 Koji Ishiya
Koji Ishiya Hideki Kosaka2
Hideki Kosaka2 Takashi Inaoka
Takashi Inaoka Nobutaka Nakashima
Nobutaka Nakashima- 1Bioproduction Research Institute, National Institute of Advanced Industrial Sciences and Technology (AIST), Sapporo, Japan
- 2Research and Development Department, Fujicco Co., Ltd., Chuo-ku, Japan
- 3Institute of Food Research, National Agriculture and Food Research Organization (NFRI/NARO), Tsukuba, Japan
We determined the whole genome sequences of three bacterial strains, designated as FNDCR1, FNDCF1, and FNDCR2, isolated from a practical nata-de-coco producing bacterial culture. Only FNDCR1 and FNDCR2 strains had the ability to produce cellulose. The 16S rDNA sequence and phylogenetic analysis revealed that all strains belonged to the Komagataeibacter genus but belonged to a different clade within the genus. Comparative genomic analysis revealed cross-strain distribution of duplicated sequences in Komagataeibacter genomes. It is particularly interesting that FNDCR1 has many duplicated sequences within the genome independently of the phylogenetic clade, suggesting that these duplications might have been obtained specifically for this strain. Analysis of the cellulose biosynthesis operon of the three determined strain genomes indicated that several cellulose synthesis-related genes, which are present in FNDCR1 and FNDCR2, were lost in the FNDCF1 strain. These findings reveal important genetic insights into practical nata de coco-producing bacteria that can be used in food development. Furthermore, our results also shed light on the variation in their cellulose-producing abilities and illustrate why genetic traits are unstable for Komagataeibacter and Komagataeibacter-related acetic acid bacteria.
Introduction
Numerous bacteria produce cellulose extracellularly. Bacterial cells are often entrapped in the cellulose fiber, forming pellicle at the air–liquid interface. Depending on the bacterial strain and culture conditions, cells consume as much as 10% of the total energy budget for cellulose biosynthesis (Ross et al., 1991; Chawla et al., 2009). Although the reason for forming pellicle remains controversial, bacterial cells in the pellicle might be able to yield oxygen more efficiently than planktonic cells in liquid (Lee et al., 2014). Bacterial cellulose is expected to offer great potential for use in biomedical and food industries because of its high water-holding capacity, ultrafine network structure, biocompatibility, and high tensile strength in a wet state (Londrina et al., 2018).
A well-known example of bacterial cellulose in industry is nata-de-coco, an indigenous jelly-like food of Southeast Asia. The fibrous base of nata-de-coco, which consists of pure bacterial cellulose, is produced industrially through fermentation of coconut water using acetic acid bacteria (AAB). Among AAB, the species of Komagataeibacter genus (also known as Gluconacetobacter genus), which are kinds of α-proteobacteria and obligately aerobic and acidophilic bacteria, are the most preferably employed practically because the bacteria of this genus have the exceptionally high capability for producing cellulose (Ryngajłło et al., 2019).
However, the productivity of nata-de-coco by these bacteria is highly unstable; AAB are well known to be genetically unstable and to lose cellulose producing ability frequently in the absence of selection pressures (Matsutani et al., 2015; Ryngajłło et al., 2019). The genetic instability of AAB is probably attributable to the complexity of genome organization. Some examples reported earlier in the literature have shown the presence of more numerous mobile genetic elements and of duplicated sequences than in ordinary bacteria (Azuma et al., 2009; Zhang et al., 2017; Ryngajłło et al., 2019).
The Komagataeibacter medellinensis NBRC 3288 strain (formerly, Gluconacetobacter xylinus NBRC 3288) was isolated from rice vinegar as a cellulose-producing strain in 1954, but complete loss of its production capability was reported in 1999 (Navarro and Komagata, 1999). Then, Matsutani et al. (2015) found crucially important gene truncation in the cellulose biosynthesis operon (bcs operon) of this strain in 2015. In Komagataeibacter bacteria, the bcs operon is typically organized as bcsZHABCD-bglX or bcsABXYC. The genes in these operons encode cyclic dimeric (3′ → 5′) guanosine monophosphate (c-di-GMP) -regulated cellulose synthase (BcsA and BcsB; inner membrane-associated), an outer membrane porin protein (BcsC), putative periplasmic protein (BcsD), cellulose-complementing protein A (BcsH), probable cellulose deacylase (BcsX), probable cellulose acylase (BcsY), endo-β-1,4-glucanase (BcsZ), and β-glucosidase (bglX). Moreover, the genes play a central role in cellulose biosynthesis (Römling and Galperin, 2015).
To elucidate the genomic characterization of nata-de-coco producing bacteria, we determined the whole genome sequences of three bacterial strains (FNDCR1, FNDCF1, FNDCR2) that had been isolated from a practical culture of nata-de-coco in Japan. This report is the first of a study of whole genome sequences of practical nata-de-coco producing bacteria. All of the strains were bacteria of Komagataeibacter genus. These data support better understanding of the physiology of Komagataeibacter bacteria and elucidate the efficient and stable production of nata-de-coco and biocellulose.
Materials and Methods
Bacterial Strains and General Techniques
The nata-de-coco producing bacteria were isolated from a nata-de-coco production facility in Japan. The FNDCR1 strain was stocked in 2017 as a strain that had shown good nata-de-coco productivity from those stored in a nata-de-coco production facility in Japan. The FNDCF1 strain was isolated in 2016 when the productivity loss occurred in the facility. FNDCR2 is a strain that was isolated in 2017 after 20 passages of culture from a stock solution stored at the facility for producing nata-de-coco. In this study, all strains were single colony isolated in 2020, followed by cellulose productivity experiments and whole genome sequencing. Unless stated otherwise, cells were cultured at 28°C using Hestrin–Schramm (HS) medium: 5 g/L yeast extract, 5 g/L peptone, 2.7 g/L Na2HPO4, 1.5 g/L citric acid, 0.1% (v/v) acetic acid, and 10 g/L sucrose or glucose. For preparation of HS-agar plates, agarose was added at 20 g/L.
Cellulose Production and Quantification
After fermentation, the cellulose amount was quantified according to the method described in an earlier report, with modifications (Ryngajłło et al., 2019). In brief, cells were spread onto an agar plate (in a 90-mm-diameter Petri dish) from a glycerol stock. After incubating the plate for 4 days, 2 mL of liquid medium was added onto the surface. The cells were scraped and collected. After the optical density of the cell suspension at 600 nm was adjusted to 0.2, 500 μL of the suspension was added to an Erlenmeyer flask containing 9.5 mL of fresh medium. The culture was incubated for 5 days without shaking. The cellulose pellicle was removed from the flask, treated with 1 M NaOH solution overnight in a Petri dish, and washed extensively with distilled water until the water pH became neutral. The washed cellulose was moved to a new and clean Petri dish, of which the weight was pre-recorded. The cellulose was dried completely, typically for 4 h, at 60°C. The Petri-dish and cellulose total weight was recorded.
Whole-Genome Sequencing
Whole-genome DNA was isolated using the neutral phenol—chloroform—isoamyl alcohol method (Nakashima and Tamura, 2018) with one modification: cellulase (Cellulase, from Aspergillus niger; Fujifilm Wako Pure Chemical Corp., Osaka, Japan) was added at 1 mg/mL and incubated for 1 h at 28°C before harvesting. The purified DNA was subjected to sequencing with PacBio RS II and Illumina Hiseq 2,500 systems (Hokkaido System Science Co., Ltd., Sapporo, Japan). The Hiseq reads were obtained with paired-end 100 base pairs (bp) sequencing. The numbers of reads over ≥ Q30 were 47,239,534, 47,458,318, and 44,626,206 bp, respectively, for FNDCR1, FNDCF1, and FNDR2 strains. In the PacBio reads, the numbers of subreads and mean subread lengths were 145,440 and 6,655 bp, 149,926 and 7,350 bp, and 175,664 and 9,362 bp, respectively, for FNDCR1, FNDCF1, and FNDR2 strains. The raw sequencing reads have been deposited in NCBI SRA under their accession numbers SRR15903111-SRR15903116.
The Hiseq reads were trimmed before assembly (TrimGalore! v 0.4.4).1 Both the trimmed Hiseq and PacBio reads were used to construct the genomes using the Unicycler software (v. 0.4.6; Wick et al., 2017) with default parameters. The assembly genome completeness was assessed using BUSCO (v.5.2.2; rhodospirillales_odb10) (Manni et al., 2021). The assembled genomes were classified into plasmids and chromosomes using PlasFlow (v. 1.1.0; Krawczyk et al., 2018) with default parameters, respectively. Genome annotation was performed using the prokka (v1.13.7) software (Seemann, 2014) and web-based RAST (v.2.0; Aziz et al., 2008)2 with preset parameters, respectively. The CGview server beta was used to illustrate the genome maps3 (Grant and Stothard, 2008; Grant et al., 2012). The assembled sequences were deposited in the NCBI Genbank under the BioProject number PRJNA763499.
