
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 22 November 2021
Sec. Microbe and Virus Interactions with Plants
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.783633
 Haijie Ma1,2,3†
Haijie Ma1,2,3† Lei Li2†
Lei Li2† Yunpeng Gai2
Yunpeng Gai2 Xiaoyan Zhang2
Xiaoyan Zhang2 Yanan Chen2
Yanan Chen2 Xiaokang Zhuo3
Xiaokang Zhuo3 Yingzi Cao2
Yingzi Cao2 Chen Jiao2
Chen Jiao2 Fred G. Gmitter Jr.3*
Fred G. Gmitter Jr.3* Hongye Li2*
Hongye Li2*Histone acetylation, which is critical for transcriptional regulation and various biological processes in eukaryotes, is a reversible dynamic process regulated by HATs and HDACs. This study determined the function of 6 histone acetyltransferases (HATs) (Gcn5, RTT109, Elp3, Sas3, Sas2, Nat3) and 6 histone deacetylases (HDACs) (Hos2, Rpd3, Hda1, Hos3, Hst2, Sir2) in the phytopathogenic fungus Alternaria alternata by analyzing targeted gene deletion mutants. Our data provide evidence that HATs and HDACs are both required for mycelium growth, cell development and pathogenicity as many gene deletion mutants (ΔGcn5, ΔRTT109, ΔElp3, ΔSas3, ΔNat3, ΔHos2, and ΔRpd3) displayed reduced growth, conidiation or virulence at varying degrees. In addition, HATs and HDACs are involved in the resistance to multiple stresses such as oxidative stress (Sas3, Gcn5, Elp3, RTT109, Hos2), osmotic stress (Sas3, Gcn5, RTT109, Hos2), cell wall-targeting agents (Sas3, Gcn5, Hos2), and fungicide (Gcn5, Hos2). ΔGcn5, ΔSas3, and ΔHos2 displayed severe growth defects on sole carbon source medium suggesting a vital role of HATs and HDACs in carbon source utilization. More SNPs were generated in ΔGcn5 in comparison to wild-type when they were exposed to ultraviolet ray. Moreover, ΔRTT109, ΔGcn5, and ΔHos2 showed severe defects in resistance to DNA-damaging agents, indicating the critical role of HATs and HDACs in DNA damage repair. These phenotypes correlated well with the differentially expressed genes in ΔGcn5 and ΔHos2 that are essential for carbon sources metabolism, DNA damage repair, ROS detoxification, and asexual development. Furthermore, Gcn5 is required for the acetylation of H3K4. Overall, our study provides genetic evidence to define the central role of HATs and HDACs in the pathological and biological functions of A. alternata.
The fundamental structural subunit of the chromatin is the nucleosome, which consists of approximately 200 bp of DNA wrapped around an octamer of core histone proteins H2A, H2B, H3, and H4 (Kornberg and Lorch, 1999). The reversible acetylation of specific lysine residues in N-terminal tails of core histones is maintained by histone acetyltransferases (HATs) and histone deacetylases (HDACs) and has been found to play a pivotal role in chromatin-regulated DNA events, such as DNA replication, damage repair, recombination, and gene expression in eukaryotes (Lee and Workman, 2007; Yang and Seto, 2008). HATs are classified into five families, including MYST (Ybf2/Sas3, Sas2, MOZ, Tip60), GNAT (Gcn5-related N-acetyltransferases), p300/CBP, nuclear receptor cofactors, and basal transcription factors (Jeon et al., 2014). HDACs are grouped into four groups based on phylogenetic analysis and sequence homology, including Class I and II (Rpd3, Hda1), Class III (Sir2 or Sir2-like protein) and Class IV (HDAC11) (Gregoretti et al., 2004; Jeon et al., 2014).
In yeast, many HATs or HDACs have been found to be involved in transcriptional regulation and many cellular functions. Gcn5 belongs to the GNAT family and acts as a core subunit of several histone acetyltransferase (HAT) complexes, such as ADA, SAGA, or SALSA (Lee and Young, 2000). Gcn5 acetylates N-terminal lysine (K) residues of histones H2B (K11/16) or H3 (K14/18/23/27/36), and the loss of its function leads to defects in gene transcription, sexual differentiation and multiple stresses resistance (Helmlinger et al., 2008; Johnsson et al., 2009; Nugent et al., 2010). H3K9/27/56 can be acetylated by RTT109, a HAT of p300/CBP family (Fillingham et al., 2008; Das et al., 2009; Burgess et al., 2010; Da Rosa et al., 2010). In yeast, H3K56ac regulates expression homeostasis and resistance to DNA-damaging agents (Masumoto et al., 2005; Ozdemir et al., 2005; Han et al., 2007; Voichek et al., 2016). Both Sas3 and Esa1 belong to MYST family, which function as the subunits of Nucleosome Acetyltransferase of histone H3 (NuA3) and H4 (NuA4) complexes, respectively (Lee and Workman, 2007). Studies revealed that Esa1 is essential for cell cycle progression in yeast (Clarke et al., 1999). In S. cerevisiae, the transcription of FLO1 was significantly reduced in the absence of both Sas3 and Ada2 (Church et al., 2017). Rpd3 is a founding member of class I and comprises an N-terminal deacetylase domain (Vidal and Gaber, 1991). In yeast, Rpd3 functions as a catalytic subunit in sin3 complexes and has been linked to many biological functions, such as transcriptional regulation, DNA repair, replication, ROS resistance, or azole resistance (Vogelauer et al., 2002; Jazayeri et al., 2004; Baker et al., 2013; Li et al., 2015; Bosio et al., 2017). Hos2 and Hda1 belong to class II, and both of them also contain an N-terminal deacetylase domain (Yang and Seto, 2008). In yeast, Hda1 and Rpd3L contribute to the repair of replication-born DSBs (double strand breaks) by facilitating cohesin loading thus preventing genome instability (Ortega et al., 2019). Hos2 acts as a key subunit of Set3 complex which deacetylates H4 and H3 by counteracting Esa1 (Pijnappel et al., 2001; Torres-Machorro et al., 2015). The yeast Hos2 is involved in various biological functions, such as these involved in cell integrity pathway, cell resistance to multiple stresses, DNA damage repair, and expression of growth-related genes (Pijnappel et al., 2001; Wirén et al., 2005; Cohen et al., 2008; Kim et al., 2012; Tkach et al., 2012). The best-characterized member in Class III is the NAD-dependent histone deacetylase (HDAC) Sir2. Yeast Sir2 acts as anti-aging gene required for cellular lifespan regulation by caloric restriction (Lin et al., 2000; Dang et al., 2009).
In filamentous fungi, HATs and HDACs have been found to be involved in growth, development, virulence, synthesis of secondary metabolites and multi-stress resistance. Studies in Aspergillus nidulans showed that GcnE (ortholog of Gcn5) controls asexual reproduction, and the biosynthesis of sterigmatocystin, penicillin and terrequinone A (Nützmann et al., 2011; Cánovas et al., 2014). Genetic deletion of Gcn5 leads to severe defects in virulence, sexual or asexual development, and/or virulence in Ustilago maydis, Beauveria bassiana, and Trichoderma reesei (Xin et al., 2013; González-Prieto et al., 2014; Cai et al., 2018b). In B. bassiana, RTT109 deletion mutant shows attenuated virulence, and increased sensitivity to the stress of oxidation and DNA-damaging agents (Cai et al., 2018c). Many other HATs also have been found to be involved in cell growth and pathogenicity in M. oryzae (Sas3), F. graminearum (Elp3), and so on (Lee et al., 2014; Dubey et al., 2019). HDAC Hos2 acts as a vital subunit of Tig1 complex crucial for conidiation and infectious growth in M. oryzae, and is required for pathogenesis and dimorphic switch in U. maydis (Ding et al., 2010; Elías-Villalobos et al., 2015). Although many HATs and HDACs have been identified to be critical for an array of biological processes in many fungi, the specific function of each gene is not completely the same among different fungi.
Alternaria alternata tangerine pathotype is a disastrous fungal pathogen which causes Citrus Brown Spot on young leaves, shoots and fruits of many citrus cultivars. Previous studies revealed that ROS detoxification-related genes such as Ap1 and Skn7 (Lin et al., 2009; Chen et al., 2012) or host-selective ACT-toxin biosynthesis genes such as ACTT5 and ACTT6 (Miyamoto et al., 2009), are critical for A. alternata to cause lesions on host tissues. Although numerous studies of HATs or HDACs indicate that many of these genes are involved in ROS detoxification, synthesis of secondary metabolites, and various biological functions in many pathogenic fungi, the roles of these genes in A. alternata have not been elucidated. In this study, we identified all the HATs and HDACs orthologs in A. alternata and clarified their roles in growth and development, carbon source utilization, multiple stresses resistance, DNA damage repair and virulence using genetic and biological analyses. We also performed a transcriptome analysis to explore the regulatory role of Gcn5 and Hos2 in this important citrus pathogen.
The wild-type Z7 strain of A. alternata (Fr.) Keissler used in the mutagenesis experiments was isolated from an infected citrus (Citrus suavissima Hort. Ex Tanaka) in Zhejiang, China (Huang et al., 2015; Wang et al., 2016). Fungal strains were cultured on potato dextrose agar (PDA) at 26°C and conidia were collected after incubating on V8 medium for 8 days. Mycelium cultured in liquid potato dextrose broth (PDB) was collected by passing through cheesecloth and used for purification of DNA or RNA.
ΔSas2, ΔSas3, ΔElp3, ΔGcn5, ΔRTT109, ΔNat3, ΔRpd3, ΔHos2, ΔHos3, ΔHda1, ΔHst2, and ΔSir2 strains were created by deleting Sas2, Sas3, Elp3, Gcn5, RTT109, Nat3, Rpd3, Hos2, Hos3, Hda1, Hst2, and Sir2, respectively, by integrating a bacterial HYG cassette under control of TrpC gene promoter and terminator in the genome of Z7 using a split marker approach mediated by protoplasts transformation as described previously (Chung et al., 2002; Ma et al., 2018). Fungal transformants were recovered from PDA containing 100 μg/ml hygromycin and examined by PCR with primers specific to its targeted gene. Oligonucleotide primers used in this study are listed in Supplementary Table 1.
Morphological observation of conidia and hyphae was carried using a Nikon microscope equipped with a LV100ND image system (Nikon, Japan).
Fungal virulence was assessed on detached Dancy (Citrus reticulata Blanco) leaves inoculated by placing a 5-mm dia. agar plug covered with fungal mycelium on each spot, and those inoculated leaves were kept in a plastic box at 26°C for 2–4 days for lesion development.
All the phenotypes of mutants and wild-type were evaluated. To examine the vegetative growth under multiple stresses, mutants and wild-type were inoculated on PDA containing 10 mM H2O2, 1 mM cumyl hydroperoxide (CHP), 1 M sorbitol, 1 M KCl, 1 M NaCl, 250 mM CaCl2, 1 mM CuSO4, 0.01% sodium dodecyl sulfate (SDS), 0.2 mg/ml congo red (CR), 10 mg/L chlorothalonil, 2 mg/L Mancozeb, 3 mg/L Boscalid, 5 mM hydroxyurea (HU), 0.1% methyl methanesulfonate (MMS), or 5 μM camptothecin (CPT). To examine the carbon utilization ability, mutants and wild-type were inoculated on MM medium using 20 g/L glucose, 10 g/L sucrose, 10 g/L starch, or 10 g/L lactose as sole carbon source. All tests were repeated at least twice with three replicates of each treatment.
