 Antón Ambroa1,2
Antón Ambroa1,2 Lucia Blasco1,2
Lucia Blasco1,2 María López1,2,3
María López1,2,3 Olga Pacios1,2
Olga Pacios1,2 Inés Bleriot1,2
Inés Bleriot1,2 Laura Fernández-García1,2
Laura Fernández-García1,2 Manuel González de Aledo1
Manuel González de Aledo1 Concha Ortiz-Cartagena1,2
Concha Ortiz-Cartagena1,2 Andrew Millard4†
Andrew Millard4† María Tomás1,2,3*†
María Tomás1,2,3*†
- 1Microbiology Department-Research Institute Biomedical A Coruña (INIBIC), Hospital A Coruña (CHUAC), University of A Coruña (UDC), A Coruña, Spain
- 2Study Group on Mechanisms of Action and Resistance to Antimicrobials (GEMARA) the Behalf of the Spanish Society of Infectious Diseases and Clinical Microbiology (SEIMC), Madrid, Spain
- 3Spanish Network for Research in Infectious Diseases (REIPI), Infectious Diseases Network Biomedical Research Center (CIBERINFEC), Carlos III Health Institute, Madrid, Spain
- 4Department of Genetics and Genome Biology, University of Leicester, Leicester, United Kingdom
To optimize phage therapy, we need to understand how bacteria evolve against phage attacks. One of the main problems of phage therapy is the appearance of bacterial resistance variants. The use of genomics to track antimicrobial resistance is increasingly developed and used in clinical laboratories. For that reason, it is important to consider, in an emerging future with phage therapy, to detect and avoid phage-resistant strains that can be overcome by the analysis of metadata provided by whole-genome sequencing. Here, we identified genes associated with phage resistance in 18 Acinetobacter baumannii clinical strains belonging to the ST-2 clonal complex during a decade (Ab2000 vs. 2010): 9 from 2000 to 9 from 2010. The presence of genes putatively associated with phage resistance was detected. Genes detected were associated with an abortive infection system, restriction–modification system, genes predicted to be associated with defense systems but with unknown function, and CRISPR-Cas system. Between 118 and 171 genes were found in the 18 clinical strains. On average, 26% of these genes were detected inside genomic islands in the 2000 strains and 32% in the 2010 strains. Furthermore, 38 potential CRISPR arrays in 17 of 18 of the strains were found, as well as 705 proteins associated with CRISPR-Cas systems. A moderately higher presence of these genes in the strains of 2010 in comparison with those of 2000 was found, especially those related to the restriction–modification system and CRISPR-Cas system. The presence of these genes in genomic islands at a higher rate in the strains of 2010 compared with those of 2000 was also detected. Whole-genome sequencing and bioinformatics could be powerful tools to avoid drawbacks when a personalized therapy is applied. In this study, it allows us to take care of the phage resistance in A. baumannii clinical strains to prevent a failure in possible phage therapy.
Introduction
As part of the ESKAPE pathogens, Acinetobacter baumannii is frequently isolated from infections in clinical environments, and its resistance against multiple antibiotics is increasingly common (Boral et al., 2019). For this reason, it is necessary to opt for alternative treatments, such as phage therapy. However, the ability of bacteria to develop resistance mechanisms against phages is possible, even when there is no previous treatment with phage therapy due to the constant coevolutionary interactions (Stern and Sorek, 2011). The spread of phage resistance presents a significant challenge to the efficacy of the therapy (Schooley et al., 2017; Oechslin, 2018).
It is important to know and characterize the phage resistance mechanisms of a certain species, clone, or strain, before phage treatment to minimize treatment failure. Whole-genome sequencing (WGS) has been demonstrated to be a powerful tool in the detection of phage resistance mechanisms, as well as the evolution of CRISPR-Cas arrays in bacteria subjected to phage pressure (Bishop-Lilly et al., 2012; Broniewski et al., 2020). WGS is increasingly becoming a cheaper and faster technology; thus, it is implemented progressively in routine hospital diagnostics and research (Mintzer et al., 2019).
Recently, new or modified phage resistance mechanisms have been discovered and characterized (Labrie et al., 2010). Although a large part of defense systems against phages are maintained over generations, there is a continuous emergence of resistance mechanisms due to spontaneous mutations as a consequence of the coexistence of phage and bacteria. Most of these mutations occur in the phage receptor proteins, used by the phages to adhere to the cell (Labrie et al., 2010). Moreover, recent publications have demonstrated that variations in genes often associated with mediating biosynthesis on the surface of bacteria are also related to phage resistance in A. baumannii, such as glucosyltransferases (Gordillo Altamirano et al., 2021; Wang et al., 2021). In recent years, phage resistance mechanisms have been attracting an increasing interest due to the rising knowledge in phage interactions with bacteria. This leads to the discovery and characterization of new phage resistance mechanisms such as Zorya, Druantia, or Thoeris (Doron et al., 2018). Phage resistance mechanisms are typically clustered in genomic “defense islands.” mobile genetic evolutionary elements that contain genes associated with phage defense systems (Krupovic et al., 2014; Doron et al., 2018).
The main resistance mechanisms are related to the inhibition of the phage adsorption, blocking of phage DNA injection, cutting of the injected DNA, inhibition of the phage DNA replication, interference in the phage assembly, and bacterial suicide (Azam and Tanji, 2019). In Figure 1, we summarized all the characterized phage resistance mechanisms. In this study, we focused on those which could be bioinformatically detected without any experimental process:
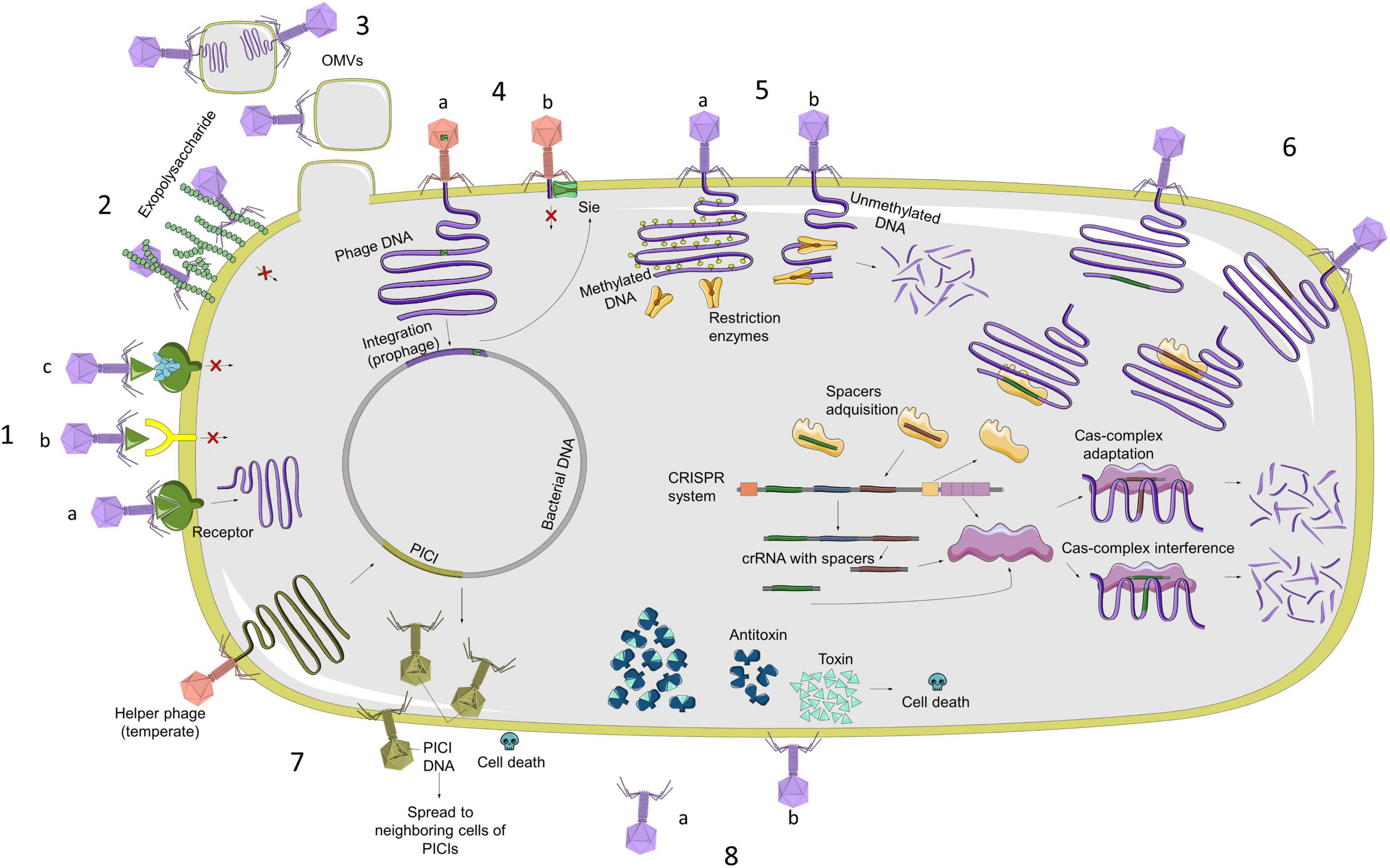
Figure 1. Representation of main mechanisms of bacterial resistance against phage infection (from left in a clockwise sense). 1. (A) Phage recognizes bacterial membrane receptor and can carry out infection; (B) alterations in receptors are produced by mutations and prevent phage from recognizing receptor, so it Will not infect bacteria; (C) bacteria can block recognition by producing inhibitors that bind to receptors. 2. Production of exopolysaccharide or extracellular matrix. 3. OMVs are composed of membrane lipids, membrane proteins, and periplasmic components. Some bacterial species use them as a decoy against phages as a defense mechanism. 4. (A) Bacteria block injection of DNA from other phages, acquiring Sie systems through prophages with this type of protein; (B) once bacterium has prophage in its genome with proteins that code for Sie system, it will be able to block entry of DNA from other phages. 5. (A) R–M system distinguishes between methylated and unmethylated DNA. Restriction enzymes cannot cut methylated DNA, which is also a protection system for bacterial DNA; (B) if phage DNA is not methylated, this system can cut injected DNA. 6. CRISPR-Cas system is an adaptative defense system, which recognizes phage DNA sequences, incorporates them into system, and then produces enzymes that are capable of recognizing these sequences to cut them. 7. PICI system is found in bacterial genome and induced by helper prophages to produce mature phage particles that assemble PICI system itself to kill infected cells and to be able to spread this system to adjacent cells. 8. (A) One of most characterized Abi systems is toxin–antitoxin system. Under normal conditions, bacterium expresses both proteins equally, so cell death does not occur; (B) When organism is subjected to stress situations, such as phage infection, toxin is highly expressed in comparison with antitoxin, causing cell death, which consequently produces a reduction in phage replication.
