 Ratthaphol Charlermroj
Ratthaphol Charlermroj Manlika Makornwattana
Manlika Makornwattana Sudtida Phuengwas
Sudtida Phuengwas Nitsara Karoonuthaisiri
Nitsara Karoonuthaisiri- 1National Center for Genetic Engineering and Biotechnology (BIOTEC), National Science and Technology Development Agency (NSTDA), Pathum Thani, Thailand
- 2International Joint Research Center on Food Security, Pathum Thani, Thailand
- 3Institute for Global Food Security, Queen’s University Belfast, Belfast, United Kingdom
Specific antibodies are essential components of immunoassay, which can be applied for the detection of pathogens. However, producing an antibody specific to live bacterial pathogens by the classical method of immunizing animals with live pathogens can be impractical. Phage display technology is an effective alternative method to obtain antibodies with the desired specificity against selected antigenic molecules. In this study, we demonstrated the power of a microarray-based technique for obtaining specific phage-derived antibody fragments against Salmonella, an important foodborne pathogen. The selected phage-displayed antibody fragments were subsequently employed to develop a lateral flow test strip assay for the detection of live Salmonella. The test strips showed specificity to Salmonella Enteritidis without cross-reactivity to eight serovars of Salmonella or other bacteria strains. The test strip assay requires 15 min, whereas the conventional biochemical and serological confirmation test requires at least 24 h. The microarray screening technique for specific phage-based binders and the test strip method can be further applied to other foodborne pathogens.
Introduction
Salmonella bacterial species are causative agents of foodborne illness in humans and animals, which are commonly found in many types of food such as pork, eggs, poultry, seafood, unpasteurized dairy products, and vegetables (Jackson et al., 2013; Gu et al., 2018). The standard methods for detecting Salmonella are based on culturing techniques including pre-enrichment, selective-enrichment, and confirmation with biochemical tests, following procedures outlined by the International Organization for Standardization (ISO 6579) or Bacteriological Analytical Manual (BAM). These methods can detect low numbers or injured viable Salmonella; however, they are time-consuming and laborious. Thus, rapid and accurate methods are required for detecting foodborne pathogens (Law et al., 2015).
Methods have been developed to detect viable bacterial cells using fluorescent dyes such as SYTO 9 and propidium iodide (Ou et al., 2019), and mammalian cell-based immunoassay (Xu et al., 2020). Although they can detect or differentiate live cells from dead cells, these methods require many steps and special equipment. These shortcomings could be addressed by immuno-based lateral flow assays, which are more rapid, simple, and affordable. However, to our knowledge, there is no report of an immuno-based lateral flow method capable of discriminating viable from dead bacteria cells. The major challenge for applying lateral flow assays for detecting viable bacteria is the requirement for an antibody that binds specifically to viable bacterial cells of interest, but not to injured or non-viable cells.
The traditional method of producing an antibody for immunoassay relies on an in vivo immune response from an antigen. The success of antibody production depends on the antigen characteristics such as types of immunogens, antigenicity, and antigen dosing. Alternatively, antibodies can be produced by phage display technology, which can identify binders to antigens regardless of their immunogenic properties, thus allowing the selection of binders against self-antigens, toxic, unstable, and non-immunogenic antigens (Frenzel et al., 2016). This technology also facilitates genetic engineering of the binding sites to improve affinity and specificity. Its advantages over the traditional in vivo antibody production method have fostered applications ranging from epitope mapping (Spillner et al., 2003; Youn et al., 2004), the detection of bacteria and viruses (Ferrer and Harrison, 1999; Yang et al., 2003; Morton et al., 2013b; Karoonuthaisiri et al., 2014; Wang et al., 2014; Niyomdecha et al., 2018), protein domains (Christ and Winter, 2006), and small molecules (Zhao et al., 2005; Qi et al., 2008).
Given the power of phage technology, this study aimed to (1) develop a bacterial microarray method to speed up the process of screening and selecting phage clones expressing specific antibody fragments and (2) utilize the selected phage clones for developing a rapid lateral flow detection method for live Salmonella Enteritidis.
Materials and methods
Bacteria, antibodies, and phage clones
All bacteria in Table 1, except for Campylobacter spp., were inoculated from a single colony grown in a LB agar plate and cultured in 10 mL of 2xYT medium (16 g/L tryptone, 10 g/L yeast extract, and 5 g/L NaCl) at 37°C, 250 rpm for 16–18 h. Campylobacter spp. were cultured in 10 mL of Campylobacter Enrichment Broth (CEB) supplemented with 20 mg/L cefoperazone, 20 mg/L vancomycin, 20 mg/L trimethoprim, and 25 mg/L natamycin (#X132, Lab M, UK) at 41.5°C, in microaerophilic conditions (5% CO2 and 10% O2) for 48 h.
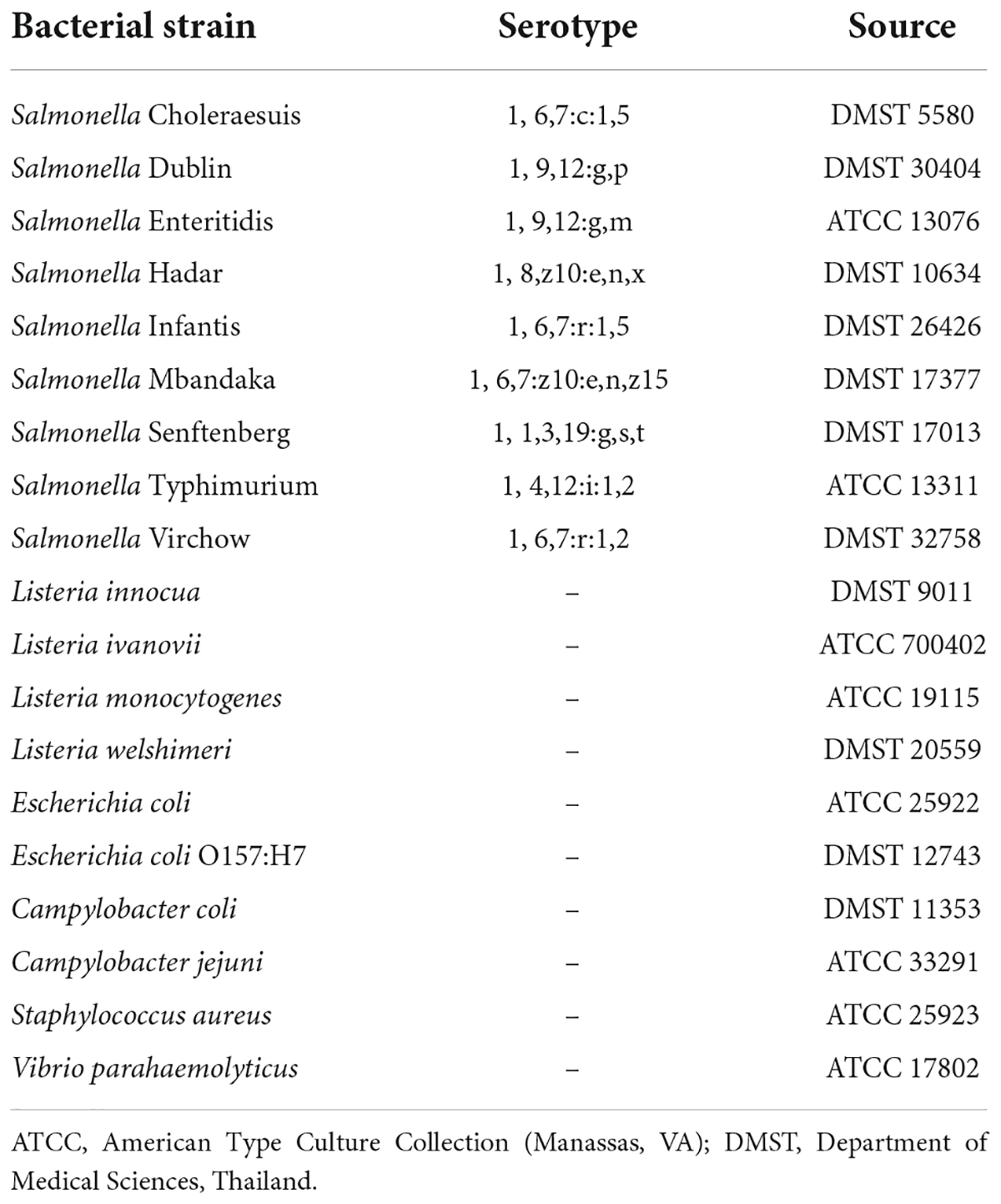
Table 1. Bacteria strains used in this project.