Wild-type, ΔGcn5 and ΔSas3 each with two replicates were cultured in PDB (120 rpm, 28°C) and were harvested after 36 h. The samples were ground and homogenized in 1 ml lysis buffer composed of 100 mM NaCl, 1% Triton X-100, 5 mM EDTA, 10 μl protease inhibitor cocktail (P8215, Sigma-Aldrich), 2 mM PMSF (P7626, Sigma-Aldrich), and 50 mM Tris-HCl, pH 7.5. The homogenates were centrifuged at 14,000 rpm, 20 min, 4°C. The Supernatants were collected as total protein. Antibody used in this study is H3K4ac (Abcam, ab176799).
Wild-type, ΔGcn5 and ΔHos2 each with two replicates were cultured in PDB (120 rpm, 28°C) and were harvested after 36 h. RNA was extracted using Axygen RNA purification kit (Capital Scientific, Union City, CA, United States). The libraries were produced using an IlluminaTruSeq RNA Sample Preparation Kit, and they were sequenced on an Illumina HIseq 2500 platform, generating 150 bp paired-end reads. Trimmomatic Ver 0.36 (Bolger et al., 2014) was used to remove adaptors and low-quality reads. TopHat2 (Kim et al., 2013) was used to map sequences to the reference genome and the mapped reads on each gene were determined by HTSeq (Anders et al., 2015). Differential expression analysis was determined using DESEQ2 (Anders and Huber, 2012) based on the overall transcript counts after normalization. Transcripts with an adjusted FDR less than 0.01 and the absolute value of log2FC (log2 fold change) greater than 2 were considered to be differentially expressed genes (DEGs). DEGs were annotated by searching against the NCBI nr databases. GO and KEGG pathway were performed using clusterProfiler v3.6 (Yu et al., 2012). anti-SMASH 4.0 (Blin et al., 2017) was used to predict gene clusters associated with secondary metabolites. Protein domains were predicted in SMART database available at http://smart.embl-heidelberg.de/. Carbohydrate-active enzymes (CAZymes) were predicted using dbCAN meta server.1 The transcriptomes raw data (SRS6969981, SRS6969980, SRS6969978, SRS6969979, SRS6969977, SRS6969976) have been deposited in NCBI’s Sequence Read Archive.
A paired-end library of each sample was constructed and sequenced by Hiseq2500, with insert sizes of approximately 350 bp. Trimmomatic was used to remove adaptors and low-quality reads. The high-quality resequencing reads was mapped to the Alternaria alternata reference genome with Bowtie2 (Langmead and Salzberg, 2012) using the default parameters. The aligned results were further filtered for unmapped reads and duplicated reads using SAMtools (Li et al., 2009) and Picard packages, respectively. SNP calling was carried out using Freebayes. Genomic variation analysis was performed using VCFtools (Danecek et al., 2011). IGV (Helga et al., 2013) was used to visualize reads on the genome. MapChart (Voorrips, 2002) was used to visualize SNPs on the genome. The raw data of whole-genome resequencing (SRS6971885, SRS6971887, SRS6971884, SRS6971886) have been deposited in NCBI’s Sequence Read Archive.
Quantitative Real-time PCR (qRT-PCR) was carried out on a 7300 Real Time PCR system (Applied Biosystems, Carlsbad, CA, United States) to validate the transcriptome data. RNA was extracted with an Axygen RNA purification kit (Capital Scientific) and the cDNA was synthesized from RNA using a PrimeScript RT regent kit (Takara, Shiga, Japan). Actin-coding gene (KP341672) was used as an internal control and the resulting data were normalized using the comparative Cτ method as described previously (Sun et al., 2011).
The statistical significance of treatments was determined by analysis of variance and means separated by Duncan’s test (P < 0.05).
Seven HATs and six HDACs were identified combining the prediction results of dbHiMo2 and hmm analysis, and the families to which these genes belong are also listed in Table 1. Nucleotide sequence length of corresponding predicted genes ranged from 654 to 3,450 bp, the number of introns contained varied from 0 to 8, and the length of proteins varied from 193 to 1,082 aa (Figures 1A,B). Functional domain prediction of amino acid sequences encoded by these genes revealed that all 13 proteins contain functional domains unique to HAT or HDAC, indicating these gene predictions are highly reliable. Mapping these HATs and HDACs to chromosomes revealed that all 13 genes are localized in the essential chromosomes, whereas no HATs or HDACs were identified in conditionally dispensable chromosomes (CDC) (Supplementary Figure 1). Phylogenetic analysis using the neighbor-joining method revealed that all HATs and HDACs predicted in A. alternata were highly similar to their corresponding genes in other fungi (Figure 1C).
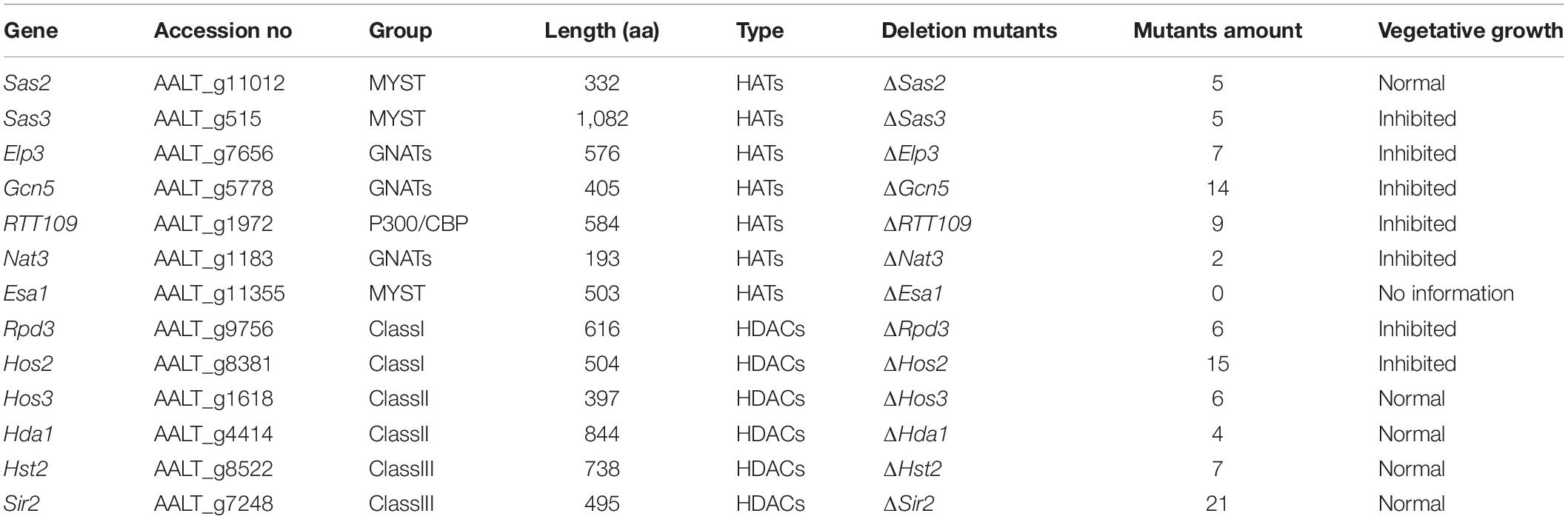
Table 1. HATs and HDACs in A. alternata.

Figure 1. Identification and phylogenetic analysis of HATs and HDACs in A. alternata. (A) Schematic depiction of ORFs of HATs and HDACs. (B) Schematic depiction of conserved domain of HATs and HDACs. (C) Phylogenetic trees were constructed by comparing amino acid sequences between different species using the neighbor-joining algorithm of MEGA6. Bar, 5% sequence divergence. Accession number of amino acid sequences are listed in Supplementary Table 10.
To investigate the function of HATs and HDACs in A. alternata, mutants defective for Gcn5 (ΔGcn5), Elp3 (ΔElp3), RTT109 (ΔRTT109), Nat3 (ΔNat3), Sas3 (ΔSas3), Sas2 (ΔSas2), Hos2 (ΔHos2), Rpd3 (ΔRpd3), Hos3 (ΔHos3), Hda1 (ΔHda1), Hst2 (ΔHst2), and Sir2 (ΔSir2) were generated by the homologous recombination method. The mutants were verified by PCR analysis (Supplementary Figure 2). However, no Esa1 deletion mutants were obtained after multiple attempts.
In HATs disrupted mutants, vegetative growth rate of ΔGcn5, ΔElp3, ΔRTT109, ΔNat3, and ΔSas3 was reduced by 66, 44, 12, 71, and 41% compared to wild-type on potato dextrose agar (PDA), indicating that most HATs play a vital role on the growth of A. alternata (Figure 2A and Supplementary Figure 3). In addition, ΔGcn5 and ΔNat3 produced fewer aerial hyphae than wild-type on PDA. Microscopic examination revealed that the impacts of different HATs on the development of A. alternata varied greatly. The ΔGcn5 strain produced swollen and stubby hyphae and more hyphae branches, but failed to produce any spores (Figures 2B,C and Supplementary Figure 4). No spores were observed in ΔNat3 after 6 days of incubation, but a few spores appeared as incubation time increased. The hyphal tip of ΔNat3 was irregularly twisted and the hyphae were frequently branched. The conidiation of ΔElp3, ΔSas3, and ΔRTT109 decreased significantly, but the mycelium morphology remained the same as the wild-type level. Only ΔSas2 displayed wild-type levels of vegetative growth, hyphal development, and conidiation.

Figure 2. HATs and HDACs were required for growth and development. (A) Radial growth of the wild-type strain (Z7) and mutants. (B) Hyphae morphology of Z7 and mutants. (C) Conidiation of Z7 and mutants.
Unlike HATs, few HDACs were involved in vegetative growth and conidiation. Only ΔHos2 and ΔRpd3 grew significantly slower (with inhibition of 37 and 67%, respectively) than the wild-type (Figure 2A). Microscopic examination revealed that conidiation was inhibited significantly (>90%) in ΔHos2 and ΔRpd3. Similar to ΔGcn5, ΔRpd3 also produced stubby and swollen hyphae (Figure 2B). ΔHos3, ΔHda1, ΔHst2, and ΔSir2 displayed wild-type levels of vegetative growth and conidiation (Figure 2C).
To determine the effect of HATs and HDACs on the pathogenicity of A. alternata, virulence assays were performed on detached Dancy leaves. For HATs deletion mutants, ΔGcn5, ΔSas3, and ΔNat3 failed to induce visible lesion on citrus leaves. ΔRTT109 and ΔElp3 induced smaller necrotic lesions than those induced by the wild type 2 days post inoculation (dpi) (Figure 3 and Supplementary Figure 5). Only ΔSas2 induced necrotic lesions at a rate and magnitude comparable to those for the wild-type on Dancy leaves 2 dpi. For HDACs deletion mutants, ΔRpd3 and ΔHos2 induced smaller necrotic lesions compared to those induced by the wild type at 2 dpi. ΔHos3, ΔSir2, and ΔHda1 produced necrotic lesions similar to those induced by Z7 (Figure 3 and Supplementary Figure 5). Quantitative analysis revealed that the area of lesion induced by the ΔRTT109, ΔElp3, ΔSas2, ΔHos2, ΔRpd3, ΔHda1, ΔHos3, ΔHst2, and ΔSir2 were about 79, 32, 92, 18, 11, 97, 104, 96, and 103%, respectively, of those induced by Z7.

