 Yongshu Wu
Yongshu Wu Zhidong Zhang
Zhidong Zhang Yanmin Li
Yanmin Li Yijing Li2
Yijing Li2
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol., 11 February 2022
Sec. Virology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.814635
This article is part of the Research TopicThe Viral Evasion of Antiviral Innate ImmunityView all 71 articles
The integrated stress response (ISR) is an adaptational signaling pathway induced in response to different stimuli, such as accumulation of unfolded and misfolded proteins, hypoxia, amino acid deprivation, viral infection, and ultraviolet light. It has been known that viral infection can activate the ISR, but the role of the ISR during viral infection is still unclear. In some cases, the ISR is a protective mechanism of host cells against viral infection, while viruses may hijack the ISR for facilitating their replication. This review highlighted recent advances on the induction of the ISR upon viral infection and the downstream responses, such as autophagy, apoptosis, formation of stress granules, and innate immunity response. We then discussed the molecular mechanism of the ISR regulating viral replication and how viruses antagonize this cellular stress response resulting from the ISR.
The integrated stress response (ISR) is an intricate signaling pathway in eukaryotic cells that is activated through the phosphorylation of eukaryotic translation initiation factor 2 alpha (eIF2α) in response to different physiological changes and pathological conditions. Activation of the ISR results in the decrease in global protein synthesis and induction of selected genes, such as activating transcriptional factor 4 (ATF4). It is speculated that the ISR’s ultimate destiny is determined by the intensity and duration of stress, the level of eIF2α phosphorylation, and the activation of ATF4 (Pakos-Zebrucka et al., 2016). A pro-survival effect is activated, and short-term stress reconstructs intracellular homeostasis. However, a cellular death program is initiated when cells are exposed to prolonged and severe stress (Harding et al., 2003; Pakos-Zebrucka et al., 2016). It has been well known that viral infection could induce the ISR, but the role of the ISR is still less defined. In some cases, the ISR is a protective mechanism against virus replication, while in other cases, the ISR may be hijacked by the virus to facilitate its replication. In this review, we summarized current knowledge of the molecular mechanism of the ISR with an emphasis on how cells initiate the ISR and the downstream cellular responses, how viral factors modulate the ISR, as well as cell prognosis upon viral infection.
In physiological conditions, eIF2 consisting of eIF2α, eIF2β, and eIF2γ possess phosphorylation and RNA binding sites. eIF2 forms a ternary complex with GTP and Met-tRNAi and then binds the 40S ribosome subunit, resulting in the formation of the 43S pre-initiation complex (PIC) with two small initiation factors (eIF1 and eIF1A) (Aitken and Lorsch, 2012; Lomakin and Steitz, 2013). The PIC is recruited to the 5′methylguanine cap of mRNA through the eIF4F complex, and the latter contains eIF4G and eIF4E. The PIC migrates to the AUG start codon and binds the Met-tRNAi anti-codon, facilitating protein synthesis. AUG recognition causes the arrest of the scanning PIC and triggers the conversion of the eIF2 GDP-bound state via gated phosphate (Pi) release and GTPase-activating (GAP) factor eIF5. The eIF2-GDP complex dissociates from the 40S ribosomal complex and transforms to GTP with the help of the eIF2B complex and enters another recycling of initiation of mRNA translation (Jackson et al., 2010; Hinnebusch and Lorsch, 2012). Under stress conditions, phosphorylated eIF2 can fully form an initiation-competent eIF2-TC, phosphorylated eIF2-GDP tightly binds to and sequesters the guanine nucleotide exchange factor eIF2B to abrogate its activity after its release, and most mRNA translation is reduced during eIF2α phosphorylation. However, translation from certain mRNAs with at least two upstream open-reading frames (uORFs) of appropriate type and position can be upregulated, such as ATF4, ATF5, and C/EBP-homologous protein (CHOP). Upregulation of ATF4, ATF5, and CHOP function activates chaperon and protease to promote cellular recovery or activate cellular death pathways under sustained stress.
Integrated stress response kinases act as an early responder in mammalian cells to restore cellular homeostasis upon different stimuli. There are four members of the ISR family: general control non-derepressible 2 (GCN2), PKR-like ER kinase (PERK), the heme-regulated inhibitor (HRI), and the interferon (IFN)-induced double-stranded RNA-dependent protein kinase (PKR). Each kinase can sense distinct stresses because each kinase possesses unique regulatory domains, although these kinases share homological catalytic domains (Donnelly et al., 2013; Lavoie et al., 2014). ISR kinases are activated in response to various stress stimuli, GCN2 is sensitive to amino acid starvation, PERK is induced by the accumulation of unfolded or misfolded proteins in the endoplasmic reticulum (ER), HRI is activated in response to heme deficiency, and PKR is activated by double-stranded RNA (dsRNA) (Lussignol et al., 2013). A schematic diagram of the protein structure of mammalian four eIF2α kinases is shown in Figure 1.
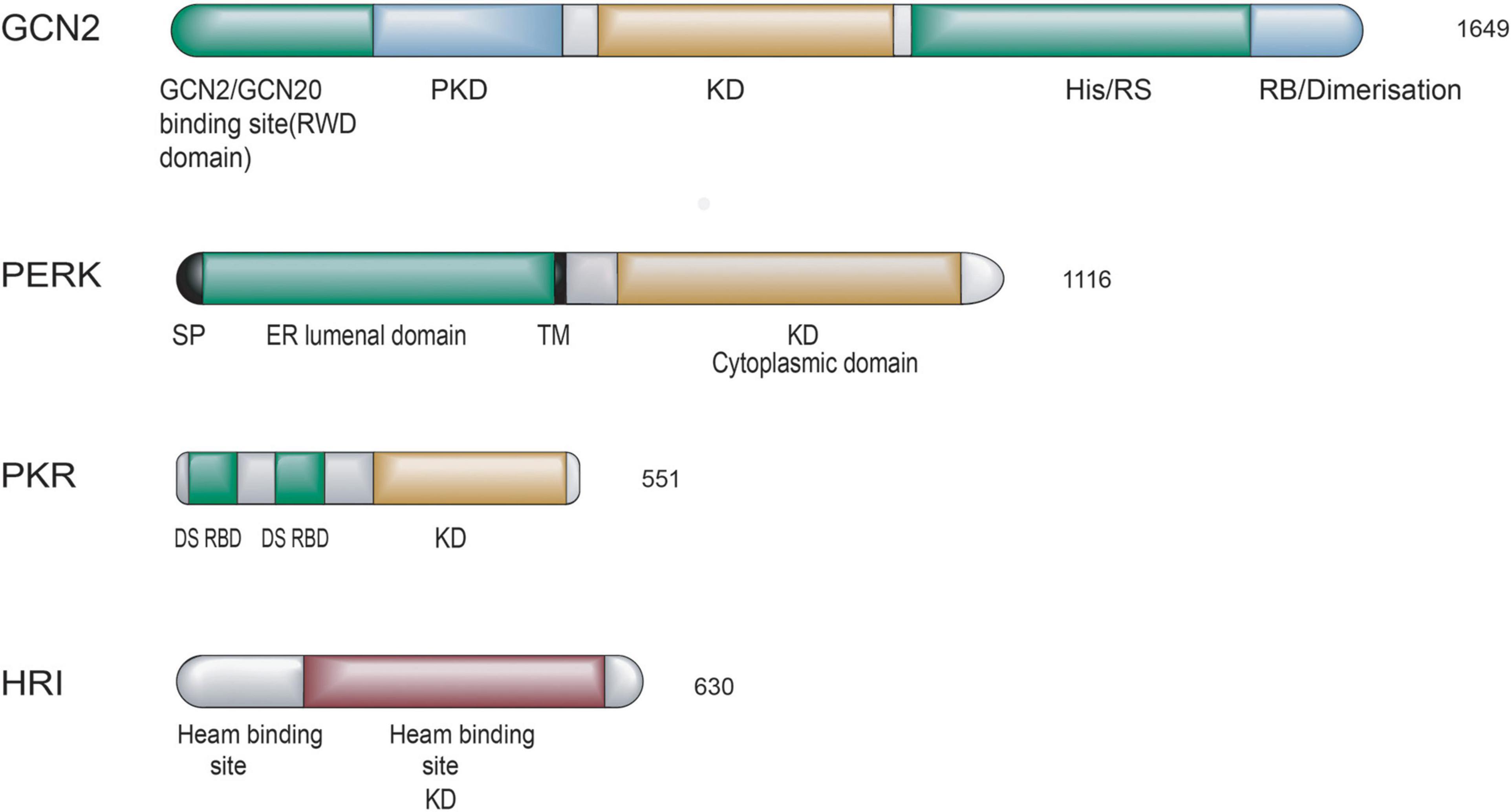
Figure 1. Schematic diagram of the domain organization of the four mammalian eIF2α kinases. Polypeptides are boxes running from N- to C-terminal domains from left to right. Length in amino acids is of proteins. The abbreviations of domains are listed: SP, signal peptide; TM, transmembrane domain; KD, kinase domain; DS RBD, double-stranded RNA binding domain; PKD, pseudokinase domain; His/Rs, histidyl-tRNA synthetase-related domain; RB, ribosome binding. Domains involved in sensing stress signals/activation are in green and black. Kinase domains are yellow and brown, other domains are colored blue, and domains are drawn to scale.
Dephosphorylation of eIF2α is a terminal signal of the ISR, and cells return to normal protein translation. The process is mediated by growth arrest and DNA damage-inducible protein (GADD34) that is a constitutive repressor of eIF2α phosphorylation, which interacts with protein phosphatase 1 (PP1) to restore protein synthesis and normal cellular function. GADD34 is a downstream production of eIF2α phosphorylation and ATF4, so GADD34 plays a pivotal role as a negative feedback loop in attenuating ISR signaling (Novoa et al., 2001; Pakos-Zebrucka et al., 2016).