The sources and reactivity of all antibodies and phage clones used in this study were reported in Supplementary Table 1.
Biopanning and individual phage clone amplification
A phage-displayed human domain antibody library displaying a single human VH framework (V3-23/D47) with diversity introduced in the antigen-binding site and short complementarity-determining region 3 (CDR3) of the heavy chains was used in this study (Lee et al., 2007) (Source Bioscience). Biopanning steps were performed according to the library instructions and modified with a suspension method previously employed (Paoli et al., 2004; Morton et al., 2013a; Figure 1A). Briefly, sterilized protein low binding tubes were blocked with 1 mL of 5% skimmed milk in phosphate buffered saline (PBS, pH 7.4 containing 1 mM KH2PO4, 0.15 mM Na2HPO4, 3 mM NaCl) overnight at 4°C. The blocked tubes were washed twice with PBS. For the first round of biopanning, a cocktail of nine serovars (Choleraesuis, Dublin, Enteritidis, Hadar, Infantis, Mbandaka, Senftenberg, Typhimurium, and Virchow) of Salmonella (5 × 109 colony forming units (CFU)/mL for each Salmonella serovar) and the phage library (5 × 1011 plaque forming unit, pfu/mL) were mixed in PBS (total volume 1 mL) in the blocked tube, 20 rpm at RT for 1 h. Unbound phages were removed by centrifuging at 3,200 × g for 10 min. The pellet of phage-bound bacterial cells was washed five times by resuspending in PBS containing 0.1% Tween 20 and separation of phage-bound bacterial cell pellet by centrifugation at 3 200 × g for 10 min. To elute phages from the bacterial target, a trypsin solution (1 mL of 100 μg/mL Trypsin in Tris-buffered saline calcium chloride) was added, and the suspension was incubated at RT for 1 h. The eluted phages were used to infect a mid-log phase culture of E. coli TG1 TR strain (OD600 = 0.5) at 37°C for 1 h. The non-infecting phages were separated by centrifugation at 3,200 × g for 5 min. To enumerate the phage-infected E. coli TG1, the pellet was resuspended in 1 mL of 2xYT medium, and the bacterial cell suspension was serially diluted and plated on TYE ampicillin glucose agar plates (10 g/L bacto-tryptone, 5 g/L yeast extract, 8 g/L NaCl, 100 mg/L ampicillin and 40 g/L glucose).
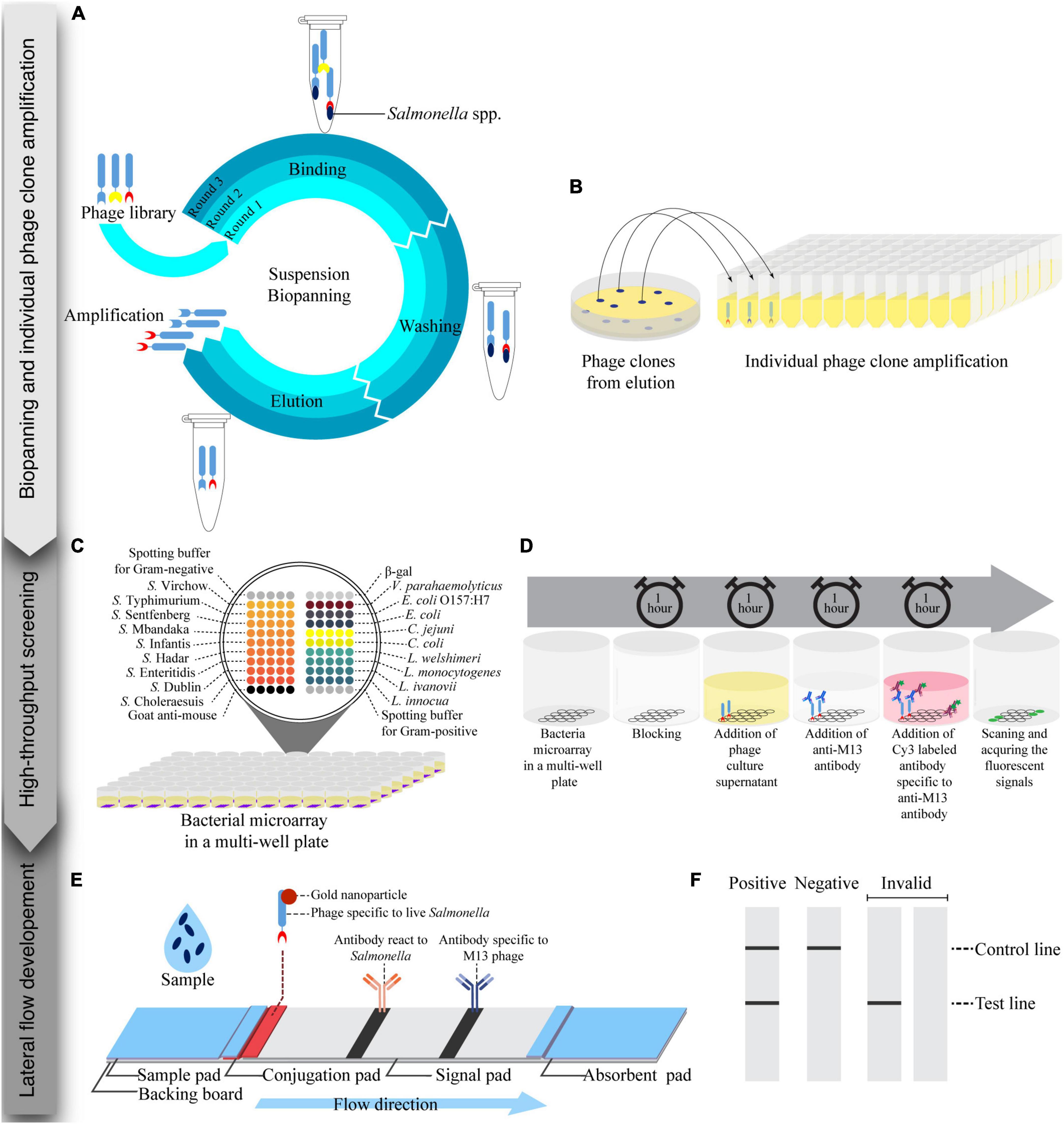
Figure 1. Schematic of the process for the development of a lateral flow strip test assay for live Salmonella detection consisting of three steps: biopanning and individual phage clones amplification, high-throughput screening, and lateral flow development. (A) Suspension biopanning was performed against a mixture of nine Salmonella serovars adapted from Lee et al. (2007). (B) Phage clones were amplified in a 96-well plate and screened by a bacterial microarray method. (C) A bacterial microarray in a 96-well plate format was developed and constructed. Each well was spotted with target and non-target bacteria. (D) Phage supernatant was tested with a bacterial microarray. (E) Lateral flow strip test was developed using a gold nanoparticle-labeled phage as a biorecognition element and signal reporter. Anti-Salmonella and anti-M13 phage antibodies were printed at the test line and control line, respectively. (F) Readouts of lateral flow strip test were visualized by the naked eye and images of the strip were captured by a smartphone.
