 Emily A. Halajian
Emily A. Halajian Emmanuelle V. LeBlanc†
Emmanuelle V. LeBlanc† Katrina Gee
Katrina Gee Che C. Colpitts
Che C. Colpitts
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 29 September 2022
Sec. Virology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.1007081
This article is part of the Research Topic Virus Induced Innate Immune Response and Inflammation View all 8 articles
Recognition of viral infection by pattern recognition receptors is paramount for a successful immune response to viral infection. However, an unbalanced proinflammatory response can be detrimental to the host. Recently, multiple studies have identified that the SARS-CoV-2 spike protein activates Toll-like receptor 4 (TLR4), resulting in the induction of proinflammatory cytokine expression. Activation of TLR4 by viral glycoproteins has also been observed in the context of other viral infection models, including respiratory syncytial virus (RSV), dengue virus (DENV) and Ebola virus (EBOV). However, the mechanisms involved in virus-TLR4 interactions have remained unclear. Here, we review viral glycoproteins that act as pathogen-associated molecular patterns to induce an immune response via TLR4. We explore the current understanding of the mechanisms underlying how viral glycoproteins are recognized by TLR4 and discuss the contribution of TLR4 activation to viral pathogenesis. We identify contentious findings and research gaps that highlight the importance of understanding viral glycoprotein-mediated TLR4 activation for potential therapeutic approaches.
During the COVID-19 pandemic, a common feature of many severe SARS-CoV-2 cases has been an aggressive inflammatory response characterized by the uncontrollable release of high levels of proinflammatory cytokines, increased activation of immune cells, and harmful hyperinflammation (Mahmudpour et al., 2020; Ragab et al., 2020; Manik and Singh, 2021). This overabundant inflammatory response is termed “cytokine storm,” where a dysfunctional immune response leads to excessive amounts of inflammatory cytokines entering circulation, resulting in organ damage and potentially multi-organ failure and death (Fajgenbaum and June, 2020). Cytokine storms can be triggered from infections with various pathogens, including viruses such as SARS-CoV-2, Ebola virus (EBOV; Younan et al., 2017), dengue virus (DENV; Srikiatkhachorn et al., 2017; Dayarathna et al., 2020), and respiratory syncytial virus (RSV; Rosenberg and Domachowske, 2012).
A shared feature of these cytokine-storm inducing viruses mentioned above, as well as the vesicular stomatitis virus (VSV), is the production of virion-bound or released glycoproteins (Table 1; Figure 1). RSV fusion protein (F), VSV glycoprotein (G), EBOV glycoprotein (GP), and SARS-CoV-2 spike (S) are membrane-associated viral glycoproteins that mediate fusion of viral envelopes with host cell membranes (Olejnik et al., 2018), while DENV non-structural protein 1 (NS1) is a glycoprotein that is secreted from cells in a hexameric soluble form (sNS1) during infection and has exposed hydrophobic domains for membrane interaction (Akey et al., 2014; Figure 1). These proteins share conserved features, such as hydrophobic domains for membrane interactions (NS1 β-roll or RSV F, VSV G, EBOV GP and SARS-CoV-2 S fusion peptides) and glycosylation sites (Figure 1). Notably, each of these viral glycoproteins has been shown to activate Toll-like receptor 4 (TLR4; Olejnik et al., 2018), which plays a role in the induction of cytokine storms (Kaushik et al., 2021; Manik and Singh, 2021), although precisely how these viral proteins activate TLR4 is still unknown. Recently, similarities between systemic inflammation during viral infections and bacterial sepsis (Mahanty and Bray, 2004; Mehedi et al., 2011; Escudero-Pérez et al., 2014; Wolf et al., 2015; Sohn et al., 2020) have been noted. Bacterial sepsis involves an overwhelming and dysregulated host immune response and is characterized by high levels of bacterial lipopolysaccharide (LPS) interacting with TLR4, leading to the overexpression of inflammatory mediators (Kuzmich et al., 2017). Similarly, viral infections leading to overwhelming cytokine responses are often characterized by high levels of viral particles or viral proteins in patients (Mahanty and Bray, 2004; Okumura et al., 2010; Mehedi et al., 2011; Wolf et al., 2015; Guzman et al., 2016; Fajgenbaum and June, 2020; Sohn et al., 2020). Therefore, parallels between LPS-driven bacterial sepsis and virus-induced cytokine storm can be drawn, with a central role for TLR4 in leading to overwhelming systemic inflammation.

Table 1. Viral glycoproteins capable of TLR4 activation.
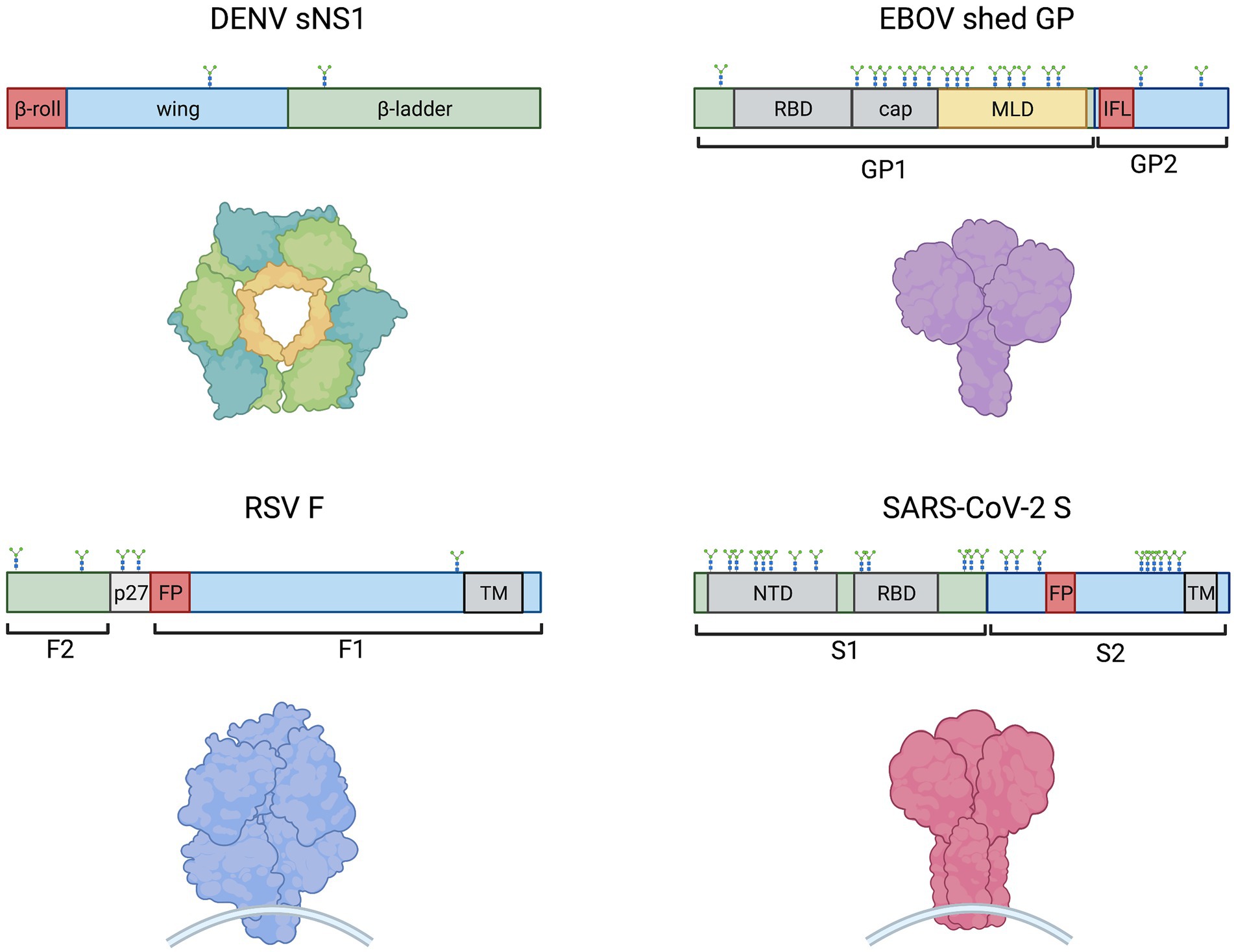
Figure 1. Schematic representations of monomeric DENV sNS1, EBOV shed GP, SARS-CoV-2 S and RSV F (clockwise) showing protein domain structure (above) and models of oligomeric proteins (below). Hydrophobic regions, such as the NS1 β-roll, the GP internal fusion loop (IFL) and fusion peptides (FP) are denoted in red. Other domains are indicated including receptor binding domains (RBD), glycan cap (cap), mucin-like domain (MLD), N-terminal domain (NTD), transmembrane domains (TM), and the RSV peptide 27 (p27). N-linked glycosylation sites are shown. The EBOV MLD shown in yellow also contains ~80 O-linked glycosylation sites (Cook and Lee, 2013).
TLR4 belongs to the Toll-like receptor family of transmembrane proteins that function as pattern recognition receptors (PRRs) recognizing pathogen- and damage-associated molecular patterns (PAMPs; DAMPs) to induce innate immune responses via downstream signaling pathways (Lester and Li, 2014; Olejnik et al., 2018). Structurally, TLR4 is comprised of an extracellular leucine rich repeat (LRR) domain, a transmembrane domain, and an intracellular Toll/Interleukin-1 receptor like (TIR) domain with which adaptor proteins TIR domain-containing adaptor protein (TIRAP) and TRIF-related adaptor molecule (TRAM) interact to trigger downstream signaling cascades (Kuzmich et al., 2017). The TLR4 signaling complex consists of cluster of differentiation 14 (CD14), myeloid differentiation factor-2 (MD-2), TLR4, and various adaptor proteins that initiate downstream signaling pathways in a dynamic manner.
Classically, CD14 presents monomers of LPS to MD-2 (Kuzmich et al., 2017; Olejnik et al., 2018). Binding of LPS or other ligands to the deep hydrophobic pocket of MD-2 causes TLR4/MD-2 to dimerize and activates the TLR4 signaling complex at the plasma membrane (Park and Lee, 2013; Kuzmich et al., 2017; Olejnik et al., 2018), resulting in recruitment of intracellular adaptor protein TIRAP to then recruit MyD88 (Park et al., 2009). The resulting MyD88-dependent downstream signaling cascade ultimately results in early activation of nuclear factor kappa B (NF-κB) and proinflammatory cytokine secretion (Akira and Takeda, 2004; Figure 2) consistent with cytokine storm. MyD88-dependent signaling activates proinflammatory cytokines such as interleukin (IL)-6, tumour necrosis factor alpha (TNFα), chemokine (C-X-C motif) ligand 1 (CXCL1), IL-1α, IL-1β, and IL-12 (Yamamoto et al., 2003; Björkbacka et al., 2004; Meissner et al., 2013). Experiments in MyD88-deficient cells have shown that lack of MyD88 involves decreased or abolished inflammatory mediator production in response to LPS stimulation (Kawai and Akira, 2007; Yamamoto and Takeda, 2010), although interferon regulatory factor 3 (IRF3) activation and interferon (IFN)-induced gene expression remain unaffected (Kawai et al., 2001; Hoshino et al., 2002).