Infection with many viruses can activate the ISR signaling pathway via eIF2α kinases. The ISR is generally triggered by virulent or pathogenic viruses instead of inactivated viruses, suggesting that ISR activation is associated with viral replication (Neerukonda et al., 2018). The ISR is mainly induced through PERK/PKR kinases upon single-stranded positive-sense RNA virus infection, and eIF2α phosphorylation inhibits overall protein translation, including viral replication. Furthermore, activation of the ISR initiates downstream signaling such as autophagy, formation of stress granules (SGs), apoptosis, and innate immune response to restore cellular homeostasis. In contrast, viruses can modulate ISR signaling to promote viral replication, activate the ISR signaling pathway, and control viruses upon viral infection, which are summarized in Table 1.
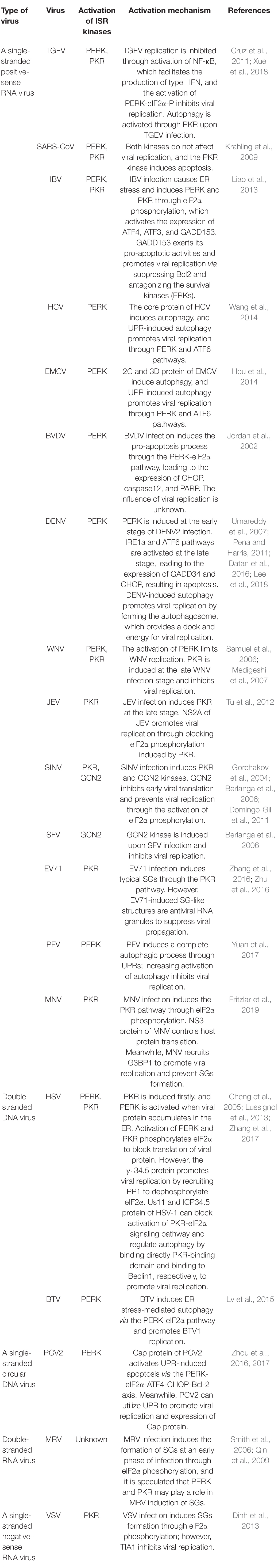
Table 1. Activation mechanism of ISR signaling pathway upon viral infection.
To date, there is no evidence that the HRI can be activated with virus infection in mammal cells. The HRI of Epinephelus coioides (EcHRI), a homolog gene in fish, is changed at the transcription level upon red-spotted grouper nervous necrosis virus (RGNNV) infection and inhibits viral replication through upregulating the expression of IFN-related cytokines, which indicates the potential role of the HRI in antiviral response (Zang et al., 2019).
The study of GCN2 against RNA viruses is not very common. GCN2 was specifically induced through eIF2α phosphorylation at an early stage of sindbis virus (SINV) infection, two non-adjacent regions of SINV genomics bonded to the histidyl-tRNA synthetase-related domain of GCN2 during this process, and GCN2 blocks early viral replication of SINV through eIF2α phosphorylation (Berlanga et al., 2006; Krishnamoorthy et al., 2008).
Autophagy is a conserved cellular lysosomal degradation process and is very important for cell survival and homeostasis. Many studies showed that the activation of unfolded protein response (UPR) regulates autophagy and controls viral replication during viral infection, although the functions between UPR and autophagy are independent. ER transmembrane receptors initiate UPRs: ATF6, inositol-requiring enzyme1 (IRE1), and PERK, a member of ISR kinases (Zhang et al., 2017). PERK-eIF2α-ATF4-ATG12 and IRE1α-JNK-Beclin1 signaling pathways were induced through autophagy to promote viral replication during dengue virus2 (DENV2) infection, IRE1α-JNK released Beclin1 via Bcl-2 phosphorylation, which triggered autophagic activity, and the PERK-eIF2α-ATF4-ATG12 signaling pathway partly had an effect on autophagy at the early stage of DENV infection (Lee et al., 2018). Another report showed that PERK participated in DENV-induced autophagy to enhance viral replication by forming autophagosomes in dog madin-darby canine kidney (MDCK) and mouse embryonic fibroblasts (MEFs), which provides a dock and energy for viral replication (Datan et al., 2016). This phenomenon is common in other members of the family Flaviviruses, such as hepatitis C virus (HCV), Japanese encephalitis virus (JEV), and West Nile virus (WNV) (Fukasawa, 2010; Syed et al., 2010; Martin-Acebes et al., 2011), which indicates that DENV and other members of the family Flaviviruses are ER-tropic viruses that accomplish translation, replication, and package in the ER.
Other viruses also induced autophagy through UPRs to enhance viral replication. Bluetongue virus (BTV) infection induced autophagy through the PERK-eIF2α-ATF4 pathway facilitates viral replication (Lv et al., 2015). Similarly, the duck enteritis virus (DEV) activated autophagy to benefit its replication through the PERK-eIF2α-ATF4 and IRE1-XBP1 signaling pathways (Yin et al., 2017). Autophagy was induced during Newcastle disease virus (NDV) infection promoting viral replication, and P and NP proteins of NDV induced autophagy via PERK and ATF6 pathways (Cheng et al., 2016). The core protein of HCV induced complete autophagy, and CHOP played a vital role in UPR-induced autophagy signaling (Ke and Chen, 2011). In addition, UPRs associated autophagy has been found to promote viral replication through PERK-eIF2α-ATF4 and ATF6 signaling pathways, and the activation of ATF4 and CHOP through PERK enhanced the expression of ATG12 and LC3B, which benefits the autophagic process (Tardif et al., 2002; Pavio et al., 2003; Shrivastava et al., 2012). 2C and 3C proteins of EMCV infection induced autophagy through PERK and ATF6 pathways facilitating viral replication (Zhang et al., 2011; Hou et al., 2014). Autophagy was induced via ER stress during coxsackievirus (CV) B3 infection, and three branches of UPRs participated in regulating autophagy (Luo et al., 2018). Capsid protein VP2 of the foot-and-mouth disease virus (FMDV) induced autophagy through the eIF2α-ATF4-AKT-MTOR signaling pathway and enhanced FMDV replication by VP2 protein interacting with heat shock protein family B small member1 (HSPB1) in mammalian cells. However, which kinase participates in remains unknown (Sun et al., 2018). These results suggest that viral infection can induce autophagy through UPRs to promote viral replication.
However, prototype foamy virus (PFV) infection induced a complete autophagic process through ER stress containing PERK, IRE1, and ATF6 branches and increased the activation of autophagy to inhibit PFV replication, which implies that PFV-induced autophagy has a novel mechanism and plays an antiviral role in viral replication (Yuan et al., 2017).
Altogether, we concluded that UPRs-induced autophagy facilitates viral replication excepting PFV infection, particularly the PERK-eIF2α pathway. It is speculated that viral proteins may alter ER member morphology and induce the ISR at the early stage of viral infection, phosphorylated eIF2α blocks cellular protein translation, including viral proteins, viral proteins that accumulate in the ER induced prolonged ER stress with a persistent viral infection, and viruses or viral proteins may suppress ER stress-induced cell death via modulating UPRs to promote autophagic activity and provide the replication platform and ATP energy for viral synthesis.
PKR was induced by dsRNA through eIF2α phosphorylation and was also required for viral-induced autophagy (Garcia et al., 2007; Lussignol et al., 2013). Autophagy was activated through PKR-eIF2α signaling during herpes simplex virues1 (HSV-1) infection, but the Us11 protein of HSV-1 blocked the activation of PKR-eIF2α signaling by binding directly to the PKR-binding domain (Talloczy et al., 2002; Lussignol et al., 2013), and the ICP34.5 protein of HSV-1 also regulated autophagy through the dephosphorylation of eIF2α and binding to Beclin1 to promote viral replication (Talloczy et al., 2006; Alexander et al., 2007; Zhu and Zheng, 2020). Viral infection activated autophagy through the ISR, and the viral protein hijacking this process is summarized in Figure 2 (red color).
