 Jintao Hu
Jintao Hu Yuhan Wu3
Yuhan Wu3 Jidong Lang
Jidong Lang Geng Tian
Geng Tian Bo Meng
Bo Meng- 1Faculty of Engineering and Information Technology, The University of Melbourne, Parkville, VIC, Australia
- 2Genesis (Beijing) Co., Ltd., Beijing, China
- 3College of Life Sciences, University of Chinese Academy of Sciences, Beijing, China
- 4Department of Gynecology, Sanmenxia Central Hospital of Henan University of Science and Technology, Sanmenxia, Henan, China
Background: Human papillomavirus (HPV) infection is the leading cause of cervical cancer. More and more studies discovered that cervical microbiota (CM) composition correlated with HPV infection and the development of cervical cancer. However, more studies need to be implemented to clarify the complex interaction between microbiota and the mechanism of disease development, especially in a specific area of China.
Materials and methods: In this study, 16S rDNA sequencing was applied on 276 Thin-prep Cytologic Test (TCT) samples of patients from the Sanmenxia area. Systematical analysis of the microbiota structure, diversity, group, and functional differences between different HPV infection groups and age groups, and co-occurrence relationships of the microbiota was carried out.
Results: The major microbiota compositions of all patients include Lactobacillus iners, Escherichia coli, Enterococcus faecalis, and Atopobium vaginae at species level, and Staphylococcus, Lactobacillus, Gardnerella, Bosea, Streptococcus, and Sneathia in genus level. Microbiota diversity was found significantly different between HPV-positive (Chao1 index: 98.8869, p < 0.01), unique-268 infected (infections with one of the HPV genotype 52, 56, or 58, 107.3885, p < 0.01), multi-268 infected (infections with two or more of HPV genotype 52, 56, and 58, 97.5337, p = 0.1012), other1 (94.9619, p < 0.05) groups and HPV-negative group (83.5299). Women older than 60 years old have higher microbiota diversity (108.8851, p < 0.01, n = 255) than younger women (87.0171, n = 21). The abundance of Gardnerella and Atopobium vaginae was significantly higher in the HPV-positive group than in the HPV-negative group, while Burkholderiaceae and Mycoplasma were more abundant in the unique-268 group compared to the negative group. Gamma-proteobacteria and Pseudomonas were found more abundant in older than 60 patients than younger groups. Kyoto Encyclopedia of Genes and Genomes (KEGG) and Clusters of Orthologous Groups (COG) analysis revealed the effects on metabolism by microbiota that the metabolism of cells, proteins, and genetic information-related pathways significantly differed between HPV-negative and positive groups. In contrast, lipid metabolism, signal transduction, and cell cycle metabolism pathway significantly differed between multi-268 and negative groups.
Conclusion: The HPV infection status and age of women were related to CM’s diversity and function pathways. The complex CM co-occurrent relationships and their mechanism in disease development need to be further investigated.
Introduction
Cervical cancer is one of the most common malignant tumours among women. Clinical and epidemiological studies have determined that persistent Human Papillomavirus (HPV) infection is the leading risk factor for developing cervical cancer (Khan et al., 2020). The average time interval from carcinogenic HPV infection to cervical cancer progression is 25–30 years. The Thin-prep Cytologic Test (TCT) and HPV DNA were recommended to be used for HPV infection and cervical cancer status determination (Zhang et al., 2020). TCT detects the morphology of the cells and analyses the bacterial population in the sample through 16S rDNA (Deoxyribonucleic Acid) (Liu et al., 2020), which makes it possible to perform large-scale testing of samples.
Recent research revealed that microbiota might be a significant factor in the relationship between HPV and cervical cancer. Klein et al. found that changes in the cervical microbiota (CM) are related to cervical cancer (Kunene and Mahlangu, 2017). Some studies have shown that cervical/vaginal Lactobacilli can produce lactic acid that inhibits the growth of bacteria associated with bacterial vaginosis (BV) and viral infections (Polatti, 2012). The change in the proportion of microorganisms is related to pathological changes in the reproductive tract. In addition, recent studies have shown a clear correlation between microbiota and HPV infection (Libby et al., 2008; Anahtar et al., 2015). A report also explained a positive correlation between cervical HPV infection and BV-related microbiota (Gillet et al., 2011; Lee et al., 2013). Therefore, microbiota may play an important role in between, which implies that the reveal of the mechanism microbiota play is beneficial to comprehend the HPV infection and cancer evolution. However, the current research results have not clarified the mutual influence (Libby et al., 2008; Anahtar et al., 2015). So far, there are relatively few studies on the association amongst CM, cervical cancer and HPV infection, especially in China, which prompted this research to be conducted.
Therefore, samples from 276 patients were obtained to conduct microbiota research for further analysis. This research aimed to explore the relationship between the HPV infection and CM changes by analysing microbiota changes and the HPV infection concerning HPV infection group, different genotype HPV groups, and the impact of microbiota on cell and metabolic functions, as well as to explore the microbiota changes amongst the group divided by ages in a cohort of populations in Sanmenxia, Henan Province. Aside from that, this research also provides a new reference basis for further understanding the CM’s overall characteristics.
Materials and methods
Study population and specimen collection
A total of 276 cervical lesion samples were collected from patients at Sanmenxia Central Hospital for high-grade squamous intraepithelial lesion (HSIL) screening. The hospital’s medical ethics committee approved this study, and all experiments were carried out following the relevant guidelines and regulations. A fluorescent HPV genotyping kit (Bioperfectus Technologies, Jiangsu, China) was used to analyse the samples for confirmation and subsequent HPV typing. Women who came to Sanmenxia central hospital to do cervical tests in 2019, including TCT test and HPV genotype test, were enrolled in this study. Those samples were not qualified for further study, or low-quality sequencing results were excluded from the study.
DNA extraction
After Pap Smear preparation, 1-ml of the remaining fluid sample was used for DNA isolation. According to the manufacturer’s instructions, the total Genomic DNA sample was extracted using TIANamp Micro DNA Kit (TIANGEN, Beijing, China). The double-stranded (ds) DNA was quantified using a Nanodrop 2000 and Qubit dsDNA HS assay kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The average fragment size of DNA (> 5 Kbp) was measured (identified by comparison to a DL2000 PLUS DNA Ladder, Life Technologies, Carlsbad, CA, USA) on a 1.0% agarose gel in 1×TAE buffer on a Bio-Rad CHEF DRII system.
Sequencing and bioinformatic processing
To build a sequencing library, use PCR primers to amplify the V3–V4 hypervariable region of the 16S rDNA gene. This area provides sufficient information for the taxonomic classification of microbial communities in specimens related to human microbiota research and is used by the Human Microbiota Project.
Then use Agencourt AMPure XP (Beckman Coulter, Indianapolis, Indiana) to select the product size in a ratio of 0.9 and group in equal moles. Then, a Qubit 2.0 fluorometer (Life Technologies) was used to quantify the pool of the selected size and loaded into the Illumina HiSeq flow cell (Illumina, Inc., San Diego, CA, USA) 2 × 250. Mix the library with the Illumina-generated PhiX control library and our genomic library, and use fresh NaOH for denaturation. Perform image analysis, base calls, and data quality assessment on the MiSeq instrument.
