 Gaku Ueki
Gaku Ueki Sheng-Nan Zhang2†
Sheng-Nan Zhang2† Xiu-Jun Wen
Xiu-Jun Wen Kôhei Kubota
Kôhei Kubota
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 14 December 2021
Sec. Microbial Symbioses
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.794904
To deepen understanding the evolutionary process of lucanid–yeast association, the lateral transmission process of yeast symbionts among stag beetle genera Platycerus and Prismognathus around the border between Japan and South Korea was estimated based on molecular analyses and species distribution modelings. Phylogenetic analyses were based on yeast ITS and IGS sequences and beetle COI sequences using Prismognathus dauricus from the Tsushima Islands and Pr. angularis from Kyushu, Japan, as well as other sequence data from our previous studies. The range overlap based on the species distribution model (SDM) and differentiation in ecological space were analyzed. Based on the IGS sequences, Clade II yeast symbionts were shared by Platycerus hongwonpyoi and Pr. dauricus in South Korea and the Tsushima Islands, and Platycerus viridicuprus in Japan. Clade III yeasts were shared by Pr. dauricus from the Tsushima Islands and Pr. angularis in Japan. During the Last Interglacial period when the land bridge between Japan and the Korean Peninsula existed, range overlap was predicted to occur between Pl. viridicuprus and Pr. dauricus in Kyushu and between Pr. dauricus and Pr. angularis in Kyushu and the Tsushima Islands. The ecological space of Pl. hongwonpyoi was differentiated from that of Pl. viridicuprus and Pr. angularis. We demonstrated the paleogeographical lateral transmission process of Scheffersomyces yeast symbionts among lucanid genera and species: putative transmission of yeasts from Pr. dauricus to Pl. viridicuprus in Kyushu and from Pr. angularis to Pr. dauricus in Kyushu or the Tsushima Islands. We also found that the yeast symbionts are likely being replaced in Pr. dauricus on the Tsushima Islands. We present novel estimates of the lateral transmission process of microbial symbionts based on phylogenetic, SDM and environmental analyses among lucanid beetles.
In studies of insect–fungus symbiotic associations, particular attention has been paid to the mode of fungal transmission, because it is deeply related to the dependency between partners and the evolution of symbiotic systems. Vertical transmission (parent to offspring) is common in insect–fungus mutualism, whereas lateral transmission may occur between neighboring nests (Biedermann and Vega, 2020). Generally, the presence of fungus-carrying organs in insects indicates obligate dependencies and fungus productivity emphasizes genetic homogeneity (ants: Mueller et al., 1998; termites: Aanen et al., 2009). Meanwhile, lateral fungal transmission increases diversity and rarely results in obligate dependency evolution in insect-fungus symbiotic systems (West et al., 2015; Biedermann and Vega, 2020).
Stag beetles belonging to the family Lucanidae feed on decaying wood during the larval stage (Tanahashi et al., 2010, 2018; Tanahashi and Kubota, 2013). Adult lucanid females commonly possess a mycangium, an exoskeletal organ on the dorsal side of the abdominal tip carrying symbiotic microorganisms (Tanahashi et al., 2010; Tanahashi and Hawes, 2016; Kubota et al., 2020). The yeasts belong to the genus Scheffersomyces Kurtzman and Suzuki, 2010, a xylose-fermenting yeast group (Du Preez and Prior, 1985; Jeffries and Kurtzman, 1994; Olsson and Hahn-Hägerdal, 1996; Jeffries et al., 2007) including S. stipitis (Pignal) Kurtzman and Suzuki, 2010, S. segobiensis (Santa María and C. García) Kurtzman and Suzuki, 2010 or closely related species (Tanahashi et al., 2010; Kubota et al., 2020). It is nearly the only fungus found in almost all female mycangia of Platycerus Geoffroy, 1762; Prismognathus Motschulsky, 1860; Dorcus MacLeay, 1819; Lucanus Scopoli, 1763; and other lucanid species that feed on white rot wood (Tanahashi et al., 2010, 2017; Kubota et al., 2020). Although these Scheffersomyces yeast symbionts appear to serve an important role for the host stag beetles, their exact symbiont function remains unknown (Kubota et al., 2020).
Kubota et al. (2020) documented the co-evolutionary relationship between host lucanid beetles and their yeast symbionts based on a phylogeographic analysis of Platycerus. All yeast colonies from individual females appear to exhibit little to no genetic variation, and colonies from a particular lucanid population are closely related to one other (Tanahashi et al., 2017; Kubota et al., 2020). However, the symbiont and host beetle phylogenies are not at all congruent with each other, suggesting the occurrence of lateral transmissions and replacements of yeast symbionts between lucanid populations or species. Although the Scheffersomyces yeasts are temporarily found in the larval gut of wood-feeding beetles of the genera of Passalidae, Cerambycidae, Scarabaeidae, and Buprestidae (Santa-María and García-Aser, 1977; Suh et al., 2003, 2005), Scheffersomyces are not easily found in decaying wood except for larval galleries of lucanid species based on our observations and Roets and Oberlander (2020). Meanwhile, Scheffersomyces species, including S. stipitis, S. segobiensis, and their relatives, are almost always found in mycangium and gut of the many lucanid taxa (e.g., Platycerus, Prismognathus, Dorcus, and Lucanus, which are common in East Asia). Therefore, this group of Scheffersomyces appears to have essentially diverged along with lucanid beetles (Kubota et al., 2020).
Here, we focus on the yeast symbionts of Platycerus and Prismognathus lucanid species in Japan and South Korea. For Platycerus, only one species, Platycerus hongwonpyoi Imura and Choe, 1989, is known in Korea, whereas 10 species forming a monophyletic clade are recognized in Japan (Imura, 2010; Kubota et al., 2011). For Prismognathus, one species, Prismognathus dauricus (Motschulsky, 1860) occurs in Korea, and three species, Prismognathus angularis Waterhouse, 1874; Pr. tokui Kurosawa, 1975; and Pr. dauricus are found in Japan. Prismognathus tokui, which is endemic to Yakushima Island, is often treated as a subspecies of Pr. angularis (Fujita, 2010). In Japan, Pr. dauricus in Japan only occurs on the Tsushima Islands, located between the Japanese Archipelago and the Korean Peninsula (Figure 1). Prismognathus angularis is not found on the Tsushima Islands. These lucanid species are adapted to mature cool temperate deciduous broad-leaved forests, of which some species (e.g., Pr. dauricus) are also found in warm temperate evergreen broad-leaved forests.
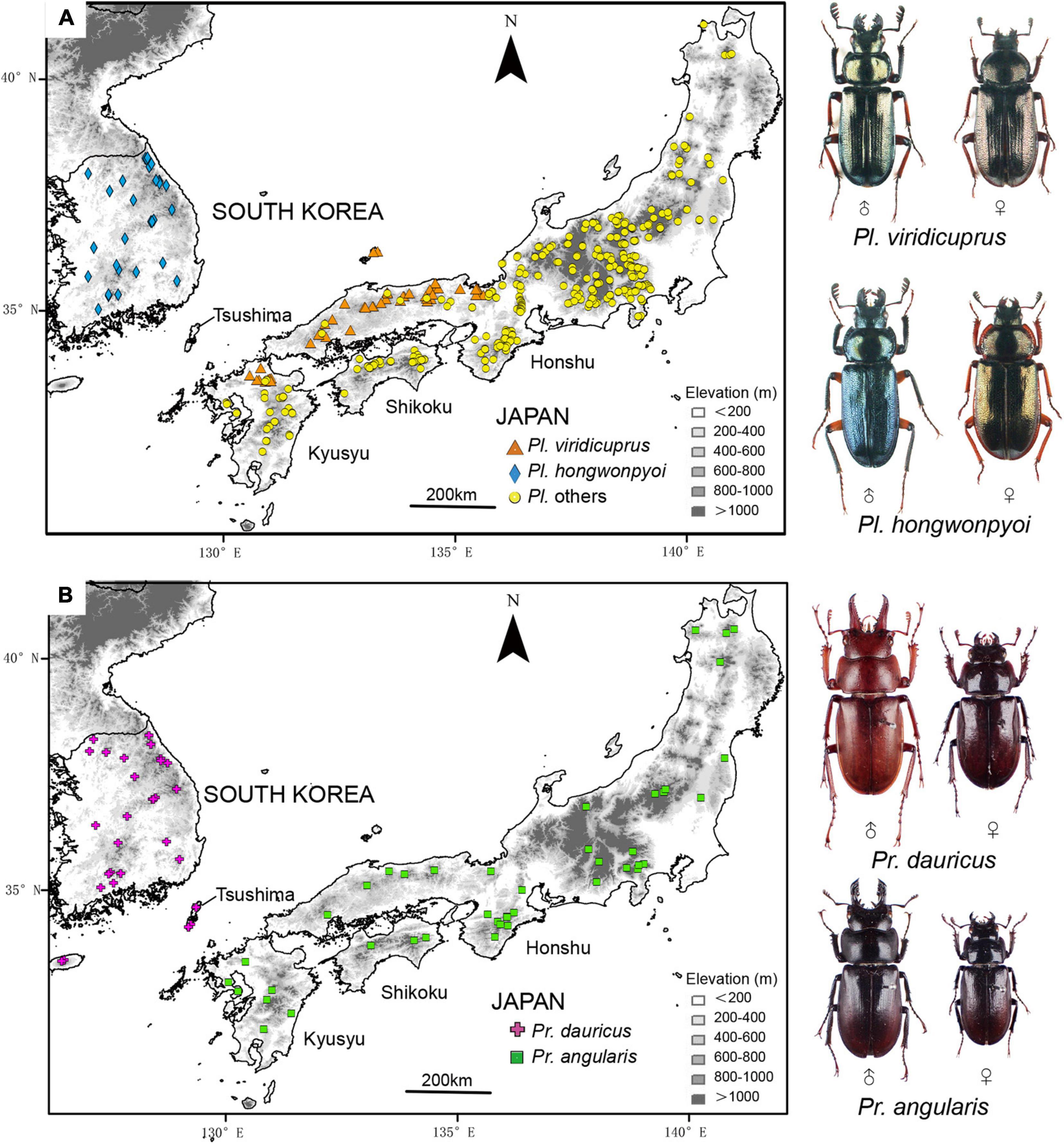
Figure 1. Distribution and occurrence data in Japan and South Korea for (A) Platycerus viridicuprus and Pl. hongwonpyoi; and (B) Prismognathus dauricus and Pr. angularis. Background map shows elevation, where higher intensity of the gray scale represents higher altitude.