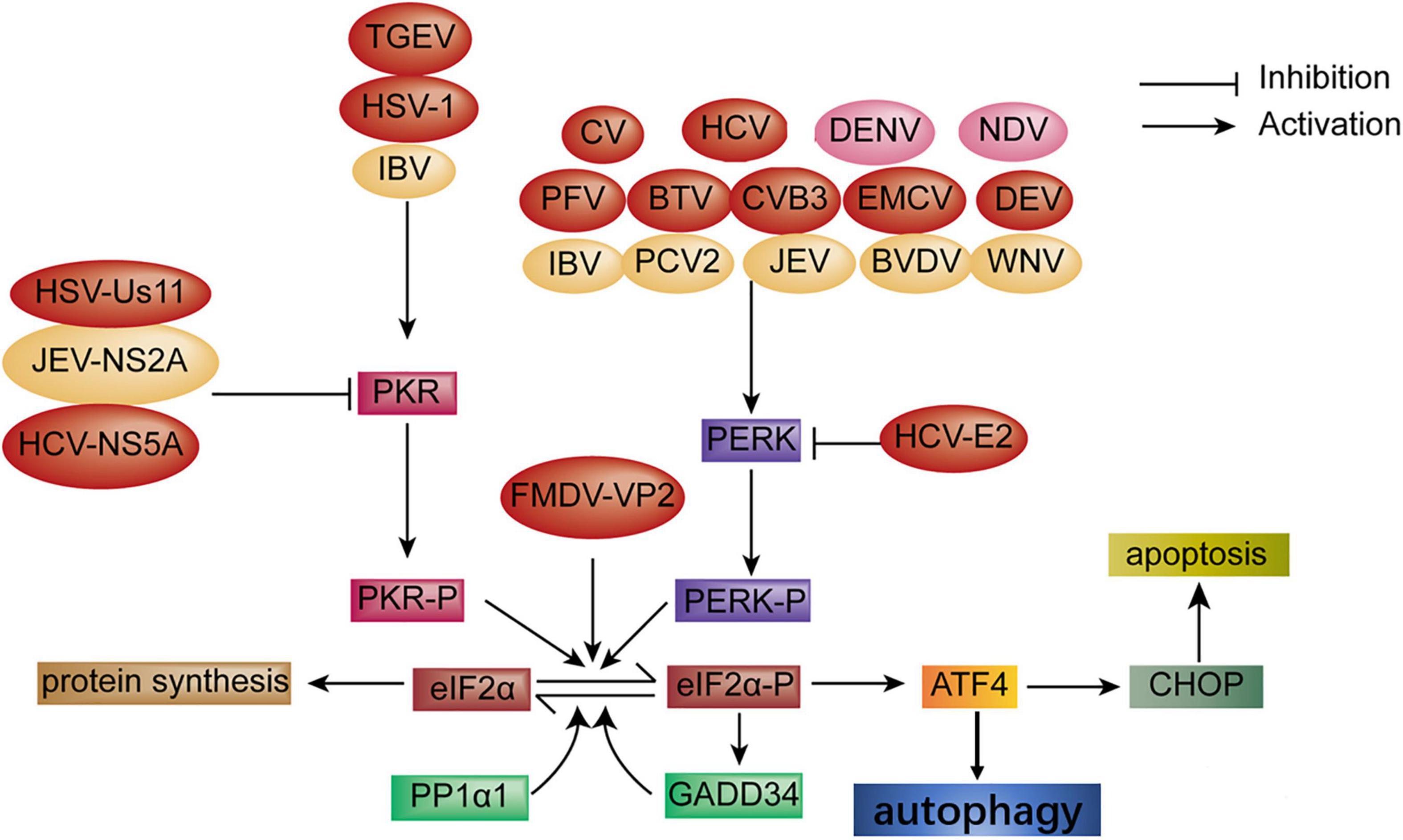
Figure 2. Diagram of the activation of autophagy and apoptosis via the ISR signaling pathway during viral infection. Autophagy: Autophagy is activated through the PKR-eIF2α pathway upon infection with transmissible gastroenteritis virus (TGEV) and HSV-1 infection; CV, HCV, PFV, BTV, CVB3, encephalomyocarditis virus (EMCV) and DEV infection, respectively, induces autophagy through the PERK-eIF2α pathway. FMDV-VP2 induces autophagy through interaction with HSPB1 and activation of the eIF2α-ATF4 pathway. In turn, HSV-Us11, HCV-NS5A, and HCV-E2 protein block autophagy (red color). Apoptosis: Apoptosis is induced via the PERK and PKR-eIF2α pathway under infection with IBV, PCV2, BVDV, JEV, and WNV, respectively. Oppositely, the JEV-NS2A protein inhibits apoptosis (yellow color). In addition, autophagy and apoptosis simultaneously are induced through the PERK-eIF2α pathway during DENV and NDV infection, respectively (pink color).
Apoptosis, a programming cell death, is a hosting strategy to combat viral infection. The PERK and PKR-eIF2α-ATF4 pathway was activated at the early stage of infectious bronchitis virus (IBV) infection in Vero cells and H1299 cells, which results in the expression of ATF4, ATF3, and growth arrest and DNA damage-inducible153 (GADD153), which is also known as CHOP. Activation of GADD153 induced the ER stress-mediated pro-apoptotic pathway through suppressing Bcl2 and antagonizing the survival kinases (ERKs) that induce tribbles homolog3 (TRIB3) (Liao et al., 2013). However, studies have shown that ER stressor IRE1α was activated in IBV-infected cells and serves as a survival factor during coronavirus infection (Wang et al., 2009; Liao et al., 2013; Fung et al., 2014). HCV triggered apoptosis through the induction of GADD153 and ER calcium depletion (Benali-Furet et al., 2005; Chan and Egan, 2005; Ciccaglione et al., 2005, 2007). JEV infection triggered UPR and apoptosis through GADD153 and p38 kinase expression. However, which branch was induced remains unknown (Su et al., 2002). The Cap protein of porcine circovirus2 (PCV2) induced UPR, resulting in apoptosis through the PERK-eIF2α-ATF4-CHOP-Bcl-2 signaling pathway, which reduces Bcl2 expression and increases caspase3 to enhance viral replication (Zhou et al., 2016, 2017). Some viruses of the family Flaviviridae also induced apoptosis via induction of a pro-apoptosis response through the PERK-eIF2α pathway, which leads to the expression of CHOP, caspase 12, and poly ADP ribose polymerase (PARP) and the downregulation of Bcl2, such as the NS protein of WNV, bovine viral diarrhea (BVDV), and DENV infection (Jordan et al., 2002; Medigeshi et al., 2007; Umareddy et al., 2007; Pena and Harris, 2011). Three branches of UPRs were involved in NDV-induced apoptosis. Meanwhile, CHOP was initiated by PERK/PKR-eIF2α signaling via downregulating BCL-2/MCL-1 to support NDV proliferation (Li et al., 2019). It is speculated that the virus may utilize translational blocking caused by PERK/PKR-eIF2α signaling for the preferential synthesis of viral proteins. Hence, CHOP may serve as a pro-apoptosis or pro-survival function depending on the condition of stress. PERK/PKR signaling pathways were induced in response to severe acute respiratory syndrome-coronavirus (SARS-CoV) infection, leading to sustained eIF2α phosphorylation, which did not inhibit viral replication indicating that SARS-CoV overcame the inhibition of eIF2α phosphorylation through a new mechanism. Furthermore, the activation of PKR induced apoptosis independent of eIF2α phosphorylation (Krahling et al., 2009). UPR-induced apoptosis is summarized in Figure 2 (yellow color).
Stress granules are formed by cytoplasmic non-membrane structures of mRNA-binding proteins (mRNPs) and related proteins in response to stress stimuli. It has been proposed that ISR kinases initiate the formation of SGs through eIF2α phosphorylation. However, SGs formation were also independent of eIF2α phosphorylation, such as the disruption of eIF4A helicase by Pateamine A treatment and the eIF4F complex by H2O2 treatment, which implies that the composition and assembly of SGs differ from that in a stress-dependent manner (Low et al., 2005; Anderson and Kedersha, 2006; Emara et al., 2012). SGs were formed in response to various stresses in mammalian cells, including oxidative stress, energy depletion, UV irradiation, hypoxia, ER stress, and viral infection (Anderson and Kedersha, 2006; Emara et al., 2012; Onomoto et al., 2012).
The virus requires cellular translation machinery to synthesize its proteins in host cells. However, SGs formation results from global translation repression of mRNAs, including the block of viral gene expression during viral infection. Thus, SGs formation may play a role in innate immune response (McCormick and Khaperskyy, 2017). Moreover, the virus also takes measures to confront these adverse conditions and maximizes replication efficiency by inhibiting SGs formation and disrupting processing bodies (PBs) assembly (Zhang et al., 2019). Therefore, the illumination of the relationship between SGs and viruses is very important to understand the interaction of the host and viruses.
The PKR branch of the ISR was activated through eIF2α phosphorylation during murine norovirus (MNV) infection, causing a stoppage of protein translation except the viral replication because MNV can suppress the formation of SGs via cytokine translation to promote viral replication, and MNV recruited SGs nucleating protein G3BP1 to enhance viral replication and prevent SGs formation, suggesting that MNV promotes viral replication through the inhibition of SGs formation and evades innate immune response (Fritzlar et al., 2019). Enteroviruses71 (EV71) infection induced the formation of typical SGs (tSGs) via the PKR-eIF2α pathway. SGs-like structures were also induced, a different canonical SGs and an antiviral structure to suppress EV71 propagation (Zhu et al., 2016). However, 2Apro of EV71 blocked tSGs formation and transformed from tSGs to atypical SGs (aSGs) through cleaving eIF4GI. 2Apro regulated SGs formation, common in picornaviruses (Ye et al., 2018). Several studies demonstrated that the composition of SGs was different from FMDV Lpro, but the exact composition of aSGs remained unclear (White et al., 2007; Zhang et al., 2016; Zhu et al., 2016).
Non-structural protein1-deficient influenza A virus (IAV-NS1–/–)-induced cytoplasmic granules are termed antiviral stress granules (avSGs), different from the canonical SGs. IAV-NS1 infection inhibited the formation of avSGs and production of IFN through the PKR-eIF2α signaling pathway (Khaperskyy et al., 2012). SINV, encephalomyocarditis virus (EMCV), Adenovirus, HCV, and NDV also triggered avSGs, implying that avSGs may play an important role in detecting viruses to initiate antiviral signaling. The NS1 protein of IAV blocked the formation of SGs via the activation of IFN genes (Onomoto et al., 2012).
Encephalomyocarditis virus was able to transiently induce SGs formation through the PKR signaling at the early stage of infection. However, the 3C protein of EMCV was found to inhibit SGs formation via cleaving G3BP1 at the late stage of infection. Similarly, the 3C protein of poliovirus (PV) and the L protein of Theiler’s murine encephalomyelitis virus (TMEV) could inhibit SGs formation (White et al., 2007; Borghese and Michiels, 2011; Ng et al., 2013). These findings indicate that picornaviruses also use the same strategy to evade the immune response by targeting G3BP1, which is essential for the efficient induction of IFN-β.