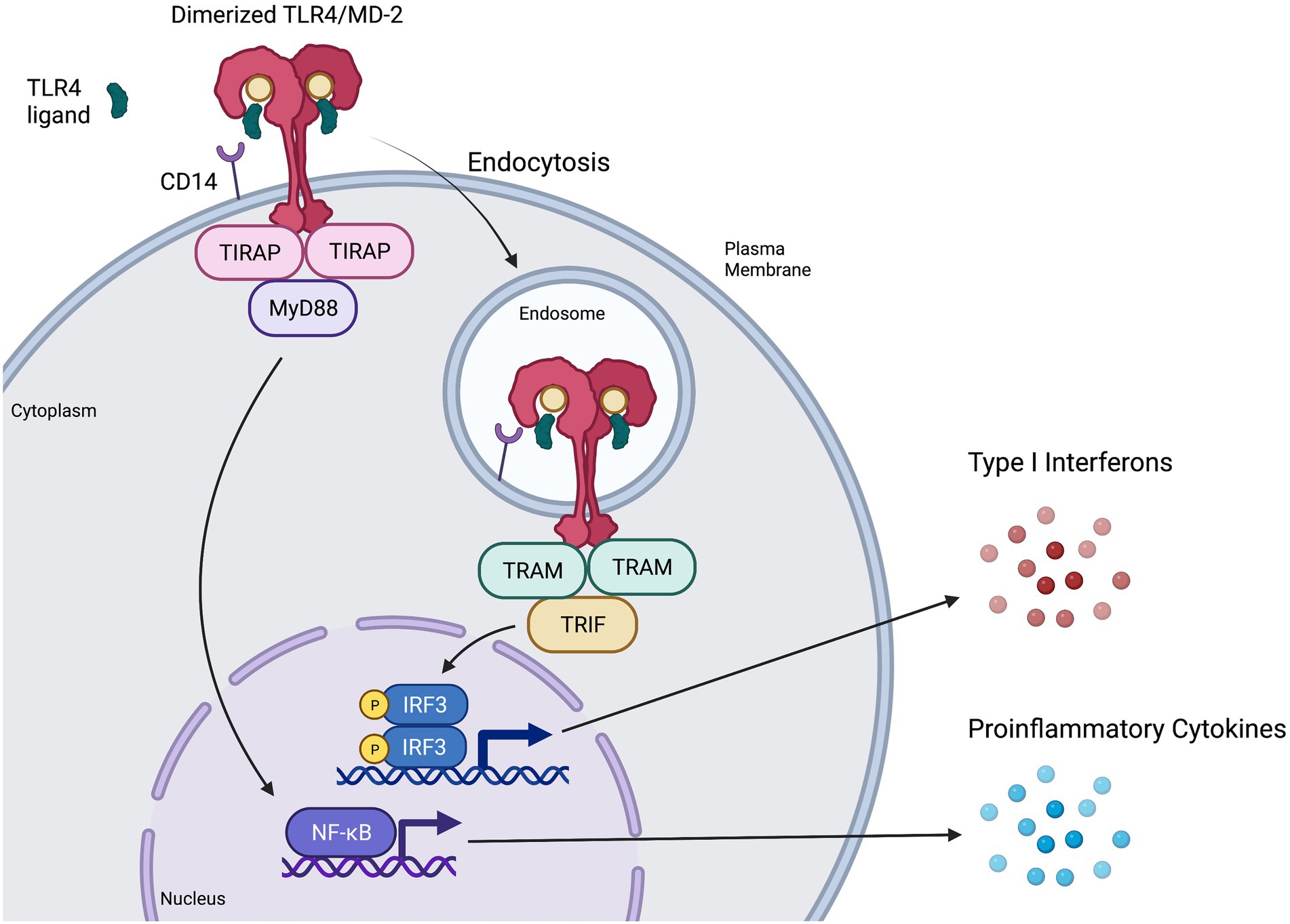
Figure 2. Representation of MyD88-dependent and-independent TLR4 signaling pathways. CD14 presents ligands such as LPS or RSV F protein to individual TLR4/MD-2 complexes, triggering them to dimerize and activate (Park and Lee, 2013; Kuzmich et al., 2017). The TIR domains of the dimerized TLR4/MD-2 complex interacts with TIR domains of the adaptor protein TIRAP (Kuzmich et al., 2017), which then recruits MyD88 to initiate the MyD88-dependent signaling pathway. This pathway results in NF-κB activation and proinflammatory cytokine induction. Once the MyD88-dependent pathway has completed, the dimerized TLR4/MD-2 complex is internalized into an endosome (Kagan et al., 2008; Zanoni et al., 2011), a process that is thought to be at least partially mediated by CD14 and MD-2. Upon internalization, the adaptor protein TRAM interacts with the TIR domains of the dimerized TLR4/MD-2 complex and recruits TRIF, initiating the MyD88-independent (or TRIF-dependent) signaling pathway. The MyD88-independent pathway activates interferon regulatory factor (IRF3) leading to the production of type I interferons (IFN) and activation of interferon-stimulated genes (ISGs).
TLR4 internalization via endocytosis is associated with the initiation of MyD88-independent signaling (Kagan et al., 2008; Tanimura et al., 2008; Zanoni et al., 2011; Goulopoulou et al., 2016; Ciesielska et al., 2021). CD14 is thought to have a major role both in regulating the endocytosis of TLR4 (Zanoni et al., 2011; Tan et al., 2015; Marongiu et al., 2019) and in activating the MyD88-independent pathway (Jiang et al., 2005). MD-2 has also shown a potential role in promoting TLR4 transport to the endosome, as well as promoting ligand transport to the TLR4 receptor (Tan et al., 2015). Adaptor protein TRAM is thought to aid in regulating endocytosis of the TLR4 signaling complex (Zanoni et al., 2011). Furthermore, TRAM recruits TIR-domain-containing adapter-inducing IFN-β (TRIF; Tanimura et al., 2008) to induce MyD88-independent signaling (Figure 2), leading to late-wave activation of NF-κB and the induction of some antiviral cytokines and chemokines such as CXCL10 (Yamamoto et al., 2002; Björkbacka et al., 2004). Importantly, MyD88-independent signaling also leads to the activation of IRF3, resulting in the production of type I IFN (Akira and Takeda, 2004; Kagan et al., 2008), induction of IFN-stimulated genes (Kawai et al., 2001) and an antiviral state (O’Neill and Bowie, 2007).
TLR4 signaling is dynamic, with activation of MyD88-dependent and MyD88-independent pathways leading to inflammatory or antiviral responses, respectively (Lester and Li, 2014). MyD88-independent signaling leads to delayed NF-κB activation, IRF3 activation, IFN induction and a beneficial antiviral response. Consistently, studies on MyD88-deficient mice finding reduced proinflammatory cytokine induction but increased activation of IRF3 and IFN-β (Kawai et al., 2001; Hoshino et al., 2002; Yamamoto et al., 2003; Takeda and Akira, 2004). In contrast, triggering of the MyD88-dependent pathway leads to early NF-κB activation and induction of proinflammatory cytokines such as TNFα and IL-6 (Kagan and Medzhitov, 2006), the overproduction of which can have a detrimental effect on the host (Molteni et al., 2016). In the context of bacterial sepsis or viral infection, it is possible that high levels of circulating LPS or viral proteins could continually engage TLR4 on the cell surface, activating MyD88-dependent signaling, while the rate-limiting step of endocytosis may lessen the extent of MyD88-independent signaling in comparison, shifting the balance towards a proinflammatory response consistent with cytokine storm.
While the precise mechanisms explaining how viral glycoproteins may interact with and activate TLR4 and its downstream signaling cascades are still unclear, several potential mechanisms have been proposed. There is some evidence to suggest that physical and hydrophobic interactions occur between these viral glycoproteins and the TLR4 signaling complex, likely through the hydrophobic pocket of MD-2 and hydrophobic fusion peptides or other domains on viral protein surfaces. It has also been proposed that glycosylation may play a role, as all of the viral proteins thought to activate TLR4 are glycosylated. Finally, there is the possibility that a particular oligomeric state of the viral proteins enables interactions with the TLR4 signaling complex. For example, DENV NS1 occurs in both dimeric or hexameric forms during infection, while viral fusion proteins such as EBOV GP, SARS-CoV-2 S, and RSV F are class I fusion proteins that are homotrimers and require proteolytic activation for fusion, which could potentially result in the release of shed fragments of these proteins. Furthermore, the downstream signaling pathways that are induced by specific viral protein-TLR4 interactions still remain to be elucidated. Further understanding of these mechanisms is necessary to determine whether the interaction of viral glycoproteins with TLR4 leads predominantly to an antiviral response or to a proinflammatory cytokine response, or both.
In this review, we summarize the current understanding of TLR4-viral protein interactions, emphasizing the similarities and differences between different viral glycoproteins, including SARS-CoV-2 S. We highlight future directions for investigation and potential therapeutic approaches, which could be employed to ameliorate cytokine storms induced by viruses such as SARS-CoV-2, DENV, EBOV and RSV.
Several viral glycoproteins have been shown to activate TLR4 (Table 1), with experimental evidence for viral glycoprotein-TLR4 interactions ranging from physical interactions between glycoproteins and the TLR4 signaling complex to the induction of downstream signaling pathways and the release of proinflammatory cytokines. In several cases, expression of these cytokines following activation by viral glycoproteins is blocked by TLR4 inhibitors or abrogated in TLR4 deficient models, consistent with a specific role for TLR4. However, the mechanisms underlying TLR4 activation by these proteins is still poorly understood. It should be noted that influenza virus infection has also been associated with TLR4 activation, but this has been attributed to the production of DAMPs such as high mobility group box 1 (HMGB1) from infected cells, rather than a viral glycoprotein (Shirey et al., 2021). While it is possible that DAMPs also contribute to TLR4 activation in the context of other viral infections, we have focused this review on the current literature implicating a specific role for viral glycoproteins in activation of TLR4.
