipes, Desulfovibrio, Butyricimonas, and Parabacteroides were over-expressed (p < 0.05 or p < 0.01) among the 22 substantially shifted genus-level OTUs in NJ. NM and NJ could also be classified from each other, although the variations in relative abundance were not as pronounced as in the case NF versus NJ.
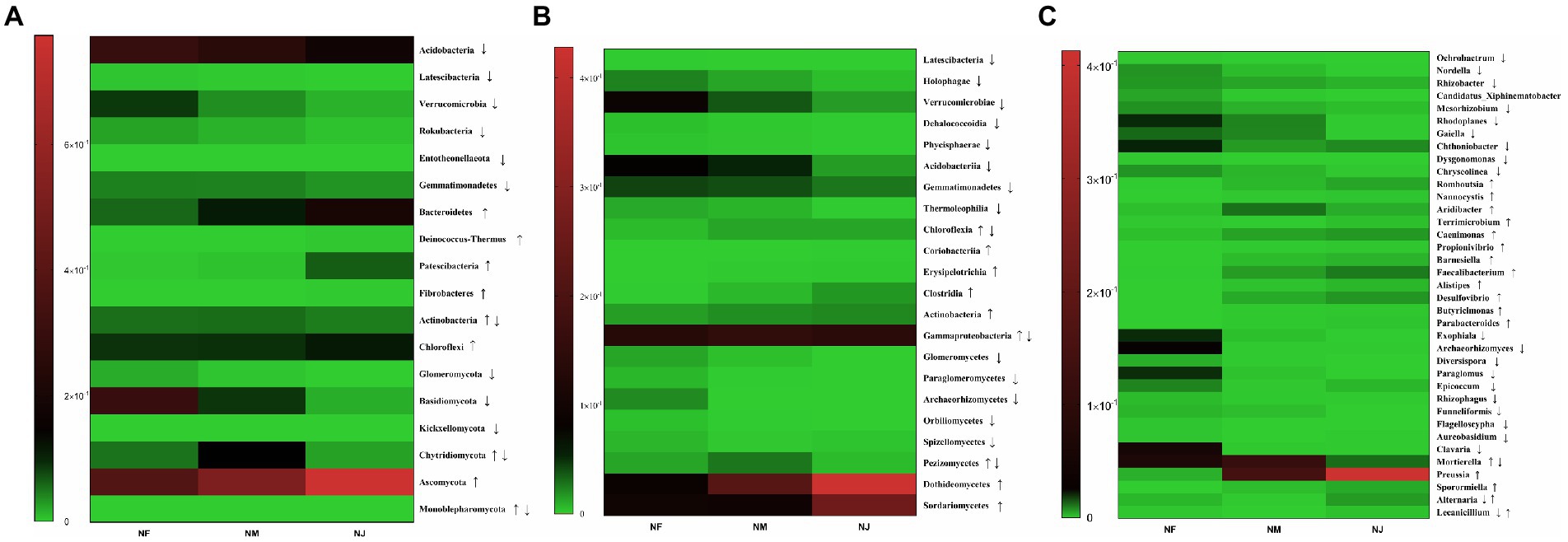
Figure 5. Heatmap of significant alterations in soil microorganisms (bacteria and fungi) at multiple classification levels as a function of excrement contamination. (A) Phylum level; (B) class level; (C) genus level. ↑/↓ represented that the abundance of taxa continued to significantly increase/decrease in NF, NM, NJ groups; ↑↓ represented that the abundance of taxa significantly increased firstly and decreased later in NF, NM, NJ groups; ↓↑ represented that the abundance of taxa significantly decreased firstly and increased later in NF, NM, NJ groups.
Significant alterations in fungal-microbiota composition and dominant species under the influence of yaks’ excrement contamination
The phylum Ascomycota accounts for the overwhelming majority of the dominant soil microbiome in fungal communities, accounting for more than 38.19 percent of all stands. There were also three dominant phyla: Basidiomycota (NF: 30.25%, NM: 8.84%, NJ: 1.72%), Mortierellomycota (NF: 8.15%, NM: 11.44%, NJ: 0.46%), Chytridiomycota (NF: 5.18%, NM: 12.23%, NJ: 2.58%) within the fungal communities (Figure 4D). At the class level, Sordariomycetes (9.45%) and Agaricomycetes (21.86%) fungi were the most predominant in the NF, while Dothideomycetes (21.92%) and Leotiomycetes (13.91%) were found in the NM. Meanwhile, the class Dothideomycetes (43.18%) was the most abundant fungi in the NJ, followed by the Sordariomycetes (25.64%; Figure 4E). Following the genus-level classification results, the most abundant genera were Mortierella (6.65%), Clavaria (5.37%), Ramariopsis (4.35%), and Hygrocybe (3.41%) in the NF, Preussia (14.42%), Mortierella (11.36%), Thelebolus (2.38%), Articulospora (5.10%), and Tetracladium (2.96%) in the NM, while Preussia (41.74%), Podospora (14.96%), Thelebolus (3.75%), and Schizothecium (2.72%) were identified as the predominant in the NJ (Figure 4F).