i) Abortive Infection (ABI) systems, characterized by the fact that the phage enters the cell, but its development is interrupted in any phase (replication, transcription, or translation). The mechanism of action is not entirely clear, either because of their complexity or because they are widely varied from one species to another (Lopatina et al., 2020).
ii) Toxin/antitoxin (TA) systems are a specific type of ABI system, but they are well-characterized and widespread through diverse species (Harms et al., 2018). In this system, a toxin is produced by the cell and is neutralized by an antitoxin. The expression of these molecules is highly controlled and varies from one system to another. When the balance between one molecule and the other is disturbed, the toxin is released, and the bacteria die (Song and Wood, 2020).
iii) Restriction–Modification (R–M) systems consist of a restriction endonuclease and a methyltransferase. This type of system distinguishes the DNA of the host from foreign DNA to recognize and destroy phage DNA after its injection into the cell. When unmethylated phage DNA enters a bacterium that possesses the R–M system, it will be cleaved by the restriction endonuclease or methylated by the methyltransferase to escape the restriction (Vasu and Nagaraja, 2013).
iv) CRISPR-Cas (clustered regularly interspaced short palindromic repeats—CRISPR-associated proteins) system is an adaptative immune system that bacteria develop against phage DNA/RNA and other foreign DNA (Mojica and Rodriguez-Valera, 2016). The typical structure of the CRISPR-Cas locus is a leader sequence, followed by the repeat-spacer array and the cas genes operon (Guan et al., 2016). The adaptation of the CRISPR-Cas system is due to the acquisition of the spacer sequences, which are small fragments of foreign nucleic acids, between the repeats of the CRISPR locus (Guan et al., 2016). The functioning of the CRISPR-Cas system, usually divided into three steps (adaptation, processing, and guidance of the crRNA-CRISPR RNA- and targeting and interference of the foreign DNA/RNA), is carried out by the Cas (CRISPR-associated) proteins (Barrangou, 2013). CRISPR-Cas systems are classified according to their conserved cas genes and the architecture of the cas operon (Makarova et al., 2011). Until recently, little data existed about CRISPR-Cas systems in A. baumannii. The pangenome analysis of A. baumannii has shown CRISPR-Cas systems in the species (Mangas et al., 2019). One of the most characterized systems in A. baumannii is the CRISPR I-Fb system (Karah et al., 2015). However, most of the Cas-related genes and CRISPR arrays are yet not identified and characterized.
In this study, we searched for putative genes associated with phage resistance, and we focused on CRISPR-Cas systems by studying the CRISPR arrays and Cas protein presence through a bioinformatic approach in 18 genomes of clinical strains of A. baumannii isolated in the “II Spanish Study of A. baumannii GEIH-REIPI 2000–2010.”
Methodology
Genome Database
Eighteen clinical A. baumannii genomes previously sequenced and annotated (II Spanish Multicenter Study, GEIH-REIPI Acinetobacter baumannii 2000–2010; Umbrella Bioproject PRJNA422585) (López et al., 2016, 2017) have been studied. Nine strains were from 2000 and nine from 2010. All of the strains belong to the ST-2 clone (López et al., 2016).
Search for General Genes Associated With Bacteriophage Resistance and Their Presence in Genomic Islands
To analyze the presence of genes putatively associated with phage resistance systems, a custom database based on genes from the ‘‘PADS Arsenal database’’1 was created (Zhang et al., 2020). The genes were grouped in five systems: ABI systems related (not belonging to toxin/antitoxin system), TA systems related, R–M system related, CRISPR-Cas-associated proteins, and newly (NEW) characterized systems-related genes. In this last category, we included those genes which hit against known phage-resistant genes but were associated with genes predicted to be associated with phage-resistant functions and whose function in A. baumannii is not clear yet, such as newly characterized systems (e.g., Zorya, Druantia, and Thoeris). In this category, it is also included those genes related to known and characterized systems but without a complete functional characterization in A. baumannii (e. g. BREX). A blast search of the complete genomes against this database and filtered out those hits with an e-value > 1E-04 was made. Statistical significance was determined by comparing the absolute number of each system in the nine genomes of Ab_GEIH-2000 collection against the absolute numbers of genes of Ab_GEIH-2010 collection with the Student’s t-test. The percentage of the genes involved in resistance was calculated by dividing the genes predicted to be associated with phage resistance by the total number of genes in the bacteria genome.
To locate genomic islands (GIs), three different approaches were used: IslandViewer with default settings (Bertelli et al., 2017), blast search with default settings and cutoff of e-value < 1e-03 against a previously constructed ICEberg database (Liu et al., 2019), and checking the GC content of the contigs of each genome (Zhang R. et al., 2014). The previously detected phage-resistant genes were localized in the GIs detected per genome, and an average percentage of genes by collection was calculated. Statistical significance was determined by comparing the absolute number of each system present in GIs in the nine genomes of Ab_GEIH-2000 collection against the absolute numbers of genes of Ab_GEIH-2010 collection with Student’s t-test.
Search and Characterization of Clustered Regularly Interspaced Short Palindromic Repeats Arrays
On a first try, CRISPRCasFinder (Couvin et al., 2018) was used, but no putative CRISPR-Cas system was found. For this reason, CRISPR arrays were found using the CRISPR Recognition Tool (CRT) (Bland et al., 2007). The modification proposed by Rho et al. (2012) for whole-metagenomic assembled genomes called metaCRT was used with the following parameters: minimum number of repeats: 3, minimum repeat length: 12, maximum repeat length: 70, minimum spacer length: 18, maximum spacer length: 80, and with a search window: 6. This search pretends to be as flexible as possible because genomes are in contig format. Most existing CRISPR identification tools use 11 as the minimum word size for the repeat, as it does not compromise the finding of potential short repeats with multiple base mismatches (Biswas et al., 2016). Regarding spacer length, a new type of V-C CRISPR-Cas has a preference for short (17–19 bp) DNA fragments to incorporate as spacers (Wright et al., 2019); thereby, a minimum spacer length of 18 nt was covenanted.
To filter and validate the CRISPR arrays, a similar procedure to the first step in the protocol developed by Shmakov et al. (2020) was followed (Figure 2). In the first step, CRISPR arrays separated by no more than six open reading frames (ORFs) to a putative Cas protein identified before were considered to be part of a putative CRISPR-Cas system. Those which were not part of a putative CRISPR-Cas system were considered to be single possible CRISPR arrays. To validate these arrays, we made a short-blastn search of the spacers of the possible CRISPR arrays against all phage genomes using the INPHARED database (Cook et al., 2021). Results that were > 95% in identity and those arrays whose query hit was larger than 20 were considered putative CRISPR arrays.
