 Xizhao Zhang1
Xizhao Zhang1 Congping Ying2Min Jiang1Danqing Lin1Lei You1Denghua Yin1Jialu Zhang1
Congping Ying2Min Jiang1Danqing Lin1Lei You1Denghua Yin1Jialu Zhang1 Kai Liu1,2*
Kai Liu1,2* Pao Xu1,2*
Pao Xu1,2*- 1Key Laboratory of Freshwater Fisheries and Germplasm Resources Utilization, Ministry of Agriculture and Rural Affairs, Freshwater Fisheries Research Center, Chinese Academy of Fishery Sciences, Wuxi, China
- 2Wuxi Fisheries College, Nanjing Agricultural University, Wuxi, China
Bacteria play an essential role in the health of marine mammals, and the bacteria of marine mammals are widely concerned, but less is known about freshwater mammals. In this study, we investigated the bacteria of various body sites of Yangtze finless porpoise (Neophocaena asiaeorientalis asiaeorientalis) and analyzed their association with freshwater environmental bacteria. The bacterial community and function of Yangtze finless porpoise showed apparent site-specificity. Various body sites have distinct differences in bacteria and have their dominant bacteria. Romboutsia, Plesiomonas, Actinobacillus, Candidatus Arthromitus dominated in the intestine (fecal and rectal samples). Fusobacterium, Streptococcus, and Acinetobacter dominated in the oral. The dominant genera in the blowhole include Suttonella, Psychrobacter, and two uncultured genera. Psychrobacter, Flavobacterium, and Acinetobacter were dominant in the skin. The alpha diversity of intestinal (fecal and rectal) bacteria was the lowest, while that of skin was the highest. The oral and skin bacteria of Yangtze finless porpoise significantly differed between the natural and semi-natural conditions, but no sex difference was observed. A clear boundary was found between the animal and the freshwater environmental bacteria. Even the skin bacteria, which are more affected by the environment, are significantly different from the environmental bacteria and harbor indigenous bacteria. Our results provide a comprehensive preliminary exploration of the bacteria of Yangtze finless porpoise and its association with bacteria in the freshwater environment.
Introduction
Microbiota broadly affects the growth and development of aquatic mammals, playing a role in nutrition, immunity, health, etc (Sanders et al., 2015; Mootapally et al., 2017; Di Guardo et al., 2019). In the more extensive study of marine mammals, researchers describe in detail the microbial composition and structure of different body sites of various marine mammals. Body sites, feeding habits, habitats, and phylogeny are the main factors affecting the microbial communities of marine mammals (Nelson et al., 2015; Sanders et al., 2015; Bik et al., 2016; Apprill et al., 2020). The study of marine mammalian microbiota provides valuable insights into the health status of marine mammals. In contrast to marine mammals, research on the diversity and composition of freshwater mammalian microbiota is relatively limited. The lack of microbial knowledge of freshwater mammals is detrimental to the health monitoring and conservation activities of freshwater mammals.
Yangtze finless porpoise (Neophocaena asiaeorientalis asiaeorientalis, YFP) inhabits the Yangtze River basin for life, is the only freshwater porpoise in the world and is endangered (Turvey et al., 2007; Mei et al., 2014; Huang et al., 2020). YFP occupies the top ecological niche of the Yangtze River ecosystem, is carnivorous, and is a unique species for studying freshwater mammals (Huang et al., 2017; Zhou et al., 2018). Currently, the studies related to the bacteria of YFP mainly focus on the fecal bacteria, mainly in the capture and semi-natural environments (McLaughlin et al., 2011, 2012, 2013; Wan et al., 2016b; You et al., 2020). In addition, Aeromonas veronii associated with the YFP disease has also been reported (Liu et al., 2018, 2020). Other body sites have been studied primarily in culture-based studies of respiratory tract microbiota (Wan et al., 2016a), with fewer reports about skin and oral bacteria. The lack of research on the microbiota of YFP limits our understanding of the health condition of YFP.
Environmental microbiota is one of the driving forces to shape marine mammalian bacteria, and it is also a potential source of marine mammal bacteria (Bik et al., 2016; Liu et al., 2018, 2020; Tian et al., 2022). Related studies have shown that environmental bacteria can colonize marine mammals and impact marine mammals’ health (Raverty et al., 2017; Huggins et al., 2020). Although there is a close relationship between environmental bacteria and marine mammalian bacteria, the boundary between them is also apparent (Apprill et al., 2014; Bik et al., 2016; Chiarello et al., 2017; You et al., 2020). Even the skin bacteria in close contact with the water environment differ from the environmental bacteria (Chiarello et al., 2017; Apprill et al., 2020). This association between marine mammals and environmental bacteria has been proven in many marine mammals. However, the association between freshwater mammalian and environmental bacteria needs further demonstrated.
This study collected the intestinal (rectal and fecal), blowhole, oral, and skin samples from 15 YFPs, including animal-related freshwater environmental samples. We analyzed the bacterial diversity and composition of different body sites of YFP and characterized the specific bacteria of different body sites. Differences in the YFP bacteria between the natural and semi-natural conditions were assessed. The functional specificity of the YFP bacteria was then analyzed. Finally, the association between YFP and freshwater environmental bacteria was investigated. These results provide an increased understanding of the diversity and composition of YFP bacteria, explore the characteristics of YFP bacterial communities and the influence of the freshwater environment on them, and offer a new reference for health monitoring and population conservation of YFP.
Materials and methods
Sample collection
The subjects included 15 YPFs from the Anqing section of the Yangtze River (natural) and the ex-situ conservation base of YFP in Xijiang, Anqing City (semi-natural). The animals were obtained in the same method as described in the previous studies (Yin et al., 2021). All the YFPs involved in the study were not treated with antibiotics or other drugs before sampling. The sampling process follows the general practice of veterinary nursing of marine mammals (Dierauf and Gulland, 2001). Sampling was carried out after the YFP was lifted from the water and placed on a soft sponge pad. All sterile cotton swabs were moistened with sterile saline, the gingival sulcus of the mandible was wiped with sterile cotton swabs to obtain oral samples, and the forehead skin of YFP was wiped with sterile cotton swabs to obtain skin samples. Each sample contains three swabs. The blowhole samples were collected by non-invasive method, and the 50 ml sterile polypropylene centrifuge tube was wetted with sterile saline, which was placed 10 cm above the blowhole, and the samples were collected after three breaths. The Fecal and rectal samples were collected through a sterile plastic tube with a diameter of 4 mm, and the front end was lubricated with Vaseline. Insert the fecal tube into the rectum 20 cm deep, take it out after 30 s, cut the tube into segments, collect the part containing feces as fecal samples, and collect the plastic tube in contact with the rectum (that is, the end of the insertion part of the tube) as rectal samples. Freshwater samples were collected with a 50 ml sterile polypropylene centrifuge tube in the animal activity area and filtered with fiberglass filter paper with a diameter of 47 mm and pore diameter of 0.22um. The filter paper is stored in a 1.5 ml sterile centrifuge tube containing alcohol. In addition, there are two stool samples and two water environment samples from the previous study (You et al., 2020). Detailed sample collection information can be found in Supplementary Table S1.
DNA extraction, amplification, and sequencing
The sample is transferred to a new 2 ml aseptic centrifuge tube, and the filter paper is cut into pieces before transfer. An appropriate amount of guanidine thiocyanate and N-lauroyl sarcosine were added to each tube at 70°C and transferred to a tissue lytic apparatus for oscillation after 1 h. After centrifuging the sample, the supernatant was removed and transferred to the 2 ml centrifuge tube. 500 μL temp was added to the residual sample, stirred, swirled, and centrifuged. Use the pipette to absorb 500 μL of supernatant and repeat the previous step 3 times. Once again, the supernatant was centrifuged at high speed, transferred to two isopropanol tubes, and stored overnight at 4°C. The sample was centrifuged at high speed, and the supernatant was discarded in next day. Phosphate buffer and potassium acetate are added to one sample sedimentation tube, transferred to another tube, and mixed. It was placed on ice for 1.5 h and centrifuged at 4°C for 30 min. Add anhydrous ethanol and NaAc and let it stand at −20°C for several hours. After centrifugation, the supernatant was removed, and 70% ethanol was added to separate the washing solution by centrifugation. After repeated washing and adding TE solution, DNA was obtained after dissolving. Remove samples with low DNA quality (total mass < 1.0 μg, concentration < 10 ng/μL, moderately or severely degraded). The V3-V4 region of 16SrDNA was amplified by primers 341F (ACTCCTACGGGAGGCAGCAG) and 806R (GGACTACHVGGGTWTCTAAT). The conditions were pre-denaturation at 94°C for 3 min, followed by 30 cycles of denaturation at 94°C for 30s, annealing at 55°C for 30s, extension at 72°C for 30s, and then at 72°C for 5 min to complete the reaction. It was excised from agarose gel and purified by the Universal DNA purification kit (Tiangen, China). The purified product was subjected to high-throughput sequencing on the Illumina Miseq 2000 platform (BGI, Shenzhen, China).