Tanahashi et al. (2017); Kubota et al. (2020), and Zhu et al. (2020a) analyzed the sequences of the internal transcribed spacer (ITS) region including the initial portion of the 26S ribosomal RNA gene (26S rRNA) (about 650 bp), and the inter-genetic spacer (IGS) region including the 26S rRNA, IGS1, 5S ribosomal RNA gene (5S rRNA), IGS2, and the 5S ribosomal RNA gene (18S rRNA) (>2,000 bp), using all of the Platycerus and Prismognathus species, except for Japanese Pr. tokui. The ITS sequences of the yeast symbionts of Platycerus and Prismognathus in Japan and Korea are the same or little varied. Based on the IGS sequences, which are suitable for detecting strain-level genetic differences, the yeast symbionts form three distinct major clades (Tanahashi et al., 2017; Kubota et al., 2020). Of these, Korean Pl. hongwonpyoi and Pr. dauricus, and most populations of Japanese Platycerus viridicuprus Kubota et al., 2008, share an IGS lineage of yeast symbionts, which suggests recent lateral yeast transmission among these species when Japan and Korea were connected by land (Zhu et al., 2020a). This is the only report that phylogenetically distant lucanid taxa (Platycerus and Prismognathus) share yeasts of the same lineage.
The presence of the mycangium and co-evolutionary relationships in lucanid–yeast symbiotic systems suggest obligate dependency. The symbiotic relationship of Platycerus and Prismognathus with yeasts around the border of Japan and Korea offers the opportunity to estimate the rare lateral transmission process. Pr. dauricus is also distributed within the Tsushima Islands between Japan and Korea, where no Platycerus species are found. However, until now, no studies have examined the yeast symbionts of Pr. dauricus on the Tsushima Islands.
Species distribution models (SDMs) are statistical models that use observed distributional occurrence data to infer species’ ecological requirements and to map their potential habitat (Austin, 2002). SDM approaches have become a widely applied tool in not only macroecological studies, conservation strategies and the identification of niche evolution, but also in quantifying ecological divergence in closely related species (Zimmermann et al., 2010; Biber et al., 2020; Boys et al., 2021). These SDMs have been paired with paleoclimate data to hindcast past distributions and to understand how they shape current population structures (Forester et al., 2013; Yannic et al., 2014; Costa and Schlupp, 2020). Here, we focus on quantifying the range overlap and niche divergence among Platycerus and Prismognathus species using the Maximum Entropy Algorithm (MaxEnt). This approach is a high-performing and highly popular method for SDMs (Phillips et al., 2006) and provides the opportunity to explore the transmission process of yeast symbionts among lucanid taxa.
In the present study, to deepen understanding the evolutionary process of lucanid–yeast association, we examined the possible yeast lateral transmission or incomplete lineage sorting following ancestral polymorphism among the stag beetle genera Platycerus and Prismognathus, using Pr. dauricus from the Tsushima Islands and additional samples from a paleogeological perspective. We analyzed the evolutionary relationships among their yeast symbionts based on molecular analyses and predicted the past range and niche overlap among genera based on SDMs. Lastly, we discuss the lateral transmission process of yeast symbionts among species and genera.
Host insect sample collection sites in this and previous studies are presented in Supplementary Table 1 and Supplementary Figure 1. Isolated yeast strains from this and previous studies are shown in Supplementary Table 2. Prismognathus dauricus were obtained as adults at sites 34 and 35 (Supplementary Figures 1, 2). Prismognathus angularis were collected as larvae from decaying wood at sites 30 and 31. These larvae were reared individually to adulthood using the same wood that they inhabited in the field. Finally, we obtained seven adult females of all Prismognathus species collected from four sites in Japan.
The protocols of yeast isolation and determining of yeast DNA sequences were the same as in Kubota et al. (2020). They are described in Supplementary Appendices 1, 2. The primer information is shown in Supplementary Table 3. We used the MAFFT online service (Katoh et al., 2019) to align the sequences and determined 15 ITS and 15 IGS sequences of 56 yeast colonies from five P. dauricus and two P. angularis females (eight colonies per female) in total.
For the ITS analysis, we aimed to determine the phylogenetic positions of the yeast symbionts of Platycerus and Prismognathus within those of lucanid species and Scheffersomyces species. Thus, we used 46 yeast ITS sequences, of which 38 were symbionts of lucanid beetles reported in previous studies (one from Pr. dauricus of South Korea, two from Pr. angularis, 14 from Japanese Platycerus species, eight from Pl. hongwonpyoi of South Korea, and 13 from other lucanid species) (Tanahashi et al., 2017; Kubota et al., 2020; Zhu et al., 2020a; Supplementary Table 2).
For the IGS analysis, we focused on the phylogenies of the yeast symbionts of the four target species within those of Platycerus and Prismognathus species. We determined 15 IGS sequences from 56 symbiotic yeast isolates from five Pr. dauricus females and two Pr. angularis females. We also used 73 yeast ITS sequences, of which 72 were symbionts of lucanid beetles reported in previous studies (one from Pr. dauricus of South Korea, two from Pr. angularis, 61 from Japanese Platycerus species, and eight from Pl. hongwonpyoi of South Korea) (Tanahashi et al., 2017; Kubota et al., 2020; Zhu et al., 2020a; Supplementary Table 2). The IGS sequences of S. segobiensis were used as the outgroup of the yeast symbionts of Platycerus and Prismognathus species, because the former is closely related to, but distinct from the latter clade (Tanahashi et al., 2017).
Notably, both mitochondria and symbiotic yeasts are essentially transmitted from mother to offspring. Because it is reasonable to compare evolutionary patterns between the cytochrome oxidase subunit I (COI) gene of the host beetles and the IGS gene of the yeast symbionts, we sequenced the COI gene of the host beetles. Most sequence data for COI used in this study were from previous studies, as listed in Supplementary Table 2 (Kubota et al., 2010, 2011, 2020; Kubota and Kubota, 2011; Tanahashi et al., 2017; Zhu et al., 2020a).
The protocol of determining of host beetle COI sequences was the same as in Kubota et al. (2020). They are described in Supplementary Appendix 3. The primer information is shown in Supplementary Table 3. To align the sequences, we used the same methods as with the yeast DNA sequences. For the COI analysis, we focused on the phylogenetic relationship between Platycerus and Prismognathus species in Japan and South Korea. We determined seven COI sequences (five Pr. dauricus females and two Pr. angularis females).
We constructed a haplotype matrix for each of the ITS and IGS regions of yeasts and the COI gene of beetles using the DnaSP v. 6.12.01 software package (Rozas et al., 2017). Aligned haplotype sequences were used to construct phylogenetic trees based on maximum likelihood (ML) and Bayesian inference (BI) methods. ML trees were constructed using RAxML v. 8.2.9 (Stamatakis, 2014) under the best-fit substitution model selected using Kakusan4 (Tanabe, 2007) based on Schwarz’s Bayesian information criterion (BIC; Schwarz, 1978). Confidence at each node was assessed using 1,000 bootstrap replications. BI trees were constructed using MrBayes5D v. 3.2.6 (Tanabe, 2008), a modified version of MrBayes (Huelsenbeck and Ronquist, 2001), under the best-fit substitution model by Kakusan4, for 10 million generations (samplefreq = 1,000), with the first 1,000,000 generations discarded as “burn-in.” The Bayesian log likelihood trace files, burn-in times, and summary statistics of estimated parameters were visualized using Tracer ver. 1.6 (Rambaut and Drummond, 2007). Tree information was visualized and edited using FigTree v. 1.3.1 (Rambaut, 2009).
Considering the known distribution of the four beetle species, we selected southern Japan and Korea (126°E–136°E, 31°N–38°N) as the geographical extent of our study (Figure 2). The occurrence data for Platycerus viridicuprus (n = 53), Pl. hongwonpyoi (n = 31), Prismognathus dauricus (n = 31), and Pr. angularis (n = 27) were obtained from field surveys and previous studies (Fujita, 2010; Imura, 2010; Kubota et al., 2011; Zhu et al., 2019, 2020a; Zhang and Kubota, 2021a; Figure 2).
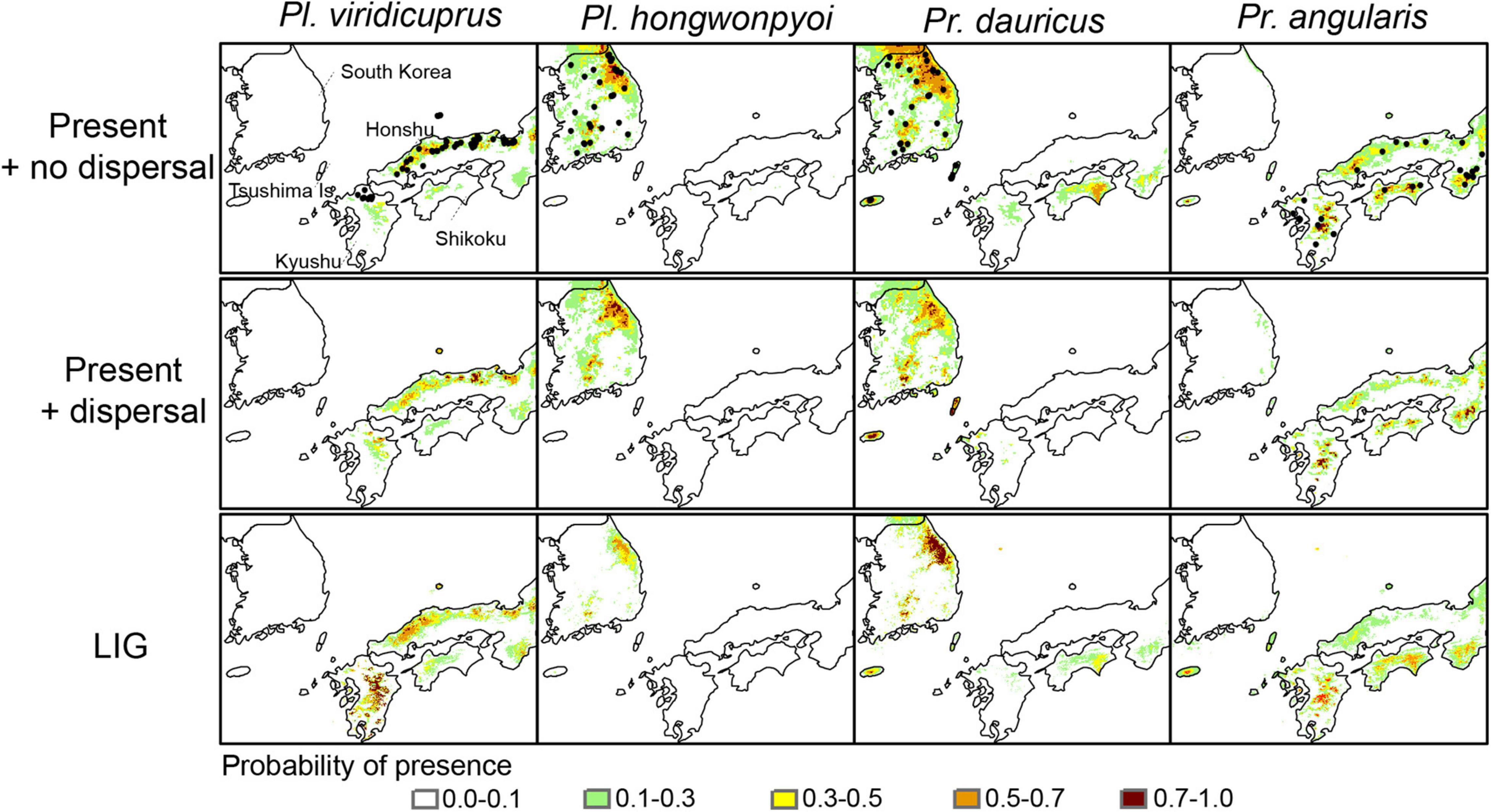
Figure 2. Locations of occurrences (black circles) and potentially suitable habitat for Platycerus viridicuprus, Pl. hongwonpyoi, Prismognathus dauricus, and Pr. angularis under present climate conditions (no dispersal, dispersal), the Last Interglacial (LIG) period in the study area. The colored areas represent moderate-to-high probability of species presence (>0.1).