Due to the role of DENV NS1 in contributing to vascular leakage in dengue hemorrhagic fever/dengue shock syndrome, researchers examined NS1-TLR4 interactions on platelets. Chao et al. (2019) observed decreased levels of NS1 binding to platelets in the presence of anti-TLR4 antibodies, or to platelets from TLR4 knockout mice, indicating that DENV NS1 binds to platelets through TLR4. The same study also compared the binding of NS1 to TLR4 and to TLR2 using enzyme-linked immunosorbent assays and concluded that NS1 had a stronger affinity for TLR4. Quirino-Teixeira et al. (2020) demonstrated that NS1 competed with LPS for binding to human platelets. While it is unlikely that platelets play a role in cytokine storm induction, these studies demonstrated an interaction between NS1 and TLR4. Consistently, studies demonstrated that NS1 and TLR4 colocalized on the surface of peripheral blood mononuclear cells (PBMCs; Modhiran et al., 2015). Similarly, flow cytometry and confocal microscopy approaches showed that EBOV GP binds to TLR4 on T cells (Iampietro et al., 2017), macrophages, and dendritic cells (Escudero-Pérez et al., 2014). Both EBOV GP and SARS-CoV-2 S were observed to interact directly with transfected TLR4 in co-immunoprecipitation studies in HEK 293 T cells (Okumura et al., 2010; Iampietro et al., 2017; Negron et al., 2021). For SARS-CoV-2, it was shown that the S1 subunit (containing the receptor binding domain) but not the S2 subunit (containing the hydrophobic fusion machinery; Figure 1) of the S protein interacted with TLR4 (Negron et al., 2021). Furthermore, the LRR but not TIR domain of TLR4 was required, indicating that the spike S1 subunit binds to the extracellular domain of TLR4. The interaction between TLR4 and the recombinant SARS-CoV-2 spike timer has also been confirmed by surface plasmon resonance (SPR; Zhao et al., 2021). Notably, in the same study, proinflammatory cytokine induction was observed in response to the spike trimer but not to the receptor binding domain (RBD) nor the N-terminal domain (NTD) alone. Based on computational modeling, the authors proposed that TLR4 may interact with a conformational concave constructed by RBD and NTD, both of which are found within the S1 subunit of spike (Figure 1).
In the context of RSV, the purified fusion (F) protein has not yet been shown to directly interact with TLR4, but was observed to co-immunoprecipitate with recombinant MD-2 of the TLR4 signaling complex (Rallabhandi et al., 2012). To identify which portion of the F protein mediated this interaction, the authors made four overlapping polypeptides spanning the length of F1 and observed that only the N-terminal peptide of F1, which contains the hydrophobic fusion peptide (Figure 1), was pulled down in an MD-2/TLR4-dependent manner.
It is still unclear how these viral glycoproteins are recognized by the TLR4 signaling complex. Given that the canonical ligand LPS binds to a hydrophobic pocket of MD-2 to induce TLR4 activation (Park et al., 2009), it is possible that hydrophobic regions of viral proteins may mimic this effect. Indeed, the glycoproteins of VSV, RSV, EBOV and SARS-CoV-2 mediate fusion with host cell membranes using a hydrophobic fusion peptide, and DENV NS1 contains exposed hydrophobic domains. One possibility, therefore, is that viral glycoproteins may mediate TLR4 activation through hydrophobic interactions.
It was previously shown that NS1 competes with LPS for binding to platelets (Quirino-Teixeira et al., 2020), suggesting that these two ligands may bind TLR4 in a similar manner. Consequently, the TLR4 inhibitor LPS-RS, a potent LPS antagonist which competes with LPS for binding to MD-2 of the TLR4/MD-2 complex (Tam et al., 2021), blocks DENV NS1-induced activation of platelets and murine RAW 264.7 macrophages in vitro (Chao et al., 2019; Coelho et al., 2021) and downstream cytokine induction and immune cell activation (Modhiran et al., 2015, 2017). Similarly, LPS-RS prevented EBOV GP-induced NF-κB activation (Olejnik et al., 2017) and reduced GP-induced cytokine production and immune cell recruitment (Lai et al., 2017).
In addition to LPS-RS, Eritoran (a lipid A analog also called E5564), which binds the deep hydrophobic pocket of MD-2 (Kim et al., 2007), inhibited RSV F-mediated TLR4 activation (Rallabhandi et al., 2012), again suggesting a shared binding site on MD-2. The N-terminal fragment peptide of F1, which contains the hydrophobic fusion peptide, reduced LPS-induced NF-κB transcriptional activity, further supporting that it requires the same binding site of MD-2 (Rallabhandi et al., 2012). Similarly, a recent study identified the hydrophobic fusion loop of EBOV GP as essential for TLR4/MD-2 signaling, with docking analysis predicting that the hydrophobic fusion loop binds into the MD-2 pocket (Scherm et al., 2022). Activation of TLR4 by exposed hydrophobic fusion peptides suggests that TLR4 may recognize post-fusion (rather than pre-fusion) conformations of viral glycoproteins, although further experiments are required to explicitly test this possibility.
Consistent with these findings, induction of TNFα expression in THP-1-derived macrophages by SARS-CoV-2 S was also suppressed by LPS-RS (Shirato and Kizaki, 2021). While molecular docking studies have provided some insight, structural analyses to understand the molecular basis for the recognition of these viral proteins by the TLR4 signaling complex are lacking. Notably, molecular binding models of the SARS-CoV-2 S trimer to the TLR4/MD-2 dimer have been proposed (Zhao et al., 2021), showing the protein surface representations and glycans. TLR4 and MD-2 glycosylation are thought to play an important role in the LPS recognition and signal transduction for cell activation (da Silva Correia and Ulevitch, 2002). However, the roles of glycans on the viral glycoproteins in TLR4 activation are still under investigation.
Considering that EBOV GP, DENV NS1, RSV F, VSV G, and SARS-CoV-2 S proteins are glycosylated, and that the core saccharides of LPS may modulate endotoxin activity (Cochet and Peri, 2017), it has been hypothesized that glycosylation of the viral proteins could have a role in TLR4-viral glycoprotein interactions. N-linked glycans on cellular proteins are highly processed into hybrid and complex-type glycan structures, whereas those on viral glycoproteins are often under-processed, resulting in oligomannose-type glycans not typically found on cellular proteins (Watanabe et al., 2019). It is possible that aberrant glycosylation patterns could activate TLR4. To date, however, only glycosylation of EBOV GP has been demonstrated to have a role in TLR4 activation, although these findings remain controversial. By treating EBOV shed GP with a combination of deglycosylases, researchers observed a reduction in TLR4-dependent TNFα secretion by both dendritic cells and macrophages, suggesting that the glycosylation pattern of EBOV shed GP is important for the recognition by TLR4 on immune cells (Escudero-Pérez et al., 2014). Contrarily, Scherm et al. (2022) designed EBOV GP mutants for individual N-glycosylation sites identified by glycosylation prediction software and found that all of these mutants could still activate TLR4 similarly to wild-type EBOV GP. While global deglycosylation of shed EBOV GP by glycosidase treatment significantly reduced TLR4 activation, consistent with the findings of Escudero-Pérez et al. (2014), Scherm et al. (2022) attributed this phenotype to instability or degradation of GP, rather than a specific role for N-linked or O-linked glycans on GP.
EBOV GP has a mucin-like domain that is rich in N- and O-glycans. Co-immunoprecipitation studies revealed that a mucin domain deletion mutant of GP retained the ability to interact with TLR4 (Okumura et al., 2010), suggesting that interaction with TLR4 is not dependent on glycosylation. Consistently, deglycosylated GP was still capable of binding both TLR4 and MD-2 (Scherm et al., 2022). However, EBOV virus-like particles (VLPs) with the mucin domain of GP deleted did not activate NF-κB reporter activity (Okumura et al., 2010), which could reflect instability or degradation of GP, consistent with the findings of Scherm et al. (2022). While the current evidence suggests that glycosylation does not play a direct role in activation of TLR4 by EBOV GP, future studies are needed to evaluate the role of glycans for other viral glycoproteins that activate TLR4.
Lectins, ubiquitously expressed carbohydrate-binding proteins, have been reported to interact with TLRs and have immunomodulatory properties. For example, mannan-binding lectin (MBL) can bind to TLR4 and suppress LPS-induced proinflammatory cytokine production (Wang et al., 2011), although multiple other lectins are reported to act as potent TLR4 agonists (Ricci-Azevedo et al., 2017). It has been proposed that lectin binding to N-linked glycans on TLRs can directly or indirectly activate receptors and induce cell signaling (Ricci-Azevedo et al., 2017). It is interesting to note that several lectins described to activate TLR4 (Park et al., 2010; Unitt and Hornigold, 2011) have specificity for terminal galactose or N-acetylgalactosamine. The spike protein of certain coronaviruses, including SARS-CoV-2, possess a “galectin-fold” with structural homology to human galectin-3, which may have potential relevance for monocyte activation by SARS-CoV-2 (Schroeder and Bieneman, 2022). However, further experiments are required to assess the relevance of the galectin fold for activation of TLR4 by SARS-CoV-2 spike.
Viral glycoproteins such as EBOV GP and DENV NS1 occur in multiple forms (e.g., shed, secreted, cell-associated or virion-associated) during infection. The question of whether the glycoproteins must occur in a specific form such as shed or secreted, virion- or non-virion associated, to interact with TLR4 remains to be fully addressed. Unlike other TLR4-activating viral glycoproteins, DENV NS1 is a non-structural viral protein that can be secreted as a hexamer formed by a trimer of stable NS1 dimers from DENV-infected insect or mammalian cells (Modhiran et al., 2017). N-linked glycosylation of sNS1 is required for its stability (Somnuke et al., 2011), allowing sNS1 to circulate at high levels in the blood of dengue patients for the duration of illness (Libraty et al., 2002; Guzman et al., 2016). Interestingly, sNS1 produced from insect and mammalian cells activated immune cells in a TLR4-dependent manner, whereas NS1 derived from E. coli did not (Modhiran et al., 2017). E. coli-derived NS1 lacks glycosylation and is produced only as a monomer (Modhiran et al., 2017), implicating NS1 glycosylation or quaternary structure in activation of TLR4.
In vitro and in vivo studies have demonstrated that secreted NS1 plays a crucial role in severe dengue immunopathology (Libraty et al., 2002; Carvalho et al., 2014), with levels of circulating sNS1 correlating with risk of developing dengue hemorrhagic fever, a more severe form of dengue disease. TLR4 recognizes and interacts with sNS1 and is a mediator for some of the key roles associated with sNS1 in DENV pathogenesis, including elicitation of inflammatory cytokine production (Modhiran et al., 2015), increased DENV platelet activation (Quirino-Teixeira et al., 2020), and enhanced DENV attachment to host cells (Coelho et al., 2021), as well as endothelial glycocalyx layer disruption (Puerta-Guardo et al., 2016).
EBOV GP is found on the virion surface as a trimer, but is also present in the form of a non-structural soluble GP (sGP) dimer secreted from infected cells, and as shed GP, which is a truncated form of GP cleaved from the plasma membrane by cellular proteases. Early studies on EBOV compared the authentic virus with VLPs or purified GP. EBOV GP alone or VLPs containing GP, but not VLPs without GP, were found to induce TLR4-mediated NF-κB activation (Okumura et al., 2010; Olejnik et al., 2017) and induce suppressor of cytokine signaling 1 (SOCS1) RNA expression (Okumura et al., 2010), highlighting the role of GP as the trigger for TLR4 activation. Escudero-Pérez et al. (2014) demonstrated that shed GP but not secreted sGP activates dendritic cells and macrophages, resulting in a substantial upregulation of several cytokines in a TLR4-dependent manner. Iampietro et al. (2018) later observed that cell supernatant containing shed GP was sufficient to induce monocyte differentiation, an effect requiring functional TLR4, and ultimately resulting in increased infection and cell death. Recently, Scherm et al. (2022) demonstrated that cleavage by TACE protease is required for activation of TLR4/MD-2 by EBOV GP.