To amplify and purify phages for the next rounds of biopanning, the phagemid-carrying E. coli TG1 bacterial cells (phage clones) from the TYE agar plates were scraped from plates using 5 mL of 2xYT medium per plate. The bacterial suspension was diluted to an OD600 value of 0.1 in 500 mL of 2xYT medium supplemented with 100 μg/mL ampicillin and 4% glucose. Bacteria were cultured at 37°C and 250 rpm to an OD600 value of 0.5. The bacterial culture was then infected with KM13 helper phages (2 × 1012 pfu) and incubated at 37°C for 1 h. The supernatant was removed by centrifuging at 3,200 × g for 10 min, and the cell pellets were resuspended in 500 mL of 2xYT medium supplemented with 0.1% glucose, 100 μg/mL ampicillin and 50 μg/mL kanamycin, and incubated at 25°C, 250 rpm for 16-20 h. Phages in the supernatant were precipitated with 20% polyethylene glycol 6,000 (20% PEG6000, 2.5 M NaCl) and reconstituted in 1 mL PBS. The phage titer was estimated using the following equation (Lee et al., 2007).
From a phage clone collection, 188 individual colonies of phage-infected cells from each biopanning round were picked from 10-fold serial dilution plates of 2xYT agar supplemented with 100 μg/mL ampicillin and 4% glucose (Figure 1B). Each colony was transferred into a well of 96-well plates (Costar) containing 200 μL of 2xYT medium supplemented with 100 μg/mL ampicillin and 4% glucose, covered with a breathable sealing film (Axygen) and incubated at 37°C, 250 rpm overnight. These phage clones were kept at 4°C until use.
To screen for specific phage and to test cross-reactivity against other relevant bacteria, 600 μL of 2xYT supplemented with 100 μg/mL ampicillin and 4% glucose was inoculated with 30 μL of a suspension of individual phage clone stock and cultured at 37°C, 250 rpm for 3 h. After 3 h of incubation, KM13 helper phages (150 μL of 8 × 109 pfu/mL) were added to infect the cells in the culture, which was then incubated at 37°C without shaking for 1 h. To remove the helper phage, the bacterial cells were collected by centrifuging at 2,000 rpm for 30 min. The cell pellets were resuspended in 600 μL of 2xYT supplemented with 100 μg/mL ampicillin, 50 μg/mL kanamycin, and 0.1% glucose and cultured at 25°C, 250 rpm for 16-24 h. The phages were separated from bacterial cells by centrifugation at 2,000 rpm for 30 min. The phage supernatants were screened by the optimized bacterial microarray method described below.
Bacterial microarray development in a multi-well plate
To develop a bacterial microarray method in a multi-well plate (Figures 1C,D), bacterial cells (1 × 1011 CFU/mL) were suspended in carbonate buffer (44 mM NaHCO3 and 6 mM Na2CO3, pH 9.6) with Tween 20 (0.05%) and glycerol (0.5%) for Gram-negative bacteria, and in a carbonate buffer with glycerol (0.5%) for Gram-positive bacteria. The bacteria suspension was spotted (5 replicates) onto microplate wells (Corning) using a NanoPrint 210 microarrayer equipped with 946NS pins (TeleChem). Relevant spotting buffers were used as negative controls, and an anti-mouse antibody was used as a positive control and to indicate spot positions. The bacterial microarrays were kept at 4°C until use.
Screening of phage-displayed antibody fragments using a bacterial microarray technique
To identify phage clones specific to Salmonella spp., bacterial microarrays were used to screen the phage clones (Figure 1D). The bacterial microarray plates were blocked with 5% skimmed milk in PBST (300 μL/well) for 1 h at RT, then washed with PBST three times by an automatic microplate washer (BIO-RAD). Phage supernatant (100 μL) was added to each well and incubated for 1 h at RT before being washed again as before. An antibody specific to bacteriophage M13 (100 μL/well, 5 μg/mL) was added and the plates were incubated for 1 h at RT. After washing again, a Cy3 labeled anti-mouse antibody (100 μL/well, 5 μg/mL) was added and incubated in the dark for 1 h at RT. The microplates were washed as before and dried by centrifuging at 200 rpm for 5 min. The plates were then scanned using a fluorescence scanner (TECAN), and the fluorescent intensities of spots were determined using the Array-Pro Analyzer software version 4.5.1.73 (TECAN). Background noise in each experiment was determined as the mean value taken from wells containing spotting buffer with no phage added. Normalized signals were determined as the ratio of mean fluorescent intensity of bacterial spots (5 technical replicates/bacterial strain) to background. Mean normalized signals from 5 spots greater than or equal to two were considered positive. To visualize microarray data, a heat map was created from the mean normalized signals with the GraphPad Prism software version 9.4.1 (681).
Plate-trapped antigen-ELISA
A plate-trapped antigen (PTA)-ELISA method was used to validate the microarray results of the selected phage clones. Bacterial cells (109CFU/mL) were heat inactivated at 100°C for 15 min. Each of the 18 inactivated bacterial strains was diluted in carbonate buffer pH 9.6 (1 × 108 CFU/mL, 100 μL/well) and coated onto plate wells overnight at 4°C. The plates were then washed by an automatic washer machine (BIO-RAD) with 300 μL/well PBS containing 0.1% Tween 20 (PBST) three times before being blocked with 5% skimmed milk (300 μL/well, Difco laboratory) in PBST for 1 h at RT. The washing step was repeated before 100 μL of phage suspension was added into each well. Plates with phage added were incubated for 1 h at RT. After washing as before, a horseradish peroxidase (HRP)-labeled anti-M13 antibody (diluted 5,000-fold in 5% skimmed in PBST; GE Healthcare) was added and the plate incubated for 1 h at RT. The plate was washed as before and a substrate solution for HRP (TMB: 3,3’,5,5’-Tetramethylbenzidine; Invitrogen) was added (100 μL/well). The plate was then incubated for 5–30 min at RT. The reaction was stopped by adding 0.5 M H2SO4 (50 μL/well) and the signal was measured at 450 nm absorbance using a SpectraMax M5 microplate reader (Molecular device). Each experiment was repeated three times. A signal three times above the value of background reading was considered positive.
Limit of detection of phage clones by plate-trapped antigen-ELISA
Twelve different bacteria titers (0, 1 × 104, 5 × 104, 1 × 105, 5 × 105, 1 × 106, 5 × 106, 1 × 107, 5 × 107, 1 × 108, 5 × 108, and 1 × 109 CFU/mL) were prepared by dilution of bacterial culture in carbonate buffer. The bacteria were coated onto plate wells. The assay was performed in triplicate using the same steps as described above in the PTA-ELISA method. Limit of detection (LOD) values were calculated as the minimal titer with a signal greater than three times background (negative control). The ELISA signal data were fitted to the following dose-response equation with three parameters and confidence level at 95% (Iturria, 2005; Charlermroj et al., 2014).
where B and B0 were absorbance values at 450 nm of a phage clone binding in the presence (B) or absence (B0) of bacterial cells, respectively, in which% B/B0 response was measured at varying titers of bacteria (X), and EC50 is the bacterial titer that produced a 50% response between B and B0.