Predictor variables were obtained from WorldClim with a spatial resolution of about 2.5 arc-min (v. 1.4; Hijmans et al., 2005).1 We then calculated pairwise Pearson’s correlation coefficients (r) and retained one predictor when two or more were highly correlated (i.e., | r| <0.80) (Lobo et al., 2010). When there were alternative candidate predictors to choose either, we gave priority to the predictor used in Homburg et al. (2013); Bosso et al. (2018), and Zhang and Kubota (2021a,b), which includes important predictors for Platycerus and other beetle species. In addition, we avoided Bio14 and Bio15 for past prediction, as it is biased when projected to past climate scenarios (Varela et al., 2015). Thus, we selected the following seven predictors for modeling: mean diurnal range (Bio2), isoterhermality (Bio3), maximum temperature of warmest month (Bio5), mean temperature of wettest quarter (Bio8), mean temperature of coldest quarter (Bio11), precipitation of wettest month (Bio13), and precipitation of driest quarter (Bio17) (Table 1 and Supplementary Table 4). We generated rasters for these variables at 2.5 arc-min for the Present time and at 30 arc-sec for the Last Interglacial (LIG; ∼120–140 ka) period. Notably, the last land-bridge between Japan and the Korean Peninsula disappeared in the beginning of LIG, they have not since been connected, including the Last Glacial Maximum (LGM) (Ohshima, 1990; Guedes et al., 2016).

Table 1. List of environmental variables used in the study.
We used SDMs for the four study species using MaxEnt v. 3.4.1, a machine learning presence−background algorithm, which is particularly suitable when absence data are not available (Phillips et al., 2006). The 10,000 background points were selected using the default setting. In MaxEnt, we used 70% of the occurrence data for model training and the remaining 30% of occurrence data for model evaluation. Ten replications were then chosen to run the model. The logistic output format was used for the model results, which indicates the habitat suitability of target species, and the resulting ASCII file was depicted using ArcGIS v. 10.4.1. Model performance was evaluated using the threshold independent area under the curve (AUC). AUC values of 1 indicate a perfect model based on the presence data of the samples, while a value of 0.5 indicates no difference from random model predictions (Phillips et al., 2006).
We also applied the null model approach for testing if the model performed significantly better than random expectation in the “dismo” package (Raes and ter Steege, 2007; Hijmans et al., 2017). A total of 100 different data sets with the same number of random points as the species occurrences in this study region was used to calculate the AUC for each species. We compared the AUC values with the ones calculated for a null model using Mann-Whitney U-tests.
Given that dispersal ability plays a key role in determining the distributional areas of species, we then conducted SDMs at the present time by incorporating the mid-range of locations (MR) dispersal constraint using the method of Zhang and Kubota (2021a). Here we used Euclidean distance to parameterize the dispersal kernel from the source point to the occurrence points of each species. A source point was defined by the mid-range longitude and latitude of the locations of each species (MR scenario). Although a SDM incorporating this scenario is effective to predict the species range of lucanid species in the present and future (Zhang and Kubota, 2021a), this modeling cannot be used for predictions in the past due to the possibility of recent local extinction.
In addition, we compared the raw MaxEnt outputs to evaluate niche similarity in their predicted niche distribution using ENMTools v. 1.4.4 (Warren et al., 2010). We calculated values of range overlap (Warren et al., 2008), Schoener’s D (Schoener, 1968), and Warren’s I index (Warren et al., 2008) among members of Pl. viridicuprus vs. Pl. hongwonpyoi, Pl. viridicuprus vs. Pr. dauricus, and Pr. dauricus vs. Pr. angularis. These indices range between 0 and 1, and values close to 1 indicate high similarity between the two compared groups.
To detect differentiation of the four study species in environmental space, we performed principal component analysis (PCA) to calculate principal components using the seven bioclimatic variables (Table 1) available at the Worldclim website. Then we extracted environmental values at the occurrence localities using ArcGIS v. 10.4.1 for each species. The statistical analysis was conducted using the R “stats” package v. 3.6.3 (R Core Team, 2013) with prcomp function and the result was visualized by function ggbiplot of “ggbiplot” package (Vu, 2011) and “ggplot2” (Wickham, 2007).
We obtained yeast symbiont colonies from mycangial homogenates prepared from all Prismognathus females collected in Japan. The colonies on PDA plates were uniform, white, circular and protuberant, with a smooth edge and matte surface. The yeast CFU values ranged from 1.2 × 102 to 2.7 × 104 for Prismognathus dauricus and Pr. angularis.
None of the ITS sequences of the yeast symbionts from the two Prismognathus angularis collected in Japan exhibited any sequence variation (containing partial 18S rRNA, ITS1, 5.8S rRNA, ITS2, and partial 26S rRNA; 651-bp; n = 6). These sequences were completely identical to sequences previously reported from two Pr. angularis specimens from Japan (Tanahashi et al., 2017; Zhu et al., 2020a). The ITS sequences for the yeast symbionts from three Pr. dauricus females (GU32, GU36, and GU37) collected in Tsushima also shared identical sequences (n = 7) with the yeast symbionts from Pr. angularis. By contrast, two sequences (652-bp) from a Pr. dauricus female (GU34) in Tsushima contained one nucleotide substitution and one insertion compared to those of the yeast symbionts from Pr. angularis. The latter sequences were identical to one Korean individual of Pr. dauricus (Zhu et al., 2020a) and the Korean and Japanese Platycerus species (Tanahashi et al., 2017; Kubota et al., 2020). Furthermore, the ITS sequences of eight yeast colonies isolated from one individual Pr. dauricus collected in Tsushima included both haplotypes: three yeast colonies were identical to the former, and the remaining five were identical to the latter. These sequences of yeast symbionts from Prismognathus and Platycerus species were similar to that of Scheffersomyces segobiensis (Santa-María and García-Aser, 1977), an already described yeast species that was first isolated from a jewel beetle Chalcophora mariana massiliensis (Coleoptera: Buprestidae) (Santa-María and García-Aser, 1977). In addition, the phylogenetic analysis based on the ITS sequences revealed that the yeast symbionts of the Prismognathus and Platycerus species constitute a distinct lineage to the clade of yeast symbionts of all other lucanid species, including environmental Scheffersomyces (Figure 3 and Supplementary Table 2). In total, 42 nucleotide substitutions and 12 insertions in the ITS were recognized among the yeast symbionts of Lucanid species and Scheffersomyces species.
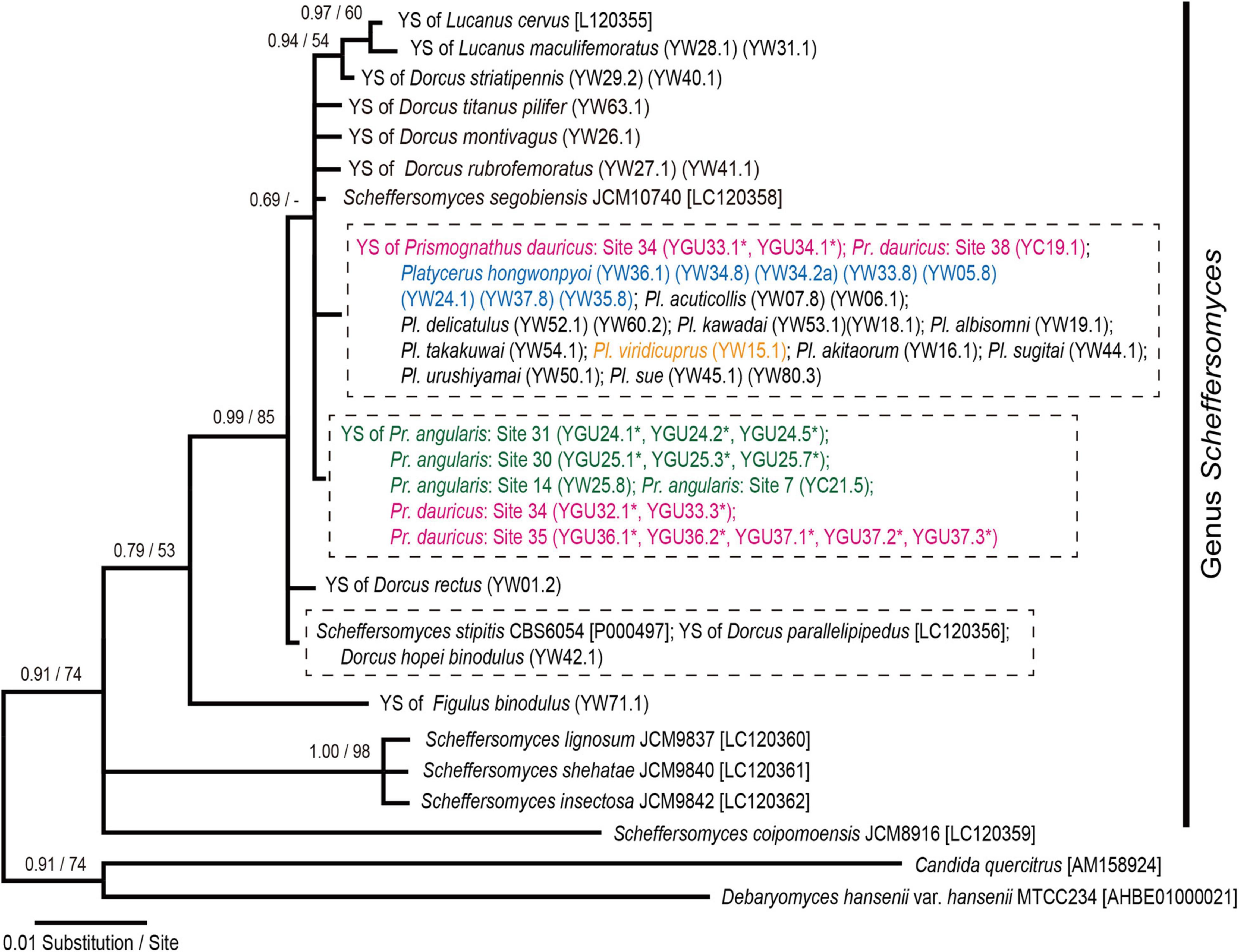
Figure 3. Bayesian inference (BI) phylogeny of the yeast symbionts of Platycerus, Prismognathus, and other stag beetles, and some yeast species based on ITS sequences. Numbers at the nodes indicate posterior probability for BI phylogeny (>50%)/bootstrap probability for maximum likelihood (ML) phylogeny (>50%). YS indicates yeast symbionts. Numbers following YW, YGU, or YC indicate the female code number and colony number (see Supplementary Table 2). Dashed boxes indicate ITS haplotypes shared by more than one species of the host stag beetles. Asterisks indicate the sequences determined in this study. HKY 85 + Gamma model (BI) and GTR + Gamma model (ML) were selected as the best-fit substitution models by Kakusan4. The BI and ML phylogenies are identical except for peripheral branches.