While DENV sNS1 and EBOV shed GP are likely the major forms associated with TLR4 activation, it is unclear if there are analogous counterparts for RSV and SARS-CoV-2 glycoproteins. Although many studies have reported that recombinant RSV F protein mediates TLR4 activation, Marr and Turvey (2012) observed no NF-κB transcriptional activity in HEK293 reporter cells transfected with TLR4, MD-2 and CD14 in response to three strains of authentic RSV. One explanation could be that it is not virion-associated F that triggers TLR4, but rather a soluble secreted or truncated form. While F2 remains covalently attached to membrane-associated F1 through disulfide bonds, there is a small segment, p27, between two furin cleavage sites that must be released, as both cleavage events are required for fusion (González-Reyes et al., 2001). Nevertheless, Lizundia et al. (2008) found that NF-κB activity was induced in response to the A2 strain of RSV but not purified F protein, using the same HEK293 reporter system. Thus, there is still no consensus as to which form of F activates TLR4 in a physiologically relevant context. Notably, a secreted form of the RSV attachment protein G has been described (Roberts et al., 1994). Secreted RSV G acts as an antigen decoy (Bukreyev et al., 2008) and has also been proposed to modulate cellular immunity by interacting with TLRs. One study proposed that secreted soluble G inhibits TLR3/TLR4-mediated ISG activation via the TRIF pathway, providing an explanation for the lack of IFN-β induction in dendritic cells in response to RSV (Shingai et al., 2008). However, RSV G may have a role in mediating the proinflammatory response against RSV, as a recent study suggested a role for RSV G in binding to and activating the TLR2/TLR6 heterodimer, with a potential partial role in activating TLR4 also described (Alshaghdali et al., 2021).
Khan et al. (2021) observed that SARS-CoV-2 S1 or S2 peptides induced proinflammatory cytokines in lung epithelial A549 and Calu-3 cells, but transfecting these cells with a plasmid encoding S did not result in induction, suggesting that cell-associated S does not induce proinflammatory cytokines. Since SARS-CoV-2 S1 is sufficient to mediate interaction and activation of TLR4, it has been suggested that circulating S1, which may be released from the viral surface following proteolytic cleavage, could be the trigger for a TLR4-mediated inflammatory response (Negron et al., 2021). While the exact form or epitope of viral glycoproteins required to interact with TLR4 remain under investigation, the current knowledge of the resulting signaling and implications for pathogenesis are explored below.
Upon recognition of specific ligands, the TLR4 signaling complex induces downstream signaling pathways. Activation of these pathways begins with the recruitment of adaptor proteins such as MyD88 or TRIF. The MyD88-dependent pathway results in the activation of NF-κB and the production of proinflammatory cytokines, while the MyD88-independent pathway results in the production of IFNs (Figure 2). Determining which signaling pathway is activated and which proinflammatory cytokines and chemokines are produced from each viral glycoprotein-TLR4 interaction will aid in understanding the mechanisms underlying how each viral glycoprotein activates TLR4 and the role of this interaction in viral pathogenesis.
VSV G was shown to activate the TLR4 signaling pathway in a TRAM-dependent MyD88-independent manner that does not activate NF-κB (Georgel et al., 2007). In response to VSV G, myeloid dendritic cells produced type-I IFN in a CD14- and TLR4-dependent manner. Consequently, TLR4- or CD14-deficient murine macrophages were much more susceptible to VSV infection. In the context of in vivo VSV infection, TLR4-mutant mice known to be defective for TLR4 responses had a higher mortality rate than their wildtype counterparts, highlighting the importance of TLR4 in antiviral defense against VSV (Georgel et al., 2007). While no studies to date have investigated the mechanisms underlying TLR4 activation by VSV G, it may be that its ligand for TLR4 is presented only in endosomes (e.g., the hydrophobic fusion peptide exposed in the post-fusion conformation), thus bypassing activation of cell-surface MyD88-dependent TLR4 signaling that would otherwise lead to proinflammatory responses.
In contrast to VSV G, TLR4 activation by other viral glycoproteins appears to lead to a disproportionate proinflammatory response. DENV sNS1 induces cytokine release upon interaction with TLR4 (Modhiran et al., 2017). In one study, cytokine responses to sNS1 were lost in mice lacking TLR4, MyD88, or TRIF, suggesting that DENV NS1 interaction with TLR4 triggers both the MyD88-dependent and-independent signaling pathways (Modhiran et al., 2015). EBOV GP has also been found to activate both the MyD88-dependent and-independent downstream signaling pathways (Iampietro et al., 2017; Olejnik et al., 2017). Reporter assays in TLR4/MD-2 expressing cells revealed that EBOV GP induced the expression of genes driven by both NF-κB and IFN-β promoters (Okumura et al., 2010). Consequently, EBOV GP stimulates increased cytokine (Lai et al., 2017; Wagstaffe et al., 2020) and SOCS1 or SOCS3 expression (Okumura et al., 2010, 2015). Multiple studies have shown that NF-κB activation by EBOV GP is dependent on TLR4 (Okumura et al., 2010; Iampietro et al., 2017; Olejnik et al., 2017). Consequently, blocking TLR4 activity with the anti-TLR4 antibodies or TLR4 inhibitors prevented EBOV GP-induced release of various cytokines (Escudero-Pérez et al., 2014; Lai et al., 2017; Wagstaffe et al., 2020), recruitment of antigen-presenting cells (Lai et al., 2017), cell death (Okumura et al., 2015; Iampietro et al., 2017, 2018), and the differentiation and activation of various immune cell types (Iampietro et al., 2017, 2018; Wagstaffe et al., 2020). Thus, unlike in the context of VSV infection, where TLR4 plays a clearly protective role, dampening signaling mediated by TLR4 in response to EBOV or DENV may be beneficial. While some TLR4 activation may be crucial for the activation of some immune cell types, the proinflammatory response may be disproportionately detrimental.
Interestingly, the in vivo role of TLR4 in response to RSV infection remains controversial although, like EBOV GP and DENV NS1, RSV F has been shown to trigger the increased secretion of proinflammatory cytokines via TLR4/MD-2/CD14 recognition and signaling (Kurt-Jones et al., 2000). In vitro studies have confirmed the importance of both MD-2 and CD14, the same components of the signaling complex involved in LPS-induced TLR4 activation (Kurt-Jones et al., 2000; Rallabhandi et al., 2012). The physiological relevance of the resulting signaling and effect on pathogenesis has been well explored in vivo. Inoculation of mice with RSV leads to rapid increase in NF-κB DNA-binding activity in nuclear extracts of lung tissue, which was dependent on alveolar macrophages and required functional TLR4 (Haeberle et al., 2002). Notably, the pioneering study in this area found that TLR4 deficient mice infected intranasally with RSV had a higher viral burden in their lungs, suggesting that TLR4 is important in RSV clearance (Kurt-Jones et al., 2000). However, it was later noted that the mice strain used (C57BL10/ScCr) not only had a TLR4 deletion, but also a defect in the IL-12 receptor. However, Haynes et al. (2001) used C57BL/10ScNCr mice (which are distinct from the C57BL10/ScCr strain in that they carry wild-type IL-12R) to confirm that the lack of TLR4 impaired RSV clearance (Haynes et al., 2001). In contrast, Ehl et al. (2004) leveraged several C57BL/10 and BALB/c mouse strains lacking TLR4, IL-12R or both to conclude that TLR4 had no impact on RSV elimination in vivo (Ehl et al., 2004). Thus, the role of TLR4 in RSV infection appears variable, depending on the mouse strain used.
As with RSV F, the response to SARS-CoV-2 S is proinflammatory. Both S1 and S2 peptides have been shown to increase proinflammatory cytokine expression in THP-1 derived macrophages while having no effect on IFN-α, IFN-β or INF-γ induction (Chiok et al., 2021; Khan et al., 2021). Reporter assays in HEK293T cells revealed that, like RSV F, SARS-CoV-2 S stimulates NF-κB transcriptional activity (Negron et al., 2021). SARS-CoV-2 S1-mediated activation of NF-κB and MAPK pathways has now been demonstrated in human PBMCs, murine peritoneal macrophages, and murine microglial cells (Olajide et al., 2021, 2022; Shirato and Kizaki, 2021). Proinflammatory cytokine induction in response to S1, S trimer, S pseudoparticles or authentic virus was decreased in the presence of inhibitors of NF-κB, c-Jun N-terminal kinase (JNK), TLR4, MD-2, or by treatment with anti-CD14 antibody (Shirato and Kizaki, 2021; Zhao et al., 2021). The proinflammatory response to SARS-CoV-2 S was also abrogated in bone marrow derived macrophages from TLR4−/− mice (Zhao et al., 2021) or with siRNA targeting TLR4 (Shirato and Kizaki, 2021; Olajide et al., 2022), further confirming the importance of TLR4 in the induction of proinflammatory cytokines. Nonetheless, further research is needed to understand whether TLR4 activation has a protective or detrimental role in viral infection, and the relative role of TLR4 compared to other PRRs.
Many studies identified a role for SARS-CoV-2 S in the activation of TLR4 using recombinant purified S trimer or S1, either produced in house or purchased commercially. Cinquegrani et al. (2022) investigated multiple sources of recombinant SARS-CoV-2 S1, checking each for levels of endotoxin contamination, and observed a varied response to these purified proteins in monocyte-derived macrophages, and ultimately proposed that the activation of macrophages correlated with endotoxin contamination or with lack of glycosylation for S1 produced in E. coli as opposed to mammalian cells. However, some studies have confirmed their findings with S pseudotyped lentiviral particles or authentic virus (Chiok et al., 2021; Zhao et al., 2021), indicating that endotoxin contamination is unlikely to be the sole driver for proinflammatory responses. Still, others also report a lack of authentic activation in their model systems. For instance, monocyte-derived dendritic cells, which abundantly express TLR4 and other TLRs, were not activated by authentic SARS-CoV-2 particles, S pseudoparticles or recombinant S protein (van der Donk et al., 2022). While the evidence points towards S1 or S2 as the trigger for a proinflammatory response, and TLR4 or TLR2 as the primary mediator of this response (Khan et al., 2021; Shirato and Kizaki, 2021; Zhao et al., 2021; Frank et al., 2022), another study suggested a role for the SARS-CoV-2 E protein in the production of inflammatory cytokines (Zheng et al., 2021). The interplay between SARS-CoV-2 and TLRs is clearly complex and worthy of further investigation, as it is crucial to understand the immunopathology of COVID-19.