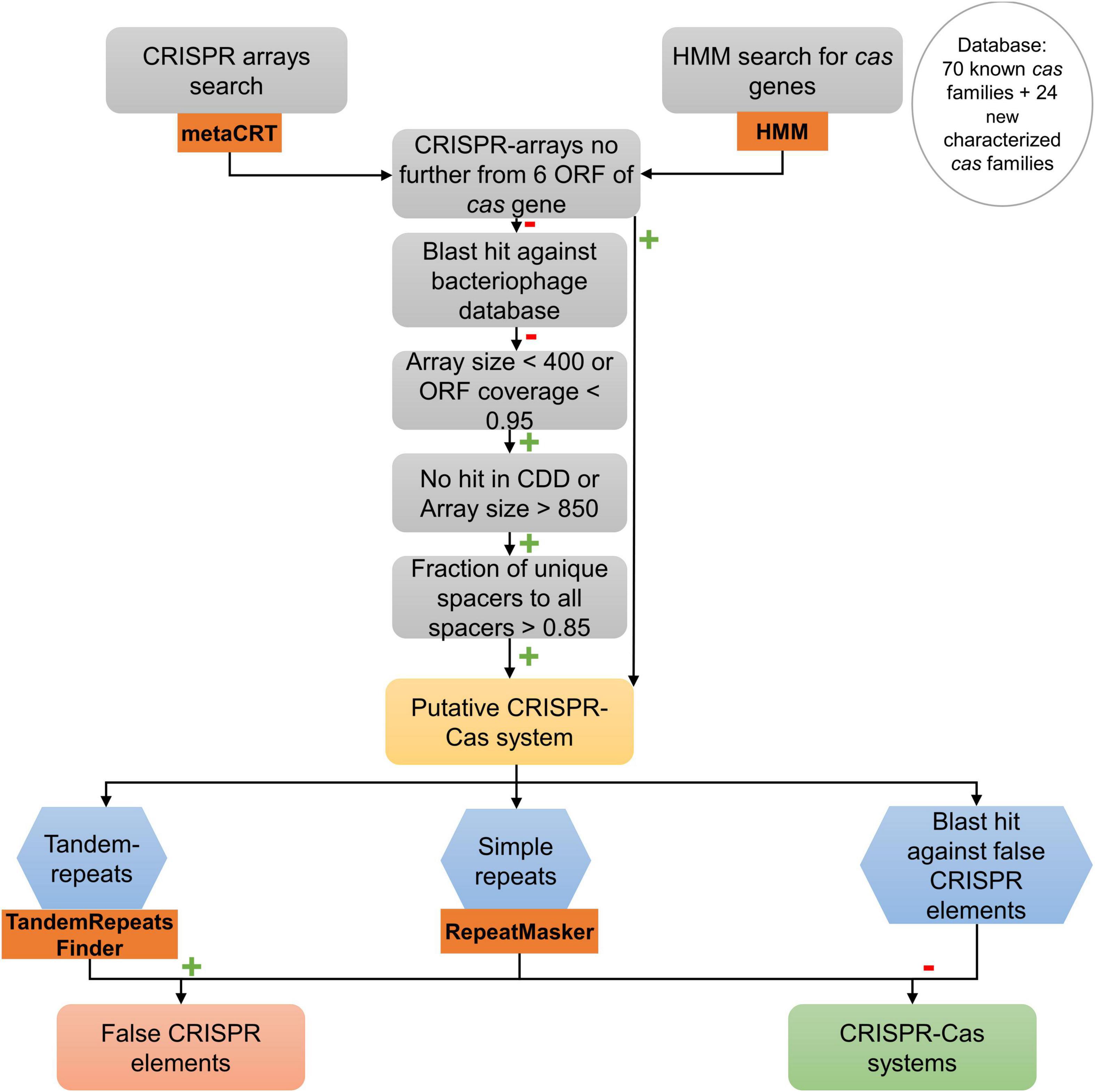
Figure 2. Search procedure of CRISPR-Cas systems in 18 genomes of A. baumannii clinical strains. Orange rectangle represents bioinformatics programs used for that task. Green plus marks represent a positive result for each operation. Red minus marks represent a negative result for each operation.
Negative blast hits of the spacers of the CRISPR arrays against bacteriophage follow the procedure described by Shmakov et al. (2020) for isolated arrays: first, arrays > 400 bp and with an ORF coverage > 0.95 were filtered out. Second, all arrays with < 850 bp that had the domain in CDD search were filtered out (Marchler-Bauer et al., 2015; Lu et al., 2020). Then, pairwise distances between spacers of each array were calculated (number of matches in the longest blastn hit between them, divided by the length of the smaller spacer in the pair). The spacers of each array were clustered using single linkage clustering following the same procedure as Shmakov et al. (2020) with a cutoff of 0.3. A spacer similarity index was calculated for each CRISPR array as the number of clusters formed divided by the number of spacers in the array (1 means that all the spacers are different). Those arrays whose spacer similarity index was < 0.85 were filtered out. The rest were considered putative CRISPR arrays.
To complement the procedure made by Shmakov et al. (2020), a search about common special low-complexity sequences that may be confused as CRISPR arrays was made, known as false-CRISPR elements (Zhang and Ye, 2017). The presence of tandem repeats, potentially hypermutable regions that enable bacteria to adapt to evolving environments without increasing their mutation rate, was checked with Tandem Repeat Finder (Benson, 1999; Rando and Verstrepen, 2007; Zhou et al., 2014). The presence of short low-complexity repeats was also examined with RepeatMasker (Chen, 2004). To test the results and to complete the search of low-complexity sequences, a blast search against an existing false-CRISPR elements database was obtained from the CRISPRone website (Zhang and Ye, 2017).
The search for possible Cas-related proteins was made, based on the method of Zhang Q. et al. (2014), but adding a search in the “HMMCAS” website of all of their available HMM models, performed with a cutoff of reported e-values of Chai et al. (2019). The 18 genomes were examined using hmmscan with all of the pfam HMM profiles based on National Center for Biotechnology Information entries of known Cas protein families searching in the pfam database 70 Cas-related protein families and other CRISPR-associated proteins in the pfam database (e.g., DEAD/DEAH box helicase), 93 families that were in the TIGRFAMS resource, and the 24 newly characterized families (Haft et al., 2003; Finn et al., 2011, 2014; Zhang Q. et al., 2014). For the TIGRFAMS database proteins, it was necessary to build an HMM profile with hmmbuild with default settings (Finn et al., 2011), making a previous alignment for each compound of proteins with Clustal Omega version 1.2.4 (ClustalO) (Sievers and Higgins, 2014).
All of the input and output data for the searches (genes associated with phage resistance, genomic islands, CRISPR arrays, and Cas proteins) were processed with Python2 and BioPython (19304878; Chapman and Chang, 2000; Cock et al., 2009).
To establish the evolution and to compare the presence of the CRISPR arrays among the same clonal complex of the 18 clinical strains of A. baumannii, a phylogenetic tree was made using the CRISPR spacers detected. Trees were built using MEGA7 with CLUSTALW alignment (MEGA version 7.9.26) (Kumar et al., 1994, 2016; Thompson et al., 1994).
Results
Genes Putatively Associated With Phage Resistance in Acinetobacter baumannii Clinical Strains and Their Presence in Genomic Islands
Between 118 and 171 genes were detected per genome; those could be putatively associated with bacterial defense against bacteriophages (Supplementary Table 1). The frequency (%) of each resistance system was calculated with the number of genes of each group divided by the total genes per genome. It was observed that the genes related to R–M and CRISPR-Cas systems showed a slightly significantly higher prevalence in 2010 strains (Figure 3A) (p < 0.024 and p < 0.0144, respectively). The frequency of the genes related to ABI, TA, and new systems remained constant in both collections. The presence of putative phage-resistant genes in GIs was also predicted (Supplementary Table 2), and it was found that GIs represent in strains of the year 2010 approximately 25% on average and approximately 19% in the strains of the year 2000 On average (Figure 3B) (p < 0.012). The observed increase was produced especially in genes related to the R–M system and to those related to new phage resistance mechanisms and the CRISPR-Cas system (p < 0.0119 and p < 0.0144, respectively).