Data analysis
First, the primers and barcodes were removed from the raw data of high-throughput sequencing. Microbiome bioinformatics analysis was performed using QIIME 22022.2 (Bolyen et al., 2019). Raw sequence data were demultiplexed and quality filtered using the q2-demux plugin, then denoising using DADA2 (Callahan et al., 2016). SILVA 13 8 99% reference sequences (Quast et al., 2013) were assigned to amplicon sequence variants (ASVs) using the classify-sklearn naïve Bayesian classifier (McDonald et al., 2012) of the q2 feature classifier (Bokulich et al., 2018). Removal of non-bacterial ASVs and chloroplast-associated ASVs. All ASVs were aligned to mafft (Katoh et al., 2002) and used to construct a phylogeny tree using fasttree2 (Price et al., 2010). Alpha diversity metrics (ACE, Chao1, Shannon, and Faith’s phylogenetic diversity) and beta diversity metric (Bray Curtis dissimilarity) were calculated after all samples were sparsed (subsampling without replacement) to the lowest sequences number of all samples. Non-metric multidimensional scaling (NMDS) and Permutational multivariate analysis of variance (PERMANOVA) are all implemented with the Vegan v2.5–7 package (Oksanen et al., 2020). The unweighted pair-group method with arithmetic means (UPGMA) clustering by ggtree v3.6.1 package (Yu et al., 2017). All heatmaps are drawn with ComplexHeatmap v2.3.1 package (Gu et al., 2016). Shared genera were created by the UpSetR v1.4.0 package (Conway et al., 2017). Source track analysis was performed by SourceTrack2 (Knights et al., 2011). The LefSef analysis (Segata et al., 2011) performed via Galaxy (Afgan et al., 2018). Bacterial function was analyzed by PICRUSt2 (Douglas et al., 2020). All visualizations in this study were implemented with R v4.1.1 (R Core Team, 2021).
Results
Bacterial composition of Yangzte finless porpoise
The four phyla, Proteobacteria, Firmicutes, Bacteroidota, and Fusobacteriota, were dominant in YFP (Figure 1A). Moreover, Actinobacteria were more abundant in the intestinal samples (fecal: 6.0 ± 2.9%, rectal: 3.5 ± 1.4%, Mean ± Standard Error). Patescibacteria was dominant in the oral (7.6 ± 1.6%) and blowhole samples (8.3 ± 1.7%, Figure 1A). The dominant bacteria in the fecal and rectal samples at the family level were Peptostreptococcaceae (fecal: 32 ± 7.3%, rectal: 24 ± 6.7%), Clostridiaceae (fecal: 17 ± 6.2%, rectal: 15 ± 6.7%), and Enterobacteriaceae (fecal: 13 ± 6.1%, rectal: 14 ± 7.5%, Figure 1B). In addition, Mycoplasmataceae (10 ± 3.7%) was abundant in the fecal samples, and Pasteurellaceae (18 ± 8.6%) was abundant in the rectal samples (Figure 1B). The bacteria of oral samples were dominated by Fusobacteriaceae (17 ± 2.7%), Moraxellaceae (13 ± 4.1%), and Streptococcaceae (9.9 ± 1.9%, Figure 1B). Cardiobacteriaceae (13 ± 2.3%), Moraxellaceae (11 ± 3%), Flavobacteriaceae (9.2 ± 0.7%), and Saccharospirillaceae (8.9 ± 2.9%) were abundant in the blowhole samples (Figure 1B). In the skin samples, Moraxellaceae (26 ± 5.3%), Flavobacteriaceae (11 ± 3.6%), and Weeksellaceae (5.5 ± 2%) were dominant bacteria at the family level (Figure 1B).
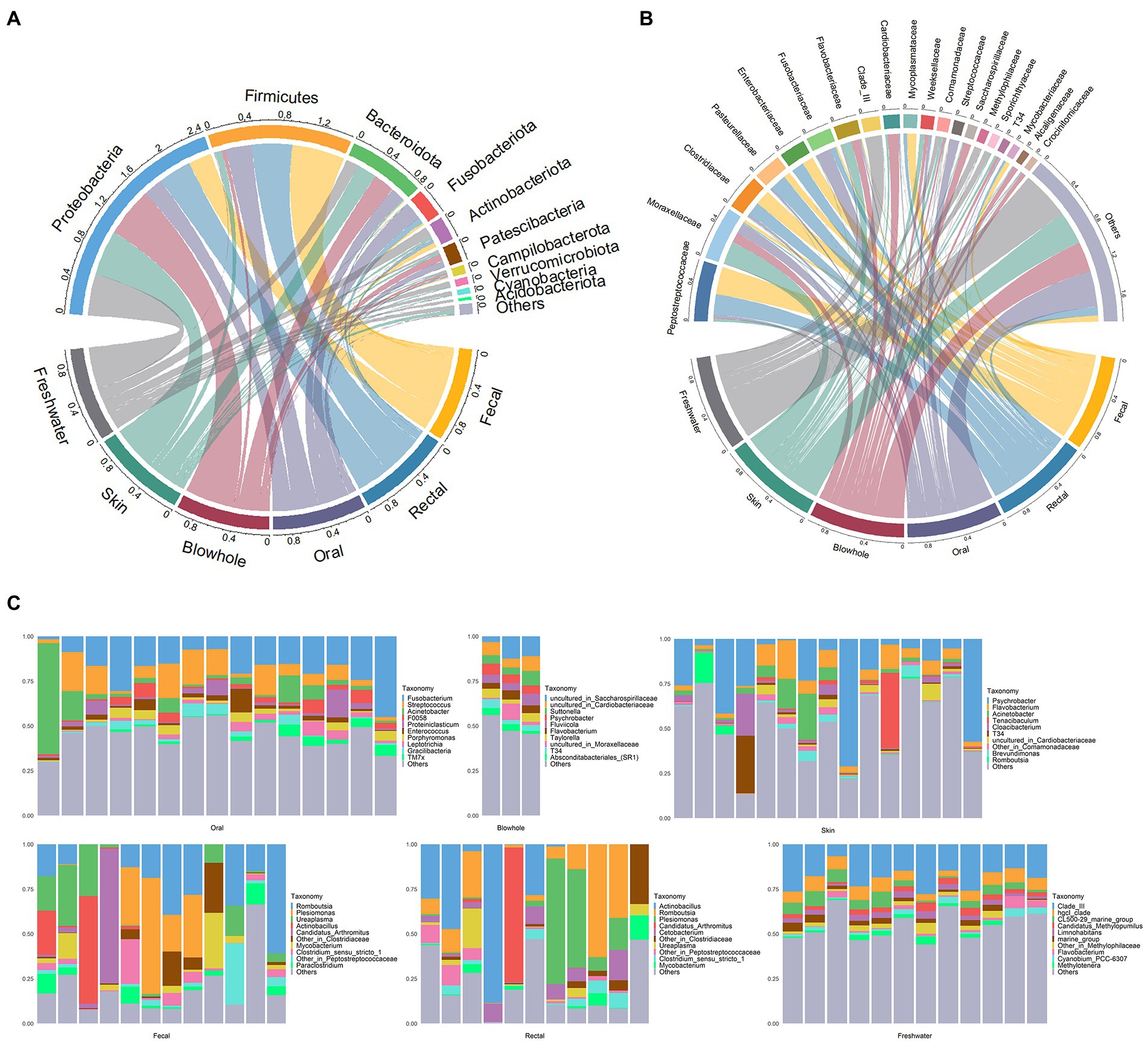
Figure 1. Bacterial composition of YFP and the freshwater environment. (A) The circos plot of bacterial composition at the phylum level. The top 10 phyla with an average scale are displayed. (B) The circos plot of bacterial composition at the family level. The top 20 families with an average scale are displayed. (C) The bar plot of bacterial composition at the genus level. The top 10 genera with an average scale are displayed in each sample.
At the genus level, Romboutsia (fecal: 20 ± 5.3%, rectal: 16 ± 6%), Plesiomonas (fecal: 13 ± 6.1%, rectal: 14 ± 7.5%), Actinobacillus (fecal: 7.3 ± 5.2%, rectal: 18 ± 8.6%), Candidatus Arthromitus (fecal: 6.8 ± 6.2%, rectal: 7 ± 6.8%) dominated in the fecal and rectal samples (Figure 1C). Ureaplasma (9.9 ± 3.5%) in the fecal samples and Cetobacterium (5.6 ± 1.6%) in the rectal samples were abundant (Figure 1C). The oral bacteria was dominated by Fusobacterium (17 ± 2.7%), Streptococcus (9.8 ± 1.9%), and Acinetobacter (8.7 ± 4.0%, Figure 1C). The dominant genera in blowhole include Suttonella (5.5 ± 1.3%), Psychrobacter (5.2 ± 0.52%), and two uncultured genera assigned to Saccharospirillaceae (8.9 ± 2.9%) and Cardiobacteriaceae (6.9 ± 0.98%, Figure 1C). Psychrobacter was dominant in the skin samples, accounting for nearly 20% (20 ± 5.6%). Other dominant genera in the skin samples were Flavobacterium (6.9 ± 1.4%) and Acinetobacter (4.7 ± 1.9%, Figure 1C).