Figure 3. HATs and HDACs were required for A. alternata pathogenicity. Pathogenicity was assayed on detached Dancy leaves by placing 5 mm agar plug covering fungal mycelium on the bottom of leaves (left, mutants; right, Z7). The leaves were incubated in a plastic box for lesion development and the necrotic lesions was observed at 2 days post inoculation (dpi). The percent changes of necrotic lesions appearing on the leaves calculated in relation to those induced by Z7 are also indicated.
To examine whether HATs or HDACs were required for the utilization of carbon sources, mycelial radical growth assays were performed on MM medium supplemented with sucrose, starch, glucose, or lactose as sole carbon sources (Figure 4). In HATs disruption mutants, the vegetative growth inhibition rate of ΔGcn5 on all tested sole carbon source media for 4 days was 100%. At the same time, the growth inhibition ratio of ΔSas3 on the MM medium supplemented with glucose, sucrose, lactose, and starch was increased by 28, 41, 39, and 41%, compared to the wild type. In contrast, ΔSas2, ΔElp3, and ΔRTT109 displayed wild-type sensitivity to all tested sole carbon source media. Most HDACs disruption mutants, including ΔSir2, ΔHos3, ΔHda1, and ΔHst2, exhibited wild-type sensitivity to all test medium. Only ΔHos2 showed slightly increased sensitivity on carbon sources of glucose and starch.
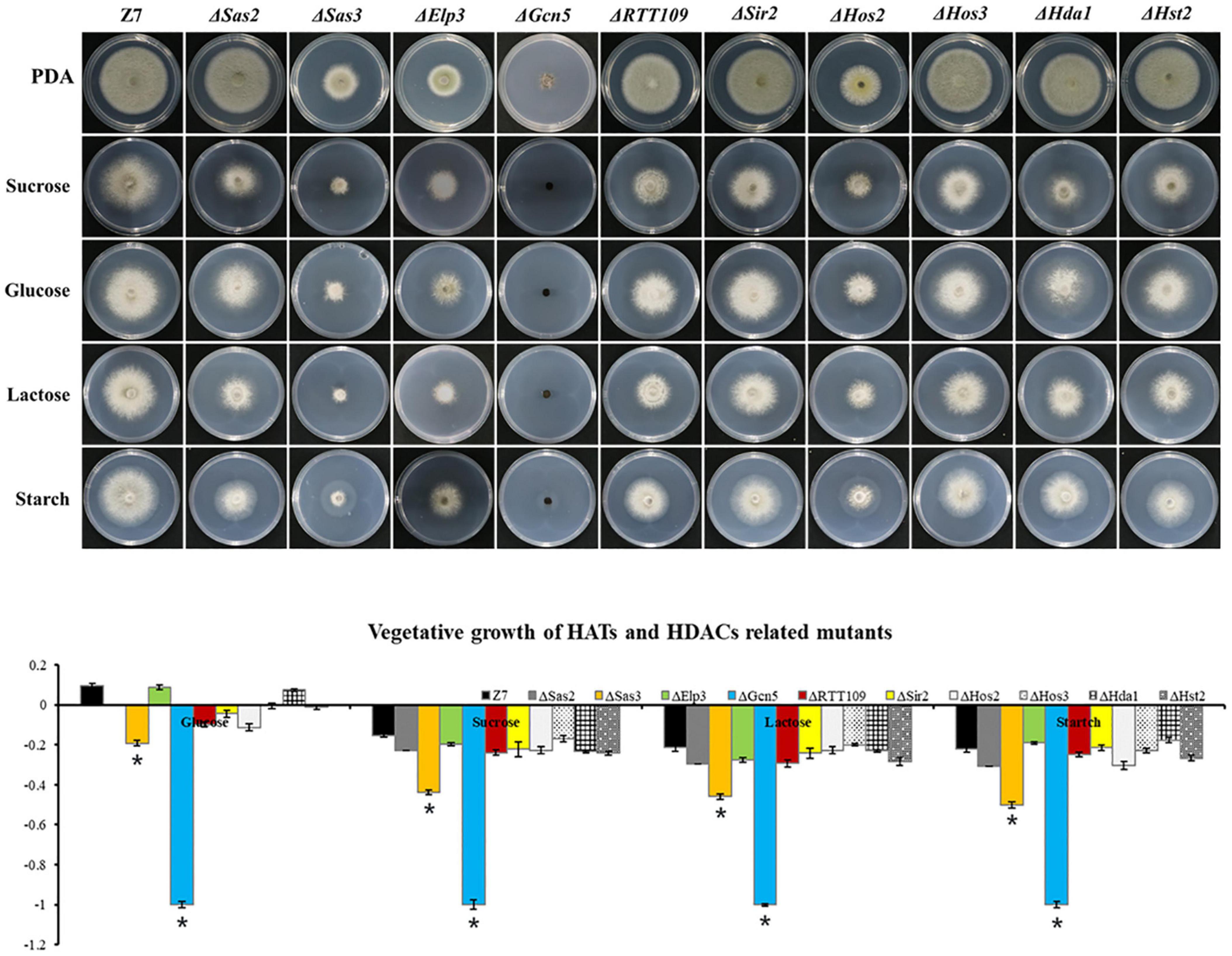
Figure 4. HATs and HDACs were involved in carbon source utilization. Z7, and mutants with impaired HATs or HDACs cultured on MM medium amended with sucrose, glucose, lactose, or starch as sole carbon source. The percentage of growth reduction determined by comparing a cumulative percentage of the growth of Z7 and mutants grown on same medium is also shown (*, p < 0.01).
To prove further that the severe growth defects of ΔGcn5 on sole carbon source medium was not caused by its slow vegetative growth, the incubation time was extended to 10 days. Growth examination indicated that the growth inhibition ratio of ΔGcn5 on the medium with lactose as sole carbon source was still 100%, and only a few hyphae of ΔGcn5 appeared on carbon sources of sucrose, starch, or glucose (the growth inhibition ratio is close to 100%), confirming that the Gcn5 was required for the utilization of carbon sources (Supplementary Figure 6).
In mutants defective for HATs, both ΔElp3 and ΔGcn5 were more sensitive to H2O2 and CHP (cumyl-H2O2), while ΔSas3 and ΔRTT109 just showed increased sensitivity to H2O2, compared with wild-type. In addition, ΔSas3 showed elevated sensitivity to cell wall-targeting agents such as Congo red (CR) and sodium dodecyl sulfonate (SDS). At the same time, ΔElp3, ΔGcn5, and ΔRTT109 just displayed increased sensitivity to CR. ΔSas3, ΔGcn5, and ΔRTT109 displayed increased sensitivity to NaCl, KCl, and CuSO4. Only ΔRTT109 displayed increased sensitivity to CaCl2. However, ΔSas3, ΔElp3, and ΔGcn5 displayed enhanced resistance to CaCl2 compared to wild-type. Both ΔSas3 and ΔElp3 also showed increased tolerance to V8 medium compared to wild-type. Only ΔSas2 showed wild-type resistance to all test chemicals (Figure 5).
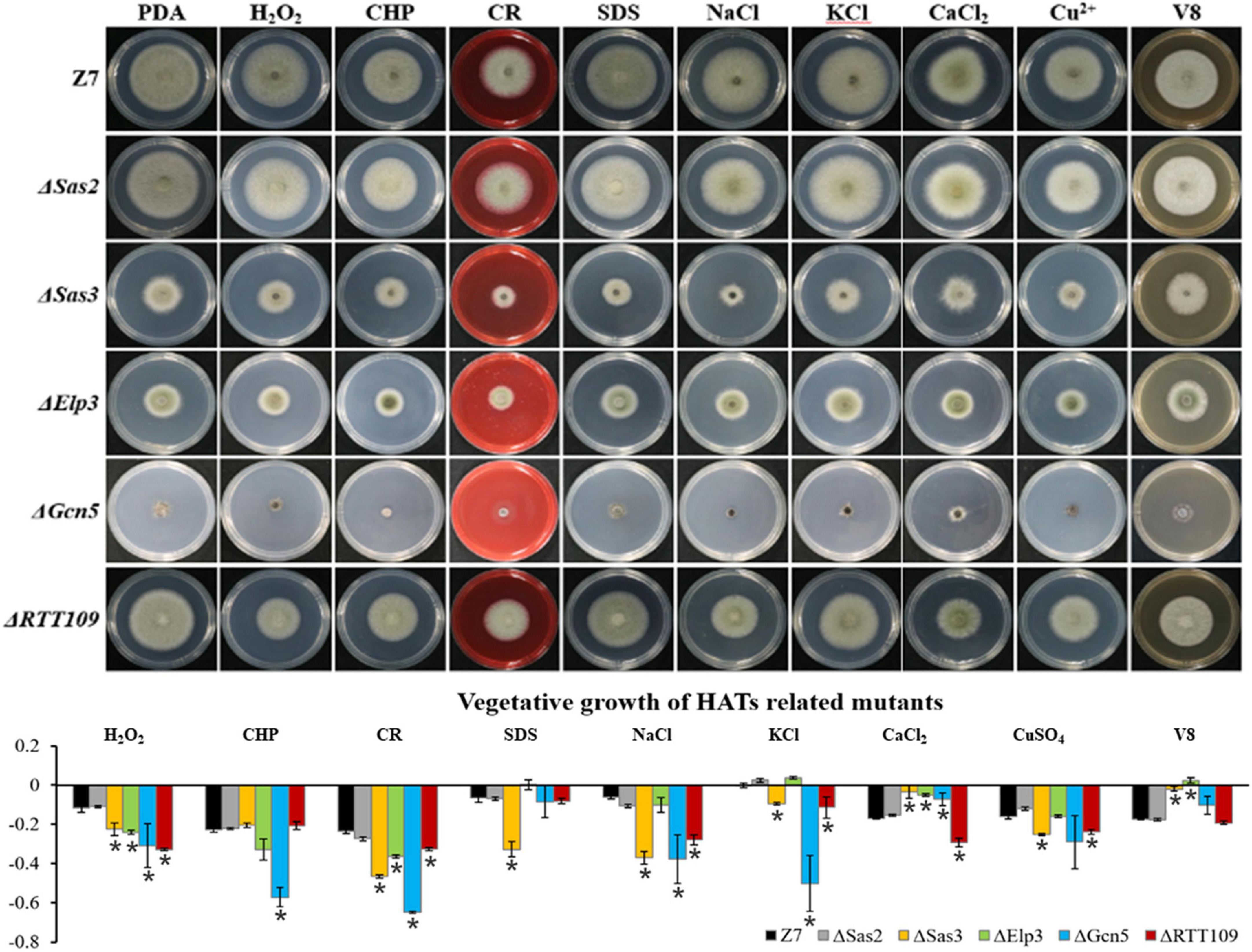
Figure 5. Chemical sensitivity assays. Z7 and HATs related mutants were grown on PDA with or without the indicated chemicals [10 mM H2O2, 1 mM cumyl hydroperoxide (CHP), 1 M KCl, 1 M NaCl, 250 mM CaCl2, 1 mM CuSO4, 0.01% sodium dodecyl sulfate (SDS), 0.2 mg/ml congo red (CR)]. The percentage of growth reduction determined by comparing a cumulative percentage of the growth of Z7 and mutants grown on same medium is also shown (*, p < 0.01).
