 Xueyan Sun1
Xueyan Sun1 Aimin Ma
Aimin Ma- 1College of Food Science and Technology, Huazhong Agricultural University, Wuhan, China
- 2Key Laboratory of Agro-Microbial Resources and Utilization, Ministry of Agriculture, Wuhan, China
Pleurotus tuber-regium is an edible and medicinal sclerotium-producing mushroom. The sclerotia of this mushroom also serve as food and folk medicine. Based on the description of its monokaryon genome, sequenced with Illumina and PacBio sequencing technologies, comparative transcriptomic analysis using RNA sequencing (RNA-seq) was employed to study its mechanism of sclerotium formation. The de novo assembled genome is 35.82 Mb in size with a N50 scaffold size of 4.29 Mb and encodes 12,173 putative proteins. Expression analysis demonstrated that 1,146 and 1,249 genes were upregulated and downregulated with the formation of sclerotia, respectively. The differentially expressed genes were associated with substrate decomposition, the oxidation-reduction process, cell wall synthesis, and other biological processes in P. tuber-regium. These genomic and transcriptomic resources provide useful information for the mechanism underlying sclerotium formation in P. tuber-regium.
Introduction
Sclerotia of certain fungal species have important medicinal values and are popular in Chinese traditional medicine. A well-known, nutritious, and medicinal mushroom, Pleurotus tuber-regium (Fr.) Singer, also known as the tiger milk mushroom, produces sclerotia that also have a variety of edible and medicinal values, including serving as components of pork sausage; agents for thickening and flavoring soups; and remedies for ailments, such as headache, stomach pain, fever, and cold (Oso, 1977; Akobundu and Eluchie, 1992; Ohiri, 2018). Because of the health-promoting potential, research involving the sclerotia of P. tuber-regium focuses on the nutritive values, bioactive compounds, and clinical potential (Isikhuemhen et al., 2005; Wong and Cheung, 2008; Ohiri, 2018). However, omics studies to assess the molecular mechanisms underlying sclerotium formation in P. tuber-regium are lacking.
With the development of next-generation sequencing technology (Ansorge, 2009), omics technology provides a valid and comprehensive method for us to widely study the genetic basis in several sclerotium-producing mushrooms, such as Wolfiporia cocos (Zhang et al., 2016), Lignosus rhinocerotis (Yap et al., 2014), Ophiocordyceps sinensis (Li et al., 2016), and Cordyceps guangdongensis (Zhang C. et al., 2018). Genome and transcriptome dissection offer powerful and effective approaches that result in the identification and characterization of diverse genes in mushrooms related to metabolites, growth and development, and response to environments (Yap et al., 2014; Kramer and Nodwell, 2017; Gupta et al., 2018).
To acquire abundant information to understand molecular biological and genetic information related to sclerotium formation in P. tuber-regium, its genome was sequenced with Illumina and PacBio sequencing platforms. The genome features and annotation and the genome-wide distribution of non-coding RNAs (ncRNAs) and repetitive elements were stated intuitively. Some studies reveal that a series of functional genes that participate in sclerotial development are differentially expressed at early stages of sclerotium formation (Xing et al., 2013; Zhang et al., 2016). Thus, the newly formed sclerotia and vegetative mycelia of P. tuber-regium were used in transcriptome sequencing to authentically provide information for identifying functional genes regulating sclerotium formation. These genome and transcriptome resources supply new insights to understand the genetic basis and gene functions associated with this sclerotium-producing mushroom.
Materials and Methods
Pleurotus tuber-regium Strains and Culture Conditions
The P. tuber-regium strain ACCC 50657 was kept in the Laboratory of Food Microbiology, Huazhong Agricultural University. Monokaryotic strains 2 and 18 were a pair of compatible single-spore isolates from the fruiting body of P. tuber-regium strain ACCC 50657, and dikaryotic strain 218 was obtained by pairing the two monokaryotic strains. These strains were maintained and subcultured on solid potato dextrose agar (PDA) medium (potato 200 g/L, glucose 20 g/L, and agar 20 g/L) at room temperature and 32°C, respectively.
To induce formation of sclerotia, culture bags were prepared that were composed of cottonseed shell, bran, gypsum, lime, and H2O. These 300-g sterilized culture bags were inoculated with each P. tuber-regium strain and incubated at 32°C for 15 d to ensure complete colonization. Then, each culture bag was buried in 2.5 L of nutrient soil using a plastic vase (approximately 5.5 L) with a rounded bottom and incubated at room temperature with proper humidity.
Genomic Analysis
Monokaryotic strain 18 was selected for genome sequencing. It was grown on PDA medium, covered with cellophane, and incubated at 32°C for 7 d in darkness. Genomic DNA was extracted using the CTAB extraction protocol (Cota-Sánchez et al., 2006). The DNA quality and quantity were determined using a Qubit dsDNA HS Assay Kit (Invitrogen, United States) and an Agilent Bioanalyzer 2100 (Agilent Technologies, United States).
After filtering low-quality reads, the genome survey was performed by counting the frequency of 17-mers from 5.7 Gb of data. Ten thousand randomly picked read pairs were blasted to the NCBI non-redundant nucleotide (nt) database to check for obvious sample contamination. Subsequently, 10 μg genomic DNA was used for 20 kb template library preparation using the BluePippin Size Selection system (Sage Science, United States) according to the manufacturer’s protocol (Pacific Biosciences, United States). The library was sequenced on the Pacific Biosciences Sequel platform. Assembly was performed using the Canu software with default parameters (Koren et al., 2017), and then, Pilon software was used to correct the preassembled structure according to NGS reads to obtain the final assembly result (Walker et al., 2014).
The protein-coding genes were annotated using predicted proteins to compare with seven major databases (NR, Swiss-Prot, KOG, KEGG, GO, Pfam, and TrEMBL), and the functional prediction of proteins was performed to combine the functional information of the proteins in the databases. Genes with putative CAZymes were annotated by BLASTP analysis according to the Carbohydrate Active Enzymes database1 (Cantarel et al., 2009).
There are two types of repetitive elements in the genome: tandem (e.g., microsatellites and minisatellites) and interspersed repeats (e.g., transposable elements or TEs). The presence of tandem repeats was investigated in all contigs using Tandem Repeats Finder (Benson, 1999). TE detection was performed with RepeatMasker2 based on the Repbase database3. The presence of snRNA and snoRNA was sought by aligning our genome assembly to the Rfam database4 using the cmscan program (Nawrocki and Eddy, 2013). tRNAs were analyzed using tRNAscan-SE (Chan and Lowe, 2019). rRNA was identified as combined Rfam with RNAMMER (Lagesen et al., 2007).
Transcriptomic Analysis
Mycelia of dikaryotic strain 218 were grown on 60-g culture substrate resting in 250-mL glass beakers that had been covered with sterilized polyethylene film and incubated at 32°C. The aerial mycelia that climbed onto the beaker walls (10–14 days) were harvested. The sclerotia were harvested when they reached about 1 cm in diameter.
Total RNA was extracted from each mycelia and sclerotia sample using Trizol reagent, according to the manufacturer’s instructions (TaKaRa, Japan). The quality and quantity of extracted RNA were measured using 1.5% RNAase-free agarose gel electrophoresis, a NanoDrop 2000 spectrophotometer (NanoDrop Technologies, United States), and a Bioanalyzer 2100 system (Agilent Technologies, United States). The construction of cDNA library and RNA-seq of the eligible six RNA samples were carried out by Frasergen, Inc (Wuhan, China) using Illumina HiSeq sequencing.
The sequenced reads were aligned to our sequenced P. tuber-regium genome using Hisat2 (Kim et al., 2015) with default parameters. The aligned records from the aligners in the BAM/SAM format were further checked to remove potential duplicate molecules. Subsequently, gene expression levels were estimated using FPKM values (fragments per kilobase of exon per million fragments mapped) by Cufflinks software (Trapnell et al., 2010).
