 Jon Y. Takemoto
Jon Y. Takemoto Guillermo A. Altenberg
Guillermo A. Altenberg Naveena Poudyal3
Naveena Poudyal3 Yagya P. Subedi
Yagya P. Subedi Cheng-Wei T. Chang
Cheng-Wei T. Chang- 1Department of Biology, Utah State University, Logan, UT, United States
- 2Department of Cell Physiology and Molecular Biophysics, Center for Membrane Protein Research, Texas Tech University Health Sciences Center, Lubbock, TX, United States
- 3Department of Chemistry and Biochemistry, Utah State University, Logan, UT, United States
Widely-used Streptomyces-derived antibacterial aminoglycosides have encountered challenges because of antibiotic resistance and toxicity. Today, they are largely relegated to medicinal topical applications. However, chemical modification to amphiphilic aminoglycosides can revive their efficacy against bacterial pathogens and expand their targets to other pathogenic microbes and disorders associated with hyperactive connexin hemichannels. For example, amphiphilic versions of neomycin and neamine are not subject to resistance and have expanded antibacterial spectra, and amphiphilic kanamycins are effective antifungals and have promising therapeutic uses as connexin hemichannel inhibitors. With further research and discoveries aimed at improved formulations and delivery, amphiphilic aminoglycosides may achieve new horizons in pharmacopeia and agriculture for Streptomyces aminoglycosides beyond just serving as topical antibacterials.
Introduction
Naturally occurring aminoglycosides (AGs) isolated primarily from Streptomyces sp. are important broad spectrum antibacterial agents that have been used clinically for decades (Figure 1; Umezawa and Hooper, 1982; Gonzalez et al., 1998; Arya, 2007; Krause et al., 2016). Besides streptomycin, neomycin and kanamycin have been two of the most studied and employed classes of AG antibiotics. Neomycin belongs to a group of AGs containing a 4,5-disubstituted 2-deoxystreptamine core (ring II), while kanamycin contains a 4,6-disubstituted 2-deoxystreptamine core. Other AG groups include gentamicin (Weinstein et al., 1963) and sisomicin (Weinstein et al., 1970), which structurally resemble kanamycin antibiotics but are produced by Micromonospora sp. Structurally, gentamicin and sisomicin usually contain deoxygenation at ring I, and have a different ring III aminosugar from kanamycin. The emergence of antibiotic resistant bacteria has significantly hampered the use of naturally occurring AGs.
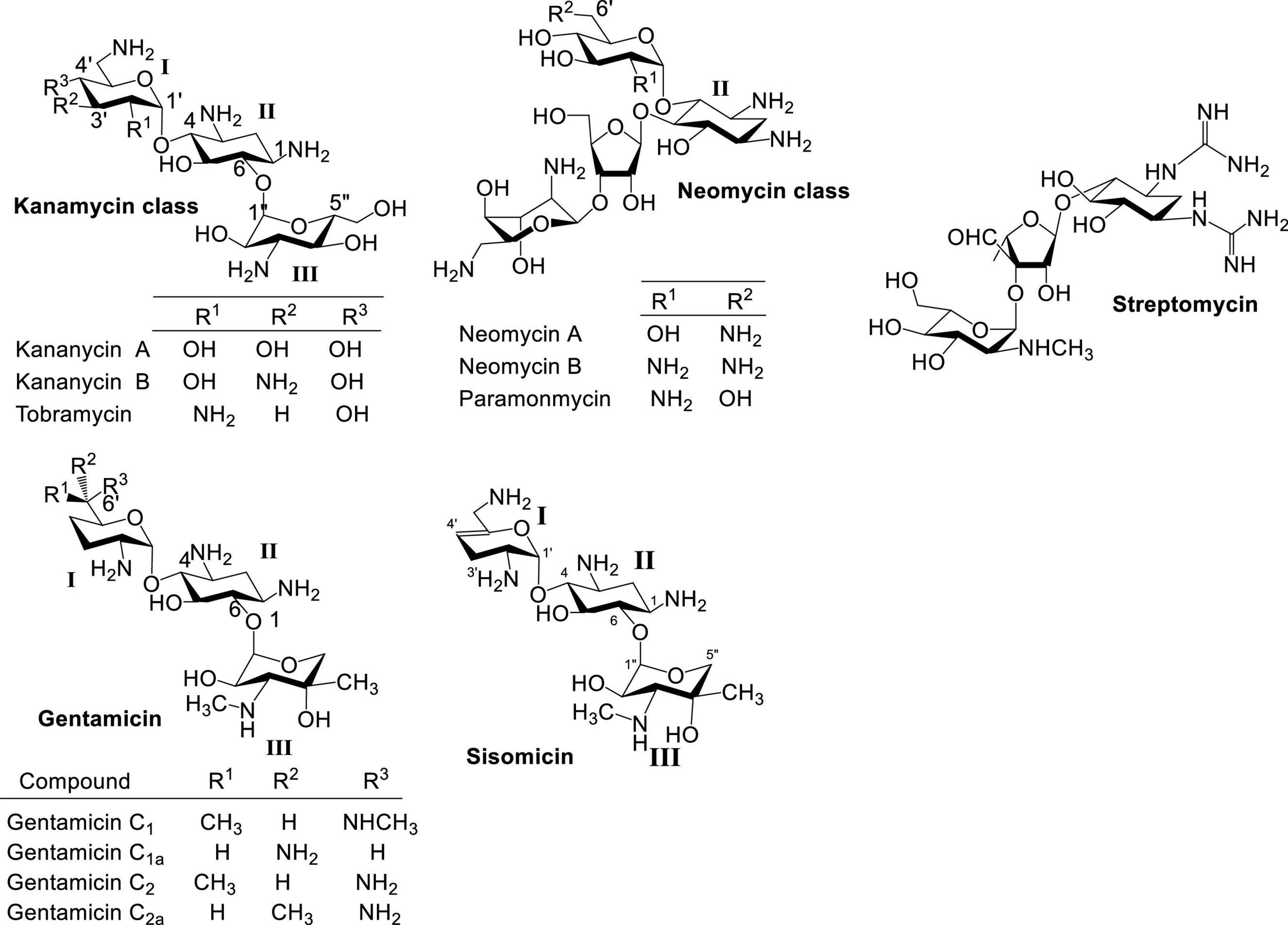
Figure 1. Structures of natural AGs.
During the 1970s, several semisynthetic AGs with improved activity against resistant bacteria were developed, such as amikacin (Kawaguchi et al., 1972), netilmicin (Campoli-Richards et al., 1989), isepamicin (Miller et al., 1978; Nagabhushan et al., 1978), arbekacin (Kobayashi et al., 1995), and most recently plazomicin (Aggen et al., 2010; Figure 2). However, these new AGs were limited when confronted with bacterial resistance arising from overexpression of AG-modifying enzymes. Over a hundred different AG-modifying enzymes are known to inactivate AGs by introducing structural modifications at various AG sites (Ramirez and Tolmasky, 2010; Garneau-Tsodikova and Labby, 2016). Some display substrate promiscuity and modify both neomycin and kanamycin classes (Fong and Berghuis, 2002). In addition, ototoxicity and nephrotoxicity often associated with AGs persist despite efforts devoted to lowering these side effects (Xie et al., 2011; Kovacik et al., 2021). Finally, as a result of the prevalence of AG-modifying enzymes, more structural motifs are needed at specific positions of AGs which adds to the challenges and cost of synthesizing new AGs. As an example, an expensive AG, sisomicin, was used as starting material for the development of plazomicin to circumvent part of the problem of regioselective chemical modification, which even at a relatively concise synthesis resulted in a cost >$3,000 US dollars per 100 mg.
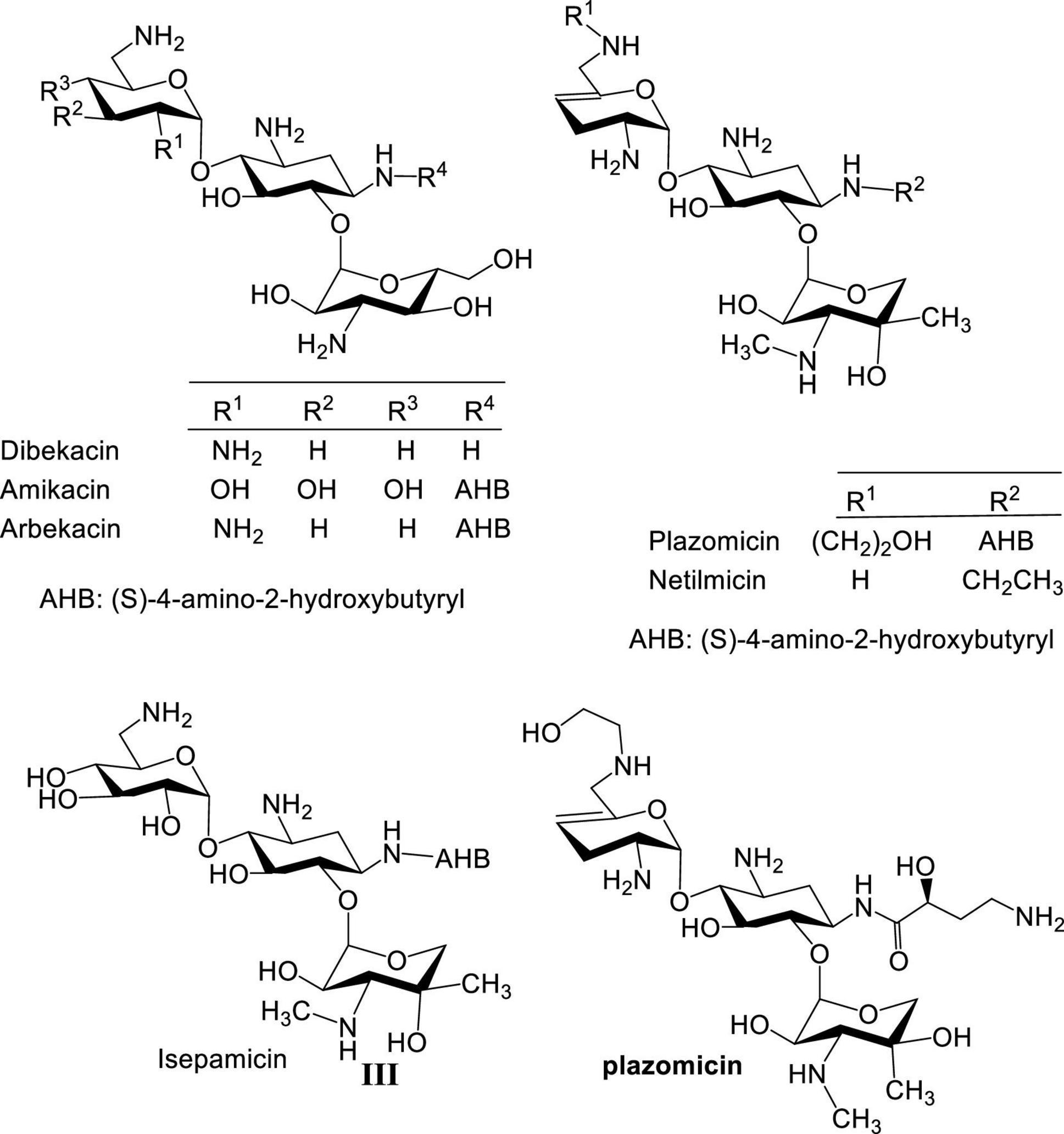
Figure 2. Structures of semisynthetic aminoglycosides.