The IGS sequences covering partial 26S rRNA, IGS1, 5S rRNA, IGS2, and partial 18S rRNA (2,184–2,195-bp; n = 16) were determined for Prismognathus angularis and Pr. dauricus. In total, 75 nucleotide substitutions and 23 insertions in the IGS were recognized. In Pr. angularis, the IGS sequences of eight colonies isolated from a host female exhibited 1–5 nucleotide substitutions and one insertion for the two females examined (GU24 and GU25), suggesting that genetic variation exists among the yeast symbionts in the mycangium of a single female. In Pr. dauricus, the IGS sequences of the eight colonies isolated from one host female were identical for the two females (GU32 and GU34) and exhibited two nucleotide substitutions and two insertions for the two females (GU36 and GU37) examined. Exceptionally, two different IGS sequences each contained 67 nucleotide substitutions and 10 insertions among the eight yeast colonies from one female (GU33) of Pr. dauricus.
In the IGS phylogeny, all yeast symbionts from all Platycerus species, one Pr. dauricus female in Tsushima and one Pr. dauricus female in South Korea, as well as a portion of the yeast symbionts from one Pr. dauricus female in Tsushima, formed a monophyletic clade (Figure 4). In this clade, two major subclades were recognized: Clade I contained primarily the yeast symbionts of the Japanese Platycerus species, whereas Clade II contained primarily those of the Pl. viridicuprus populations in Japan, all populations of Pl. hongwonpyoi in South Korea, two Pr. dauricus females in Tsushima and one Pr. dauricus female in South Korea. The yeast sequences from all Pr. angularis females collected in Japan and the four Pr. dauricus females in Tsushima formed another monophyletic clade (Clade III).
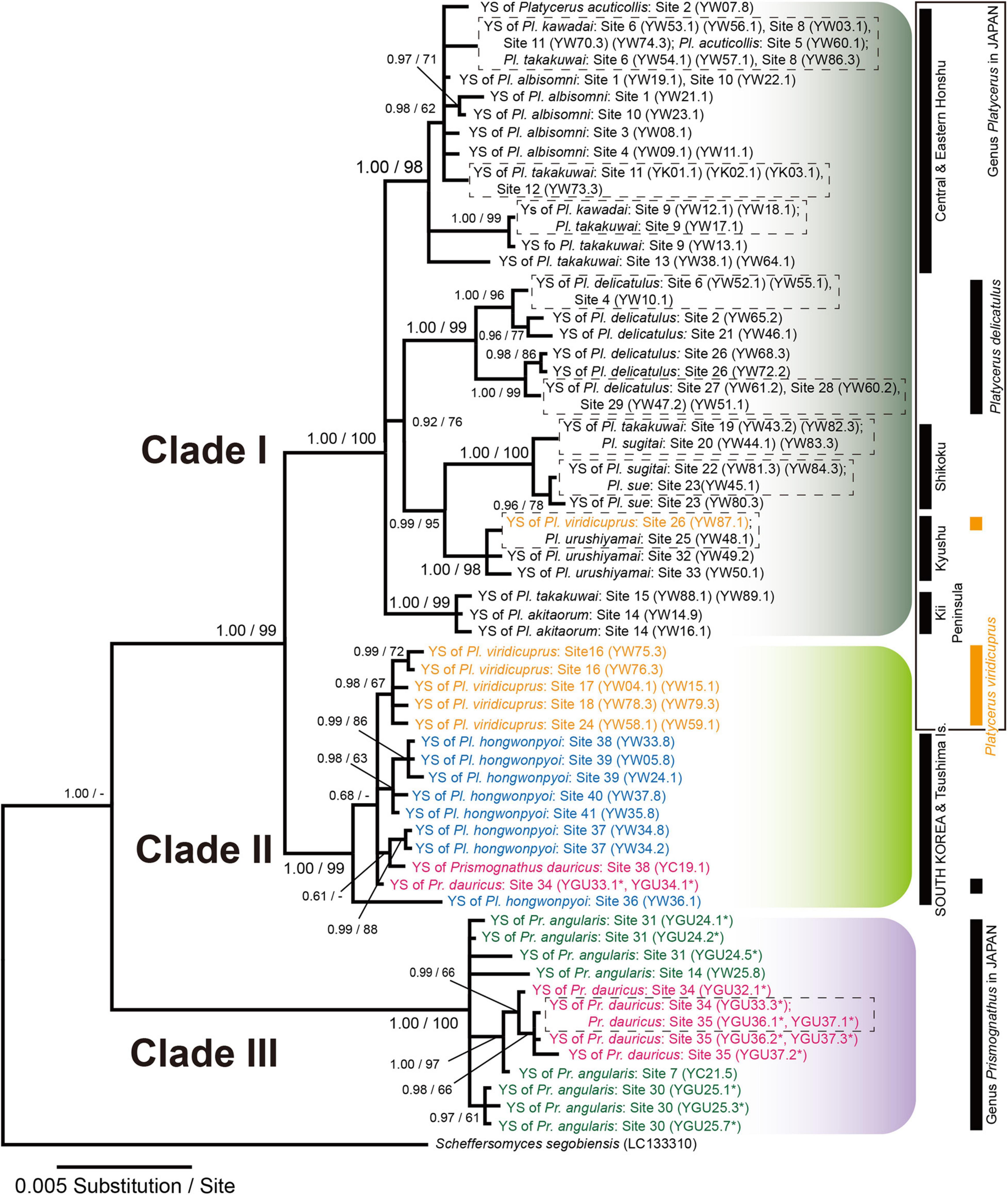
Figure 4. Bayesian inference (BI) phylogeny of the yeast symbionts of Platycerus and Prismognathus stag beetles based on IGS sequences. Scheffersomyces segobiensis was treated as the outgroup. Numbers at the nodes indicate posterior probability for BI phylogeny (>50%)/bootstrap probability for maximum likelihood (ML) phylogeny (>50%). YS indicates yeast symbionts. Numbers following YGU, YW, or YC indicate the female code number and colony number (see Supplementary Table 2). Dashed boxes indicate IGS haplotypes shared by more than one species of the host stag beetles. Asterisks indicate the sequences determined in this study. HKY 85 + Gamma model (BI) and GTR + Gamma model (ML) were selected as the best-fit substitution models by Kakusan4. The BI and ML phylogenies are identical except for peripheral branches.
The yeast symbionts of Clades II (GU33, 34) and III (GU32, 33, 36, 37) were isolated from Pr. dauricus females in Tsushima. GU33 was the only female that possessed symbionts belonging to both Clades II and III (Figure 4 and Supplementary Table 2).
We constructed the COI phylogeny of the host insects (Figure 5) and compared it to the IGS phylogeny of the yeast symbionts. Essentially, the host phylogeny and the symbiont phylogeny were not congruent, indicating that no strict host-symbiont co-speciation has been established in the evolutionary course of the Prismognathus and Platycerus stag beetles. The genera Prismognathus and Platycerus constituted an independent distant monophyletic group. Furthermore, Pr. dauricus constituted a different clade from that of Pr. angularis. Although 10 Japanese Platycerus species are distinctly identified by morphological features including genital organs (Kubota et al., 2009), introgressive hybridizations have occurred between some species combinations (e.g., Pl. takakuwai) (Figure 5). Such species always possess the Clade I yeasts in the IGS. Pl. viridicuprus forms a distinct monophyletic clade (Figure 5).