Preparation of whole phage-gold nanoparticles (phage-AuNPs) conjugates
To prepare phage-AuNPs, AuNPs solution (1 mL, 40 nm, #KP-05120003, Kestrel Bioscience, Thailand) was adjusted to pH 8.0 with 100 mM K2CO3 before purified whole phage (100 μL of 1012 pfu/mL) suspension was added. The mixture was incubated at RT for 10 min. The unconjugated AuNPs were subsequently blocked with bovine serum albumin (BSA, 110 μL of 10% (w/v) in distilled water adjusted to pH 7.0) at 4°C overnight. The excess of phage was removed by centrifugation at 12,000 rpm at 4°C for 30 min. The pellet was resuspended in 50 μL of conjugate buffer (PBS containing 10% sucrose, and 5% trehalose, pH 7.4). The phage-AuNPs suspension was kept at 4°C until use.
Preparation of lateral flow test strips
The composition of lateral flow test strips is shown in Figure 1E. A sample pad (CF3, GE Healthcare, USA) was impregnated with PBS containing 0.4% Tween 20, and 2% (w/v) BSA, pH 7.4, before drying overnight at 37°C. The suspension of whole phage-AuNPs conjugates (10 μL) was applied on a conjugated pad (4 mm × 1 cm., GF33, Kestrel Bioscience, Thailand), and dried at 37°C for 30 min. An antibody specific to Salmonella (1 mg/mL, ab35156, AbCAM, UK) and an antibody specific to M13 phage (0.5 mg/mL) were prepared in carbonate buffer, pH 9.6. The antibodies were dispensed (0.8 μL/cm) on the signal pad (CN95, Kestrel Bioscience, Thailand) at a test line (TL) for Salmonella detection and a control line (CL) for positive control, respectively, using a non-contact microarray dispenser equipped with Biojet Elite dispenser (AD1520, BioDot, USA). The signal pad was then dried at RT for 30 min before blocking with treating buffer (10 mM di-sodium tetraborate containing 1% (w/v) BSA, 0.5% (w/v) polyvinyl pyrrolidone (PVP40), and 0.15% (v/v) Triton X-100, pH 8.0) and dried at 37°C overnight. The sample pad, conjugate pad, and absorbent pad (CF5, GE Healthcare, USA) were assembled onto a backing board, and cut into 4 mm wide strips with a guillotine cutter (CM5000, BioDot, USA).
Lateral flow assay procedure
The lateral flow test strips were tested as follows. Bacteria were cultured in Luria-Bertani broth (LB, Difco, USA) at 37°C, 250 rpm for 16-18 h. Test sample (100 μL) was applied onto the sample pad, and incubated for 15 min at RT. Signals were captured at 11 cm above the strip by a smart phone (Samsung Note 20) under the white light condition for qualitative assessment of the result (Figure 1F). To test the specificity of the lateral flow test strips, nine serovars of Salmonella, heat-killed Salmonella Enteritidis, and three other bacteria strains (E. coli O157:H7, L. monocytogenes, and S. aureus) were tested at 108 CFU/mL with three replications. For sensitivity of detection, ten different titers of Salmonella Enteritidis (0, 1 × 106, 5 × 106, 1 × 107, 5 × 107, 1 × 108, 5 × 108, 1 × 109, 5 × 109, 1 × 1010 CFU/mL were tested with three replications. To analyze optical density of test line (TL) and control line (CL), the images were converted to grayscale using the Adobe Photoshop Software version 23.5.0, and band intensities were subsequently measured using the Quantity One Software version 4.6.8. The TL/CL ratio values were calculated and used for a statistical analysis.
Statistical analysis
All data are express as the mean values ± standard deviations (mean ± SD) of three replicates, except a specificity test by an ELISA method. All analyses were conducted using the GraphPad Prism 9 (681) software. The experiments were compared using an analysis of variance (ANOVA) and Tukey’s post-test. P-value of < 0.05 was considered to indicate a significant difference.
Results
Phage high-throughput screening and characterization
To facilitate the screening of phage binders specific to bacteria of interest, a bacterial microarray was developed. Microarray spotting buffers were first optimized using Salmonella Typhimurium and Listeria monocytogenes as model cells. We found that a carbonate buffer containing Tween 20 and glycerol (CBTG) and a carbonate buffer containing glycerol (CBG) was suitable for Gram-negative (Supplementary Figure 1A) and Gram-positive bacteria, respectively (Supplementary Figure 1B). These spotting buffers were validated with 18 different bacterial strains using antibodies and a phage with known specificities (Supplementary Table 1). The antibodies and phage showed accurate detection of their corresponding bacterial targets when the bacterial cells were prepared in the selected spotting buffers, except in case of SalKPL and ListKPL antibodies (Supplementary Figure 1C). SalKPL antibody reacted strongly with Salmonella spp. with some cross reactivity with E. coli in the bacterial microarray. This result is in agreement with cross reactivity to related Enterobacteriaceae reported by the antibody’s supplier. ListKPL showed minor cross reactivity to Salmonella Infantis on the microarray, which agrees with the cross-reactivity information from the antibody supplier. The results showed that they were suitable for the production of bacterial microarray.
The bacterial microarray was used to screen a total of 564 phage clones (188 from each round of biopanning, 3 rounds in total) and to test the specificity of the phage-displayed antibody against 18 different bacterial strains. The number of Salmonella-specific phage clones increased after three rounds of biopanning (20.2, 20.2, and 35.6% for the 1st–3rd biopanning rounds, respectively), while the number of phage clones that could bind to Salmonella and cross-react with other bacteria dramatically increased (8.5, 22.3, and 57.4% for the 1st–3rd biopanning rounds, respectively) (Figure 2). We sought phage clones that can detect the nine serovars of Salmonella with no cross-reactivity to other bacterial species. Unfortunately, no phage clones were isolated that bound to all nine serovars of Salmonella after the third round of biopanning. Moreover, most of the phage clones detecting one or more serovars of Salmonella also cross-reacted with Vibrio parahaemolyticus. However, we were able to identify phage clones specific to Salmonella Enteritidis. Thus, we randomly selected four phage clones from the third round of biopanning for further characterization using a PTA-ELISA method against 18 different bacterial strains (Figure 3A). All randomly selected phage clones could specifically bind to Salmonella Enteritidis, except for clone 03P1D05 that showed some cross-reactivity to V. parahaemolyticus (Figure 3A). Of the remaining three Salmonella-specific phage clones, clone 03P2H03 exhibited the highest signal (Figure 3A) and was selected for its ability to discriminate between live and dead Salmonella. 03P2H03 was able to distinguish between live and dead (heat-killed) Salmonella Enteritidis cells, while the commercial antibodies could not (Figure 3B). The limit of detection (LOD) of the 03P2H03 phage clone in detecting Salmonella was found to be within the same order of magnitude (3.0 × 106 CFU/mL) as that of the Salmonella-specific antibody (4.1 × 106 CFU/mL) (Figure 3C).