An alternative approach is to revive and repurpose AGs using cost-effective AGs like neomycin or kanamycin as feedstocks for the synthesis of amphiphilic AGs (AAGs) (Chang et al., 2010; Chang and Takemoto, 2014; Domalaon et al., 2018; Subedi et al., 2018; Dezanet et al., 2020). AGs are hydrophilic, and the attachment of hydrophobic groups creates AAGs. In general, AAGs have two distinct features that make them different from AGs: broader antimicrobial profile and altered mode of action. AGs are active against aerobic Gram-positive and Gram-negative bacteria but inactive against anaerobic bacteria and fungi. In contrast, AAGs have antibacterial—aerobic and facultative anaerobic—and antifungal activities. Traditional AGs display antibacterial activity by binding to 16S rRNA thereby interfering with protein synthesis, whereas AAGs exert bioactivity by primarily targeting microbial membranes (Udumula et al., 2013). The latter bioactivity allows AAGs to be active against bacteria that are resistant to AGs, and also against fungi.
In contrast to the semisynthesis of AGs, where regioselective incorporation of structural motifs is essential, the synthesis of AAGs can be performed at the sites that are most amenable to modification. This drastically alleviates the burden of chemical modification and the cost of production. The most cost-effective feedstocks of AGs are neomycin B and kanamycin A, and this review focuses on the applications and biological activities of AAGs derived from neomycin B, kanamycin A and neamine, a derivative from neomycin B. Particular attention is directed toward synthetic approaches as they dictate the costs of production and the feasibility of offering marketable products.
Amphiphilic neamines and neomycins
Several groups have reported the synthesis and antibacterial investigations of amphiphilic neomycins (Figure 3; Bera et al., 2008, 2010a; Zhang et al., 2008, 2009). The lead amphiphilic neomycins show broad spectrum and unusual biological activities, especially against bacteria resistant to AGs. For example, 5″-aminoneomycin with attached linear acyl groups displays prominent antibacterial activities against a panel of bacteria, including methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE). The activity against VRE is of particular interest since facultative enterococci are intrinsically resistant against traditional AGs such as neomycin, amikacin, gentamicin, tobramycin and kanamycin due to the lack of AG-uptake mechanisms. Therefore, the unusual activities of AAGs, especially against VRE, strongly imply that the attachment of hydrophobic groups results in alteration of the traditional antibacterial mode of action.
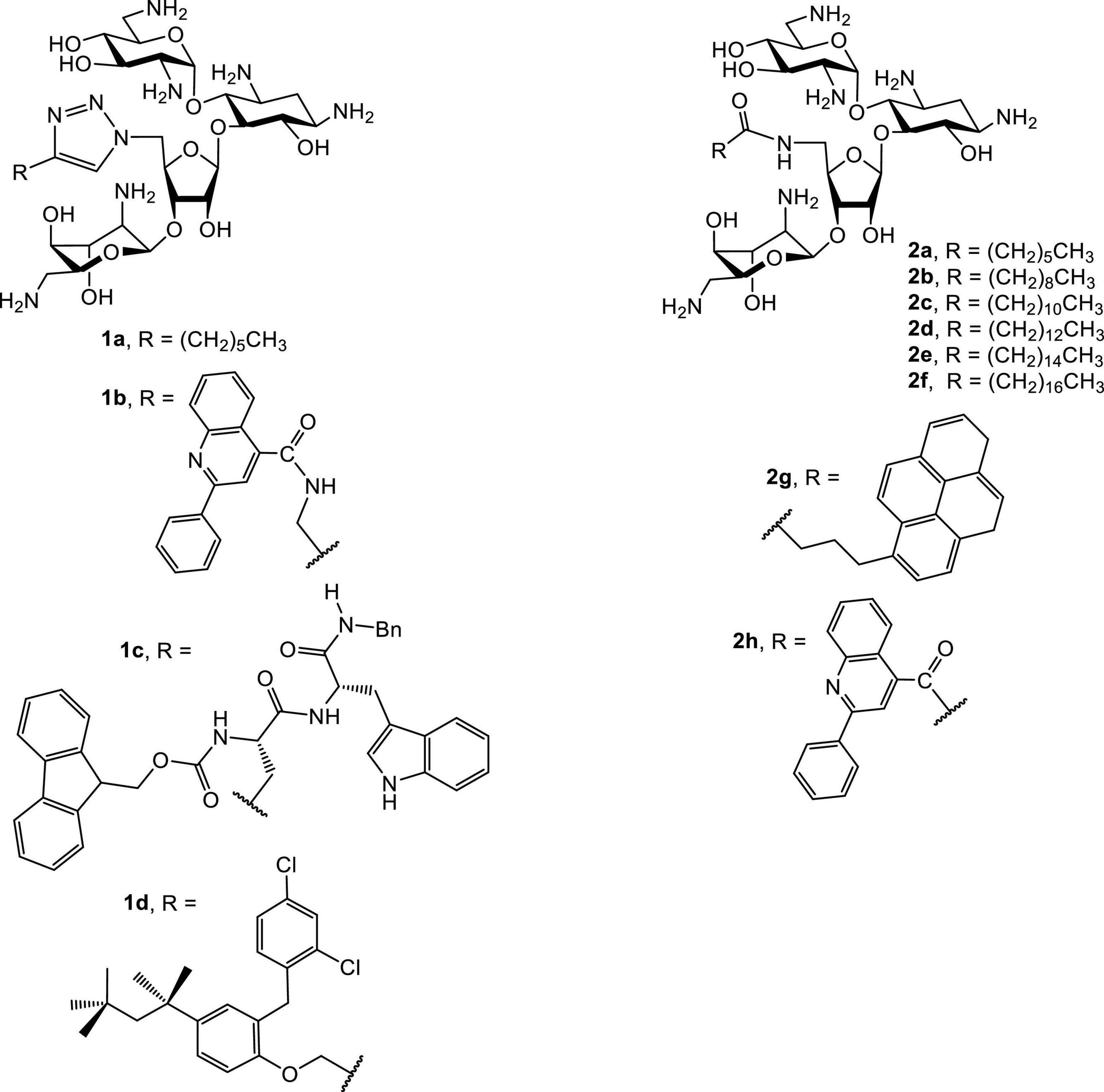
Figure 3. Structures of representative amphiphilic neomycins.
The antibacterial minimum inhibitory concentrations (MICs) of amphiphilic neomycins are summarized in Tables 1, 2. In general, the amphiphilic neomycin derivatives in Table 1 are less active against Gram-negative bacteria. There is no clear structure-activity relationship (SAR) for the derivatives with aromatic motifs attached (1b,c, 2g,h), but the activity of 1c against MRSA is of particular interest (Table 1). For the derivatives with linear alkyl chains attached (1a, 2a-f), increasing the hydrophobicity (lipophilicity) appears to enhance antibacterial activity, as 2e (C16, hexadecyl) is the most active. Nevertheless, extending the alkyl chain further to C18 (octadecyl) decreases the activity slightly. The most significant findings are the unusual activities of 2e against MRSA (Tables 1, 2) and VRE (Table 2). Traditional AGs are inactive or much less active against these two pathogenic bacterial strains. Amphiphilic neomycins with hydrophobic groups attached at the N-6‴ position have also been investigated (Mével et al., 2012), and trends of biological activities similar to those of other amphiphilic neomycins were observed.
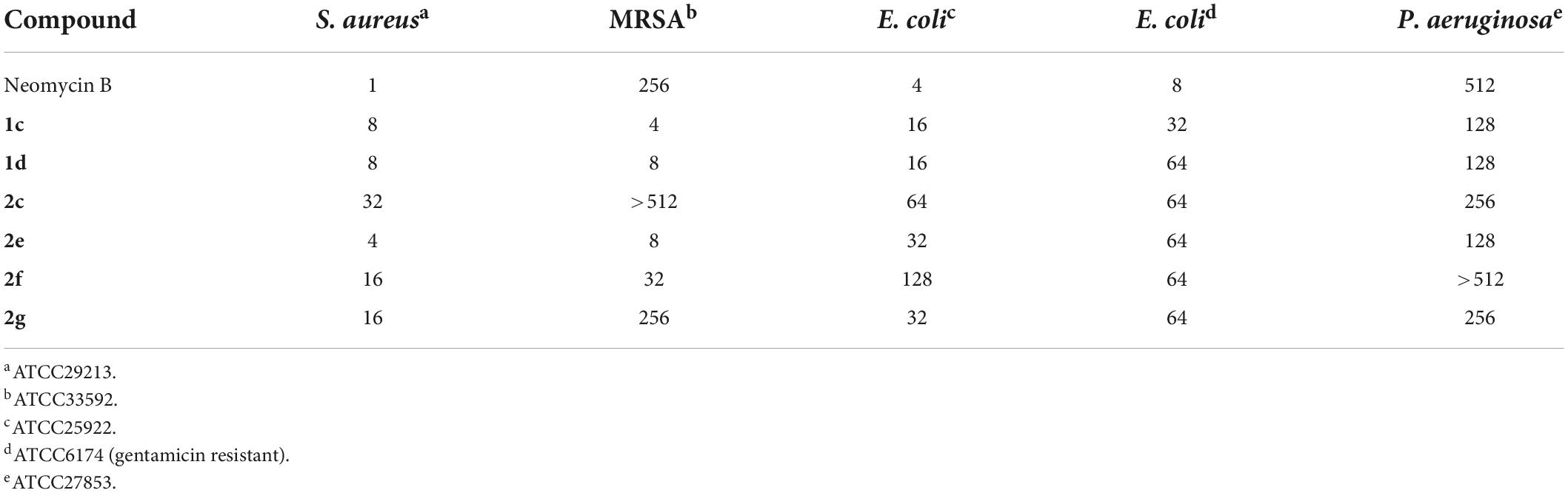
Table 1. MICs (μg/mL) of 5″-modified neomycin AAGs against MRSA (Bera et al., 2008, 2010a).