The EdgeR (v3.6.8) package method was used to evaluate differential gene expression between mycelial and sclerotial samples (Robinson et al., 2010). False discovery rate (FDR) values < 0.05 and | log2Fold Change| ≥ 1 were used as thresholds to assess the significance of gene expression differences.
Functional analysis of the transcriptome genes was carried out using GO (Ashburner et al., 2000; Young et al., 2010) and KEGG pathways (Kanehisa et al., 2004).
Reverse transcription quantitative polymerase chain reaction (RT-qPCR) was employed to analyze the eight upregulated and eight downregulated candidate genes among transcriptome samples to verify the quality of RNA-seq results. The gene-specific primers used for RT-qPCR are shown in Supplementary Table 1. The reactions were executed in QuantStudio™ 6 Flex System (Thermo Fisher Scientific, United States) using the following parameters: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, and 60°C for 1 min, and the melt curve for each product was detected. Three biological and three technical replicates were performed for each sample using the combination of E3upl and Tif-5a as the internal reference genes (Sun et al., 2021). The log2Fold Change of sclerotia relative to mycelia was calculated using the 2–ΔΔCt method.
Results
Genome Features and Annotation
In total, the final genome assembly of P. tuber-regium monokaryotic strain 18 yielded a total size of 35.82 Mb, 65 contigs, 47.5% G + C content, and a contig N50 of 4.29 Mb. Approximately, 527 × coverage data were acquired. The monokaryon genome is bigger in size than the dikaryon genome of this previously sequenced mushroom using the Illumina platform (Lam et al., 2018). The genome sequenced in our study is similar in size to the genomes of several other Agaricales species, including L. rhinocerotis (34.31 Mb) (Yap et al., 2014) and P. ostreatus (34.9 Mb) (Qu et al., 2016) but smaller than that of W. cocos (50.48 Mb) (Floudas et al., 2012), P. tuoliensis (48.2 Mb), and P. eryngii (49.9 Mb) (Zhang Z. et al., 2018). The general statistics and view of the sequenced P. tuber-regium genome are shown in Table 1 and Figure 1, respectively.

Table 1. Genome statistics of P. tuber-regium.
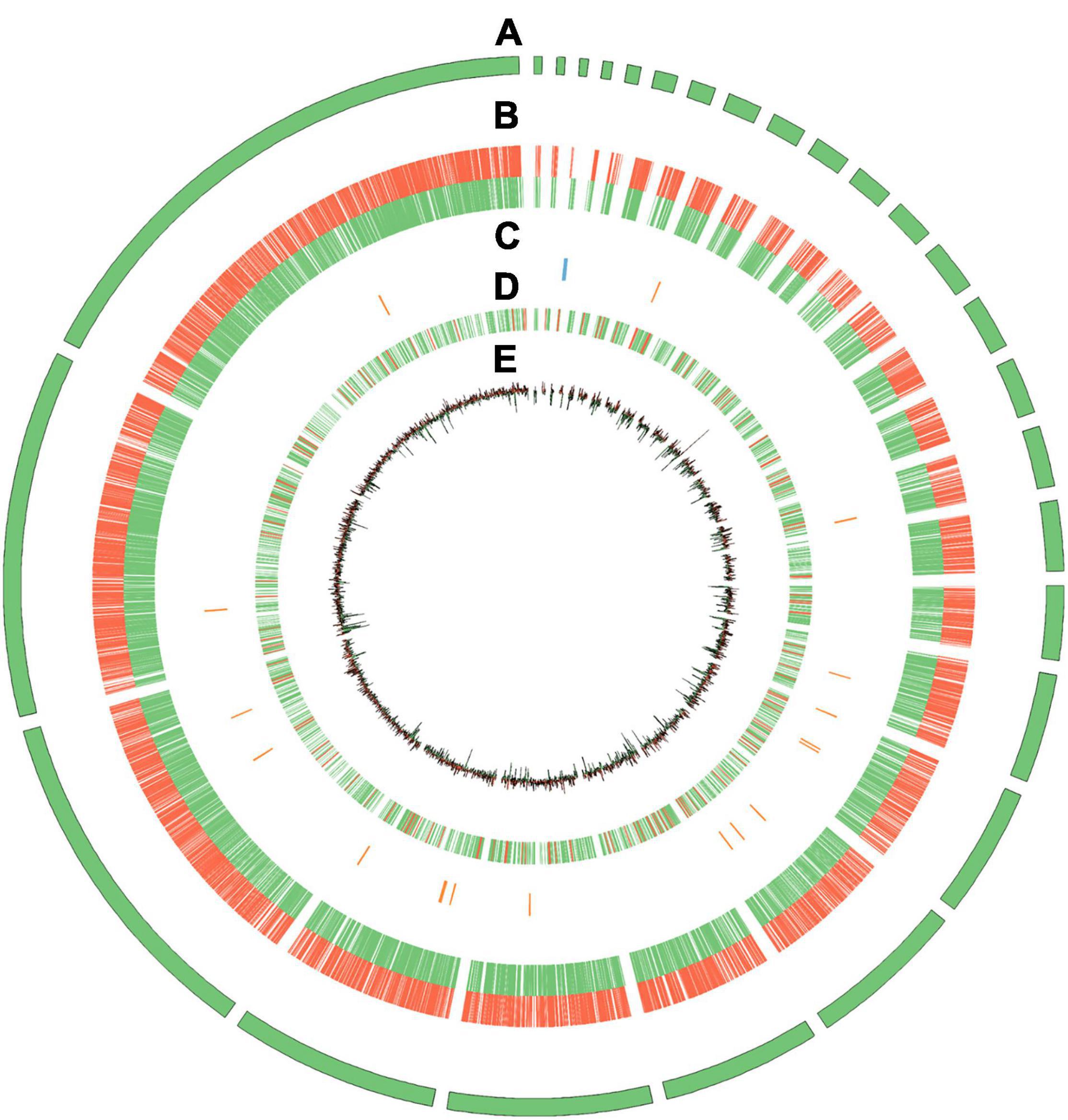
Figure 1. The general view of P. tuber-regium genome. (A) Contigs longer than 100 kb. (B) Gene density, represented as the number of genes in 100 kb non-overlapping windows. (C) Distribution of rRNA, tRNA, snRNA, and snoRNA. (D) Distribution of coverage of repetitive sequences. (E) GC content, increased in color from green to red.
A large portion of ncRNAs are functional and can produce various regulatory activities, such as signal molecules, ligands, and enzymes, and they are widely expressed in both prokaryotes and eukaryotes (Fu, 2014). The structural ncRNAs, including rRNA, tRNA, snRNA, and snoRNA, and regulatory ncRNAs were identified and mapped to the P. tuber-regium genome (Figure 1). Splicing catalysis by spliceosome is an essential step in the process of gene expression. The spliceosome is formed by the stepwise integration of five snRNPs composed of U1, U2, U4, U5, and U6 snRNAs and more than 150 proteins binding sequentially to pre-mRNA (Stark and Lührmann, 2006). Besides U1, the U2, U4, U5, and U6 snRNAs were also identified in our fungal strain. It is worth noting that U1 was also not identified in other basidiomycetous mushrooms, including P. ostreatus (Qu et al., 2016). With regard to tRNAs, 173 were annotated along with their loci, lengths, and transported amino acids. In addition, six snoRNAs, five 5s rRNAs, five 5.8s rRNAs, four 18s rRNAs, and four 28s rRNAs were identified in the genome.
Repetitive elements are essential for organismal development, including genome organization, evolution, epigenetic diversity, and plasticity (Foulongne-Oriol et al., 2013; Freire-Benéitez et al., 2016). Tandem repeats accounted for 1.13% of the genome assembly with an average length of 127 bp. The same as Agaricus bisporus, microsatellites appeared widely and distributed evenly over the whole genome sequence (Figure 1). The TEs, composed of Class I retrotransposons and Class II DNA transposons, accounted for 2.45% of the genome assembly with a total length of 876,249 bp. For class I, 339 sequences had homologies with long terminal repeats (LTRs), two sequences were observed having long interspersed nuclear elements (LINEs), and none had short interspersed elements (SINEs). For class II, 58 DNA transposons were identified in the genome assembly of P. tuber-regium.