Bacterial diversity of Yangtze finless porpoise
Shannon rarefaction curves showed sufficient coverage of diversity in all samples (Supplementary Figure S1). The bacterial alpha diversity metrics (ACE, Chao1, Shannon, Faith PD) between various body sites of YFP showed that the skin had the highest diversity, significantly higher than other body sites. On the contrary, the intestinal bacteria (fecal and rectal) was the lowest alpha diversity (Figure 2A). No significant alpha diversity difference was found between fecal and rectal samples (Wilcoxon test, p > 0.05, Figure 2A). There were no significant differences in alpha diversity between oral and blowhole samples, except for the Shannon metric.
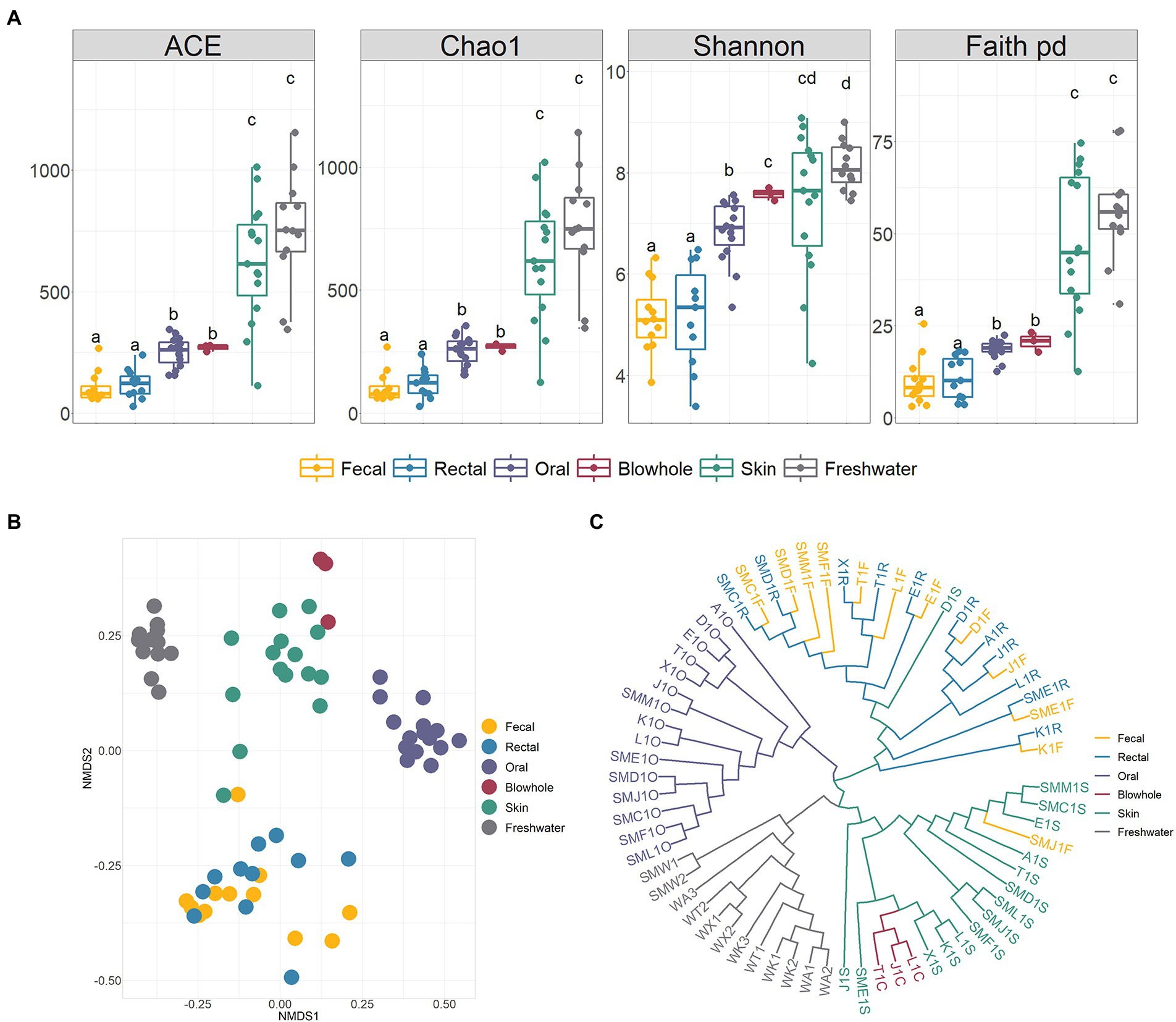
Figure 2. Alpha and beta diversity of bacteria in the YFP and the freshwater. (A) Alpha diversity (ACE, Chao1, Shannon, Faith PD). Wilcoxon test calculates statistical significance between samples, with different letters indicating significant differences (p < 0.05). (B) NMDS ordination based on Bray Curtis dissimilarity across various body sites and the freshwater. (C) The unweighted pair-group method with arithmetic means (UPGMA) clustering analysis base on Bray Curtis dissimilarity. The meaning of the sample corresponding to the label is shown in Supplementary Table S1.
The NMDS ordination, UPGMA clustering, and PERMANOVA analysis based on Bray Curtis dissimilarity demonstrated significant divergence of bacteria in various body sites (PERMANOVA, p < 0.01, Figures 2B,C; Supplementary Table S2). The intestinal, oral, blowhole and skin bacteria showed an evident dissimilarity to each other and constructed four distinct clusters (PERMANOVA, p < 0.001, Figures 2B,C; Supplementary Table S2). In particular, no significant differences between the fecal and rectal samples (PERMANOVA, p > 0.05, Figure 2B), and the fecal and rectal samples were gathered in the same cluster (Figure 2C).
Bacteria in different conditions
Compare the bacterial differences of YFP in different sexes and conditions. The results showed no differences in the bacterial communities between sexes (PERMANOVA, p > 0.05, Supplementary Table S2). The YFP in natural and semi-natural conditions also did not show significant community differences in alpha and beta diversity of the intestinal (fecal and rectal) bacteria (PERMANOVA, p > 0.05, Supplementary Table S2 and Supplementary Figures S3A,B). However, the skin and oral bacteria were different between the two conditions. The Faith PD metric of skin bacteria in the semi-natural is significantly more than that in the natural condition (Wilcoxon test, p < 0.05, Figure 3B). A significant difference exists between the oral bacterial communities of the YFP in natural and semi-natural conditions (PERMANOVA, oral: p < 0.001, skin: p < 0.05, Figure 3A; Supplementary Table S2). Enhydrobacter, JGI 0000069-P22, and Bacteroides were significantly abundant in the oral bacteria of the YFP in natural conditions. In contrast, Chryseobacterium, Proteiniphilum, TM7x, Tannerella, and Flavobacterium were abundant in the semi-natural YFP (Wilcoxon test, p < 0.05, Figure 3C). Four significantly different genera of skin bacteria were found. Acinetobacter was abundant in the oral of the natural YFP, and Luteolibacter, CL500-29 marine group, hgcl clade were higher in the oral of the semi-natural YFP (Wilcoxon test, p < 0.05, Figure 3C). By comparing the bacterial differences in the freshwater environments between different conditions, 18 differential bacteria were found, of which 14 were abundant in the semi-natural conditions (Supplementary Figure S2).