The soil fungal-microbiota composition and dominant species assembly were then estimated for the three samples. According to the heatmap, the phyla Glomeromycota, Basidiomycota, and Kickxellomycota are the most abundant in NF soil microbiome (Figure 5A), Chytridiomycota, Monoblepharomycota were dominated in NM, while NJ had a large proportion of Ascomycota (p < 0.05 or p < 0.01). The class-level findings revealed a continuing enrichment in the relative abundances of the phyla Dothideomycetes and Sordariomycetes with increasing degrees of excrement contamination (Figure 5B), whereas Glomeromycetes, Paraglomeromycetes, Archaeorhizomycetes, Orbiliomycetes, Spizellomycetes, Pezizomycetes had the opposite trend (p < 0.05 or p < 0.01). Exophiala, Archaeorhizomyces, Diversispora, Paraglomus, Epicoccum, Rhizophagus, Funneliformis, Flagelloscypha, Aureobasidium, and Clavaria were the most prevalent genera in NF group. On the other hand, Preussia, Sporormiella, Alternaria, and Lecanicillium were relatively abundant in NJ group. Furthermore, Mortierella remained the predominant population in NM group (Figure 5C).
Discussion
The plateau of Aba Autonomous Prefecture, China, only inhabits the traditional Tibetan people and a few Han Chinese due to the unique geographical and survival conditions. The abundant pasture resources allow these locals to herd grazing for their livelihood. Increasing evidence suggests that soil microbiome promotes ecosystem multi-functionality and regulates resistance to land-surface’s common disturbance practices. However, interference from fecal deposition excrement caused by extensive grazing may disrupt this context dependence throughout complex and harmoniously balanced ecosystems (Jing et al., 2015; Delgado-Baquerizo et al., 2016). The pressures associated with excrement contamination might deteriorate soil micro-ecology with time, affecting herbage yields substantially (Shah et al., 2017). So, the research into the mechanistic understanding of the connections between interfering ecological processes and variations in plateau soil microbial diversity is very important. This is the first in-depth study of bacterial and fungal taxonomic changes in excrement contamination soils in the plateau. According to our findings, excrement contamination significantly reduces soil bacterial and fungal diversity and richness, indicating that environmental filtering substantially impacts microbial community formation. We noticed significant differences in soil microbiome composition across groups, with NF soil communities separating the most from NM and NJ soil communities.
Grazing activities reduce soil microbial activity in three main ways: (1) soil compaction decreases aggregate stability index, nutrient availability, and porosity, which leads to the reduction of soil health (Shah et al., 2017), (2) it lowers plant performance via reduced root growth (Lai and Kumar, 2020), (3) excremental contamination (Wegrzyn et al., 2018). Based on this data, we hypothesized that excrement contamination, a kind of land-surface disturbance, might disrupt a complex and harmoniously balanced ecosystem in the plateau, affecting both the quality and diversity of below-ground microorganisms. Current data may support this hypothesis since NF group had significantly higher alpha diversity than both NM and NJ, with NJ being the worst of the three. Previous studies have shown that excrement contamination negatively impacts soil characteristics and microbial communities, suggesting strong soil microbiota response-specific patterns under the influence of land-surface disturbance practices (Dombos, 2001). Furthermore, the scatter plot represents the cluster of similarities and branches in the evolutionary tree, sequestering into three clusters with a higher degree of excrement contamination, indicating that this type of contamination drove the microbial composition and structural segregation in plateau soil.
Most soil microbiome studies have focused on a single counterpart (bacteria or fungi) without considering the multi-kingdom state of the soil ecosystem. Furthermore, many previous studies that have identified soil microbial variations focused on natural conditions such as stages of soil microecology development and plant community succession (Da et al., 2009; Rime et al., 2015). However, few studies have investigated soil microbial responses to land-surface disturbance practices. Here, we demonstrated clear alterations in bacteria and fungi communities driven by animal excrement contamination in the plateau. Identifying these processes aided us in understanding the soil microbial system resilience and contributed to the increasing evidence that soil microbial communities can change dramatically in response to soil ecological processes (Fierer et al., 2007).