Transcriptome Sequencing, Assembly, and Functional Annotation
To better elucidate the molecular mechanisms of sclerotium formation in P. tuber-regium, RNA-seq was performed to compare gene expression differences between its mycelia and newly formed sclerotia. After data filtering and trimming, high-quality reads ranging from 29 to 37 Mb (mycelia) and 23 to 31 Mb (sclerotia) were obtained from the six examined libraries (Table 2). The Q30 of the six samples was more than 92%, indicating that the overall sequencing quality was satisfactory, and the data were suitable for further assembly analysis. Hisat2 software mapped over 89.47% of our clean reads to the genome, which was used for further analysis (Table 2). Annotation and functional classification results of 9,815 genes were obtained using the eight data sets. A total of 9,781 (77%) genes associated with known proteins in the Nr database, followed by TrEMBL database (9,769: 77%), Pfam database (6,623: 52%), SwissProt database (5,318: 41%), KOG database (4,264: 33%), GO database (4,210: 33%), KEGG database (2,922: 23%), and COG database (2,746: 21%).
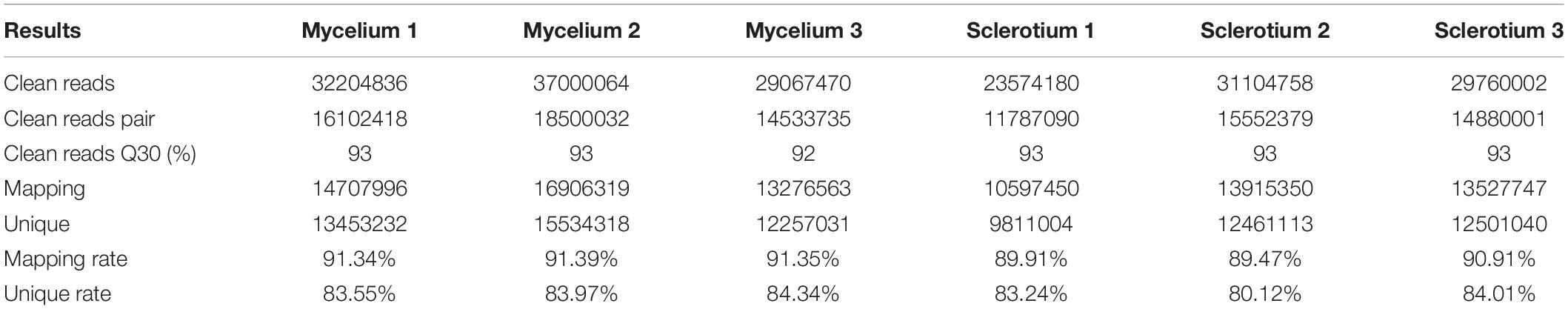
Table 2. Transcriptome output and assembly data of mycelia and sclerotia samples in triplicate.
According to DEG analysis, we found conspicuous differences in the gene expression levels between mycelia and newly formed sclerotia. We compared libraries for gene expression differences that were ≥ 2 or ≤ −2 and detected 1,146 upregulated genes and 1,249 downregulated genes in the newly formed sclerotia compared with mycelia. To verify the reliability of the transcriptomic data, RT-qPCR of 16 DEGs that had been observed through RNA-seq showed that our profiling of these DEGs were consistent with RNA-seq results (Figure 2).
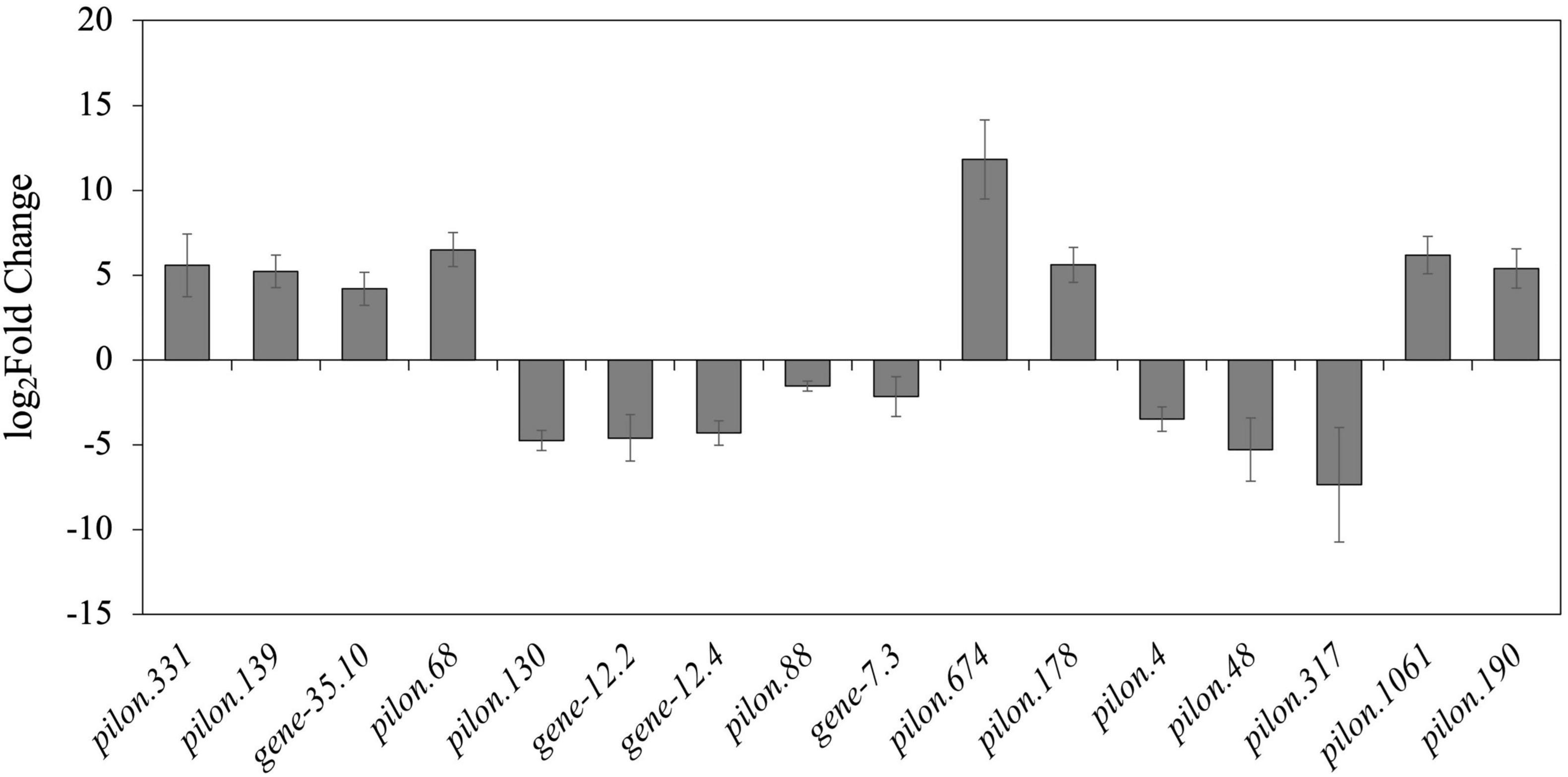
Figure 2. Bar chart based on RT-qPCR validation of observed expression levels for eight upregulated or downregulated DEGs. The log2Fold change values of the RT-qPCR assay were calculated by the 2–ΔΔCt method and shown with standard deviations from the mean.