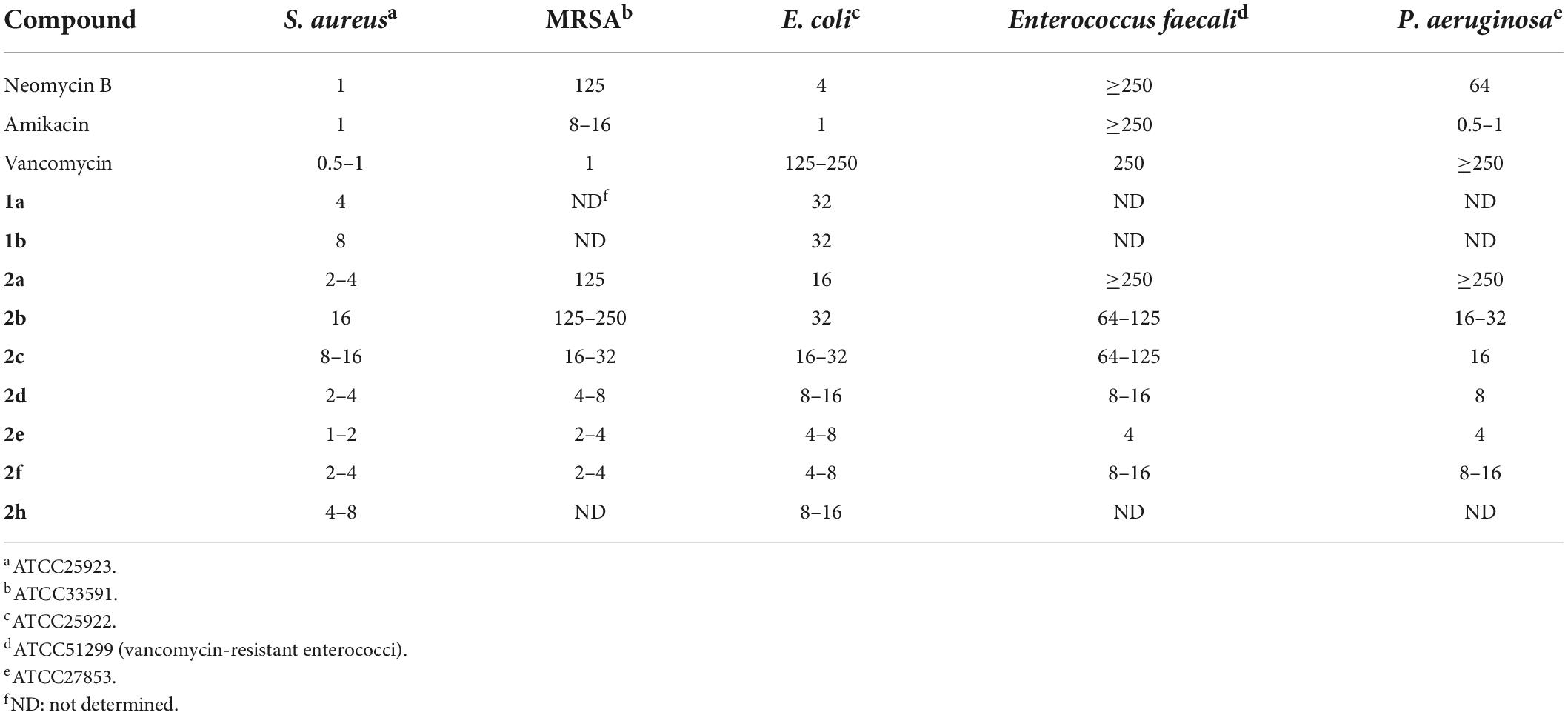
Table 2. MICs (μg/mL) of 5″-modified neomycin AAGs against MRSA and VRE (Zhang et al., 2008, 2009).
Neamine is obtained by acid-hydrolysis of neomycin. Modification to amphiphilic neamine usually begins with the protection or masking of amino groups, for example tritylation followed with alkylation of hydroxyl groups and global deprotection (Figure 4; Benhamou et al., 2015; Mingeot-Leclercq and Décout, 2016; Zimmermann et al., 2016). In general, amphiphilic neamines have been noted for their unique activities especially against Pseudomonas sp (Mingeot-Leclercq and Décout, 2016; Zimmermann et al., 2016). This is significant as Pseudomonas sp. are known for their unusual membrane compositions and efflux resistance that compromise the activity of many antibiotics. Additionally, amphiphilic neamines with di-N-methylation have improved activities against Gram-negative bacteria (Benhamou et al., 2015).
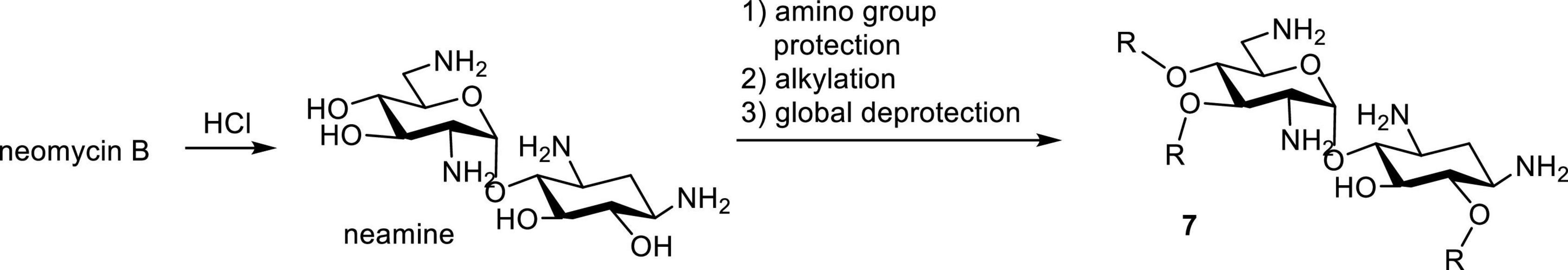
Figure 4. Synthesis of amphiphilic neamine.
Amphiphilic paromomycins have also been synthesized and employed as inhibitors against AG-modifying enzymes (Szychowski et al., 2011) and antibacterials toward Gram-positive pathogens (Berkov-Zrihen et al., 2013a). AAGs derived from streptomycin, gentamicin and sisomicin have not been reported, probably due to cost considerations and the challenges of site-selective modifications.
Since AAGs have been shown to have mode of antibacterial action different from tradidional AGs, several groups have engaged in investigations on their mode of action. Using atomic force microscopy, Mingeot-Leclercq and co-workers have reported that amphiphilic neamine derivatives bind to the lipopolysaccharides of P. aeruginosa and induce membrane depolarization (Mingeot-Leclercq and Décout, 2016). More evidence supporting this mode of action comes from studies using the fluorogenic dye SYTOX (Udumula et al., 2013). SYTOX cannot penetrate the intact bacterial membrane, but enters cells through pores formed in the bacterial membrane and binds nucleic acids which results in strong fluorescence. Following exposure to 6e, Escherichia coli and S. aureus strains showed high levels of green fluorescence, whereas S. aureus cells treated with neomycin and SYTOX did not, as pore formation with traditional AGs is not expected.
It is interesting to note the similarities between the antibacterial activities of AAGs and lipopeptides, another class of amphiphilic compounds. Addition of acyl chains to peptides alters membrane lipid composition and packing. The antimicrobial activity of lipopeptides is associated with membrane modifications, including changes in its curvature, surface charge and dipole potential, as well response to changes in ionic strength (Balleza et al., 2019; Shahane et al., 2019; Juhi et al., 2021). Although details of the mode of action of AAG are still being deciphered, it is likely that they will resemble those of antimicrobial lipopeptides.
Among the reported investigations of amphiphilic neomycin and neamine, several studies found these compounds less toxic based on hemolysis experiments (Zhang et al., 2009; Benhamou et al., 2015). Compounds 2a, 2e and 2f were found to be weakly antifungal with MICs ranging from 62.5 to 125 μg/mL against the wheat and barley pathogen Fusarium graminearum (Takemoto and Chang, 2010). However, little is known regarding the bioactivity of these compounds beyond their antibacterial activity.
Amphiphilic kanamycins
Early successes in the synthesis of amphiphilic kanamycins (AK) employed kanamycin B as the starting material (Schepdael et al., 1991). Following the protection of Boc groups, the 6″-OH (the only primary hydroxyl group) can be selectively modified using a nucleophilic substitution approach (Figure 5). Derivatives with various functional groups, such as thioether, alkoxyl, alkylamino, alkylamido and esters, have been synthesized and evaluated for their antibacterial activities. Although these compounds manifest good antibacterial activity against AG susceptible strains, they do not display significant antibacterial activity against AG-resistant bacteria such as Streptococcus faecalis and P. aeruginosa (Schepdael et al., 1991).
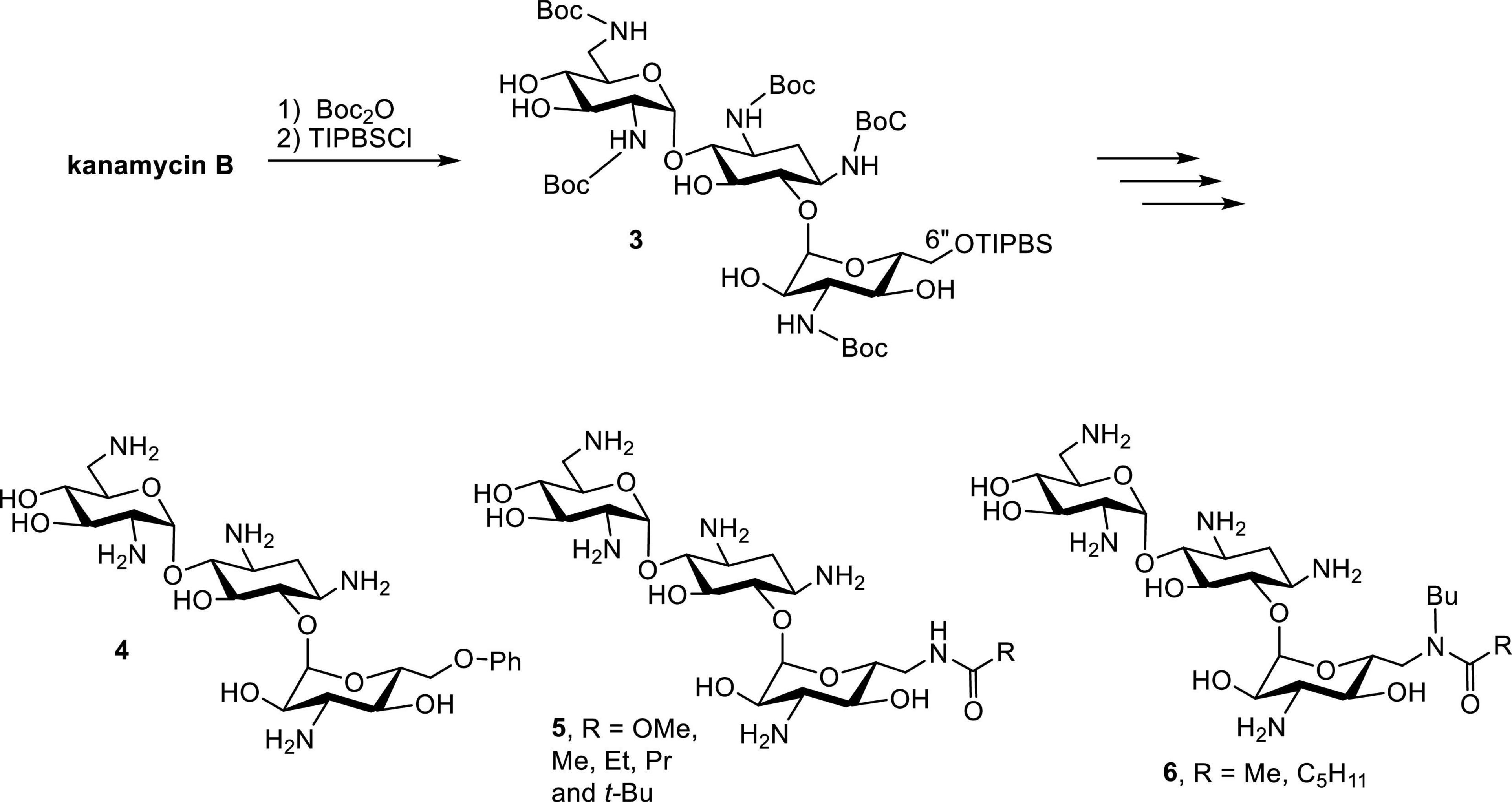
Figure 5. Synthesis of AKs.