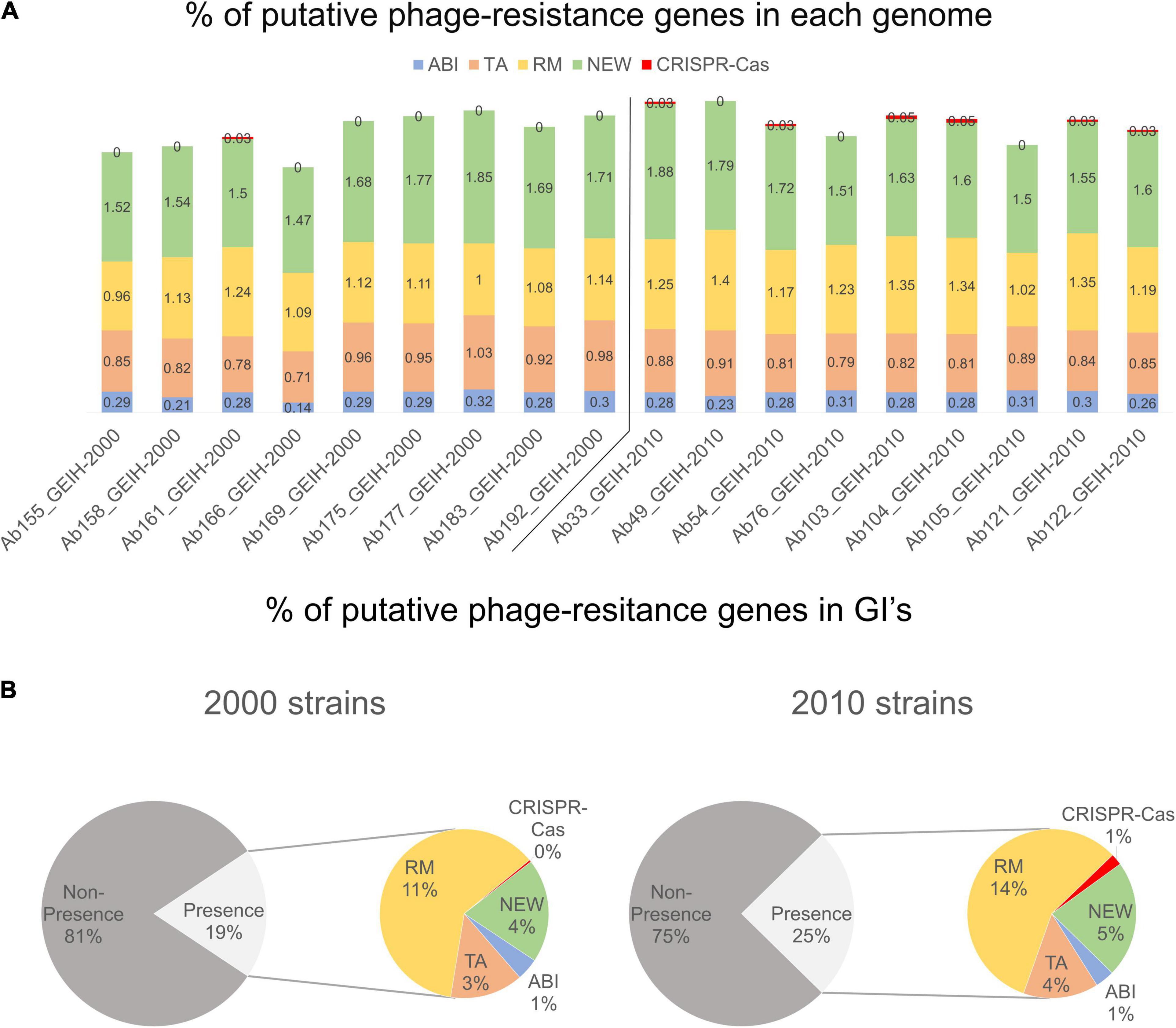
Figure 3. (A) Frequency (%, rounded to two decimal numbers) of each group of genes in each genome with respect to total of genes: abortive infection (ABI) system, toxin/antitoxin (TA) system, restriction–modification (R–M) system, and genes associated with newly phage resistance bacterial mechanisms (NEW), e.g., Zorya, Hachiman, and Druantia. (B) Presence and non-presence of putative phage-resistant genes in GIs. Presence (%, rounded without decimal numbers) section is divided into different groups of genes.
Clustered Regularly Interspaced Short Palindromic Repeats Arrays
One hundred eighty putative predicted arrays were found (without filtering) using metaCRT in the 18 genomes of ST-2 A. baumannii clinical strains (Supplementary Table 3). After the complete process of filtering designed by Shmakov et al. (2020) and removing the low-complexity sequences (Zhang and Ye, 2017), only 40 CRISPR arrays were selected (Table 1): 18 CRISPR arrays were present in the 2000 strains and 22 in the 2010 strains. All the strains, except in the Ab161_GEIH-2000 strain, presented at least one CRISPR array. Surrounding proteins nearby the selected CRISPR arrays are also attached (Supplementary Table 4).
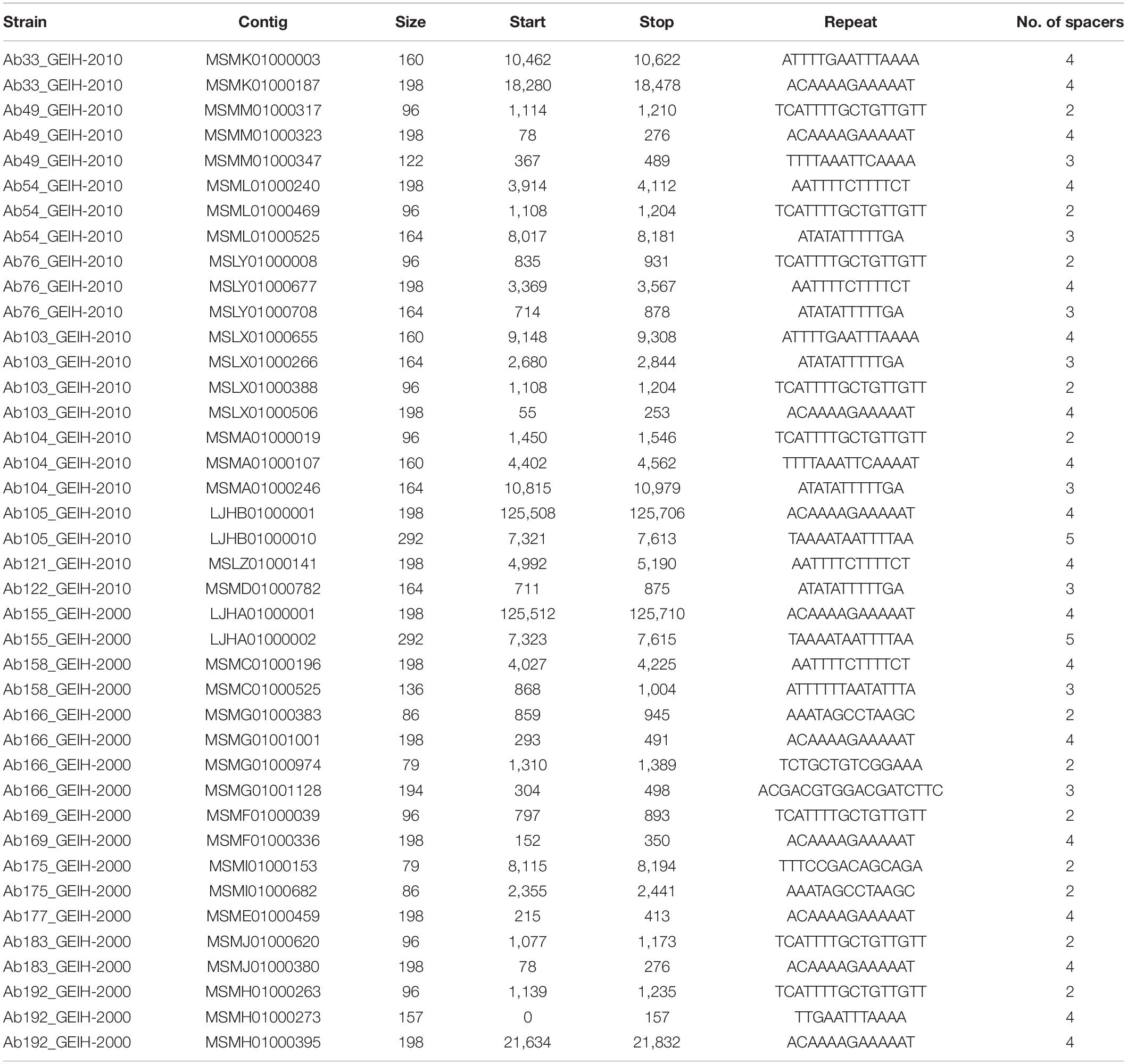
Table 1. CRISPR arrays present in genomes of 18 A. baumannii clinical strains.