GO enrichment was conducted for the genes obtained from each group of samples, which were classified into the three primary GO categories annotated to biological process (GO-BP), cellular component (GO-CC), and molecular function (GO-MF) (Figure 3A). As shown from the results of GO-BP analysis, the most highly enriched term was metabolic processes, followed by cellular processes, illustrating that these processes are of functional importance for mycelial growth and sclerotial development in P. tuber-regium. In the category of GO-CC, membrane and cell were significantly enriched in the transcriptome genes of mycelia and sclerotia. In the GO-MF category, the most highly enriched term was binding. The second highly enriched GO-MF category was catalytic activity. Overall, the GO terms for the transcriptome genes of mycelia and sclerotia, which included metabolic process, membrane, and catalytic activity might potentially account for the formation of sclerotia in P. tuber-regium.
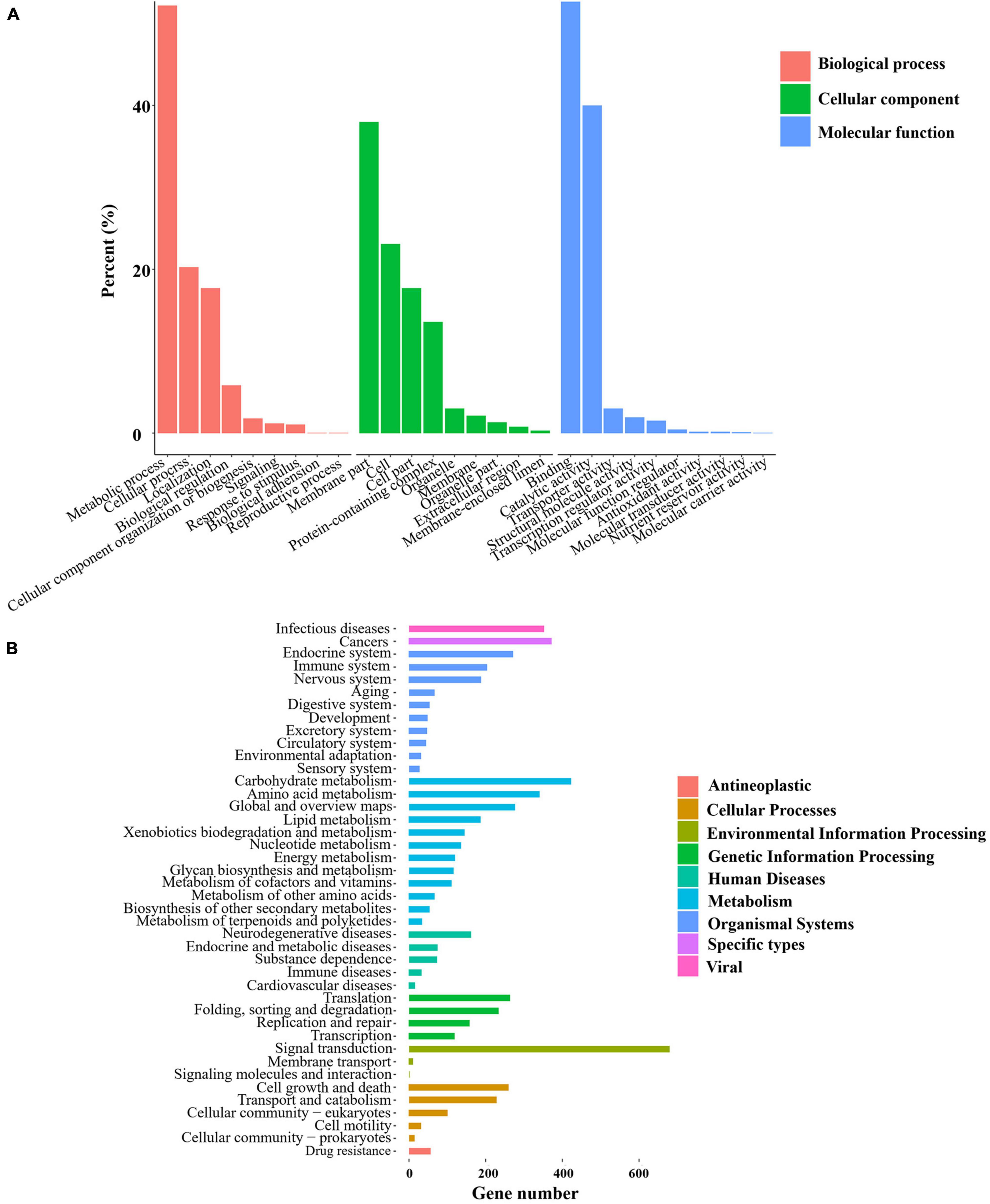
Figure 3. Statistics of GO and KEGG annotation results. (A) GO enrichment analysis of the P. tuber-regium transcriptome. The Y-axis indicates the percentage of a specific category. (B) KEGG enrichment analysis of pathways of the P. tuber-regium transcriptome. The X-axis indicates the number of genes in each function.
For better understanding of the functional pathways involved in sclerotia differentiation, we matched the transcriptome genes of mycelia and sclerotia to the KEGG database and displayed several metabolic and signal pathways (Figure 3B). We also conducted the KEGG enrichment analysis of DEGs, the KEGG results from our two groups of libraries revealed that DEGs listed in differentiation of mycelia to sclerotia were primarily enriched in the carbon metabolism; lysosome, cysteine, and methionine metabolism; and arginine and proline metabolism, etc (p < 0.05).
Differentially Expressed Genes Related to Sclerotium Formation
Some studies revealed that a series of metabolic process participated in sclerotium formation (Yap et al., 2015; Zhang et al., 2016). The analysis of these DEGs mainly focused on carbohydrate metabolism, oxidation-reduction process, and cell wall synthesis. In addition, some gene clusters speculated to participate in the sclerotia development were further analyzed, such as carbohydrate active enzymes (CAZymes), oxidoreductases, chitin synthases, and hydrophobins.
The differential expression of several genes in the CAZymes correlated well with our transcriptomic data. The CAZymes, including glycoside hydrolases (GHs), carbohydrate esterases (CEs), auxiliary activities (AAs), carbohydrate-binding modules (CBMs), glycosyl transferases (GTs), and polysaccharide lyases (PLs), are involved in carbohydrate metabolism. We analyzed the DEGs between mycelia and sclerotia and identified 76 and 97 CAZymes genes that were significantly upregulated and downregulated during the early stage of sclerotial development, respectively (Figure 4). Among these DEGs, more than half of them belonged to GH (33.53%) and AA (30.64%) families, indicating the importance of P. tuber-regium GHs and AAs families for sclerotium formation. In CAZymes, the CBMs encoded by maker-contig_6_pilon-exonerate_protein2genome-gene-13.1 and evm.TU.contig_17_pilon.35 were the most upregulated and downregulated in scletotia, respectively. It was reported that the CAZymes genes were not only involved in substrate decomposition, but also in sclerotium formation in W. cocos (Zhang et al., 2016). Our findings suggested that the CAZymes genes identified in P. tuber-regium might also participate in sclerotium formation.