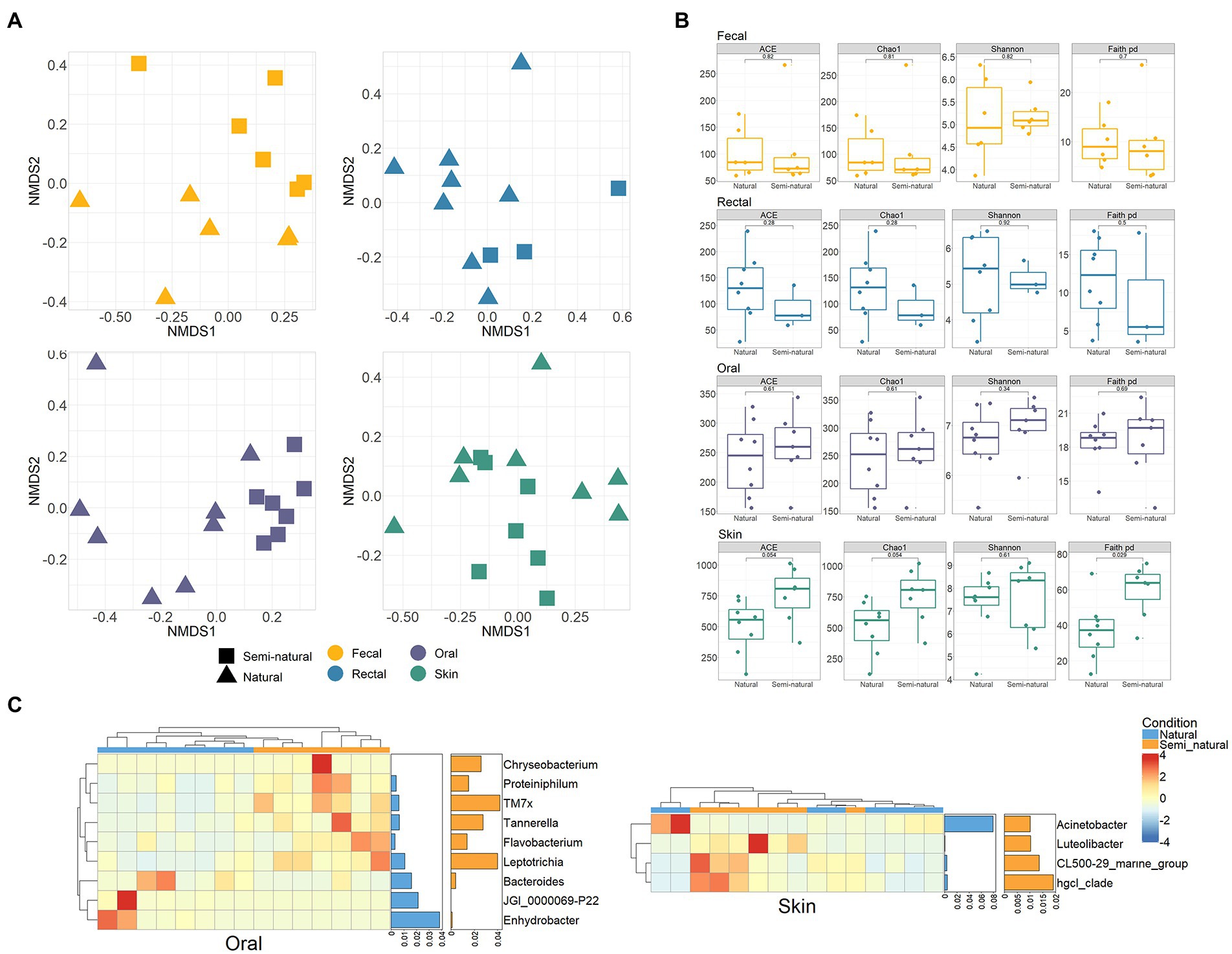
Figure 3. Bacterial differences in the natural and semi-natural condition. (A) NMDS ordination based on Bray Curtis dissimilarity between the natural and semi-natural condition in four body sites of YFP. (B) Comparison of alpha diversity differences of YFP bacteria under two conditions. Wilcoxon test calculates statistical significance between samples. (C) Heatmap of the oral and skin differential bacteria (genus level) between the natural and semi-natural conditions. Each row and column of the heatmap corresponds to genus and samples, respectively. The row data for each genus were z-score transformed. The bar plot on the right represents the average proportion of bacteria in two conditions. Wilcoxon test was used to test the significance (p < 0.05), and the genera whose proportion was less than 1% were excluded.
Bacterial function of Yangtze finless porpoise
The NMDS ordination and PERMANOVA analysis show significant differences in bacterial functions in different body sites of YFP (PERMANOVA, p < 0.001, Figure 4A; Supplementary Table S3). In particular, the function of fecal and rectal bacteria did not differ significantly, nor did the function of blowhole and skin bacteria (PERMANOVA, p > 0.05, Figure 4A; Supplementary Table S3). The bacterial function of each body site showed no significant difference in different sexes and conditions (PERMANOVA, p > 0.05, Supplementary Table S3).
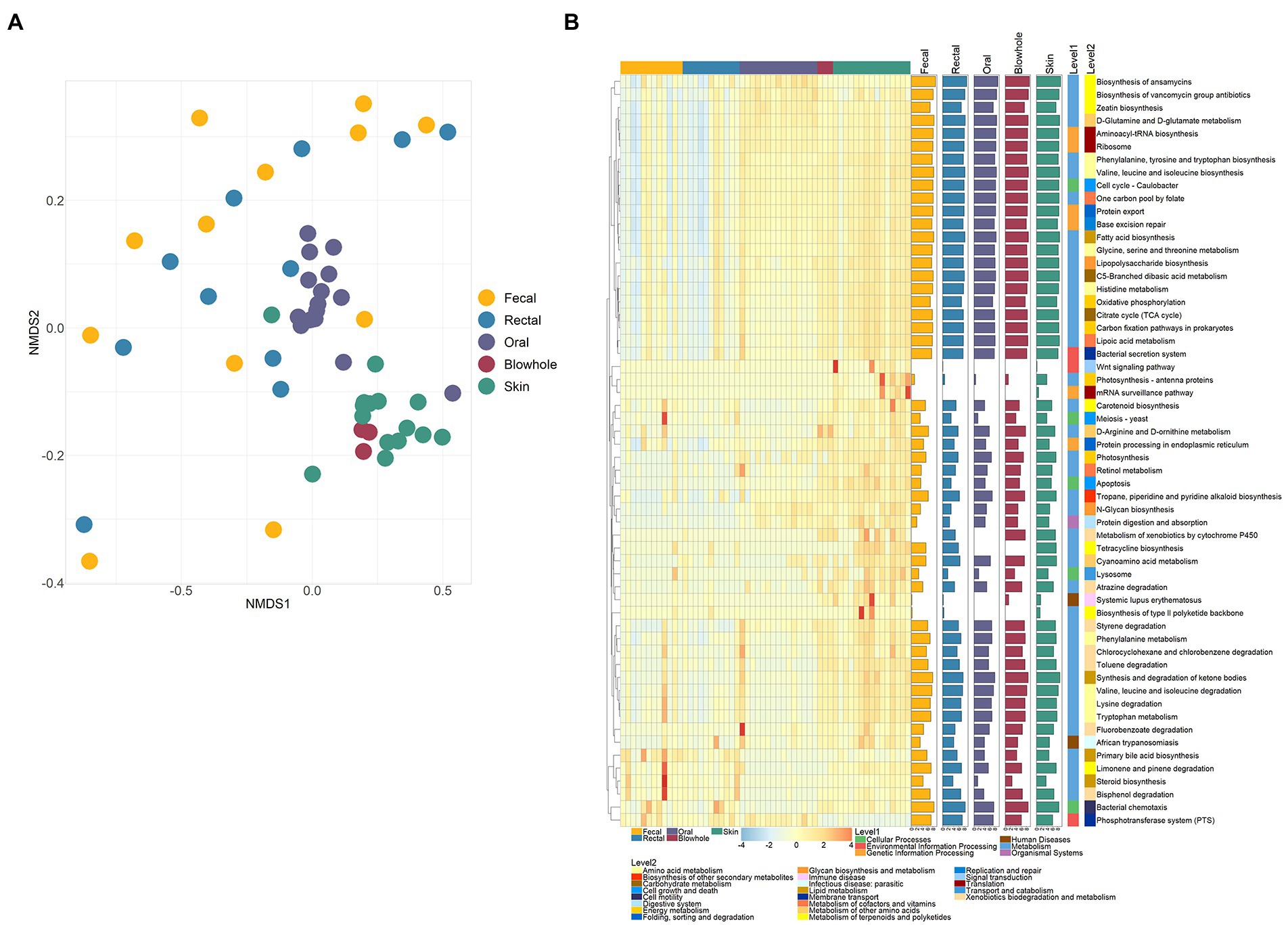
Figure 4. Bacterial functions of various body sites. (A) NMDS ordination of PICRUSt2 predicted functions using KEGG orthology (KO) based on Bray Curtis dissimilarity. (B) Heatmap of significant pathways in various body sites. Each row and column of the heatmap corresponds to a pathway and samples, respectively. The row data for each pathway were z-score transformed. The bar plot on the right represents the average abundance of the pathway (data is logarithmically converted). Kruskal–Wallis test was used to calculate the significance and correction by Bonferroni (p < 0.05).