A phylogenetic tree of the complete CRISPR array sequences was constructed (Figure 4) and showed an equal distribution of the spacers between the strains of the 2 years. Some of the spacers were predicted to be the same even in strains of different year collections. Few of the arrays were unique with respect to the other, such as the present in the 2000 strains Ab158_GEIH-2000_MSMC01000525, Ab166_MSMG01000383, Ab166_MSMG01000974, Ab166_MSMG01001128, Ab175_MSMI01000153, or Ab175_ MSMI01000682. However, there were five CRISPR arrays grouped that only were represented in the 2010 strains (Figure 4).
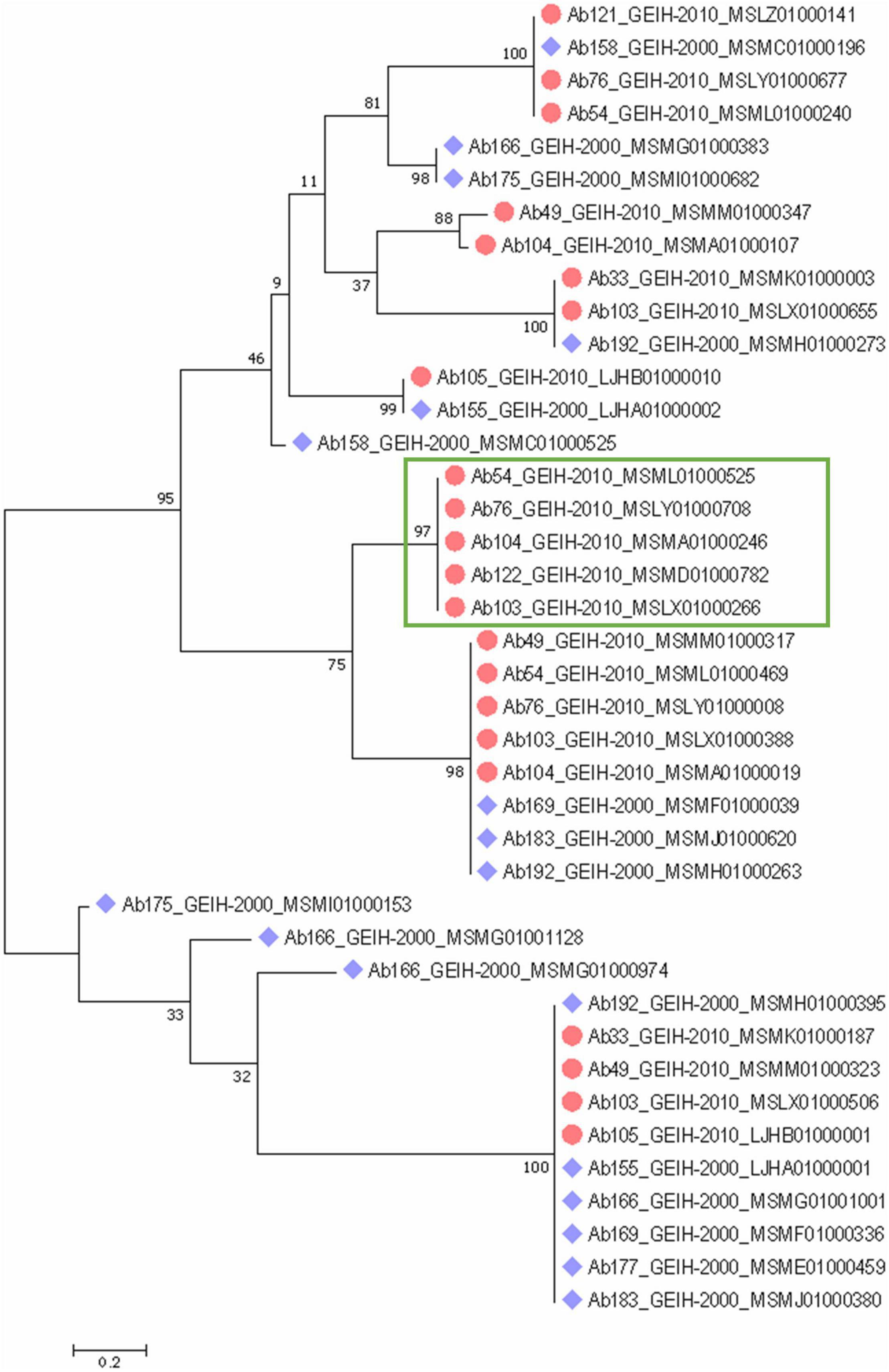
Figure 4. Phylogenetic classification of CRISPR arrays detected in 18 genomes of A. baumannii ST-2 clinical strains through a maximum-likelihood tree with suggested model by MEGA analysis Tamura 3-parameter with uniform rates among sites and a bootstrap of 100. Blue rhomboid indicates that strain belongs to 2000 collection. Red circles to 2010 collection. Green rectangle indicates five CRISPR arrays only detected in 2010 strains.
Cas-Related Proteins
When HMM against Cas-known, Cas-related, and CRISPR-associated protein families was used, 705 Cas-related proteins were identified in the 18 genomes: 341 Cas-related proteins were detected in the 2000 strains and 364 in the 2010 strains (Supplementary Table 5). Most of them were identified as DEAD/DEAH box helicase (207 of the total) and as type III Restriction Unit Res III (195 of 705). The vast majority of them were located next to proteins whose predicted function does not match with a Cas protein function or to proteins whose function was unknown. Other Cas-related proteins were close in the same contig, thereby giving us a clue to help identify a functional Cas cluster. For example, in the contig MSLX01000260 from the Ab103_GEIH-2010 strain, a putative Helicase_C protein (OLV37994.1) and a Cas_St_Csn2 protein (OLV37998.1) were only of 2 ORF distance between them. However, the function of the surrounding proteins was hypothetical, thus hindering the identification process as a Cas cluster.
Discussion
In clinical laboratories, genomics is rapidly being developed and utilized to track antibiotic resistance. As a result, it is critical to explore how to detect and avoid phage-resistant strains if a treatment based on phages was going to be applied, using WGS metadata analysis. In this study, we looked for genes linked to phage resistance in 18 clinical strains of A. baumannii. We constructed a database with genes based in the public PADS database, as it is a complete database about prokaryotic antiviral defense systems so far, as well as being collecting newly discovered types of defense systems to the BIG Data Center (BIG Data Center Members, 2019; Zhang et al., 2020). As mutations in bacterial membrane proteins or LPS is a primary defense due to causing a lower fitness cost to the bacteria (van Houte et al., 2016), and they should potentially also be included to track phage resistance in WGS because they proved appearance in the application of cocktail of phages (Hesse et al., 2020; Yang et al., 2020), the aim was to identify defense genes whose function were putatively only related to phage resistance mechanisms. In this case, the high number of genes made us establish groups to simplify the results of the blast hits. We also tried to identify the presence of CRISPR-Cas systems by separating the search in CRISPR arrays and Cas proteins.
A difference between the presence of phage-resistant genes in the 2010 strains and the 2000 strains was observed, with a higher presence of genes related to the R–M system and CRISPR-Cas system and lower TA-related genes. The natural reciprocal selection pressure between host bacteria and phage increases the infectivity of the phage and the phage resistance in the bacterium side (Hampton et al., 2020). In fact, phage populations are ubiquitous at body surfaces such as lungs, intestines, or skin, and they outnumber bacteria at least 10-fold (Batinovic et al., 2019). The study of the mobilome (plasmids and bacteriophages) in these same 18 genomes belonging to the A. baumannii clonal group ST-2 in 2018 (López et al., 2018) connects the presence of functional quorum-sensing/quorum-quenching network in those cells with a higher presence of complete prophages in the 2010 collection. As it is pointed out, the representative strain Ab105_GEIH-2010 (representative of the Ab_GEIH-2010 group of strains) owns two complete prophages: Ab105-1Φ and Ab105-2Φ. One of them, Ab105-2Φ, was turned into a mutant lytic phage, and its phage infectivity was tested as host range and efficiency of plating against some A. baumannii clinical strains, in which some of the strains of this study are included (Blasco et al., 2019). By contrast, none of the Ab_GEIH-2000 group possess any complete prophage. For this reason, the acquisition of phage-resistant genes is correlated with a higher presence of complete prophages in the strains of 2010 in comparison with those of 2000.