As previously mentioned, the consensus is that the loss of functional microorganisms’ biodiversity residing below-ground reduces the functions of terrestrial ecosystems and destroys their long-term stability (Bardgett and van der Putten, 2014). This knowledge conveyed that the reduction in potential functional microbiota exacerbates the terrestrial ecological imbalance or that soil property harmed by land-surface disturbance practices causes a decrease in beneficial and functional microorganisms. The total of 12 bacterial phyla showed a significant change in relative abundance among three groups in our data, supported by previous work. The bacterial phyla Acidobacteria and Latescibacteria had previously been shown to be more abundant in grassland soils than in grazing soil, which the trend might associate with their functional roles in ecosystems [e.g., Acidobacteria had been proven to have a beneficial role in recovering soils to improve plant growth and nutrient cycling after surface disturbance (Huang et al., 2015); and Latescibacteria mediated the turnover of niche specialization in organic carbon that reaches deeper anoxic/microoxic habitats (Youssef et al., 2015)]. What data suggests differs from the multimetal resistance pathway in phylum Entotheonellaeota, phylum Verrucomicrobia including class Verrucomicrobiae, as metal-resistant bacteria could secrete EPS-proteins and EPS-polysaccharides to detoxify Cr, were associated with C/N cycling processes (Zhang et al., 2021). Heavy metals (including As, Cr, Cd, Hg, Cu, etc., in polluted soils) are ubiquity in various environmental mediums and attract attention in China and around the world for their toxicity, bioaccumulation, and carcinogenicity (Stefanowicz et al., 2008; Zhao et al., 2015). The interaction between microbiota and metal became important in contaminated field soils due to the various functions of soil microbes’ capacity to produce metal carbonate precipitation along with nitrogen cycling (Satapute et al., 2019). The significantly decreased abundance of microorganisms related to metal detoxification in our data might be attributed to the decreasing self-repairing ability of the soil ecosystem with the increasing degree of excrement contamination. So, the current conclusion is consistent with previously published results (Abdalla et al., 2018). Additionally, phylum Chloroflexi and class Chloroflexia were frequently dominant in methanogenic reactors, and they were also more abundant in NM and NJ than NF (Bovio et al., 2019). Similar to earlier studies, our findings revealed a negative response of soil pathogens to animal excrement contamination (Gagliardi and Karns, 2000). The decline in soil microbial diversity and beneficial microorganisms shifted dominance to soil pathogen-associated phylotypes, such as Coriobacteriia (Gardiner et al., 2015), Erysipelotrichia (Singh et al., 2020), Clostridia and Actinobacteria (Miao and Davies, 2010; Moore and Lacey, 2019). However, grazing was not the sole reason for increasing class-level pathogenic bacterium. Our data at the genus level further support animal excrement contamination that is further connected with significantly increased pathogens in the soil. Abundant Desulfovibrio is the inducer in intestinal pathogenesis, which can induce intestinal epithelial cells abnormal proliferation and metabolism and even impair intestinal barrier function (Gill et al., 2006). The relative abundance of Romboutsia, Butyricimonas, and Parabacteroides in soil significantly enriched with the increasing degree of excrement contamination, which might be dangerous information because these three genera pose a big threat to host health (Ricaboni et al., 2016; Kierzkowska et al., 2018; De Donder et al., 2020). More than 90% of the yaks in the world live in the plateaus of China (> 3,000 m), facing harsh environmental conditions, especially at calve stage (Wang et al., 2018). According to statistical research, diarrheal disease epidemic in calves in April (Li et al., 2018). The pathogenic bacteria in the host intestines can infiltrate the soil, even underground water bodies. As a result, excrement is a significant vector for disease transportation and has even become a serious public health issue (Lipp et al., 2001). Hence, we hypothesized a linkage between the significant abundance of soil pathogens and the susceptibility to intestinal diseases in calves’ yak. While suggesting that such soil pathogens impact the etiology of intestinal diseases in the host that needs further study.
The specimen collection in the current study avoided other surface disturbance activities, e.g., cultivation, man-made sabotage, ensuring that animal excrement was the main driver for altering the bacterial and fungal communities in this area. A common view is that fungi utilize the Carbon from the soil more effectively than other microbial groups. The main information to emerge is that the fungal community is highly sensitive to changes in environmental condition (Valentin-Vargas et al., 2014). Basidiomycota represents the dominant phylum of the fungal kingdom, which plays a significant role in medicinal properties, ecosystem functioning, and carbon recycling (Schmidt-Dannert, 2016). Ecologists began exploring soil fungi’s role in soil nutrient dynamics in the 1980s, revealing the function of microorganisms with an important role in stimulating nutrient mineralization and decomposition processes, e.g., Chytridiomycota and Kickxellomycota. The latter is positively associated with soil carbon, phosphorous, nitrogen, and potassium (Tarin et al., 2021). The significant decrease in the above-mentioned functional fungi in NM and NJ was of great concern since it conveyed that animal excrement contamination in the plateau gradually destroyed the soil ecosystem’s nutrient cycling.
Furthermore, previous evidence demonstrated that intensive grazing could lower biomass and decomposition rate of plant roots, which reduce the soil Nitrogen (N) nutrients (NO3- and NH4-), subsequently reducing soil total N (He et al., 2012). Animal excretes increased N in the soil. However, the deposition rate is minor (Macdiarmid and Watkin, 1972; BALL et al., 1979). Therefore, the loss of nitrogen driven by grazing was more significant than nitrogen deposition in manure, ultimately leading to an overall reduction of soil N. Besides the impacts of fungi in soil nutrient dynamics, several beneficial microorganisms, i.e., phylum Glomeromycota along with classes Glomeromycetes and Paraglomeromycetes, and genera Diversispora and Paraglomus could be used in rhizostabilisation strategies to reduce soil waste heaps and extreme metal contamination, which pollution is a permanent threat for nearby surroundings (Sanchez-Castro et al., 2017). The genus Epicoccum has produced multi metabolites with potential biotechnological applications, e.g., anticancer, antioxidant, and antimicrobial compounds. It is mainly known as biocontrol agent against phytopathogens (Braga et al., 2018). Arbuscular mycorrhizal fungi, obligatory plant symbionts, e.g., Rhizophagus and Funneliformis, have a beneficial impact on plants in contaminated conditions (Malicka et al., 2020); while bacterial genera Rhodoplanes and Gaiella are predominant in the rhizospheric soils, accelerates the degradation of herbicide (atrazine) residues (Aguiar et al., 2020). The relationships of pathogens-pollutants-microbes might be described as a disease triangle, and soil pollution indirectly or directly interacts with pathogens in soil through containing impacts on the host plant (Dean et al., 2012). It was also demonstrated that amount of soil-borne pathogens differed significantly between NM and NJ groups, with the highest abundance in NJ group, i.e., phylum Monoblepharomycota classes Dothideomycetes and Sordariomycetes, and genera Alternaria and Lecanicillium, among which vegetable foods infected by Alternaria conveyed host-specific toxins to the human diet (Van Rooij et al., 2015; Weber et al., 2015; Pinto and Patriarca, 2017; Haridas et al., 2020; Zhou et al., 2020). When emphasizing on soil community variability, the opposite had to include soil system resilience. It is a resistance/tolerance of soil immune responses against biotic infections such as microbiome-mediated degradation of pollutants (Raaijmakers and Mazzola, 2016). Soil pollution, as an invasive element wreaking on the soil ecology, may be mitigated by the soil microbiome (Wang and Li, 2019). However, in the current study, the strong resilience of the soil microbial community did not conceal the detrimental impacts of surface disturbance, suggesting that the destructive effect of excremental contamination exceeds resilience in plateau soil (Figure 6). With many selection pressures imposed by animal excrement contamination and soil-borne diseases, we predicted that a limited soil ecological niche with more pathogens and a lower functioning microbiome would emerge (Wood et al., 2016).