Data analysis
Paired-end sequencing (2 × 250) was performed on Illumina HiSeq. The FASTQ conversion of the original data file is completed after demultiplexing with MiSeq Reporter. The quality assessment of FASTQ files is carried out using FASTQC,1 then quality filtering is performed using the FASTX toolkit.2 The high-quality reads used for analysis (where 80% of the base Q scores > 20 reads) and reads with unknown bases (“N”) are discarded. The remaining steps are performed using the Quantitative Analysis of Microbial Ecology (QIIME) software package version 1.8. Use UCHIME to filter chimeric sequences and use UCLUST to group sequences into the Operational Taxa Unit (OTU) with a similarity threshold of 97%. The Ribosomal Database Program (RDP) classifier trained using Greengenes 16S rDNA database (v13.8) assigns all OTUs to all OTUs with a confidence threshold of 80%. OTUs with an average abundance of less than 0.005% are eliminated. Use PyNAST v1.2 for multiple sequence alignment and FastTree v2.1 to construct a phylogenetic tree. The alpha and beta diversity indicators are calculated according to the method implemented in QIIME. A Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) is used to predict the orthologs from the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Clusters of Orthologous Groups (COG) (i.e., the count of functional genes) for each sample and inferred genes. The count is allocated to the KEGG and COG channels.
Statistic analysis
The numeric values representing the relative abundance of the OTU were analysed for statistical significance by performing a t-test. The statistical software in Sigma Plot version 11.0 was used (Systat Software Inc., San Jose, CA, USA). A total of 95% confidence intervals were estimated for sensitivity and specificity with a binomial test. Differences in means with p-values less than 0.05 were considered statistically significant. When tests for normality failed, the non-parametric data were analysed using the Mann–Whitney Rank Sum test and median values were determined.
Results
Patient cohort sociodemographic and characteristics
Complete data were available for 276 cervical smear samples taken from eligible women. The patients’ characteristics of the study population were summarised in Table 1, and detailed information about each patient was shown in Supplementary Table 1. The mean age of the patient’s cohort was 44.68 ± 10.94, from 17 to 80 years old. There were 83 HPV-infected patients in the 255 candidates under 60 years old and 11 HPV-infected patients in the 21 candidates over 60. The infection status of the subjects, including HPV single-type infection and multi-type infection. Five majors HPV types were detected, including HPV16 (n = 10), HPV39 (n = 7), HPV52 (n = 8), HPV56 (n = 7), and HPV58 (n = 12), and 36 subjects were infected by other HPV genotypes. Several cases were detected as unique-268 infections (52, 56, or 58 genotype infections, n = 27) among the 80 single infections and other related co-infections were multi-268 infections (52, 56, and 58 genotypes infecting two or more, n = 13) among the 14 multiple infections. Hence, the number of infections excluding 52, 56, and 58 were 54 infections, denoting as other1. Among the 21 subjects diagnosed with Cervical Intraepithelial Neoplasms (CIN), thirteen patients were infected by HPV and eight were not. While 81 HPV-positive and 174 (255 totally) HPV-negative subjects were identified in subjects without CIN status.
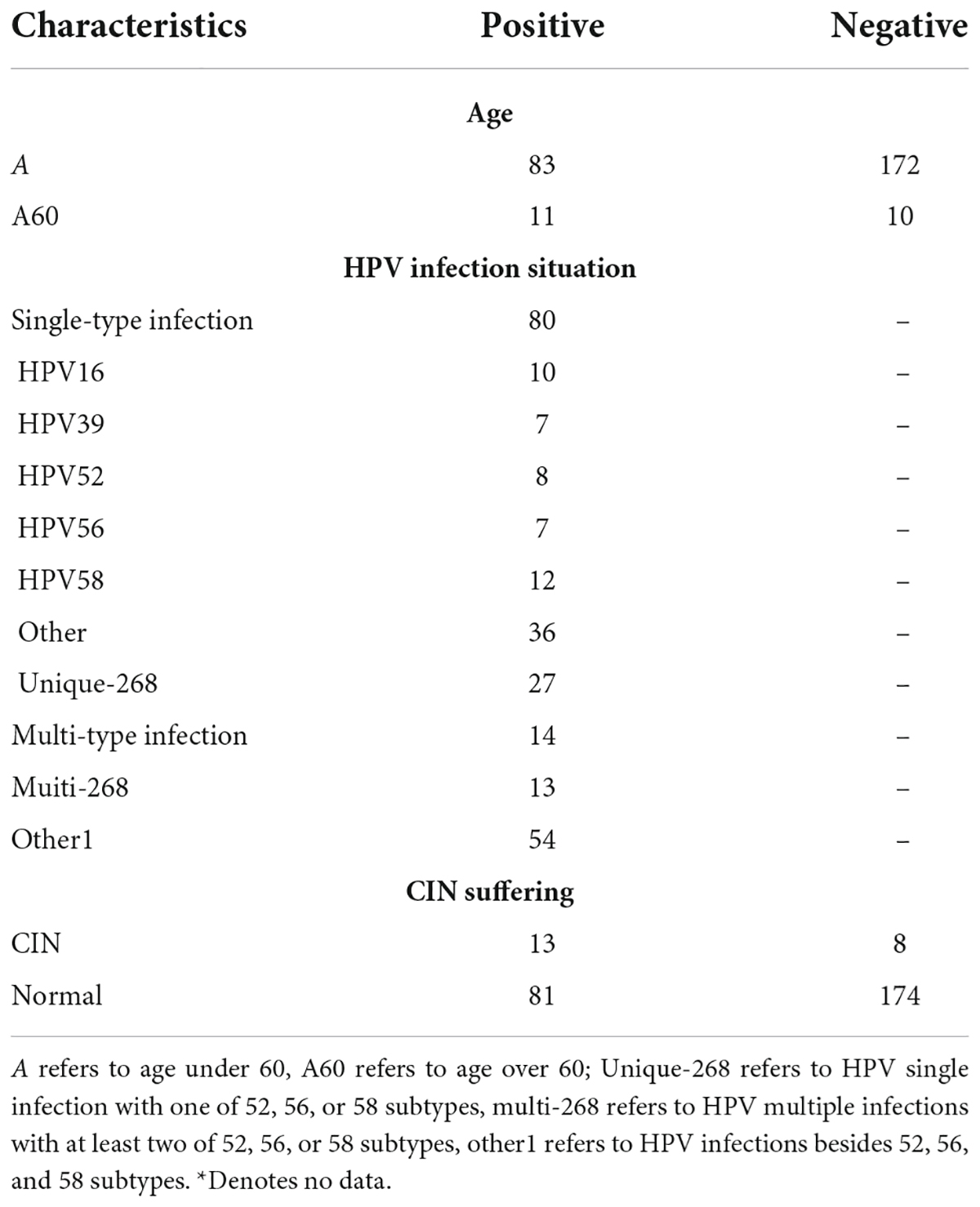
Table 1. Patients age and infection status.
Baseline composition similar but the structure of the cervical microbiota variant between samples
The average length of the PCR product was about 465 bps from V3 to V4 segments of the 16S rDNA genes. After sequencing, the data amount and quality of each sample were evaluated by GC content (averagely 52.8%), Q20 value (averagely 96.2%), Q30 value (averagely 92%), and effectiveness (averagely 73.3). The detailed information of each sample was summarised in Supplementary Table 1. After removing the low-quality sequencing reads, a total number of 14,435,817 clean tags and an average of 79,095 tags of each specimen (each specimen generating at least 8,807 clean tags) were obtained. After the removal of singletons and rare OTUs (species abundance less than 0.005%), a total of 11 phyla, 17 classes, 30 orders, 53 families, 99 genera, and 132 species (Supplementary Table 2) were identified from the study cohort sequencing data.