Phylogenetic Analysis
From the assembled genomes, 16S rRNA gene sequences were extracted (barrnap v.0.9).4 Phylogenetic classification was then performed from the 16S rRNA gene sequences using the Alignment, Classification and Tree Service5 of SILVA (Release 138; Quast et al., 2013).
Based on the classification results, the public genome of an assigned genus (Komagataeibacter genus) was obtained from NCBI Genbank6 using NCBI-genome-download v.0.3.0.7 To ensure the genome assembly levels, the downloaded genomes were limited to “complete” whole genomes.
To identify the core gene sets from three new assembly genomes and downloaded genomes, a pan-genomic analysis was performed (Roary v.3.11.2; Page et al., 2015). Using these core gene sets, a maximum-likelihood phylogenetic tree was constructed with IQ-TREE v.2.0.3 (bootstrap: 1,000 replicates; best-fitting substitution model: GTR + F + R6 model) (Nguyen et al., 2015). The phylogenetic tree was visualized using iTOL (v.6.3)8 (Letunic and Bork, 2021).
Identification of Synteny Blocks and Duplicated Regions
To detect synteny blocks, the three assembled genomes were subjected to pairwise comparisons to themselves and other genomes (Sibelia v.3.0.7; Minkin and Medvedev, 2020). The detected synteny blocks were visualized as paths between genomes (Circa v.1.2.2; OMGenomics). Duplicate regions within the genome were sought (RepSeek v.6.6; Achaz et al., 2006) with minimum length thresholds of 1,000, 2,000, and 3,000 bp. Moreover, correlation tests and linear regression analyses were conducted using implemented functions in R language (R v.4.1.1; R Development Team, 2021) to assess the relation between the duplicated lengths and the number of mobile genetic elements. The R script are available on https://github.com/omics-tools/natagenome_rscript.
Results
Validation of Cellulose Productivity
Cellulose productivity of three strains was quantified as described in section “Materials and Methods” (Figure 1). As carbon sources, glucose or sucrose was fed to the cells. The FNDCR2 strain produced more cellulose than the FNDCR1 strain, although the FNDCF1 strain did not produce at all. This result indicates that the nata-de-coco producing bacterial culture contained both cellulose producing and non-producing cells.
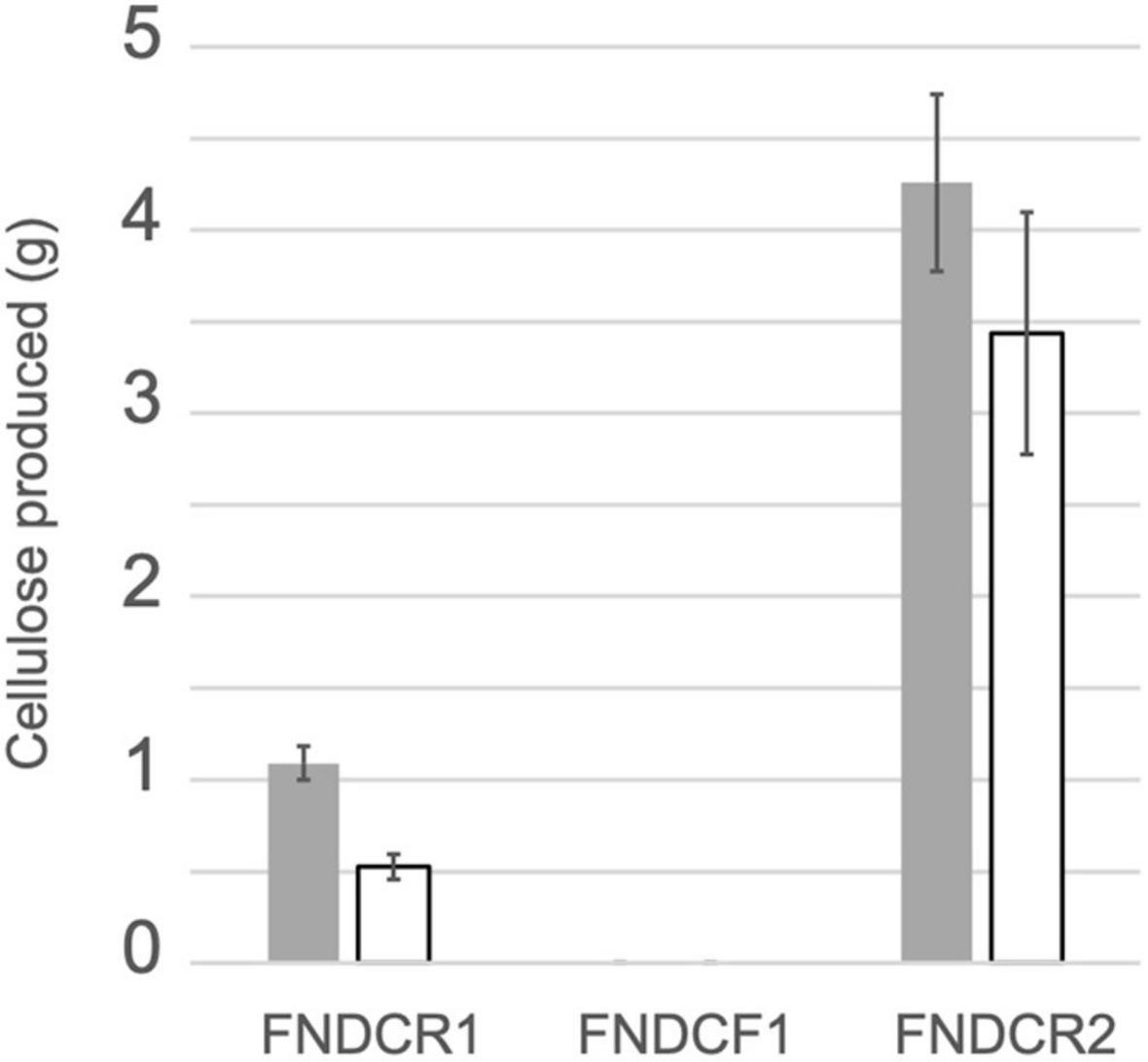
Figure 1. Cellulose productivity of the present strains. Cellulose was produced from glucose (gray bars) or sucrose (open bars) using the designated strains. Quantification results of cellulose from three replicate experiments are shown as mean ± SEM.
Whole Genome Sequence Determination
To identify the taxonomy and to investigate cellulose biosynthesis mechanisms of the present strains, the genome sequences were obtained with shotgun sequencing. For constructing complete genome sequences, both short-read and long-read sequencing were performed. After sequencing, the whole genome sequences were constructed with hybrid assembly (section “Materials and Methods”). The assembled genomes of FNDCR1, FNDCF1, and FNDCR2, respectively, showed high completeness of 99.5, 99.6, and 99.7%. All the strains were found to harbor multiple circular plasmids in addition to circular chromosomes (Figures 2–4). There was no shared plasmid in these three strains (Supplementary Figure 1). The relative value of sequencing coverage of each plasmid is expected to reflect the plasmid copy number. Two CRISPR sequences were found on the pKFR1-1 plasmid.