Defense systems are regularly obtained by bacteria and archaea through horizontal gene transfer (HGT) owing to an environmental adaptation of the bacterial communities (van Houte et al., 2016; Koonin et al., 2017). We found a major average of genes acquired by HGT in the 2010 strains (25% were in GIs regions) rather than in the 2000 ones (19% were in GIs regions), especially those genes related to R–M system and CRISPR-Cas. It was demonstrated that only ∼4% of R–M systems are in the core genomes of prokaryotic species, suggesting they are commonly transferred (Oliveira et al., 2014). CRISPR-Cas systems display weak consistency within the core genome, demonstrating the prevalence of the HGT spreading this system (Oliveira et al., 2014; Makarova et al., 2015). The R–M system and the CRISPR-Cas system commonly coexist with an elevated contribution to bacterial immunity, and they rarely operate on their own (Dupuis et al., 2013; Oliveira et al., 2014). However, they are far from being perfect in the bacterial resistance, and phage can escape these systems in many different ways, for example, the anti-CRISPR proteins (Labrie et al., 2010; Bondy-Denomy et al., 2013). We also observed a decreasing number of TA-related genes through the years; even their presence in GIs is higher in the 2000 strains than in the 2010s. This could be because the counteradaptation of the phage may be reached by developing antitoxin in the phage genome that inhibits the cell death and thus promote the infection of the phage (Otsuka and Yonesaki, 2012; Wei et al., 2016) or because they could have evolved into Cas proteins of the CRISPR-Cas system, as the TA proteins are considered as ancestors of Cas2 proteins (Makarova et al., 2020). In A. baumannii, little is known about defense mechanisms against phage. Another recent genomic bioinformatics analysis, in this case in the sequence type ST374, revealed the presence of a defense island with genes involved in Abi, R–M, BREX, or TA systems (Fedrigo et al., 2021).
Furthermore, we found the CRISPR-Cas genes blast hit results incomplete due to the separation in contig assembly of the genomes, which prevented us from identifying proteins or arrays related to the CRISPR-Cas proteins identified in small contigs (data not shown), and also due to the high diversity of the Cas proteins and the little knowledge about these proteins in clinical strains of A. baumannii, which increases the difficulty of identifying these type of proteins (Mangas et al., 2019; Pinilla-Redondo et al., 2020). As a consequence, we examined the presence of CRISPR arrays and Cas proteins separately. We establish a methodology to discard false-CRISPR elements based on the method of Shmakov et al. (2020) and posteriorly completed with a full evaluation of the quality of the CRISPR arrays filtered based on the search of tandem repeats, simple repeats, and their presence on phage genomes (Zhang and Ye, 2017). Secondly, another reason for developing an alternative method is the nature of the multiresistant pathogens, their constant adaptation to different environments, and thus the continuous acquisition of different mobile elements, which provokes the appearance of new CRISPR-Cas yet to be identified (Kamruzzaman and Iredell, 2019). This also fosters and extends the variability in the Cas proteins, complicating their characterization.
Forty CRISPR arrays were found in the 18 A. baumannii clinical strains from the ST-2 clone. All of the strains presented at least one CRISPR array except one, Ab161_GEIH-2000. The vast majority of the arrays are shared between the clone ST-2 in both collections, with some exceptions such as the five arrays only found in the 2010 strains. It has been shown that the distribution of the CRISPR-Cas system is MLST-dependent and non-random and thought to be a better discriminating tool than classical MLST in discriminating different Klebsiella pneumoniae (Shen et al., 2017; Liao et al., 2020). On the other hand, the detection of different unique CRISPR arrays only in the 2000 strains demonstrates the dynamic interaction of these arrays throughout the years.
All of the CRISPR arrays in this study were without any Cas or putative Cas protein near to them. It was described that these “orphan” arrays belong to unknown CRISPR-Cas systems due to an extremely evolutionarily remote type of CRISPR-Cas (Shmakov et al., 2020). This existence of isolated CRISPR arrays could be explained for four reasons. First, the contig format of the studied genomes could provoke that some arrays are detected in small or incomplete contigs. Secondly, some Cas endonucleases such as Cas1 and/or Cas6 can recognize remote CRISPR arrays (Reimann et al., 2020; Hoikkala et al., 2021). Third, the possibility of some of the unique isolated arrays forming part of an undescribed CRISPR-Cas cluster extremely distant from the ones already characterized may occur (Shmakov et al., 2020). Fourth, the strains may have lost the cas genes, thus leaving the isolated arrays (Shmakov et al., 2020). The Cas distribution observed in this work would correspond and complete any of the hypotheses about the explanation of “orphan” CRISPR arrays mentioned before, as the putative Cas proteins hit through the HMM search could form part of a complete Cas cluster. However, as it was said at the end of Results, it was impossible to determine in silico if the putative Cas detected form part of complete and functional Cas loci.
The localization and characterization of defense systems against phages are necessary steps when designing an effective phage therapy. The WGS combined with an effective bioinformatics strategy would allow us to know what mechanisms the clinical strains have and potentially discard those phages to which bacteria may possess resistance to the treated strain. This study shows the wide presence of genes associated with resistance against phages and their acquisition by GIs for 10 years in clinical A. baumannii strains from the same clonal complex ST-2 and the CRISPR arrays present on them. Additional investigations are needed to characterize and discover the molecular mechanisms of phage-resistant genes discovered in this study in A. baumannii.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
AA, LB, ML, OP, IB, LF-G, MG, and CO-C analyzed the results and wrote the manuscript. AM revised the results and the manuscript. MT obtained the research funding and supervised the writing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by grants PI16/01163 and PI19/00878 awarded to MT within the State Plan for R + D + I 2013–2016 (National Plan for Scientific Research, Technological Development and Innovation 2008–2011) and co-financed by the ISCIII-Deputy General Directorate of evaluation and Promotion of Research-European Regional Development Fund “A way of Making Europe” and Instituto de Salud Carlos III FEDER. MT was financially supported by the Miguel Servet Research Programme (SERGAS and ISCIII).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to the following organizations and researchers who participated in the study: GEIH-GEMARA (SEIMC, http://www.seimc.org) and the REIPI (Spanish Network for the Research in Infectious Disease-REIPI, RD12/0015/0010 and RD16/0016/0001).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.784949/full#supplementary-material
Supplementary Table 3 | Putative predicted arrays (without filtering) using metaCRT in the 18 genomes of ST-2 A. baumannii clinical strains (.xls file).
Supplementary Table 4 | Adjacent predicted proteins of CRISPR arrays detected.
Supplementary Table 5 | Cas-related proteins identified with HMM against a prebuilt database with known Cas proteins and proteins with functions related with Cas proteins (.xls file).