The abundance of the ten most abundant bacteria families was summarised and listed in Supplementary Table 3. Lactobacillaceae, Enterobacteriaceae, Staphylococcaceae, Enterococcaceae, Bifidobacteriaceae, Beijerinckiaceae, Streptococcaceae, Leptotrichiaceae, Burkholderiaceae, and Corynebacteriaceae were the top 10 most abundant bacteria families in all the samples. The corresponding distribution figure is shown in Supplementary Figure 1. Results showed that the microbiota structure variant obviously between samples, but there were still pattern similarities between parts of the samples. As previously published papers mentioned, the CM was classified into five clusters (Zhou et al., 2020). Our results showed sample clusters with highly abundant Escherichia-Shigella, Lactobacillus, Enterococcus, Staphylococcus, and Lactobacillus in each cluster, respectively (Supplementary Figure 1). Moreover, some samples showed different characteristics with more bacterium types in the structure. Sample alpha diversity, including Chao1, ACE, Shannon, and Simpson indexes of all the samples, was calculated and summarised in Supplementary Table 4, indicating that the diverse samples differentiated significantly between samples.
Differences in human papillomavirus infection status or age are highly relevant to the change of microbiota structure and diversity
This study explored the difference in microbiota composition and diversity between sample groups. Two group pairs showed significant differences in HPV infection status and age. The relative abundance results of different HPV infection status groups (Figure 1A and Supplementary Table 4) showed the following conclusions: (1) Both normal sample groups with HPV infection (n = 78) and non-infection (n = 187) were predominated by the following types of bacteria, including Lactobacillus iners (HPV-positive/HPV-negative: 0.185/0.104), Escherichia coli (0.112/0.143), Enterococcus faecalis (0.071/0.108), and Atopobium vaginae (0.033/0.013) in species level, and Staphylococcus (0.116/0.117), Lactobacillus (excluding Lactobacillus iners AB1, 0.078/0.069), Gardnerella (0.076/0.048), Bosea (0.026/0.049), Streptococcus (0.015/0.043), and Sneathia (0.031/0.020) in genus level; (2) In the microbiota structure (relative abundance), which may illustrate a structure change before and after the HPV infection (Figure 1A and Supplementary Table 4). Further study on the unique-268 (sum of 52, 56, and 58 genotype HPV infection cases, n = 27) and multi-268 (co-infections with one or two HPV 52, 56, and 58 genotypes, n = 13) types of HPV infected samples, the microbiota detected was almost the same as the HPV-positive except for the composition proportion (Figure 1B and Supplementary Table 4). Additionally, the age group under 60 (n = 255) and above 60 (n = 31) detected a similar microbiota with a different proportion of composition (Figure 1C and Supplementary Table 4).
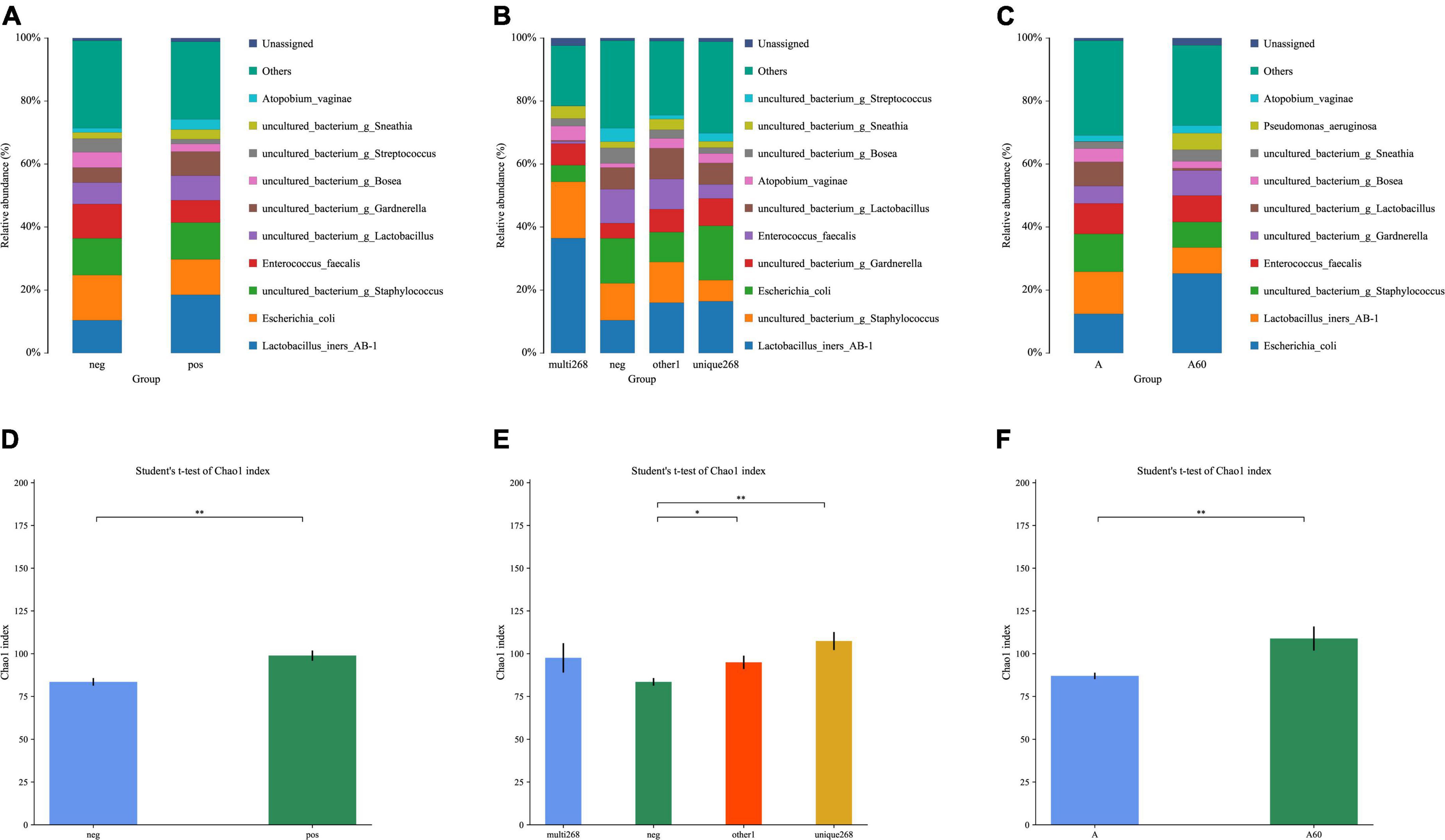
Figure 1. Relative abundance and alpha-diversity analysis of this study. (A) Microbiota relative abundance distribution of HPV-positive and HPV-negative patient groups; (B) microbiota relative abundance distribution of HPV-positive unique-268 infection and multiple 268 infection, and HPV-negative patient groups; (C) microbiota relative abundance analysis of the elder (age > 60) and the younger (age ≤ 60) patient groups; alpha-diversity analysis; a-diversity comparison bar diagram, (D) Chao1 index for normal (without CIN) HPV-positive and HPV-negative patient groups, (E) Chao1 index for HPV-positive unique-268, and multiple 268 infections, HPV-negative patient groups, (F) Chao1 index for the elder (age > 60) and the younger (age ≤ 60) patient groups. neg: HPV-negative without CIN, pos: HPV-positive without CIN, multi-268: HPV 52, 56, and 58 genotypes multiple infection (regardless of CIN), unique268: HPV 52, 56, or 58 genotypes unique (single) infection (regardless of CIN), other1: other HPV infection situations (regardless of CIN). A: age under 60, A60: age above 60, *p-value < 0.05, **p-value < 0.01.