Hepatitis C virus infection triggered SGs formation via the PKR-eIF2α-P signaling pathway (Garaigorta et al., 2012). In addition, SGs formation was induced through the PKR-P-eIF2α-SGs pathway with respiratory syncytial virus (RSV), vaccinia virus (VV), measles virus (MeV) and human immunodeficiency virus (HIV), C protein-deficient Sendai virus (SeV), tick-borne encephalitis virus (TBEV), SINV, EV71, and PV infection (Takeuchi et al., 2008; Heinicke et al., 2009; Venticinque and Meruelo, 2010; Lindquist et al., 2011; Simpson-Holley et al., 2011; Okonski and Samuel, 2013; Yang et al., 2018). It is suggested that the PKR-P-eIF2α-SGs pathway is essential for SGs formation. SGs formation was activated through eIF2α phosphorylation upon reovirus infection. However, which kinase induced this process remains unknown (Smith et al., 2006). Hence, PKR kinase is mainly involved in SGs formation during viral infection, and the formation of SGs plays an important role in antiviral defense and restoring cell homeostasis.
Recent studies have demonstrated that some viruses induced the formation of SGs at the early stage of viral infection but inhibited the formation of SGs at later stages by blocking the phosphorylation of eIF2α or cleaving SGs scaffold proteins like G3BP1; other viruses inhibited the formation of SGs by altering from SGs proteins to atypical granules to promote viral replication (Raaben et al., 2007; Montero et al., 2008; Qin et al., 2009; Abrahamyan et al., 2010; Rojas et al., 2010; Lindquist et al., 2011; Linero et al., 2011; Ruggieri et al., 2012), such as HCV, RSV, rotavirus, mammalian orthoreovirus (MRV), and mouse hepatitis coronavirus (MHV). Furthermore, SGs formation was induced or inhibited at a different stage of a viral replication cycle or via different signaling pathways, such as Semliki Forest virus (SFV), HCV, and RSV (Poblete-Duran et al., 2016), which indicates that it is a game process between SGs formation and antagonism of the virus. The summary of SGs formation through eIF2α phosphorylation during viral infection is shown in Figure 3.
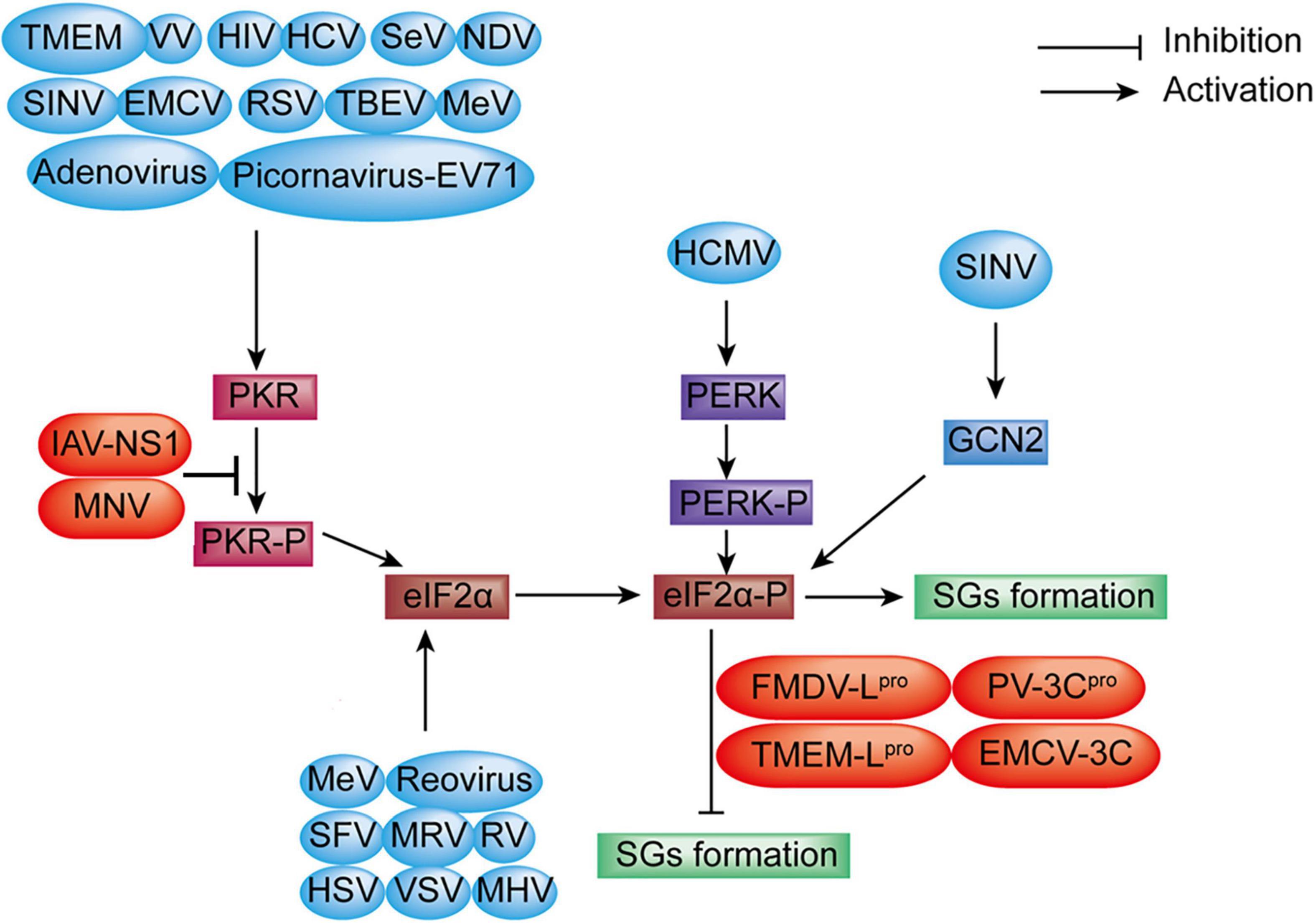
Figure 3. Diagram of SGs formation under viral infection. SGs formation: EV71, VV, HIV, HCV, SeV, EMCV, RSV, TBEV, MeV, TMEV, Adenovirus, NDV, and SINV infection induce SGs formation through the PKR-eIF2α-P signaling pathway. MeV, Reovirus, SFV, rotavirus (RV), HSV, VSV, MHV, and MRV benefit SGs formation via direct eIF2α phosphorylation (blue color). However, MNV, nervous necrosis virus (NNV) infection, and the expression of IAV-NS1, FMDV-Lpro, PV-3Cpro, TMEMV-Lpro, and EMCV-3C inhibit SGs formation through blocking eIF2α phosphorylation (red color). SGs formation is increased via the PERK-eIF2α-P signaling during human cytomegalovirus (HCMV) infection. SINV infection enhances SGs formation through the GCN2-eIF2α-P signaling pathway.
Apart from the importance of the ISR on controlling cellular homeostasis, ISR kinases also play a vital role in innate immunity during viral infection, which is thought to function as an antiviral pathway (McCormick and Khaperskyy, 2017; Ma et al., 2018). ISR kinases block overall protein translation through eIF2α phosphorylation, including viral protein. Hence, this process is an antiviral response, and PKR plays an important role in this process because it can directly recruit the formation of SGs. Thus, SGs formation is also an innate immune response to viral infection (McCormick and Khaperskyy, 2017). Transmissible gastroenteritis virus (TGEV) infection was found to activate all three UPRs through the upregulation of GRP78, but the PERK-eIF2α branch mainly suppressed viral replication through inducing IFN-I production and eIF2α phosphorylation-mediated global attenuation of protein translation during TGEV infection in vitro and in vivo (Xue et al., 2018), which suggests the key role of the PERK-eIF2α-P branch of the ISR in innate immunity. However, another study showed that TGEV infection could only induce the PKR-eIF2α signaling pathway (Cruz et al., 2011). This discrepancy was still unclear, but it might be due to the different TGEV strains used in those two studies. GCN2 also plays a novel role in the antiviral response to certain RNA viruses. GCN2 blocked the translation of viral proteins and further prevented the replication of SINV through eIF2α phosphorylation (Berlanga et al., 2006). The antiviral response of the ISR is summarized in Figure 4.