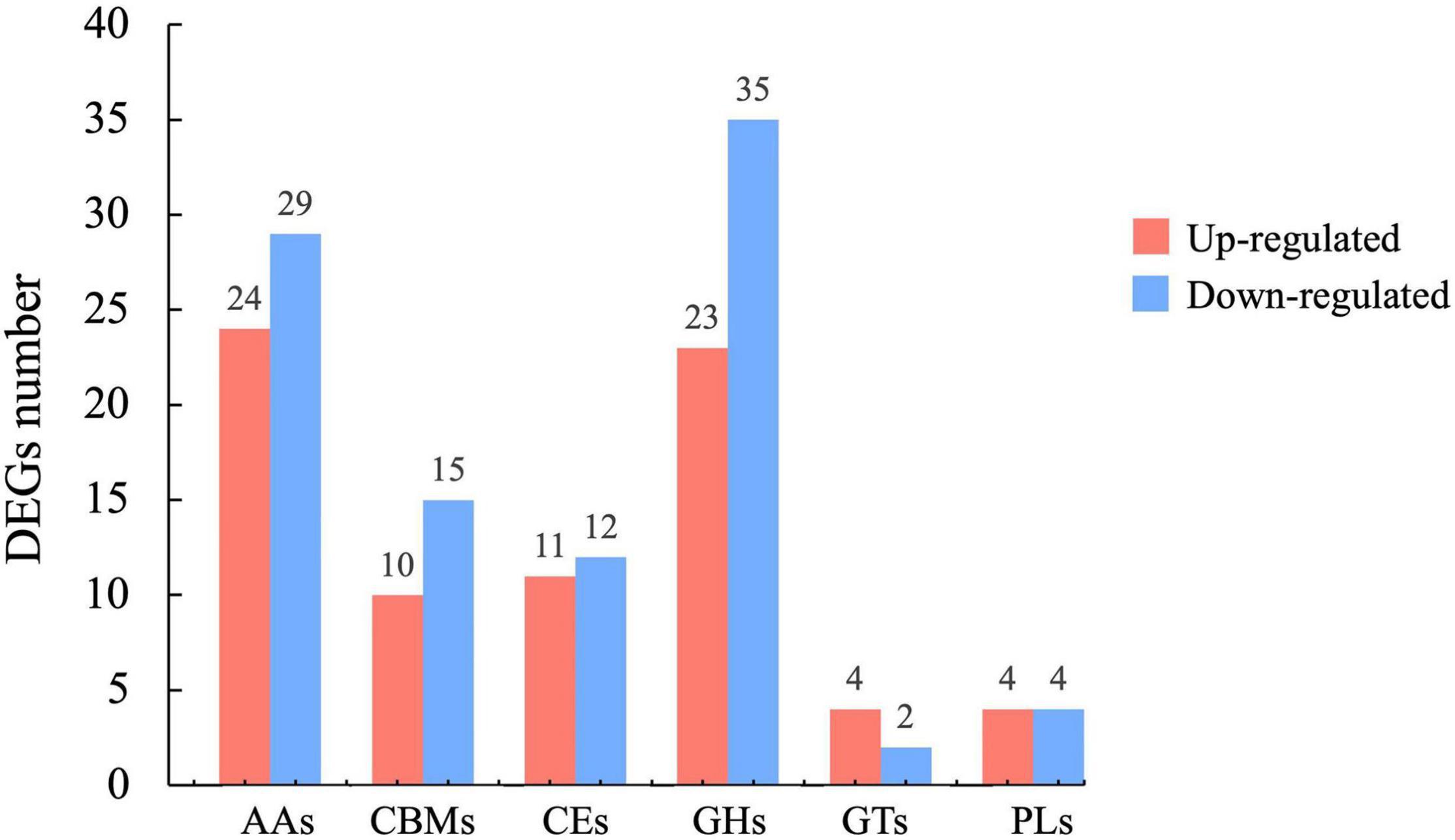
Figure 4. Histogram of upregulated (red bars) and downregulated (blue bars) CAZyme DEGs in newly formed sclerotia.
We analyzed the D-glucose, the monomer of glucan and 1, 3-ß-glucan related pathway derived from the starch and sucrose metabolism pathway in KEGG. The beta-glucosidase genes (evm.TU.contig_30_pilon.59 and evm.TU.contig_5_pilon.88) and glucan 1, 3-beta-glucosidase genes (evm.TU.contig_3_pilon.1871 and maker-contig_32_pilon-exonerate_protein2genome-gene-7.3) involved in the D-glucose pathway were downregulated with the exception of gene maker-contig_6_pilon-exonerate_protein2genome-gene-7.10. This may account for the fact that sclerotia may only have early stage growth needs and typically exist in relatively dormant states during other growth periods when requirements of energy for life cycle activities are reduced. Additionally, with the exception of evm.TU.contig_93_pilon.104, most of the chitinase genes (evm.TU.contig_13_pilon.236, evm.TU.contig_15_pilon.689, and evm.TU.contig_3_pilon.1375) were downregulated in our P. tuber-regium sclerotia.
Genes related to the oxidation-reduction process were differentially expressed during sclerotium formation in P. tuber-regium. Oxidation -reduction–related genes were differentially expressed in mycelium vs. sclerotium production in other mushroom-producing species, such as Polyporus umbellatus, W. cocos, and L. rhinocerotis (Song et al., 2014; Yap et al., 2015; Wu et al., 2016; Li et al., 2017). The molecular mechanisms controlling sclerotium formation might include oxidative stress, signal transduction, and gene expression regulation (Sun et al., 2020). In this study, 155 genes involved in oxidation-reduction progress were significantly and differentially expressed in GO-BP (Figure 5A). Among them, 31 Cytochrome P450 (CYP) monooxygenases, demonstrating differential transcriptional abundances, deserved further attention. CYP monooxygenases have the unique ability to catalyze regio-, chemo-, and stereospecific oxidation of a wide range of substrates under mild reaction conditions, thereby addressing a significant challenge in chemocatalysis (Durairaj et al., 2016). Among the DEGs, the genes (evm.TU.contig_17_pilon.68 and evm.TU.contig_17_pilon.69) encoding laccases were dramatically upregulated in sclerotia. Compared with the mycelia stage, the lipoxygenase encoded by evm.TU.contig_25_pilon.317 in the sclerotia decreased significantly.
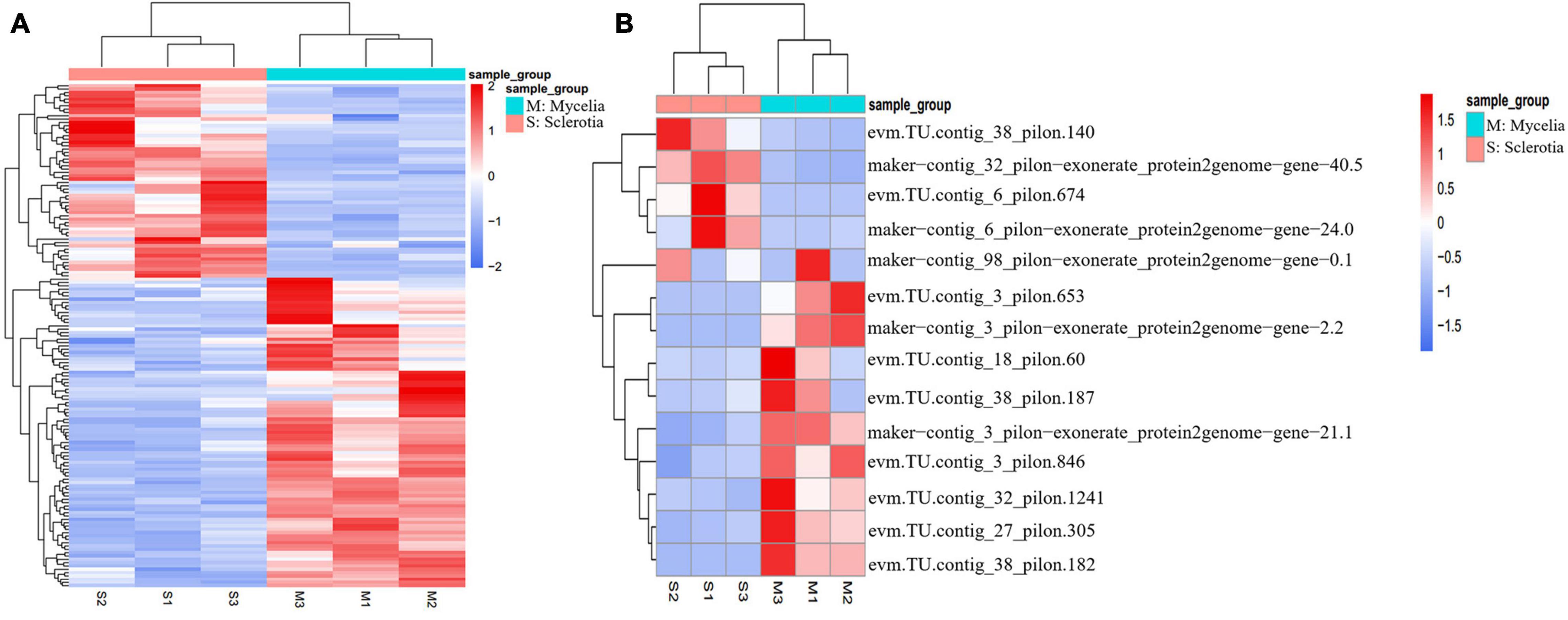
Figure 5. Heat map demonstration of expression patterns analysis in DEGs (p value < 0.05, | log2Fold Change| > 1) between sclerotia and mycelia using the FPKM. (A) Genes involved in oxidation-reduction progress show significant different expression in GO-BP. (B) Hydrophobins had more significant differential expression in sclerotia than in mycelia.