The KO results were further inferred to the KEGG pathway level, and 58 pathways with significant differences were identified. Of these pathways, 41 pathways are categorized as Metabolism, five as Cellular Processes, and six as Genetic Information Processing (Figure 4B). The intestinal bacteria were significantly more abundant in Limonene and pinene degradation, Bisphenol degradation, Tetracycline biosynthesis, Carotenoid biosynthesis, Steroid biosynthesis than in other sites (Figure 4B). The six pathways, D-Glutamine and D-glutamate metabolism, Aminoacyl-tRNA biosynthesis, Biosynthesis of vancomycin group antibiotics, Ribosome, Phosphotransferase system (PTS), and Zeatin biosynthesis, were abundant in the oral bacteria (Figure 4B). The blowhole and skin bacterial function were abundant in Biosynthesis of ansamycins, D-Glutamine and D-glutamate metabolism, Phenylalanine, tyrosine and tryptophan biosynthesis, Tropane, piperidine and pyridine alkaloid biosynthesis, etc (Figure 4B).
Freshwater environmental bacteria
The bacterial community in the freshwater environment was significantly different from that of YFP body sites (PERMANOVA, p < 0.01, Figure 2B; Supplementary Table S2). The dominant bacteria in the freshwater environment were Clade III (19 ± 1.8%), hgcI clade (6.5 ± 0.48%), and CL500-29 marine group (4.6 ± 0.79%, Figure 1C). However, the similarity between freshwater environment bacteria and skin bacteria is higher than that of other body sites (PERMANOVA, p > 0.05, Figure 2B, Supplementary Table S2). Bray Curtis distance between skin and freshwater is 0.97 ± 0.0021, and the mean distance between freshwater and other body sites is above 0.99 (Figure 5A). The alpha diversity of bacteria in the skin and freshwater environments did not show significant differences (Wilcoxon test, p > 0.05, Figure 2A). SourceTrack analysis showed that an average of 2.4% of the skin bacteria was sourced from freshwater environment, while the freshwater environmental associated bacteria in other body sites were less than 0.1% (Figure 4B). In addition, the shared bacterial genera between the skin and freshwater environments reached 285, significantly higher than other body sites (Figure 5C). The shared bacteria accounted for 30% of the total genus of skin and 41% of the total genus of the freshwater environment (Figure 5C). Considering the proximity of freshwater environmental bacteria to skin bacteria, the indigenous bacteria on the skin of YFP were further analyzed by LefSe. A total of nine genus-level bacteria significantly enriched in the skin (LDA >3, mean > 1%) were identified. Psychrobacter was significantly more abundant on the skin than in freshwater environments (skin: 20 ± 5.6%, freshwater: 0.045 ± 0.028%, LDA =5.0, Wilcoxon Test, p < 0.01, Figures 4D,E). Flavobacterium was also enriched on the skin (skin: 6.9 ± 1.4%, LDA = 4.4, Wilcoxon Test, p < 0.01, Figures 5D,E). In particular, T34 and Tenacibaculum were not present in freshwater environments.
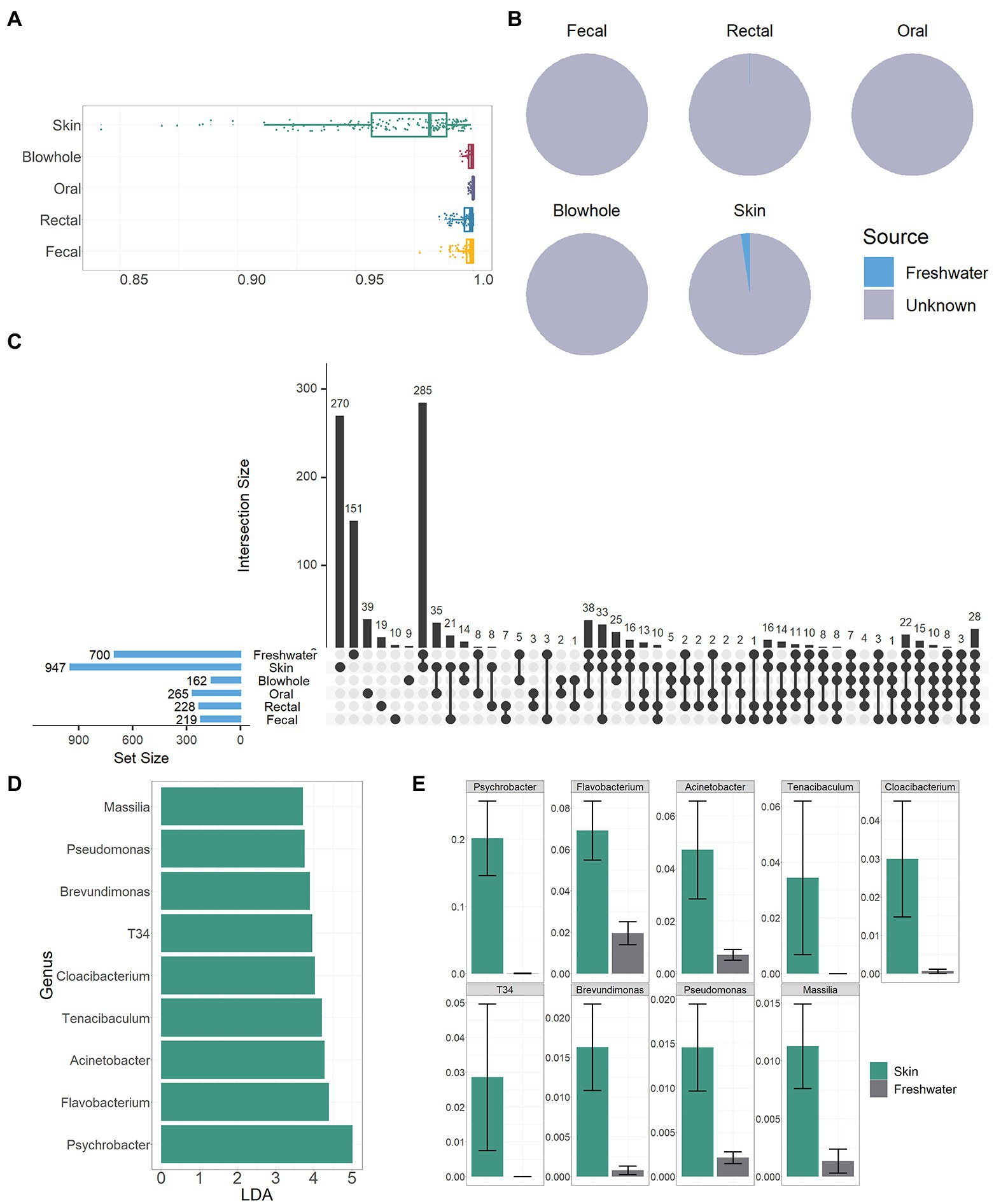
Figure 5. Association of freshwater environmental bacteria with YFP. (A) Bray Curtis dissimilarity between the freshwater environmental and YFP bacteria, the closer the value is to one, the greater the dissimilarity. (B) The pie chart demonstrates the source of the YFP bacteria. (C) Vertical bars of the upper plot show number of intersecting genera among the YFP and freshwater environments, denoted by connected black circles below the histogram. Horizontal bars show genus set size. (D) LDA value of nine genera. (E) The proportion of nine bacterial genera in the skin and freshwater environment.
Discussion
This study used 16 s-rRNA high-throughput sequencing technology to characterize the bacterial diversity and composition of various body sites of YFP. The bacteria of YFP show apparent body site-specificity, the boundaries of bacteria in different body sites are obvious, and each body site has its dominant bacteria. This site-specific feature is also evidenced in the bacterial function of YFP. Our results are consistent with previous studies on mammals such as humans (Lloyd-Price et al., 2016), macaques (Chen et al., 2018), dolphins, and sea lions (Bik et al., 2016), where body sites are a significant factor in constructing mammalian microbiota. Previous studies have suggested that body sites often act as environmental filters, hindering the colonization of bacteria not equipped to survive in their respective environments, allowing body sites to form distinct microbial communities (Pereira and Berry, 2017; García-Bayona and Comstock, 2018; Rojas et al., 2020). The site-specificity of YFP bacteria inspired us to consider the influence of microbiota in studying the structural characteristics and physiology of various body sites of YFP.
The intestinal bacteria of YFP was the first to attract the attention of researchers (McLaughlin et al., 2011, 2012, 2013; Wan et al., 2016b; You et al., 2020). We identified some dominant genera not characterized in previous studies, including Romboutsia, Candidatus Arthromitus, Plesiomonas, and Actinobacillus. Romboutsia is highly adapted to the intestinal environment and has the ability to ferment amino acids (Gerritsen, 2015; Gerritsen et al., 2019). Plesiomonas is a bacteria that mainly lives in various water environments (Janda et al., 2016), can cause gastrointestinal diseases in humans, and have a certain degree of antibiotic resistance (Hong et al., 2019; Cortés-Sánchez et al., 2021). Although there are no studies of this bacteria causing disease in YFP, it is worth paying attention to. Cetobacterium was found in the intestines of a variety of marine mammals (Bik et al., 2016; Suzuki et al., 2021; Bai et al., 2022) and can produce vitamin B12 (Suzuki et al., 2022), which may be a potential probiotic for YFP. In this study, there was no significant difference in the diversity and composition of fecal and rectal bacteria, which indicated that fecal and rectal samples were equivalent in characterizing intestinal microbiota. This result reminds us that we can choose a less invasive sampling method according to the actual situation to obtain intestinal bacteria samples of YFP.