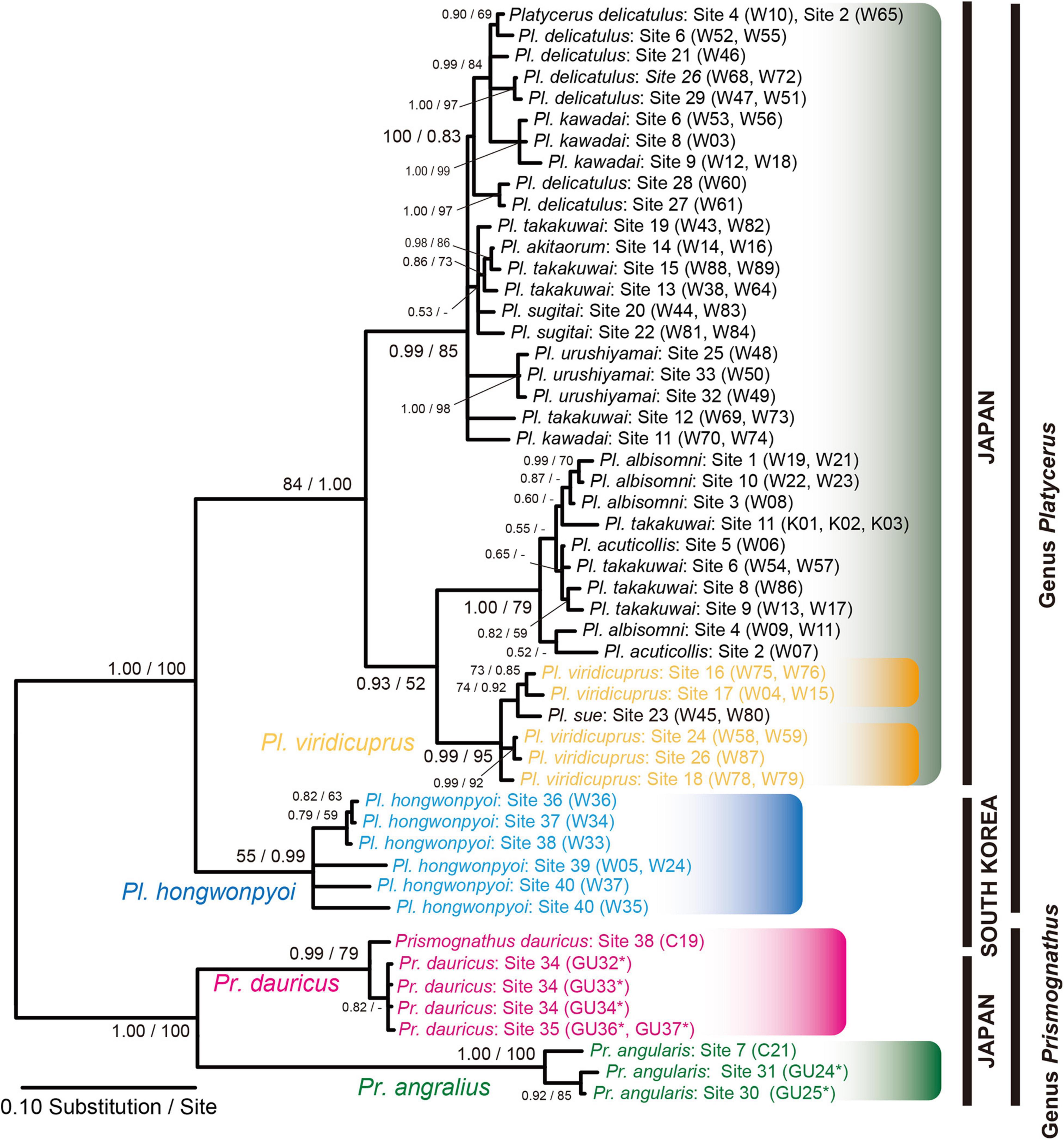
Figure 5. Bayesian inference (BI) phylogeny of Platycerus and Prismognathus stag beetles based on COI gene sequences. Numbers at the nodes indicate posterior probability for BI phylogeny (>50%)/bootstrap probability for maximum likelihood (ML) phylogeny (>50%). Asterisks indicate the sequences determined in this study. GU, W, or C indicate the female code number (see Supplementary Table 2). Asterisks indicate the sequences determined in this study. HKY 85 + Gamma Invariant model (BI) and GTR + Gamma model (ML) were selected as the best-fit substitution models by Kakusan4. The BI and ML phylogenies are identical except for peripheral branches.
The mean AUC values for all the models and target species were 0.902–0.977; these models were significantly better than the null models, indicating that the models performed well (p < 0.001; Mann–Whitney U-test; Supplementary Table 5). The potential distributions of the four study species were well aligned with the known species’ ranges under present models with their respective dispersal constraints, although the potential area based on the models with no dispersal was larger than the areas for models incorporating dispersal constraints (Figure 2). For Pl. hongwonpyoi and Pr. angularis, the potential ranges during the LIG were smaller than those based on present climate conditions. By contrast, the MaxEnt models indicated a large potential range for Pl. viridicuprus in the LIG. In that time, the occurrences of Pl. viridicuprus in the Korean Peninsula and Pl. hongwonpyoi in Japan were not supported, whereas those of both Pr. dauricus and Pr. angularis in Kyushu, Japan, were predicted (Figure 2).
Range overlap and niche overlap analysis under present climatic conditions with no dispersal in the study region exhibited a pattern of little overlap for the SDMs of Pl. hongwonpyoi vs. Pl. viridicuprus and for Pr. viridicuprus vs. Pr. dauricus, but larger overlap for Pr. dauricus vs. Pr. angularis (Schoener’s D = 0.261, Warren’s I = 0.540, Range overlap = 0.318) (Table 2). In addition, the models that incorporated dispersal ability predicted the smallest overlap for all examined species pairs. During the LIG period, the lowest amount of overlap occurred between Pl. hongwonpyoi and Pl. viridicuprus (Schoener’s D = 0.029, Warren’s I = 0.111, Range overlap = 0), while the highest amount of overlap was between Pr. dauricus and Pr. angularis (Schoener’s D = 0.208, Warren’s I = 0.438, Range overlap = 0.306) (Table 2).
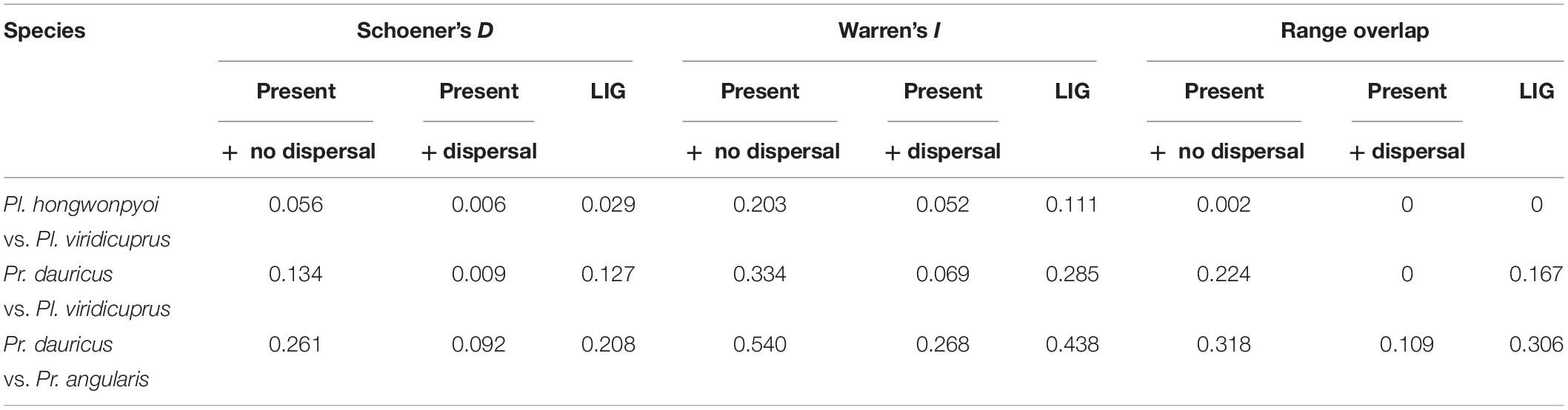
Table 2. Niche overlap (Schoener’s D, Warren’s I) values and range overlap for species-pair comparisons at Present and the Last interglacial (LIG) between Platycerus and Prismognathus species.
The potential distribution of the four species and their associated environmental variables can be estimated using the environmental PCA space (Figure 6 and Supplementary Table 6). The first two axes explained more than 78% of the variation in distribution. The climatic variables that contributed most to the first two principal components were mean temperature of coldest quarter (Bio11, 0.506 to PC1) and maximum temperature of warmest month (Bio5, 0.536 to PC2). In the environmental space, we found differentiation between Pl. viridicuprus, Pl. hongwonpyoi, and Pr. dauricus. However, the environment for Pl. viridicuprus was similar to that of Pr. dauricus, but more restricted. In addition, Pr. dauricus exhibited the largest environmental range.
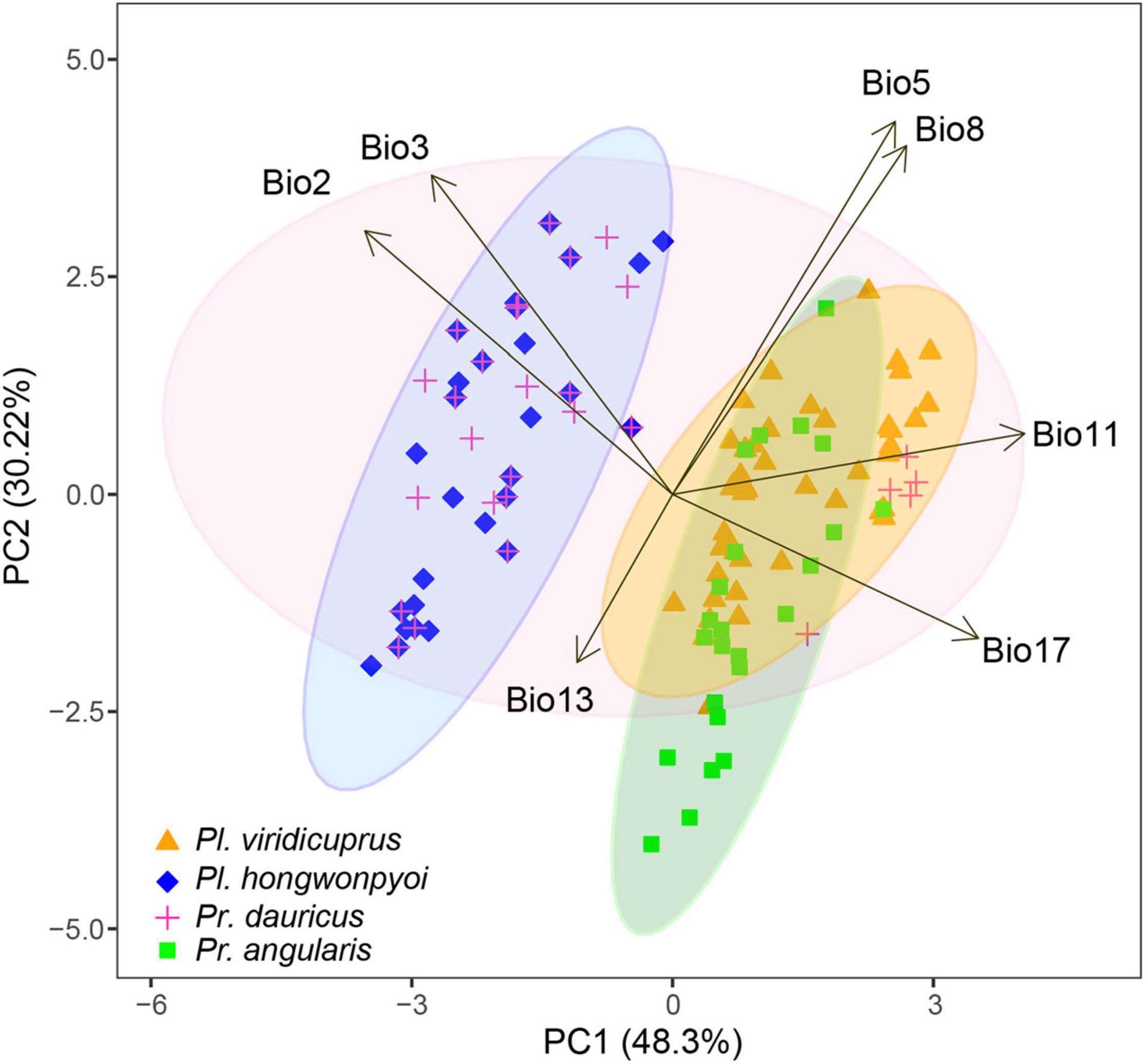
Figure 6. Principal component analysis (PCA) plots of the study species based on seven bioclimatic variables. Different colors indicate different species. Ellipses define 95% confidence intervals.