Differentially expressed genes involved in the cell wall system might participate in sclerotium formation in P. tuber-regium. Cell wall biogenesis is a very complicated process that incorporates not only multiple gene families directly involved in the polysaccharide biosynthesis and the remodeling of different cell wall components, but also structural proteins involved in cell wall assembly and rearrangement (Hu et al., 2017). We further analyzed genes with upregulated or downregulated expression in the formation of sclerotia that associated with GO cell composition terms related to “wall.” Notably, 16 cell wall–associated genes were screened, and 14 of them were hydrophobins (HPs), the structural constituents of cell walls. HPs are surface-active proteins unique in fungal cells and have roles in fungal growth as structural components and in the interaction of fungi with their environment (Linder et al., 2005; Kulkarni et al., 2017). In P. tuber-regium sclerotia, evm.TU.contig_38_pilon.140 had the highest expression abundance, with more than 50,000 FPKM (Figure 5B). In particular, expression level of HP gene evm.TU.contig_6_pilon.674 in sclerotia was hundreds of times greater than in mycelia. It is reported that HPs can affect the cell wall composition and protect emergent structures against adverse environmental conditions (Wetter et al., 2000; Wösten, 2001). We also observed an upregulated PII-type proteinase homologous to Lactococcus lactis subsp. Cremoris and a downregulated minor extracellular protease vpr homologous to Bacillus subtilis (strain 168) related to serine-type endopeptidase activity. Cell walls of sclerotia were thicker than those of mycelia in P. tuber-regium (Sun et al., 2020). This is likely because of the accumulation of cell wall components in addition to HPs, such as chitin, glycan, and xylan, as reported for P. umbellatus sclerotia (Li et al., 2021).
Discussion
The interpretation of monokaryon genome and transcriptome information permitted us to research the gene function and biogenesis of sclerotia in P. tuber-regium. Compared with the early published genome, the improvements in sequencing and analysis methodology were reflected in the lower contig numbers and higher N50 length of the monokaryon genome. In the current study, genome sequencing results demonstrated fewer scaffolds (65), more protein-coding genes (12,173), and a longer N50 size (4.29 Mb). In KOG analysis, the three highest KOG annotations in descending order were general function prediction only; posttranslational modification, protein turnover, chaperones; and signal transduction mechanisms. In terms of KEGG analysis, the pathway was primarily enriched in signal transduction followed by carbohydrate metabolism. In the analysis of CAZymes, more CAZymes genes (776) were acquired compared with the published genome. They were composed of 202 AAs, 82 CBMs, 103 CEs, 251 GHs, 110 GTs, and 28 PLs. In addition to the different sequencing and analysis methods, the reasons for these differences might also be caused by the different sources of P. tuber-regium strains and the differences in monokaryon and dikaryon strains. Dikaryon not only added a complete and different set of genomes, but also interfered with each other between the two sets of monokaryon genomes, which affected the accuracy of sequencing results. Monokaryon could reduce the complexity of genome analysis. Monokaryotic genome sequencing provided advantages for the analysis of genes related to sclerotium formation and other biological characteristics without duplications. In other mushrooms, monokaryons were also used for genome sequencing, such as Postia (Rhodonia) placenta, Dichomitus squalens, and Hericium erinaceus (Gaskell et al., 2017; López et al., 2019; Gong et al., 2020). The high-quality monokaryon genome sequences of P. tuber-regium ACCC 50657-18 were used in this study to mine genes involved in the sclerotium formation. For example, genes coding CAZymes, oxidoreductases, and hydrophobins were analyzed. The actual functions of these genes during sclerotium formation in P. tuber-regium is still unknown though they are shown to play important roles in other sclerotium-producing mushrooms.
Orthology is used to accurately describe differences and similarities in the composition of genomes among different species because orthologs share an ancestral gene that was present in a common ancestor of the compared species (Gabaldón and Koonin, 2013). Ortholog analysis was conducted for P. tuber-regium and the 22 other fungal species. A total of 50 single-copy orthologous genes were identified and were sequentially used to infer a phylogenetic tree (Figure 6) based on the neighbor-joining (NJ) method. Phylogenetic analysis revealed a close evolutionary relationship of P. tuber-regium to P. ostreatus and P. eryngii, supporting the classification of this fungus in the genus Pleurotus rather than Lentinus (Isikhuemhen et al., 2005). It should be noted that, although P. tuber-regium shares the same genus with P. ostreatus and P. eryngii, it is relatively distant from them and exhibits distinct physiological features, such as producing sclerotia. In addition, unlike other Pleurotus mushrooms, P. tuber-regium is a tropical mushroom and can grow in equatorial areas (Isikhuemhen et al., 2005; Wong and Cheung, 2008). P. tuber-regium also represents a unique intersterility group in the genus Pleurotus (Isikhuemhen et al., 2000).

Figure 6. Phylogenetic tree of P. tuber-regium demonstrating the evolutionary distance with other fungal species. Time scale along the bottom indicates millions of years ago. The inferred divergence time of fungal species is located on the left side of each node.
Although the ability of P. tuber-regium to produce sclerotia was well-known, the potential molecular mechanisms and related functional genes in sclerotium formation were unknown. In this study, sclerotium morphogenesis was researched using a cotton seed hull cultivation substrate assay and constructed two libraries to acquire the DEGs and pathways related to sclerotium formation using high-throughput RNA-seq.
Mitogen-activated protein kinases (MAPKs) are key mediators of signaling in fungi, participating in the response to diverse stresses and developmental processes (Liu D. et al., 2019). We screened MAPK signaling pathway - yeast genes and observed upregulated expression of a catalase gene (evm.TU.contig_3_pilon.1115) and downregulated expression of Pheromone B alpha 3 receptor (evm.TU.contig_25_pilon.509) and GTP-binding protein rhoA (evm.TU.contig_32_pilon.1268) genes, and so on. CAZymes degrade the culture matrix to make the material available to fungi. Sixty-nine CAZymes genes were significantly upregulated during the early stage of sclerotial development compared with that of mycelia in W. cocos (Zhang et al., 2016). The CAZyme transcripts showed significant differences at the three stages of sclerotium development in Morchella importuna (Liu W. et al., 2019). In terms of oxidative stress response, mutants generated by deletion of AflrsmA in Aspergillus flavus displayed less sensitivity to the oxidative reagent tert-Butyl hydroperoxide and decreased production of sclerotia (Wang et al., 2020). Deletion of a sclerotial regulator in A. niger, ansclR, increased its susceptibility to oxidative stress (Lv et al., 2015). Antioxidant activity associated with glycolysis was critical for sclerotia growth in P. umbellatus (Li et al., 2017). DEGs were also enriched in oxidative stress–related genes and pathways, so these genes might play key roles in sclerotial biogenesis in P. tuber-regium. As for cell wall proteins, they play vital roles in different morphological stages, including mycelium, fruiting body, and sclerotium production in mushrooms (Chen et al., 2012). Although the P. tuber-regium sclerotia cell wall had high levels of protein content, the number of its identified cell wall proteins was much lower than those found in mycelia or fruiting bodies (Chen et al., 2012). Additionally, the sclerotial cell wall was significantly thicker than that of the mycelial cell wall in P. tuber-regium (Sun et al., 2020). Thus, DEGs associated with cell wall proteins deserve further verification and functional research.