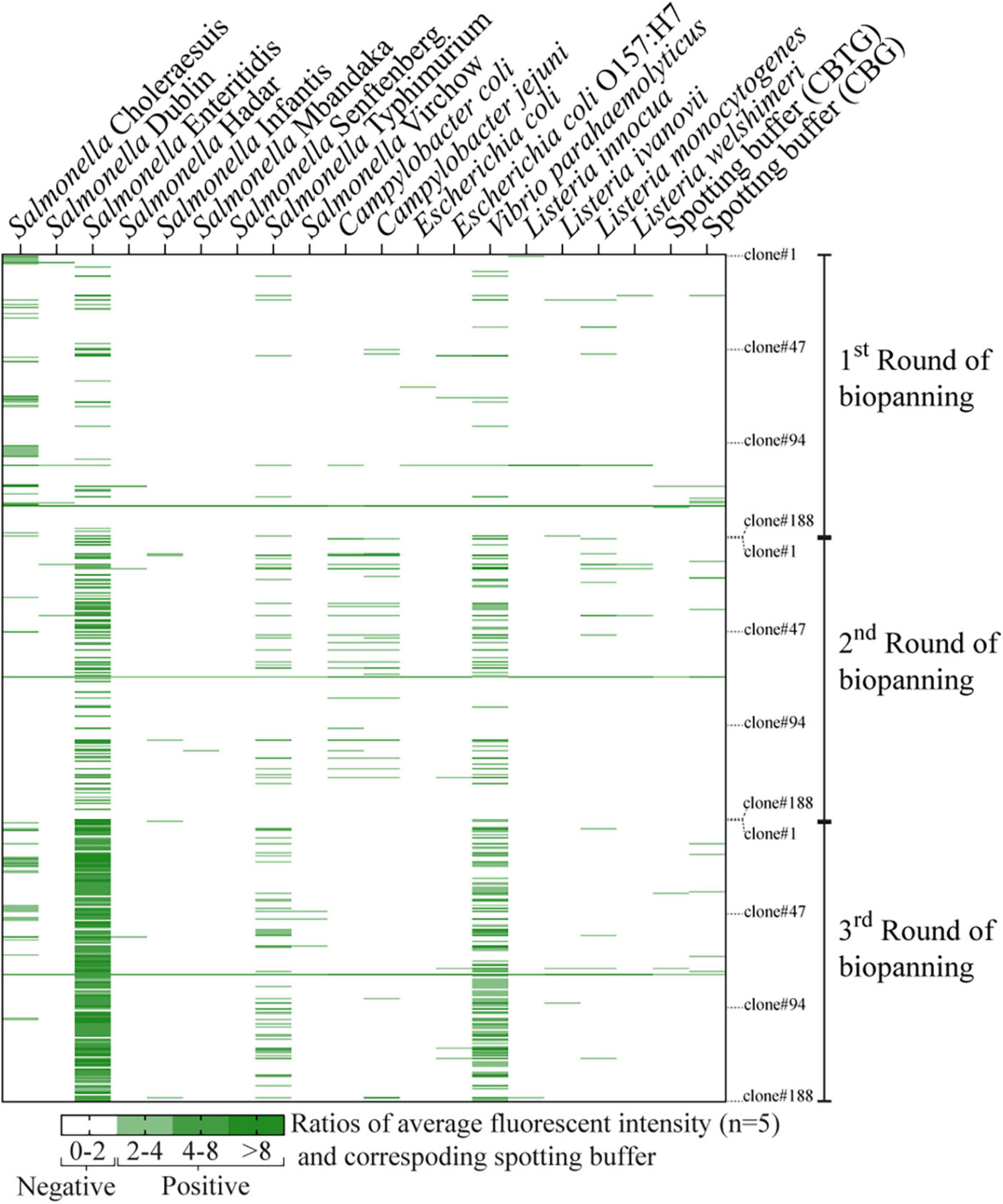
Figure 2. Heat map of individual phage clones (rows) tested against 18 different bacteria strains (columns). A total of 564 phage clones (188 clones for each round of biopanning, 3 rounds total) were tested for their binding specificity using bacterial microarray. Fluorescent intensity values were averaged from five spots and normalized with the signal from their corresponding spotting buffers (CBTG for Gram-negative bacteria, and CBG for Gram-positive bacteria). Normalized fluorescent signals greater than or equal to two were considered as positive results and are indicated by the green color gradient. Normalized signals (negative results) below the threshold are indicated in white.
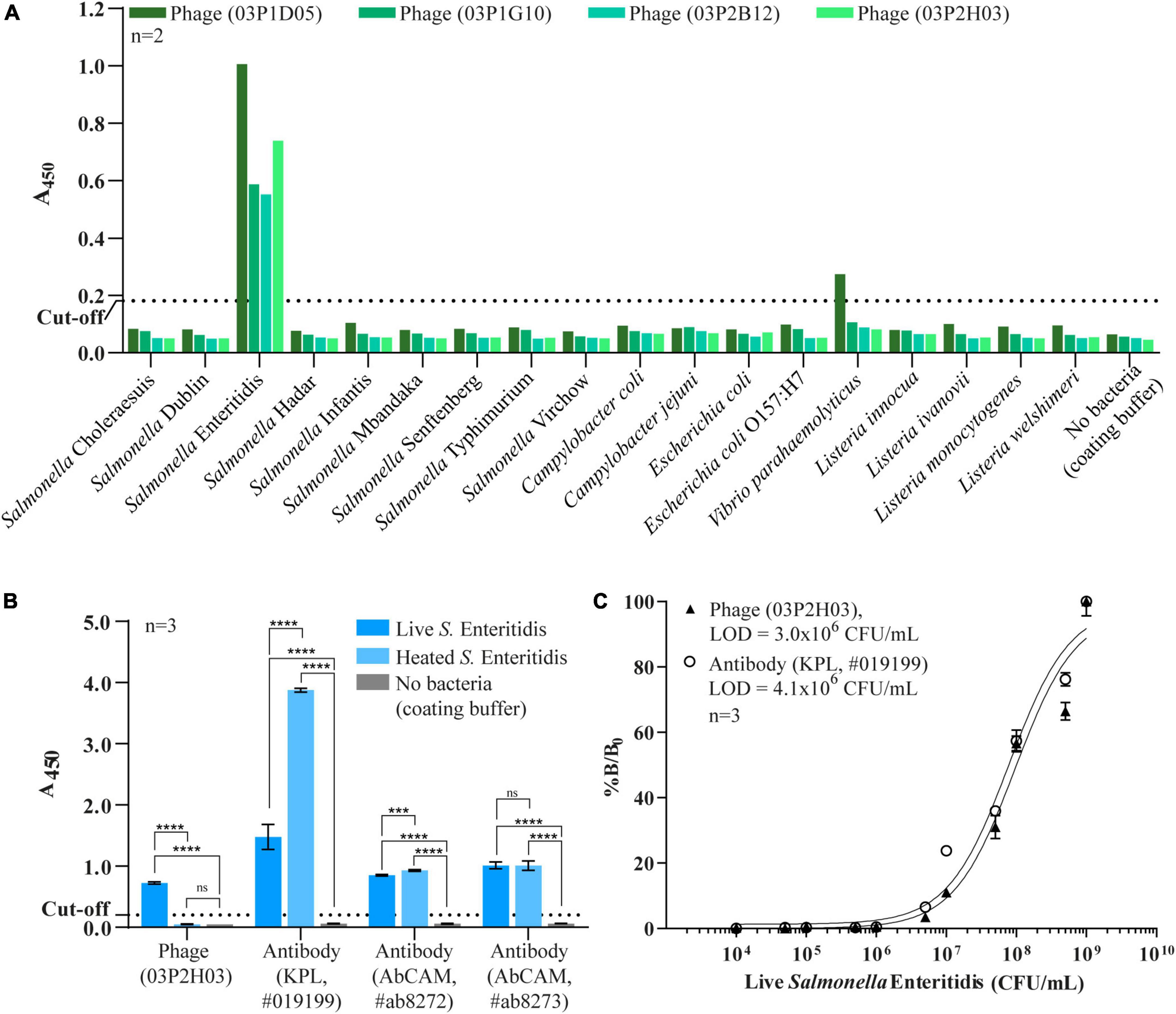
Figure 3. Characterization of randomly selected phage clones by a PTA-ELISA method against 18 different strains of bacteria. (A) Specificity of the four phage clones previously selected from bacterial microarray data. (B) Comparison of specificity against live/dead cells of the selected phage clone 03P2H03 and the three commercial antibodies specific to Salmonella. Each bar is represented and mean ± standard derivation (SD) which was compared to no bacteria (one-way ANOVA for indicate comparison, Tukey’s multiple comparison test, ***p < 0.005, ****p < 0.0001, and ns = no significance). (C) Limit of detection (LOD) for live Salmonella Enteritidis of phage clone (03P2H03) and a commercial Salmonella antibody by PTA-ELISA. The dotted line represents the cut-off value three times that of the background (LB medium) used to determine the limit of detection. Error bars indicate standard derivation (SD) (n = 3). Solid lines indicate curve fits to the data using a three-parameter dose-response model.