Unlike HATs which were widely involved in the adaption to various stresses, most of HDACs such as Hos3, Hda1, Hst2, and Sir2 were not involved in the resistance to most tested chemicals. Sensitivity assays revealed that ΔHos2 showed increased sensitivity to H2O2, CHP, CR, SDS, NaCl, KCl, CaCl2, and V8 medium, but not sorbitol and CuSO4 (Figure 6).
Figure 6. Chemical sensitivity assays. Z7 and HDACs related mutants were grown on PDA with or without the indicated chemicals [10 mM H2O2, 1 mM cumyl hydroperoxide (CHP), 1 M sorbitol, 1 M KCl, 1 M NaCl, 250 mM CaCl2, 1 mM CuSO4, 0.01% sodium dodecyl sulfate (SDS), 0.2 mg/ml congo red (CR)]. The percentage of growth reduction determined by comparing a cumulative percentage of the growth of Z7 and mutants grown on same medium is also shown (*, p < 0.01).
Assays for fungicide sensitivity indicated that both ΔGcn5 and ΔHos2 displayed increased sensitivity to chlorothalonil, but not mancozeb and boscalid. All the other HATs or HDACs mutants displayed wild-type sensitivity to the test fungicides (Supplementary Figure 7).
The results described above indicated that both Gcn5 and Hos2 played vital roles in vegetative growth, development, multiple stresses resistance, carbon sources utilization, ROS detoxification, and virulence. To explore the transcriptional regulatory mechanism of Gcn5 and Hos2, transcriptome analyses of Gcn5 and Hos2 compared to wild type were performed. Overall, 1,503 up-regulated and 1,051 down-regulated genes were identified in ΔGcn5 (Supplementary Table 2). In ΔHos2, 469 up-regulated and 536 down-regulated genes were identified (Supplementary Table 3). Gcn5 and Hos2 were one of the genes with lowest transcription level in ΔGcn5 and ΔHos2, respectively, indicating the genetic deletion of Gcn5 and Hos2 was reliable (Supplementary Tables 2, 3). In addition, RT-qPCR results of 14 randomly selected differentially expressed genes (DEGs) was consistent with transcriptome data of ΔGcn5 and ΔHos2, indicating that the transcriptome data were reliable. Although the DEGs in ΔGcn5 and ΔHos2 accounted for up to 20 and 8% of all genes in the genome, respectively, the transcription levels of the other 12 HATs and HDACs in ΔGcn5 or ΔHos2 did not change significantly, indicating that Gcn5 and Hos2 are not required for the transcription of the other HATs and HDACs.
COG (Clusters of Orthologous Groups of proteins) analysis of ΔGcn5 and ΔHos2 revealed that many DEGs were enriched in the metabolism category, such as Carbohydrate transport and metabolism (G), Amino acid transport and metabolism (E), Lipid transport and metabolism (I), and Inorganic ion transport and metabolism (P). There are also many DEGs in ΔGcn5 or ΔHos2 enriched in Transcription (K) and Replication, recombination and repair (L) (Supplementary Figures 8, 9). KEGG enrichment analysis revealed that the most affected metabolism-related pathways in ΔGcn5 or ΔHos2 were mainly related to carbon and nitrogen sources metabolism, such as starch and sucrose metabolism, galactose metabolism, Fructose and mannose metabolism, Methane metabolism, and Nitrogen metabolism. In addition, many DEGs in ΔGcn5 or ΔHos2 were enriched in genetic information processing, such as DNA damage repair, RNA transport, and tRNA biosynthesis (Supplementary Figures 8, 9). Although many DEGs in ΔGcn5 and ΔHos2 were enriched in the same pathways, the amount and expression level of those genes varied greatly.
Gcn5 and Hos2 are involved in regulating the transcription of many genes involved in carbon (sugar) source utilization. Analysis of DEGs in Glycolysis/Gluconeogensis (ko00010), Fructose and Mannose Metabolism (ko00051), Galactose Metabolism (ko00052), Starch and Sucrose metabolism (ko00500), Pentose Phosphate Pathway (ko00030), Citrate cycle (ko00020), and Pentose and Glucuronate interconversions (ko00040) in ΔGcn5 and ΔHos2 revealed that the number of down-regulated genes (ΔGcn5, 22; ΔHos2, 15) was significantly greater than the up-regulated genes (ΔGcn5, 22; ΔHos2, 15) (Figure 7). In yeast, invertase and hexokinase are critical for the utilization of sucrose, lactose, glucose, or fructose (Carlson and Botstein, 1982). In A. alternata, AALT_g6039 encodes hexokinase, and AALT_g5797, AALT_g10082 and AALT_g2401 encode invertases. All four genes were significantly down-regulated in ΔHos2, but only AALT_g6039 and AALT_g2401 were significantly down-regulated in ΔGcn5.
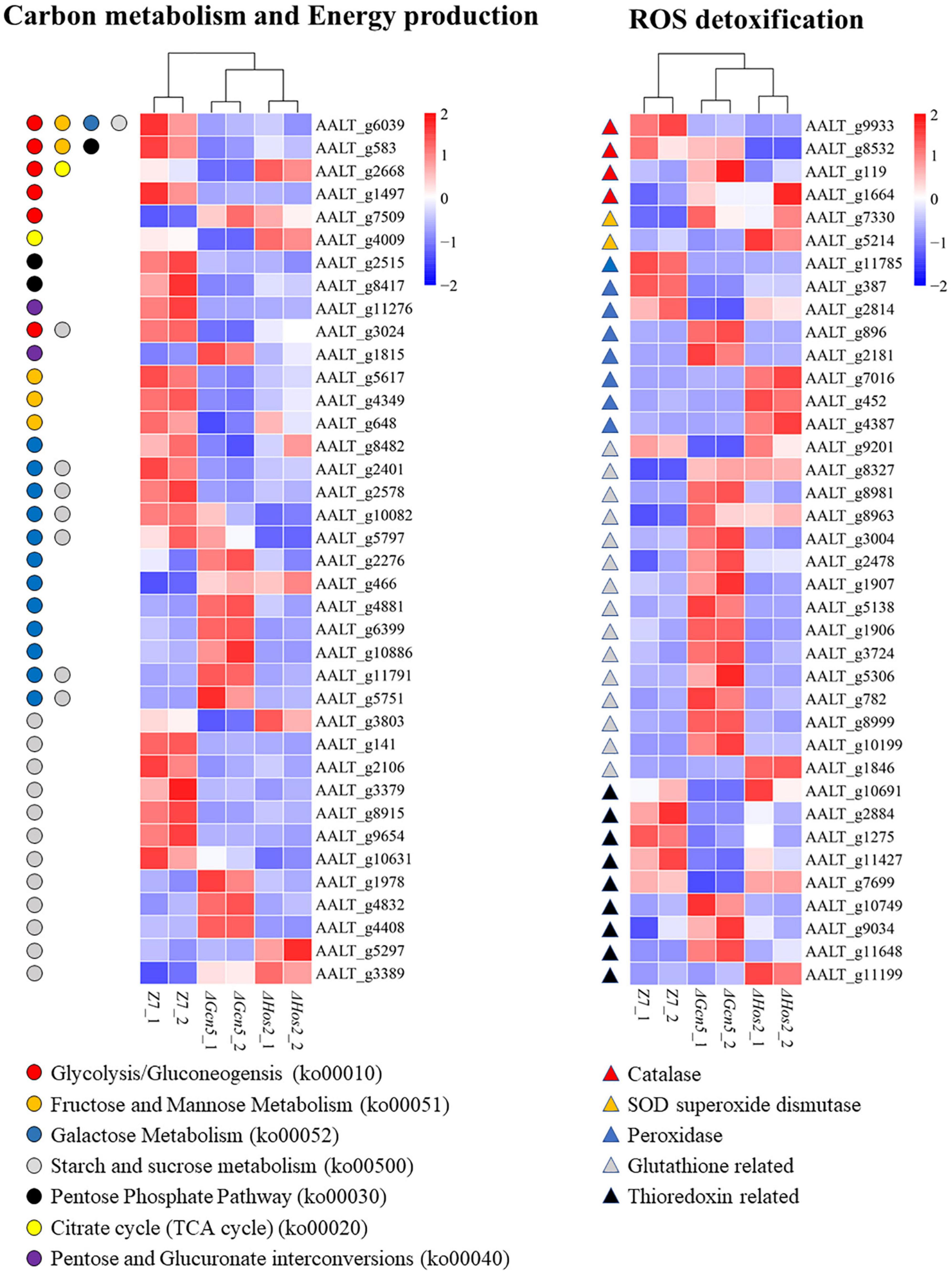
Figure 7. Heatmap of differentially expressed genes enriched in carbon metabolism and energy production, and ROS detoxification.
Gcn5 and Hos2 affect the expression of ROS detoxification genes. In view of the increased sensitivity of ΔGcn5 and ΔHos2 to oxidative stress, we analyzed the transcription level of 96 ROS detoxification genes, including 7 catalases, 7 SOD superoxide dismutases, 42 glutathione system-related genes, 20 thioredoxin system-related genes, and 20 peroxidases (Supplementary Table 4). Although ΔGcn5 and ΔHos2 were more sensitive to oxidants, most of DEGs in these 96 ROS detoxification genes were up-regulated. In ΔGcn5, 21 DEGs were up-regulated and 10 DEGs were down-regulated (Figure 7). In ΔHos2, 9 DEGs were up-regulated and 4 DEGs were down-regulated.
Gcn5 and Hos2 mediate the expression of genes required for DNA damage repair. Base excision repair (BER), Nucleotide excision repair (NER), Mismatch repair (MMR), Homologous recombination (HR), and Non-homologous end-joining (NHEJ) play important roles in the repair of damaged DNA in organisms. Transcriptome analysis revealed that BER, NER, MMR, or HR-related genes including UDG (Uracil-DNA glycosylase, AALT_g5833), Polδ (DNA polymerase delta, AALT_g8966), and TFIIH (transcription factor b, AALT_g7297) were down-regulated in ΔGcn5 (Supplementary Figure 10 and Supplementary Tables 2, 3). In ΔHos2, BER and NER-related gene PARP [poly (ADP-ribose) polymerase, AALT_g9674], and NER related gene XPA (DNA-repair protein, AALT_g6731) were down-regulated (Supplementary Table 3). In addition, AALT_g76 (BER, endonuclease), AALT_g10858 (NER, RING-box protein 1) in ΔGcn5, and AALT_g15 (NHEJ, DNA ligase 4) in ΔHos2 were up-regulated (Supplementary Table 3).