Tobramycin, a kanamycin class AG, has been converted to 6″-modified amphiphilic derivatives using similar methods (Figure 6; Herzog et al., 2012). From the selected MICs, a long linear alkyl chain (C14 and C16) seems to improve antibacterial activity (Table 3). The most active compounds,8a and 8b, inhibit growth of VRE and tobramycin-resistant E. coli. On the other hand, oxidation of alkylthio to sulfones or sulfoxides slightly decreases antibacterial activities.
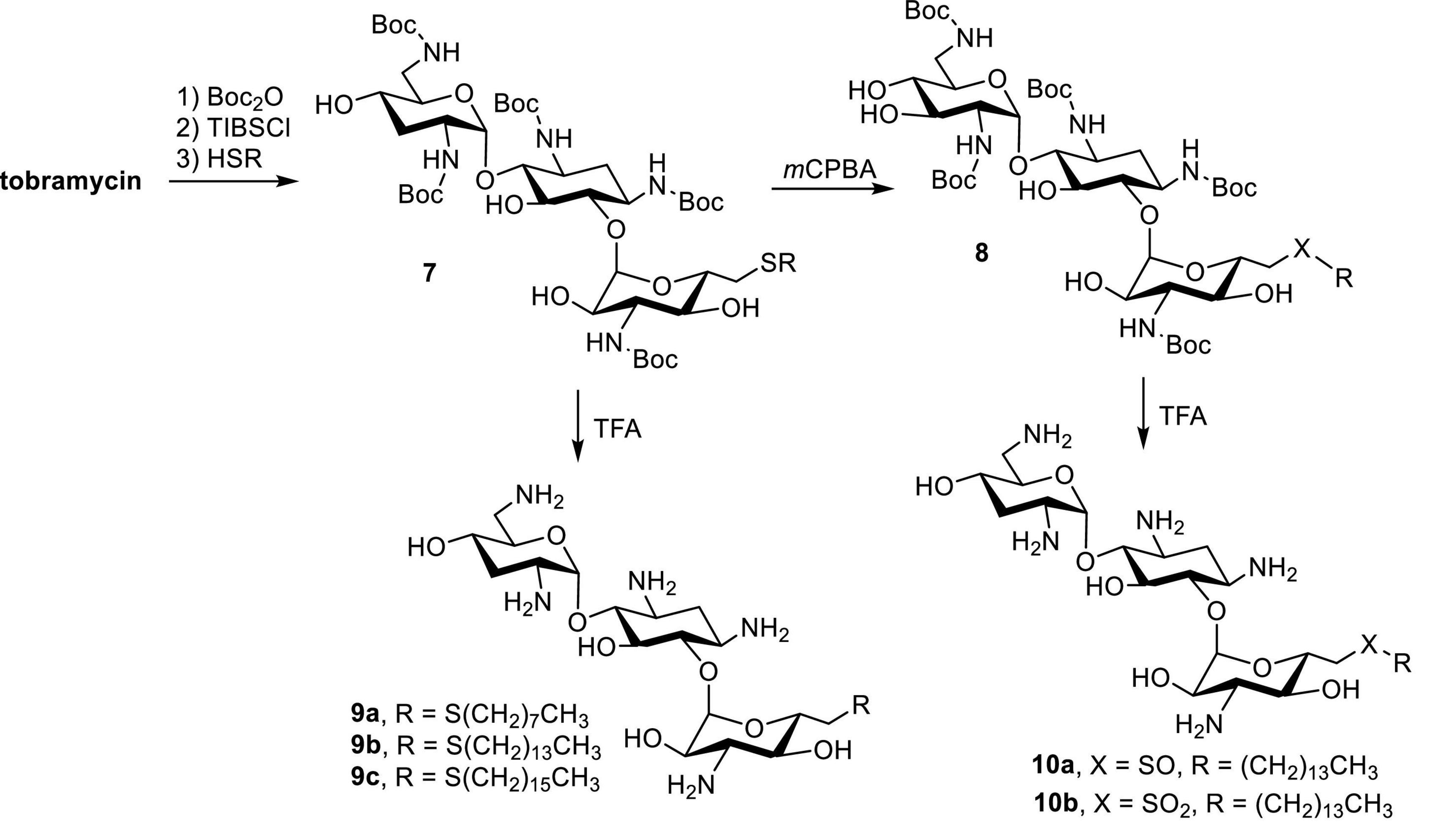
Figure 6. Synthesis of amphiphilic tobramycin.
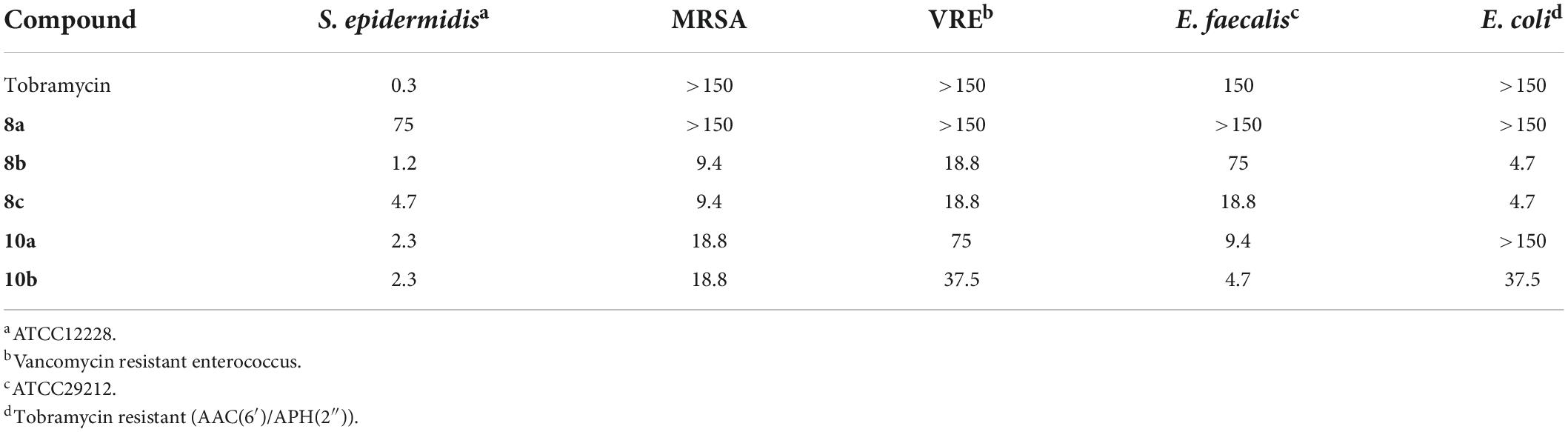
Table 3. MICs of 6″-modified amphiphilic tobramycin derivatives (in μg/mL) (Herzog et al., 2012).
Other AK designs have been reported, including kanamycin A with a 6″-alkyltriazole group (Bera et al., 2010b), 4′,6″-dialkylthio tobramycin (Berkov-Zrihen et al., 2013b) and 6′-chelostolcarbonyl kanamycin A (Sainlos et al., 2003). In general, these compounds manifest broad spectrum antibacterial activities at low micromolar concentrations.
In 2006, it was reported that AGs such as neomycin, paromomycin, ribostamycin and streptomycin possess antifungal activities against Phytophthora and Pythium species (Lee et al., 2005). Inspired by this article, screening of libraries of neomycin, kanamycin and pyranmycin derivatives led to the identification of AK FG08 as the most potent antifungal AG tested (Figure 7; Chang et al., 2010). Interestingly, FG08 was inactive against bacteria, a discovery pointing to a new and significant bioactivity of AAGs and AKs with potential for broader applications in medicine and agriculture. Furthermore, the antifungal activity of FG08 involved formation of pores in the plasma membrane (Shrestha et al., 2013). Therefore, attachment of hydrophobic groups to make AAGs not only expanded the antibacterial target species spectrum, but also uncovered a “switch” that adds antifungal activity to the AG antimicrobial profile.
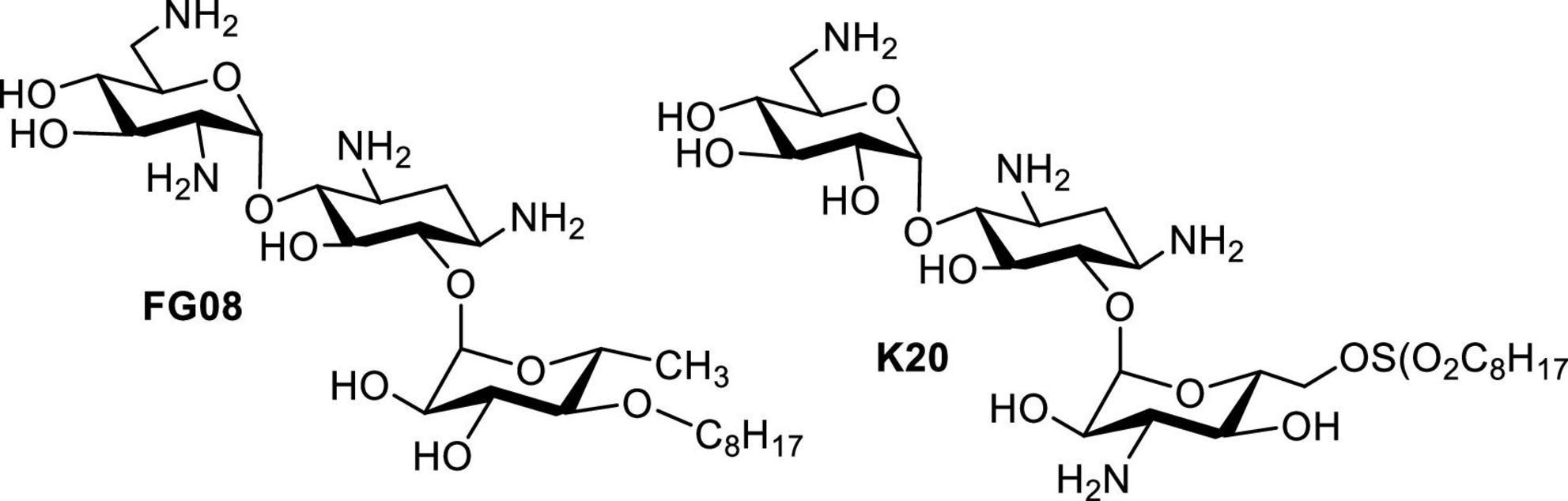
Figure 7. Structures of FG08 and K20.