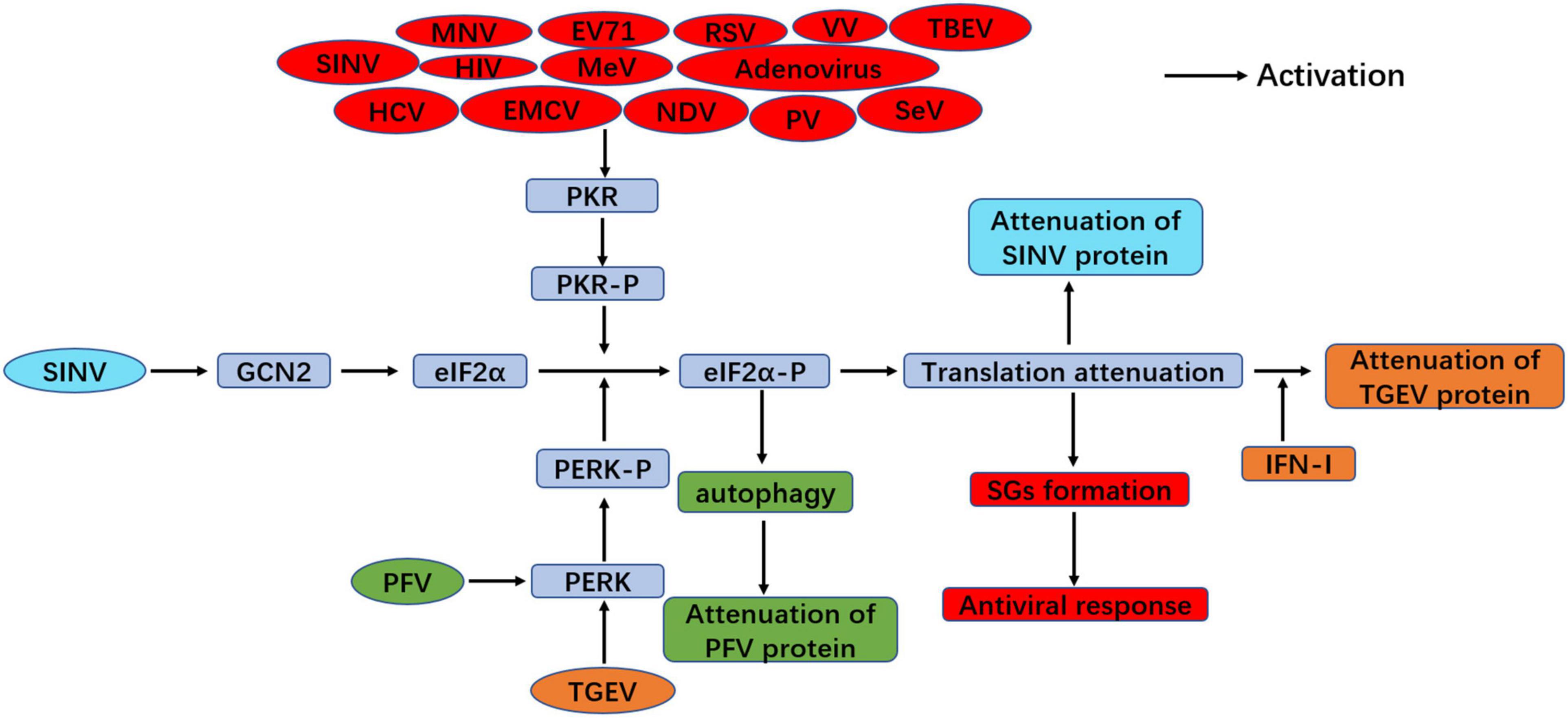
Figure 4. Antiviral response of the ISR during viral infection. PERK-eIF2α signaling suppresses viral replication through inducing IFN-I production and eIF2α phosphorylation-mediated translation attenuation with TGEV infection (orange color). PFV infection inhibits viral replication through PERK-mediated autophagy (green color). In addition, viral infection induces SGs formation through PKR-eIF2α phosphorylation and plays antiviral response, such as MNV, EV71, RSV, VV, TBEV, SINV, HIV, MeV, Adenovirus, HCV, EMCV, NDV, PV and SeV (red color). GCN2-eIF2α signaling inhibit viral replication upon SINV infection (blue color).
The ISR signaling pathway is activated with a different viral infection. Meanwhile, viruses develop different mechanisms to manipulate the ISR signaling pathway to promote viral translation and persistence during viral infection (Ambrose and Mackenzie, 2013b; Green et al., 2014). To facilitate replication, proteins encoded by viruses regulate the ISR pathway selectively and enhance ER protein folding capacity and metabolic regulation of cells. The M protein of vesicular stomatitis virus (VSV) is responsible for counteracting the antiviral response of eIF2α phosphorylation (Connor and Lyles, 2005). The NS5A protein and E2 protein of HCV were found to interfere with PKR and PERK kinase, respectively, which leads to the inhibition of downstream eIF2α phosphorylation and helps viral replication (Pavio et al., 2003; Jheng et al., 2014), whereas the NS2A protein of JEV was found to inhibit PKR-induced eIF2α phosphorylation (Tu et al., 2012; Jheng et al., 2014). IAV-NS1 limited eIF2α phosphorylation through hampering PKR dimerization and autophosphorylation (Lu et al., 1995). Upon DENV infection, PERK-induced eIF2α phosphorylation is suppressed through upregulating the expression of GADD34, which interacts with PP1 to dephosphorylate eIF2α (Pena and Harris, 2011). The protein 7 of TGEV and the M of VSV antagonize eIF2α phosphorylation during viral infection (Connor and Lyles, 2005; Cruz et al., 2011). PERK/PKR was induced through eIF2α phosphorylation at the early stage of HSV infection. However, the γ134.5 protein of HSV inhibited PERK phosphorylation for promoting viral replication. Meanwhile, the expression of GADD34 bonded in an eIF2α-independent mechanism to PP1 to dephosphorylate eIF2α. It is speculated that the γ134.5 protein may recruit PP1 to dephosphorylate eIF-2α and antagonize the activities of both PKR and PERK (Cheng et al., 2005; Zhang et al., 2017). PKR was induced through eIF2α phosphorylation upon MNV infection. At the same time, the expression of IFN-α, IFN-β, and IL-6 was suppressed through eIF2α phosphorylation to promote MNV replication, which is also an immune evasion strategy (Fritzlar et al., 2019).
Generally, viruses hijack cellular protein synthesis mechanisms for the synthesis of viral proteins and disrupt ER homeostasis (Fung et al., 2015). Meanwhile, viruses also develop mechanisms that manipulate the host ISR signaling pathway to promote viral translation and persistence during viral infection (Ambrose and Mackenzie, 2013a; Green et al., 2014).
The ISR is induced through eIF2α phosphorylation by different stresses, leading to the inhibition of overall protein translation and the preferential transcription of targeting genes to restore cellular homeostasis. The ISR was firstly observed in 2002. Recently, the ISR has been concerning because it is a hub for many signaling pathways that converge on eIF2α phosphorylation, which initiates downstream signalings, such as autophagy, apoptosis, SGs formation, cell homeostasis, and innate immunity response.
PERK, an ER kinase, is activated through eIF2α phosphorylation with the accumulation of misfolded and unfolded proteins in the ER. The PERK-eIF2α pathway plays an important role in regulating viral replication via UPR-induced autophagy. Other branches of UPRs are also involved, indicating that the synthesis of viral proteins is an ER tropism. We find that UPR-induced autophagy mainly promotes viral replication excepting PFV infection. It is speculated that autophagy has a dual role in regulating viral replication, a survival process is too short to be detected, and a cellular death program is a primary response during viral infection.
PKR plays a vital role in virus-induced SGs formation. It inhibits viral replication through eIF2α phosphorylation and provides a platform to promote the production of the IFN gene (Ruggieri et al., 2012; Albornoz et al., 2014), which means that the formation of SGs is an antiviral response. However, the leader protein of EMCV can inhibit IFN gene activation (Borghese and Michiels, 2011), and some viruses can disturb the formation of SGs (Ng et al., 2013). Altogether, the formation SGs is mainly an antiviral response and provides a platform to inhibit viral replication through the PKR-eIF2α pathway, although the virus confronts this process.
Apoptosis occurs through ISR signaling pathways during viral infection. CHOP-mediated apoptosis is activated through the PERK/PKR-eIF2α signaling pathway for supporting viral replication, which is a complicated process, and other mechanisms are also involved. It is speculated that viruses confront translational shut-off resulting from eIF2α phosphorylation and allow themselves to translate preferentially. However, the role of UPR-mediated apoptosis in viral prognosis needs further elaboration.
Recently, emerging evidence showed that the ISR, autophagy, and apoptosis are induced simultaneously during viral infection (Chiramel et al., 2013; Jheng et al., 2014). It was reported that complete autophagy could be induced during HCV infection. Meanwhile, CHOP played a pivotal role in the ISR-induced apoptosis (Ke and Chen, 2011). The core protein of HCV activated autophagy through PERK-eIF2α-ATF4 and ATF6 pathways to facilitate the expression of ATG12 and LC3 via the activation of ATF4 and CHOP (Wang et al., 2014). Hence, the ISR is a complicated and integrated signaling response to different stimuli.
On the contrary, viruses take different strategies for promoting the synthesis of viral proteins. For example, the M protein of VSV can counteract antiviral response. Both Chikungunya virus (CHIKV) and VSV antagonize eIF2α phosphorylation (Connor and Lyles, 2005; Rathore et al., 2013). In addition, some viruses switch translation mode from an eIF2-dependent to an eIF2-independent process to ensure efficient replication, such as PV and enterovirus (EV) (de Breyne et al., 2008; Redondo et al., 2011). It was reported that DENV infection inhibits PERK-mediated eIF2α phosphorylation by elevating the expression of GADD34, which interacts with PP1 to dephosphorylate eIF2α (Pena and Harris, 2011).
As discussed above, the ISR is a complicated, integrated, and adaptational response, and eIF2α phosphorylation is the core of the ISR and blocks overall protein translation to restore cellular homeostasis upon viral infection, suggesting that eIF2α phosphorylation plays an antiviral defense response. However, the decrease in viral proteins resulting from eIF2α phosphorylation is detected at an early stage, and the synthesis of viral proteins is increased at a later stage; it suggests that the ISR is an early sponsor, and the antagonism of virus favoring itself replication is a primary factor at a later stage. A pro-survival response is induced early with short and mild stress to restore cell homeostasis, but cell death signaling is activated at a later stage with prolonged and severe stress during viral infection. However, the shift mechanism between pro-survival and cell death signaling must be further illuminated. It is adverse for viruses with persistent replication, and how virus balances the replication and cell survival for the propagation of progeny.
In conclusion, the role of the ISR is becoming more and more important during viral infection. The ISR is a complicated, integrated, pro-survival cellular response that converges on eIF2α phosphorylation. A PERK-eIF2α signaling pathway is vital in enhancing viral replication via UPR-induced autophagy. The PKR-eIF2α signaling pathway involves mainly in the formation of SGs and UPR-induced apoptosis through CHOP expression. Meanwhile, other branches of UPRs are also involved. The virus also modulates ISR signaling pathways to favor its replication, which is vital to illuminating the interaction between the host and viruses and as a therapeutic targeting to enhance host defense against viruses.
YmL and YjL conceived the idea and revised and edited the manuscript. YW collected information and drafted the manuscript. YW and ZZ performed the structural analysis. All authors read and made final approval of the manuscript.