Gcn5 and Hos2 regulate expression of genes involved in protein processing and degradation. In ΔGcn5, 7 DEGs enriched in “protein processing in endoplasmic reticulum (ko04141)” were up-regulated, including protein glycosyltransferase (AALT_g5706), Mannosyl-oligosaccharide glucosidase (AALT_g11241), Mannosyl-oligosaccharide alpha-1,2-mannosidase (AALT_g2989), HSP20 (AALT_g9536), UBC5 (AALT_g6888), UBC7 (AALT_g619), and RBX1 (AALT_g4961), which are involved in protein processing and degradation. Furthermore, eight up-regulated genes were enriched in “Ubiquitin mediated proteolysis (ko4120)” and “Regulation of Autophagy (ko04140)” (Supplementary Figure 11 and Supplementary Table 2). Only 4 genes were down-regulated in the above three pathways. In ΔHos2, all four DEGs enriched in “protein processing in endoplasmic reticulum” (AALT_g173 and AALT_g6960), “Ubiquitin mediated proteolysis” (AALT_g6960), and “Regulation of Autophagy” were up-regulated (AALT_g11308) (Supplementary Table 3).
In addition, deletion of Gcn5 or Hos2 also affected the transcription of genes involved in Carbohydrate-active enzymes (CAZymes), secondary metabolite cluster, CYP450, and conidiation (Supplementary Tables 5–8 and Supplementary Figure 12). The transcription of all ACT toxin synthesis-related genes did not change significantly in ΔGcn5 and ΔHos2 compared to wild-type. However, AALT_g11758 (CYP450), located in the ACT toxin gene cluster and required for pathogenicity but plays no role for ACT toxin synthesis (Wang et al., 2019), was significantly down-regulated in both ΔGcn5 (FC = −25) and ΔHos2 (FC = −6).
Previous studies showed that Gcn5, Hos2, and RTT109 are required for DNA damage repair in B. bassiana and S. cerevisiae (Burgess et al., 2010; Cai et al., 2018a, c). Therefore, we tested whether RTT109, Gcn5, and Hos2 were involved in the resistance to DNA-damaging agents such as camptothecin (CPT), hydroxyurea (HU) and methyl methanesulfonate (MMS) in A. alternata. Compared with wild-type, growth of ΔRTT109 was significantly suppressed by 40 and 89% on PDA containing 5 μM CPT or 0.1% MMS, respectively (Figure 8). However, ΔRTT109 displayed wild-type radial growth on PDA amended with 5 mM HU. Unlike ΔRTT109, the growth of ΔHos2 on HU-added PDA was inhibited by 20% compared to wild-type. At the same time, ΔHos2 showed wild-type resistance to 5 μM CPT or 0.1% MMS. In addition, ΔGcn5 displayed increased sensitivity to 5 mM HU and 0.1% MMS (the growth inhibition rate is 29 and 19%, respectively, compared to wild-type) but displayed wild-type resistance to 5 μM CPT (Figure 8).
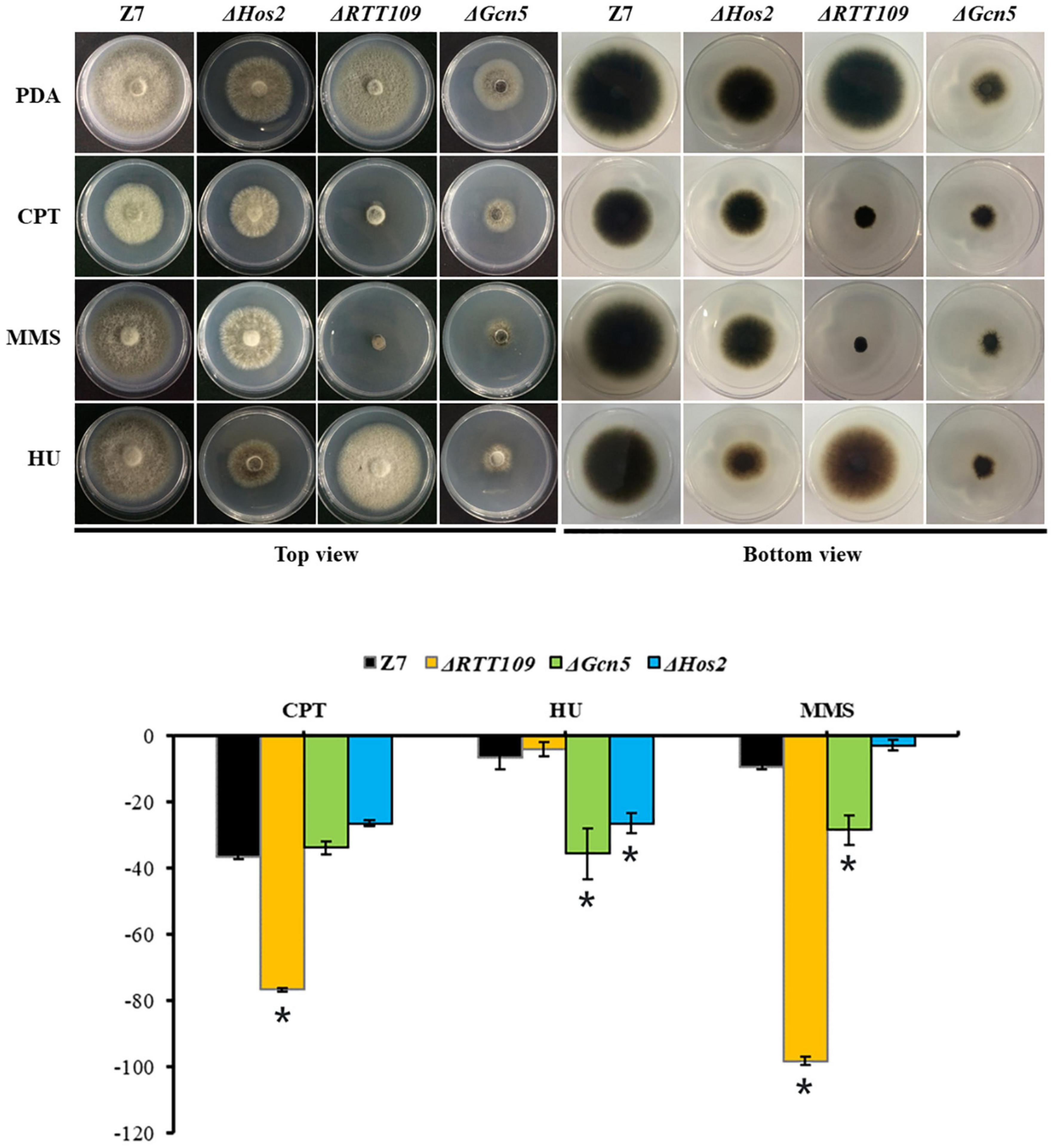
Figure 8. HATs and HDACs were involved in the resistance to DNA-damaging agents. Z7, ΔHos2, ΔRTT109, and ΔGcn5 were grown on PDA with or without the indicated chemicals [5 mM hydroxyurea (HU), 0.1% methyl methanesulfonate (MMS), or 5 μM camptothecin (CPT)]. The percentage of growth reduction determined by comparing a cumulative percentage of the growth of Z7 and mutants grown on same medium is also shown (*, p < 0.01).
In order to analyze the DNA damage repair ability of Gcn5, we performed genome resequencing using ultraviolet ray (UV) irradiated ΔGcn5 and wild-type. In the Gcn5 coding region, no reads were detected in ΔGcn5, but many reads were found in wild-type, which proves once again that the knock-out experiment of Gcn5 was successful (Figure 9B). In ΔGcn5, 949 unique SNPs were detected after UV irradiation. However, in UV irradiated wild-type, only 298 unique SNPs were found, which accounted for 31% of ΔGcn5, indicating that Gcn5 is involved in the repair of DNA damage caused by UV (Figure 9A).
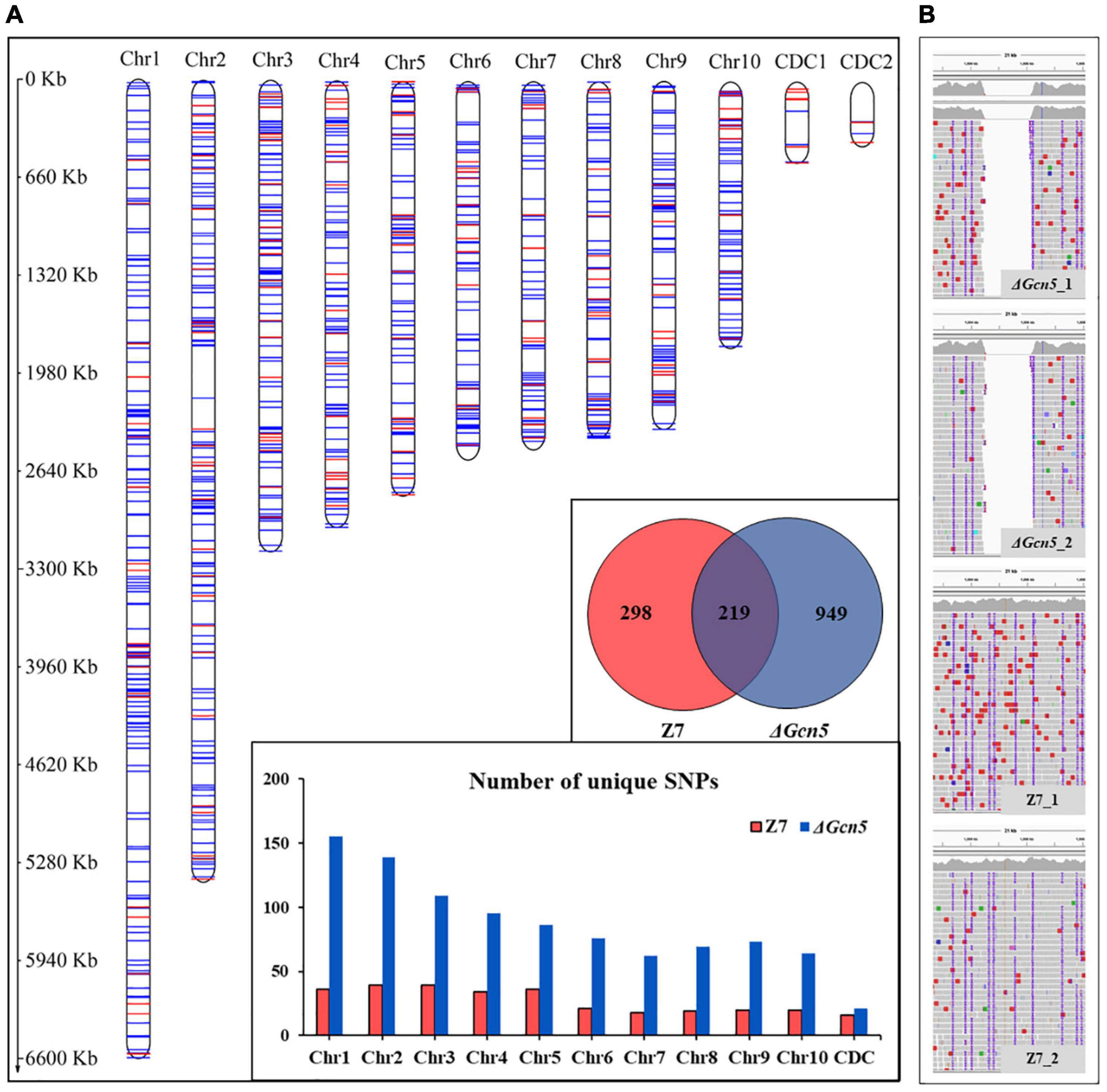
Figure 9. SNPs analysis of ΔGcn5 and wild-type irradiated by UV. ΔGcn5 and Z7 grown on PDA were irradiated with UV for 10 s every 24 h, and repeated 4 times. Hyphae were taken using sterilized toothpicks and incubated in PDB (150 rpm, 2 days). DNA was extracted from mycelium collected from PDB for whole-genome resequencing. The protocol for SNPs analysis is described in Experimental procedures. (A) Statistical analysis of SNP number in ΔGcn5 and Z7. (B) The mapping result of ΔGcn5 and Z7 genome sequencing data on the reference genome.