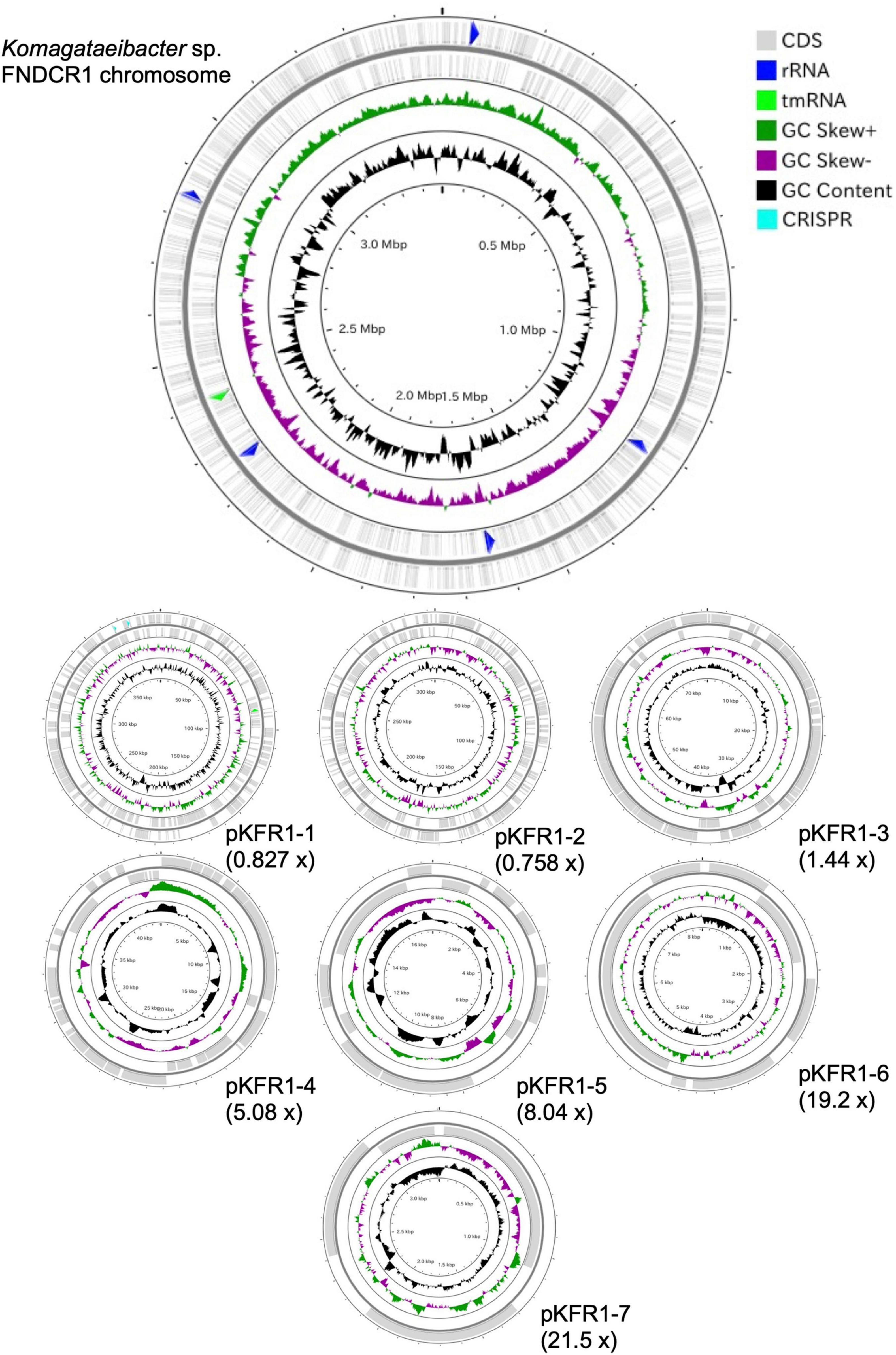
Figure 2. Genome structure of the FNDCF1 strain. One circular chromosome and seven putative plasmids (pKFR1-1, pKFR1-2, pKFR1-3, pKFR1-4, pKFR1-5, pKFR1-6, and pKFR1-7) are shown. The value presented in parentheses represents the sequencing coverage of a plasmid relative to that of chromosome. Smaller contigs below 5,000 bp and non-circularized contigs are not shown in this figure.
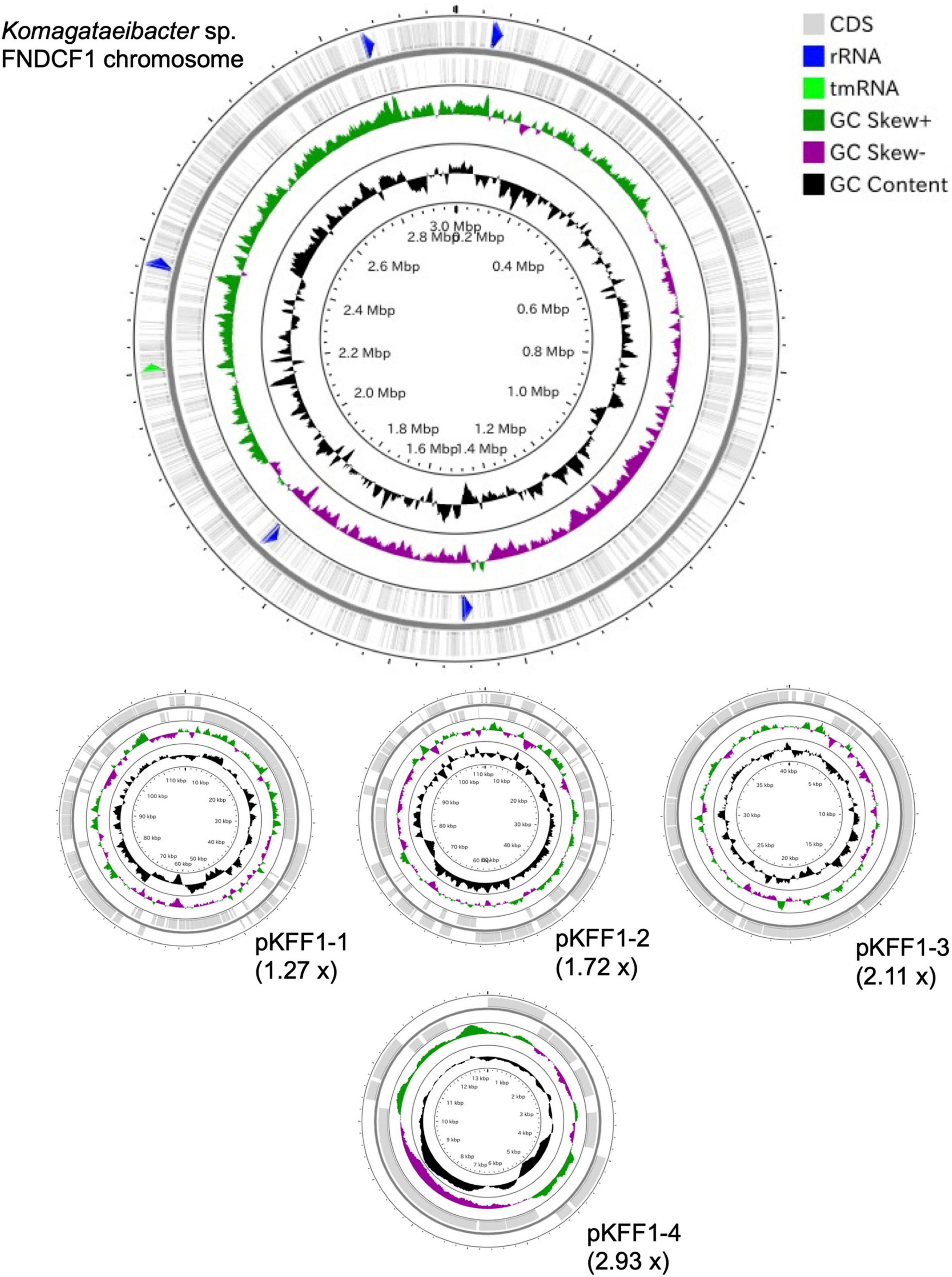
Figure 3. Genome structure of the FNDCR1 strain. One circular chromosome and four putative plasmids (pKFF1-1, pKFF1-2, pKFF1-3, and pKFF1-4 pKFR1-7) are shown. The value given in parentheses represents the sequencing coverage of a plasmid relative to that of chromosome. Smaller contigs below 5,000 bp and non-circularized contigs are not shown in this figure.