Lateral flow test strip for detection of live Salmonella Enteritidis
To test the specificity and sensitivity of the constructed lateral flow test strip using whole phage-AuNPs as a detecting molecule, different bacterial strains and varying titers of live Salmonella Enteritidis were tested. The lateral flow test strip could specifically detect Salmonella Enteritidis but showed no reaction to eight Salmonella serovars and other relevant bacterial strains (Figures 4A,B). For sensitivity, the test line signal indicating the presence of Salmonella Enteritidis was apparent to the naked eye for cell titers 1 × 107–1 × 1010CFU/mL, indicating a LOD of 1 × 107 CFU/mL for the test strip. At the highest titer tested (1 × 1010CFU/mL), the test line showed slightly lower intensity than lower titers (Figures 4C,D). The assay time of the lateral flow test strip was only 15 min. These results demonstrated the usefulness of whole phage as a biorecognition element for a lateral flow detection method of live Salmonella Enteritidis.
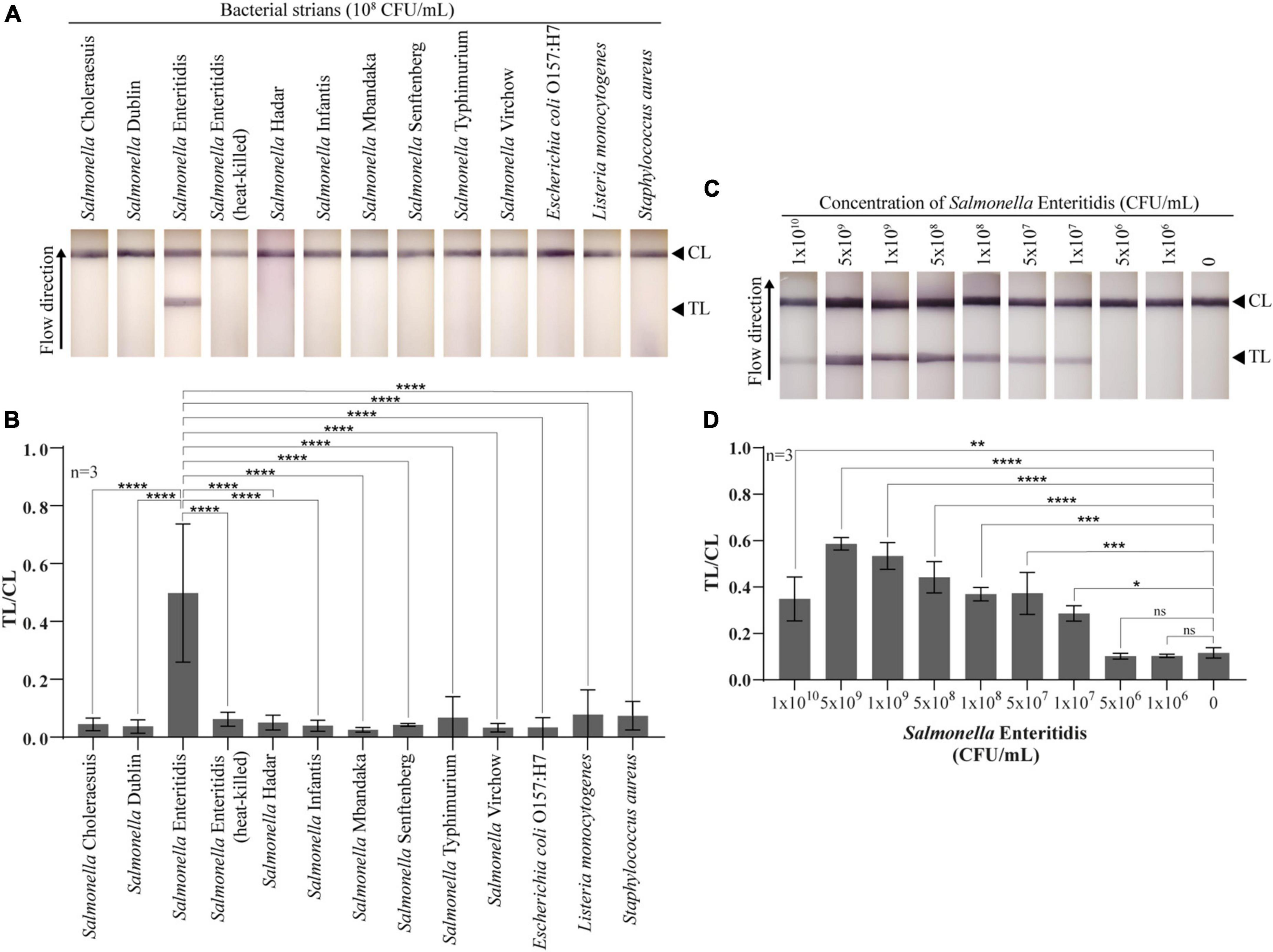
Figure 4. Specificity and sensitivity of the developed lateral flow test strip for Salmonella Enteritidis detection. (A) Photographs of the lateral flow test strips with different 12 bacterial strains and heat-killed Salmonella Enteritidis at 108 CFU/mL in LB medium. (B) Quantitative of lateral flow test strips for specificity test. Test line (TL) and control line (CL) ratios were compared with Salmonella Enteritidis using a statistical analysis. (C) Photographs of the lateral flow test with ten different titers of Salmonella Enteritidis. (D) Quantitative of lateral flow test strips for sensitivity test. Test line (TL) and control line (CL) ratios were compared with no bacteria using a statistical analysis. All experiments were tested with three independent test strips (one-way ANOVA for indicate comparison, Tukey’s multiple comparison test, *p < 0.05, **p < 0.001, ***p < 0.005, ****p < 0.0001, and ns = no significance).
Discussion
While phage technology can be applied for pathogen detection, its full potential has yet to be realized owing to the difficulty in obtaining specific phage clones for downstream applications. In this study, a high-throughput microarray method was developed to screen and characterize the binding specificity of phage clones. From prior experience, we found that selection of appropriate spotting buffers was the key factor for success. For instance, we found that a carbonate-based buffer was suitable for producing an antibody array (Charlermroj et al., 2014). Unlike the antibody array, the bacterial microarray in this study is more complicated because the major components of the bacterial cell wall for each Gram stain reactive group are different, i.e., lipopolysaccharide for Gram-negative bacteria and peptidoglycan for Gram-positive bacteria. Thus, spotting buffers needed to be optimized for each type of bacterial cell.
The bacterial microarray in multi-well plate format provides several advantages. First and foremost, the microarray was able to identify phages specific to Salmonella from the first round of biopanning. Generally, most protocols recommend 3–5 rounds of biopanning to obtain specific phage clones (Song et al., 2011; Hamzeh-Mivehroud et al., 2013). However, when a phage-antibody library is selected against highly complex targets such as whole cells, a strong bias for binders against abundant proteins has been reported. Therefore, the ability to characterize binders early in the biopanning process will result in a wider diversity of binders that can be used for diagnostics development (de Wildt et al., 2000).