The Platycerus species and their yeast symbionts co-evolved to some extent but incompletely based on their genetic phylogenies (Kubota et al., 2020). These results strongly suggest that lateral transfers and/or replacements of Scheffersomyces yeast symbionts are essentially rare but have occurred repeatedly during the evolutionary history of Platycerus (and probably also in other lucanid taxa).
To date, IGS sequence analyses have indicated that the yeast symbionts of Japanese and Korean Platycerus and Prismognathus possess the same monophyletic yeast symbionts [Clades (I + II + III), Figure 4]. This clade of symbionts is distinct from any other known yeast symbionts of other lucanid genera in East Asia (Aegus, Dorcus, Prosopocoilus, Lucanus, Neolucanus, Figulus, Nigidius, Aesalus, Nicagus, and Ceruchus: Tanahashi et al., 2010; Kubota et al., 2020; Zhu et al., 2020a; Figure 3), as stag beetles generally possess species- or genus-specific lineages of yeasts. That is, the lateral transmission of yeast symbionts between lucanid taxa is rare. The ancestor of all Japanese Platycerus species diverged from the ancestor of Asian continental Platycerus species, including Pl. hongwonpyoi, about 10 Mya (Zhu et al., 2020b). Compared to it, Platycerus and Prismognathus diverged earlier than 10 Mya (Figure 5). On the other hand, the yeast symbionts of Japanese Pl. viridicuprus covering a large area of species range (Sites 16, 17, 18, and 24: Supplementary Table 1) form a monophyletic clade, which is in a derived position compared to those of the Korean species in Clade II (Figures 4, 5). All other yeast symbionts are those of the Japanese Platycerus species belonging to Clade I (Figure 4). Therefore, to explain the phylogenetic discordance between the lucanid species and their yeast symbionts by the incomplete lineage sorting following ancestral polymorphism, the yeast IGS sequences within Clade II are too close (Figure 4). These findings suggest that the yeast symbionts were laterally transmitted from the Korean Platycerus or Prismognathus to Pl. viridicuprus.
This study presents novel results that a lucanid population (and a female) possesses two lineages of yeasts of different origins (Figures 3, 4). Based on IGS sequences at both the population and individual level (GU33), Pr. dauricus of the Tsushima Islands possesses yeast symbionts belonging to Clades II and III. Of these, the yeasts belonging to Clade III were closely related to and derived from those of Japanese Pr. angularis (Figure 4) and these two lucanid species seem to diverged earlier than 10 Mya (Figure 5), suggesting that the yeast symbionts (Clade III) were laterally transmitted from Japanese Pr. angularis to Pr. dauricus around the Tsushima Islands.
Scheffersomyces yeast symbionts can survive outside the symbiotic organ of the insect hosts and are cultivable (Supplementary Figure 2). These yeasts are commonly found in and have been isolated from the galleries of decaying wood inhabited by lucanid larvae based on our observations. The Scheffersomyces yeasts are involved in xylose fermentation and are thus likely to be adapted to the use of woody materials (Jeffries et al., 2007; Tanahashi et al., 2010). Meanwhile, the genetic lineages of the yeast symbionts corresponded to their host lucanid taxa (i.e., species or genus) (Figures 3, 4). These local host-symbiont compatibilities are likely due to the mycangium-mediated vertical transmission of the yeast symbionts generally found in stag beetles (Tanahashi et al., 2010). We have observed that Platycerus and Prismognathus species prefer similar mature temperate deciduous broad-leaved forests and similar white-rot wood. In fact, the adults or larvae of the two genera are often found in the same pieces of decaying wood in Japan and South Korea (Zhu et al., 2020a), which may lead to the lateral transmission of yeast symbionts among these taxa. It is unlikely that the yeast symbionts of Clades (I + II + III) were transmitted from other sympatric lucanid taxa to Platycerus or Prismognathus species, since the other taxa exclusively possess their own yeast lineages not but Clades (I + II + III) (Tanahashi et al., 2010; Kubota et al., 2020). Insects other than lucanid species may temporally possess and bring the yeast symbionts of Lucanid species. Even if yeast transmission between lucanid individuals via such other insects occurs, it is considered the same as transmission from a lucanid to a lucanid.
For yeast transmission between Japanese and Korean species to have occurred, they were likely sympatric in the past. However, Pl. viridicuprus and Pr. angularis in Japan are currently allopatrically distributed relative to Pl. hongwonpyoi (Korea) and Pr. dauricus (Korea and Tsushima in Japan, Figure 1). Therefore, estimations of the distributions of these species in the past are critical to the discussion of yeast transmission. Since Platycerus and Prismognathus prefer mature broad-leaved forests and showed fine geographical genetic variations due to their low dispersal ability (Figure 5; Kubota et al., 2011; Zhu et al., 2020a), they are unlikely to disperse across the sea. Because the last connection between Korea and Japan via the Tsushima land-bridge disappeared approximately 130 ka (the end of the Riss ice age) (Ohshima, 1990; Guedes et al., 2016), SDM projections for the target species in the LIG (∼120–140 ka) estimate the last sympatric possibilities.
Based on SDM projections, there is no potential range overlap between Pl. viridicuprus and Pl. hongwonpyoi both at present and in the LIG. Moreover, the environmental spaces did not overlap between these two species (Figure 2). These estimations suggest minimal possibility of lateral yeast transmission from Pl. hongwonpyoi to Pl. viridicuprus. Conversely, the SDM predicted larger range and niche overlaps between Pr. dauricus and Pl. viridicuprus, especially in the LIG (Figure 2 and Table 2). Their environmental spaces also largely overlapped (Figure 6). In the LIG, the range of Pr. dauricus was expected to extend to Japan, whereas that of Pl. viridicuprus was not expected to extend to the Tsushima Islands or South Korea (Figure 2). These findings suggest the possibility of lateral yeast transmission from Pr. dauricus to Pl. viridicuprus in Japan. The larger overlap of ecological space between these two species based on the PCA is consistent with this estimation (Figure 6). The ecological space of Pr. dauricus are separated to that in the Tsushima Islands and South Korea. Since the genetic differentiation of P. dauricus between both areas is less than those of P. angularis, Pl. hongwonpyoi, and Pl. viridicuprus within species (Figure 5), it is likely that Pr. dauricus has larger niche range than other species.
Additionally, the yeast symbionts of Clade III are likely to have been transmitted from Pr. angularis to Pr. dauricus (Figure 4). For these two species, the potential range and niche overlaps were relatively high all in the present, LIG (Figure 2 and Table 2). Because Pr. angularis and Pr. dauricus are actually allopatric, range exclusion appears to exist between them, perhaps caused by interspecific competition or reproductive interference. If so, it is possible that these two species have been in contact with each other in Tsushima or Kyushu, which is close to the current boundary. Since Pl. viridicuprus hardly reached Tsushima in the LIG, Pr. dauricus likely existed in Kyushu then. Our hypotheses are summarized in Figure 7: the Clade II yeasts were likely transmitted from Pr. dauricus to Pl. viridicuprus in Kyushu, and the Clade III yeasts were transmitted from Pr. angularis to Pr. dauricus around Tsushima. For the yeast symbionts of Pr. dauricus in Tsushima, replacement from Clade II to Clade III may be occurring.
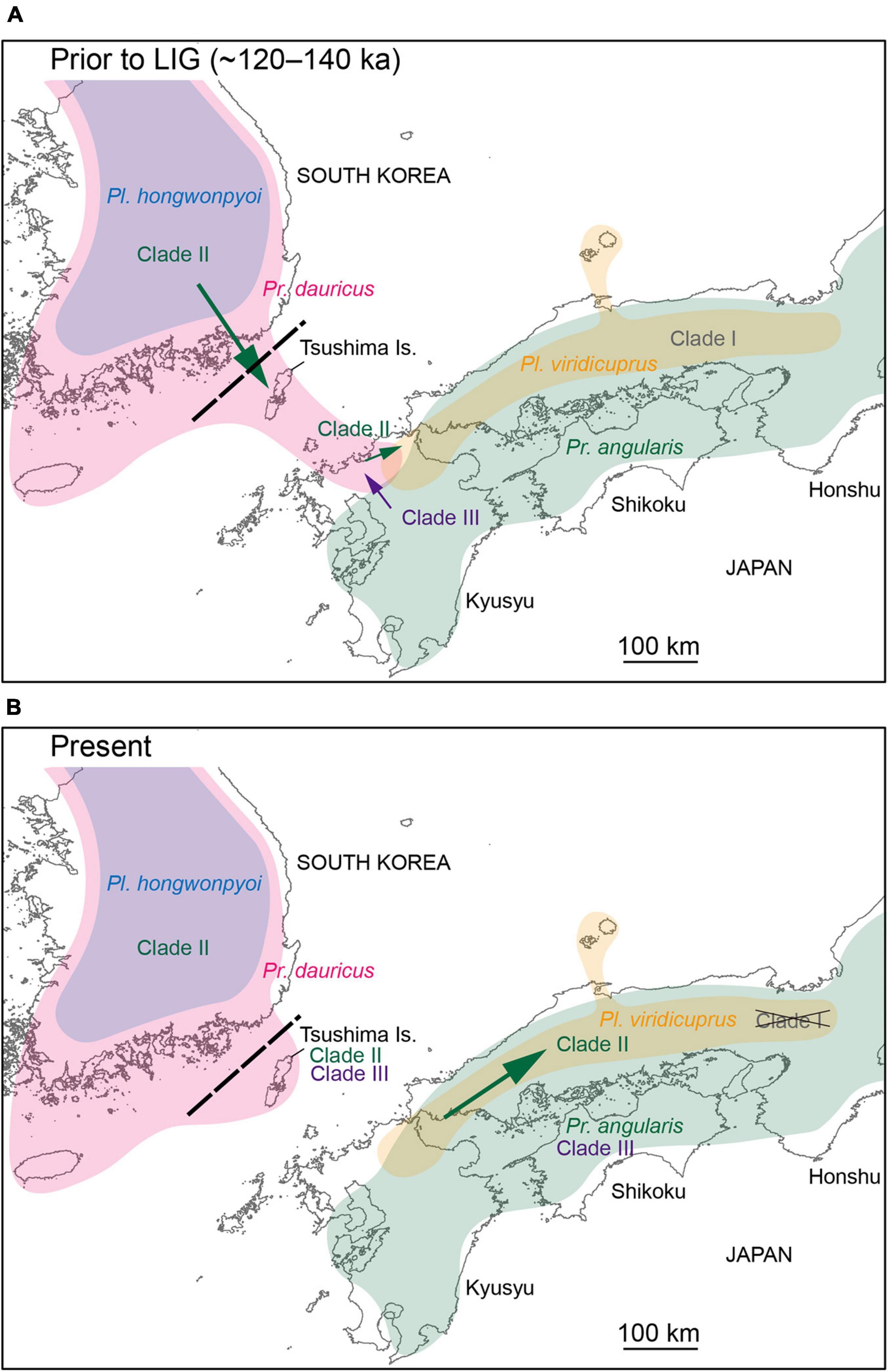
Figure 7. Schematic diagram of the lateral transmission of yeast symbionts among Platycerus and Prismognathus species in Japan and South Korea. During a certain period prior to the last interglacial (LIG) period, Platycerus hongwonpyoi and Prismognathus dauricus shared Clade II yeasts in South Korea. Platycerus viridicuprus and Prismognathus angularis possessed Clade I and Clade III yeasts, respectively, and did not share yeast symbionts in Japan. (A) Prior to or during the LIG, Pr. dauricus dispersed from Korea to Kyushu through the Tsushima Islands. Clade II yeasts were laterally transmitted from Pr. dauricus to Pl. viridicuprus in Kyushu. Meanwhile, Clade III yeasts were transmitted from Pr. angularis to Pr. dauricus in Kyushu (or on the Tsushima Islands, if Pr. angularis had reached there at that time). (B) At the present time, Clade I yeasts have been nearly replaced by Clade II yeasts in Pl. viridicuprus in Kyushu and western Honshu. On the Tsushima Islands, Clade II yeasts are being replaced by Clade III yeasts, or they coexist in Pr. dauricus.