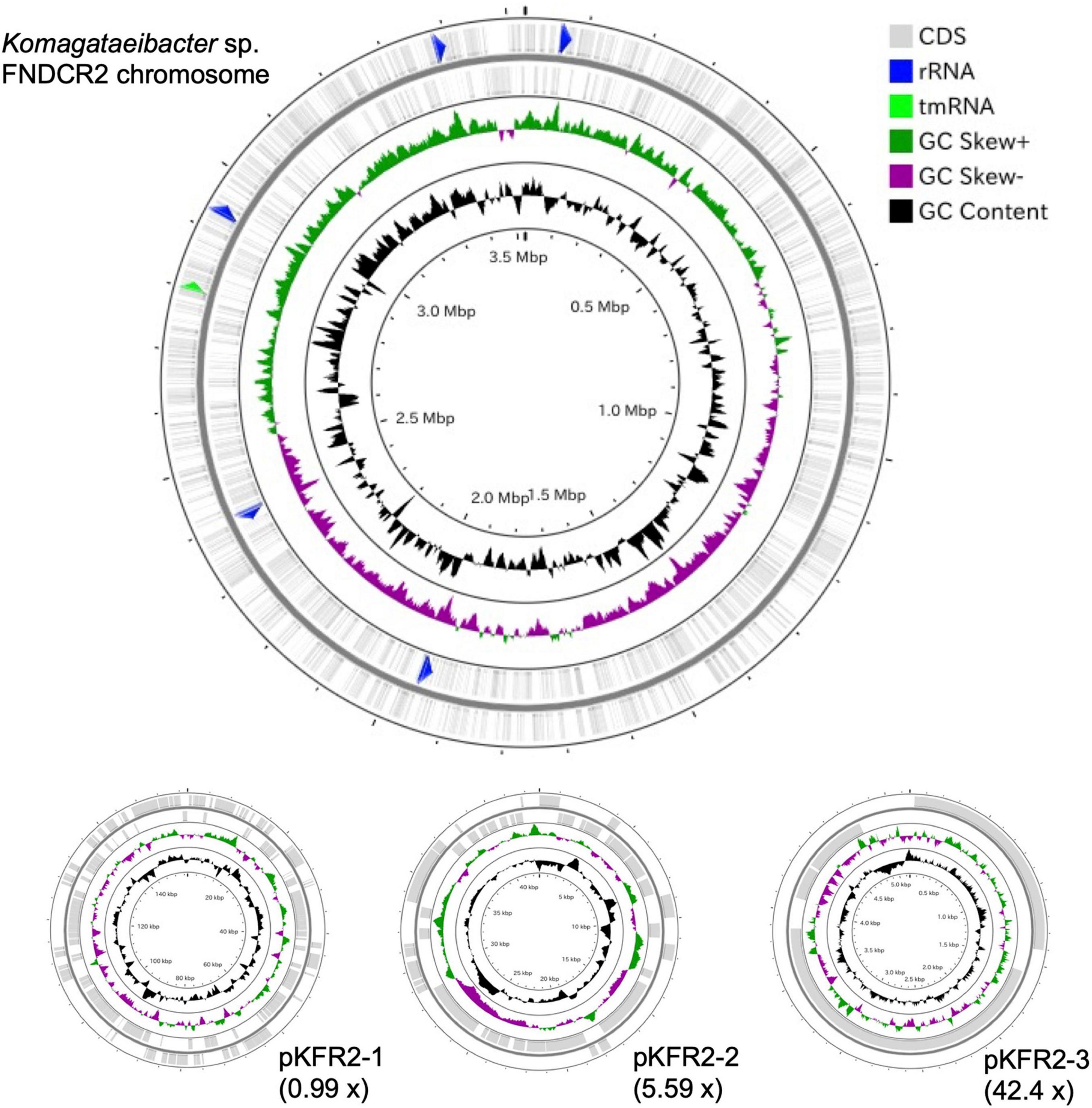
Figure 4. Genome structure of the FNDCR2 strain. One circular chromosome and three putative plasmids (pKFR2-1, pKFR2-2, pKFR2-3) are shown. The value given in parentheses represents the sequencing coverage of a plasmid relative to that of chromosome. Smaller contigs below 5,000 bp and non-circularized contigs are not shown in this figure.
Phylogenetic Relation Analysis
Each strain harbored five 16 rRNA genes on the chromosome. All 16S rRNA sequences of the FNDCR1, FNDCF1, and FNDCR2 strains were assigned to those of Komagataeibacter with > 99% similarities (Supplementary Table 1). The phylogenetic tree constructed based on the core gene set derived from the pan-genome analysis of Komagateibacter (Figure 5) shows that the non-cellulose synthesizing type, FNDCF1, is in a clade close to the root in the phylogenetic tree of Komagateibacter bacteria. This strain is close to the Komagataeibacter kakiaceti JCM25156 strain isolated from traditional Japanese fruit vinegar (Iino et al., 2012). The cellulose-producing type FNDCR1 is located close to the vinegar-producing acetic acid bacterium Komagataeibacter maltaceti LMG 1529 (Zhang et al., 2018). The cellulose-producing type FNDCR2 is represented as a single clade, with the larger clade being closer to the Komagataeibacter rhaeticus lineage. However, none of the present genomes is the same as known strain genomes. These results indicate that all the present strains belong to Komagataeibacter genus, but all might be novel strain genomes.
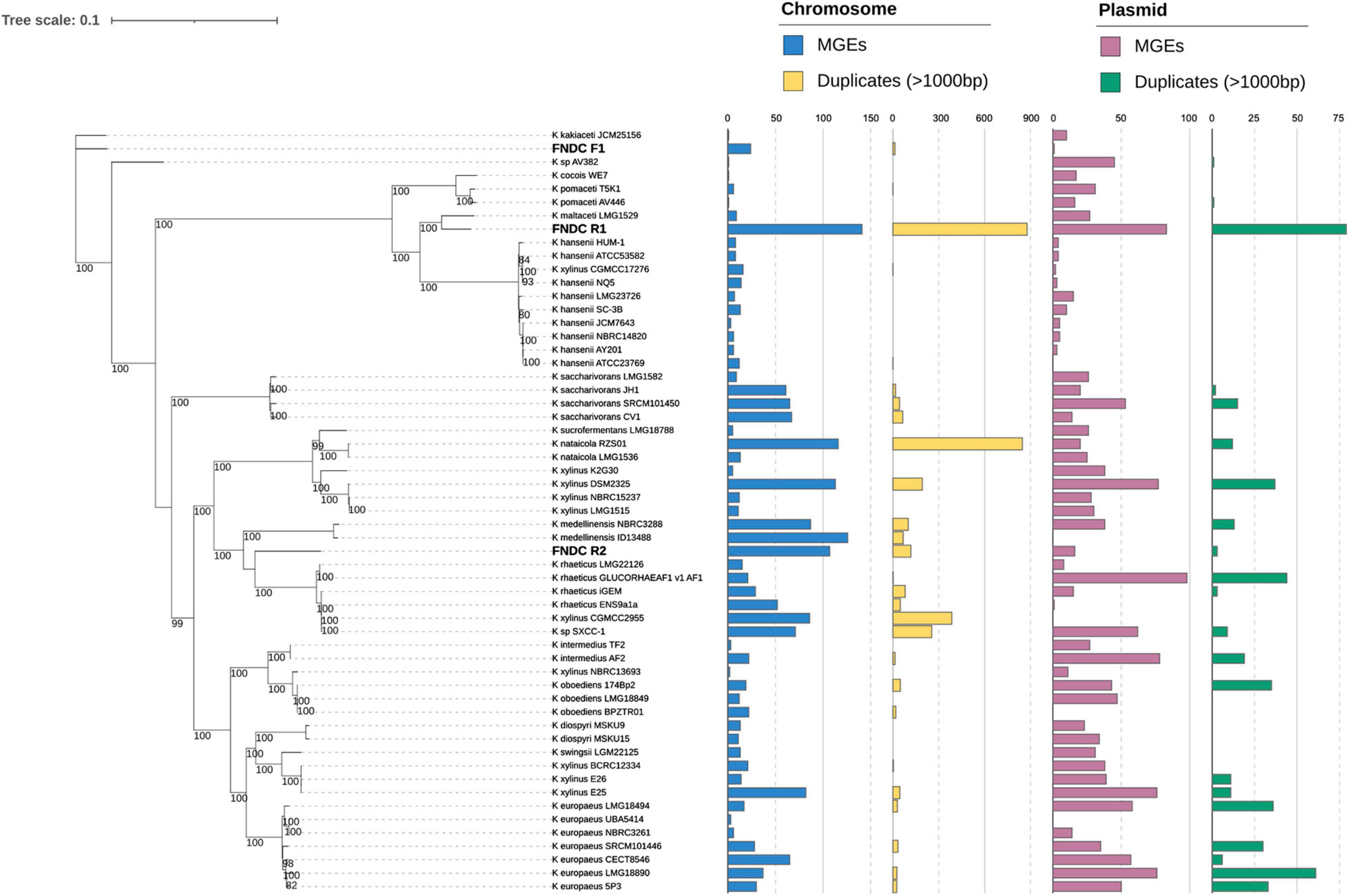
Figure 5. Maximum-likelihood phylogenetic tree in Komagataeibacter genus. A maximum-likelihood phylogenetic tree was built using IQ-TREE based on the public Komagataeibacter genomes and the present strain genomes (FNDCR1, FNDCF1, and FNDCR2). The numbers at nodes are the bootstrap values > 80% as a percentage of 1,000 replications. The bar plot on the right shows the number of mobile genetic elements (MGEs) and Duplicates > 1,000 bp, respectively, on the chromosome and plasmid. The term “Duplicates” refers to homologous sequence pairs in the genome, as detected by RepSeek. MGEs and Duplicates on chromosomes, respectively, correspond to cyan and yellow, whereas MGEs and Duplicates on plasmids, respectively, correspond to pink and green.
Analysis of Genome Structure and Genome Complexity in Cellulose Producing Strains
To compare the genome structures of the respective strains, the three assembled genomes were searched for synteny blocks between themselves and other genomes. Although homologous synteny blocks were found between the three genomes, the overall genome structure did not closely match each other (Figure 6). Results show that the plasmids in each strain harbor regions with DNA homologous to the chromosome (Figure 7). For the plasmids in the FNDCR1 and FNDCR2 strains, many homologous regions were found in their chromosomes, although few such regions were found in the FNDCF1 chromosome.