Although FG08 showed promising fungicidal activity against crop disease caused by Fusarium head blight (FHB), its complex synthesis makes it unsuitable for scale-up production, and therefore, practical applications for FG08 in medicine and agriculture are limited. Based on the SAR investigation of FG08 and related compounds (Fosso M. et al., 2015), a simplified protocol for the production of a 2nd generation antifungal AK (K20) was explored (Shrestha et al., 2014). In addition to showing broad spectrum antifungal activity, K20 can be synthesized in 200–300 g scale batch (Table 4). Furthermore, K20 also exerts strong synergism with commercially available crop fungicides (Shrestha et al., 2014, 2015b) and these K20 combinations demonstrated efficacy in wheat field trials against FHB (Takemoto et al., 2018). In short, a combination of K20 and crop fungicides can reduce FHB and production of the toxin deoxynivalenol that is associated with FHB. These findings suggest the possible use of K20 in combination with established crop fungicides at lower concentrations, thus reducing costs, toxicities and environmental impacts of crop protection strategies.
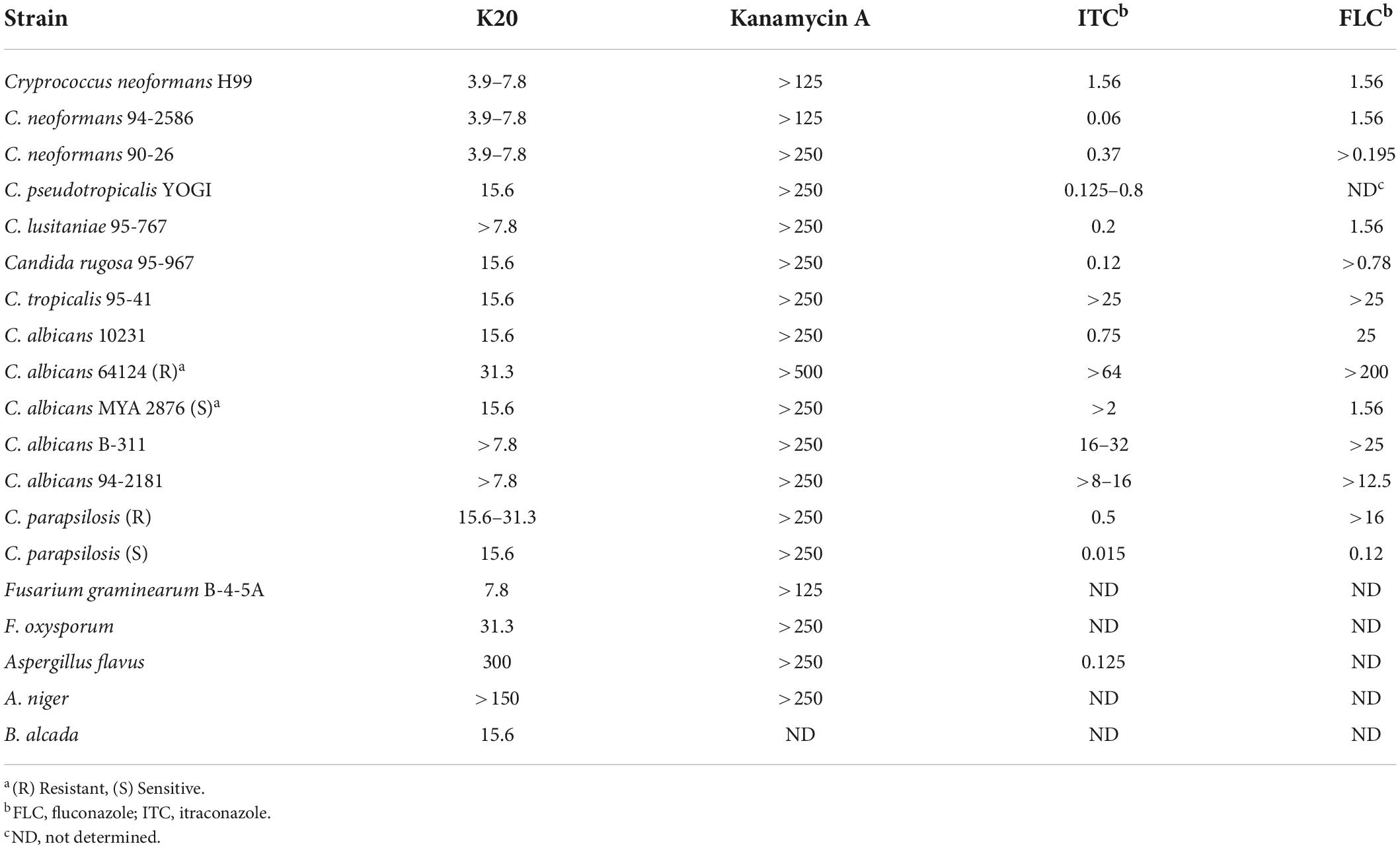
Table 4. MICs (μg/mL) of K20 and kanamycin A against bacteria and fungia.
Newer and improved antifungal AK designs have been reported since the discovery of K20 (Fosso M. Y. et al., 2015; Shrestha et al., 2015b; Steinbuch et al., 2018; Subedi et al., 2019a,b; Logviniuk and Fridman, 2020). A one-step synthesis of AK was developed to better compete with the commercially available crop fungicides and therapeutic antifungal agents (Figure 8; Subedi et al., 2019a). Among the compounds synthesized, the lead compounds 11a and 11b show strong antifungal activities, including activities against clinical strains of C. auris and Aspergilla fumigatus (Table 5). In pharmacokinetic studies in a mouse model, 11b did not show signs of toxicity (e.g., body weight loss) up to the highest doses tested (52 mg/kg) (Subedi et al., 2020b). The cost of 11a and 11b production is lower than those of K20, making them more competitive than the widely used azole-based antifungal agents. As a group, the antifungal AKs represent a significant breakthrough for the classical AGs as they allow for the repurposing of large stockpiles of kanamycin sulfate, which is otherwise clinically obsolete as an antibacterial agent.
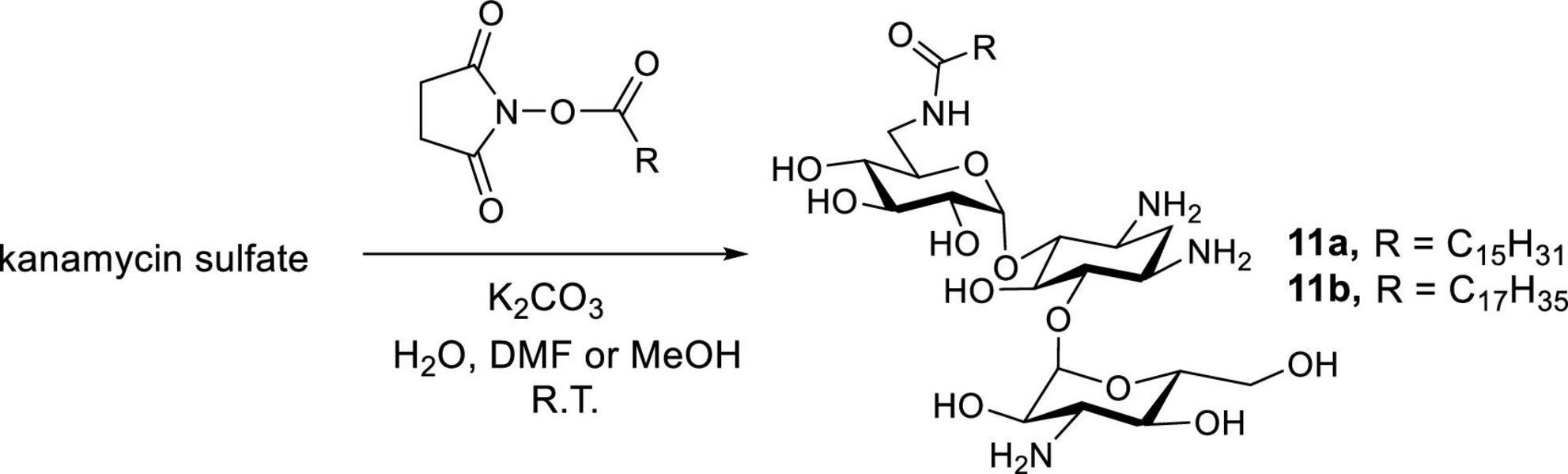
Figure 8. One-step synthesis of AKs 11a and 11b.
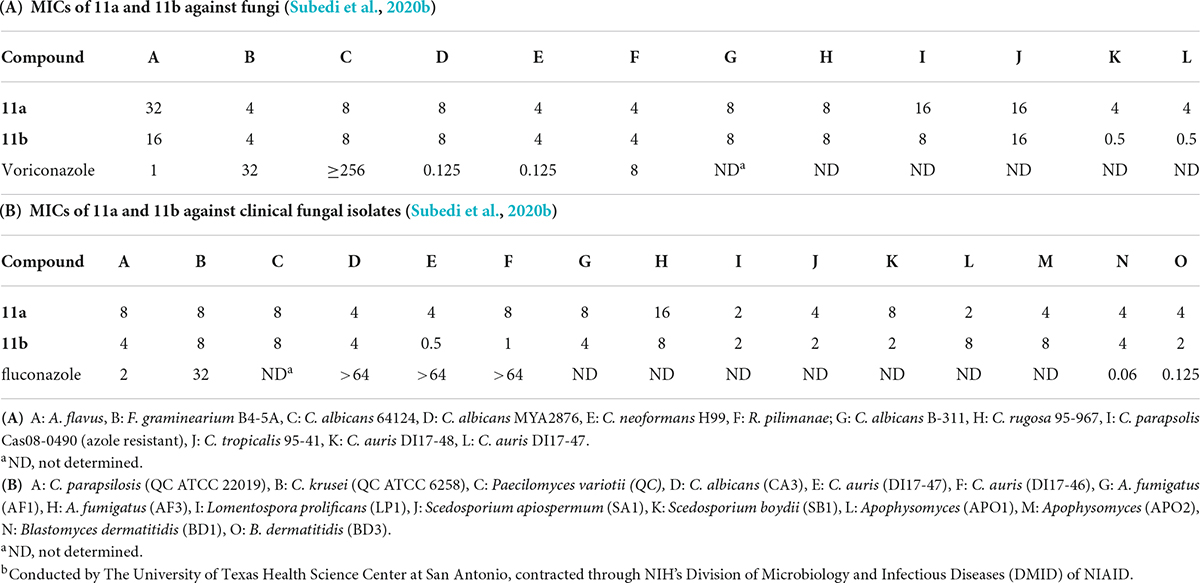
Table 5. Summarized MICs (μg/mL) of 11a and 11bb.
Amphiphilic kanamycins as connexin hemichannel inhibitors
Large stockpiles of AGs, mainly neomycin sulfate and kanamycin sulfate are available worldwide, but their clinical use is limited due to the prevalence of bacterial resistance. Thus, efforts have been devoted to repurposing AGs for other applications. These include AGs as therapeutic options for spinal muscular atrophy (SMA) (Mattis et al., 2006, 2009) and disorders associated with hyperactivity of connexin hemichannels (HCs) (Fiori et al., 2017). Following these examples, AKs have also been investigated as connexin HC inhibitors with the goal of identifying leads that do not have antibacterial activity, show reduced cytotoxicity, and target HCs formed by specific connexin isoforms.