The study was supported by Southwest Minzu University Research Startup Fund (2021115 and 16011211013) and Gansu Province Science and Technology Planning Project (20YF3WA008).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abrahamyan, L. G., Chatel-Chaix, L., Ajamian, L., Milev, M. P., Monette, A., Clement, J. F., et al. (2010). Novel staufen1 ribonucleoproteins prevent formation of stress granules but favour encapsidation of HIV-1 genomic RNA. J. Cell Sci. 123, 369–383. doi: 10.1242/jcs.055897
Aitken, C. E., and Lorsch, J. R. (2012). A mechanistic overview of translation initiation in eukaryotes. Nat. Struct. Mol. Biol. 19, 568–576. doi: 10.1038/nsmb.2303
Albornoz, A., Carletti, T., Corazza, G., and Marcello, A. (2014). The stress granule component TIA-1 binds tick-borne encephalitis virus RNA and is recruited to perinuclear sites of viral replication to inhibit viral translation. J. Virol. 88, 6611–6622. doi: 10.1128/JVI.03736-13
Alexander, D. E., Ward, S. L., Mizushima, N., Levine, B., and Leib, D. A. (2007). Analysis of the role of autophagy in replication of herpes simplex virus in cell culture. J. Virol. 81, 12128–12134. doi: 10.1128/JVI.01356-07
Ambrose, R. L., and Mackenzie, J. M. (2013a). ATF6 signaling is required for efficient west nile virus replication by promoting cell survival and inhibition of innate immune responses. J. Virol. 87, 2206–2214. doi: 10.1128/JVI.02097-12
Ambrose, R. L., and Mackenzie, J. M. (2013b). Flaviviral regulation of the unfolded protein response: can stress be benefcial? Future Virol. 8, 1095–1109. doi: 10.2217/fvl.13.100
Anderson, P., and Kedersha, N. (2006). RNA granules. J. Cell Biol. 172, 803–808. doi: 10.1083/jcb.200512082
Benali-Furet, N. L., Chami, M., Houel, L., De Giorgi, F., Vernejoul, F., Lagorce, D., et al. (2005). Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene 24, 4921–4933. doi: 10.1038/sj.onc.1208673
Berlanga, J. J., Ventoso, I., Harding, H. P., Deng, J., Ron, D., Sonenberg, N., et al. (2006). Antiviral effect of the mammalian translation initiation factor 2alpha kinase GCN2 against RNA viruses. EMBO J. 25, 1730–1740. doi: 10.1038/sj.emboj.7601073
Borghese, F., and Michiels, T. (2011). The leader protein of cardioviruses inhibits stress granule assembly. J. Virol. 85, 9614–9622. doi: 10.1128/JVI.00480-11
Chan, S. W., and Egan, P. A. (2005). Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. FASEB J. 19, 1510–1512. doi: 10.1096/fj.04-3455fje
Cheng, G., Feng, Z., and He, B. (2005). Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2alpha dephosphorylation by the gamma(1)34.5 protein. J. Virol. 79, 1379–1388. doi: 10.1128/JVI.79.3.1379-1388.2005
Cheng, J. H., Sun, Y. J., Zhang, F. Q., Zhang, X. R., Qiu, X. S., Yu, L. P., et al. (2016). Newcastle disease virus NP and P proteins induce autophagy via the endoplasmic reticulum stress-related unfolded protein response. Sci. Rep. 6:24721. doi: 10.1038/srep24721
Chiramel, A. I., Brady, N. R., and Bartenschlager, R. (2013). Divergent roles of autophagy in virus infection. Cells 2, 83–104. doi: 10.3390/cells2010083
Ciccaglione, A. R., Costantino, A., Tritarelli, E., Marcantonio, C., Equestre, M., Marziliano, N., et al. (2005). Activation of endoplasmic reticulum stress response by hepatitis C virus proteins. Arch. Virol. 150, 1339–1356. doi: 10.1007/s00705-004-0487-4
Ciccaglione, A. R., Marcantonio, C., Tritarelli, E., Equestre, M., Vendittelli, F., Costantino, A., et al. (2007). Activation of the ER stress gene gadd153 by hepatitis C virus sensitizes cells to oxidant injury. Virus Res. 126, 128–138. doi: 10.1016/j.virusres.2007.02.006
Connor, J. H., and Lyles, D. S. (2005). Inhibition of host and viral translation during vesicular stomatitis virus infection. eIF2 is responsible for the inhibition of viral but not host translation. J. Biol. Chem. 280, 13512–13519. doi: 10.1074/jbc.M501156200
Cruz, J. L. G., Sola, I., Becares, M., Alberca, B., Plana, J., Enjuanes, L., et al. (2011). Coronavirus gene 7 counteracts host defenses and modulates virus virulence. PLoS Pathogens 7:e1002090. doi: 10.1371/journal.ppat.1002090
Datan, E., Roy, S. G., Germain, G., Zali, N., McLean, J. E., Golshan, G., et al. (2016). Dengue-induced autophagy, virus replication and protection from cell death require ER stress (PERK) pathway activation. Cell Death Dis. 7:e2127. doi: 10.1038/cddis.2015.409
de Breyne, S., Bonderoff, J. M., Chumakov, K. M., Lloyd, R. E., and Hellen, C. U. (2008). Cleavage of eukaryotic initiation factor eIF5B by enterovirus 3C proteases. Virology 378, 118–122. doi: 10.1016/j.virol.2008.05.019
Dinh, P. X., Beura, L. K., Das, P. B., Panda, D., Das, A., and Pattnaik, A. K. (2013). Induction of stress granule-like structures in vesicular stomatitis virus-infected cells. J. Virol. 87, 372–383. doi: 10.1128/JVI.02305-12
Domingo-Gil, E., Toribio, R., Najera, J. L., Esteban, M., and Ventoso, I. (2011). Diversity in viral anti-PKR mechanisms: a remarkable case of evolutionary convergence. PLoS One 6:e16711. doi: 10.1371/journal.pone.0016711
Donnelly, N., Gorman, A. M., Gupta, S., and Samali, A. (2013). The eIF2alpha kinases: their structures and functions. Cell Mol. Life Sci. 70, 3493–3511. doi: 10.1007/s00018-012-1252-6
Emara, M. M., Fujimura, K., Sciaranghella, D., Ivanova, V., Ivanov, P., and Anderson, P. (2012). Hydrogen peroxide induces stress granule formation independent of eIF2alpha phosphorylation. Biochem. Biophys. Res. Commun. 423, 763–769. doi: 10.1016/j.bbrc.2012.06.033
Fritzlar, S., Aktepe, T. E., Chao, Y. W., Kenney, N. D., McAllaster, M. R., Wilen, C. B., et al. (2019). Mouse norovirus infection arrests host cell translation uncoupled from the stress granule-PKR-eIF2alpha axis. MBio 10, e960–e919. doi: 10.1128/mBio.00960-19
Fukasawa, M. (2010). Cellular lipid droplets and hepatitis C virus life cycle. Biol. Pharm. Bull. 33, 355–359. doi: 10.1248/bpb.33.355
Fung, T. S., Liao, Y., and Liu, D. X. (2014). The endoplasmic reticulum stress sensor IRE1alpha protects cells from apoptosis induced by the coronavirus infectious bronchitis virus. J. Virol. 88, 12752–12764. doi: 10.1128/JVI.02138-14
Fung, T. S., Torres, J., and Liu, D. X. (2015). The emerging roles of viroporins in ER stress response and autophagy Induction during Virus Infection. Viruses 7, 2834–2857. doi: 10.3390/v7062749
Garaigorta, U., Heim, M. H., Boyd, B., Wieland, S., and Chisari, F. V. (2012). Hepatitis C virus (HCV) induces formation of stress granules whose proteins regulate HCV RNA replication and virus assembly and egress. J. Virol. 86, 11043–11056. doi: 10.1128/JVI.07101-11
Garcia, M. A., Meurs, E. F., and Esteban, M. (2007). The dsRNA protein kinase PKR: virus and cell control. Biochimie 89, 799–811. doi: 10.1016/j.biochi.2007.03.001
Gorchakov, R., Frolova, E., Williams, B. R., Rice, C. M., and Frolov, I. (2004). PKR-dependent and -independent mechanisms are involved in translational shutoff during Sindbis virus infection. J. Virol. 78, 8455–8467. doi: 10.1128/JVI.78.16.8455-8467.2004
Green, A. M., Beatty, P. R., Hadjilaou, A., and Harris, E. (2014). Innate immunity to dengue virus infection and subversion of antiviral responses. J. Mol. Biol. 426, 1148–1160. doi: 10.1016/j.jmb.2013.11.023
Harding, H. P., Zhang, Y., Zeng, H., Novoa, I., Lu, P. D., Calfon, M., et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633. doi: 10.1016/s1097-2765(03)00105-9
Heinicke, L. A., Wong, C. J., Lary, J., Nallagatla, S. R., Diegelman-Parente, A., Zheng, X., et al. (2009). RNA dimerization promotes PKR dimerization and activation. J. Mol. Biol. 390, 319–338. doi: 10.1016/j.jmb.2009.05.005
Hinnebusch, A. G., and Lorsch, J. R. (2012). The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harb Perspect Biol. 4:a011544. doi: 10.1101/cshperspect.a011544
Hou, L., Ge, X. N., Xin, L. X., Zhou, L., Guo, X., and Yang, H. C. (2014). Nonstructural proteins 2C and 3D areinvolved in autophagy as induced by the encephalomyocarditis virus. Virol. J. 11:156. doi: 10.1186/1743-422X-11-156
Jackson, R. J., Hellen, C. U., and Pestova, T. V. (2010). The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 11, 113–127. doi: 10.1038/nrm2838