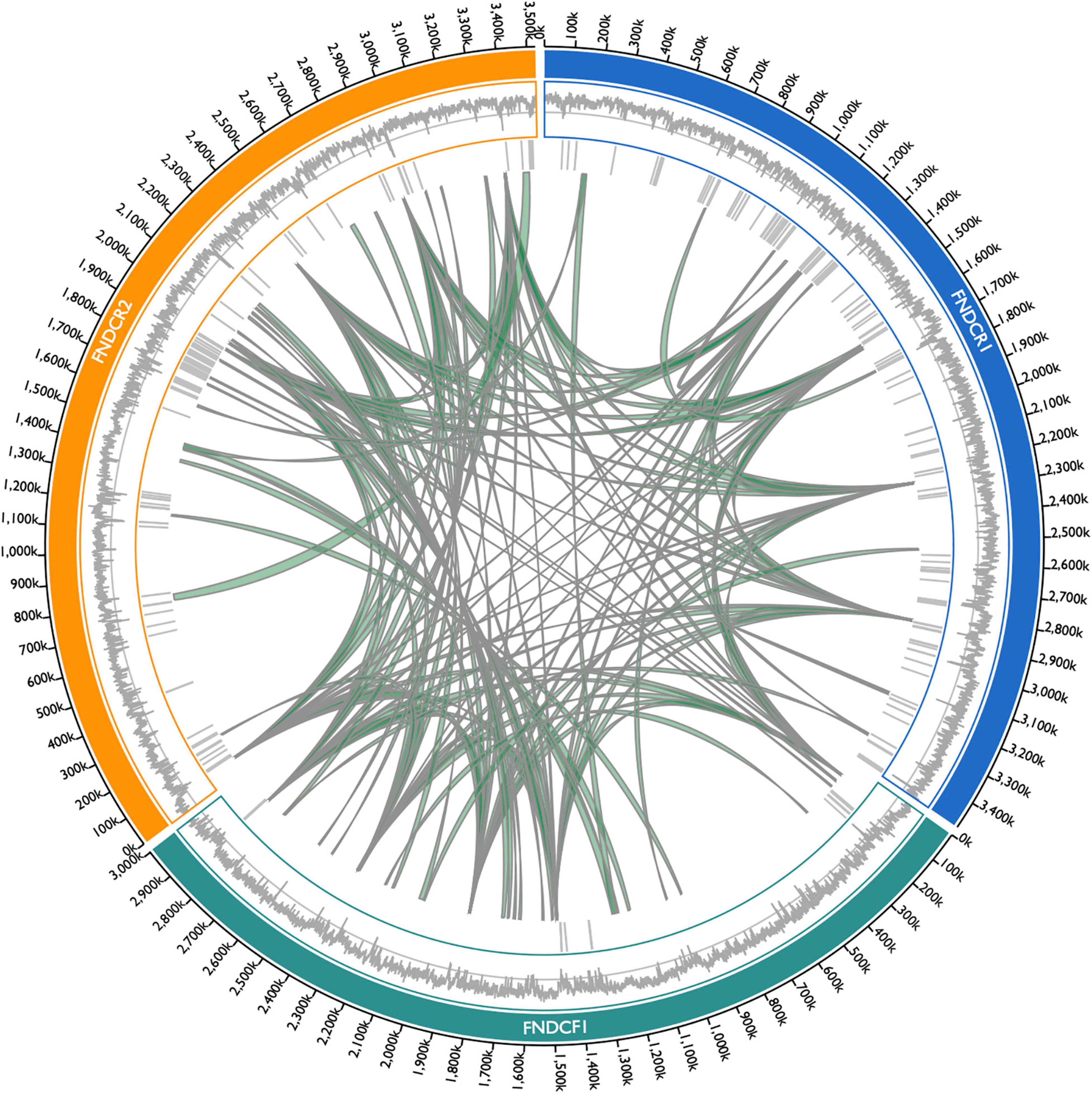
Figure 6. Synteny blocks among the present strain chromosomes. Synteny blocks > 5,000 bp within/among chromosome genomes of FNDCR1 (cyan), FNDCF1 (light green), and FNDCR2 (orange) are shown as gray-colored paths. Synteny blocks were detected using Siberia. The barcode plot outside the synteny block shows where the MGEs are located in the genome. The outer line plot shows the GC content in the genome. The middle line shows the 50% GC content. Numbers on the outside represent the chromosome positions.
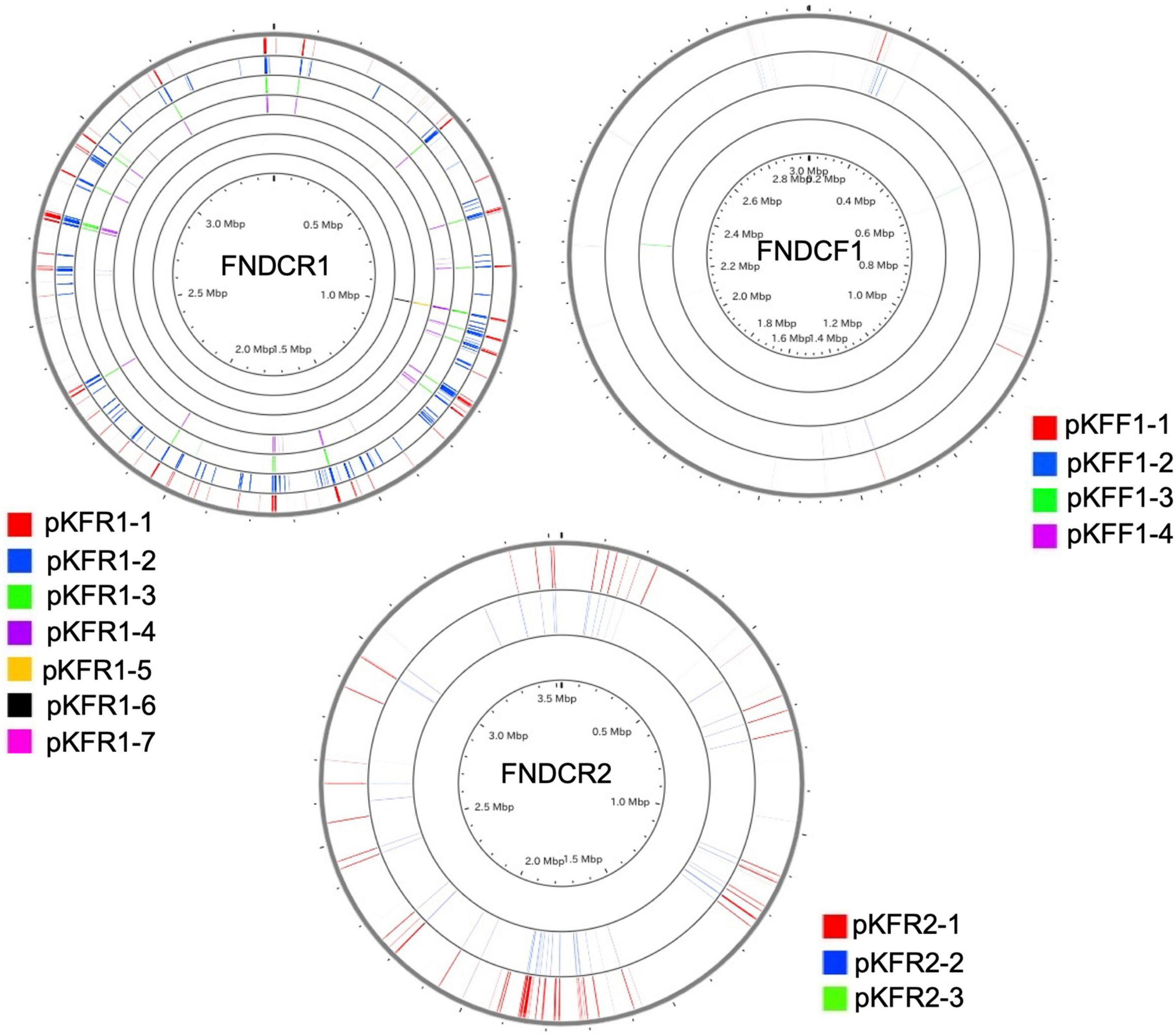
Figure 7. Sequences shared with the chromosome and the plasmids. Homology search (blast algorithm) between each chromosome and plasmids in each strain (other circles) was performed on the CGview server beta. Each plasmid sequence was used as a query. Homologous sequences found on the chromosome are marked with colored lines.