Connexins are transmembrane proteins that constitute the key building blocks of connexin HCs. Gap-junction channels (GJCs) formed by the head-to-head docking of two HCs, one from each of two adjacent cells, play diverse and pivotal roles in development, intercellular communication, and regulation (Meşe et al., 2007; Nielsen et al., 2012). Abnormal increases and decreases in the activity of HCs and GJCs have been linked to numerous genetic and acquired disorders such as deafness (Lee and White, 2009), heart arrhythmias and infarcts (Shintani-Ishida et al., 2007; Schulz et al., 2015; Leybaert et al., 2017), and cancer (Naus and Laird, 2010; Banerjee, 2016). Connexin HCs are mostly closed, but abnormally increased HC activity in inherited or acquired disorders results in cell damage. However, there are more than twenty different connexin isoforms, which have different functional properties and regulation, and are expressed differentially in various cells, tissues and organs (Beyer et al., 2009). Thus, desirable inhibitors should be selective not only toward connexin HCs but also the HCs of interest.
Among the connexin isoforms, connexin 43 (Cx43) and connexin 26 (Cx26) have attracted particular attention. Cx43 is expressed in many organs, including heart, brain, liver, kidney, skin, and myometrium. Mutations of Cx43 have been reported to associate with acute and chronic diseases occurred in these organs (Delmar and Makita, 2012; Prakoura et al., 2018; Slavi et al., 2018; Xu et al., 2019; Yang et al., 2019; Ma et al., 2020), whereas Cx26 mutations can lead to deafness and/or skin disorders (Bruzzone et al., 2003; Lee and White, 2009).
Following the recent discovery that natural AGs inhibit connexin HCs without affecting GJCs (Figueroa et al., 2014), various classes of AKs were screened against Cx43 and Cx26 HCs (Alfindee et al., 2018; Subedi et al., 2020b). These AKs manifested inhibitory effects, and further SAR investigations revealed that several 6″- and 6′-modified AKs display inhibitory activities that can be tuned toward Cx43 HCs (Figure 9; Subedi et al., 2020a,2021). The results showed that compounds 12a and 13c were most selective toward Cx43 when compared to Cx26 HCs, with calculated selectivities (Cx26 IC50/Cx43 IC50) of ∼7 and ∼8, respectively (Table 6). These selectivities are ∼30-fold higher than that of kanamycin A (0.24). In addition, the AKs selected had no antibacterial activity and were not cytotoxic to HeLa cells at concentrations of up to 100 μM.
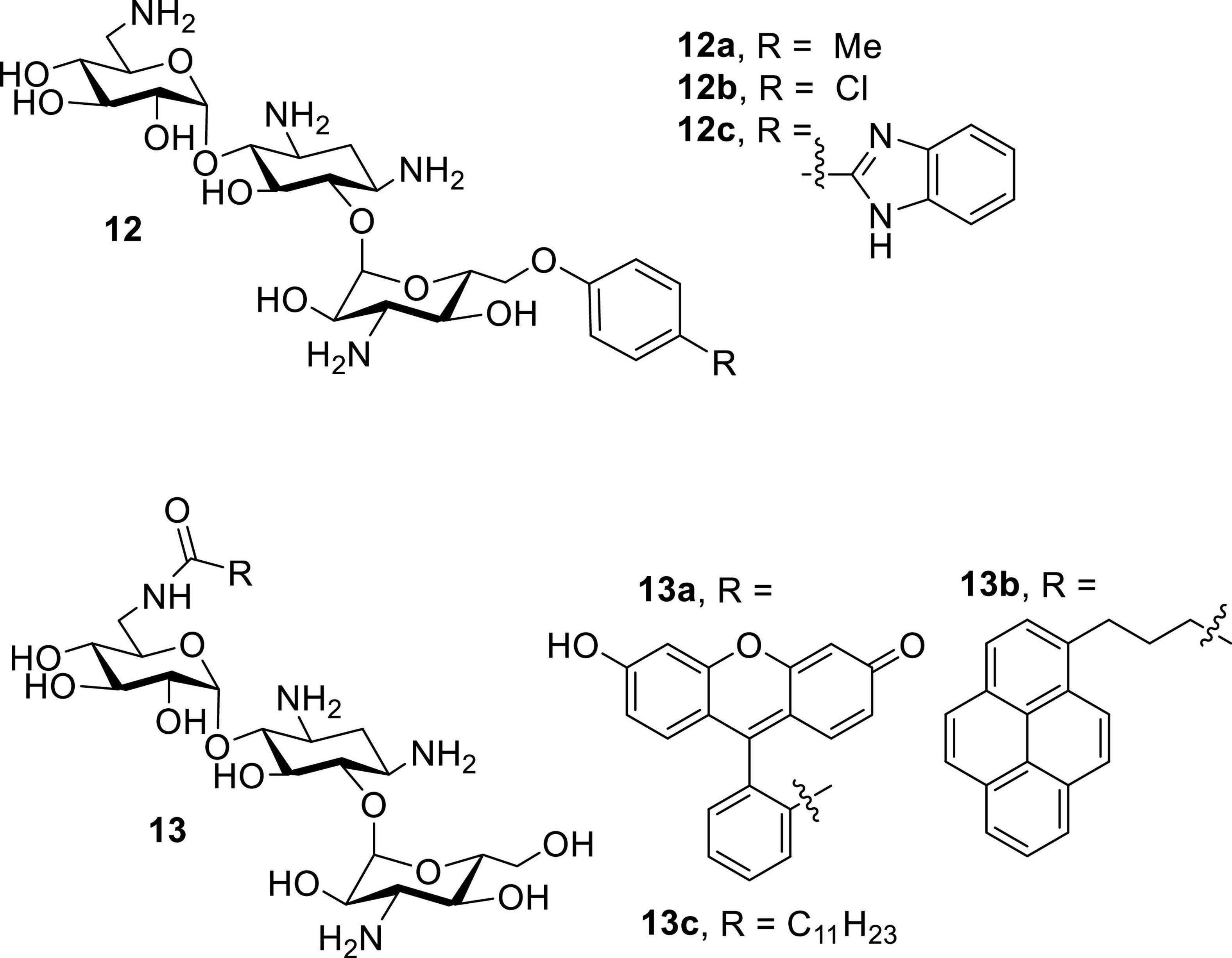
Figure 9. Amphiphilic kanamycin as connexin inhibitor.

Table 6. IC50 of selected 6″ - and 6′-modified AKs toward Cx26 and Cx43 HCs.
Conclusions and future perspectives
In developed countries, AGs are often considered as a “last resort” for the treatment of serious bacterial infections. Semisynthetic approaches have been effective to modify naturally occurring AGs, yielding new AGs with revived antibacterial activity against AG-resistant bacteria. However, due to the rampage of bacteria equipped with diverse mechanisms of resistance such as AG-modifying enzymes, efflux pumps, decreases in membrane permeability and RNA target modifications, the semisynthetic approach still faces major challenges. The obvious problem is the cost of production. To combat the constantly evolving resistance mechanisms and complicated synthetic efforts, rare or expensive natural AGs have been employed as starting material. As a result, the end products are economically costly. Furthermore, these semisynthetic AGs still act by the same mechanisms as traditional AGs, binding to the 16S rRNA and interfering with protein synthesis, and thus, resistance evolves readily and soon after development of these new AGs. Finally, these AGs are likely to be less effective against bacteria such as Enterococcus or Pseudomonas sp., which are intrinsically resistant to AGs or equipped with flexible efflux resistance mechanisms, respectively.
AAGs offer an alternative solution to combat resistant pathogens. They can be prepared from cost effective AG feedstocks such as neomycin and kanamycin sulfates, which are mass produced by fermentation but have few clinical uses due to the resistance problem. Since AG-modifying enzymes are cellular enzymes, AAGs that target the bacterial membrane can be bactericidal without entering the cells, circumventing bacterial resistance conferred by AG-modifying enzymes. These two features, cost effective synthesis and bypassing a predominant bacterial mode of AG resistance, enable the potential uses of AAGs as effective antibacterials and for AG-based therapeutic uses previously limited by AG resistance. Additionally, AAGs exert no or much reduced cytotoxicity when compared to traditional AGs.
Highlights about AAGs in this review are that: (1) amphiphilic neomycins and neamine demonstrate potent antibacterial activities; (2) AKs have antibacterial as well as antifungal activities. The antifungal activity of AKs is of particular interest because traditional AGs are generally inactive against fungi and AKs employ novel membrane pore forming modes of action; (3) there are strong synergies between AKs and commercially used antifungal agents; (4) methods of cost-effective synthesis of lead AKs have been developed; and (5) AKs are connexin HC inhibitors. As such, AKs are potential therapeutics and experimental tools for deciphering the functions of HCs in human physiology and pathophysiology. AKs offer potential selective inhibition of HCs such as those formed by Cx43, and do not affect GJCs, thus paving the way for therapeutic use against disorders specifically due to hyperactive HCs.
Despite progress on the development and testing of AAGs, demonstration of efficacy in vivo is a crucial step toward the systemic applications of these compounds. To date, most of the reported AAGs have been studied only in vitro and a recent study showed that serum could prevent interactions between antimicrobial AAGs and plasma membranes (Logviniuk and Fridman, 2020). This report suggests limited use of AAGs as non-systemic drugs or for topical application. However, progress on formulations and carrier/delivery vehicles for AAGs or AAG modifications to reduce binding to serum proteins could improve clinical efficacy.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Funding
The authors acknowledge research grant support from the USDA-National Institute of Food and Agriculture, United States (UTA01271), National Science Foundation (STTR Award 1521060) and (CHE Award 1429195), United States, Utah Science, Technology and Research (USTAR) initiative, Department of Chemistry and Biochemistry, Utah State University (graduate student support), and Baicor, LC, Logan Utah, United States.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aggen, J. B., Armstrong, E. S., Goldblum, A. A., Dozzo, P., Linsell, M. S., Gliedt, M. J., et al. (2010). Synthesis and spectrum of the neoglycoside ACHN-490. Antimicrob. Agents Chemother. 54, 4636–4642. doi: 10.1128/AAC.00572-10
Alfindee, M., Subedi, Y. P., Fiori, M. C., Krishnan, S., Kjellgren, A., Altenberg, G. A., et al. (2018). Inhibition of connexin hemichannels by new amphiphilic amino-glycosides without antibiotic activity. ACS Med. Chem. Lett. 9, 697–701. doi: 10.1021/acsmedchemlett.8b00158
Arya, D. P. (ed.) (2007). Aminoglycoside Antibiotics: From Chemical Biology to Drug Discovery. Hoboken: John Wiley & Sons, Inc.