Although 10 Platycerus species, including Pl. viridicuprus and Pr. angularis are widely sympatric in Japan, there is no Platycerus species possessing Clade III yeasts (n = 67) or Pr. angularis possessing Clade I or II yeasts (n = 7) (Tanahashi et al., 2010, 2017; Kubota et al., 2020; Zhu et al., 2020a). Therefore, yeast transmission between Platycerus and Pr. angularis is unlikely to occur. In addition, Pl. viridicuprus is distributed sympatrically with Pl. delicatulus and parapatrically with Pl. urushiyamai and Pl. takakuwai in western Japan, whereas Japanese Platycerus other than Pl. viridicuprus always possess the Clade I yeasts and not Clade II yeasts, even if they are collected from the same wood piece. By contrast, some Japanese Platycerus species share the same lineage of yeasts. These facts indicate that the presence or absence of the yeast transmission is varied even among closely related species. However, the benefit of yeast replacement for the host lucanid species and yeasts is still unclear.
Applications of SDMs to paleobiology have grown substantially during the last decade due to their accessibility and the improved availability of paleoclimate simulations (Svenning et al., 2011; Gavin et al., 2015). Specifically, previous research using SDM typically had several main objectives: to understand past shifts of species with climate; to identify glacial refugia, particularly during the cold period of the Last Glacial Maximum (LGM, ∼21 ka); and to determine the effect of climate and interspecific interactions in moving hybrid zones (Engler et al., 2013; Rödder et al., 2013; Sosa-Pivatto et al., 2017; Coelho et al., 2019). However, the LIG projections in the present study have provided insight into microbial transmission among the study species. We also present novel estimates of the lateral transmission process of microbial symbionts based on phylogenetic, SDM and environmental analyses among lucanid beetles. Such elucidation of the yeast transmission as in this study will contribute to our understanding of the evolutionary dynamics of insect–microbial symbiotic systems, especially the impacts to co-evolutionary relationships.
The sequence datasets presented in this study can be found in the online repository of DNA Data Bank of Japan (DDBJ). The accession numbers can be found in Supplementary Table 2.
GU, S-NZ, and X-JZ were interested in evolutionary ecology of stag beetles and their yeast symbionts. GU contributed to the data generation of this study and performed the genetic analyses. S-NZ performed the SDM and ecological space analysis. KK contributed to the study design. GU and S-NZ wrote the manuscript under the supervising of KT and KK. All authors approved the final version of the manuscript.
This study was partly supported by the Sasakawa Scientific Research Grant from The Japan Science Society, the Shimabara Peninsula UNESCO Geopark Academic Research Encouragement Project to GU, a research grant from the Institute of Mountain Science, Shinshu University to KT, and the Japan Society for the Promotion of Science KAKENHI Grant Nos. 25292082 and 21H04736 to KK.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are grateful to the Kyushu Regional Environmental Office of the Ministry of the Environment and the Kyushu Forest Management Bureau for allowing us to collect protected beetles. Specimens from Mt. Unzendake area of the Unzen-Amakusa National Park were collected with permits issued to GU and KT of Shinshu University from Agency for Cultural Affairs.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.794904/full#supplementary-material
Aanen, D. K., de Fine Licht, H. H., Debets, A. J., Kerstes, N. A., Hoekstra, R. F., and Boomsma, J. J. (2009). High symbiont relatedness stabilizes mutualistic cooperation in fungus-growing termites. Science 326, 1103–1106. doi: 10.1126/science.1173462
Austin, M. (2002). Spatial prediction of species distribution: an interface between ecological theory and statistical modelling. Ecol. Model. 157, 101–118. doi: 10.1016/S0304-3800(02)00205-3
Biber, M. F., Voskamp, A., Niamir, A., Hickler, T., and Hof, C. (2020). A comparison of macroecological and stacked species distribution models to predict future global terrestrial vertebrate richness. J. Biogeogr. 47, 114–129. doi: 10.1111/jbi.13696
Biedermann, P. H., and Vega, F. E. (2020). Ecology and evolution of insect–fungus mutualisms. Annu. Rev. Entomol. 65, 431–455. doi: 10.1146/annurev-ento-011019-024910
Bosso, L., Smeraldo, S., Rapuzzi, P., Sama, G., Garonna, A. P., and Russo, D. (2018). Nature protection areas of Europe are insufficient to preserve the threatened beetle Rosalia alpina (Coleoptera: Cerambycidae): evidence from species distribution models and conservation gap analysis. Ecol. Entomol. 43, 192–203. doi: 10.1111/een.12485
Boys, W. A., Siepielski, A. M., Smith, B. D., Patten, M. A., and Bried, J. T. (2021). Predicting the distributions of regional endemic dragonflies using a combined model approach. Insect Conserv. Divers. 14, 52–66. doi: 10.1111/icad.12444
Coelho, M. T. P., Rodrigues, J. F. M., Diniz-Filho, J. A. F., and Rangel, T. F. (2019). Biogeographical history constrains climatic niche diversification without adaptive forces driving evolution. J. Biogeogr. 46, 1020–1028. doi: 10.111/jbi.13553
Costa, G. C., and Schlupp, I. (2020). Placing the hybrid origin of the asexual Amazon molly (Poecilia formosa) based on historical climate data. Biol. J. Linn. Soc. 129, 835–843. doi: 10.1093/biolinnean/blaa010
Du Preez, J., and Prior, B. (1985). A quantitative screening of some xylose-fermenting yeast isolates. Biotechnol. Lett. 7, 241–246. doi: 10.1007/BF01042370
Engler, J. O., Rödder, D., Elle, O., Hochkirch, A., and Secondi, J. (2013). Species distribution models contribute to determine the effect of climate and interspecific interactions in moving hybrid zones. J. Evol. Biol. 26, 2487–2496. doi: 10.1111/jeb.12244
Forester, B. R., DeChaine, E. G., and Bunn, A. G. (2013). Integrating ensemble species distribution modelling and statistical phylogeography to inform projections of climate change impacts on species distributions. Divers. Distrib. 19, 1480–1495. doi: 10.1111/ddi.12098
Gavin, D. G., Fitzpatrick, M. C., Gugger, P. F., Heath, K. D., Rodríguez-Sánchez, F., Dobrowski, S. Z., et al. (2015). Climate refugia: joint inference from fossil records, species distribution models and phylogeography. New Phytol. 204, 37–54. doi: 10.1111/nph.12929
Guedes, J. D., Austermann, J., and Mitrovica, J. X. (2016). Lost foraging opportunities for East Asian hunter-gatherers due to rising sea level since the last glacial maximum. Geoarchaeology 31, 255–266. doi: 10.1002/gea.21542
Hijmans, R. J., Cameron, S. E., Parra, J. L., Jones, P. G., and Jarvis, A. (2005). Very high resolution interpolated climate surfaces for global land areas. Int. J. Climatol. 25, 1965–1978. doi: 10.1002/joc.1276
Hijmans, R. J., Phillips, S., Leathwick, J., Elith, J., and Hijimans, M. R. J. (2017). Package ‘dismo’. Circles 9, 1–68.
Homburg, K., Drees, C., Gossner, M. M., Rakosy, L., Vrezec, A., and Assmann, T. (2013). Multiple glacial refugia of the low-dispersal ground beetle Carabus irregularis: molecular data support predictions of species distribution models. PLoS One 8:e61185. doi: 10.1371/journal.pone.0061185
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Imura, Y. (2010). The genus Platycerus of East Asia. Tokyo: Roppon-Ashi Entomological Books. (in Japanese, partly in English and Chinese).
Jeffries, T., and Kurtzman, C. (1994). Strain selection, taxonomy, and genetics of xylose-fermenting yeasts. Enzyme Microb. Technol. 16, 922–932. doi: 10.1016/0141-0229(94)90001-9
Jeffries, T. W., Grigoriev, I. V., Grimwood, J., Laplaza, J. M., Aerts, A., Salamov, A., et al. (2007). Genome sequence of the lignocellulose-bioconverting and xylose-fermenting yeast Pichia stipitis. Nat. Biotechnol. 25, 319–326.
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kubota, K., and Kubota, N. (2011). Gene flow between two closely related species in the acuticollis species group (Coleoptera: lucanidae: genus Platycerus) near their distribution border based on morphology and mitochondrial gene. Entomol. Sci. 14, 198–202. doi: 10.1111/j.1479-8298.2010.00424.x
Kubota, K., Kubota, N., and Otobe, H. (2010). Mitochondrial gene diversity of Platycerus sue (Coleoptera, Lucanidae), a temporarily designated endangered species based on the law for the conservation of endangered species of wild fauna and flora. Bull. Biogeogr. Soc. Jpn. 65, 151–158. (In Japanese, with English title and Abstract),Google Scholar
Kubota, K., Nagahata, Y., Ikeda, H., Kubota, N., Otobe, H., and Umetsu, K. (2011). Diversification process of stag beetles belonging to the genus Platycerus Geoffroy (Coleoptera: lucanidae) in Japan based on nuclear and mitochondrial genes. Entomol. Sci. 14, 411–427.