Due to the role of TLR4 in modulating an immune response, the use of TLR4 agonists as potential vaccine adjuvants for DENV, EBOV and RSV have been proposed and are under investigation (Sunay et al., 2019; Zheng et al., 2020; Kayesh et al., 2021). However, in the context of infection, dampening TLR4 activation may be beneficial. Thus, research into the use of TLR4 inhibitors in the treatment of EBOV infection has been recommended (Denner, 2015). In animal models of sepsis, dampening TLR-induced inflammation led to reduced sepsis progression. Specifically, reducing or blocking MyD88 led to decreased systemic hyperinflammation (Weighardt et al., 2002; Weighardt and Holzmann, 2008). These findings could potentially be applied to virus-induced TLR4 activation, as overwhelming activation of TLR4 by LPS in sepsis may be similar to the induction of cytokine storm by virus glycoprotein-TLR4 interactions (Perrin-Cocon et al., 2017; Sohn et al., 2020).
As reviewed here, in vitro studies have demonstrated that TLR4 inhibitors dampen MyD88-dependent signal transduction and the release of inflammatory cytokines in response to EBOV GP, RSV F and SARS-CoV-2 S (Rallabhandi et al., 2012; Iampietro et al., 2017, 2018; Lai et al., 2017; Olejnik et al., 2017; Shirato and Kizaki, 2021; Zhao et al., 2021). Notably, a Japanese pharmaceutical company is participating in collaborative COVID-19 clinical trials to test the efficacy of a TLR4 antagonist, Eritoran (E5564), in reducing cytokine storm (Tsukahara, 2022). Consistently, Eritoran had been found to confer significant benefit in the context of influenza infection in mice by blocking TLR signaling (Shirey et al., 2013). Beyond specific TLR4 inhibitors, dexamethasone was shown to significantly reduce SARS-CoV-2 S1-dependent cytokine induction in human PBMCs (Olajide et al., 2021). Although the mechanism has not been fully elucidated, it was observed that dexamethasone pre-treatment reduced activation of NF-κB and MAPK pathways in PBMCs in response to S1, or in human umbilical vein endothelial cells infected with authentic SARS-CoV-2, further supporting the physiological relevance of this inhibitory activity (Ma et al., 2022). Finally, most recently, SARS-CoV-2 S-binding DNA aptamers that selectively disrupt the SARS-CoV-2 S-TLR4 interaction have been identified (Yang et al., 2022). Aptamer treatment of S trimer or authentic SARS-CoV-2 prevented proinflammatory cytokine production by monocytes and neutrophils, while maintaining responsiveness to LPS stimulation (Yang et al., 2022). The specific anti-inflammatory activity in vitro and demonstrated low immunogenicity in mice warrant further investigations into the potential therapeutic utility of these aptamers.
Overall, further in vitro and in vivo studies will be important to elucidate the molecular mechanisms and better understand the physiological relevance of viral glycoprotein-TLR4 activation. These studies are required to explore the therapeutic potential of targeting TLR4 in the context of viral infection to ameliorate cytokine storm.
Further characterization of the mechanisms underlying TLR4 recognition of viral glycoproteins is critical to understand unbalanced inflammatory responses leading to cytokine storm during viral infection. Structural and biochemical studies may elucidate a common molecular basis for viral glycoprotein-TLR4 complex interaction and activation. Further research to identify specific domains of viral glycoproteins that interact with TLR4, as well as to determine how the viral glycoprotein-TLR4 interaction leads to uncontrolled inflammatory cytokine responses, as opposed to a controlled anti-viral response (Olejnik et al., 2018), will be instrumental in understanding viral disease pathogenesis and identifying targets for therapeutic intervention.
The outcomes of viral glycoprotein-TLR4 interactions are complex. Multiple studies on DENV NS1 and EBOV GP demonstrated that these viral glycoproteins induce differentiation and activation of various immune cells (Iampietro et al., 2017, 2018; Olejnik et al., 2017; Wagstaffe et al., 2020; Coelho et al., 2021), emphasizing a broad role for viral glycoprotein-TLR4 interactions in immune responses. NS1-TLR4 interactions modulate the pathogenesis of DENV through a variety of mechanisms, including lipid raft accumulation for cell attachment (Coelho et al., 2021), disruption of the endothelial glycocalyx layer (Puerta-Guardo et al., 2016), platelet activation (Quirino-Teixeira et al., 2020), endocan (a biomarker for endothelial cell activation) expression (Domínguez-Alemán et al., 2021), thrombocytopenia and hemorrhage (Chao et al., 2019). The NS1-TLR4 interaction has also been implicated in vascular leakage (Modhiran et al., 2015), although further studies are necessary to elucidate the relative importance of TLR4 activation (Glasner et al., 2017). A potential mediator of the signal transduction and immune response mediated by TLR4 is its localization at the plasma membrane or in endosomes. Researchers have raised the possibility of EBOV GP interacting with internal TLR4 as well as surface TLR4 (Iampietro et al., 2017). The localization of TLR4 interaction and activation remains to be characterized for DENV NS1, RSV F, and SARS-CoV-2 S, particularly since recent findings indicate that immune cells can be non-productively infected with SARS-CoV-2 (Junqueira et al., 2022; Sefik et al., 2022).
The role of TLR4 in viral infections could be further explored and informed by population studies of TLR4 polymorphisms. Two TLR4 polymorphisms, D299G and T399I, have been associated with hyporesponsiveness to LPS and increased incidence of bacterial sepsis (Arbour et al., 2000; Lorenz et al., 2002). However, there have been conflicting studies on the association of common human TLR4 polymorphisms (D299G and T399I) with symptomatic RSV infection in children (Tal et al., 2004; Awomoyi et al., 2007; Paulus et al., 2007). Studies in the context of dengue infection have yielded similarly conflicting findings (Djamiatun et al., 2011; Sharma et al., 2016; Posadas-Mondragón et al., 2020), while TLR4 polymorphisms D299G and T399I were associated with COVID-19 severity and cytokine storm (Taha et al., 2021). The conflicting results from these studies may reflect small cohort sizes and differences in study populations. It is also possible that indirect effects of viral infection modulate TLR4 activation. For example, one study evaluating the link between RSV infection, airway inflammation and asthma provided evidence that RSV infection leads to a heightened responsiveness to LPS, mediated by increased TLR4 mRNA production and protein membrane localization in lung epithelial cells after RSV infection (Monick et al., 2003). More generally, viral infections are associated with alterations to the respiratory microbiome and increased colonization of potentially pathogenic bacteria in the upper respiratory tract (Hanada et al., 2018). Such changes in the bacterial community during viral infection could modulate TLR4 activation. Further studies in animal models are necessary to characterize whether viral infection-induced changes to the microbiome alter immune responses and TLR4 activation.
In general, in vivo models will be crucial to determine the therapeutic potential of targeting TLR4. For example, the role of TLR4 in RSV infection clearance remains unresolved. As for SARS-CoV-2, S1 was demonstrated to have a role in vivo, outside the context of authentic virus infection (Frank et al., 2022). To support the investigation of S1 independent of viral infection it is important to note that a most recent study identified circulating S as a blood biomarker for post-acute sequalae of COVID-19, while detection of SARS-CoV-2 nucleoprotein was lesser, providing some indication that freely circulating S may be a trigger for symptoms (Swank et al., 2022). In the in vivo study of Frank et al., it was shown that intra-cisterna magna (ICM) injection of S1 in mice resulted in behavioral changes, including reduced total activity and increased social avoidance, that are consistent with the sickness response to infection. The authors identified that S1 was sufficient to modulate neuroimmune gene expression in several brain regions and increase proinflammatory cytokine secretion in hippocampal tissue, confirming cytokine induction in vivo. While the role of specific PRRs remained unaddressed, these findings provide a model for investigating inhibitors that specifically counteract the effects of S1 that are most likely mediated by TLR4 or other PRRs. Further in vivo studies of EBOV and DENV infection are warranted to directly test the potential of TLR4 inhibitors as therapeutics for viral infections that result in TLR4 activation and an excessive immune response. Overall, further insight into the interactions of viral glycoproteins with TLR4 is critical for understanding viral pathogenesis and identifying therapeutic interventions to prevent cytokine storm during severe SARS-CoV-2, EBOV, DENV, and RSV infections.
CC conceptualized the review. CC and KG supervised the review and edited the manuscript. EH and EL conducted the literature review, wrote the first draft, and designed the figures. All authors contributed to the article and approved the submitted version.
The laboratories of CC and KG are supported by Discovery Grants from the Natural Sciences and Engineering Research Council of Canada, and the Faculty of Health Sciences Spear-Start grant for Pulmonary and Respiratory Diseases Research (Queen’s University). CC is supported by the Canadian Foundation for Innovation John R. Evans Leaders Fund, the Banting Research Foundation, the J.P. Bickell Foundation for Medical Research, and Queen’s University Research Initiation Grant. EVL is supported by a Vanier Canada Graduate scholarship.