Footnotes
References
Azam, A. H., and Tanji, Y. (2019). Bacteriophage-host arm race: an update on the mechanism of phage resistance in bacteria and revenge of the phage with the perspective for phage therapy. Appl. Microbiol. Biotechnol. 103, 2121–2131. doi: 10.1007/s00253-019-09629-x
Barrangou, R. (2013). CRISPR-Cas systems and RNA-guided interference. Wiley Interdiscip. Rev. RNA 4, 267–278. doi: 10.1002/wrna.1159
Batinovic, S., Wassef, F., Knowler, S. A., Rice, D. T. F., Stanton, C. R., Rose, J., et al. (2019). Bacteriophages in natural and artificial environments. Pathogens 8:100. doi: 10.3390/pathogens8030100
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bertelli, C., Laird, M. R., Williams, K. P., Lau, B. Y., Hoad, G., Winsor, G. L., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
BIG Data Center Members (2019). Database resources of the BIG data center in 2019. Nucleic Acids Res. 47, D8–D14. doi: 10.1093/nar/gky993
Bishop-Lilly, K. A., Plaut, R. D., Chen, P. E., Akmal, A., Willner, K. M., Butani, A., et al. (2012). Whole genome sequencing of phage resistant Bacillus anthracis mutants reveals an essential role for cell surface anchoring protein CsaB in phage AP50c adsorption. Virol. J. 9:246. doi: 10.1186/1743-422X-9-246
Biswas, A., Raymond, H. J. S., Morales, S. E., Fineran, P. C., and Brown, C. M. (2016). CRISPRDetect: a flexible algorithm to define CRISPR arrays. BMC Genomics 17:356. doi: 10.1186/s12864-016-2627-0
Bland, C., Ramsey, T. L., Sabree, F., Lowe, M., Brown, K., Kyrpides, N. C., et al. (2007). CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8:209. doi: 10.1186/1471-2105-8-209
Blasco, L., Ambroa, A., Lopez, M., Fernandez-Garcia, L., Bleriot, I., Trastoy, R., et al. (2019). Combined use of the Ab105-2phiDeltaCI lytic mutant phage and different antibiotics in clinical isolates of multi-resistant Acinetobacter baumannii. Microorganisms 7:556. doi: 10.3390/microorganisms7110556
Bondy-Denomy, J., Pawluk, A., Maxwell, K. L., and Davidson, A. R. (2013). Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493, 429–432. doi: 10.1038/nature11723
Boral, B., Unaldi, Ö, Ergin, A., Durmaz, R., Eser, Ö, and Acinetobacter Study Group (2019). A prospective multicenter study on the evaluation of antimicrobial resistance and molecular epidemiology of multidrug-resistant Acinetobacter baumannii infections in intensive care units with clinical and environmental features. Ann. Clin. Microbiol. Antimicrob. 18:19. doi: 10.1186/s12941-019-0319-8
Broniewski, J. M., Meaden, S., Paterson, S., Buckling, A., and Westra, E. R. (2020). The effect of phage genetic diversity on bacterial resistance evolution. ISME J. 14, 828–836. doi: 10.1038/s41396-019-0577-7
Chai, G., Yu, M., Jiang, L., Duan, Y., and Huang, J. (2019). HMMCAS: a web tool for the identification and domain annotations of CAS proteins. IEEE/ACM Trans. Comput. Biol. Bioinform. 16, 1313–1315. doi: 10.1109/TCBB.2017.2665542
Chapman, B., and Chang, J. (2000). Biopython: python tools for computational biology. SIGBIO Newsl. 20, 15–19. doi: 10.1145/360262.360268
Chen, N. (2004). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics Chapter 4:Unit4.10. doi: 10.1002/0471250953.bi0410s25
Cock, P. J., Antao, T., Chang, J. T., Chapman, B. A., Cox, C. J., Dalke, A., et al. (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. doi: 10.1093/bioinformatics/btp163
Cook, R., Brown, N., Redgwell, T., Rihtman, B., Barnes, M., Clokie, M., et al. (2021). INfrastructure for a PHAge REference database: identification of large-scale biases in the current collection of phage genomes. bioRxiv [preprint]. doi: 10.1101/2021.05.01.442102
Couvin, D., Bernheim, A., Toffano-Nioche, C., Touchon, M., Michalik, J., Néron, B., et al. (2018). CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 46, W246–W251. doi: 10.1093/nar/gky425
Doron, S., Melamed, S., Ofir, G., Leavitt, A., Lopatina, A., Keren, M., et al. (2018). Systematic discovery of antiphage defence systems in the microbial pangenome. Science 359:eaar4120. doi: 10.1126/science.aar4120
Dupuis, M., Villion, M., Magadán, A. H., and Moineau, S. (2013). CRISPR-Cas and restriction-modification systems are compatible and increase phage resistance. Nat. Commun. 4:2087. doi: 10.1038/ncomms3087
Fedrigo, N. H., Xavier, D. E., Cerdeira, L., Fuga, B., Marini, P. V. B., Shinohara, D. R., et al. (2021). Genomic insights of Acinetobacter baumannii ST374 reveal wide and increasing resistome and virulome. Infect. Genet. Evol. 97:105148. doi: 10.1016/j.meegid.2021.105148
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Gordillo Altamirano, F., Forsyth, J. H., Patwa, R., Kostoulias, X., Trim, M., Subedi, D., et al. (2021). Bacteriophage-resistant Acinetobacter baumannii are resensitized to antimicrobials. Nat. Microbiol. 6, 157–161. doi: 10.1038/s41564-020-00830-7
Guan, L., Han, Y., Zhu, S., and Lin, J. (2016). Application of CRISPR-Cas system in gene therapy: pre-clinical progress in animal model. DNA Repair (Amst.) 46, 1–8. doi: 10.3389/fonc.2020.01387
Haft, D. H., Selengut, J. D., and White, O. (2003). The TIGRFAMs database of protein families. Nucleic Acids Res. 31, 371–373. doi: 10.1093/nar/gkg128
Hampton, H. G., Watson, B. N. J., and Fineran, P. C. (2020). The arms race between bacteria and their phage foes. Nature 577, 327–336. doi: 10.1038/s41586-019-1894-8
Harms, A., Brodersen, D. E., Mitarai, N., and Gerdes, K. (2018). Toxins, targets, and triggers: an overview of toxin-antitoxin biology. Mol. Cell 70, 768–784. doi: 10.1016/j.molcel.2018.01.003
Hesse, S., Rajaure, M., Wall, E., Johnson, J., Bliskovsky, V., Gottesman, S., et al. (2020). Phage resistance in multidrug-resistant Klebsiella pneumoniae ST258 evolves via diverse mutations that culminate in impaired adsorption. mBio 11:e02530-19. doi: 10.1128/mBio.02530-19
Hoikkala, V., Ravantti, J., Díez-Villaseñor, C., Tiirola, M., Conrad, R. A., Mcbride, M. J., et al. (2021). Cooperation between different CRISPR-cas types enables adaptation in an RNA-targeting system. mBio 12:e03338-20. doi: 10.1128/mBio.03338-20
Kamruzzaman, M., and Iredell, J. R. (2019). CRISPR-Cas system in antibiotic resistance plasmids in Klebsiella pneumoniae. Front. Microbiol. 10:2934. doi: 10.3389/fmicb.2019.02934
Karah, N., Samuelsen, Ø, Zarrilli, R., Sahl, J. W., Wai, S. N., and Uhlin, B. E. (2015). CRISPR-cas subtype I-Fb in Acinetobacter baumannii: evolution and utilization for strain subtyping. PLoS One 10:e0118205. doi: 10.1371/journal.pone.0118205
Koonin, E. V., Makarova, K. S., and Wolf, Y. I. (2017). Evolutionary genomics of defence systems in archaea and bacteria. Annu. Rev. Microbiol. 71, 233–261. doi: 10.1146/annurev-micro-090816-093830
Krupovic, M., Makarova, K. S., Forterre, P., Prangishvili, D., and Koonin, E. V. (2014). Casposons: a new superfamily of self-synthesizing DNA transposons at the origin of prokaryotic CRISPR-Cas immunity. BMC Biol. 12:36. doi: 10.1186/1741-7007-12-36
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kumar, S., Tamura, K., and Nei, M. (1994). MEGA: molecular evolutionary genetics analysis software for microcomputers. Comput. Appl. Biosci. 10, 189–191. doi: 10.1093/bioinformatics/10.2.189
Labrie, S. J., Samson, J. E., and Moineau, S. (2010). Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 8, 317–327. doi: 10.1038/nrmicro2315
Liao, W., Liu, Y., Chen, C., Li, J., Du, F., Long, D., et al. (2020). Distribution of CRISPR-Cas systems in clinical carbapenem-resistant Klebsiella pneumoniae strrains in a Chinese Tertiary hospital and its potential relationship with virulence. Microb. Drug Resist. 26, 630–636. doi: 10.1089/mdr.2019.0276
Liu, M., Li, X., Xie, Y., Bi, D., Sun, J., Li, J., et al. (2019). ICEberg 2.0: an updated database of bacterial integrative and conjugative elements. Nucleic Acids Res. 47, D660–D665. doi: 10.1093/nar/gky1123
Lopatina, A., Tal, N., and Sorek, R. (2020). Abortive infection: bacterial suicide as an antiviral immune strategy. Annu. Rev. Virol. 7, 371–384. doi: 10.1146/annurev-virology-011620-040628
López, M., Mayer, C., Fernández-García, L., Blasco, L., Muras, A., Ruiz, F. M., et al. (2017). Quorum sensing network in clinical strains of A. baumannii: AidA is a new quorum quenching enzyme. PLoS One 12:e0174454. doi: 10.1371/journal.pone.0174454