Alpha diversity was applied to analyse the complexity of species diversity. Regarding the analysis, the HPV-positive group had higher diversity (Chao1 index: 98.8869, p < 0.01) compared to the negative group (Chao1 index: 83.5299, Figures 1C,D and Supplementary Table 5). This disparity in microbiota diversity resulted from the significant differences between the two cases and evidenced structural change. Unique-268 (Chao1 index: 107.3885, p < 0.01), multi-268 (Chao1 index: 97.53) and other1 (Chao1 index: 94.9619, p < 0.05) had a higher microbiota diversity compared to the HPV-negative groups (Figure 1E and Supplementary Table 5). In addition, compared to younger patients, the elder group (age > 60, n = 31) has a higher diversity with statistical significance (Chao1 index: 108.8851, p < 0.01) than the younger group (age ≤ 60, n = 255, Chao1 index: 87.0171, Figure 1F and Supplementary Table 5), which also demonstrates the difference in microbiota structure. Hence, this diversity analysis indicates the following conclusions: (1) The normal HPV-positive groups and (2) unique-268 HPV and other1 infections were more diverse in microbiota than the HPV-negative groups, while (3) the age group over 60 had higher diversity concerning the microbiota.
Bacteria biomarkers were identified in different subject groups
Linear discriminant analysis (LDA) score was used to compare the different bacteria of each group. The results showed that Bifidobacteriales (order), Bifidobacteriaceae (family), Gardnerella (genus), Coriobacteriia (class), Atopobium vaginae (species), and Clostridia (class) were higher in HPV-infected group compared with the negative (Figure 2A). Between multi-268 and unique-268 groups, Betaproteobacteriales (order), Burkholderiaceae (family), Weeksellaceae (family), Flavobacteriales (family), Gardnerella (genus), Pseudomonas aeruginosa (species), and Mycoplasma (genus) were found with higher relative abundance in unique-268. In contrast, Saccharimonadales (order), Saccharimonadia (class), Patescibacteria (phylum), Bifidobacteriales (order), and Bifidobacteriaceae (family) were higher in the multi-268 group (Figure 2B). Among them, Bifidobacteriaceae was the most significantly different between the two groups, indicating its strong association with multi-268 infection (Figure 2B). Corynebacterium (genus), Lactobacillus iners AB-1 (species), Bacilli (class), and Firmicutes (phylum) were identified higher in the group with age younger than 60 and Gamma-proteobacteria (class) and Pseudomonas (genus) higher in the group older than 60 years old (Figure 2C).
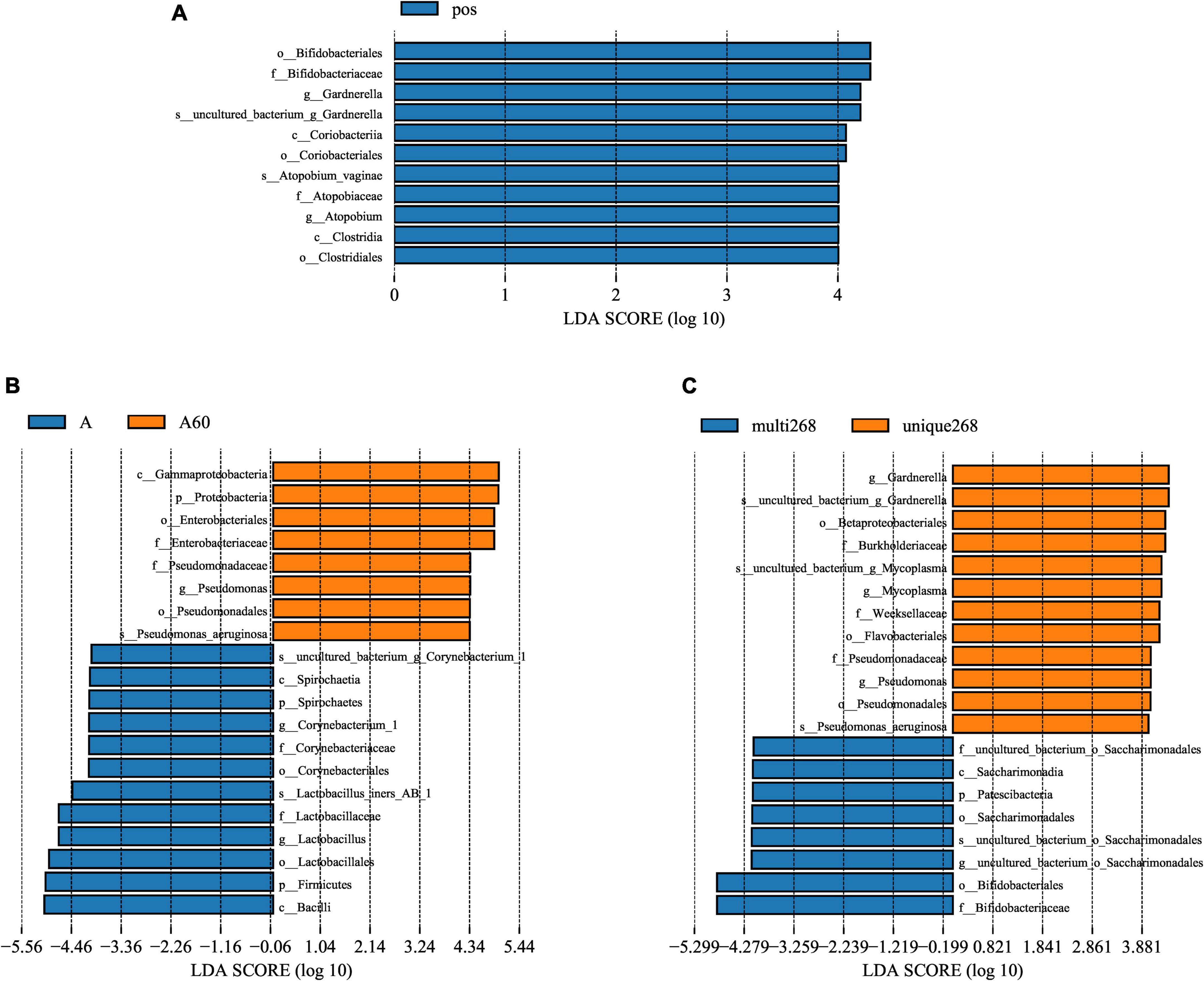
Figure 2. Microbiota significant difference analysed by LEfSe. (A) HPV-positive and HPV-negative patient groups, (B) HPV-positive single infection and multiple infection patient groups, and (C) age above and below 60 years old patient groups. LDA score threshold set as 4, above 4 will be shown in charts. LDA, linear discriminant analysis; LEfSe, LDA effective size. HPV, Human Papillomavirus. k_: kingdom, p_: phylum, c_: class_, o_: order, f_: family_, g_: genus.