Balleza, D., Alessandrini, A., and Beltrán García, M. J. (2019). Role of Lipid Composition, Physicochemical Interactions, and Membrane Mechanics in the Molecular Actions of Microbial Cyclic Lipopeptides. J Membr. Biol. 252, 131–157. doi: 10.1007/s00232-019-00067-4
Banerjee, D. (2016). Connexin’s Connection in Breast Cancer Growth and Progression. Int. J. Cell Biol. 2016:9025905. doi: 10.1155/2016/9025905
Benhamou, R. I., Shaul, P., Herzog, I. M., and Fridman, M. (2015). Di-N-methylation of anti-gram-positive aminoglycoside-derived membrane disruptors improves antimicrobial potency and broadens spectrum to Gram-negative bacteria. Angew. Chem. Int. Ed Engl. 54, 13617–13621. doi: 10.1002/anie.201506814
Bera, S., Zhanel, G. G., and Schweizer, F. (2008). Design, Synthesis, and Antibacterial Activities of Neomycin-Lipid Conjugates: Polycationic Lipids with Potent Gram-Positive Activity. J. Med. Chem. 51, 6160–6164. doi: 10.1021/jm800345u
Bera, S., Zhanel, G. G., and Schweizer, F. (2010a). Antibacterial activities of aminoglycoside antibiotics-derived cationic amphiphiles. Polyol-modified neomycin B-, kanamycin A-, amikacin-, and neamine-based amphiphiles with potent broad spectrum antibacterial activity. J. Med. Chem. 53, 3626–3631. doi: 10.1021/jm1000437
Bera, S., Zhanel, G. G., and Schweizer, F. (2010b). Evaluation of amphiphilic aminoglycoside–peptide triazole conjugates as antibacterial agents. Bioorg. Med. Chem. Lett. 20, 3031–3035. doi: 10.1016/j.bmcl.2010.03.116
Berkov-Zrihen, Y., Herzog, I. M., Feldman, M., and Fridman, M. (2013a). Site-selective displacement of tobramycin hydroxyls for preparation of antimicrobial cationic amphiphiles. Org. Lett. 15, 6144–6147. doi: 10.1021/ol4030138
Berkov-Zrihen, Y., Herzog, I. M., Feldman, M., Sonn-Segev, A., Roichmanand, Y., and Fridman, M. (2013b). Di-alkylated paromomycin derivatives: Targeting the membranes of Gram positive pathogens that cause skin infections. Bioorg. Med. Chem. 21, 3624–3631. doi: 10.1016/j.bmc.2013.03.046
Beyer, E. C., Berthoud, V. M., Harris, A. L., and Locke, D. (2009). Connexins: A Guide. New York, NY: Humana Press.
Bruzzone, R., Veronesi, V., Gomes, D., Bicego, M., Duval, N., Marlin, S., et al. (2003). Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS Lett. 533, 79–88. doi: 10.1016/S0014-5793(02)03755-9
Campoli-Richards, D. M., Chaplin, S., Sayce, R. H., and Goa, K. L. (1989). Netilmicin. A review of its antibacterial activity, pharmacokinetic properties and therapeutic use. Drugs 38, 703–756. doi: 10.2165/00003495-198938050-00003
Chang, C.-W. T., Fosso, M., Kawasaki, Y., Shrestha, S., Bensaci, M. F., Wang, J., et al. (2010). Antibacterial to antifungal conversion of neamine aminoglycosides through alkyl modification. Strategy for reviving old drugs into agrofungicides. J. Antibiot. 63, 667–672. doi: 10.1038/ja.2010.110
Chang, C.-W. T., and Takemoto, J. Y. (2014). Antifungal amphiphilic aminoglycosides. Med. Chem. Comm. 5, 1048–1057. doi: 10.1039/c4md00078a
Delmar, M., and Makita, N. (2012). Cardiac connexins, mutations and arrhythmias. Curr. Opin. Cardiol. 27, 236–241. doi: 10.1097/HCO.0b013e328352220e
Dezanet, C., Kempf, J., Mingeot-Leclercq, M.-P., and Décout, J.-L. (2020). Amphiphilic aminoglycosides as medicinal agents. Int. J. Mol. Sci. 21:7411. doi: 10.3390/ijms21197411
Domalaon, R., Idowu, T., Zhanel, G. G., and Schweizer, F. (2018). Antibiotic hybrids: The next generation of agents and adjuvants against gram-negative pathogens? Clin. Microbiol. Rev. 31:e00077–17. doi: 10.1128/CMR.00077-17
Figueroa, V. A., Retamal, M. A., Cea, L. A., Salas, J. D., Vargas, A. A., Verdugo, C. A., et al. (2014). Extracellular gentamicin reduces the activity of connexin hemichannels and interferes with purinergic Ca2+ signaling in HeLa cells. Front. Cell. Neurosci. 8:265. doi: 10.3389/fncel.2014.00265
Fiori, M. C., Krishnan, S., Kjellgren, A., Cuello, L. G., and Altenberg, G. A. (2017). Inhibition by commercial aminoglycosides of human connexin hemichannels expressed in bacteria. Molecules 22:2063. doi: 10.3390/molecules22122063
Fong, D. H., and Berghuis, A. M. (2002). Substrate promiscuity of an aminoglycoside antibiotic resistance enzyme via target mimicry. EMBO J. 21, 2323–2331. doi: 10.1093/emboj/21.10.2323
Fosso, M., AlFindee, M. N., Zhang, Q., Nziko, V. P. N., Kawasaki, Y., Shrestha, S. K., et al. (2015). Structure–Activity Relationships for Antibacterial to Antifungal Conversion of Kanamycin to Amphiphilic Analogues. J. Org. Chem. 80, 4398–4411. doi: 10.1021/acs.joc.5b00248
Fosso, M. Y., Shrestha, S. K., Green, K. D., and Garneau-Tsodikova, S. (2015). Synthesis and bioactivities of kanamycin b-derived cationic amphiphiles. J. Med. Chem. 58, 9124–9132. doi: 10.1021/acs.jmedchem.5b01375
Garneau-Tsodikova, S., and Labby, K. J. (2016). Mechanisms of resistance to aminoglycoside antibiotics: Overview and perspectives. Med. Chem. Commun. 7, 11–27. doi: 10.1039/c5md00344j
Gonzalez, L. S. III., Pharm, D., and Spencer, J. P. (1998). Aminoglycosides: A practical review. Am. Fam. Physician 58, 1811–1820.
Herzog, I. M., Green, K. D., Berkov-Zrihen, Y., Feldman, M., Vidavski, R. R., Eldar-Boock, A., et al. (2012). 6″-Thioether tobramycin analogues: Towards selective targeting of bacterial membranes. Angew. Chem. Int. Ed 51, 5652–5656. doi: 10.1002/anie.201200761
Juhi, D. W., Glattard, E., Aisenbrey, C., and Bechinger, B. (2021). Antimicrobial peptides: Mechanism of action and lipid-mediated synergistic interactions within membranes. Faraday Discuss. 232, 419–434. doi: 10.1039/d0fd00041h
Kawaguchi, H., Naito, T., Nakagawa, S., and Fujisawa, K. I. (1972). BB-K8, a new semisynthetic aminoglycoside antibiotic. J. Antibiot. 25, 695–708. doi: 10.7164/antibiotics.25.695
Kobayashi, Y., Uchida, H., and Kawakami, Y. (1995). Arbekacin. Intl. J. Antimicrobial. Agents 5, 227–230. doi: 10.1016/0924-8579(95)00014-Y
Kovacik, A., Tvrda, E., Jambor, T., Fulopova, D., Kovacikova, E., Hleba, L., et al. (2021). Cytotoxic effect of aminoglycoside antibiotics on the mammalian cell lines. J. Environ. Sci. Health Part A 56, 1–8. doi: 10.1080/10934529.2020.1830653
Krause, K. M., Serio, A. W., Kane, T. R., and Connolly, L. E. (2016). Aminoglycosides: An overview. Cold Spring Harb. Perspect. Med. 6:a027029. doi: 10.1101/cshperspect.a027029
Lee, H. B., Kim, Y., Kim, J. C., Choi, G. J., Park, S.-H., Kim, C.-J., et al. (2005). Activity of some aminoglycoside antibiotics against true fungi, Phytophthora and Pythium species. J. Appl. Microbiol. 99, 836–843. doi: 10.1111/j.1365-2672.2005.02684.x
Lee, J. R., and White, T. W. (2009). Connexin-26 mutations in deafness and skin disease. Expert Rev. Mol. Med. 11:e35. doi: 10.1017/S1462399409001276
Leybaert, L., Lampe, P. D., Dhein, S., Kwak, B. R., Ferdinandy, P., Beyer, E. C., et al. (2017). Connexins in cardiovascular and neurovascular health and disease: Pharmacological implications. Pharmacol. Rev. 69, 396–478. doi: 10.1124/pr.115.012062
Logviniuk, D., and Fridman, M. (2020). Serum prevents interactions between antimicrobial amphiphilic aminoglycosides and plasma membranes. ACS Infect. Dis. 6, 3212–3223. doi: 10.1021/acsinfecdis.0c00588
Ma, J.-W., Ji, D.-D., Li, Q.-Q., Zhang, T., and Luo, L. (2020). Inhibition of connexin 43 attenuates oxidative stress and apoptosis in human umbilical vein endothelial cells. BMC Pulm. Med. 20:19. doi: 10.1186/s12890-019-1036-y
Mattis, V., Wang, J., Rai, R., Chang, C.-W. T., and Lorson, C. L. (2006). Novel aminoglycosides increase SMN levels in spinal muscular atrophy fibroblasts. Hum. Genet. 120, 589–601. doi: 10.1007/s00439-006-0245-7
Mattis, V. B., Fosso, M. Y., Chang, C.-W. T., and Lorson, C. L. (2009). Subcutaneous administration of TC007 reduces disease severity in an animal model of SMA. BMC Neurosci. 10:142. doi: 10.1186/1471-2202-10-142
Meşe, G., Richard, G., and White, T. (2007). Gap junctions: Basic structure and function. J. Invest. Dermatol. 127, 2516–2524. doi: 10.1038/sj.jid.5700770
Mével, M., Sainlos, M., Chatin, B., Oudrhiri, N., Hauchecorne, M., Lambert, O., et al. (2012). Paromomycin and neomycin B derived cationic lipids: Synthesis. and transfection studies. J. Control. Release 158, 461–469. doi: 10.1016/j.jconrel.2011.12.019
Miller, G. H., Chiu, P. J. S., and Waitz, J. A. (1978). Biological activity of sch 21420, the 1-n-s-α-hydroxy-β-aminopropionyl derivative of gentamicin B. J. Antibiot. 31, 688–690. doi: 10.7164/antibiotics.31.688
Mingeot-Leclercq, M.-P., and Décout, J.-L. (2016). Bacterial lipid membranes as promising targets to fight antimicrobial resistance, molecular foundations and illustration through the renewal of aminoglycoside antibiotics and emergence of amphiphilic aminoglycosides. Med. Chem. Commun. 7, 586–611. doi: 10.1039/C5MD00503E