Jheng, J. R., Ho, J. Y., and Horng, J. T. (2014). ER stress, autophagy, and RNA viruses. Front. Microbiol. 5:388. doi: 10.3389/fmicb.2014.00388
Jordan, R., Wang, L., Graczyk, T. M., Block, T. M., and Romano, P. R. (2002). Replication of a cytopathic strain of bovine viral diarrhea virus activates PERK and induces endoplasmic reticulum stress-mediated apoptosis of MDBK cells. J. Virol. 76, 9588–9599. doi: 10.1128/jvi.76.19.9588-9599.2002
Ke, P. Y., and Chen, S. S. (2011). Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Invest 121, 37–56. doi: 10.1172/JCI41474
Khaperskyy, D. A., Hatchette, T. F., and McCormick, C. (2012). Influenza A virus inhibits cytoplasmic stress granule formation. FASEB J. 26, 1629–1639. doi: 10.1096/fj.11-196915
Krahling, V., Stein, D. A., Spiegel, M., Weber, F., and Muhlberger, E. (2009). Severe acute respiratory syndrome coronavirus triggers apoptosis via protein kinase R but is resistant to its antiviral activity. J. Virol. 83, 2298–2309. doi: 10.1128/JVI.01245-08
Krishnamoorthy, J., Mounir, Z., Raven, J. F., and Koromilas, A. E. (2008). The eIF2alpha kinases inhibit vesicular stomatitis virus replication independently of eIF2alpha phosphorylation. Cell Cycle 7, 2346–2351. doi: 10.4161/cc.6323
Lavoie, H., Li, J. J., Thevakumaran, N., Therrien, M., and Sicheri, F. (2014). Dimerization-induced allostery in protein kinase regulation. Trends Biochem. Sci. 39, 475–486. doi: 10.1016/j.tibs.2014.08.004
Lee, Y. R., Kuo, S. H., Lin, C. Y., Fu, P. J., Lin, Y. S., Yeh, T. M., et al. (2018). Dengue virus-induced ER stress is required for autophagy activation, viral replication, and pathogenesis both in vitro and in vivo. Sci. Rep. 8:489. doi: 10.1038/s41598-017-18909-3
Li, Y. R., Jiang, W. Y., Niu, Q. N., Sun, Y. J., Meng, C. C., Tan, L., et al. (2019). eIF2α-CHOP-BCl-2/JNK and IRE1α-XBP1/JNK signaling promote apoptosis and inflammation and support the proliferation of Newcastle disease virus. Cell Death Dis. 10:891. doi: 10.1038/s41419-019-2128-6
Liao, Y., Fung, T. S., Huang, M., Fang, S. G., Zhong, Y., and Liu, D. X. (2013). Upregulation of CHOP/GADD153 during coronavirus infectious bronchitis virus infection modulates apoptosis by restricting activation of the extracellular signal-regulated kinase pathway. J. Virol. 87, 8124–8134. doi: 10.1128/JVI.00626-13
Lindquist, M. E., Mainou, B. A., Dermody, T. S., and Crowe, J. E. Jr. (2011). Activation of protein kinase R is required for induction of stress granules by respiratory syncytial virus but dispensable for viral replication. Virology 413, 103–110. doi: 10.1016/j.virol.2011.02.009
Linero, F. N., Thomas, M. G., Boccaccio, G. L., and Scolaro, L. A. (2011). Junin virus infection impairs stress-granule formation in Vero cells treated with arsenite via inhibition of eIF2alpha phosphorylation. J. Gen. Virol. 92, 2889–2899. doi: 10.1099/vir.0.033407-0
Lomakin, I. B., and Steitz, T. A. (2013). The initiation of mammalian protein synthesis and mRNA scanning mechanism. Nature 500, 307–311. doi: 10.1038/nature12355
Low, W. K., Dang, Y., Schneider-Poetsch, T., Shi, Z., Choi, N. S., Merrick, W. C., et al. (2005). Inhibition of eukaryotic translation initiation by the marine natural product pateamine A. Mol. Cell 20, 709–722. doi: 10.1016/j.molcel.2005.10.008
Lu, Y., Wambach, M., and Krug, R. M. (1995). Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phosphorylates the eIF-2 translation initiation factor. Virology 214, 222–228. doi: 10.1006/viro.1995.9937
Luo, X. N., Yao, H. L., Song, J., Song, Q. Q., Shi, B. T., Xia, D., et al. (2018). Coxsackievirus B3 Infection Triggers Autophagy through 3 Pathways of Endoplasmic Reticulum Stress. Biomed. Environ. Sci. 31, 867–875. doi: 10.3967/bes2018.115
Lussignol, M., Queval, C., Bernet-Camard, M. F., Cotte-Laffitte, J., Beau, I., Codogno, P., et al. (2013). The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J. Virol. 87, 859–871. doi: 10.1128/JVI.01158-12
Lv, S., Sun, E. C., Xu, Q. Y., Zhang, J. K., and Wu, D. L. (2015). Endoplasmic reticulum stress-mediated autophagy contributes to bluetongue virus infection via the PERK-eIF2alpha pathway. Biochem. Biophys. Res. Commun. 466, 406–412. doi: 10.1016/j.bbrc.2015.09.039
Ma, Y., Wang, C., Xue, M., Fu, F., Zhang, X., Li, L., et al. (2018). The Coronavirus Transmissible Gastroenteritis Virus Evades the Type I Interferon Response through IRE1alpha-Mediated Manipulation of the MicroRNA miR-30a-5p/SOCS1/3 Axis. J. Virol 92, e728–e718. doi: 10.1128/JVI.00728-18
Martin-Acebes, M. A., Blazquez, A. B., Jimenez, de Oya, N., Escribano-Romero, E., and Saiz, J. C. (2011). West Nile virus replication requires fatty acid synthesis but is independent on phosphatidylinositol-4-phosphate lipids. PLoS One 6:e24970. doi: 10.1371/journal.pone.0024970
McCormick, C., and Khaperskyy, D. A. (2017). Translation inhibition and stress granules in the antiviral immune response. Nat. Rev. Immunol. 17, 647–660. doi: 10.1038/nri.2017.63
Medigeshi, G. R., Lancaster, A. M., Hirsch, A. J., Briese, T., Lipkin, W. I., Defilippis, V., et al. (2007). West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J. Virol. 81, 10849–10860. doi: 10.1128/JVI.01151-07
Montero, H., Rojas, M., Arias, C. F., and Lopez, S. (2008). Rotavirus infection induces the phosphorylation of eIF2alpha but prevents the formation of stress granules. J. Virol. 82, 1496–1504. doi: 10.1128/JVI.01779-07
Neerukonda, S. N., Katneni, U. K., Bott, M., Golovan, S. P., and Parcells, M. S. (2018). Induction of the unfolded protein response (UPR) during Marek’s disease virus (MDV) infection. Virology 522, 1–12. doi: 10.1016/j.virol.2018.06.016
Ng, C. S., Jogi, M., Yoo, J. S., Onomoto, K., Koike, S., Iwasaki, T., et al. (2013). Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J. Virol. 87, 9511–9522. doi: 10.1128/JVI.03248-12
Novoa, I., Zeng, H. Q., Harding, H. P., and Ron, D. (2001). Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 153, 1011–1021. doi: 10.1083/jcb.153.5.1011
Okonski, K. M., and Samuel, C. E. (2013). Stress granule formation induced by measles virus is protein kinase PKR dependent and impaired by RNA adenosine deaminase ADAR1. J. Virol. 87, 756–766. doi: 10.1128/JVI.02270-12
Onomoto, K., Jogi, M., Yoo, J. S., Narita, R., Morimoto, S., Takemura, A., et al. (2012). Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One 7:e43031. doi: 10.1371/journal.pone.0043031
Pakos-Zebrucka, K., Koryga, I., Mnich, K., Ljujic, M., Samali, A., and Gorman, A. M. (2016). The integrated stress response. EMBO Rep. 17, 1374–1395. doi: 10.15252/embr.201642195
Pavio, N., Romano, P. R., Graczyk, T. M., Feinstone, S. M., and Taylor, D. R. (2003). Protein Synthesis and Endoplasmic Reticulum Stress Can Be Modulated by the Hepatitis C Virus Envelope Protein E2 through the Eukaryotic Initiation Factor 2 Kinase PERK. J. Virol. 77, 3578–3585. doi: 10.1128/jvi.77.6.3578-3585.2003
Pena, J., and Harris, E. (2011). Dengue virus modulates the unfolded protein response in a time-dependent manner. J. Biol. Chem. 286, 14226–14236. doi: 10.1074/jbc.M111.222703
Poblete-Duran, N., Prades-Perez, Y., Vera-Otarola, J., Soto-Rifo, R., and Valiente-Echeverria, F. (2016). Who Regulates Whom? An Overview of RNA Granules and Viral Infections. Viruses 8:180. doi: 10.3390/v8070180
Qin, Q. S., Hastings, C., and Miller, C. L. (2009). Mammalian orthoreovirus particles induce and are recruited into stress granules at early times postinfection. J. Virol. 83, 11090–11101. doi: 10.1128/JVI.01239-09
Raaben, M., Groot Koerkamp, M. J. A., Rottier, P. J. M., and de Haan, C. A. M. (2007). Mouse hepatitis coronavirus replication induces host translational shutoff and mRNA decay, with concomitant formation of stress granules and processing bodies. Cell Microbiol. 9, 2218–2229. doi: 10.1111/j.1462-5822.2007.00951.x
Rathore, A. P. S., Ng, M. L., and Vasudevan, S. G. (2013). Differential unfolded protein response during Chikungunya and Sindbis virus infection CHIKV nsP4 suppresses eIF2α phosphorylation. Virol. J. 10:36. doi: 10.1186/1743-422X-10-36