Microbiota function difference of subjects group was identified
Kyoto Encyclopedia of Genes and Genomes and COG analysis were applied, and functional difference between groups was explored. Among the three group comparisons, two group pairs were found significantly different and they are HPV-negative/positive group pair and the multi-268/negative group pair. No significant function difference was identified between Age groups. Between HPV-positive and negative groups, KEGG pathways, including Cell growth and death, Excretory system, Folding, sorting, and degradation, Endocrine and metabolic diseases, Nucleotide metabolism, Replication and repair, Immune system, and Transcription, were significantly different (Figure 3A). Moreover, the COG categories of Amino acid transport and metabolism, Cell cycle control, cell division, chromosome partitioning, Inorganic ion transport and metabolism, Translation, ribosomal structure and biogenesis and Defence mechanisms between the two groups were significantly different (Figure 3B). Comparison analysis results of multi-268 and the negative group showed that KEGG pathway Excretory system, Lipid metabolism, Signal transduction and Folding, sorting and degradation and COG categories of Cell cycle control, cell division, chromosome partitioning were significantly different (Figures 3C,D).
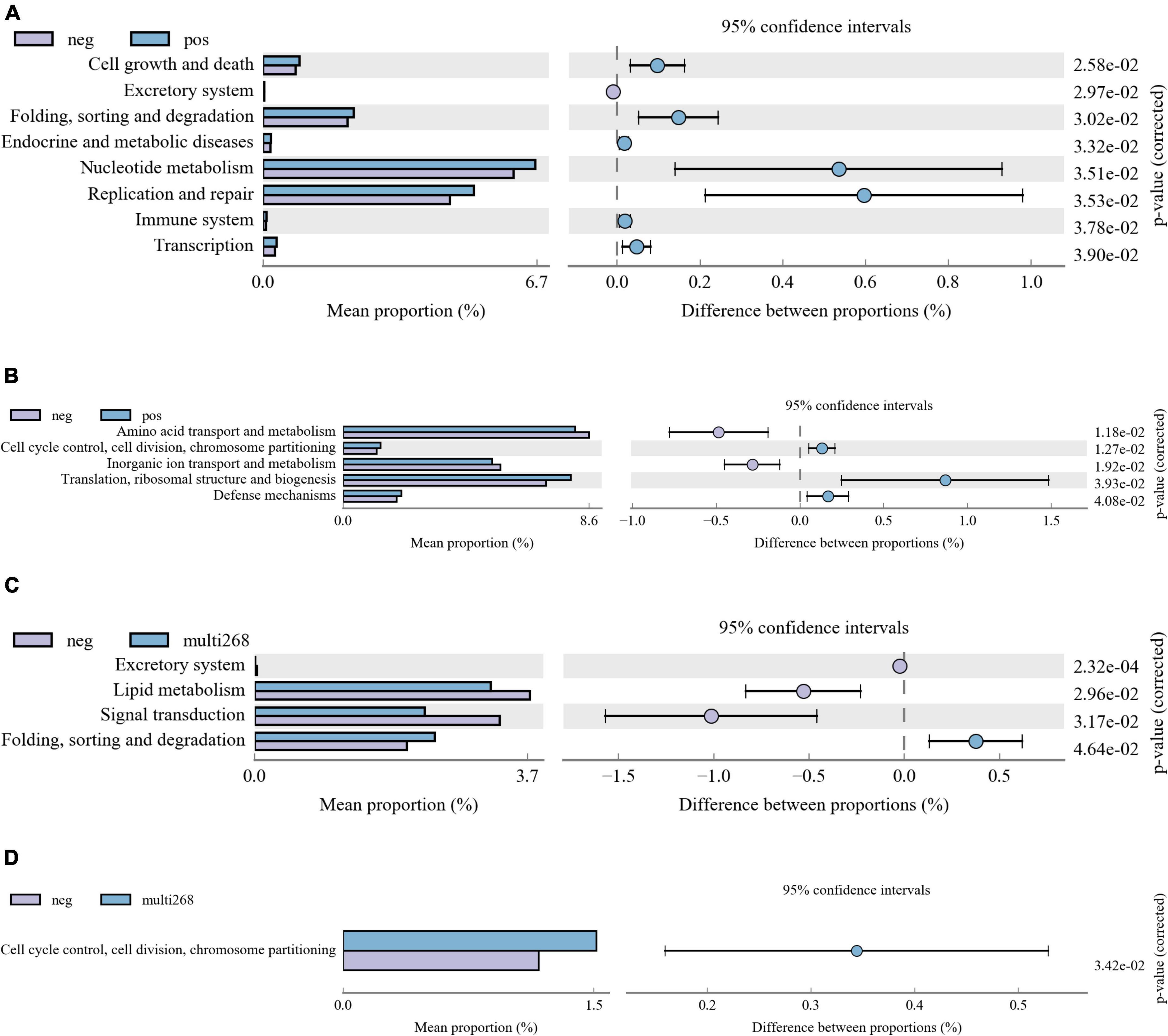
Figure 3. Microbiota KEGG and COG function difference diagrams. The picture shows the difference analysis diagram of the KEGG (and COG) metabolic pathway under the second level (also can be analysed for the third or first level): the different colours in the picture represent different groups. The figure shows the abundance ratio of different functions in the two sets of sample groups, the middle shows the difference ratio of the function abundance within the 95% confidence interval, and the rightmost value is the p-value. (A) KEGG pathway difference between HPV-positive and HPV-negative patient groups, (B) COG pathway difference between HPV-positive and HPV-negative patient groups. (C) KEGG pathway difference between multi-268 HPV infections and HPV-negative patient groups, (D) COG pathway difference between multi-268 HPV infections and HPV-negative patient groups. KEGG, Kyoto Encyclopedia of Genes and Genomes; COG, Clusters of Orthologous Groups.
Complexed cervical microbiota network relationships existed in the cervical microbiota system
Co-occurrent analysis of identified microbiota in cervical samples and results are shown in Figure 4. There were 80 genera of bacteria identified with more than seven relationships with other bacteria, and the correlation was higher than 10%, with a p-value less than 0.05. A network relationship was identified between them (Figure 4). The top 50 bacteria with high correlation were shown in the figure (Figure 4), 24 of which were correlated with two or more other genera. The abundance of the genera was different, ranging from 12.0 to 15463.4, and no significant correlation was observed between genera abundance and its correlation with other genera. The abundance of Lactobacillus (abundance of 15463.4), Escherichia-Shigella (10609.8), and Staphylococcus (9270.3) were high, but the correlation with other bacteria is relatively low, less than 0.34, 0.32, and 0.27, respectively. On the contrary, some genera’s abundance was relatively low, but the correlations with others were quite high (Supplementary Table 6). For example, Atopobium (abundance: 1510.4) is highly correlated to Dialister (correlation: 0.675, abundance: 236.2), Prevotella (0.656, 1259.3) and Fastidiosipila (0.573, 1121.7); Achromobacter (483.5) is tensely correlated to Stenotrophomonas (0.793, 305.4), Sphingobium (0.759, 241.9), and Herbaspirillum (0.648, 21.8); Gardnerella (4349.4) is highly correlated to Atopobium (0.659, 1510.4), Aerococcus (0.527, 246.2) and Sneathia (0.0.498, 2389.7); Sneathia (2389.7) is highly correlated to Fastidiosipila (0.648, 1121.7), DNF00809 (0.604, 242.4), Parvimonas (0.572, 278.8), and Atopobium (0.546, 1510.4). In summary, a complexed bacteria network relationship was existing in cervical system and the interactions between genera was not correlated with its abundance.