Figures were created using Biorender.com. We acknowledge the contributions of all our colleagues in this field and apologize for any literature we could not include due to space limitations.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Akey, D. L., Brown, W. C., Dutta, S., Konwerski, J., Jose, J., Jurkiw, T. J., et al. (2014). Flavivirus NS1 structures reveal surfaces for associations with membranes and the immune system. Science 343, 881–885. doi: 10.1126/science.1247749
Akira, S., and Takeda, K. (2004). Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511. doi: 10.1038/nri1391
Alshaghdali, K., Saeed, M., Kamal, M. A., and Saeed, A. (2021). Interaction of ectodomain of respiratory syncytial virus G protein with TLR2/TLR6 heterodimer: an in vitro and in silico approach to decipher the role of RSV G protein in proinflammatory response against the virus. Curr. Pharm. Design. 27, 4464–4476. doi: 10.2174/1381612827666210716160030
Arbour, N. C., Lorenz, E., Schutte, B. C., Zabner, J., Kline, J. N., Jones, M., et al. (2000). TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat. Genet. 25, 187–191. doi: 10.1038/76048
Awomoyi, A. A., Rallabhandi, P., Pollin, T. I., Lorenz, E., Sztein, M. B., Boukhvalova, M. S., et al. (2007). Association of TLR4 polymorphisms with symptomatic respiratory syncytial virus infection in high-risk infants and young children. J. Immunol. 179, 3171–3177. doi: 10.4049/jimmunol.179.5.3171
Björkbacka, H., Fitzgerald, K. A., Huet, F., Li, X., Gregory, J. A., Lee, M. A., et al. (2004). The induction of macrophage gene expression by LPS predominantly utilizes Myd 88-independent signaling cascades. Physiol. Genomics 19, 319–330. doi: 10.1152/physiolgenomics.00128.2004
Bukreyev, A., Yang, L., Fricke, J., Cheng, L., Ward, J. M., Murphy, B. R., et al. (2008). The secreted form of respiratory syncytial virus g glycoprotein helps the virus evade antibody-mediated restriction of replication by acting as an antigen decoy and through effects on fc receptor-bearing leukocytes. J. Virol. 82, 12191–12204. doi: 10.1128/JVI.01604-08
Carvalho, D. M., Garcia, F. G., Terra, A. P. S., Lopes Tosta, A. C., de Silva, L., Castellano, L. R., et al. (2014). elevated dengue virus nonstructural protein 1 serum levels and altered toll-like receptor 4 expression, nitric oxide, and tumor necrosis factor alpha production in dengue hemorrhagic fever patients. J. Trop. Med. 2014, 1–9. doi: 10.1155/2014/901276
Chao, C.-H., Wu, W.-C., Lai, Y.-C., Tsai, P.-J., Perng, G.-C., Lin, Y.-S., et al. (2019). Dengue virus nonstructural protein 1 activates platelets via Toll-like receptor 4, leading to thrombocytopenia and hemorrhage. PLoS Pathog. 15:e1007625. doi: 10.1371/journal.ppat.1007625
Chiok, K., Hutchison, K., Miller, L. G., Bose, S., and Miura, T. A. (2021). Proinflammatory responses in SARS-CoV-2 infected and soluble spike glycoprotein S1 subunit activated human macrophages. bioRxiv [Preprint]. doi: 10.1101/2021.06.14.448426
Ciesielska, A., Matyjek, M., and Kwiatkowska, K. (2021). TLR4 and CD14 trafficking and its influence on LPS-induced proinflammatory signaling. Cell. Mol. Life Sci. 78, 1233–1261. doi: 10.1007/s00018-020-03656-y
Cinquegrani, G., Spigoni, V., Iannozzi, N. T., Parello, V., Bonadonna, R. C., and Dei Cas, A. (2022). SARS-CoV-2 Spike protein is not proinflammatory in human primary macrophages: endotoxin contamination and lack of protein glycosylation as possible confounders. Cell Biol. Toxicol. 38, 667–678. doi: 10.1007/s10565-021-09693-y
Cochet, F., and Peri, F. (2017). The Role of Carbohydrates in the Lipopolysaccharide (LPS)/Toll-Like Receptor 4 (TLR4) Signalling. Int. J. Mol. Sci. 18:2318. doi: 10.3390/ijms18112318
Coelho, D. R., Carneiro, P. H., Mendes-Monteiro, L., Conde, J. N., Andrade, I., Cao, T., et al. (2021). Apo A1 neutralizes proinflammatory effects of dengue virus NS1 protein and modulates viral immune evasion. J. Virol. 95:e0197420. doi: 10.1128/JVI.01974-20
Cook, J. D., and Lee, J. E. (2013). The secret life of viral entry glycoproteins: moonlighting in immune evasion. PLoS Pathog. 9:e1003258. doi: 10.1371/journal.ppat.1003258
da Silva Correia, J., and Ulevitch, R. J. (2002). MD-2 and TLR4 N-linked glycosylations are Important for a functional lipopolysaccharide receptor. J. Biol. Chem. 277, 1845–1854. doi: 10.1074/jbc.M109910200
Dayarathna, S., Jeewandara, C., Gomes, L., Somathilaka, G., Jayathilaka, D., Vimalachandran, V., et al. (2020). Similarities and differences between the ‘cytokine storms’ in acute dengue and COVID-19. Sci. Rep. 10:19839. doi: 10.1038/s41598-020-76836-2
Denner, J. (2015). Treatment of Ebola virus infections with inhibitors of TLR4. Med. Hypotheses 85, 253–257. doi: 10.1016/j.mehy.2015.05.009
Djamiatun, K., Dolmans, W. M. V., Ferwerda, B., Faradz, S. M. H., Netea, M. G., and van der Ven, A. J. A. M. (2011). Toll-like receptor 4 polymorphisms in dengue virus–infected children. Am. J. Trop. Med. Hyg. 85, 352–354. doi: 10.4269/ajtmh.2011.10-0728
Domínguez-Alemán, C. A., Sánchez-Vargas, L. A., Hernández-Flores, K. G., Torres-Zugaide, A. I., Reyes-Sandoval, A., Cedillo-Barrón, L., et al. (2021). Dengue virus induces the expression and release of endocan from endothelial cells by an NS1–TLR4-dependent mechanism. Microorganisms 9:1305. doi: 10.3390/microorganisms9061305
Ehl, S., Bischoff, R., Ostler, T., Vallbracht, S., Schulte-Mönting, J., Poltorak, A., et al. (2004). The role of Toll-like receptor 4 versus interleukin-12 in immunity to respiratory syncytial virus. Eur. J. Immunol. 34, 1146–1153. doi: 10.1002/eji.200324449
Escudero-Pérez, B., Volchkova, V. A., Dolnik, O., Lawrence, P., and Volchkov, V. E. (2014). Shed GP of ebola virus triggers immune activation and increased vascular permeability. PLoS Pathog. 10:e1004509. doi: 10.1371/journal.ppat.1004509
Fajgenbaum, D. C., and June, C. H. (2020). Cytokine storm. N. Engl. J. Med. 383, 2255–2273. doi: 10.1056/NEJMra2026131
Frank, M. G., Nguyen, K. H., Ball, J. B., Hopkins, S., Kelley, T., Baratta, M. V., et al. (2022). SARS-CoV-2 spike S1 subunit induces neuroinflammatory, microglial and behavioral sickness responses: Evidence of PAMP-like properties. Brain Behav. Immun. 100, 267–277. doi: 10.1016/j.bbi.2021.12.007
Georgel, P., Jiang, Z., Kunz, S., Janssen, E., Mols, J., Hoebe, K., et al. (2007). Vesicular stomatitis virus glycoprotein G activates a specific antiviral Toll-like receptor 4-dependent pathway. Virology 362, 304–313. doi: 10.1016/j.virol.2006.12.032
Glasner, D. R., Ratnasiri, K., Puerta-Guardo, H., Espinosa, D. A., Beatty, P. R., and Harris, E. (2017). Dengue virus NS1 cytokine-independent vascular leak is dependent on endothelial glycocalyx components. PLoS Pathog. 13:e1006673. doi: 10.1371/journal.ppat.1006673
González-Reyes, L., Ruiz-Argüello, M. B., García-Barreno, B., Calder, L., López, J. A., Albar, J. P., et al. (2001). Cleavage of the human respiratory syncytial virus fusion protein at two distinct sites is required for activation of membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 98, 9859–9864. doi: 10.1073/pnas.151098198
Goulopoulou, S., McCarthy, C. G., and Webb, R. C. (2016). Toll-like Receptors in the Vascular System: Sensing the Dangers Within. Pharmacol. Rev. 68, 142–167. doi: 10.1124/pr.114.010090
Guzman, M. G., Gubler, D. J., Izquierdo, A., Martinez, E., and Halstead, S. B. (2016). Dengue infection. Nat. Rev. Dis. Primers. 2:16055. doi: 10.1038/nrdp.2016.55
Haeberle, H. A., Takizawa, R., Casola, A., Brasier, A. R., Dieterich, H.-J., van Rooijen, N., et al. (2002). Respiratory syncytial virus–induced activation of nuclear factor–κB in the lung involves alveolar macrophages and toll-like receptor 4–dependent pathways. J. Infect. Dis. 186, 1199–1206. doi: 10.1086/344644
Hanada, S., Pirzadeh, M., Carver, K. Y., and Deng, J. C. (2018). Respiratory viral infection-induced microbiome alterations and secondary bacterial pneumonia. Front. Immunol. 9:2640. doi: 10.3389/fimmu.2018.02640
Haynes, L. M., Moore, D. D., Kurt-Jones, E. A., Finberg, R. W., Anderson, L. J., and Tripp, R. A. (2001). Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J. Virol. 75, 10730–10737. doi: 10.1128/JVI.75.22.10730-10737.2001
Hoshino, K., Kaisho, T., Iwabe, T., Takeuchi, O., and Akira, S. (2002). Differential involvement of IFN-b in Toll-like receptor-stimulated dendritic cell activation. Int. Immunol. 14, 1225–1231. doi: 10.1093/intimm/dxf089
Iampietro, M., Santos, R. I., Lubaki, N. M., and Bukreyev, A. (2018). Ebola virus shed glycoprotein triggers differentiation, infection, and death of monocytes through Toll-like receptor 4 activation. J. Infect. Dis. 218, S327–S334. doi: 10.1093/infdis/jiy406
Iampietro, M., Younan, P., Nishida, A., Dutta, M., Lubaki, N. M., Santos, R. I., et al. (2017). Ebola virus glycoprotein directly triggers T lymphocyte death despite of the lack of infection. PLoS Pathog. 13:e1006397. doi: 10.1371/journal.ppat.1006397
Jiang, Z., Georgel, P., Du, X., Shamel, L., Sovath, S., Mudd, S., et al. (2005). CD14 is required for MyD88-independent LPS signaling. Nat. Immunol. 6, 565–570. doi: 10.1038/ni1207
Junqueira, C., Crespo, Â., Ranjbar, S., de Lacerda, L. B., Lewandrowski, M., Ingber, J., et al. (2022). Fcγ R-mediated SARS-CoV-2 infection of monocytes activates inflammation. Nature 606, 576–584. doi: 10.1038/s41586-022-04702-4
Kagan, J. C., and Medzhitov, R. (2006). Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cells 125, 943–955. doi: 10.1016/j.cell.2006.03.047
Kagan, J. C., Su, T., Horng, T., Chow, A., Akira, S., and Medzhitov, R. (2008). TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat. Immunol. 9, 361–368. doi: 10.1038/ni1569