The selected phage clone reacts to Salmonella Enteritidis, but did not react to eight serovars of Salmonella. While Salmonella Enteritidis and Salmonella Dublin belong to the same group of O antigen (D1 group), these two serovars have different H antigens: g and m for Salmonella Enteritidis, and g and p for Salmonella Dublin (Grimont and Weill, 2007). It might be that this phage clone bind to flagella “m” antigen of S. Enteritidis, resulting in specificity to only Salmonella Enteritidis among the nine Salmonella serovars, which should be investigated further in the next research.
The phage-derived antibody fragments identified by the microarray method showed similar characteristics (LODs and specificity) to those of the commercial antibodies. Moreover, the selected phage clone was able to distinguish between live and dead cells, whereas commercial antibodies cannot. Many publications reported the use of phages as biorecognition elements for the diagnosis of Salmonella (Morton et al., 2013b; Karoonuthaisiri et al., 2014); however, to our knowledge, there is no report on phages that can discriminate live from dead Salmonella Enteritidis.
The fact that our phage-derived antibody fragments were able to discriminate between viable and dead cells can be instrumental for many applications, especially for food safety. Molecular techniques have been developed to discriminate between viable and dead cells, including PCR-based methods such as reverse transcription (RT)-PCR and nucleic acid sequence-based amplification (NASBA) (Morin et al., 2004), and viability dyes coupled with DNA amplification (Elizaquivel et al., 2013). These molecular techniques are often more tedious and laborious than immunoassay-based methods and are typically only employed in a laboratory. Therefore, the phage-derived antibody fragments that can distinguish viable from dead foodborne pathogens can open a new horizon for more rapid and simpler diagnostics for food safety, which can be applied in different settings outside of the laboratory.
We developed a lateral flow test strip using the selected phage as a biorecognition element for the detection of live Salmonella Enteritidis and proved the specificity of the assay for detecting Salmonella Enteritidis. In addition, although this phage clone was not previously screened against Staphylococcus aureus during the screening process, the test strips using this phage showed no cross-reactivity with S. aureus. The low intensity observed at the highest titer of Salmonella (1 × 1010 CFU/mL) could be explained by the hook effect from excessively high antigen concentration in lateral flow immunoassays (Ross et al., 2020). Although sensitivities of the phage clone and antibody were around 106 CFU/mL by the PTA-ELISA method, the sensitivity for the phage-based lateral flow test strip was found to be 107 CFU/mL. The greater sensitivity of the ELISA method compared with that of the lateral flow method has previously been reported (Serrano et al., 2020). However, the sensitivity of the phage-based lateral flow test strip in this study is lower than that of a previous report of an immuno-lateral flow assay for Salmonella Enteritidis and Typhimurium at 106 and 104CFU/mL in culture media, respectively (Moongkarndi et al., 2011). The sensitivity of the test strip can be further improved by exploring other reporting molecules such as luminescent nanoparticles (Xie et al., 2014), carbon nanoparticles (Noguera et al., 2011), and streptavidin-labeled AuNPs (Chen et al., 2022).
In summary, a lateral flow test strip assay developed using phage from microarray screening was able to distinguish live from dead Salmonella Enteritidis. The assay time of 15 min is much shorter than that of any culture-based method which usually requires at least 24 h. The microarray-based screening method presented here is not limited to the selection of Salmonella-specific phage-derived binders, but it can also be readily adapted for phage-based binder selection against other to other pathogen targets, obviating the need for animal immunization. In addition, phage-based lateral flow test strip assay is not limited to foodborne pathogens and could be applied to other targets of interest, e.g., viruses.
Data availability statement
The original contributions presented in this study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s.
Author contributions
RC and NK conceived the idea, designed the experiments, and wrote the manuscript. RC, MM, and SP carried out the experiments. All authors contributed to the data analysis and reviewed the manuscript.
Funding
This project was financially supported by the National Center for Genetic Engineering and Biotechnology (BIOTEC, Thailand) (grant agreement nos. P1450542 and P1950903) and the Thailand Science Research and Innovation (grant agreement nos. TRG5780014 and RSA6280033).
Acknowledgments
Antibodies specific to Salmonella spp. and Listeria monocytogenes were kindly supplied by Oraprapai Gajanandana, Orawan Himananto, and Mallika Kumposiri, Monoclonal Antibody Production and Application Research Team, BIOTEC, Thailand. We are also grateful to David Brandon, USDA, for supplying the antibodies specific to Campylobacter (C818Ab), and Linda Stewart for her invaluable comments and suggestions to improve the manuscript. We thank Philip J. Shaw for manuscript editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1008817/full#supplementary-material
References
Charlermroj, R., Himananto, O., Seepiban, C., Kumpoosiri, M., Warin, N., Gajanandana, O., et al. (2014). Antibody array in a multiwell plate format for the sensitive and multiplexed detection of important plant pathogens. Anal. Chem. 86, 7049–7056. doi: 10.1021/ac501424k
Chen, X., Zhou, Q., Tan, Y., Wang, R., Wu, X., Liu, J., et al. (2022). Nanoparticle-based lateral flow biosensor integrated with loop-mediated isothermal amplification for rapid and visual identification of Chlamydia trachomatis for point-of-care use. Front. Microbiol. 13:914620. doi: 10.3389/fmicb.2022.914620
Christ, D., and Winter, G. (2006). Identification of protein domains by shotgun proteolysis. J. Mol. Biol. 358, 364–371. doi: 10.1016/j.jmb.2006.01.057
de Wildt, R. M., Mundy, C. R., Gorick, B. D., and Tomlinson, I. M. (2000). Antibody arrays for high-throughput screening of antibody-antigen interactions. Nat. Biotechnol. 18, 989–994. doi: 10.1038/79494
Elizaquivel, P., Azizkhani, M., Sanchez, G., and Aznar, R. (2013). Evaluation of Zataria multiflora Boiss. essential oil activity against Escherichia coli O157:H7, Salmonella enterica and Listeria monocytogenes by propidium monoazide quantitative PCR in vegetables. Food Control 34, 770–776. doi: 10.1016/j.foodcont.2013.06.036
Ferrer, M., and Harrison, S. C. (1999). Peptide ligands to human immunodeficiency virus type 1 gp120 identified from phage display libraries. J. Virol. 73, 5795–5802. doi: 10.1128/JVI.73.7.5795-5802.1999
Frenzel, A., Schirrmann, T., and Hust, M. (2016). Phage display-derived human antibodies in clinical development and therapy. MAbs 8, 1177–1194. doi: 10.1080/19420862.2016.1212149
Grimont, P. A. D., and Weill, F.-X. (2007). Antigenic Formulae of the Salmonella Serovars. Paris: WHO.