Many studies have investigated the oral microbiota of marine mammals and discovered many novel bacteria in the oral cavity of dolphins (Bik et al., 2016; Dudek et al., 2017; Soares-Castro et al., 2019), but little known about the oral bacteria of YFP. The high abundance of Fusobacterium found in the oral samples of YFP is consistent with previous findings in several species of dolphins (Bik et al., 2016; Soares-Castro et al., 2019), indicating that Fusobacterium is a common microbe in the oral cavity of cetaceans. Streptococcus is considered the dominant genus in the healthy human oral cavity and plays a vital role in the assembly of the oral microbiota (Abranches et al., 2018). Characterizing the oral bacteria of YFP enhanced our understanding of the oral health status of YFP.
Skin is an essential organ of aquatic mammals and is colonized by a rich microbial community (Apprill et al., 2011, 2014; Bik et al., 2016; Chiarello et al., 2017; Li et al., 2019). The skin of YFP has the highest bacterial diversity and has unique indigenous bacteria. Flavobacterium, Psychrobacter, and Acinetobacter predominate in the skin of YFP. Combined with previous studies (Apprill et al., 2011, 2020), these genera may be a class of microbiota broadly associated with the skin of aquatic mammals. Flavobacterium often plays the role of pathogenic bacteria in freshwater aquaculture systems (Duchaud et al., 2018; Pérez-Pascual et al., 2021), but its function and pathogenicity on the skin of YFP have not been studied. In particular, The Tenacibaculum and T34 were only found in the skin but not in the freshwater environment. Tenacibaculum is a bacteria that causes skin ulcers in fish (Olsen et al., 2022). T34 is a class of uncultivated bacteria belonging to Burkholderiales (Kim et al., 2022). The function of these bacteria on the animal is unknown. Presently, the potential sources of Tenacibaculum and T34 on the skin have not been determined, which is worthy of further analysis of their sources and functions.
Blowhole microbiota is a crucial index to evaluate the health status of aquatic mammals, and researchers collect qualified samples of blowhole microbiota through drones and other methods (Nelson et al., 2019; Centelleghe et al., 2020; Vendl et al., 2021). Considering the low extraction rate of blowhole samples in this study (only three blowhole samples were successfully extracted), we thought that perhaps the number of breaths collected should be increased at the time of sample collection to increase the amount of microbial DNA in the samples. Our findings are similar to those of a study in the blowhole of bottlenose dolphins (Lima et al., 2012), where the Saccharospirillaceae and Cardiobacteriaceae were the dominant bacteria. However, other studies on cetaceans have different results (Apprill et al., 2017; Nelson et al., 2019; Centelleghe et al., 2020), which may require further comprehensive cross-ocean mammalian blowhole microbial studies to explain. Some species in Suttonella can cause avian pneumonia (Peniche et al., 2017; Fischer et al., 2021), and it has not been reported that such bacteria can cause respiratory disease in YFP, but it needs attention.
Based on the body site-specific, we explored the differences between sexes and between natural and semi-natural conditions in YFP bacteria. There is no unanimous conclusion on the effect of sex on marine mammal microbiota (Apprill et al., 2014; Bik et al., 2016; Chiarello et al., 2017; Rojas et al., 2020). In this study, the operation mode of sex on the YFP bacteria was not found, indicating that bacteria differences between male and female YFP need to be further studied. This study initially showed that porpoises with different living conditions differed in oral and skin bacteria, while no differences were shown in intestinal bacteria. The differences in freshwater environmental bacteria could explain some of the reasons for the differences in skin and oral bacteria, such as Flavobacterium and Luteolibacter. The difference in microbiota in aquatic mammals under different living conditions is one of the crucial concerns in conservation biology (Bik et al., 2016; Wan et al., 2016b; Trevelline et al., 2019). Natural and semi-natural YFP may differ in the food source, water environment, and population size. While referring to previous studies of aquatic mammals (Bik et al., 2016; Wan et al., 2016b; Tian et al., 2022), these factors may be the potential factors affecting the bacteria of the YFP. Further research on the natural and semi-natural microbiota of YFP is of great significance for the ex situ conservation assessment of YFP.
Our study shows that the association between the YFP bacteria and freshwater environmental bacteria is consistent with previous findings on marine mammals that a clear boundary is maintained between animal and environmental bacteria (Apprill et al., 2014; Bik et al., 2016; Chiarello et al., 2017; You et al., 2020). There are apparent differences in the community structure and composition between YFP and the freshwater environment. Of course, the difference between YFP and environmental bacteria does not mean there is no mutual communication between the animal and environment. SourceTrack analysis showed that freshwater environmental bacteria were one of the sources of YFP. There are more bacteria from the freshwater environment in the skin, and very few in other parts, indicating that the freshwater environment bacteria have different effects on the bacteria of different body sites of YFP. The microbial environment background in this study only focuses on freshwater environment, and both food and air may be the source of bacteria in various body sites of aquatic mammals (Bik et al., 2016; Vendl et al., 2019). The role of YFP and the bacteria in diverse environmental contexts deserves more research to improve our insights into habitat selection and assessment of YFP from a microbial perspective.
In summary, we provide a more comprehensive bacterial map of an endangered natural animal, Yangtze finless porpoise. This study characterized the bacterial composition and diversity of the intestine (fecal and rectal), oral, blowhole, and skin of YFP. The bacteria differed significantly between different body sites and had their major bacteria, showing clear body site specificity. Meanwhile, the bacterial function of YFP also showed site-specificity. The bacterial communities of YFP in natural and semi-natural conditions were significantly different. There is a clear boundary between freshwater environmental bacteria and the YFP bacteria. The skin bacteria are relatively more affected by the freshwater environment bacteria. Our study shows the basic information on the YFP bacteria and the influence of the freshwater environment and provides a reference for the research on the YFP bacteria. Further research on the bacteria of YFP is of great significance for protecting YFP.
Data availability statement
The raw data of this study have been submitted to the NCBI database under the project number PRJNA857500. The code in the study has been uploaded to Github (https://github.com/CesarZhang/YZFP-16s).
Ethics statement
The animal study was reviewed and approved by the Wildlife Protection Law of the People’s Republic of China China’s Laboratory animal—Guideline for Ethical Review of Animal Welfare (GB/T 35892-2018) Office of Fisheries Law Enforcement for Yangtze River Basin of Ministry of Agriculture and Rural Affairs of the People's Republic of China (2017 [185]) and the Fisheries Bureau of Anqing City, Anhui Province, respectively.