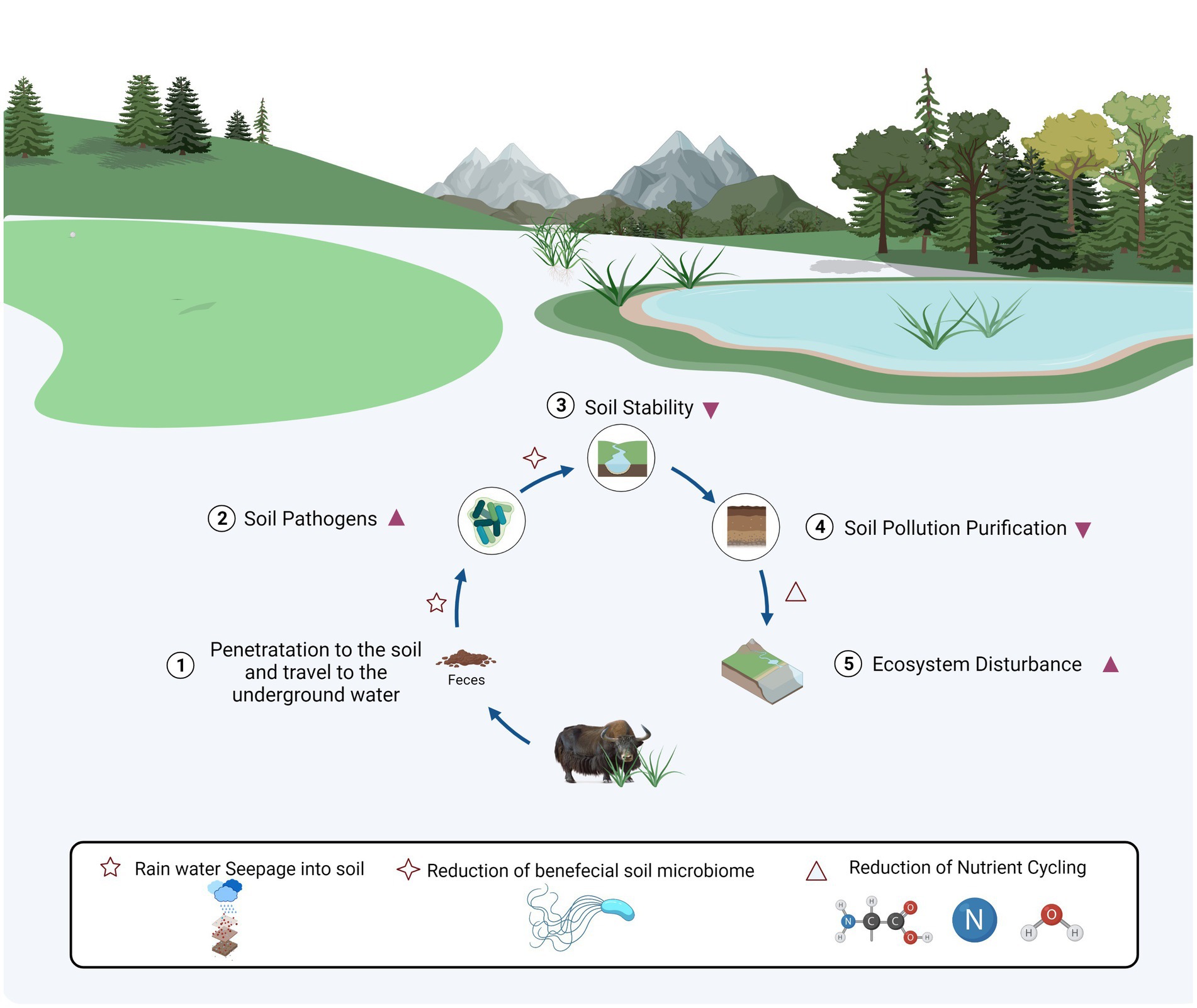
Figure 6. The mechanism insight of soil pathogens due to excrement contamination at plateau.