For the whole genomes in the FNDCR1 and FNDCR2 strains, many duplicated sequences were observed, but these characteristic structures were not found in FNDCF1 strain (Figures 5, 8). Furthermore, to investigate relationships between these duplications and mobile genetic elements, the comparative genome analysis was performed with published other Komagataeibacter genomes. Detected duplicates with minimum length of 1,000 bp were significantly and positively correlated with the number of mobile genetic elements in the genome. This relation was more apparent for plasmids than for chromosomes (Figure 8). However, for duplicates with 2,000 bp or greater length, no positive correlation was found either for chromosomes or for plasmids. These results suggest that mobile genetic elements can be associated with the transposition of sequences less than 2,000 bp on their genomes. In addition, the assembled genome of FNDCR1 clearly has more duplicated sequences than the other strains, suggesting the existence of a strain-specific evolutionary event related with accelerated sequence duplication. The coding sequences on these repeats and their neighbors have been summarized as Supplementary Table 2 and Supplementary Figure 2. As a result, about 30 and 19% of the repeated coding sequences in chromosomes and plasmids were annotated as genes related to mobile genetic elements, respectively (Supplementary Figure 2). This result supports the association between the number of mobile genetic elements in the genome and the number of repeats as shown in Figures 5, 8. Most of the other repeated coding sequences were annotated as hypothetical proteins, and their functions and effects are not well understood. Interestingly, some cellulose synthesis-related genes (cellulose synthase 1, cellulose synthase operon protein C and D) were also observed in the repeated and repeated-neighbor regions in chromosomes (Supplementary Table 2). Transposons (ISs) were also observed in the neighboring region of the cellulose synthesis operon of the three strains in this study (Figure 9). These results suggest that mobile genetic elements in Komagataeibacter genomes may also affect the loss and gains of cellulose synthesis-related genes.
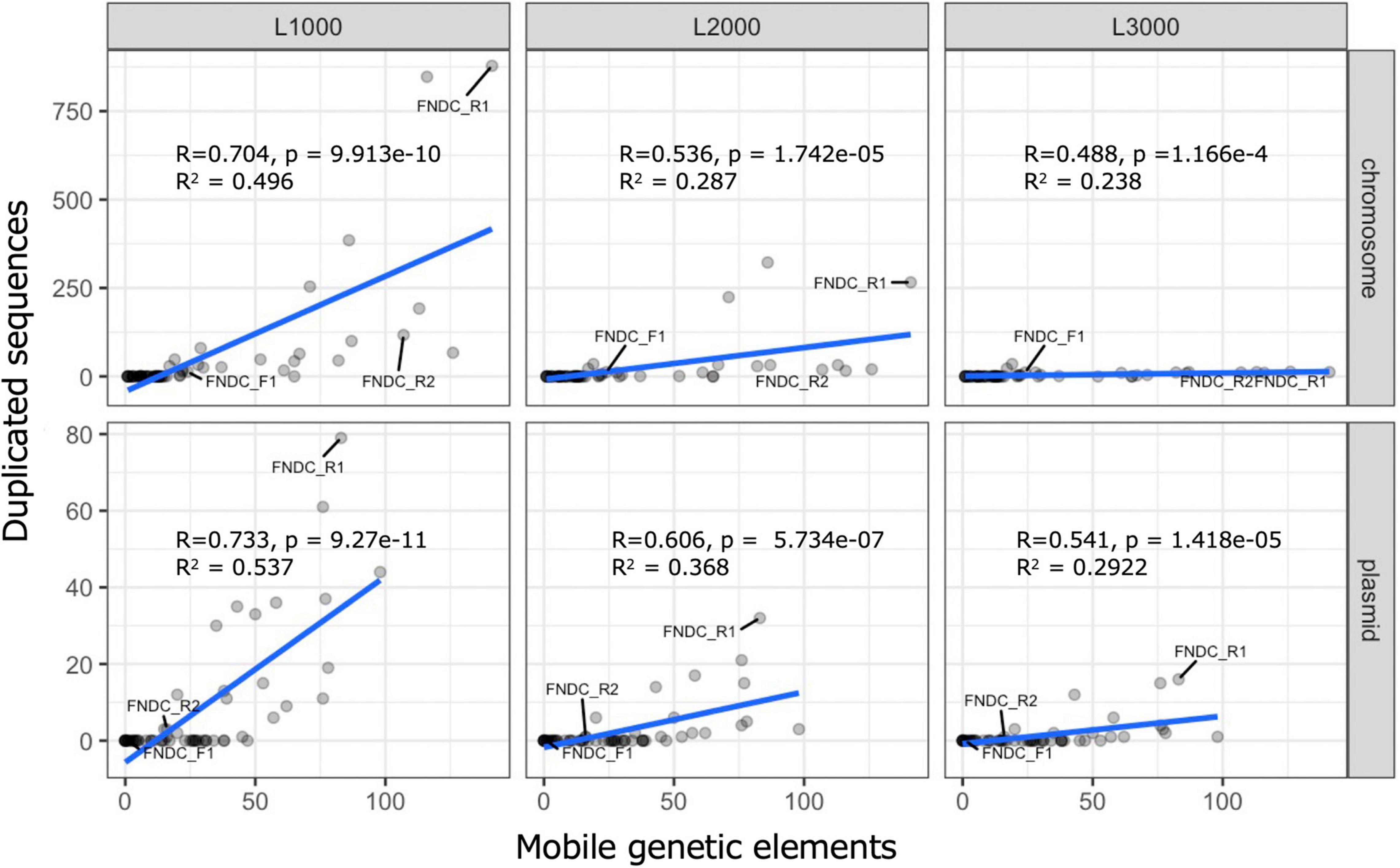
Figure 8. Scatter plots for duplicated sequences and mobile genetic elements. These scatter plots show results of correlation and regression analysis of the number of duplicated sequences and mobile genetic elements in the genome. Correlation analysis and regression analysis were performed with functions implemented in R. The top and bottom rows, respectively, present results for chromosomes and plasmids. Columns L1000, L2000, and L3000, respectively, correspond to the conditions that set 1,000, 2,000, and 3,000 bp as the minimum length threshold during the duplicate sequence search. The line written in blue is the regression line. R, p, and R2, respectively, denote the correlation coefficient, the p-value of the uncorrelation test, and the multiple correlation coefficient.
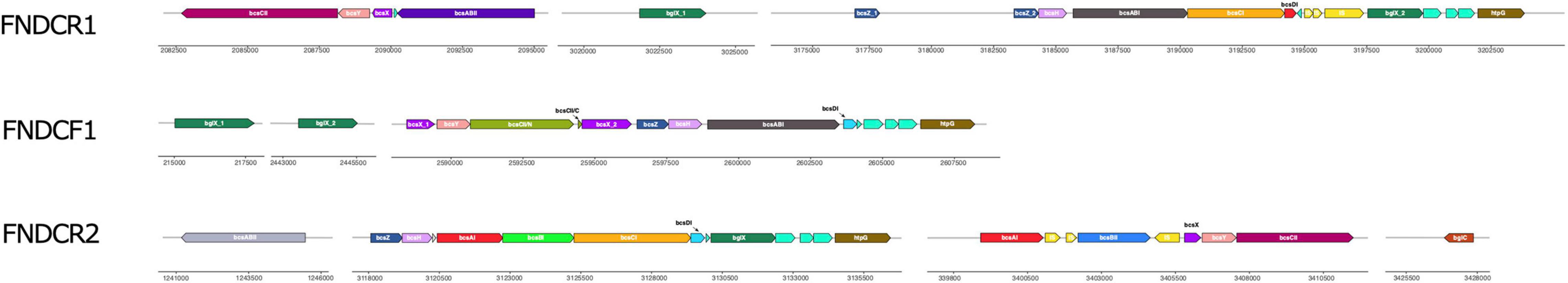
Figure 9. Cellulose biosynthesis operons of the present strains. The figure portrays the operon structure for genes involved in cellulose biosynthesis. The structure was drawn based on results obtained for gene annotation (Supplementary Table 1). Rectangles with arrowheads represent regions that encode genes, open reading frames (ORFs), or transposons (ISs). The arrowhead direction shows the DNA strand orientation (forward/reverse). The numbers represent the genomic positions in the chromosomes. Colors correspond to the encoded genes or ISs. Those without names are the predicted ORFs.
The sequence of each plasmid was compared to those of all other plasmids presented herein, as well as those of plasmids found previously in K. xylinus E25 strain (Supplementary Figure 1). Actually, pKFR1-2, pKFR2-1, and pGX5 (K. xylinus E25) were mutually similar. Also, pKFR1-4 was similar to pKFR2-2 throughout the plasmids. Moreover, pKFF1-4, pKFR1-4, pKFR2-1, pGX4 (K. xylinus E25), and pGX3 (K. xylinus E25) shared partial homology: an approximately 3 kb part of pKFR1-7 (total length of 3.4 kb) was highly homologous to the part of pKFR1-6 and pGX1 (K. xylinus E25). In addition, a consensus sequence of approximately 3.7 kb in pKFR1-1, pKFR1-2, pKFR1-3, pKFR1-4, pKFR1-5, pKFR1-6, pKFR2-1, and pKFR2-2 was identified. On this sequence, only transposase and resolvase were coded. Therefore, the same transposable elements were present on these plasmids.