To test whether histone acetylation levels were altered in ΔSas3 and ΔGcn5, western blot was carried out using specific antibodies, directed against H3K4ac, while antibody against H3 was used as a loading control. As shown in Figure 10, the level of H3K4ac was significantly decreased (P < 0.01) in the ΔGcn5 compared to Z7. No reduction of the signal for H3K4ac was observed in ΔSas3.
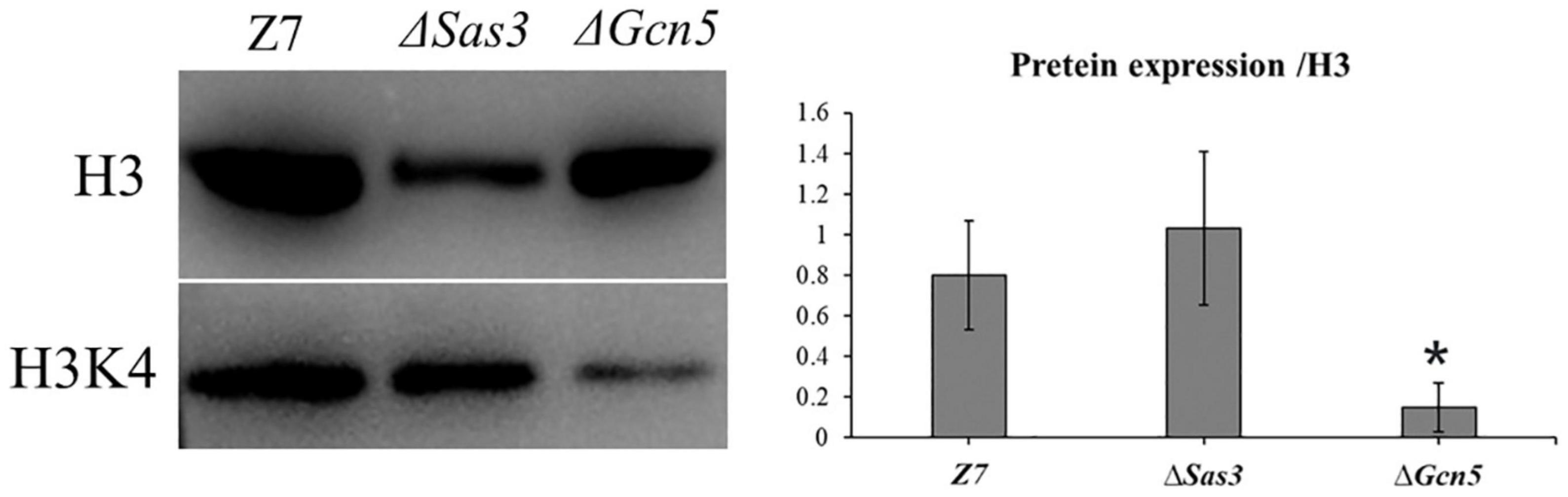
Figure 10. Western blotting analysis of proteins extracted from Z7, ΔSas3, and ΔGcn5, respectively. The respective strains were grown for 36 h in PDB. The anti-acetyl H2K4 (H3K4ac) and anti-acetyl H3K18 (H3K18ac) were used for detecting alteration of acetylation levels. Antibody H3 was used as a loading reference. (*, p < 0.01).
Histone acetylation, which is important for multiple cellular processes, occurs in different organisms ranging from fungi to mammals, and the acetylation level manifested by HATs and HDACs dynamically changes over time during development and differentiation (Lee and Workman, 2007; Kong et al., 2018). In plant pathogenic fungi, several HATs and HDACs have been found to be required for growth, virulence and adaptation to environmental stress (Maeda et al., 2017; Kong et al., 2018; Dubey et al., 2019). Although HATs and HDACs are conserved in cells, the functions of these genes are various in different species. In this study, a total of 13 HATs and HDACs were identified in A. alternata. To systematically elucidate the functions of HATs and HDACs in A. alternata, we created 6 HATs and 6 HDACs deletion mutants. Our studies showed that HATs and HDACs were widely involved in biological processes of mycelial growth and development, conidial production, multiple stresses resistance, carbon source utilization, DNA damage repair, and pathogenicity in A. alternata.
As shown in the present study, HATs and HDACs are involved in developmental processes because ΔGcn5, ΔHos2, ΔRpd3, ΔNat3, ΔSas3, and ΔRTT109 produce no or fewer conidia. In addition, ΔGcn5, ΔRpd3, and ΔNat2 produces more hyphal branches than wild-type. In some filamentous fungi like N. crassa and A. nidulans, the central development pathway relies upon the key activator genes BrlA, AbaA, and WetA (Etxebeste et al., 2010; Park and Yu, 2012). Six upstream developmental activators (FlbA, FlbB, FlbC, FlbD, FlbE, and FluG) are required for the activation of BrlA and the initiation of conidiation (Adams et al., 1988; Wieser et al., 1994). Our transcriptome data revealed that FlbA, FlbC, and FlbD were down-regulated in ΔGcn5 (Supplementary Table 8). Previous studies have revealed the conidiation of A. alternata is closely regulated by the calcium-mediated signaling pathway, Fus3 and Slt2 MAPK signaling pathway, as well as the cAMP dependent protein kinase A (PKA) (Lin et al., 2010; Wang et al., 2010; Yago et al., 2011; Tsai et al., 2013). Other factors including NADPH oxidase (Nox), thioredoxin reductase (Trr1), glutathione reductase (Glr1), nascent polypeptide associated complex α subunit (Nac1), and skn7 response regulator are also required for the developmental processes in A. alternata (Chen et al., 2012; Yang and Chung, 2013; Ma et al., 2018; Wang et al., 2020). Whether or not those regulators interact with HATs or HDACs remains to be determined. In addition to conidia formation, HATs and HDACs are required for vegetative growth. ΔGcn5, ΔNat3, ΔSas3, ΔRTT109, ΔElp3, ΔHos2, and ΔRpd3 displayed growth reduction compared with wild-type, consistent with the findings in B. bassiana (Gcn5, RTT109, Hos2, and Rpd3), F. graminearum (Gcn5, RTT109, Elp3, and Sas3), and S. cerevisiae (Nat3) (Polevoda and Sherman, 2003; Lee et al., 2014; Cai et al., 2018a,b,c,d; Kong et al., 2018).
Experiments further demonstrated that ΔGcn5, ΔSas3, ΔRTT109, and ΔHos2 are required for cellular resistance to osmotic stress. In yeast, SAGA (Spt-Ada-Gcn5) is required for the transcription of Hog1-mediated genes under severe osmostress (Zapater et al., 2007). Chip-on-chip experiments revealed that acetylation at lysines 9 and 12 of histone H3 increases in osmostress up-regulated genes and decreases in repressed genes (Magraner-Pardo et al., 2014). The Hog1 deletion mutant in A. alternata displayed reduced vegetative growth rate and increased sensitivity to KCl and NaCl, which is consistent with the phenotype of ΔGcn5, ΔSas3, ΔRTT109, and ΔHos2, indicating that transcription of some genes regulated by Hog1, Gcn5, Sas3, and Hos2 may be similar (Lin and Chung, 2010). Similar results were observed in F. graminearum, in which the Hog1, Gcn5, Sas3, RTT109 are also involved in vegetative growth and resistance to osmotic stress (Nguyen et al., 2012; Kong et al., 2018). In addition, ΔSas3 and ΔHos2 are more sensitive to cell wall-disturbing compounds including CR and SDS, indicating both of these genes play a positive role in cell wall assembly.
Carbon source (sugar) metabolism, which is one of the most important biological processes in organisms, is involved in growth, development, and multiple stress resistance (Koch, 2004; Ene et al., 2012). This study found that Sas3, Hos2, and especially Gcn5 displayed severe growth defects on sole carbon source medium, suggesting an involvement of these genes in carbon metabolism in A. alternata. In C. albicans, acetyl-CoA and acetate metabolism play a central role in growth on both glucose and non-glucose carbon sources (Carman et al., 2008). In B. bassiana, deletion of Rpd3 and Gcn5 resulted in growth defects on medium modified with different carbon sources, confirming the important role of histone acetylation in carbon source utilization (Cai et al., 2018b, d). In addition, most DEGs enriched in carbon source metabolism pathways, such as “Glycolysis/Gluconeogensis,” Fructose and Mannose Metabolism,” “Galactose Metabolism,” “Starch and Sucrose Metabolism,” etc., are down-regulated in ΔGcn5 and ΔHos2, making it not surprising to see the reduced carbon source utilization ability in ΔGcn5 and ΔHos2. In microorganisms, glucose metabolites would lead to carbon catabolite repression (CCR) and inhibit the utilization of other carbon sources (Celenza and Carlson, 1986; Kayikci and Nielsen, 2015). Sucrose non-fermenting 1 (SNF1) protein kinase complex is required for the utilization of non-fermentable carbon sources in fungi and enable them to adapt to the adversity of glucose deficiency (Alepuz et al., 1997). In Fusarium virguliforme, the growth of FvSnf1 deletion mutants on medium amended with galactose as sole carbon source is completely inhibited (Islam et al., 2017). SNF1 complex-related genes (AaSnf1, AaSip2, and AaSnf4) were not significantly differentially expressed in ΔGcn5 and ΔHos2 compared to wild-type. However, the transcription of SUC2 (AALT_g2401), which is regulated by Snf1 and is essential for sucrose utilization in S. cerevisiae (Carlson et al., 1981; Oezcan et al., 1997), was down-regulated significantly in ΔGcn5 and ΔHos2. Previous studies have shown that Snf1, which regulates Gcn5 occupancy and H3 acetylation at a specific sequence, works in concert with Gcn5 to regulate transcription (Lo et al., 2001; Abate et al., 2012). In addition, similar with Gcn5 and Hos2, Snf1 also plays an important role in the carbon source utilization in A. alternata (Wang et al., 2020). Therefore, we speculate that the interaction relationship between Snf1 and these two genes may also exist.