Conclusion
Our study first demonstrated the dynamic dispersion of soil microbiomes, which comprised bacterial and fungal communities in soils disturbed by land-surface practices (excrement contamination induced by grazing) on the plateau. Specifically, our findings that excrement contamination reduced soil diversity and richness of bacterial and fungal communities indicated that land-surface disturbance practices had a major impact on microbial community formation. Furthermore, a continuous decrease in the relative abundances of microorganisms associated with ecological nutrient cycling, soil pollution purification, and root soil stability continue to decline with the increasing degree of excrement contamination, whereas soil pathogens had the opposite trend in grazing. The alterations in the dominant species and composition of the soil microbiome supported the concept that animal excrement contamination induced a low diversity and richness with more pathogens and lower functioning microbiome in the soil microbiome. Such colonization and succession of the soil excrement contamination induce microbiome may offer an important potential management basis for microbiome-based soil health in the plateau. However, the limitation of current research is the lack of exploration and validation of the effects of excrement contamination on soil functionality in the plateau, and further experiments are still needed.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA772904.
Author contributions
ZS, YW, MA, KL, and SL provided the research idea. ZT and XC contributed reagents, materials, and analysis tools. YW wrote the manuscript. MK revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by Start-up fund of Nanjing Agricultural University (804131), Start-up Fund for Distinguished Scholars of Nanjing Agricultural University (80900219), Tibet Regional Science and Technology Collaborative Innovation Project (QYXTZX-NQ2021-01) and The Central Government supports the special fund project for the reform and development of local universities (KY2022ZY-01).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1016852/full#supplementary-material
References
Abdalla, M., Hastings, A., Chadwick, D. R., Jones, D. L., Evans, C. D., Jones, M. B., et al. (2018). Critical review of the impacts of grazing intensity on soil organic carbon storage and other soil quality indicators in extensively managed grasslands. Agric. Ecosyst. Environ. 253, 62–81. doi: 10.1016/j.agee.2017.10.023
Aguiar, L. M., Souza, M. F., de Laia, M. L., de Oliveira, M. J., Da, C. M., Goncalves, J. F., et al. (2020). Metagenomic analysis reveals mechanisms of atrazine biodegradation promoted by tree species. Environ. Pollut. 267:115636. doi: 10.1016/j.envpol.2020.115636
Ball, R., Keeney, D. R., Theobald, P. W., and Nes, P. (1979). Nitrogen-balance in urine-affected areas of a New-Zealand pasture. Agron. J. 71, 309–314. doi: 10.2134/agronj1979.00021962007100020022x
Bardgett, R. D., and van der Putten, W. H. (2014). Below-ground biodiversity and ecosystem functioning. Nature 515, 505–511. doi: 10.1038/nature13855
Bovio, P., Cabezas, A., and Etchebehere, C. (2019). Preliminary analysis of chloroflexi populations in full-scale uasb methanogenic reactors. J. Appl. Microbiol. 126, 667–683. doi: 10.1111/jam.14115
Braga, R. M., Padilla, G., and Araujo, W. L. (2018). The biotechnological potential of epicoccum spp.: diversity of secondary metabolites. Crit. Rev. Microbiol. 44, 759–778. doi: 10.1080/1040841X.2018.1514364
Cardinale, B. J. (2012). Biodiversity loss and its impact on humanity (vol 486, pg 59, 2012). Nature 489:326. doi: 10.1038/nature11373
Chase, J. M. (2010). Stochastic community assembly causes higher biodiversity in more productive environments. Science 328, 1388–1391. doi: 10.1126/science.1187820
Da, C. J. E., Marsh, T. L., Tiedje, J. M., and Moreira, F. M. (2009). Changes in land use alter the structure of bacterial communities in western amazon soils. ISME J. 3, 1004–1011. doi: 10.1038/ismej.2009.47
de Boer, W., Folman, L. B., Summerbell, R. C., and Boddy, L. (2005). Living in a fungal world: impact of fungi on soil bacterial niche development. FEMS Microbiol. Rev. 29, 795–811. doi: 10.1016/j.femsre.2004.11.005
De Donder, L., Uyttebroek, O., Van Cleven, S., and Berrevoet, F. (2020). A subcutaneous infection mimicking necrotizing fasciitis due to butyricimonas virosa. Acta Chir. Belg. 120, 425–428. doi: 10.1080/00015458.2019.1631612