Figure 4. Microbiota network diagram. The top 50 most relevant genera were shown here. The circle in the figure represents the species, and the size of the circle represents the abundance; the line represents the relation between the two species, the thickness of the line represents the strength of the relation, and the colour of the line: orange represents positive correlation, and the green represents negative correlation.
Discussion
Data analysis showed that changes in the cervical microbiome, especially anaerobic bacteria, were significantly correlated with HPV infection status. Gardnerella, Atopobium vaginae, and Sneathia were the three most increased amongst microbiota, and these microorganisms form pathogenic biofilms through close cooperation. Gardnerella acts as a “scaffold” for biofilms (Harwich et al., 2010; Fethers et al., 2012; Curty et al., 2017; Khan et al., 2020), promoting the growth of Atopobium vaginae (Libby et al., 2008; Anahtar et al., 2015), Sneathia and other related pathogens by altering the microenvironment (Lee et al., 2013; Zhou et al., 2020). The growth of these pathogens has led to a rise in microbial diversity. Not only that, the cellular pro-inflammatory responses that these pathogenic microorganisms elicited will affect cellular metabolisms (Kacerovsky et al., 2015; Mitra et al., 2015, 2020; Onderdonk et al., 2016; Gosmann et al., 2017; Khan et al., 2020), such as amino acid transport (Mitra et al., 2020) and inorganic ion transport, and even cell shedding (Harwich et al., 2012; Africa et al., 2014). This deteriorates the immune response and leads to a defection of the microenvironment (Anahtar et al., 2015), making cervix HPV susceptible and possibly leading to the cervical cancer. Therefore, these three microorganisms are of great significance as biomarkers in the clinical identification of HPV infection.
In addition to the correlation with HPV, there is also a relationship between CM and age status. Lee et al. (2013) suggested that the decline in oestrogen and progesterone levels in the female reproductive tract after menopause is associated with an increased proportion of anaerobic bacteria. Several studies have also confirmed that older women have a higher proportion of anaerobic bacteria, and the biofilm produced by them is an important factor in HPV susceptibility (Singh et al., 2015; Zhou et al., 2020). Besides, Lee’s experiment also evidenced that due to the presence of oestrogen and progesterone, the proportion of Lactobacillus in young women is higher to maintain the homeostasis of the reproductive tract microenvironment (Lee et al., 2013; Oh et al., 2015). In contrast, the proportion of Lactobacillus in older women is lower, so it insufficiently maintains the homeostasis of the internal environment, making the proportion of γ-proteobacteria and Pseudomonas species increase as biomarkers.
However, this study also showed some results that differed from the prevailing view. In this study, the proportion of Lactobacillus in the HPV-positive group was higher than that in the normal group. It contradicts the mainstream ideas that the presence of Lactobacillus can maintain pH stability (Larsson et al., 1991; Brotman et al., 2014) and homeostasis in the reproductive tract (Mitra et al., 2016; Borgogna et al., 2020) and is therefore reduced in the HPV-positive group. However, a report from Iran showed the same results as this research and concluded that the proportion change of Lactobacillus was not strongly correlated with HPV infection status but did not rule out the influence of factors such as customs on sample interference (Ghaniabadi et al., 2020). Therefore, the influence of other factors, such as personal habits, could not be ruled out for the presence of interference with the sample microbiota. The more detailed mechanisms still need more experiments to verify and analyse.
Aside from that, this study also presented some new findings. A higher abundance of Bifidobacterium was also found in the multi-268 group than in the unique-268 group when comparing the two case samples. Under such circumstances, the same bacteria have different environmental adaptations in different case samples. Besides, by analysing the low-abundance flora of different groups, it was found that there were differences in the microbial composition of different HPV infection states. Comparing the LEfSe analysis of unique-268 and multi-268, it could be seen that some low-abundance bacterial groups play important roles in different HPV-infected samples. In the unique-268 group of patients, Burkholderiaceae, a pathogenic bacteria, could sensitise cells to HPV (Brenner et al., 2005). Mycoplasma could promote HPV penetration, survival and persistence, and it is frequently present in high-risk HPV patients (Biernat Sudolska et al., 2011; Ye et al., 2018; Wei et al., 2021). Pseudomonas aeruginosa is a prevalent factor in high-risk HPV samples, especially in the cancerous cervix (Werner et al., 2012; Di Paola et al., 2017; Zhang et al., 2021). The low-abundance species Saccharimonadales, Saccharimonadia, and Patescibacteria in the multi-268 group were all related to the synthesis of compound elements (Herrmann et al., 2019; Lemos et al., 2019; Tian et al., 2020; Hosokawa et al., 2021; Mason et al., 2021; Zhou et al., 2021; Wang et al., 2022). The results of the above microbiota under different HPV infection statuses have clinical implications for biomarkers for identifying cases.
In addition to the above microorganisms, this study also found that the microbiota (especially pathogenic microorganisms) significantly impacted metabolic function. Apart from the abnormal cellular metabolism mentioned above, differences in genetic metabolism, lipid metabolism, signal transduction and cell cycle metabolism were also detected between the HPV-positive group and the multi-268 group. Abnormalities in these functions are likely associated with increased microbial diversity and an increased proportion of pathogenic microorganisms (Mitra et al., 2020). However, there are few studies in this regard, so further experiments are needed to explore their relationship.
This study analysed the possible effects of cervical microbiome changes from different aspects. However, due to the limited number of statistical samples in the research process, we could not perform significant statistics for some more refined HPV genotypes. In addition, the lack of clinical information about patients (such as smoking, eating and other behaviours that may cause cervical cancer) also interfered with the experiment to a certain extent. However, the data analysis of this experiment still provides a sufficient factual basis and data support for clinical examination. Meanwhile, the microbial changes of single-infection and multi-infection case samples and the differences in metabolic functions under different HPV infection conditions were compared from a new perspective.
Conclusion
Overall, the characteristics of cervical samples microbiota were explored in this study. Escherichia coli, Enterococcus faecalis, and Atopobium vaginae in species level, Staphylococcus, Lactobacillus (excluding Lactobacillus iners AB1), Gardnerella, Bosea, Streptococcus, and Sneathia in genus level were found as high abundant bacteria in studied samples. Microbiota composition was related to HPV infection status and age, which further influenced the diversity. Specific bacteria were identified with significantly different abundance between groups. For instance, compared with unique-268, Bifidobacteriaceae impacted more on the multi-268 group. Moreover, some low abundance bacteria also play a vital role in specific HPV infections, such as Saccharimonadales, Saccharimonadia, and Patescibacteria in multi-268, Burkholderiaceae Mycoplasma, and Pseudomonas aeruginosa in unique-268. Besides, the different composition of microbiota also affected the disparities of function pathways to the metabolism of the cell, protein and genetic information between HPV infection and HPV-negative groups, and the metabolism of lipid, signal transduction and cell cycle between multi-268 infection and HPV-negative groups. In summary, our study descriptively explored the microbiota characteristics of cervical samples from Sanmenxia area patients. The analysis of single infections was not developed due to the sample size. The research concerns specific single infections, and CIN could be further investigated into their microbiota in future works.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA795603.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee, Sanmenxia Central Hospital. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author contributions
JH contributed to the manuscript writing, revision, and partial statistical data analysis. YW contributed to the initial draft discussing, editing and the general ideas of the manuscript, and collection of literature. LQ contributed to all the sample collection, ethic letter application, and results discussion. JL contributed to the part of the data analysis and data subscribing. WY did the experiments of 16S rDNA sequencing. GT contributed to the conceptualization of the study, study fund support, and results discussion. BM contributed to the conceptualization of the study, data generation and analysis, and draft revision. All authors read and revised the manuscript.