Kaushik, D., Bhandari, R., and Kuhad, A. (2021). TLR4 as a therapeutic target for respiratory and neurological complications of SARS-CoV-2. Expert. Opin. Ther. Tar. 25, 491–508. doi: 10.1080/14728222.2021.1918103
Kawai, T., and Akira, S. (2007). Signaling to NF-κB by Toll-like receptors. Trends Mol. Med. 13, 460–469. doi: 10.1016/j.molmed.2007.09.002
Kawai, T., Takeuchi, O., Fujita, T., Inoue, J., Mühlradt, P. F., Sato, S., et al. (2001). Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J. Immunol. 167, 5887–5894. doi: 10.4049/jimmunol.167.10.5887
Kayesh, M. E. H., Kohara, M., and Tsukiyama-Kohara, K. (2021). Recent insights into the molecular mechanism of Toll-like receptor response to dengue virus infection. Front. Microbiol. 12:744233. doi: 10.3389/fmicb.2021.744233
Khan, S., Shafiei, M. S., Longoria, C., Schoggins, J. W., Savani, R. C., and Zaki, H. (2021). SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway. elife 10:e68563. doi: 10.7554/eLife.68563
Kim, H. M., Park, B. S., Kim, J.-I., Kim, S. E., Lee, J., Oh, S. C., et al. (2007). Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist eritoran. Cells 130, 906–917. doi: 10.1016/j.cell.2007.08.002
Kurt-Jones, E. A., Popova, L., Kwinn, L., Haynes, L. M., Jones, L. P., Tripp, R. A., et al. (2000). Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 1, 398–401. doi: 10.1038/80833
Kuzmich, N., Sivak, K., Chubarev, V., Porozov, Y., Savateeva-Lyubimova, T., and Peri, F. (2017). TLR4 signaling pathway modulators as potential therapeutics in inflammation and sepsis. Vaccine 5:34. doi: 10.3390/vaccines5040034
Lai, C.-Y., Strange, D. P., Wong, T. A. S., Lehrer, A. T., and Verma, S. (2017). Ebola virus glycoprotein induces an innate immune response in vivo via TLR4. Front. Microbiol. 8:1571. doi: 10.3389/fmicb.2017.01571
Lester, S. N., and Li, K. (2014). Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 426, 1246–1264. doi: 10.1016/j.jmb.2013.11.024
Libraty, D. H., Young, P. R., Pickering, D., Endy, T. P., Kalayanarooj, S., Green, S., et al. (2002). High circulating levels of the dengue virus nonstructural protein NS1 early in dengue illness correlate with the development of dengue hemorrhagic fever. J. Infect. Dis. 186, 1165–1168. doi: 10.1086/343813
Lizundia, R., Sauter, K.-S., Taylor, G., and Werling, D. (2008). Host species-specific usage of the TLR4-LPS receptor complex. Innate Immunol. 14, 223–231. doi: 10.1177/1753425908095957
Lorenz, E., Mira, J. P., Frees, K. L., and Schwartz, D. A. (2002). Relevance of mutations in the TLR4 receptor in patients with Gram-negative septic shock. Arch. Intern. Med. 162, 1028–1032. doi: 10.1001/archinte.162.9.1028
Ma, Z., Li, X., Fan, R. L. Y., Yang, K. Y., Ng, C. S. H., Lau, R. W. H., et al. (2022). A human pluripotent stem cell-based model of SARS-CoV-2 infection reveals an ACE2-independent inflammatory activation of vascular endothelial cells through TLR4. Stem. Cell. Rep. 17, 538–555. doi: 10.1016/j.stemcr.2022.01.015
Mahanty, S., and Bray, M. (2004). Pathogenesis of filoviral haemorrhagic fevers. Lancet Infect. Dis. 4, 487–498. doi: 10.1016/S1473-3099(04)01103-X
Mahmudpour, M., Roozbeh, J., Keshavarz, M., Farrokhi, S., and Nabipour, I. (2020). COVID-19 cytokine storm: the anger of inflammation. Cytokine 133:155151. doi: 10.1016/j.cyto.2020.155151
Manik, M., and Singh, R. K. (2021). Role of toll-like receptors in modulation of cytokine storm signaling in SARS-CoV-2-induced COVID-19. J. Med. Virol. 94, 869–877. doi: 10.1002/jmv.27405
Marongiu, L., Gornati, L., Artuso, I., Zanoni, I., and Granucci, F. (2019). Below the surface: the inner lives of TLR4 and TLR9. J. Leukoc. Biol. 106, 147–160. doi: 10.1002/JLB.3MIR1218-483RR
Marr, N., and Turvey, S. E. (2012). Role of human TLR4 in respiratory syncytial virus-induced NF-κB activation, viral entry and replication. Innate Immunol. 18, 856–865. doi: 10.1177/1753425912444479
Mehedi, M., Groseth, A., Feldmann, H., and Ebihara, H. (2011). Clinical aspects of Marburg hemorrhagic fever. Futur. Virol. 6, 1091–1106. doi: 10.2217/fvl.11.79
Meissner, F., Scheltema, R. A., Mollenkopf, H.-J., and Mann, M. (2013). Direct proteomic quantification of the secretome of activated immune cells. Science 340, 475–478. doi: 10.1126/science.1232578
Modhiran, N., Watterson, D., Blumenthal, A., Baxter, A. G., Young, P. R., and Stacey, K. J. (2017). Dengue virus NS1 protein activates immune cells via TLR4 but not TLR2 or TLR6. Immunol. Cell Biol. 95, 491–495. doi: 10.1038/icb.2017.5
Modhiran, N., Watterson, D., Muller, D. A., Panetta, A. K., Sester, D. P., Liu, L., et al. (2015). Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci. Transl. Med. 7:304ra142. doi: 10.1126/scitranslmed.aaa3863
Molteni, M., Gemma, S., and Rossetti, C. (2016). The role of Toll-like receptor 4 in infectious and noninfectious inflammation. Mediat. Inflamm. 2016, 1–9. doi: 10.1155/2016/6978936
Monick, M. M., Yarovinsky, T. O., Powers, L. S., Butler, N. S., Carter, A. B., Gudmundsson, G., et al. (2003). respiratory syncytial virus up-regulates TLR4 and sensitizes airway epithelial cells to endotoxin. J. Biol. Chem. 278, 53035–53044. doi: 10.1074/jbc.M308093200
Negron, S. G., Kessinger, C. W., Xu, B., Pu, W. T., and Lin, Z. (2021). Selectively expressing SARS-CoV-2 Spike protein S1 subunit in cardiomyocytes induces cardiac hypertrophy in mice. bioRxiv [Preprint]. doi: 10.1101/2021.06.20.448993
O’Neill, L. A. J., and Bowie, A. G. (2007). The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 7, 353–364. doi: 10.1038/nri2079
Okumura, A., Pitha, P. M., Yoshimura, A., and Harty, R. N. (2010). Interaction between ebola virus glycoprotein and host Toll-like receptor 4 leads to induction of proinflammatory cytokines and SOCS1. J. Virol. 84, 27–33. doi: 10.1128/JVI.01462-09
Okumura, A., Rasmussen, A. L., Halfmann, P., Feldmann, F., Yoshimura, A., Feldmann, H., et al. (2015). Suppressor of cytokine signaling 3 is an inducible host factor that regulates virus egress during ebola virus infection. J. Virol. 89, 10399–10406. doi: 10.1128/JVI.01736-15
Olajide, O. A., Iwuanyanwu, V. U., Adegbola, O. D., and Al-Hindawi, A. A. (2022). SARS-CoV-2 spike glycoprotein S1 induces neuroinflammation in BV-2 microglia. Mol. Neurobiol. 59, 445–458. doi: 10.1007/s12035-021-02593-6
Olajide, O. A., Iwuanyanwu, V. U., Lepiarz-Raba, I., and Al-Hindawi, A. A. (2021). Induction of exaggerated cytokine production in human peripheral blood mononuclear cells by a recombinant SARS-CoV-2 spike glycoprotein S1 and its inhibition by dexamethasone. Inflammation 44, 1865–1877. doi: 10.1007/s10753-021-01464-5
Olejnik, J., Forero, A., Deflubé, L. R., Hume, A. J., Manhart, W. A., Nishida, A., et al. (2017). Ebolaviruses associated with differential pathogenicity induce distinct host responses in human macrophages. J. Virol. 91, e00179–e00117. doi: 10.1128/JVI.00179-17
Olejnik, J., Hume, A. J., and Mühlberger, E. (2018). Toll-like receptor 4 in acute viral infection: Too much of a good thing. PLoS Pathog. 14:e1007390. doi: 10.1371/journal.ppat.1007390
Park, H., Hong, J., Kwon, H., Kim, Y., Lee, K., Kim, J., et al. (2010). TLR4-mediated activation of mouse macrophages by Korean mistletoe lectin-C (KML-C). Biochem. Biophys. Res. Commun. 396, 721–725. doi: 10.1016/j.bbrc.2010.04.169
Park, B. S., and Lee, J.-O. (2013). Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 45:e66. doi: 10.1038/emm.2013.97
Park, B. S., Song, D. H., Kim, H. M., Choi, B.-S., Lee, H., and Lee, J.-O. (2009). The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature 458, 1191–1195. doi: 10.1038/nature07830
Paulus, S. C., Hirschfeld, A. F., Victor, R. E., Brunstein, J., Thomas, E., and Turvey, S. E. (2007). Common human Toll-like receptor 4 polymorphisms—Role in susceptibility to respiratory syncytial virus infection and functional immunological relevance. Clin. Immunol. 123, 252–257. doi: 10.1016/j.clim.2007.03.003
Perrin-Cocon, L., Aublin-Gex, A., Sestito, S. E., Shirey, K. A., Patel, M. C., André, P., et al. (2017). TLR4 antagonist FP7 inhibits LPS-induced cytokine production and glycolytic reprogramming in dendritic cells, and protects mice from lethal influenza infection. Sci. Rep. 7:40791. doi: 10.1038/srep40791
Posadas-Mondragón, A., Aguilar-Faisal, J. L., Zuñiga, G., Magaña, J. J., Santiago-Cruz, J. A., Guillén-Salomón, E., et al. (2020). Association of genetic polymorphisms in TLR3, TLR4, TLR7, and TLR8 with the clinical forms of dengue in patients from veracruz. Mexico. Viruses 12:1230. doi: 10.3390/v12111230
Puerta-Guardo, H., Glasner, D. R., and Harris, E. (2016). Dengue virus NS1 disrupts the endothelial glycocalyx, leading to hyperpermeability. PLoS Pathog. 12:e1005738. doi: 10.1371/journal.ppat.1005738
Quirino-Teixeira, A. C., Rozini, S. V., Barbosa-Lima, G., Coelho, D. R., Carneiro, P. H., Mohana-Borges, R., et al. (2020). Inflammatory signaling in dengue-infected platelets requires translation and secretion of nonstructural protein 1. Blood Adv. 4, 2018–2031. doi: 10.1182/bloodadvances.2019001169
Ragab, D., Salah Eldin, H., Taeimah, M., Khattab, R., and Salem, R. (2020). The COVID-19 cytokine storm; what we know so far. Front. Immunol. 11:1446. doi: 10.3389/fimmu.2020.01446
Rallabhandi, P., Phillips, R. L., Boukhvalova, M. S., Pletneva, L. M., Shirey, K. A., Gioannini, T. L., et al. (2012). Respiratory syncytial virus fusion protein-induced toll-like receptor 4 (TLR4) signaling is inhibited by the TLR4 antagonists Rhodobacter sphaeroides lipopolysaccharide and eritoran (E 5564) and requires direct interaction with MD-2. MBio 3, e00218–e00212. doi: 10.1128/mBio.00218-12