Dean, R., Van Kan, J. A., Pretorius, Z. A., Hammond-Kosack, K. E., Di Pietro, A., Spanu, P. D., et al. (2012). The top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 13, 414–430. doi: 10.1111/j.1364-3703.2011.00783.x
Delgado-Baquerizo, M., Maestre, F. T., Reich, P. B., Jeffries, T. C., Gaitan, J. J., Encinar, D., et al. (2016). Microbial diversity drives multifunctionality in terrestrial ecosystems. Nat. Commun. 7:10541. doi: 10.1038/ncomms10541
Di, H. J., Cameron, K. C., Milne, J., Drewry, J. J., Smith, N. P., Hendry, T., et al. (2001). A mechanical hoof for simulating animal treading under controlled conditions. New Zeal. J. Agr. Res. 44, 111–116. doi: 10.1080/00288233.2001.9513465
Dombos, M. (2001). Collembola of loess grassland: effects of grazing and landscape on community composition. Soil Biol. Biochem. 33, 2037–2045. doi: 10.1016/S0038-0717(01)00125-0
Fierer, N., Bradford, M. A., and Jackson, R. B. (2007). Toward an ecological classification of soil bacteria. Ecology 88, 1354–1364. doi: 10.1890/05-1839
Gagliardi, J. V., and Karns, J. S. (2000). Leaching of escherichia coli o157:h7 in diverse soils under various agricultural management practices. Appl. Environ. Microbiol. 66, 877–883. doi: 10.1128/AEM.66.3.877-883.2000
Gardiner, B. J., Tai, A. Y., Kotsanas, D., Francis, M. J., Roberts, S. A., Ballard, S. A., et al. (2015). Clinical and microbiological characteristics of eggerthella lenta bacteremia. J. Clin. Microbiol. 53, 626–635. doi: 10.1128/JCM.02926-14
Geisen, S., Tveit, A. T., Clark, I. M., Richter, A., Svenning, M. M., Bonkowski, M., et al. (2015). Metatranscriptomic census of active protists in soils. ISME J. 9, 2178–2190. doi: 10.1038/ismej.2015.30
Gill, S. R., Pop, M., Deboy, R. T., Eckburg, P. B., Turnbaugh, P. J., Samuel, B. S., et al. (2006). Metagenomic analysis of the human distal gut microbiome. Science 312, 1355–1359. doi: 10.1126/science.1124234
Guo, B., Liu, C., Gibson, C., and Frigon, D. (2019). Wastewater microbial community structure and functional traits change over short timescales. Sci. Total Environ. 662, 779–785. doi: 10.1016/j.scitotenv.2019.01.207
Haridas, S., Albert, R., Binder, M., Bloem, J., LaButti, K., Salamov, A., et al. (2020). 101 dothideomycetes genomes: a test case for predicting lifestyles and emergence of pathogens. Stud. Mycol. 96, 141–153. doi: 10.1016/j.simyco.2020.01.003
Harwood, V. J., Staley, C., Badgley, B. D., Borges, K., and Korajkic, A. (2014). Microbial source tracking markers for detection of fecal contamination in environmental waters: relationships between pathogens and human health outcomes. FEMS Microbiol. Rev. 38, 1–40. doi: 10.1111/1574-6976.12031
He, W., Shen, Y., and Cornelissen, J. H. C. (2012). Soil nutrient patchiness and plant genotypes interact on the production potential and decomposition of root and shoot litter: evidence from short-term laboratory experiments with triticum aestivum. Plant Soil 353, 145–154. doi: 10.1007/s11104-011-1018-1
Hu, J., Nie, Y., Chen, J., Zhang, Y., Wang, Z., Fan, Q., et al. (2016). Gradual changes of gut microbiota in weaned miniature piglets. Front. Microbiol. 7:1727. doi: 10.3389/fmicb.2016.01727
Huang, X., Liu, L., Wen, T., Zhu, R., Zhang, J., and Cai, Z. (2015). Illumina miseq investigations on the changes of microbial community in the fusarium oxysporum f.sp cubense infected soil during and after reductive soil disinfestation. Microbiol. Res. 181, 33–42. doi: 10.1016/j.micres.2015.08.004
Imhoff, S., Da Silva, A. P., and Tormena, C. A. (2000). Applications of the resistance curve in the control of the physical quality of soils under grass. Pesqui. Agropecu. Bras. 35, 1493–1500. doi: 10.1590/S0100-204X2000000700025
Jing, X., Sanders, N. J., Shi, Y., Chu, H., Classen, A. T., Zhao, K., et al. (2015). The links between ecosystem multifunctionality and above- and below-ground biodiversity are mediated by climate. Nat. Commun. 6:8159. doi: 10.1038/ncomms9159
Kardol, P., and Wardle, D. A. (2010). How understanding aboveground-belowground linkages can assist restoration ecology. Trends Ecol. Evol. 25, 670–679. doi: 10.1016/j.tree.2010.09.001
Kierzkowska, M., Majewska, A., Szymanek-Majchrzak, K., Sawicka-Grzelak, A., Mlynarczyk, A., and Mlynarczyk, G. (2018). In vitro effect of clindamycin against bacteroides and parabacteroides isolates in Poland. J Glob Antimicrob Resist. 13, 49–52. doi: 10.1016/j.jgar.2017.11.001
Lai, L., and Kumar, S. (2020). A global meta-analysis of livestock grazing impacts on soil properties. PLoS One 15:e236638:e0236638. doi: 10.1371/journal.pone.0236638