Mutants lacking Gcn5, Sas3, Elp3, RTT109, or Hos2 in A. alternata were defective in ROS detoxification at varying degrees, indicating all of these five genes are involved in resistance to oxidative stress. RTT109, Gcn5, and Hos2 in B. bassiana, Elp3 and Sas3 in F. graminearum also play important roles in the resistance to oxidative stress (Lee et al., 2014; Cai et al., 2018a,b,c; Kong et al., 2018). However, Gcn5 in Aspergillus flavus is not required for ROS resistance (Lan et al., 2016). In addition, Hos2 plays a negative role in H2O2 resistance in M. oryzae, indicating that the function of specific genes in HATs and HDACs have been differentiated in fungi (Lee et al., 2019). In A. alternata, the ROS detoxification ability is critical for pathogenesis and survival (Chung, 2012; Ma et al., 2018; Gai et al., 2019). A. alternata strains lacking Nox, Hog1, Yap1, Skn7, Gpx3, or Nac1 all exhibit increased cellular sensitivity to ROS and decreased pathogenicity on citrus leaves (Lin and Chung, 2010; Chen et al., 2012; Yang and Chung, 2012; Chung, 2014; Yang et al., 2016; Wang et al., 2020). Virulence assays revealed that the virulence of ΔGcn5 and ΔSas3 was completely lost, which is consistent with the findings in F. graminearum (Kong et al., 2018). Furthermore, the virulence of ΔElp3, ΔRTT109, and ΔHos2 decreased significantly, supporting the requirement of ROS detoxification for successful infection by A. alternata. In addition, we observed that ΔNat3 and ΔRpd3 were significantly reduced in pathogenicity and vegetative growth rate. The severe growth defects of ΔNat3 and ΔRpd3 may contribute to its reduced virulence. Recent research shows that chlorothalonil causes a redox state change leading to oxidative stress generation in Danio rerio (Da Silva Barreto et al., 2020). Interestingly, sensitivity assays revealed that A. alternata strains with impaired Gcn5 or Hos2 displayed high sensitivity to chlorothalonil fungicide. In addition, transcriptome data of ΔGcn5 revealed that all DEGs in the sterol synthesis pathway were significantly down-regulated, including CYP51 homologous genes, which is closely related to DMI fungicide resistance. In C. albicans, the loss of Hda1 and Rpd3 both caused the pathogen to be significantly more sensitive to azole fungicides, indicating that histone acetylation levels are crucial for pathogens to resist DMI fungicides (Li et al., 2015). Transcriptome analysis also revealed that most DEGs involved in ROS detoxification were up-regulated in ΔGcn5 and ΔHos2 even without external oxidative stress, implying that the redox state in cells of these two mutants are changed. In eukaryotic cells, endoplasmic reticulum (ER) is essential for folding and trafficking of proteins that enter the secretory pathway, while the disorder of redox homeostasis of ER could result in protein misfolding (Wang and Kaufman, 2016). In ΔGcn5 and ΔHos2, many genes involved in “Protein processing in endoplasmic reticulum,” “Ubiquitin mediated proteolysis,” and “Regulation of autophagy” pathways are up-regulated, which mainly play roles in protein folding and degradation.
It is now clear that a subset of HATs and HDACs regulate the genome stability (Driscoll et al., 2007; Nagarajan et al., 2013). In B. bassiana, mutants lacking HATs (Mst2 and RTT109) or HDACs (Hos2) displayed increased sensitivity to DNA-damaging stresses, indicating that all of these genes are involved in DNA damage repair (Cai et al., 2018a, c; Wang et al., 2018). In our study, many DEGs are enriched in DNA damage repair pathways. Moreover, ΔGcn5, ΔRTT109, and ΔHos2 displayed hypersensitivity to a variety of DNA-damaging agents such as MMS, HU, or CPT, all of which induce replication fork collapse or stalling (Interthal and Heyer, 2000; Boddy et al., 2001; Doe et al., 2002), suggesting Gcn5, RTT109, and Hos2 play an important role in DNA damage repair in A. alternata. This speculation was evidenced by the result that more SNPs were generated in ΔGcn5 in comparison to wild-type when both of them were irradiated by UV.
In summary, our study revealed that HATs and HDACs are required for the vegetative growth, conidiation, pathogenicity, multiple stresses resistance, DNA damage repair, and carbon source utilization in A. alternata. In addition, Gcn5 and Hos2 play direct or indirect roles in the transcriptional regulation of genes involved in carbon source metabolism, DNA damage repair, ROS detoxification, protein processing and degrading, ergosterol synthesis, and secondary metabolite synthesis.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
HM, FG, and HL contributed to the conception of the study. HM, LL, YG, XYZ, YNC, XKZ, and YZC performed the experiments. HM, YG, and CJ performed the data analysis. HM wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (No: 31571948) and the earmarked fund for China Agriculture Research System (CARS-27) to HL.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We acknowledge Prof. Kuang-Ren Chung for helpful scientific discussion and the technical support of Xuepeng Sun and Yating Zeng.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.783633/full#supplementary-material
Abate, G., Bastonini, E., Braun, K. A., Verdone, L., Young, E. T., and Caserta, M. (2012). Snf1/AMPK regulates Gcn5 occupancy, H3 acetylation and chromatin remodelling at S. cerevisiae ADY2 promoter. Biochim. Biophys. Acta 1819, 419–427. doi: 10.1016/j.bbagrm.2012.01.009
Adams, T. H., Boylan, M. T., and Timberlake, W. E. (1988). brlA is necessary and sufficient to direct conidiophore development in Aspergillus nidulans. Cell 54, 353–362. doi: 10.1016/0092-8674(88)90198-5
Alepuz, P. M., Cunningham, K. W., and Estruch, F. (1997). Glucose repression affects ion homeostasis in yeast through the regulation of the stress-activated ENA1 gene. Mol. Microbiol. 26, 91–98. doi: 10.1046/j.1365-2958.1997.5531917.x
Anders, S., and Huber, W. (2012). Differential Expression Of RNA-Seq Data At The Gene Level–The Deseq Package. Heidelberg: European Molecular Biology Laboratory (EMBL).
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638
Baker, L. A., Ueberheide, B. M., Dewell, S., Chait, B. T., Zheng, D., and Allis, C. D. (2013). The yeast Snt2 protein coordinates the transcriptional response to hydrogen peroxide-mediated oxidative stress. Mol. Cell. Biol. 33, 3735–3748. doi: 10.1128/MCB.00025-13
Blin, K., Wolf, T., Chevrette, M. G., Lu, X., Schwalen, C. J., Kautsar, S. A., et al. (2017). AntiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 45, W36–W41. doi: 10.1093/nar/gkx319
Boddy, M. N., Gaillard, P.-H. L., Mcdonald, W. H., Shanahan, P., and Yates, 3rd, J.R, and Russell, P. (2001). Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 107, 537–548. doi: 10.1016/s0092-8674(01)00536-0
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1016/S0092-8674(01)00536-0
Bosio, M. C., Fermi, B., Spagnoli, G., Levati, E., Rubbi, L., Ferrari, R., et al. (2017). Abf1 and other general regulatory factors control ribosome biogenesis gene expression in budding yeast. Nucleic Acids Res. 45, 4493–4506. doi: 10.1093/nar/gkx058
Burgess, R. J., Zhou, H., Han, J., and Zhang, Z. (2010). A role for Gcn5 in replication-coupled nucleosome assembly. Mol. Cell 37, 469–480. doi: 10.1016/j.molcel.2010.01.020
Cai, Q., Wang, J. J., Fu, B., Ying, S. H., and Feng, M. G. (2018b). Gcn5-dependent histone H3 acetylation and gene activity is required for the asexual development and virulence of Beauveria bassiana. Environ. Microbiol. 20, 1484–1497. doi: 10.1111/1462-2920.14066
Cai, Q., Wang, J. J., Shao, W., Ying, S. H., and Feng, M. G. (2018c). Rtt109-dependent histone H3 K56 acetylation and gene activity are essential for the biological control potential of Beauveria bassiana. Pest Manag. Sci. 74, 2626–2635. doi: 10.1002/ps.5054
Cai, Q., Tong, S. M., Shao, W., Ying, S. H., and Feng, M. G. (2018a). Pleiotropic effects of the histone deacetylase Hos2 linked to H4-K16 deacetylation. H3-K56 acetylation, and H2A-S129 phosphorylation in Beauveria bassiana. Cell. Microbiol. 20:e12839. doi: 10.1111/cmi.12839
Cai, Q., Wang, Z. K., Shao, W., Ying, S. H., and Feng, M. G. (2018d). Essential role of Rpd3-dependent lysine modification in the growth, development and virulence of Beauveria bassiana. Environ. Microbiol. 20, 1590–1606. doi: 10.1111/1462-2920.14100
Cánovas, D., Marcos, A. T., Gacek, A., Ramos, M. S., Gutiérrez, G., Reyes-Domínguez, Y., et al. (2014). The histone acetyltransferase GcnE (GCN5) plays a central role in the regulation of Aspergillus asexual development. Genetics 197, 1175–1189. doi: 10.1534/genetics.114.165688
Carlson, M., and Botstein, D. (1982). Two differentially regulated mRNAs with different 5’ ends encode secreted and intracellular forms of yeast invertase. Cell 28, 145–154. doi: 10.1016/0092-8674(82)90384-1
Carlson, M., Osmond, B. C., and Botstein, D. (1981). Mutants of yeast defective in sucrose utilization. Genetics 98, 25–40. doi: 10.1093/genetics/98.1.25
Carman, A. J., Vylkova, S., and Lorenz, M. C. (2008). Role of acetyl coenzyme a synthesis and breakdown in alternative carbon source utilization in Candida albicans. Eukaryotic Cell 7, 1733–1741. doi: 10.1128/EC.00253-08
Celenza, J. L., and Carlson, M. (1986). A yeast gene that is essential for release from glucose repression encodes a protein kinase. Science 233, 1175–1180. doi: 10.1126/science.3526554
Chen, L.-H., Lin, C.-H., and Chung, K.-R. (2012). Roles for SKN7 response regulator in stress resistance, conidiation and virulence in the citrus pathogen Alternaria alternata. Fungal Genet. Biol. 49, 802–813. doi: 10.1016/j.fgb.2012.07.006
Chung, K.-R. (2012). Stress response and pathogenicity of the necrotrophic fungal pathogen Alternaria alternata. Scientifica 2012:635431. doi: 10.6064/2012/635431
Chung, K.-R. (2014). Reactive oxygen species in the citrus fungal pathogen Alternaria alternata: the roles of NADPH-dependent oxidase. Physiol.Mol. Plant Pathol. 88, 10–17. doi: 10.1016/j.pmpp.2014.08.001
Chung, K.-R., Shilts, T., Li, W., and Timmer, L. (2002). Engineering a genetic transformation system for Colletotrichum acutatum, the causal fungus of lime anthracnose and postbloom fruit drop of citrus. FEMS Microbiol. Lett. 213, 33–39. doi: 10.1016/S0378-1097(02)00757-7