Author contributions
XZ wrote the original manuscript and performed the data analysis and visualization. CY, KL, and PX reviewed and edited the article. MJ, LY, DY, and JZ participated in the investigation of this study. DL provided the necessary resources for this study. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Key R&D Program of China (No, 2021YFD1200304) and project of the Yangtze finless porpoise protection and implementation in the middle and lower reaches of the Yangtze River.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1006251/full#supplementary-material
References
Abranches, J., Zeng, L., Kajfasz, J. K., Palmer, S. R., Chakraborty, B., Wen, Z. T., et al. (2018). Biology of oral streptococci. Microbiol. Spect. 6:2018. doi: 10.1128/microbiolspec.GPP3-0042-2018
Afgan, E., Baker, D., Batut, B., van den Beek, M., Bouvier, D., Čech, M., et al. (2018). The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544. doi: 10.1093/nar/gky379
Apprill, A., Miller, C. A., Moore, M. J., Durban, J. W., Fearnbach, H., and Barrett-Lennard, L. G. (2017). Extensive Core microbiome in drone-captured whale blow supports a framework for health monitoring. mSystems 2:17. doi: 10.1128/mSystems.00119-17
Apprill, A., Miller, C. A., Van Cise, A. M., U’Ren, J. M., Leslie, M. S., Weber, L., et al. (2020). Marine mammal skin microbiotas are influenced by host phylogeny. R. Soc. Open Sci. 7:192046. doi: 10.1098/rsos.192046
Apprill, A., Mooney, T. A., Lyman, E., Stimpert, A. K., and Rappé, M. S. (2011). Humpback whales harbour a combination of specific and variable skin bacteria. Environ. Microbiol. Rep. 3, 223–232. doi: 10.1111/j.1758-2229.2010.00213.x
Apprill, A., Robbins, J., Eren, A. M., Pack, A. A., Reveillaud, J., Mattila, D., et al. (2014). Humpback whale populations share a core skin bacterial community: towards a health index for marine mammals? PLoS One 9:e90785. doi: 10.1371/journal.pone.0090785
Bai, S., Zhang, P., Zhang, X., Yang, Z., and Li, S. (2022). Gut microbial characterization of melon-headed whales (Peponocephala electra) stranded in China. Microorganisms 10:572. doi: 10.3390/microorganisms10030572
Bik, E. M., Costello, E. K., Switzer, A. D., Callahan, B. J., Holmes, S. P., Wells, R. S., et al. (2016). Marine mammals harbor unique microbiotas shaped by and yet distinct from the sea. Nat. Commun. 7:10516. doi: 10.1038/ncomms10516
Bokulich, N. A., Kaehler, B. D., Rideout, J. R., Dillon, M., Bolyen, E., Knight, R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90. doi: 10.1186/s40168-018-0470-z
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Centelleghe, C., Carraro, L., Gonzalvo, J., Rosso, M., Esposti, E., Gili, C., et al. (2020). The use of unmanned aerial vehicles (UAVs) to sample the blow microbiome of small cetaceans. PLoS One 15:e0235537. doi: 10.1371/journal.pone.0235537
Chen, Z., Yeoh, Y. K., Hui, M., Wong, P. Y., Chan, M. C. W., Ip, M., et al. (2018). Diversity of macaque microbiota compared to the human counterparts. Sci. Rep. 8:15573. doi: 10.1038/s41598-018-33950-6
Chiarello, M., Vill, E., Ger, S., Bouvier, C., Auguet, J. C., and Bouvier, T. (2017). Captive bottlenose dolphins and killer whales harbor a species-specific skin microbiota that varies among individuals. Sci. Rep. 7:15269. doi: 10.1038/s41598-017-15220-z
Conway, J. R., Lex, A., and Gehlenborg, N. (2017). UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940. doi: 10.1093/bioinformatics/btx364
Cortés-Sánchez, A. D. J., Espinosa-Chaurand, L. D., Díaz-Ramirez, M., and Torres-Ochoa, E. (2021). Plesiomonas: a review on food safety, fish-borne diseases, and tilapia. Sci. World J. 2021:e3119958, 1–10. doi: 10.1155/2021/3119958
Di Guardo, G., Criscitiello, M. F., Sierra, E., and Mazzariol, S. (2019). Comparative immunology of marine mammals. Front. Immunol. 10:2300. doi: 10.3389/fimmu.2019.02300
Dierauf, L., and Gulland, F. M. (2001). CRC handbook of marine mammal medicine: Health, disease, and rehabilitation. CRC press.
Douglas, G. M., Maffei, V. J., Zaneveld, J. R., Yurgel, S. N., Brown, J. R., Taylor, C. M., et al. (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688. doi: 10.1038/s41587-020-0548-6
Duchaud, E., Rochat, T., Habib, C., Barbier, P., Loux, V., Guérin, C., et al. (2018). Genomic diversity and evolution of the fish pathogen Flavobacterium psychrophilum. Front. Microbiol. 9:138. doi: 10.3389/fmicb.2018.00138
Dudek, N. K., Sun, C. L., Burstein, D., Kantor, R. S., Goltsman, D. S. A., Bik, E. M., et al. (2017). Novel microbial diversity and functional potential in the marine mammal oral microbiome. Curr. Biol. 27, 3752–3762.e6. doi: 10.1016/j.cub.2017.10.040
Fischer, L., Peters, M., Merbach, S., Eydner, M., Kuczka, A., Lambertz, J., et al. (2021). Increased mortality in wild tits in North Rhine-Westphalia (Germany) in 2020 with a special focus on Suttonella ornithocola and other infectious pathogens. Eur. J. Wildl. Res. 67:56. doi: 10.1007/s10344-021-01500-7
García-Bayona, L., and Comstock, L. E. (2018). Bacterial antagonism in host-associated microbial communities. Science 361:eaat 2456. doi: 10.1126/science.aat2456
Gerritsen, J. (2015). The genus Romboutsia: Genomic and functional characterization of novel bacteria dedicated to life in the intestinal tract. [thesis]. Wageningen:Wageningen University.
Gerritsen, J., Hornung, B., Ritari, J., Paulin, L., Rijkers, G. T., Schaap, P. J., et al. (2019). A comparative and functional genomics analysis of the genus Romboutsia provides insight into adaptation to an intestinal lifestyle. bioRxiv :845511. doi: 10.1101/845511
Gu, Z., Eils, R., and Schlesner, M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. doi: 10.1093/bioinformatics/btw313
Hong, Y., PengFei, M., ShuWen, T., Ying, Y., YongLiang, W., Yan Feng, C., et al. (2019). Relationship between drug resistance and antidrug gene in bacterial pathogen Plesiomonas shigelloides. Fish. Sci. 38, 266–270.
Huang, J., Mei, Z., Chen, M., Han, Y., Zhang, X., Moore, J. E., et al. (2020). Population survey showing hope for population recovery of the critically endangered Yangtze finless porpoise. Biol. Conserv. 241:108315. doi: 10.1016/j.biocon.2019.108315
Huang, S.-L., Mei, Z., Hao, Y., Zheng, J., Wang, K., and Wang, D. (2017). Saving the Yangtze finless porpoise: time is rapidly running out. Biol. Conserv. 210, 40–46. doi: 10.1016/j.biocon.2016.05.021
Huggins, J. L., Garner, M. M., Raverty, S. A., Lambourn, D. M., Norman, S. A., Rhodes, L. D., et al. (2020). The emergence of Mucormycosis in free-ranging marine mammals of the Pacific northwest. Front. Mar. Sci. 7. doi: 10.3389/fmars.2020.00555
Janda, J. M., Abbott, S. L., and McIver, C. J. (2016). Plesiomonas shigelloides revisited. Clin. Microbiol. Rev. 29, 349–374. doi: 10.1128/CMR.00103-15
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kim, D. D., Han, H., Yun, T., Song, M. J., Terada, A., Laureni, M., et al. (2022). Identification of nosZ-expressing microorganisms consuming trace N2O in microaerobic chemostat consortia dominated by an uncultured Burkholderiales. ISME J. 16, 2087–2098. doi: 10.1038/s41396-022-01260-5
Knights, D., Kuczynski, J., Charlson, E. S., Zaneveld, J., Mozer, M. C., Collman, R. G., et al. (2011). Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 8, 761–763. doi: 10.1038/nmeth.1650
Li, C., Tan, X., Bai, J., Xu, Q., Liu, S., Guo, W., et al. (2019). A survey of the sperm whale (Physeter catodon) commensal microbiome. PeerJ 7:e7257. doi: 10.7717/peerj.7257
Lima, N., Rogers, T., Acevedo-Whitehouse, K., and Brown, M. V. (2012). Temporal stability and species specificity in bacteria associated with the bottlenose dolphins respiratory system. Environ. Microbiol. Rep. 4, 89–96. doi: 10.1111/j.1758-2229.2011.00306.x
Liu, Z., Li, A., Wang, Y., Iqbal, M., Zheng, A., Zhao, M., et al. (2020). Comparative analysis of microbial community structure between healthy and Aeromonas veronii -infected Yangtze finless porpoise. Microb. Cell Factories 19, 1–12. doi: 10.1186/s12934-020-01383-4
Liu, Z. G., Zheng, A. F., Chen, M. M., Lian, Y. X., Zhang, X. K., Zhang, S. Z., et al. (2018). Isolation and identification of pathogenic Aeromonas veronii from a dead Yangtze finless porpoise. Dis. Aquat. Org. 132, 13–22. doi: 10.3354/dao03288
Lloyd-Price, J., Abu-Ali, G., and Huttenhower, C. (2016). The healthy human microbiome. Genome Med. 8:51. doi: 10.1186/s13073-016-0307-y
McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., DeSantis, T. Z., Probst, A., et al. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. doi: 10.1038/ismej.2011.139
McLaughlin, R. W., Chen, M., Zheng, J., Zhao, Q., and Wang, D. (2012). Analysis of the bacterial diversity in the fecal material of the endangered Yangtze finless porpoise, Neophocaena phocaenoides asiaeorientalis. Mol. Biol. Rep. 39, 5669–5676. doi: 10.1007/s11033-011-1375-0
McLaughlin, R. W., Zheng, J. S., Chen, M. M., Zhao, Q. Z., and Wang, D. (2011). Detection of helicobacter in the fecal material of the endangered Yangtze finless porpoise Neophocaena phocaenoides asiaeorientalis. Dis. Aquat. Org. 95, 241–245. doi: 10.3354/dao02364
McLaughlin, R. W., Zheng, J., Ruan, R., Wang, C., Zhao, Q., and Wang, D. (2013). Isolation of Robinsoniella peoriensis from the fecal material of the endangered Yangtze finless porpoise, Neophocaena asiaeorientalis asiaeorientalis. Anaerobe 20, 79–81. doi: 10.1016/j.anaerobe.2012.12.008
Mei, Z., Zhang, X., Huang, S.-L., Zhao, X., Hao, Y., Zhang, L., et al. (2014). The Yangtze finless porpoise: on an accelerating path to extinction? Biol. Conserv. 172, 117–123. doi: 10.1016/j.biocon.2014.02.033
Mootapally, C. S., Poriya, P., Nathani, N. M., Venmathi Maran, B. A., and Gadhvi, I. R. (2017). “Recent advances in the Metagenomics of marine mammals microbiome” in Understanding host-microbiome interactions - an Omics approach: Omics of host-microbiome association. eds. R. P. Singh, R. Kothari, P. G. Koringa, and S. P. Singh (Singapore: Springer), 327–336. doi: 10.1007/978-981-10-5050-3_18
Nelson, T. M., Apprill, A., Mann, J., Rogers, T. L., and Brown, M. V. (2015). The marine mammal microbiome: current knowledge and future directions. Microbiol. Australia 36, 8–13. doi: 10.1071/MA15004
Nelson, T. M., Wallen, M. M., Bunce, M., Oskam, C. L., Lima, N., Clayton, L., et al. (2019). Detecting respiratory bacterial communities of wild dolphins: implications for animal health. Mar. Ecol. Prog. Ser. 622, 203–217. doi: 10.3354/meps13055
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., et al. (2020). Vegan: Community ecology package. Available at: https://CRAN.R-project.org/package=vegan [Accessed October 11, 2022].