Ricci-Azevedo, R., Roque-Barreira, M.-C., and Gay, N. J. (2017). Targeting and recognition of Toll-like receptors by plant and pathogen lectins. Front. Immunol. 8:1820. doi: 10.3389/fimmu.2017.01820
Roberts, S. R., Lichtenstein, D., Ball, L. A., and Wertz, G. W. (1994). The membrane-associated and secreted forms of the respiratory syncytial virus attachment glycoprotein G are synthesized from alternative initiation codons. J. Virol. 68, 4538–4546. doi: 10.1128/jvi.68.7.4538-4546.1994
Rosenberg, H. F., and Domachowske, J. B. (2012). Inflammatory responses to respiratory syncytial virus (RSV) infection and the development of immunomodulatory pharmacotherapeutics. Curr. Med. Chem. 19, 1424–1431. doi: 10.2174/092986712799828346
Scherm, M. J., Gangloff, M., and Gay, N. (2022). Activation of Toll-like receptor 4 by ebolavirus shed glycoprotein is direct and requires the internal fusion loop but not glycosylation. Preprint. doi: 10.2139/ssrn.4073009
Schroeder, J. T., and Bieneman, A. P. (2022). The S1 subunit of the SARS-CoV-2 spike protein activates human monocytes to produce cytokines linked to COVID-19: relevance to galectin-3. Front. Immunol. 13:831763. doi: 10.3389/fimmu.2022.831763
Sefik, E., Qu, R., Junqueira, C., Kaffe, E., Mirza, H., Zhao, J., et al. (2022). Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature 606, 585–593. doi: 10.1101/2021.09.27.461948
Sharma, S., Singh, S. K., Kakkar, K., Nyari, N., Dhole, T. N., Kashyap, R., et al. (2016). Analysis of TLR4 (Asp 299Gly and Thr 399Ile) gene polymorphisms and mRNA level in patients with dengue infection: A case-control study. Infect. Genet. Evol. 43, 412–417. doi: 10.1016/j.meegid.2016.06.027
Shingai, M., Azuma, M., Ebihara, T., Sasai, M., Funami, K., Ayata, M., et al. (2008). Soluble G protein of respiratory syncytial virus inhibits Toll-like receptor 3/4-mediated IFN-beta induction. Int. Immunol. 20, 1169–1180. doi: 10.1093/intimm/dxn074
Shirato, K., and Kizaki, T. (2021). SARS-CoV-2 spike protein S1 subunit induces proinflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon 7:e06187. doi: 10.1016/j.heliyon.2021.e06187
Shirey, K. A., Blanco, J. C. G., and Vogel, S. N. (2021). Targeting TLR4 signaling to blunt viral-mediated acute lung injury. Front. Immunol. 12:705080. doi: 10.3389/fimmu.2021.705080
Shirey, K. A., Lai, W., Scott, A. J., Lipsky, M., Mistry, P., Pletneva, L. M., et al. (2013). The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature 497, 498–502. doi: 10.1038/nature12118
Sohn, K. M., Lee, S.-G., Kim, H. J., Cheon, S., Jeong, H., Lee, J., et al. (2020). COVID-19 patients upregulate Toll-like receptor 4-mediated inflammatory signaling that mimics bacterial sepsis. J. Korean Med. Sci. 35:e343. doi: 10.3346/jkms.2020.35.e343
Somnuke, P., Hauhart, R. E., Atkinson, J. P., Diamond, M. S., and Avirutnan, P. (2011). N-linked glycosylation of dengue virus NS1 protein modulates secretion, cell-surface expression, hexamer stability, and interactions with human complement. Virology 413, 253–264. doi: 10.1016/j.virol.2011.02.022
Srikiatkhachorn, A., Mathew, A., and Rothman, A. L. (2017). Immune-mediated cytokine storm and its role in severe dengue. Semin. Immunopathol. 39, 563–574. doi: 10.1007/s00281-017-0625-1
Sunay, M. M. E., Martins, K. A. O., Steffens, J. T., Gregory, M., Vantongeren, S. A., Van Hoeven, N., et al. (2019). Glucopyranosyl lipid adjuvant enhances immune response to Ebola virus-like particle vaccine in mice. Vaccine 37, 3902–3910. doi: 10.1016/j.vaccine.2019.05.026
Swank, Z., Senussi, Y., Alter, G., and Walt, D. R. (2022). Persistent circulating SARS-CoV-2 spike is associated with post-acute COVID-19 sequelae. medRxiv [Preprint]. doi: 10.1101/2022.06.14.22276401
Taha, S. I., Shata, A. K., Baioumy, S. A., Fouad, S. H., Anis, S. G., Mossad, I. M., et al. (2021). Toll-like receptor 4 polymorphisms (896A/G and 1196C/T) as an indicator of COVID-19 severity in a convenience sample of egyptian patients. J. Inflamm. Res. 14, 6293–6303. doi: 10.2147/JIR.S343246
Takeda, K., and Akira, S. (2004). TLR signaling pathways. Semin. Immunol. 16, 3–9. doi: 10.1016/j.smim.2003.10.003
Tal, G., Mandelberg, A., Dalal, I., Cesar, K., Somekh, E., Tal, A., et al. (2004). Association between common Toll-like receptor 4 mutations and severe respiratory syncytial virus disease. J. Infect. Dis. 189, 2057–2063. doi: 10.1086/420830
Tam, J. S. Y., Coller, J. K., Hughes, P. A., Prestidge, C. A., and Bowen, J. M. (2021). Toll-like receptor 4 (TLR4) antagonists as potential therapeutics for intestinal inflammation. Indian J. Gastroenterol. 40, 5–21. doi: 10.1007/s12664-020-01114-y
Tan, Y., Zanoni, I., Cullen, T. W., Goodman, A. L., and Kagan, J. C. (2015). Mechanisms of Toll-like receptor 4 endocytosis reveal a common immune-evasion strategy used by pathogenic and commensal bacteria. Immunity 43, 909–922. doi: 10.1016/j.immuni.2015.10.008
Tanimura, N., Saitoh, S., Matsumoto, F., Akashi-Takamura, S., and Miyake, K. (2008). Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem. Bioph. Res. Co. 368, 94–99. doi: 10.1016/j.bbrc.2008.01.061
Tsukahara, K. (2022). Contribution to development of remedies for COVID-19: focusing on Eritoran. Nihon Yakurigaku Zasshi 157, 38–40. doi: 10.1254/fpj.21041
Unitt, J., and Hornigold, D. (2011). Plant lectins are novel Toll-like receptor agonists. Biochem. Pharmacol. 81, 1324–1328. doi: 10.1016/j.bcp.2011.03.010
van der Donk, L. E. H., Eder, J., van Hamme, J. L., Brouwer, P. J. M., Brinkkemper, M., van Nuenen,, et al. (2022). SARS-CoV-2 infection activates dendritic cells via cytosolic receptors rather than extracellular TLRs. Eur. J. Immunol. 52, 646–655. doi: 10.1002/eji.202149656
Wagstaffe, H. R., Clutterbuck, E. A., Bockstal, V., Stoop, J. N., Luhn, K., Douoguih, M., et al. (2020). Ebola virus glycoprotein stimulates IL-18–dependent natural killer cell responses. J. Clin. Invest. 130, 3936–3946. doi: 10.1172/JCI132438
Wang, M., Chen, Y., Zhang, Y., Zhang, L., Lu, X., and Chen, Z. (2011). Mannan-binding lectin directly interacts with Toll-like receptor 4 and suppresses lipopolysaccharide-induced inflammatory cytokine secretion from THP-1 cells. Cell. Mol. Immunol. 8, 265–275. doi: 10.1038/cmi.2011.1
Watanabe, Y., Bowden, T. A., Wilson, I. A., and Crispin, M. (2019). Exploitation of glycosylation in enveloped virus pathobiology. Biochim. Biophys. Acta Gen. Subj. 1863, 1480–1497. doi: 10.1016/j.bbagen.2019.05.012
Weighardt, H., and Holzmann, B. (2008). Role of Toll-like receptor responses for sepsis pathogenesis. Immunobiology 212, 715–722. doi: 10.1016/j.imbio.2007.09.010
Weighardt, H., Kaiser-Moore, S., Vabulas, R. M., Kirschning, C. J., Wagner, H., and Holzmann, B. (2002). Cutting edge: myeloid differentiation factor 88 deficiency improves resistance against sepsis caused by polymicrobial infection. J. Immunol. 169, 2823–2827. doi: 10.4049/jimmunol.169.6.2823
Wolf, T., Kann, G., Becker, S., Stephan, C., Brodt, H.-R., de Leuw, P., et al. (2015). Severe Ebola virus disease with vascular leakage and multiorgan failure: treatment of a patient in intensive care. Lancet 385, 1428–1435. doi: 10.1016/S0140-6736(14)62384-9
Yamamoto, M., Sato, S., Hemmi, H., Hoshino, K., Kaisho, T., Sanjo, H., et al. (2003). Role of adaptor trif in the MYD88-independent Toll-like receptor signaling pathway. Science 301, 640–643. doi: 10.1126/science.1087262
Yamamoto, M., Sato, S., Hemmi, H., Sanjo, H., Uematsu, S., Kaisho, T., et al. (2002). Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature 420, 324–329. doi: 10.1038/nature01182
Yamamoto, M., and Takeda, K. (2010). Current Views of Toll-Like Receptor Signaling Pathways. Gastroenterol. Res. Pract. 2010, 1–8. doi: 10.1155/2010/240365
Yang, G., Zhang, S., Wang, Y., Li, L., Li, Y., Yuan, D., et al. (2022). Aptamer blocking S-TLR4 interaction selectively inhibits SARS-CoV-2 induced inflammation. Signal Transduct. Target. Ther. 7:120. doi: 10.1038/s41392-022-00968-2
Younan, P., Iampietro, M., Nishida, A., Ramanathan, P., Santos, R. I., Dutta, M., et al. (2017). Ebola virus binding to Tim-1 on T lymphocytes induces a cytokine storm. MBio 8, e00845–e00817. doi: 10.1128/mBio.00845-17
Zanoni, I., Ostuni, R., Marek, L. R., Barresi, S., Barbalat, R., Barton, G. M., et al. (2011). CD14 controls the LPS-Induced endocytosis of toll-like receptor 4. Cells 147, 868–880. doi: 10.1016/j.cell.2011.09.051
Zhao, Y., Kuang, M., Li, J., Zhu, L., Jia, Z., Guo, X., et al. (2021). SARS-CoV-2 spike protein interacts with and activates TLR4. Cell Res. 31, 818–820. doi: 10.1038/s41422-021-00495-9
Zheng, Y., Bian, L., Zhao, H., Liu, Y., Lu, J., Liu, D., et al. (2020). Respiratory syncytial virus F subunit vaccine with AS02 adjuvant elicits balanced, robust humoral and cellular immunity in BALB/c mice. Front. Immunol. 11:526965. doi: 10.3389/fimmu.2020.526965
Keywords: Toll-like receptor 4, viral glycoproteins, cytokine storm, SARS-CoV-2, dengue
Citation: Halajian EA, LeBlanc EV, Gee K and Colpitts CC (2022) Activation of TLR4 by viral glycoproteins: A double-edged sword? Front. Microbiol. 13:1007081. doi: 10.3389/fmicb.2022.1007081
Edited by:
Bin Su, Capital Medical University, ChinaReviewed by:
David Verhoeven, Iowa State University, United StatesCopyright © 2022 Halajian, LeBlanc, Gee and Colpitts. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Che C. Colpitts, Y2hlLmNvbHBpdHRzQHF1ZWVuc3UuY2E=; Katrina Gee, a2dlZUBxdWVlbnN1LmNh
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.