Gu, G., Strawn, L. K., Oryang, D. O., Zheng, J., Reed, E. A., Ottesen, A. R., et al. (2018). Agricultural practices influence Salmonella contamination and survival in pre-harvest tomato production. Front. Microbiol. 9:2451. doi: 10.3389/fmicb.2018.02451
Hamzeh-Mivehroud, M., Alizadeh, A. A., Morris, M. B., Bret Church, W., and Dastmalchi, S. (2013). Phage display as a technology delivering on the promise of peptide drug discovery. Drug Discov. Today 18, 1144–1157. doi: 10.1016/j.drudis.2013.09.001
Iturria, S. J. (2005). Statistical inference for relative potency in bivariate dose-response assays with correlated responses. J. Biopharm. Stat. 15, 343–351. doi: 10.1081/Bip-200048798
Jackson, B. R., Griffin, P. M., Cole, D., Walsh, K. A., and Chai, S. J. (2013). Outbreak-associated Salmonella enterica serotypes and food commodities, United States, 1998-2008. Emerg. Infect. Dis. 19, 1239–1244. doi: 10.3201/eid1908.121511
Karoonuthaisiri, N., Charlermroj, R., Morton, M. J., Oplatowska-Stachowiak, M., Grant, I. R., and Elliott, C. T. (2014). Development of a M13 bacteriophage-based SPR detection using Salmonella as a case study. Sens. Actuators B Chem. 190, 214–220. doi: 10.1016/j.snb.2013.08.068
Law, J. W.-F., Ab Mutalib, N.-S., Chan, K.-G., and Lee, L.-H. (2015). Rapid methods for the detection of foodborne bacterial pathogens: principles, applications, advantages and limitations. Front. Microbiol. 5:770. doi: 10.3389/fmicb.2014.00770
Lee, C. M., Iorno, N., Sierro, F., and Christ, D. (2007). Selection of human antibody fragments by phage display. Nat. Protoc. 2, 3001–3008. doi: 10.1038/nprot.2007.448
Moongkarndi, P., Rodpai, E., and Kanarat, S. (2011). Evaluation of an immunochromatographic assay for rapid detection of Salmonella enterica serovars Typhimurium and Enteritidis. J. Vet. Diagn. Invest. 23, 797–801. doi: 10.1177/1040638711408063
Morin, N. J., Gong, Z. L., and Li, X. F. (2004). Reverse transcription-multiplex PCR assay for simultaneous detection of Escherichia coli O157 : H7, Vibrio cholerae OI, and Salmonella typhi. Clin. Chem. 50, 2037–2044. doi: 10.1373/clinchem.2004.036814
Morton, J., Karoonuthaisiri, N., Charlermroj, R., Stewart, L. D., Elliott, C. T., and Grant, I. R. (2013a). Phage display-derived binders able to distinguish Listeria monocytogenes from other Listeria species. PLoS One 8:e74312. doi: 10.1371/journal.pone.0074312
Morton, J., Karoonuthaisiri, N., Stewart, L. D., Oplatowska, M., Elliott, C. T., and Grant, I. R. (2013b). Production and evaluation of the utility of novel phage display-derived peptide ligands to Salmonella spp. for magnetic separation. J. Appl. Microbiol. 115, 271–281. doi: 10.1111/jam.12207
Niyomdecha, S., Limbut, W., Numnuam, A., Kanatharana, P., Charlermroj, R., Karoonuthaisiri, N., et al. (2018). Phage-based capacitive biosensor for Salmonella detection. Talanta 188, 658–664. doi: 10.1016/j.talanta.2018.06.033
Noguera, P., Posthuma-Trumpie, G. A., van Tuil, M., van der Wal, F. J., de Boer, A., Moers, A. P., et al. (2011). Carbon nanoparticles in lateral flow methods to detect genes encoding virulence factors of Shiga toxin-producing Escherichia coli. Anal. Bioanal. Chem. 399, 831–838. doi: 10.1007/s00216-010-4334-z
Ou, F., McGoverin, C., Swift, S., and Vanholsbeeck, F. (2019). Near real-time enumeration of live and dead bacteria using a fibre-based spectroscopic device. Sci. Rep. 9:4807. doi: 10.1038/s41598-019-41221-1
Paoli, G. C., Chen, C.-Y., and Brewster, J. D. (2004). Single-chain Fv antibody with specificity for Listeria monocytogenes. J. Immunol. Methods 289, 147–155. doi: 10.1016/j.jim.2004.04.001
Qi, M., O’Brien, J. P., and Yang, J. (2008). A recombinant triblock protein polymer with dispersant and binding properties for digital printing. Biopolymers 90, 28–36. doi: 10.1002/bip.20878
Ross, G. M. S., Filippini, D., Nielen, M. W. F., and Salentijn, G. I. J. (2020). Unraveling the hook effect: a comprehensive study of high antigen concentration effects in sandwich lateral flow immunoassays. Anal. Chem. 92, 15587–15595. doi: 10.1021/acs.analchem.0c03740
Serrano, M. M., Rodríguez, D. N., Palop, N. T., Arenas, R. O., Córdoba, M. M., Mochón, M. D. O., et al. (2020). Comparison of commercial lateral flow immunoassays and ELISA for SARS-CoV-2 antibody detection. J. Clin. Virol. 129:104529. doi: 10.1016/j.jcv.2020.104529
Song, J., Matthews, A. Y., Reboul, C. F., Kaiserman, D., Pike, R. N., Bird, P. I., et al. (2011). “Chapter twelve - Predicting serpin/protease interactions,” in Methods in Enzymology, eds J. C. Whisstock and P. I. Bird (Cambridge, MA: Academic Press), 237–273. doi: 10.1016/B978-0-12-385950-1.00012-2
Spillner, E., Deckers, S., Grunwald, T., and Bredehorst, R. (2003). Paratope-based protein identification by antibody and peptide phage display. Anal. Biochem. 321, 96–104. doi: 10.1016/s0003-2697(03)00439-1
Wang, J., Morton, M. J., Elliott, C. T., Karoonuthaisiri, N., Segatori, L., and Biswal, S. L. (2014). Rapid detection of pathogenic bacteria and screening of phage-derived peptides using microcantilevers. Anal. Chem. 86, 1671–1678. doi: 10.1021/ac403437x
Xie, Q. Y., Wu, Y. H., Xiong, Q. R., Xu, H. Y., Xiong, Y. H., Liu, K., et al. (2014). Advantages of fluorescent microspheres compared with colloidal gold as a label in immunochromatographic lateral flow assays. Biosens. Bioelectron. 54, 262–265. doi: 10.1016/j.bios.2013.11.002
Xu, L., Bai, X., Tenguria, S., Liu, Y., Drolia, R., and Bhunia, A. K. (2020). Mammalian cell-Based immunoassay for detection of viable bacterial pathogens. Front. Microbiol. 11:575615. doi: 10.3389/fmicb.2020.575615
Yang, B., Gao, L., Li, L., Lu, Z., Fan, X., Patel, C. A., et al. (2003). Potent suppression of viral infectivity by the peptides that inhibit multimerization of human immunodeficiency virus type 1 (HIV-1) Vif proteins. J. Biol. Chem. 278, 6596–6602. doi: 10.1074/jbc.M210164200
Youn, J. H., Myung, H.-J., Liav, A., Chatterjee, D., Brennan, P. J., Choi, I.-H., et al. (2004). Production and characterization of peptide mimotopes of phenolic glycolipid-I of Mycobacterium leprae. FEMS Immunol. Med. Microbiol. 41, 51–57. doi: 10.1016/j.femsim.2004.01.001
Keywords: phage-derived antibody fragment, foodborne pathogen, Salmonella Enteritidis, lateral flow assay, colorimetric assay
Citation: Charlermroj R, Makornwattana M, Phuengwas S and Karoonuthaisiri N (2022) A rapid colorimetric lateral flow test strip for detection of live Salmonella Enteritidis using whole phage as a specific binder. Front. Microbiol. 13:1008817. doi: 10.3389/fmicb.2022.1008817
Received: 01 August 2022; Accepted: 15 September 2022;
Published: 29 September 2022.
Edited by:
Khamis Youssef, Agricultural Research Center, EgyptReviewed by:
Khaled A. El-Tarabily, United Arab Emirates University, United Arab EmiratesHoda S. El-Sayed, National Research Centre, Egypt
Shukho Kim, Kyungpook National University, South Korea
Copyright © 2022 Charlermroj, Makornwattana, Phuengwas and Karoonuthaisiri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ratthaphol Charlermroj, cmF0dGhhcGhvbC5jaGFAYmlvdGVjLm9yLnRo