Funding
This work was supported by Geneis (Beijing) Co., Ltd.
Acknowledgments
We would like to acknowledge Department of Gynecology, Sanmenxia Central Hospital of Henan University of Science and Technology, Sanmenxia, Henan, China.
Conflict of interest
Author LQ was employed by Sanmenxia Central Hospital. Authors WY, JL, GT, and BM were employed by Geneis (Beijing) Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1004664/full#supplementary-material
Footnotes
- ^ http://www.Bioinformatics.babraham.ac.uk/projects/fastqc/
- ^ http://hannonlab.cshl.edu/fastx_toolkit/
References
Africa, C. W., Nel, J., and Stemmet, M. (2014). Anaerobes and bacterial vaginosis in pregnancy: Virulence factors contributing to vaginal colonisation. Int. J. Environ. Res. Public Health 11, 6979–7000. doi: 10.3390/ijerph110706979
Anahtar, M. N., Byrne, E. H., Doherty, K. E., Bowman, B. A., Yamamoto, H. S., Soumillon, M., et al. (2015). Cervicovaginal bacteria are a major modulator of host inflammatory responses in the female genital tract. Immunity 42, 965–976. doi: 10.1016/j.immuni.2015.04.019
Biernat Sudolska, M., Szostek, S., Rojek Zakrzewska, D., Klimek, M., and Kosz Vnenchak, M. (2011). Concomitant infections with human papillomavirus and various mycoplasma and ureaplsasma species in women with abnormal cervical cytology. Adv. Med. Sci. 56, 299–303. doi: 10.2478/v10039-011-0028-9
Borgogna, J., Shardell, M., Santori, E., Nelson, T., Rath, J., Glover, E., et al. (2020). The vaginal metabolome and microbiota of cervical HPV-positive and HPV-negative women: A cross-sectional analysis. BJOG 127, 182–192. doi: 10.1111/1471-0528.15981
Brenner, D. J., Boone, D. R., Garrity, G. M., Goodfellow, M., Krieg, N. R., Rainey, F. A., et al. (2005). Bergey’s Manual of Systematic Bacteriology, 2nd Edition, Vol. 2 (The Proteobacteria), part C (The Alpha-, Beta-, Delta-, and Epsilonproteobacteria). New York, NY: Springer.
Brotman, R. M., Shardell, M. D., Gajer, P., Tracy, J. K., Zenilman, J. M., Ravel, J., et al. (2014). Interplay between the temporal dynamics of the vaginal microbiota and human papillomavirus detection. J. Infect. Dis. 210, 1723–1733. doi: 10.1093/infdis/jiu330
Curty, G., Costa, R. L., Siqueira, J. D., Meyrelles, A. I., Machado, E. S., Soares, E. A., et al. (2017). Analysis of the cervical microbiome and potential biomarkers from postpartum HIV-positive women displaying cervical intraepithelial lesions. Sci. Rep. 7:17364. doi: 10.1038/s41598-017-17351-9
Di Paola, M., Sani, C., Clemente, A. M., Iossa, A., Perissi, E., Castronovo, G., et al. (2017). Characterization of cervico-vaginal microbiota in women developing persistent high-risk Human Papillomavirus infection. Sci. Rep. 7:10200. doi: 10.1038/s41598-017-09842-6
Fethers, K., Twin, J., Fairley, C. K., Fowkes, F. J., Garland, S. M., Fehler, G., et al. (2012). Bacterial vaginosis (BV) candidate bacteria: Associations with BV and behavioural practices in sexually-experienced and inexperienced women. PLoS One 7:e30633. doi: 10.1371/journal.pone.0030633
Ghaniabadi, R., Hashemi, S., Bajgiran, M. S., Javadi, S., Mohammadzadeh, N., and Masjedian, F. (2020). Distribution of Lactobacillus species in Iranian women with both human papillomavirus (HPV) infection and bacterial vaginosis (BV). Meta Gene 26:100791. doi: 10.1016/j.mgene.2020.100791
Gillet, E., Meys, J. F., Verstraelen, H., Bosire, C., De Sutter, P., Temmerman, M., et al. (2011). Bacterial vaginosis is associated with uterine cervical human papillomavirus infection: A meta-analysis. BMC Infect. Dis. 11:10. doi: 10.1186/1471-2334-11-10
Gosmann, C., Anahtar, M. N., Handley, S. A., Farcasanu, M., Abu-Ali, G., Bowman, B. A., et al. (2017). Lactobacillus-deficient cervicovaginal bacterial communities are associated with increased HIV acquisition in young South African women. Immunity 46, 29–37. doi: 10.1016/j.immuni.2016.12.013
Harwich, M. D., Alves, J. M., Buck, G. A., Strauss, J. F., Patterson, J. L., Oki, A. T., et al. (2010). Drawing the line between commensal and pathogenic Gardnerella vaginalis through genome analysis and virulence studies. BMC Genom. 11:375. doi: 10.1186/1471-2164-11-375
Harwich, M. D., Serrano, M. G., Fettweis, J. M., Alves, J. M., Reimers, M. A., Buck, G. A., et al. (2012). Genomic sequence analysis and characterisation of Sneathia amnii sp. nov. BMC Genom. 13:S4. doi: 10.1186/1471-2164-13-S8-S4
Herrmann, M., Wegner, C.-E., Taubert, M., Geesink, P., Lehmann, K., Yan, L., et al. (2019). Predominance of Cand. Patescibacteria in groundwater is caused by their preferential mobilisation from soils and flourishing under oligotrophic conditions. Front. Microbiol. 10:1407. doi: 10.3389/fmicb.2019.01407
Hosokawa, S., Kuroda, K., Narihiro, T., Aoi, Y., Ozaki, N., Ohashi, A., et al. (2021). Cometabolism of the Superphylum Patescibacteria with Anammox Bacteria in a Long-Term Freshwater Anammox Column Reactor. Water 13:208.
Kacerovsky, M., Vrbacky, F., Kutova, R., Pliskova, L., Andrys, C., Musilova, I., et al. (2015). Cervical microbiota in women with preterm prelabor rupture of membranes. PLoS One 10:e0126884. doi: 10.1371/journal.pone.0126884
Khan, S., Vancuren, S. J., and Hill, J. E. (2020). A generalist lifestyle allows rare Gardnerella spp. to persist at low levels in the vaginal microbiome. Microb. Ecol. 82, 1048–1060. doi: 10.1007/s00248-020-01643-1
Kunene, V., and Mahlangu, J. (2017). “Access to Systemic Anticancer Treatment and Radiotherapy Services in Sub-Saharan Africa,” in Cancer in Sub-Saharan Africa, ed. O. Adedeji (Berlin: Springer), 175–190.