Larigauderie, A., Prieur-Richard, A. H., Mace, G. M., Lonsdale, M., Mooney, H. A., Brussaard, L., et al. (2012). Biodiversity and ecosystem services science for a sustainable planet: the diversitas vision for 2012-20. Curr. Opin. Environ. Sustain. 4, 101–105. doi: 10.1016/j.cosust.2012.01.007
Li, K., Mehmood, K., Zhang, H., Jiang, X., Shahzad, M., Dong, X., et al. (2018). Characterization of fungus microbial diversity in healthy and diarrheal yaks in gannan region of Tibet autonomous prefecture. Acta Trop. 182, 14–26. doi: 10.1016/j.actatropica.2018.02.017
Li, A., Wang, Y., Li, Z., Qamar, H., Mehmood, K., Zhang, L., et al. (2019). Probiotics isolated from yaks improves the growth performance, antioxidant activity, and cytokines related to immunity and inflammation in mice. Microb. Cell Factories 18:112. doi: 10.1186/s12934-019-1161-6
Li, L., and Zhao, X. (2015). Comparative analyses of fecal microbiota in tibetan and chinese han living at low or high altitude by barcoded 454 pyrosequencing. Sci. Rep. 5:14682. doi: 10.1038/srep14682
Lipp, E. K., Farrah, S. A., and Rose, J. B. (2001). Assessment and impact of microbial fecal pollution and human enteric pathogens in a coastal community. Mar. Pollut. Bull. 42, 286–293. doi: 10.1016/S0025-326X(00)00152-1
Macdiarmid, B. N., and Watkin, B. R. (1972). Cattle dung patch. 2. Effect of a dung patch on chemical status of soil, and ammonia nitrogen losses from patch. J. Br. Grassl. Soc. 27, 43–47. doi: 10.1111/j.1365-2494.1972.tb00684.x
Malicka, M., Magurno, F., Piotrowska-Seget, Z., and Chmura, D. (2020). Arbuscular mycorrhizal and microbial profiles of an aged phenol-polynuclear aromatic hydrocarbon-contaminated soil. Ecotoxicol. Environ. Saf. 192:110299. doi: 10.1016/j.ecoenv.2020.110299
Miao, V., and Davies, J. (2010). Actinobacteria: the good, the bad, and the ugly. Antonie Van Leeuwenhoek 98, 143–150. doi: 10.1007/s10482-010-9440-6
Mille-Lindblom, C., Fischer, H., and Tranvik, L. J. (2006). Antagonism between bacteria and fungi: substrate competition and a possible tradeoff between fungal growth and tolerance towards bacteria. Oikos 113, 233–242. doi: 10.1111/j.2006.0030-1299.14337.x
Moore, R. J., and Lacey, J. A. (2019). Genomics of the pathogenic clostridia. Microbiol. Spectr. 7. doi: 10.1128/microbiolspec.GPP3-0033-2018
Pinto, V. E., and Patriarca, A. (2017). Alternaria species and their associated mycotoxins. Methods Mol. Biol. 1542, 13–32. doi: 10.1007/978-1-4939-6707-0_2
Raaijmakers, J. M., and Mazzola, M. (2016). Ecology. Soil immune responses. Science 352, 1392–1393. doi: 10.1126/science.aaf3252
Ricaboni, D., Mailhe, M., Khelaifia, S., Raoult, D., and Million, M. (2016). Romboutsia timonensis, a new species isolated from human gut. New Microbes New Infect. 12, 6–7. doi: 10.1016/j.nmni.2016.04.001
Rime, T., Hartmann, M., Brunner, I., Widmer, F., Zeyer, J., and Frey, B. (2015). Vertical distribution of the soil microbiota along a successional gradient in a glacier forefield. Mol. Ecol. 24, 1091–1108. doi: 10.1111/mec.13051
Rosindell, J., Hubbell, S. P., and Etienne, R. S. (2011). The unified neutral theory of biodiversity and biogeography at age ten. Trends Ecol. Evol. 26, 340–348. doi: 10.1016/j.tree.2011.03.024
Ruiz, S. A., McKay, F. D., Boghi, A., Williams, K. A., Duncan, S. J., Scotson, C. P., et al. (2020). Image-based quantification of soil microbial dead zones induced by nitrogen fertilization. Sci. Total Environ. 727:138197. doi: 10.1016/j.scitotenv.2020.138197
Sanchez-Castro, I., Gianinazzi-Pearson, V., Cleyet-Marel, J. C., Baudoin, E., and van Tuinen, D. (2017). Glomeromycota communities survive extreme levels of metal toxicity in an orphan mining site. Sci. Total Environ. 598, 121–128. doi: 10.1016/j.scitotenv.2017.04.084
Satapute, P., Paidi, M. K., Kurjogi, M., and Jogaiah, S. (2019). Physiological adaptation and spectral annotation of arsenic and cadmium heavy metal-resistant and susceptible strain pseudomonas taiwanensis. Environ. Pollut. 251, 555–563. doi: 10.1016/j.envpol.2019.05.054
Schmidt-Dannert, C. (2016). Biocatalytic portfolio of basidiomycota. Curr. Opin. Chem. Biol. 31, 40–49. doi: 10.1016/j.cbpa.2016.01.002
Schutzius, G., Mi, N., and Navab-Daneshmand, T. (2019). Antibiotic resistance in fecal sludge and soil in ho Chi Minh city, Vietnam. Environ. Sci. Pollut. R. 26, 34521–34530. doi: 10.1007/s11356-019-06537-5
Shah, A. N., Tanveer, M., Shahzad, B., Yang, G., Fahad, S., Ali, S., et al. (2017). Soil compaction effects on soil health and crop productivity: an overview. Environ. Sci. Pollut. R. 24, 10056–10067. doi: 10.1007/s11356-017-8421-y