Nagabhushan, T. L., Cooper, A. B., Tsai, H., Daniels, P. J., and Miller, G. H. (1978). The syntheses and biological properties of l-n-(s-4-amino- 2-hydroxybutyryl)-gentamicin b and 1-n-(s-3-amino- 2-hydroxypropionyl)-gentamicin B. J. Antibiot. 31, 681–687. doi: 10.7164/antibiotics.31.681
Naus, C. C., and Laird, D. W. (2010). Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 10, 435–441. doi: 10.1038/nrc.2016.105
Nielsen, M. S., Axelsen, L. N., Sorgen, P. L., Verma, V., Delmar, M., and Holstein-Rathlou, N.-H. (2012). Gap junctions. Compr. Physiol 2, 1981–2035. doi: 10.1002/cphy.c110051
Prakoura, N., Kavvadas, P., and Chadjichristos, C. E. (2018). Connexin 43: A new therapeutic target against chronic kidney disease. Cell. Physiol. Biochem. 49:985. doi: 10.1159/000493230
Ramirez, M. S., and Tolmasky, M. E. (2010). Aminoglycoside modifying enzymes. Drug Resist. Updat. 13, 151–171. doi: 10.1016/j.drup.2010.08.003
Sainlos, M., Belmont, P., Vigneron, J.-P., Lehn, P., and Lehn, J.-M. (2003). Aminoglycoside-derived cationic lipids for gene transfection: Synthesis of kanamycin A derivatives. Eur. J. Org. Chem. 15, 2764–2774. doi: 10.1002/ejoc.200300164
Schepdael, A. V., Delcourt, J., Mulier, M., Busson, R., Verbist, L., Vanderhaeghe, H. V., et al. (1991). New derivatives of kanamycin B obtained by modifications and substitutions in position 6″. 1. Synthesis and microbiological evaluation. J. Med. Chem. 34, 1468–1475. doi: 10.1021/jm00108a035
Schulz, R., Görge, P. M., Görbe, A., Ferdinandy, P., Lampe, P. D., and Leybaert, L. (2015). Connexin 43 is an emerging therapeutic target in ischemia/reperfusion injury, cardioprotection and neuroprotection. Pharmacol. Ther. 153, 90–106. doi: 10.1016/j.pharmthera.2015.06.005
Shahane, G., Ding, W., Palaiokostas, M. A., Zevedo, H. S., and Orsi, M. (2019). Interaction of Antimicrobial Lipopeptides with Bacterial Lipid Bilayers. J. Membr. Biol. 252, 317–329. doi: 10.1007/s00232-019-00068-3
Shintani-Ishida, K., Uemura, K., and Yoshida, K.-I. (2007). Hemichannels in cardiomyocytes open transiently during ischemia and contribute to reperfusion injury following brief ischemia. Am. J. Physiol. Heart Circ. Physiol. 293:H1714–H1720. doi: 10.1152/AJPHEART.00022.2007
Shrestha, S., Chang, C.-W. T., Meissner, N., Oblad, J., Shrestha, J. P., Sorensen, K., et al. (2014). Antifungal amphiphilic aminoglycoside K20: Bioactivities and mechanism of action. Front. Microbiol. 5:671. doi: 10.3389/fmicb.2014.00671
Shrestha, S., Grilley, M., Fosso, M.-Y., Chang, C.-W. T., and Takemoto, J. Y. (2013). Membrane lipid-modulated mechanism of action and non-cytotoxicity of novel fungicide aminoglycoside FG08. PLoS One 8:e73843. doi: 10.1371/journal.pone.0073843
Shrestha, S. K., Fosso, M. Y., Green, K. D., and Garneau-Tsodikova, S. (2015a). Amphiphilic tobramycin analogues as antibacterial and antifungal agents. Antimicrob. Agents Chemother. 59, 4861–4869. doi: 10.1128/AAC.00229-15
Shrestha, S. K., Grilley, M., Anderson, T., Dhiman, C., Oblad, J., Chang, C.-W. T., et al. (2015b). In vitro antifungal synergy between amphiphilic aminoglycoside K20 and azoles against Candida species and Cryptococcus neoformans. Med. Mycol. 53, 837–844. doi: 10.1093/mmy/myv063
Slavi, N., Toychiev, A. H., Kosmidis, S., Ackert, J., Bloomfield, S. A., Wulff, H., et al. (2018). Suppression of connexin 43 phosphorylation promotes astrocyte survival and vascular regeneration in proliferative retinopathy. Proc. Natl. Acad. Sci. U.S.A. 115:E5934–E5943. doi: 10.1073/pnas.1803907115
Steinbuch, K. B., Benhamou, R. I., Levin, L., Stein, R., and Fridman, M. (2018). Increased degree of unsaturation in the lipid of antifungal cationic amphiphiles facilitates selective fungal cell disruption. ACS Infect. Dis. 4, 825–836. doi: 10.1021/acsinfecdis.7b00272
Subedi, Y. P., Alfindee, M., Takemoto, J. Y., and Chang, C.-W. T. (2018). Antifungal amphiphilic kanamycins: New life for an old drug. Med. Chem. Comm. 9, 909–919. doi: 10.1039/c8md00155c
Subedi, Y. P., Altenberg, G. A., and Chang, C.-W. T. (2021). Advances in the development of connexin hemichannel inhibitors selective toward Cx43. Futur. Med. Chem. 13, 379–392. doi: 10.4155/fmc-2020-0291
Subedi, Y. P., Kjellgren, A., Roberts, P., Montgomery, H., Thackeray, N., Fiori, M. C., et al. (2020a). Amphiphilic aminoglycosides with increased selectivity for inhibition of connexin 43 (Cx43) hemichannels. Eur. J. Med. Chem. 203:112602. doi: 10.1016/j.ejmech.2020.112602
Subedi, Y. P., Takemoto, J. Y., and Chang, C. W. T. (2020b). Scalable and cost-effective synthesis of antifungal amphiphilic kanamycin.
Subedi, Y. P., Pandey, U., Alfindee, M., Montgomery, H., Roberts, P., Wight, J., et al. (2019a). Scalable and cost-effective tosylation-mediated synthesis of antifungal and fungal diagnostic 6″-Modified amphiphilic kanamycins. Eur. J. Med. Chem. 182:111639. doi: 10.1016/j.ejmech.2019.111639
Subedi, Y. P., Roberts, P., Grilley, M., Takemoto, J. Y., and Chang, C.-W. T. (2019b). Development of fungal selective amphiphilic kanamycin: Cost effective synthesis and use of fluorescent analogs for mode of action investigation. ACS Infect. Dis. 5, 473–483. doi: 10.1021/acsinfecdis.8b00327
Szychowski, J., Kondo, J., Zahr, O., Auclair, K., Westhof, E., Hanessian, S., et al. (2011). Inhibition of aminoglycoside-deactivating enzymes aph(3′)-iiia and aac(6′)-ii by amphiphilic paromomycin o2″-ether analogues. ChemMedChem 6, 1961–1966. doi: 10.1002/cmdc.201100346
Takemoto, J. Y., and Chang, C. W. T. (2010). Investigation of antifungal activity of amphiphilic neomycin.
Takemoto, J. Y., Wegulo, S. N., Yuen, G. Y., Stevens, J. A., Jochum, C. C., Chang, C.-W. T., et al. (2018). Suppression of Wheat Fusarium Head Blight by Novel Amphiphilic Aminoglycoside Fungicide K20. Fungal Biol. 122, 465–470. doi: 10.1016/j.funbio.2017.12.001
Udumula, V., Ham, Y. W., Fosso, M. Y., Chen, K. Y., Rai, R., Zhang, J., et al. (2013). Investigation of antibacterial mode of action for traditional and amphiphilic aminoglycosides. Bioorg. Med. Chem. Lett. 23, 1671–1675. doi: 10.1016/j.bmcl.2013.01.073
Umezawa, H., and Hooper, I. R., (Eds.) (1982). Aminoglycoside Antibiotics. New York, NY: Springer-Verlag.
Weinstein, M. J., Luedemann, G. M., Oden, E. M., Wagman, G. H., Rosselet, J. P., Marquez, J. A., et al. (1963). Gentamicin, a new antibiotic complex from micromonospora. J. Med. Chem. 6, 463–464. doi: 10.1021/jm00340a034
Weinstein, M. J., Marquez, J. A., Testa, R. T., Wagman, G. H., Oden, E. M., and Waitz, J. A. (1970). Antibiotic 6640, a new micromonospora-produced aminoglycoside antibiotic. J. Antibiot. 23, 551–554. doi: 10.7164/antibiotics.23.551
Xie, J., Talaska, A. E., and Schacht, J. (2011). New developments in aminoglycoside therapy and ototoxicity. Hear. Res. 281, 28–37. doi: 10.1016/j.heares.2011.05.008
Xu, C.-Y., Zhang, W.-S., Zhang, H., Cao, Y., and Zhou, H.-Y. (2019). The Role of Connexin-43 in the Inflammatory Process: A New Potential Therapy to Influence Keratitis. J. Ophthalmol. 2019:9312827. doi: 10.1155/2019/9312827
Yang, L., Yan, J., Zhang, J.-A., Zhou, X.-H., Fang, C., Zeng, Er-M, et al. (2019). The important role of connexin 43 in subarachnoid hemorrhage-induced cerebral vasospasm. J. Transl. Med. 17:433. doi: 10.1186/s12967-019-02190-1
Zhang, J., Chiang, F.-I., Wu, L., Czyryca, G. P., Li, D., and Chang, C.-W. T. (2008). Surprising alteration of antibacterial activity of 5″-modified neomycin against resistant bacteria. J. Med. Chem. 51, 7563–7573. doi: 10.1021/jm800997s
Zhang, J., Keller, K., Takemoto, J. Y., Bensaci, M., Litke, A., Czyryca, P. G., et al. (2009). Synthesis and Combinational Antibacterial Study of 5″-Modified Neomycin. J. Antibiot. 62, 539–544. doi: 10.1038/ja.2009.66
Zimmermann, L., Das, I., Deìsireì, J., Sautrey, G., Vinicius Barros, R. S., Khoury, M. El, et al. (2016). New Broad-Spectrum Antibacterial Amphiphilic Aminoglycosides Active against Resistant Bacteria: From Neamine Derivatives to Smaller Neosamine Analogues. J. Med. Chem. 59, 9350–9369. doi: 10.1021/acs.jmedchem.6b00818
Keywords: amphiphilic aminoglycosides, kanamycin, neomycin, neamine, antibiotic resistance, connexin
Citation: Takemoto JY, Altenberg GA, Poudyal N, Subedi YP and Chang C-WT (2022) Amphiphilic aminoglycosides: Modifications that revive old natural product antibiotics. Front. Microbiol. 13:1000199. doi: 10.3389/fmicb.2022.1000199
Received: 21 July 2022; Accepted: 30 August 2022;
Published: 23 September 2022.
Edited by:
Jia Zeng, Thermo Fisher Scientific, United StatesReviewed by:
Daniel Balleza, Instituto Tecnológico de Veracruz, MexicoFrank Schweizer, University of Manitoba, Canada
Copyright © 2022 Takemoto, Altenberg, Poudyal, Subedi and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cheng-Wei T. Chang, dG9tLmNoYW5nQHVzdS5lZHU=