Taking these findings together, one can infer that the genome structure of FNDCR1 and FNDCR2 strains are complex, leading us to predict that the genomes of these strains are genetically unstable.
Structure of the Cellulose Biosynthesis Operon
For cellulose biosynthesis, the bcsABCD genes are the most important genes: with other accessory genes, they are involved in efficient biosynthesis (Römling and Galperin, 2015). We manually extracted information related to bcs and other genes from the gff3 annotation file created using the prokka and RAST programs (Figure 9). It is noteworthy that many strains of Komagataeibacter bacteria harbor two bcs operons (type I and II). Typically, the type I and II operons, respectively, comprise bcsZ-bcsH-bcsAI-bcsBI-bcsCI-bcsDI-bglX, and bcsAII-bcsBII-bcsX-bcsY-bcsCII (the bcsA and bcsB genes are sometimes found as a fused gene, bcsAB) (Römling and Galperin, 2015; Ryngajłło et al., 2019).
The FNDCR1 and FNDCR2 strains harbored operons of both types as intact, however, only one bcs operon was found in the FNDCF1 strain. The sole bcs operon of the FNDCF1 strain was a fused one of type I and II operons, respectively. Furthermore, no bcsCI, bcsAII, or bcsBII gene was found. A premature stop codon appeared in the C-terminal part of bcsCII in the FNDCF1 strain (Figure 9). The loss of these genes can have affected the function of guanosine monophosphate (c-di-GMP)-regulated cellulose synthase and outer membrane porin proteins in the FNDCF1 strain. This operon structure explains why this strain does not produce cellulose at all.
We also found that the genes for Tiorf138 protein, hypothetical protein of Cupin superfamily, hydrolase of NUDIX family, and chaperone protein HtpG were located immediately downstream of bcsDI in all the presented strains. These four genes were also located similarly on the chromosomes of Komagataeibacter spp. E25, NBRC3288, and ATCC23769 strains (Supplementary Table 3), implying possible involvement of these genes in cellulose biosynthesis. The “Discussion” section provides additional details.
Discussion
The genetic instability of Komagataeibacter bacteria at least partly derives from the presence of the exceptionally huge number of mobile genetic elements on the genomes (Azuma et al., 2009; Zhang et al., 2017). Furthermore, high degrees of gene duplication, hyper-mutability, and complex plasmid contents have been reported for many Komagataeibacter bacteria (Römling and Galperin, 2015). The present study revealed a similar complex genomic structure of nata-de-coco producing Komagataeibacter bacteria. These findings show many truncations or insertions of ISs within the bcs operon genes of the present and other Komagataeibacter strains (Supplementary Table 3). These genetic variations are inferred to be the result of genomic structure complexity. Additionally, this report is the first of a positive correlation found between the number of mobile genetic elements and the number of duplicated sequences on the genomes (Figure 8) and the first describing that not all genomes of Komagataeibacter strains are complex (Figures 7, 8; the FNDCF1 strain had the simple genome).
The bcsCII gene of the FNDCF1 strain had a premature stop codon. Some bcs genes were not found on the genome (Figure 9). In the Komagataeibacter medellibensis NBRC3288 strain, the loss and restoration of cellulose productivity were observed according to a loss-of-function mutation (frameshift mutation) and intragenic suppressor mutations (reversion of frameshift) in the bcsBI gene (Matsutani et al., 2015). Furthermore, Komagateeibacter europaeus LMG18890 and LMG18494 strains are non-cellulose producers. They have some impairments in either bcsBI or bcsCI genes (Matsutani et al., 2015). A non-sense mutation in the bcsCI gene of Komagataeibacter oboediens MSKU3 has been reported to engender loss of cellulose productivity (Taweecheep et al., 2019). Therefore, we infer that the loss of cellulose productivity in the FNDCF1 strain is attributed to loss of a full functional set of the bcs operon.
Expression from the bcs operon in Komagataeibacter bacteria is known to be highly stimulated by c-di-GMP (Zhang et al., 2017). Therefore, the intracellular level of c-di-GMP is important for cellulose production. However, the FNDCF1 strain harbored five intact diguanylate cyclase/phosphodiesterase genes that are necessary to synthesize c-di-GMP, suggesting that the c-di-GMP level is normal in this strain. Similarly, the FNDCR1 and FNDCR2 strains, respectively, harbored five and two diguanylate cyclase/phosphodiesterase genes.
The genes for Tiorf138 protein, hypothetical protein of Cupin superfamily, hydrolase of NUDIX family, and chaperone protein HtpG are located immediately downstream of the bcs operon in many Komagataeibacter strains (Figure 9 and Supplementary Table 3). Hydrolases of NUDIX (nucleoside diphosphates-linked moiety-X) family hydrolyzes the pyrophosphate linkage in various nucleoside diphosphates linked to another moiety X (NDP-X) such as pyridine nucleotides, coenzyme A, dinucleoside polyphosphates, and nucleotide sugars (Fisher et al., 2002). Uridine-5′-diphosphate-α-D-glucose (UDP-glucose), a substrate for cellulose polymerization in bacteria (Ryngajłło et al., 2019), is a kind of NDP-X (a nucleotide sugar). In the case of glycogen biosynthesis in which UDP-glucose is a polymerization substrate, NUDIX hydrolases are thought to influence the flux of glucose into glycogen (Heyen et al., 2009). Therefore, the NUDIX hydrolases near the bcs operons might be involved in UDP-glucose degradation and might regulate cellulose biosynthesis. In this respect, determining the substrate specificity of those NUDIX hydrolases is important. Actually, HtpG, a chaperon protein of hsp90 family, is known to interact with proteins containing tetratrico peptide repeat (TPR) domain (Scheufler et al., 2000). In fact, because the N-terminal portion of BcsC contains 17 TPR, HtpG might interact with BcsC (Du et al., 2016; Nojima et al., 2017). Additionally, in Pseudomonas aeruginosa, results demonstrated that low intracellular levels of c-di-GMP increase the intracellular HtpG level (Chua et al., 2013).
In the phylogenetic classification based on 16S rRNA genes, all three isolated strains were assigned to Komagataeibacter genus. Phylogenetic tree analysis of the conserved core genes indicated that the three strains belong to mutually different clades. None of them matches any known strain. This finding suggests that the present strains are novel strains that have not been reported in the literature as Komagataeibacter strains. Duplicated genome regions as their genomic signature were also specifically examined. In fact, FNDCR1 and FNDCR2 have many duplicated sequences within their own genomes. By contrast, FNDCF1 has only a few duplicates in its own genome. It is particularly interesting that FNDCF1 has far fewer mobile genetic elements in its genome than the other two cellulose-producing lineages (FNDCR1 and FNDCR2), which might be attributable to the cross-strain variation in the sequence duplication among Komagataeibacter strains. After applying correlation analysis between the length of these homologous duplicated sequences and the number of mobile genetic elements, duplicates with minimum length of 1,000 bp were found to be positively correlated with the number of mobile genetic elements. Nevertheless, no correlation was found for duplicates with minimum length longer than 2,000 bp. This finding suggests that mobile genetic elements are related with transposition in the genome, but they might be subject to negative selection in their duplicated sequence lengths.
Our initial motivation for this study was to understand the genetic instability of nata-de-coco producing bacteria and stabilization of nata-de-coco production yields. For industrial production of acetic acid, Acetobacter pasteurianus 386B strain is exploited frequently. Its low number of mobile genetic elements in the genome and absence of complete phage genomes makes this strain more genetically stable than other A. pasteurianus strains (Illeghems et al., 2013; Ryngajłło et al., 2019). From results of the comparative genomics study presented herein, although FNDCF1 lacks the ability to produce cellulose, it can be expected to have less influence of mobile factors on genome stability. Given these findings, after the bcs operon of FNDCR1 or FNDCR2 strains is genetically transferred to the chromosome of FNDCF1 strain, it can be anticipated for use in stable industrial nata-de-coco production. To this end, useful genetic engineering technologies including genome editing should be developed for Komagataeibacter bacteria.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA763499, PRJNA763499.
Author Contributions
KI and NN designed the study, collected the data, conducted data analyses, and drafted the manuscript. HK, TI, and KK reviewed the manuscript. All authors read and approved the manuscript.