The vegetative mycelia and newly formed sclerotia of P. tuber-regium were subjected to transcriptome sequencing analysis and assembly with reference genome, resulting in the first comprehensive transcriptome analyses related to sclerotium formation in P. tuber-regium. We screened DEGs and pathways associated with the MAPK signaling pathway, carbohydrate catabolism, cell wall system, oxidative stress response, and so on. Further studies are necessary to individually test the DEGs related to sclerotia biogenesis to better assess their developmental roles. In summary, the elucidation of genome and transcriptome data of P. tuber-regium in this manuscript provide a new reference for further studies of sclerotium formation.
Accession Number
The genome sequence has been deposited at NCBI (accession number JACFYU000000000) with a BioProject number (PRJNA644065) and a BioSample number (SAMN15447141). The raw sequencing data of transcriptome were deposited at the NCBI Sequence Read Archive under Bioproject No. PRJNA801849.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
XS and AM designed the research. XS performed the experiments, analyzed the data, and wrote the manuscript. JW, SZ, LL, CM, LS, and AM critically reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC) (No. 31772375) to AM.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.815954/full#supplementary-material
Footnotes
- ^ http://www.cazy.org
- ^ http://repeatmasker.org
- ^ http://www.girinst.org/repbase/
- ^ http://rfam.xfam.org/
References
Akobundu, E. N. T., and Eluchie, G. U. (1992). Quality characteristics of pork sausage containing mushroom (Pleurotus tuberregium) and local spices. J. Food Technol. 29, 159–161. doi: 10.1007/s13197-019-03786-7
Ansorge, W. J. (2009). Next-generation DNA sequencing techniques. Nat. Biotechnol. 25, 195–203. doi: 10.1146/annurev.genom.9.081307.164359
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2009). The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 37, 233–238. doi: 10.1093/nar/gkn663
Chan, P. P., and Lowe, T. M. (2019). tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol. Biol. 1962, 1–14. doi: 10.1007/978-1-4939-9173-0_1
Chen, L., Zhang, B. B., and Cheung, P. C. K. (2012). Comparative Proteomic analysis of mushroom cell wall proteins among the different developmental stages of Pleurotus tuber-regium. J. Agric. Food Chem. 60, 6173–6182. doi: 10.1021/jf301198b
Cota-Sánchez, J. H., Remarchuk, K., and Ubayasena, K. (2006). Ready-to-use DNA extracted with a CTAB method adapted for herbarium specimens and mucilaginous plant tissue. Plant Mol. Biol. Rep. 24, 161–167. doi: 10.1007/BF02914055
Durairaj, P., Hur, J. S., and Yun, H. (2016). Versatile biocatalysis of fungal cytochrome P450 monooxygenases. Microb. Cell Fact. 15:125. doi: 10.1186/s12934-016-0523-6
Floudas, D., Binder, M., Riley, R., Barry, K., Blanchette, R. A., Henrissat, B., et al. (2012). The paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 336, 1715–1719. doi: 10.1126/science.1221748
Foulongne-Oriol, M., Murat, C., Castanera, R., Ramírezc, L., and Sonnenberg, A. S. M. (2013). Genome-wide survey of repetitive DNA elements in the button mushroom Agaricus bisporus. Fungal. Genet. Biol. 55, 6–21. doi: 10.1016/j.fgb.2013.04.003
Freire-Benéitez, V., Price, R. J., Tarrant, D., Berman, J., and Buscaino, A. (2016). Candida albicans repetitive elements display epigenetic diversity and plasticity. Sci. Rep. 6:22989. doi: 10.1038/sre22989
Fu, X. D. (2014). Non-coding RNA: a new frontier in regulatory biology. Natl. Sci. Rev. 1, 190–204. doi: 10.1093/nsr/nwu008
Gabaldón, T., and Koonin, E. V. (2013). Functional and evolutionary implications of gene orthology. Nat. Rev. Genet. 14, 360–366. doi: 10.1038/nrg3456
Gaskell, J., Kersten, P., Larrondo, L., Canessa, P., Martínez, D., Hibbett, D., et al. (2017). Draft genome sequence of a monokaryotic model brown-rot fungus Postia (Rhodonia) placenta SB12. Genom. Data 14, 21–23. doi: 10.1016/j.gdata.2017.08.003
Gong, W., Wang, Y., Xie, C., Zhou, Y., Zhu, Z., and Peng, Y. (2020). Whole genome sequence of an edible and medicinal mushroom, Hericium erinaceus (Basidiomycota, Fungi). Genomics 112, 2393–2399. doi: 10.1016/j.ygeno.2020.01.011
Gupta, D. K., Rühl, M., Mishra, B., Kleofas, V., Hofrichter, M., Herzog, R., et al. (2018). The genome sequence of the commercially cultivated mushroom Agrocybe aegerita reveals a conserved repertoire of fruiting related genes and a versatile suite of biopolymer-degrading enzymes. BMC Genomics 19:48. doi: 10.1186/s12864-017-4430-y
Hu, R., Xu, Y., Yu, C., He, K., Tang, Q., Jia, C., et al. (2017). Transcriptome analysis of genes involved in secondary cell wall biosynthesis in developing internodes of Miscanthus lutarioriparius. Sci. Rep. 7:9034. doi: 10.1038/s41598-017-08690-8
Isikhuemhen, O. S., Moncalvo, J. M., Nerud, F., and Vilgalys, R. (2000). Mating compatibility and phylogeography in Pleurotus tuberregium. Mycol. Res. 104, 732–737. doi: 10.1017/S0953756299001926
Isikhuemhen, O. S., Stamets, P. E., and Vilgalys, R. (2005). Biology, food, medicinal, and biotechnological applications of the tropical mushroom Pleurotus tuberregium (Rumph.:Fr.) Singer. Int. J. Med. Mushrooms 7, 415–416. doi: 10.1615/IntJMedMushrooms.v7.i3.580
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y., and Hattori, M. (2004). The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, D277–D280. doi: 10.1093/nar/gkh063
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kramer, J. L., and Nodwell, J. R. (2017). Chromosome level assembly and secondary metabolite potential of the parasitic fungus Cordyceps militaris. BMC Genomics 18:912. doi: 10.1186/s12864-017-4307-0
Kulkarni, S., Nene, S., and Joshi, K. (2017). Production of Hydrophobins from fungi. Process Biochem. 61, 1–11. doi: 10.1016/j.procbio.2017.06.012
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lam, K. L., Si, K., Wu, X., Tang, S., Sun, X., Kwan, H. S., et al. (2018). The diploid genome of the only sclerotia-forming wild-type species in the genus Pleurotus – Pleurotus tuber-regium – provides insights into the mechanism of its biomass conversion from lignocellulose substrates. J. Biotechnol. 283, 22–27. doi: 10.1016/j.jbiotec.2018.07.009
Li, B., Sun, L., Xing, Y. L. L., and Guo, S. (2021). Cell wall strengthening and remodeling provide new insights on sclerotia development of medicinal fungus Polyporus umbellatus. J. Appl. Microb. Res. 4, 34–41. doi: 10.21203/rs.3.rs-122799/v1
Li, B., Tian, X., Wang, C., Zeng, X., Xing, Y., Ling, H., et al. (2017). SWATH label-free proteomics analyses revealed the roles of oxidative stress and antioxidant defensing system in sclerotia formation of Polyporus umbellatus. Sci. Rep. 7:41283. doi: 10.1038/srep41283