Redondo, N., Sanz, M. A., Welnowska, E., and Carrasco, L. (2011). Translation without eIF2 promoted by poliovirus 2A protease. PLoS One 6:e25699. doi: 10.1371/journal.pone.0025699
Rojas, M., Arias, C. F., and Lopez, S. (2010). Protein kinase R is responsible for the phosphorylation of eIF2alpha in rotavirus infection. J. Virol. 84, 10457–10466. doi: 10.1128/JVI.00625-10
Ruggieri, A., Dazert, E., Metz, P., Hofmann, S., Bergeest, J. P., Mazur, J., et al. (2012). Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe 12, 71–85. doi: 10.1016/j.chom.2012.05.013
Samuel, M. A., Whitby, K., Keller, B. C., Marri, A., Barchet, W., and Williams, B. R. (2006). PKR and RNase L contribute to protection against lethal West Nile Virus infection by controlling early viral spread in the periphery and replication in neurons. J. Virol. 80, 7009–7019. doi: 10.1128/JVI.00489-06
Shrivastava, S., Bhanja Chowdhury, J., Steele, R., Ray, R., and Ray, R. B. (2012). Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J. Virol. 86, 8705–8712. doi: 10.1128/JVI.00616-12
Simpson-Holley, M., Kedersha, N., Dower, K., Rubins, K. H. Anderson, P., Hensley, L. E., et al. (2011). Formation of antiviral cytoplasmic granules during orthopoxvirus infection. J. Virol. 85, 1581–1593. doi: 10.1128/JVI.02247-10
Smith, J. A., Schmechel, S. C., Raghavan, A., Abelson, M., Reilly, C., Katze, M. G., et al. (2006). Reovirus induces and benefits from an integrated cellular stress response. J. Virol. 80, 2019–2033. doi: 10.1128/JVI.80.4.2019-2033.2006
Su, H. L., Liao, C. L., and Lin, Y. L. (2002). Japanese encephalitis virus infection initiates endoplasmic reticulum stress and an unfolded protein response. J. Virol. 76, 4162–4171. doi: 10.1128/jvi.76.9.4162-4171.2002
Sun, P., Zhang, S., Qin, X., Chang, X., Cui, X., Li, H., et al. (2018). Foot-and-mouth disease virus capsid protein VP2 activates the cellular EIF2S1-ATF4 pathway and induces autophagy via HSPB1. Autophagy 14, 336-346. doi: 10.1080/15548627.2017.1405187
Syed, G. H., Amako, Y., and Siddiqui, A. (2010). Hepatitis C virus hijacks host lipid metabolism. Trends Endocrinol. Metab. 21, 33–40. doi: 10.1016/j.tem.2009.07.005
Takeuchi, K., Komatsu, T., Kitagawa, Y., Sada, K., and Gotoh, B. (2008). Sendai virus C protein plays a role in restricting PKR activation by limiting the generation of intracellular double-stranded RNA. J. Virol. 82, 10102–10110. doi: 10.1128/JVI.00599-08
Talloczy, Z., Jiang, W. X., Virgin, H. W. IV, Leib, D. A., Scheuner, D., Kaufman, R. L., et al. (2002). Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. PNAS 99, 190–195. doi: 10.1073/pnas.012485299
Talloczy, Z., Virgin, H. W. T., and Levine, B. (2006). PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2, 24–29. doi: 10.4161/auto.2176
Tardif, K. D., Mori, K., and Siddiqui, A. (2002). Hepatitis C Virus Subgenomic Replicons Induce Endoplasmic Reticulum Stress Activating an Intracellular Signaling Pathway. J. Virol. 76, 7453–7459. doi: 10.1128/jvi.76.15.7453-7459.2002
Tu, Y. C., Yu, C. Y., Liang, J. J., Lin, E., Liao, C. L., and Lin, Y. L. (2012). Blocking double-stranded RNA-activated protein kinase PKR by Japanese encephalitis virus nonstructural protein 2A. J. Virol. 86, 10347–10358. doi: 10.1128/JVI.00525-12
Umareddy, I., Pluquet, O., Wang, Q. Y., Vasudevan, S. G., Chevet, E., and Gu, F. (2007). Dengue virus serotype infection specifies the activation of the unfolded protein response. Virol. J. 4:91. doi: 10.1186/1743-422X-4-91
Venticinque, L., and Meruelo, D. (2010). Sindbis viral vector induced apoptosis requires translational inhibition and signaling through Mcl-1 and Bak. Mol. Cancer 9:37. doi: 10.1186/1476-4598-9-37
Wang, J., Kang, R. Y., Huang, H., Xi, X. Y., Wang, B., Wang, J. W., et al. (2014). Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy 10, 766–784. doi: 10.4161/auto.27954
Wang, X., Liao, Y., Yap, P. L., Png, K. J., Tam, J. P., and Liu, D. X. (2009). Inhibition of Protein Kinase R Activation and Upregulation of GADD34 Expression Play a Synergistic Role in Facilitating Coronavirus Replication by Maintaining De Novo Protein Synthesis in Virus-Infected Cells. J. Virol. 83, 12462–12472. doi: 10.1128/JVI.01546-09
White, J. P., Cardenas, A. M., Marissen, W. E., and Lloyd, R. E. (2007). Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2, 295–305. doi: 10.1016/j.chom.2007.08.006
Xue, M., Fu, F., Ma, Y., Zhang, X., Li, L., Feng, L., et al. (2018). The PERK Arm of the Unfolded Protein Response Negatively Regulates Transmissible Gastroenteritis Virus Replication by Suppressing Protein Translation and Promoting Type I Interferon Production. J. Virol. 92, e431–e418. doi: 10.1128/JVI.00431-18
Yang, X. D., Hu, Z. L., Fan, S. S., Zhang, Q., Zhong, Y., Guo, D., et al. (2018). Picornavirus 2A protease regulates stress granule formation to facilitate viral translation. PLoS Pathog. 14:e1006901. doi: 10.1371/journal.ppat.1006901
Ye, X., Pan, T., Wang, D., Fang, L., Ma, J., Zhu, X., et al. (2018). Foot-and-mouth disease virus counteracts on internal ribosome entry site suppression by G3BP1 and inhibits G3BP1-mediated stress granule assembly via post-translational mechanisms. Front. Immunol. 9:1142. doi: 10.3389/fimmu.2018.01142
Yin, H., Zhao, L., Jiang, X., Li, S., Huo, H., and Chen, H. (2017). DEV induce autophagy via the endoplasmic reticulum stress related unfolded protein response. PLoS One 12:e0189704. doi: 10.1371/journal.pone.0189704
Yuan, P., Dong, L., Cheng, Q., Wang, S., Li, Z., Sun, Y., et al. (2017). Prototype foamy virus elicits complete autophagy involving the ER stress-related UPR pathway. Retrovirology 14:16. doi: 10.1186/s12977-017-0341-x
Zang, S., Zhang, X., Li, C., Wang, L., Wei, J., and Qin, Q. (2019). HRI of Epinephelus coioides is a critical factor in the grouper immune response to RGNNV infection. Fish Shellfish Immunol. 87, 659–668. doi: 10.1016/j.fsi.2019.02.011
Zhang, H., Chen, N., Li, P., Pan, Z., Ding, Y., Zou, D., et al. (2016). The nuclear protein Sam68 is recruited to the cytoplasmic stress granules during enterovirus 71 infection. Microb. Pathog. 96, 58–66. doi: 10.1016/j.micpath.2016.04.001
Zhang, P., Su, C., Jiang, Z., and Zheng, C. (2017). Herpes Simplex Virus 1 UL41 Protein Suppresses the IRE1/XBP1 Signal Pathway of the Unfolded Protein Response via Its RNase Activity. J. Virol. 91, doi: 10.1128/JVI.02056-16
Zhang, Q., Sharma, N. R., Zheng, Z. M., and Chen, M. (2019). Viral Regulation of RNA Granules in Infected Cells. Virol. Sin. 34, 175–191. doi: 10.1007/s12250-019-00122-3
Zhang, Y., Li, Z., Ge, X., Guo, X., and Yang, H. (2011). Autophagy promotes the replication of encephalomyocarditis virus in host cells. Autophagy 7, 613–628.
Zhou, Y., Qi, B., Gu, Y., Xu, F., Du, H., Li, X., et al. (2016). Porcine Circovirus 2 Deploys PERK Pathway and GRP78 for Its Enhanced Replication in PK-15 Cells. Viruses 8:56. doi: 10.3390/v8020056
Zhou, Y. S., Gu, Y. X., Qi, B. Z., Zhang, Y. K., Li, X. L., and Fang, W. H. (2017). Porcine circovirus type 2 capsid protein induces unfolded protein response with subsequent activation of apoptosis. J. Zhejiang Univ. Sci. B 18, 316–323. doi: 10.1631/jzus.B1600208
Zhu, H. F., and Zheng, C. F. (2020). The Race between Host Antiviral Innate Immunity and the Immune Evasion Strategies of Herpes Simplex Virus 1. Microbiol. Mol. Biol. Rev. 84, e99–e20. doi: 10.1128/MMBR.00099-20
Keywords: integrated stress response, eIF2α phosphorylation, unfolded protein response, viral replication, host
Citation: Wu Y, Zhang Z, Li Y and Li Y (2022) The Regulation of Integrated Stress Response Signaling Pathway on Viral Infection and Viral Antagonism. Front. Microbiol. 12:814635. doi: 10.3389/fmicb.2021.814635
Received: 14 November 2021; Accepted: 15 December 2021;
Published: 11 February 2022.
Edited by:
Chunfu Zheng, University of Calgary, CanadaReviewed by:
Xingui Tian, The First Affiliated Hospital of Guangzhou Medical University, ChinaCopyright © 2022 Wu, Zhang, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanmin Li, bGl5YW5taW5AY2Fhcy5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.