López, M., Rueda, A., Florido, J. P., Blasco, L., Fernández-García, L., Trastoy, R., et al. (2018). Evolution of the Quorum network and the mobilome (plasmids and bacteriophages) in clinical strains of Acinetobacter baumannii during a decade. Sci. Rep. 8:2523. doi: 10.1038/s41598-018-20847-7
López, M., Rueda, A., Florido, J. P., Blasco, L., Gato, E., Fernández-García, L., et al. (2016). Genomic evolution of two Acinetobacter baumannii clinical strains from ST-2 clones isolated in 2000 and 2010 (ST-2_clon_2000 and ST-2_clon_2010). Genome Announc. 4:e01182-16. doi: 10.1128/genomeA.01182-16
Lu, S., Wang, J., Chitsaz, F., Derbyshire, M. K., Geer, R. C., Gonzales, N. R., et al. (2020). CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 48, D265–D268. doi: 10.1093/nar/gkz991
Makarova, K. S., Haft, D. H., Barrangou, R., Brouns, S. J., Charpentier, E., Horvath, P., et al. (2011). Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9, 467–477. doi: 10.1038/nrmicro2577
Makarova, K. S., Wolf, Y. I., Alkhnbashi, O. S., Costa, F., Shah, S. A., Saunders, S. J., et al. (2015). An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 13, 722–736. doi: 10.1038/nrmicro3569
Makarova, K. S., Wolf, Y. I., Iranzo, J., Shmakov, S. A., Alkhnbashi, O. S., Brouns, S. J. J., et al. (2020). Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 18, 67–83. doi: 10.1038/s41579-019-0299-x
Mangas, E. L., Rubio, A., Álvarez-Marín, R., Labrador-Herrera, G., Pachón, J., Pachón-Ibáñez, M. E., et al. (2019). Pangenome of Acinetobacter baumannii uncovers two groups of genomes, one of them with genes involved in CRISPR/Cas defence systems associated with the absence of plasmids and exclusive genes for biofilm formation. Microb. Genom. 5:e000309. doi: 10.1099/mgen.0.000309
Marchler-Bauer, A., Derbyshire, M. K., Gonzales, N. R., Lu, S., Chitsaz, F., Geer, L. Y., et al. (2015). CDD: NCBI’s conserved domain database. Nucleic Acids Res. 43, D222–D226. doi: 10.1093/nar/gku1221
Mintzer, V., Moran-Gilad, J., and Simon-Tuval, T. (2019). Operational models and criteria for incorporating microbial whole genome sequencing in hospital microbiology – A systematic literature review. Clin. Microbiol. Infect. 25, 1086–1095. doi: 10.1016/j.cmi.2019.04.019
Mojica, F. J., and Rodriguez-Valera, F. (2016). The discovery of CRISPR in archaea and bacteria. FEBS J. 283, 3162–3169. doi: 10.1111/febs.13766
Oechslin, F. (2018). Resistance development to bacteriophages occurring during bacteriophage therapy. Viruses 10:351. doi: 10.3390/v10070351
Oliveira, P. H., Touchon, M., and Rocha, E. P. (2014). The interplay of restriction-modification systems with mobile genetic elements and their prokaryotic hosts. Nucleic Acids Res. 42, 10618–10631. doi: 10.1093/nar/gku734
Otsuka, Y., and Yonesaki, T. (2012). Dmd of bacteriophage T4 functions as an antitoxin against Escherichia coli LsoA and RnlA toxins. Mol. Microbiol. 83, 669–681. doi: 10.1111/j.1365-2958.2012.07975.x
Pinilla-Redondo, R., Mayo-Muñoz, D., Russel, J., Garrett, R. A., Randau, L., Sørensen, S. J., et al. (2020). Type IV CRISPR-Cas systems are highly diverse and involved in competition between plasmids. Nucleic Acids Res. 48, 2000–2012. doi: 10.1093/nar/gkz1197
Rando, O. J., and Verstrepen, K. J. (2007). Timescales of genetic and epigenetic inheritance. Cell 128, 655–668. doi: 10.1016/j.cell.2007.01.023
Reimann, V., Ziemann, M., Li, H., Zhu, T., Behler, J., Lu, X., et al. (2020). Specificities and functional coordination between the two Cas6 maturation endonucleases in Anabaena sp. PCC 7120 assign orphan CRISPR arrays to three groups. RNA Biol. 17, 1442–1453. doi: 10.1080/15476286.2020.1774197
Rho, M., Wu, Y. W., Tang, H., Doak, T. G., and Ye, Y. (2012). Diverse CRISPRs evolving in human microbiomes. PLoS Genet. 8:e1002441. doi: 10.1371/journal.pgen.1002441
Schooley, R. T., Biswas, B., Gill, J. J., Hernandez-Morales, A., Lancaster, J., Lessor, L., et al. (2017). Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection. Antimicrob. Agents Chemother. 61:e00954-17. doi: 10.1128/AAC.00954-17
Shen, J., Lv, L., Wang, X., Xiu, Z., and Chen, G. (2017). Comparative analysis of CRISPR-Cas systems in Klebsiella genomes. J. Basic Microbiol. 57, 325–336. doi: 10.1002/jobm.201600589
Shmakov, S. A., Utkina, I., Wolf, Y. I., Makarova, K. S., Severinov, K. V., and Koonin, E. V. (2020). CRISPR arrays away from cas genes. CRISPR J. 3, 535–549. doi: 10.1089/crispr.2020.0062
Sievers, F., and Higgins, D. G. (2014). Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 1079, 105–116. doi: 10.1007/978-1-62703-646-7_6
Song, S., and Wood, T. K. (2020). A primary physiological role of toxin/antitoxin systems is phage inhibition. Front. Microbiol. 11:1895. doi: 10.3389/fmicb.2020.01895
Stern, A., and Sorek, R. (2011). The phage-host arms race: shaping the evolution of microbes. Bioessays 33, 43–51. doi: 10.1002/bies.201000071
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
van Houte, S., Buckling, A., and Westra, E. R. (2016). Evolutionary ecology of prokaryotic immune mechanisms. Microbiol. Mol. Biol. Rev. 80, 745–763. doi: 10.1128/MMBR.00011-16
Vasu, K., and Nagaraja, V. (2013). Diverse functions of restriction-modification systems in addition to cellular defence. Microbiol. Mol. Biol. Rev. 77, 53–72.
Wang, X., Loh, B., Gordillo Altamirano, F., Yu, Y., Hua, X., and Leptihn, S. (2021). Colistin-phage combinations decrease antibiotic resistance in Acinetobacter baumannii via changes in envelope architecture. Emerg. Microbes Infect. 10, 2205–2219.
Wei, Y., Gao, Z., Zhang, H., and Dong, Y. (2016). Structural characterizations of phage antitoxin Dmd and its interactions with bacterial toxin RnlA. Biochem. Biophys. Res. Commun. 472, 592–597. doi: 10.1016/j.bbrc.2016.03.025
Wright, A. V., Wang, J. Y., Burstein, D., Harrington, L. B., Paez-Espino, D., Kyrpides, N. C., et al. (2019). A functional mini-integrase in a two-protein-type V-C CRISPR system. Mol. Cell 73, 727–737.e723. doi: 10.1016/j.molcel.2018.12.015
Yang, Y., Shen, W., Zhong, Q., Chen, Q., He, X., Baker, J. L., et al. (2020). Development of a bacteriophage cocktail to constrain the emergence of phage-resistant Pseudomonas aeruginosa. Front. Microbiol. 11:327. doi: 10.3389/fmicb.2020.00327
Zhang, Q., and Ye, Y. (2017). Not all predicted CRISPR-Cas systems are equal: isolated cas genes and classes of CRISPR like elements. BMC Bioinformatics 18:92. doi: 10.1186/s12859-017-1512-4
Zhang, Q., Doak, T. G., and Ye, Y. (2014). Expanding the catalog of cas genes with metagenomes. Nucleic Acids Res. 42, 2448–2459. doi: 10.1093/nar/gkt1262
Zhang, R., Ou, H. Y., Gao, F., and Luo, H. (2014). Identification of horizontally-transferred genomic islands and genome segmentation points by using the GC profile method. Curr. Genomics 15, 113–121. doi: 10.2174/1389202915999140328163125
Zhang, Y., Zhang, Z., Zhang, H., Zhao, Y., and Xiao, J. (2020). PADS Arsenal: a database of prokaryotic defence systems related genes. Nucleic Acids Res. 48, D590–D598. doi: 10.1093/nar/gkz916
Keywords: bacterial, resistance, genomic island, phages, Acinetobacter baumannii, CRISPR, WGS or whole-genome sequencing
Citation: Ambroa A, Blasco L, López M, Pacios O, Bleriot I, Fernández-García L, González de Aledo M, Ortiz-Cartagena C, Millard A and Tomás M (2022) Genomic Analysis of Molecular Bacterial Mechanisms of Resistance to Phage Infection. Front. Microbiol. 12:784949. doi: 10.3389/fmicb.2021.784949
Received: 28 September 2021; Accepted: 27 December 2021;
Published: 17 February 2022.
Edited by:
Iraj Rasooli, Shahed University, IranReviewed by:
Shuang-Yong Xu, New England Biolabs, United StatesSebastian Leptihn, Zhejiang University-University of Edinburgh Institute, China
Vishvanath Tiwari, Central University of Rajasthan, India
Copyright © 2022 Ambroa, Blasco, López, Pacios, Bleriot, Fernández-García, González de Aledo, Ortiz-Cartagena, Millard and Tomás. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María Tomás, bWEuZGVsLm1hci50b21hcy5jYXJtb25hQHNlcmdhcy5lcw==
†These authors have contributed equally to this work