Singh, S. P., Chand, H. S., Banerjee, S., Agarwal, H., Raizada, V., Roy, S., et al. (2020). Acetylcholinesterase inhibitor pyridostigmine bromide attenuates gut pathology and bacterial dysbiosis in a murine model of ulcerative colitis. Digest. Dis. Sci. 65, 141–149. doi: 10.1007/s10620-019-05838-6
Stefanowicz, A. M., Niklinska, M., and Laskowski, R. (2008). Metals affect soil bacterial and fungal functional diversity differently. Environ. Toxicol. Chem. 27, 591–598. doi: 10.1897/07-288.1
Tarin, M., Fan, L., Xie, D., Tayyab, M., Rong, J., Chen, L., et al. (2021). Response of soil fungal diversity and community composition to varying levels of bamboo biochar in red soils. Microorganisms. 9. doi: 10.3390/microorganisms9071385
Valentin-Vargas, A., Root, R. A., Neilson, J. W., Chorover, J., and Maier, R. M. (2014). Environmental factors influencing the structural dynamics of soil microbial communities during assisted phytostabilization of acid-generating mine tailings: a mesocosm experiment. Sci. Total Environ. 500-501, 314–324. doi: 10.1016/j.scitotenv.2014.08.107
Van Rooij, P., Martel, A., Haesebrouck, F., and Pasmans, F. (2015). Amphibian chytridiomycosis: a review with focus on fungus-host interactions. Vet. Res. 46:137. doi: 10.1186/s13567-015-0266-0
Vellend, M. (2010). Conceptual synthesis in community ecology. Q. Rev. Biol. 85, 183–206. doi: 10.1086/652373
Wang, Y., Fu, Y., He, Y., Kulyar, M. F., Iqbal, M., Li, K., et al. (2021). Longitudinal characterization of the gut bacterial and fungal communities in yaks. J Fungi (Basel). 7. doi: 10.3390/jof7070559
Wang, L., and Li, X. (2019). Steering soil microbiome to enhance soil system resilience. Crit. Rev. Microbiol. 45, 743–753. doi: 10.1080/1040841X.2019.1700906
Wang, Y., Li, A., Jiang, X., Zhang, H., Mehmood, K., Zhang, L., et al. (2018). Probiotic potential of leuconostoc pseudomesenteroides and lactobacillus strains isolated from yaks. Front. Microbiol. 9:2987. doi: 10.3389/fmicb.2018.02987
Wardle, D. A. (2002). Communities and ecosystems: linking the aboveground and below-ground components. Monogr. Popul. Biol. 34, 1–392. doi: 10.1515/9781400847297
Weber, C. F., King, G. M., and Aho, K. (2015). Relative abundance of and composition within fungal orders differ between cheatgrass (bromus tectorum) and sagebrush (artemisia tridentate)-associated soils. PLoS One 10:e117026. doi: 10.1371/journal.pone.0117026
Wegrzyn, M. H., Wietrzyk, P., Lehmann-Konera, S., Chmiel, S., Cykowska-Marzencka, B., and Polkowska, Z. (2018). Annual variability of heavy metal content in svalbard reindeer faeces as a result of dietary preferences. Environ. Sci. Pollut. Res. Int. 25, 36693–36701. doi: 10.1007/s11356-018-3479-8
Wood, J. L., Zhang, C., Mathews, E. R., Tang, C., and Franks, A. E. (2016). Microbial community dynamics in the rhizosphere of a cadmium hyper-accumulator. Sci. Rep. 6:36067. doi: 10.1038/srep36067
Youssef, N. H., Farag, I. F., Rinke, C., Hallam, S. J., Woyke, T., and Elshahed, M. S. (2015). In silico analysis of the metabolic potential and niche specialization of candidate phylum "latescibacteria" (ws3). PLoS One 10:e127499. doi: 10.1371/journal.pone.0127499
Zhang, J., Shi, Q., Fan, S., Zhang, Y., Zhang, M., and Zhang, J. (2021). Distinction between cr and other heavy-metal-resistant bacteria involved in c/n cycling in contaminated soils of copper producing sites. J. Hazard. Mater. 402:123454. doi: 10.1016/j.jhazmat.2020.123454
Zhao, F. J., Ma, Y., Zhu, Y. G., Tang, Z., and McGrath, S. P. (2015). Soil contamination in China: current status and mitigation strategies. Environ. Sci. Technol. 49, 750–759. doi: 10.1021/es5047099
Keywords: excrement contamination, plateau, soil microbiota, bacterial and fungal communities, yaks
Citation: Shang Z, Wang Y, An M, Chen X, Kulyar M-A, Tan Z, Liu S and Li K (2022) The successional trajectory of bacterial and fungal communities in soil are fabricated by yaks’ excrement contamination in plateau, China. Front. Microbiol. 13:1016852. doi: 10.3389/fmicb.2022.1016852
Edited by:
Maria Alicja Niklińska, Jagiellonian University, PolandReviewed by:
Prerona Boruah, DY Patil Deemed to be University, IndiaAwais Ihsan, COMSATS University Islamabad, Pakistan
Copyright © 2022 Shang, Wang, An, Chen, Kulyar, Tan, Liu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaping Wang, d2FuZ3lwOTZAc2luYS5jb20=; Suozhu Liu, bGl1c3Vvemh1QHh6YS5lZHUuY24=; Kun Li, bGszMDA1QG5qYXUuZWR1LmNu
†These authors have contributed equally to this work