Larsson, P., Platz-Christensen, J., and Sundström, E. (1991). Is bacterial vaginosis a sexually transmitted disease? Int. J. STD AIDS 2, 362–364.
Lee, J. E., Lee, S., Lee, H., Song, Y.-M., Lee, K., Han, M. J., et al. (2013). Association of the vaginal microbiota with human papillomavirus infection in a Korean twin cohort. PLoS One 8:e63514. doi: 10.1371/journal.pone.0063514
Lemos, L. N., Medeiros, J. D., Dini-Andreote, F., Fernandes, G. R., Varani, A. M., Oliveira, G., et al. (2019). Genomic signatures and co-occurrence patterns of the ultra-small Saccharimonadia (phylum CPR/Patescibacteria) suggest a symbiotic lifestyle. Mol. Ecol. 28, 4259–4271. doi: 10.1111/mec.15208
Libby, E. K., Pascal, K. E., Mordechai, E., Adelson, M. E., and Trama, J. P. (2008). Atopobium vaginae triggers an innate immune response in an in vitro model of bacterial vaginosis. Microbes Infect. 10, 439–446. doi: 10.1016/j.micinf.2008.01.004
Liu, S., Guo, Y., Li, B., Zhang, H., Zhang, R., and Zheng, S. (2020). Analysis of Clinicopathological Features of Cervical Mucinous Adenocarcinoma with a Solitary Ovarian Metastatic Mass as the First Manifestation. Cancer Manag. Res. 12:8965. doi: 10.2147/CMAR.S270675
Mason, L., Eagar, A., Patel, P., Blackwood, C., and Deforest, J. (2021). Potential microbial bioindicators of phosphorus mining in a temperate deciduous forest. J. Appl. Microbiol. 130, 109–122. doi: 10.1111/jam.14761
Mitra, A., Macintyre, D. A., Marchesi, J. R., Lee, Y. S., Bennett, P. R., and Kyrgiou, M. (2016). The vaginal microbiota, human papillomavirus infection and cervical intraepithelial neoplasia: What do we know and where are we going next? Microbiome 4:58. doi: 10.1186/s40168-016-0203-0
Mitra, A., Macintyre, D. A., Paraskevaidi, M., Moscicki, A.-B., Mahajan, V., Smith, A., et al. (2020). The Vaginal Microbiota and Innate Immunity After Local Excisional Treatment for Cervical Intraepithelial Neoplasia. Genome Med. Actions 13:176. doi: 10.1186/s13073-021-00977-w
Mitra, A., Macintyre, D., Lee, Y., Smith, A., Marchesi, J. R., Lehne, B., et al. (2015). Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity. Sci. Rep. 5:16865.
Oh, H., Kim, B.-S., Seo, S.-S., Kong, J.-S., Lee, J.-K., Park, S.-Y., et al. (2015). The association of uterine cervical microbiota with an increased risk for cervical intraepithelial neoplasia in Korea. Clin. Microbiol. Infect. 21:674.e1–9.
Onderdonk, A. B., Delaney, M. L., and Fichorova, R. N. (2016). The human microbiome during bacterial vaginosis. Clin. Microbiol. Rev. 29, 223–238.
Polatti, F. (2012). Bacterial vaginosis, Atopobium vaginae and nifuratel. Curr. Clin. Pharmacol. 7, 36–40.
Singh, S., Zhou, Q., Yu, Y., Xu, X., Huang, X., Zhao, J., et al. (2015). Distribution of HPV genotypes in Shanghai women. Int. J. Clin. Exp. Pathol. 8:11901.
Tian, R., Ning, D., He, Z., Zhang, P., Spencer, S. J., Gao, S., et al. (2020). Small and mighty: Adaptation of superphylum Patescibacteria to groundwater environment drives their genome simplicity. Microbiome 8:51. doi: 10.1186/s40168-020-00825-w
Wang, G., Jin, Z., Wang, X., George, T. S., Feng, G., and Zhang, L. (2022). Simulated root exudates stimulate the abundance of Saccharimon to improve the alkaline phosphatase activity in maise rhizosphere. Appl. Soil Ecol. 170:104274. doi: 10.1016/j.apsoil.2021.104274
Wei, Z.-T., Chen, H.-L., Wang, C.-F., Yang, G.-L., Han, S.-M., and Zhang, S.-L. (2021). Depiction of vaginal microbiota in women with high-risk human papillomavirus infection. Front. Public Health 8:587298. doi: 10.3389/fpubh.2020.587298
Werner, J., Decarlo, C. A., Escott, N., Zehbe, I., and Ulanova, M. (2012). Expression of integrins and Toll-like receptors in cervical cancer: Effect of infectious agents. Innate Immun. 18, 55–69. doi: 10.1177/1753425910392934
Ye, H., Song, T., Zeng, X., Li, L., Hou, M., and Xi, M. (2018). Association between genital mycoplasmas infection and human papillomavirus infection, abnormal cervical cytopathology, and cervical cancer: A systematic review and meta-analysis. Arch. Gynecol. Obstet. 297, 1377–1387. doi: 10.1007/s00404-018-4733-5
Zhang, Y.-Y., Xu, X.-Q., Zhang, D., Wu, J., and Zhang, H.-X. (2020). Triage human papillomavirus testing for cytology-based cervical screening in women of different ages in primary hospitals: A retrospective clinical study. Medicine 99:e22320. doi: 10.1097/MD.0000000000022320
Zhang, Z., Li, T., Zhang, D., Zong, X., Bai, H., Bi, H., et al. (2021). Distinction between vaginal and cervical microbiota in high-risk human papilloma virus-infected women in China. BMC Microbiol. 21:90. doi: 10.1186/s12866-021-02152-y
Zhou, F.-Y., Zhou, Q., Zhu, Z.-Y., Hua, K.-Q., Chen, L.-M., and Ding, J.-X. (2020). Types and viral load of human papillomavirus, and vaginal microbiota in vaginal intraepithelial neoplasia: A cross-sectional study. Ann. Transl Med. 8:1408. doi: 10.21037/atm-20-622
Keywords: cervical cancer, TCT, HPV - human papillomavirus, microbiota, diversity
Citation: Hu J, Wu Y, Quan L, Yang W, Lang J, Tian G and Meng B (2022) Research of cervical microbiota alterations with human papillomavirus infection status and women age in Sanmenxia area of China. Front. Microbiol. 13:1004664. doi: 10.3389/fmicb.2022.1004664
Received: 27 July 2022; Accepted: 20 September 2022;
Published: 06 October 2022.
Edited by:
Fei Ma, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaReviewed by:
Nazan Yurtcu, Sivas Cumhuriyet University Faculty of Medicine, TurkeyAna Afonso, Universidade NOVA de Lisboa, Portugal
Yi Zhao, Institute of Computing Technology (CAS), China
Copyright © 2022 Hu, Wu, Quan, Yang, Lang, Tian and Meng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Meng, Ym8wMzI4QHFxLmNvbQ==; Geng Tian, VGlhbmdAZ2VuZWlzLmNu