Kubota, K., Nagahata, Y., Kubota, N., and Otobe, H. (2009). A morphological phylogeny of the genus Platycerus (Coleoptera, Lucanidae) in Japan. Biogeography 11, 57–72.
Kubota, K., Watanabe, K., Zhu, X.-J., Kawakami, K., Tanahashi, M., and Fukatsu, T. (2020). Evolutionary relationship between Platycerus stag beetles and their mycangium-associated yeast symbionts. Front. Microbiol. 11:1436. doi: 10.3389/fmicb.2020.01436
Lobo, J. M., Jiménez-Valverde, A., and Hortal, J. (2010). The uncertain nature of absences and their importance in species distribution modelling. Ecography 33, 103–114. doi: 10.1111/j.1600-0587.2009.06039.x
Mueller, U. G., Rehner, S. A., and Schultz, T. R. (1998). The evolution of agriculture in ants. Science 281, 2034–2038. doi: 10.1126/science.281.5385.2034
Ohshima, K. (1990). The history of straits around the Japanese Islands the late-quqternary. Quat. Res. 29, 193–208. (in Japanese with English title and abstract)
Olsson, L., and Hahn-Hägerdal, B. (1996). Fermentation of lignocellulosic hydrolysates for ethanol production. Enzyme Microb. Technol. 18, 312–331. doi: 10.1016/0141-0229(95)00157-3
Phillips, S. J., Anderson, R. P., and Schapire, R. E. (2006). Maximum entropy modeling of species geographic distributions. Ecol. Model. 190, 231–259. doi: 10.1016/j.ecolmodel.2005.03.026
R Core Team. (2013). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Raes, N., and ter Steege, H. (2007). A null-model for significance testing of presence-only species distribution models. Ecography 30, 727–736. doi: 10.1111/j.2007.0906-7590.05041.x
Rambaut, A. (2009). FigTree, Version 1.3.1. Available online at: http://tree.bio.ed.ac.uk/software/figtree/ (accessed March 8, 2021).
Rambaut, A., and Drummond, A. J. (2007). Tracer, Version 1.5. MCMC Trace Analysis Tool. Available online at: http://tree.bio.ed.ac.uk/software/tracer/ (accessed March 8, 2021).
Rödder, D., Lawing, A. M., Flecks, M., Ahmadzadeh, F., Dambach, J., Engler, J. O., et al. (2013). Evaluating the significance of paleophylogeographic species distribution models in reconstructing Quaternary range-shifts of Nearctic Chelonians. PLoS One 8:e72855. doi: 10.1371/journal.pone.0072855
Roets, F., and Oberlander, K. C. (2020). Symbiotic yeasts from the mycangium, larval gut and woody substrate of an African stag beetle Xiphodontus antilope (Coleoptera: lucanidae). Antonie Van Leeuwenhoek 113, 1123–1134. doi: 10.1007/s10482-020-01418-1
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Santa-María, J., and García-Aser, C. (1977). Pichia segobiensis, sp. nov. An. Inst. Nac. Invest. Agrar. Ser. Gen. 5, 45–50.
Schoener, T. W. (1968). The Anolis lizards of Bimini: resource partitioning in a complex fauna. Ecology 49, 704–726. doi: 10.2307/1935534
Schwarz, G. (1978). Estimating the dimension of a model. Ann. Statist. 6, 461–464. doi: 10.1214/aos/1176344136
Sosa-Pivatto, M., Cosacov, A., Baranzelli, M. C., Iglesias, M. D. R., Espíndola, A., and Sérsic, A. N. (2017). Do 120,000 years of plant-pollinator interactions predict floral phenotype divergence in Calceolaria polyrhiza? A reconstruction using species distribution models. Arthropod Plant Interact. 11, 351–361. doi: 10.1007/s11829-016-9490-4
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Suh, S. O., Marshall, C. J., Mchugh, J. V., and Blackwell, M. (2003). Wood ingestion by passalid beetles in the presence of xylose-fermenting gut yeasts. Mol. Ecol. 12, 3137–3145. doi: 10.1046/j.1365-294X.2003.01973.x
Suh, S.-O., McHugh, J. V., Pollock, D. D., and Blackwell, M. (2005). The beetle gut: a hyperdiverse source of novel yeasts. Mycol. Res. 109, 261–265. doi: 10.1017/S0953756205002388
Svenning, J.-C., Fløjgaard, C., Marske, K. A., Nógues-Bravo, D., and Normand, S. (2011). Applications of species distribution modeling to paleobiology. Quat. Sci. Rev. 30, 2930–2947. doi: 10.1016/j.quascirev.2011.06.012
Tanabe, A. S. (2007). Kakusan: a computer program to automate the selection of a nucleotide substitution model and the configuration of a mixed model on multilocus data. Mol. Ecol. Notes 7, 962–964. doi: 10.1111/j.1471-8286.2007.01807.x
Tanabe, A. S. (2008). MrBayes5D, Version 3.2.6. Available online at: https://www.fifthdimension.jp/products/mrbayes5d/ (accessed March 8, 2021)
Tanahashi, M., and Hawes, C. J. (2016). The presence of a mycangium in European Sinodendron cylindricum (Coleoptera: lucanidae) and the associated yeast symbionts. J. Insect Sci. 16:76. doi: 10.1093/jisesa/iew054
Tanahashi, M., Ikeda, H., and Kubota, K. (2018). Elementary budget of stag beetle larvae associated with selective utilization of nitrogen in decaying wood. Sci. Nat. 105:33. doi: 10.1007/s00114-018-1557-x
Tanahashi, M., Kim, J.-K., Watanabe, K., Fukatsu, T., and Kubota, K. (2017). Specificity and genetic diversity of xylose-fermenting Scheffersomyces yeasts associated with small blue stag beetles of the genus Platycerus in East Asia. Mycologia 109, 630–642. doi: 10.1080/00275514.2017.1382648
Tanahashi, M., and Kubota, K. (2013). Utilization of the nutrients in the soluble and insoluble fractions of fungal mycelium by larvae of the stag beetle, Dorcus rectus (Coleoptera: lucanidae). Eur. J. Entomol. 110, 611–615. doi: 10.14411/eje.2013.083
Tanahashi, M., Kubota, K., Matsushita, N., and Togashi, K. (2010). Discovery of mycangia and the associated xylose-fermenting yeasts in stag beetles (Coleoptera: lucanidae). Naturwissenschaften 97, 311–317. doi: 10.1007/s00114-009-0643-5
Varela, S., Lima-Ribeiro, M. S., and Terribile, L. C. (2015). A short guide to the climatic variables of the last glacial maximum for biogeographers. PLoS One 10:e0129037. doi: 10.1371/journal.pone.0129037
Vu, V. Q. (2011). ggbiplot: A ggplot2 Based Biplot. Available online at: http://github.com/vqv/ggbiplot (accessed March 3, 2021).
Warren, D. L., Glor, R. E., and Turelli, M. (2008). Environmental niche equivalency versus conservatism: quantitative approaches to niche evolution. Evolution 62, 2868–2883. doi: 10.1111/j.1558-5646.2008.00482.x
Warren, D. L., Glor, R. E., and Turelli, M. (2010). ENMTools: a toolbox for comparative studies of environmental niche models. Ecography 33, 607–611. doi: 10.1111/j.1600-0587.2009.06142.x
West, S. A., Fisher, R. M., Gardner, A., and Kiers, E. T. (2015). Major evolutionary transitions in individuality. Proc. Natl. Acad. Sci. U.S.A. 112, 10112–10119. doi: 10.1073/pnas.1421402112
Wickham, H. (2007). The ggplot package. Available online at: http://ftp.uni-bayreuth.de/math/statlib/R/CRAN/doc/packages/ggplot.pdf (accessed March 3, 2021).
Yannic, G., Pellissier, L., Ortego, J., Lecomte, N., Couturier, S., Cuyler, C., et al. (2014). Genetic diversity in caribou linked to past and future climate change. Nat. Clim. Chang. 4, 132–137. doi: 10.1038/nclimate2074
Zhang, S.-N., and Kubota, K. (2021a). Dispersal constraints on the potential distribution of cold-adapted stag beetles (genus Platycerus) in Japan and the implications of climate change. Insect Conserv. Divers. 14, 356–366. doi: 10.1111/icad.12461
Zhang, S.-N., and Kubota, K. (2021b). Integrating intraspecific variation in species distribution models by quantifying niche differentiation. Biol. J. Linn. Soc. 133, 187–201. doi: 10.1093/biolinnean/blab021
Zhu, X.-J., Jang, T.-W., Kim, J.-K., and Kubota, K. (2019). Genetic divergence of Platycerus hongwonpyoi (Coleoptera: lucanidae) in South Korea. Entomol. Sci. 22, 86–97. doi: 10.1111/ens.12337
Zhu, X.-J., Jang, T.-W., and Kubota, K. (2020a). First report of two phylogenetically distant lucanid taxa, Platycerus hongwonpyoi Imura et Choe and Prismognathus dauricus (Motschulsky) (Coleoptera, Lucanidae) in South Korea, sharing the same lineage of yeast symbionts. Elytra N. Ser. 10, 187–194.
Zhu, X.-J., Ma, T., Imura, Y., Wen, X.-J., and Kubota, K. (2020b). Molecular phylogeny and historical biogeography of the genus Platycerus (Coleoptera, Lucanidae) in East Asia. Zool. Scr. 49, 582–595. doi: 10.1111/zsc.12429
Keywords: phylogenetic analysis, species distribution model, ecological space, Scheffersomyces, Prismognathus dauricus, Prismognathus angularis, Platycerus viridicuprus, Platycerus hongwonpyoi
Citation: Ueki G, Zhang S-N, Zhu X-J, Wen X-J, Tojo K and Kubota K (2021) Lateral Transmission of Yeast Symbionts Among Lucanid Beetle Taxa. Front. Microbiol. 12:794904. doi: 10.3389/fmicb.2021.794904
Received: 14 October 2021; Accepted: 15 November 2021;
Published: 14 December 2021.
Edited by:
Chih-Horng Kuo, Academia Sinica, TaiwanReviewed by:
Jen-Pan Huang, Academia Sinica, TaiwanCopyright © 2021 Ueki, Zhang, Zhu, Wen, Tojo and Kubota. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kôhei Kubota, a29oZWlAZnIuYS51LXRva3lvLmFjLmpw
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.