Conflict of Interest
HK was employed by company Research and Development Department, Fujicco Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Masako Sato, Ayane Osanai, and Miyako Hata for their assistance. We are also grateful to members of our research group for their help and valuable discussions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.798010/full#supplementary-material
Footnotes
- ^ https://github.com/FelixKrueger/TrimGalore
- ^ https://rast.nmpdr.org/
- ^ http://cgview.ca
- ^ https://github.com/tseemann/barrnap
- ^ https://www.arb-silva.de/aligner/
- ^ https://www.ncbi.nlm.nih.gov/genbank/
- ^ https://github.com/kblin/ncbi-genome-download
- ^ https://itol.embl.de/
References
Achaz, G., Boyer, F., Rocha, E. P. C., Viari, A., and Coissac, E. (2006). Repseek, a tool to retrieve approximate repeats from large DNA sequences. Bioinformatics 23:119121. doi: 10.1093/bioinformatics/btl519
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Azuma, Y., Hosoyama, A., Matsutani, M., Furuya, N., Horikawa, H., Harada, T., et al. (2009). Whole-genome analyses reveal genetic instability of Acetobacter pasteurianus. Nucleic Acids Res. 37:57685783. doi: 10.1093/nar/gkp612
Chawla, R. P., Bajaj, B. I., Survase, A. S., and Singhal, S. R. (2009). Microbial cellulose: fermentative production and applications. Food Technol. Biotechnol. 2:107124.
Chua, S. L., Tan, S. Y.-Y., Rybtke, M. T., Chen, Y., Rice, S. A., Kjelleberg, S., et al. (2013). Bis-(3′-5′)-Cyclic dimeric GMP regulates antimicrobial peptide resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 57:20662075. doi: 10.1128/aac.02499-12
Du, J., Vepachedu, V., Cho, S. H., Kumar, M., and Nixon, B. T. (2016). Structure of the cellulose synthase complex of Gluconacetobacter hansenii at 23.4 Å Resolution. PLoS One 11:e0155886. doi: 10.1371/journal.pone.0155886
Fisher, D. I., Safrany, S. T., Strike, P., McLennan, A. G., and Cartwright, J. L. (2002). Nudix hydrolases that degrade dinucleoside and diphosphoinositol polyphosphates also have 5-phosphoribosyl 1-pyrophosphate (prpp) pyrophosphatase activity that generates the glycolytic activator ribose 1,5-bisphosphate. J. Biol. Chem. 277:4731347317. doi: 10.1074/jbc.m209795200
Grant, J. R., and Stothard, P. (2008). The CGView server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 36:W181W184. doi: 10.1093/nar/gkn179
Grant, J. R., Arantes, A. S., and Stothard, P. (2012). Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genomics 13:202. doi: 10.1186/1471-2164-13-202
Heyen, C. A., Tagliabracci, V. S., Zhai, L., and Roach, P. J. (2009). Characterization of mouse UDP-glucose pyrophosphatase, a Nudix hydrolase encoded by the Nudt14 gene. Biochem. Biophys. Res. Commun. 390:14141418. doi: 10.1016/j.bbrc.2009.11.007
Iino, T., Suzuki, R., Tanaka, N., Kosako, Y., Ohkuma, M., Komagata, K., et al. (2012). Gluconacetobacter kakiaceti sp. nov., an acetic acid bacterium isolated from a traditional Japanese fruit vinegar. Int. J. Syst. Evol. Microbiol. 62:14651469. doi: 10.1099/ijs.0.031773-0
Illeghems, K., Vuyst, L. D., and Weckx, S. (2013). Complete genome sequence and comparative analysis of Acetobacter pasteurianus 386B, a strain well-adapted to the cocoa bean fermentation ecosystem. BMC Genomics 14:526. doi: 10.1186/1471-2164-14-526
Krawczyk, P. S., Lipinski, L., and Dziembowski, A. (2018). PlasFlow: predicting plasmid sequences in metagenomic data using genome signatures. Nucleic Acids Res. 46:gkx1321. doi: 10.1093/nar/gkx1321
Lee, K., Buldum, G., Mantalaris, A., and Bismarck, A. (2014). More than meets the eye in bacterial cellulose: biosynthesis, bioprocessing, and applications in advanced fiber composites. Macromol. Biosci. 14:1032. doi: 10.1002/mabi.201300298
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293-W296. doi: 10.1093/nar/gkab301
Londrina, P. R., Gomes, R. J., Borges, M., de, F., Fortaleza, C. E., Rosa, M., et al. (2018). Acetic Acid Bacteria in the food industry: systematics, characteristics and applications. Food Technol. Biotech. 56:139151. doi: 10.17113/ftb.56.02.18.5593
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A., and Zdobnov, E. M. (2021). BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654. doi: 10.1093/molbev/msab199
Matsutani, M., Ito, K., Azuma, Y., Ogino, H., Shirai, M., Yakushi, T., et al. (2015). Adaptive mutation related to cellulose producibility in Komagataeibacter medellinensis (Gluconacetobacter xylinus) NBRC 3288. Appl. Microbiol. Biotechnol. 99:72297240. doi: 10.1007/s00253-015-6598-x
Minkin, I., and Medvedev, P. (2020). Scalable multiple whole-genome alignment and locally collinear block construction with Sibelia Z. Nat. Commun. 11:6327. doi: 10.1038/s41467-020-19777-8
Nakashima, N., and Tamura, T. (2018). Whole-Genome Sequence of Acetobacter orientalis Strain FAN1, isolated from caucasian yogurt. Genome Announc. 6, e201–e218. doi: 10.1128/genomea.00201-18
Navarro, R. R., and Komagata, K. (1999). Differentiation of Gluconacetobacter liquefaciens and Gluconacetobacter xylinus based on DNA base composition, DNA relatedness, and oxidation products from glucose. J. Gen. Appl. Microbiol. 45:715. doi: 10.2323/jgam.45.7
Nguyen, L.-T., Schmidt, H. A., Haeseler, A., and von Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32:268274. doi: 10.1093/molbev/msu300
Nojima, S., Fujishima, A., Kato, K., Ohuchi, K., Shimizu, N., Yonezawa, K., et al. (2017). Crystal structure of the flexible tandem repeat domain of bacterial cellulose synthesis subunit C. Sci. Rep. 7:13018. doi: 10.1038/s41598-017-12530-0
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31:36913693. doi: 10.1093/bioinformatics/btv421
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41:D590D596. doi: 10.1093/nar/gks1219
Römling, U., and Galperin, M. Y. (2015). Bacterial cellulose biosynthesis: diversity of operons, subunits, products, and functions. Trends Microbiol. 23:545557. doi: 10.1016/j.tim.2015.05.005
Ross, P., Mayer, R., and Benziman, M. (1991). Cellulose biosynthesis and function in bacteria. Microbiol. Rev. 55:3558. doi: 10.1128/mr.55.1.35-58.1991
Ryngajłło, M., Kubiak, K., Jêdrzejczak-Krzepkowska, M., Jacek, P., and Bielecki, S. (2019). Comparative genomics of the Komagataeibacter strains – Efficient bionanocellulose producers. Microbiol. Open 8:e00731. doi: 10.1002/mbo3.731
Scheufler, C., Brinker, A., Bourenkov, G., Pegoraro, S., Moroder, L., Bartunik, H., et al. (2000). Structure of TPR domain peptide complexes critical elements in the assembly of the Hsp70 Hsp90 multichaperone machine. Cell 101:199210. doi: 10.1016/s0092-8674(00)80830-2
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:20682069. doi: 10.1093/bioinformatics/btu153
Taweecheep, P., Naloka, K., Matsutani, M., Yakushi, T., Matsushita, K., and Theeragool, G. (2019). Superfine bacterial nanocellulose produced by reverse mutations in the bcsC gene during adaptive breeding of Komagataeibacter oboediens. Carbohydr. Polym. 226:115243. doi: 10.1016/j.carbpol.2019.115243
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Zhang, H., Xu, X., Chen, X., Yuan, F., Sun, B., Xu, Y., et al. (2017). Complete genome sequence of the cellulose-producing strain Komagataeibacter nataicola RZS01. Sci. Rep. 7:4431. doi: 10.1038/s41598-017-04589-6
Keywords: cellulose, comparative genome analysis, duplicated sequences, Komagataeibacter, nata-de-coco production
Citation: Ishiya K, Kosaka H, Inaoka T, Kimura K and Nakashima N (2022) Comparative Genome Analysis of Three Komagataeibacter Strains Used for Practical Production of Nata-de-Coco. Front. Microbiol. 12:798010. doi: 10.3389/fmicb.2021.798010
Received: 19 October 2021; Accepted: 22 December 2021;
Published: 04 February 2022.
Edited by:
Jerome Combrisson, Mars, United StatesReviewed by:
Mario Tello, University of Santiago, ChileRodrigo Bentes Kato, Federal University of Minas Gerais, Brazil
Copyright © 2022 Ishiya, Kosaka, Inaoka, Kimura and Nakashima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nobutaka Nakashima, bi1uYWthaHNpbWFAYWlzdC5nby5qcA==