Olsen, A. B., Spilsberg, B., Nilsen, H. K., Lagesen, K., Gulla, S., Avendaño-Herrera, R., et al. (2022). Tenacibaculum piscium sp. nov., isolated from skin ulcers of sea-farmed fish, and description of Tenacibaculum finnmarkense sp. nov. with subdivision into genomovars finnmarkense and ulcerans. Int. J. Syst. Evol. Microbiol. 70, 6079–6090. doi: 10.1099/ijsem.0.004501
Peniche, G., Fernandez, J. R.-R., Durrant, C., John, S. K., Macgregor, S. K., Cunningham, A. A., et al. (2017). Nested PCR for Suttonella ornithocola reveals widespread infection in British Paridae species. Eur. J. Wildl. Res. 63, 1–9. doi: 10.1007/s10344-017-1105-6
Pereira, F. C., and Berry, D. (2017). Microbial nutrient niches in the gut. Environ. Microbiol. 19, 1366–1378. doi: 10.1111/1462-2920.13659
Pérez-Pascual, D., Vendrell-Fernández, S., Audrain, B., Bernal-Bayard, J., Patiño-Navarrete, R., Petit, V., et al. (2021). Gnotobiotic rainbow trout (Oncorhynchus mykiss) model reveals endogenous bacteria that protect against Flavobacterium columnare infection. PLoS Pathog. 17:e1009302. doi: 10.1371/journal.ppat.1009302
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
R Core Team (2021). R: A language and environment for statistical computing. Vienna, Austria: R core team. Available at: https://www.R-project.org/ [Accessed October 10, 2022].
Raverty, S. A., Rhodes, L. D., Zabek, E., Eshghi, A., Cameron, C. E., Hanson, M. B., et al. (2017). Respiratory microbiome of endangered southern resident killer whales and microbiota of Surrounding Sea surface microlayer in the eastern North Pacific. Sci. Rep. 7, 394–312. doi: 10.1038/s41598-017-00457-5
Rojas, C. A., Holekamp, K. E., Winters, A. D., and Theis, K. R. (2020). Body-site specific microbiota reflect sex and age-class among wild spotted hyenas. FEMS Microbiol. Ecol. 96:fiaa007. doi: 10.1093/femsec/fiaa007
Sanders, J. G., Beichman, A. C., Roman, J., Scott, J. J., Emerson, D., McCarthy, J. J., et al. (2015). Baleen whales host a unique gut microbiome with similarities to both carnivores and herbivores. Nat. Commun. 6:8285. doi: 10.1038/ncomms9285
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Soares-Castro, P., Araújo-Rodrigues, H., Godoy-Vitorino, F., Ferreira, M., Covelo, P., López, A., et al. (2019). Microbiota fingerprints within the oral cavity of cetaceans as indicators for population biomonitoring. Sci. Rep. 9, 1–15. doi: 10.1038/s41598-019-50139-7
Suzuki, A., Akuzawa, K., Kogi, K., Ueda, K., and Suzuki, M. (2021). Captive environment influences the composition and diversity of fecal microbiota in indo-Pacific bottlenose dolphins, Tursiops aduncus. Mar. Mamm. Sci. 37, 207–219. doi: 10.1111/mms.12736
Suzuki, A., Shirakata, C., Anzai, H., Sumiyama, D., and Suzuki, M. (2022). Vitamin B12 biosynthesis of Cetobacterium ceti isolated from the intestinal content of captive common bottlenose dolphins (Tursiops truncatus). Microbiology 168:001244. doi: 10.1099/mic.0.001244
Tian, J., Sanganyado, E., Wang, Z., Kong, Z., Han, J., Lu, Z., et al. (2022). Spotted seals (Phoca largha) harbor unique gut microbiota shaped by their host habitat. Sci. Total Environ. 832:155015. doi: 10.1016/j.scitotenv.2022.155015
Trevelline, B. K., Fontaine, S. S., Hartup, B. K., and Kohl, K. D. (2019). Conservation biology needs a microbial renaissance: a call for the consideration of host-associated microbiota in wildlife management practices. Proc. R. Soc. B 286:20182448. doi: 10.1098/rspb.2018.2448
Turvey, S. T., Pitman, R. L., Taylor, B. L., Barlow, J., Akamatsu, T., Barrett, L. A., et al. (2007). First human-caused extinction of a cetacean species? Biol. Lett. 3, 537–540. doi: 10.1098/rsbl.2007.0292
Vendl, C., Ferrari, B. C., Thomas, T., Slavich, E., Zhang, E., Nelson, T., et al. (2019). Interannual comparison of core taxa and community composition of the blow microbiota from east Australian humpback whales. FEMS Microbiol. Ecol. 95:fiz102. doi: 10.1093/femsec/fiz102
Vendl, C., Nelson, T., Ferrari, B., Thomas, T., and Rogers, T. (2021). Highly abundant core taxa in the blow within and across captive bottlenose dolphins provide evidence for a temporally stable airway microbiota. BMC Microbiol. 21:20. doi: 10.1186/s12866-020-02076-z
Wan, X., McLaughlin, R. W., Zhou, J., Hao, Y., Zheng, J., and Wang, D. (2016a). Isolation of culturable aerobic bacteria and evidence of Kerstersia gyiorum from the blowhole of captive Yangtze finless porpoises. Antonie Van Leeuwenhoek 109, 1167–1175. doi: 10.1007/s10482-016-0713-6
Wan, X., Ruan, R., McLaughlin, R. W., Hao, Y., Zheng, J., and Wang, D. (2016b). Fecal bacterial composition of the endangered Yangtze finless porpoises living under captive and semi-natural conditions. Curr. Microbiol. 72, 306–314. doi: 10.1007/s00284-015-0954-z
Yin, D., Lin, D., Guo, H., Gu, H., Ying, C., Zhang, Y., et al. (2021). Integrated analysis of blood mRNAs and microRNAs reveals immune changes with age in the Yangtze finless porpoise (Neophocaena asiaeorientalis). Comp. Biochem. Physiol. B: Biochem. Mol. Biol. 256:110635. doi: 10.1016/j.cbpb.2021.110635
You, L., Ying, C., Liu, K., Zhang, X., Lin, D., Yin, D., et al. (2020). Changes in the fecal microbiome of the Yangtze finless porpoise during a short-term therapeutic treatment. Open Life Sci. 15, 296–310. doi: 10.1515/biol-2020-0032
Yu, G., Smith, D. K., Zhu, H., Guan, Y., and Lam, T. T.-Y. (2017). Ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36. doi: 10.1111/2041-210X.12628
Keywords: Yangtze finless porpoise, site-specific, freshwater environment, bacteria, bacterial community, bacterial function
Citation: Zhang X, Ying C, Jiang M, Lin D, You L, Yin D, Zhang J, Liu K and Xu P (2022) The bacteria of Yangtze finless porpoise (Neophocaena asiaeorientalis asiaeorientalis) are site-specific and distinct from freshwater environment. Front. Microbiol. 13:1006251. doi: 10.3389/fmicb.2022.1006251
Edited by:
Jinping Chen, Guangdong Academy of Science (CAS), ChinaReviewed by:
Consuelo Rubio-Guerri, Fundación Oceanogràfic de la Comunitat Valenciana, SpainChunhong Zhu, Jiangsu Institute of Poultry Science (CAAS), China
Copyright © 2022 Zhang, Ying, Jiang, Lin, You, Yin, Zhang, Liu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai Liu, bGl1a0BmZnJjLmNu; Pao Xu, eHVwQGZmcmMuY24=