Li, Y., Hsiang, T., Yang, R. H., Hu, X. D., Wang, K., Wang, W. G., et al. (2016). Comparison of different sequencing and assembly strategies for a repeat-rich fungal genome, Ophiocordyceps sinensis. J. Microbiol. Methods 128, 1–6. doi: 10.1016/j.mimet.2016.06.025
Linder, M. B., Szilvay, G. R., Nakari-Setälä, T., and Penttilä, M. (2005). Hydrophobins: the protein-amphiphiles of filamentous fungi. FEMS Microbiol. Rev. 29, 877–896. doi: 10.1016/j.femsre.2005.01.004
Liu, D., Pujiana, D., Wang, Y., Zhang, Z., Zheng, L., Chen, L., et al. (2019). Comparative transcriptomic analysis identified differentially expressed genes and pathways involved in the interaction between Tremella fuciformis and Annulohypoxylon stygium. Antonie van Leeuwenhoek 112, 1675–1689. doi: 10.1007/s10482-019-01294-4
Liu, W., Cai, Y., He, P., Chen, L., and Bian, Y. (2019). Comparative transcriptomics reveals potential genes involved in the vegetative growth of Morchella importuna. 3 Biotech 9:81. doi: 10.1007/s13205-019-1614-y
López, S. C., Peng, M., Daly, P., Andreopoulos, B., Pangilinan, J., Lipzen, A., et al. (2019). Draft genome sequences of three monokaryotic isolates of the white-rot basidiomycete fungus Dichomitus squalens. Microbiol. Resour. Announc. 8, e00264–19. doi: 10.1128/MRA.00264-19
Lv, Y., Zhou, F., Wang, B., and Pan, L. (2015). Morphological transitions under oxidative stress in response to metabolite formation in Aspergillus niger. Biotechnol. Lett. 37, 601–608. doi: 10.1007/s10529-014-1713-0
Nawrocki, E. P., and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Ohiri, R. C. (2018). Nutriceutical potential of Pleurotus tuber-regium sclerotium. Ukr. Biochem. J. 90, 84–93. doi: 10.15407/ubj90.03.084
Oso, B. A. (1977). Pleurotus tuber-regium from Nigeria. Mycologia 69, 271–279. doi: 10.1080/00275514.1977.12020058
Qu, J., Zhao, M., Hsiang, T., Feng, X., and Zhang, J. (2016). Identification and characterization of small noncoding RNAs in genome sequences of the edible fungus Pleurotus ostreatus. Biomed Res. Int. 2016:2503023. doi: 10.1155/2016/2503023
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 129–140. doi: 10.1093/bioinformatics/btp616
Song, C., Liu, M., Xing, Y., and Guo, S. (2014). ESTs analysis of putative genes engaged in Polyporus umbellatus sclerotial development. Int. J. Mol. Sci. 15, 15951–15962. doi: 10.3390/ijms150915951
Stark, H., and Lührmann, R. (2006). Cryo-electron microscopy of spliceosomal components. Annu. Rev. Biophys. Biomol. Struct. 35, 435–457. doi: 10.1146/annurev.biophys.35.040405.101953
Sun, X., Liu, D., Wang, Y., and Ma, A. (2020). Biogenesis of macrofungal sclerotia: influencing factors and molecular mechanisms. Appl. Microbiol. Biotechnol. 104, 4227–4234. doi: 10.1007/s00253-020-10545-8
Sun, X., Wu, J., Mo, C., and Ma, M. (2021). Selection and validation of endogenous reference genes for RT-qPCR normalization in different stresses and tissues of the tiger milk mushroom, Pleurotus tuber-regium. Mycoscience 62, 281–288. doi: 10.47371/mycosci.2021.05.001
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., Baren, M. J. V., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, X., Zha, W., Liang, L., Fasoyin, O. E., Wu, L., and Wang, S. (2020). The bZIP transcription factor AflRsmA regulates aflatoxin B1 biosynthesis, oxidative stress response and sclerotia formation in Aspergillus flavus. Toxins 12:271. doi: 10.3390/toxins12040271
Wetter, M. A. V., Wösten, H. A., Sietsma, J. H., and Wessels, J. G. (2000). Hydrophobin gene expression affects hyphal wall composition in Schizophyllum commune. Fungal Genet. Biol. 31, 99–104. doi: 10.1006/fgbi.2000.1231
Wong, K. H., and Cheung, P. C. K. (2008). Sclerotium of culinary-medicinal king tuber oyster mushroom, Pleurotus tuberregium (Fr.) Singer (Agaricomycetideae): its cultivation, biochemical composition, and biopharmacological effects (review). Int. J. Med. Mushrooms 10, 303–313. doi: 10.1615/IntJMedMushr.v10.i4.20
Wösten, H. A. (2001). Hydrophobins: multipurpose proteins. Annu. Rev. Microbiol. 55, 625–646. doi: 10.1146/annurev.micro.55.1.625
Wu, Y., Zhu, W., Wei, W., Zhao, X., Wang, Q., Zeng, W., et al. (2016). De novo assembly and transcriptome analysis of sclerotial development in Wolfiporia cocos. Gene 588, 149–155. doi: 10.1016/j.gene.2016.05.020
Xing, Y. M., Chen, J., Song, C., Liu, Y. Y., Guo, S. X., and Wang, C. L. (2013). Nox gene expression and cytochemical localization of hydrogen peroxide in Polyporus umbellatus sclerotial formation. Int. J. Mol. Sci. 14, 22967–22981. doi: 10.3390/ijms141122967
Yap, H. Y. Y., Chooi, Y. H., Firdaus-Raih, M., Fung, S. Y., Ng, S. T., Tan, C. S., et al. (2014). The genome of the Tiger Milk mushroom, Lignosus rhinocerotis, provides insights into the genetic basis of its medicinal properties. BMC Genomics 15:635. doi: 10.1186/1471-2164-15-635
Yap, H. Y. Y., Chooi, Y. H., Fung, S. Y., Ng, S. T., Tan, C. S., and Tan, N. H. (2015). Transcriptome analysis revealed highly expressed genes encoding secondary metabolite pathways and small cysteine-rich proteins in the sclerotium of Lignosus rhinocerotis. PLoS One 10:e0143549. doi: 10.1371/journal.pone.0143549
Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A. (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11:R14. doi: 10.1186/gb-2010-11-2-r14
Zhang, C., Deng, W., Yan, W., and Li, T. (2018). Whole genome sequence of an edible and potential medicinal fungus, Cordyceps guangdongensis. G3 Genes Genom. Genet. 8, 1863–1870. doi: 10.1101/254243
Zhang, Z., Wen, J., Li, J., Ma, X., Yu, Y., Tan, X., et al. (2018). The evolution of genomic and epigenomic features in two Pleurotus fungi. Sci. Rep. 8:8313. doi: 10.1038/s41598-018-26619-7
Zhang, S., Hu, B., Wei, W., Xiong, Y., Zhu, W., Peng, F., et al. (2016). De novo analysis of Wolfiporia cocos transcriptome to reveal the differentially expressed carbohydrate-active enzymes (CAZymes) genes during the early stage of sclerotial growth. Front. Microbiol. 7:83. doi: 10.3389/fmicb.2016.00083
Keywords: Pleurotus tuber-regium, whole genome sequencing, RNA-seq, molecular mechanisms, sclerotium formation
Citation: Sun X, Wu J, Zhang S, Luo L, Mo C, Sheng L and Ma A (2022) Genome and Comparative Transcriptome Dissection Provide Insights Into Molecular Mechanisms of Sclerotium Formation in Culinary-Medicinal Mushroom Pleurotus tuber-regium. Front. Microbiol. 12:815954. doi: 10.3389/fmicb.2021.815954
Received: 24 November 2021; Accepted: 28 December 2021;
Published: 17 February 2022.
Edited by:
Tofazzal Islam, Bangabandhu Sheikh Mujibur Rahman Agricultural University, BangladeshReviewed by:
Geromy G. Moore, Southern Regional Research Center (USDA-ARS), United StatesWei Guo, Chinese Academy of Agricultural Sciences (CAAS), China
Copyright © 2022 Sun, Wu, Zhang, Luo, Mo, Sheng and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aimin Ma, YWltaW5tYUBtYWlsLmh6YXUuZWR1LmNu