Church, M., Smith, K. C., Alhussain, M. M., Pennings, S., and Fleming, A. B. (2017). Sas3 and Ada2 (Gcn5)-dependent histone H3 acetylation is required for transcription elongation at the de-repressed FLO1 gene. Nucleic Acids Res. 45, 4413–4430. doi: 10.1093/nar/gkx028
Clarke, A. S., Lowell, J. E., Jacobson, S. J., and Pillus, L. (1999). Esa1p is an essential histone acetyltransferase required for cell cycle progression. Mol. Cell. Biol. 19, 2515–2526. doi: 10.1128/MCB.19.4.2515
Cohen, T., Mallory, M., Strich, R., and Yao, T.-P. (2008). Hos2p/Set3p deacetylase complex signals secretory stress through the Mpk1p cell integrity pathway. Eukaryotic Cell 7, 1191–1199. doi: 10.1128/EC.00059-08
Da Rosa, J. L., Boyartchuk, V. L., Zhu, L. J., and Kaufman, P. D. (2010). Histone acetyltransferase Rtt109 is required for Candida albicans pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 107, 1594–1599. doi: 10.1073/pnas.0912427107
Da Silva Barreto, J., De Melo Tarouco, F., and Da Rosa, C. E. (2020). Chlorothalonil causes redox state change leading to oxidative stress generation in Danio rerio. Aquatic Toxicol. 225:105527. doi: 10.1016/j.aquatox.2020.105527
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Dang, W., Steffen, K. K., Perry, R., Dorsey, J. A., Johnson, F. B., Shilatifard, A., et al. (2009). Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459:802. doi: 10.1038/nature08085
Das, C., Lucia, M. S., Hansen, K. C., and Tyler, J. K. (2009). CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 459, 113–117. doi: 10.1038/nature07861
Ding, S.-L., Liu, W., Iliuk, A., Ribot, C., Vallet, J., Tao, A., et al. (2010). The tig1 histone deacetylase complex regulates infectious growth in the rice blast fungus Magnaporthe oryzae. Plant Cell 22, 2495–2508. doi: 10.1105/tpc.110.074302
Doe, C. L., Ahn, J. S., Dixon, J., and Whitby, M. C. (2002). Mus81-Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks. J. Biol. Chem. 277, 32753–32759. doi: 10.1074/jbc.M202120200
Driscoll, R., Hudson, A., and Jackson, S. P. (2007). Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science 315, 649–652. doi: 10.1126/science.1135862
Dubey, A., Lee, J., Kwon, S., Lee, Y. H., and Jeon, J. (2019). A MYST family histone acetyltransferase. MoSAS3, is required for development and pathogenicity in the rice blast fungus. Mol. Plant Pathol. 20, 1491–1505. doi: 10.1111/mpp.12856
Elías-Villalobos, A., Fernández-Álvarez, A., Moreno-Sánchez, I., Helmlinger, D., and Ibeas, J. I. (2015). The Hos2 histone deacetylase controls Ustilago maydis virulence through direct regulation of mating-type genes. PLoS Pathog. 11:e1005134. doi: 10.1371/journal.ppat.1005134
Ene, I. V., Adya, A. K., Wehmeier, S., Brand, A. C., Maccallum, D. M., Gow, N. A., et al. (2012). Host carbon sources modulate cell wall architecture, drug resistance and virulence in a fungal pathogen. Cell. Microbiol. 14, 1319–1335. doi: 10.1111/j.1462-5822.2012.01813.x
Etxebeste, O., Garzia, A., Espeso, E. A., and Ugalde, U. (2010). Aspergillus nidulans asexual development: making the most of cellular modules. Trends Microbiol. 18, 569–576. doi: 10.1016/j.tim.2010.09.007
Fillingham, J., Recht, J., Silva, A. C., Suter, B., Emili, A., Stagljar, I., et al. (2008). Chaperone control of the activity and specificity of the histone H3 acetyltransferase Rtt109. Mol. Cell. Biol. 28, 4342–4353. doi: 10.1128/MCB.00182-08
Gai, Y., Liu, B., Ma, H., Li, L., Chen, X., Moenga, S., et al. (2019). The methionine biosynthesis regulator AaMetR contributes to oxidative stress tolerance and virulence in Alternaria alternata. Microbiol. Res. 219, 94–109. doi: 10.1016/j.micres.2018.11.007
González-Prieto, J. M., Rosas-Quijano, R., Domínguez, A., and Ruiz-Herrera, J. (2014). The UmGcn5 gene encoding histone acetyltransferase from Ustilago maydis is involved in dimorphism and virulence. Fungal Genet. Biol. 71, 86–95. doi: 10.1016/j.fgb.2014.09.002
Gregoretti, I., Lee, Y.-M., and Goodson, H. V. (2004). Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol. 338, 17–31. doi: 10.1016/j.jmb.2004.02.006
Han, J., Zhou, H., Horazdovsky, B., Zhang, K., Xu, R.-M., and Zhang, Z. (2007). Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 315, 653–655. doi: 10.1126/science.1133234
Helga, T., Robinson, J. T., and Mesirov, J. P. (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2, 178–192. doi: 10.1093/bib/bbs017
Helmlinger, D., Marguerat, S., Villén, J., Gygi, S. P., Bähler, J., and Winston, F. (2008). The S. pombe SAGA complex controls the switch from proliferation to sexual differentiation through the opposing roles of its subunits Gcn5 and Spt8. Genes Dev. 22, 3184–3195. doi: 10.1101/gad.1719908
Huang, F., Fu, Y., Nie, D., Stewart, J. E., Peever, T. L., and Li, H. (2015). Identification of a novel phylogenetic lineage of Alternaria alternata causing citrus brown spot in China. Fungal Biol. 119, 320–330. doi: 10.1016/j.funbio.2014.09.006
Interthal, H., and Heyer, W.-D. (2000). MUS81 encodes a novel helix-hairpin-helix protein involved in the response to UV-and methylation-induced DNA damage in Saccharomyces cerevisiae. Mol. Gen. Genet. 263, 812–827. doi: 10.1007/s004380000241
Islam, K. T., Bond, J. P., and Fakhoury, A. M. (2017). FvSNF1, the sucrose non-fermenting protein kinase gene of Fusarium virguliforme, is required for cell-wall-degrading enzymes expression and sudden death syndrome development in soybean. Curr. Genet. 63, 723–738. doi: 10.1007/s00294-017-0676-9
Jazayeri, A., Mcainsh, A. D., and Jackson, S. P. (2004). Saccharomyces cerevisiae Sin3p facilitates DNA double-strand break repair. Proc. Natl. Acad. Sci. U.S.A. 101, 1644–1649. doi: 10.1073/pnas.0304797101
Jeon, J., Kwon, S., and Lee, Y.-H. (2014). Histone acetylation in fungal pathogens of plants. Plant Pathol. J. 30:1. doi: 10.5423/PPJ.RW.01.2014.0003
Johnsson, A., Durand-Dubief, M., Xue-Franzén, Y., Rönnerblad, M., Ekwall, K., and Wright, A. (2009). HAT–HDAC interplay modulates global histone H3K14 acetylation in gene-coding regions during stress. EMBO Rep. 10, 1009–1014. doi: 10.1038/embor.2009.127
Kayikci, Ö, and Nielsen, J. (2015). Glucose repression in Saccharomyces cerevisiae. FEMS Yeast Res. 15:fov068. doi: 10.1093/femsyr/fov068
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36. doi: 10.1186/gb-2013-14-4-r36
Kim, T., Xu, Z., Clauder-Münster, S., Steinmetz, L. M., and Buratowski, S. (2012). Set3 HDAC mediates effects of overlapping noncoding transcription on gene induction kinetics. Cell 150, 1158–1169. doi: 10.1016/j.cell.2012.08.016
Koch, K. (2004). Sucrose metabolism: regulatory mechanisms and pivotal roles in sugar sensing and plant development. Curr. Opin. Plant Biol. 7, 235–246. doi: 10.1016/j.pbi.2004.03.014
Kong, X., Van Diepeningen, A. D., Van Der Lee, T. A., Waalwijk, C., Xu, J., Xu, J., et al. (2018). The Fusarium graminearum histone acetyltransferases are important for morphogenesis. DON biosynthesis, and pathogenicity. Front. Microbiol. 9:654. doi: 10.3389/fmicb.2018.00654
Kornberg, R. D., and Lorch, Y. (1999). Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98, 285–294. doi: 10.1016/S0092-8674(00)81958-3
Lan, H., Sun, R., Fan, K., Yang, K., Zhang, F., Nie, X. Y., et al. (2016). The Aspergillus flavus histone acetyltransferase AflGcnE regulates morphogenesis, aflatoxin biosynthesis, and pathogenicity. Front. Microbiol. 7:1324. doi: 10.3389/fmicb.2016.01324
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lee, J., Lee, J.-J., and Jeon, J. (2019). A histone deacetylase, MoHOS2 regulates asexual development and virulence in the rice blast fungus. J. Microbiol. 57, 1115–1125. doi: 10.1007/s12275-019-9363-5
Lee, K. K., and Workman, J. L. (2007). Histone acetyltransferase complexes: one size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 8, 284–295. doi: 10.1038/nrm2145
Lee, T. I., and Young, R. A. (2000). Transcription of eukaryotic protein-coding genes. Annu. Rev. Genet. 34, 77–137. doi: 10.1146/annurev.genet.34.1.77
Lee, Y., Min, K., Son, H., Park, A. R., Kim, J.-C., Choi, G. J., et al. (2014). ELP3 is involved in sexual and asexual development, virulence, and the oxidative stress response in Fusarium graminearum. Mol. Plant Microbe Interact. 27, 1344–1355. doi: 10.1094/MPMI-05-14-0145-R
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, X., Cai, Q., Mei, H., Zhou, X., Shen, Y., Li, D., et al. (2015). The Rpd3/Hda1 family of histone deacetylases regulates azole resistance in Candida albicans. J. Antimicrob. Chemother. 70, 1993–2003. doi: 10.1093/jac/dkv070
Lin, C.-H., and Chung, K.-R. (2010). Specialized and shared functions of the histidine kinase-and HOG1 MAP kinase-mediated signaling pathways in Alternaria alternata, a filamentous fungal pathogen of citrus. Fungal Genet. Biol. 47, 818–827. doi: 10.1016/j.fgb.2010.06.009
Lin, C.-H., Yang, S. L., and Chung, K.-R. (2009). The YAP1 homolog–mediated oxidative stress tolerance is crucial for pathogenicity of the necrotrophic fungus Alternaria alternata in citrus. Mol. Plant Microbe Interact. 22, 942–952. doi: 10.1094/MPMI-22-8-0942
Lin, C.-H., Yang, S. L., Wang, N.-Y., and Chung, K.-R. (2010). The FUS3 MAPK signaling pathway of the citrus pathogen Alternaria alternata functions independently or cooperatively with the fungal redox-responsive AP1 regulator for diverse developmental, physiological and pathogenic processes. Fungal Genet. Biol. 47, 381–391. doi: 10.1016/j.fgb.2009.12.009
Lin, S.-J., Defossez, P.-A., and Guarente, L. (2000). Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 289, 2126–2128. doi: 10.1126/science.289.5487.2126
Lo, W.-S., Duggan, L., Tolga, N., Belotserkovskya, R., Lane, W. S., Shiekhattar, R., et al. (2001). Snf1–a histone kinase that works in concert with the histone acetyltransferase Gcn5 to regulate transcription. Science 293, 1142–1146. doi: 10.1126/science.1062322
Ma, H., Wang, M., Gai, Y., Fu, H., Zhang, B., Ruan, R., et al. (2018). Thioredoxin and glutaredoxin systems required for oxidative stress resistance, fungicide sensitivity, and virulence of Alternaria alternata. Appl. Environ. Microbiol. 84, e86–e118. doi: 10.1128/AEM.00086-18
Maeda, K., Izawa, M., Nakajima, Y., Jin, Q., Hirose, T., Nakamura, T., et al. (2017). Increased metabolite production by deletion of an HDA 1-type histone deacetylase in the phytopathogenic fungi, Magnaporthe oryzae (Pyricularia oryzae) and Fusarium asiaticum. Lett. Appl. Microbiol. 65, 446–452. doi: 10.1111/lam.12797
Magraner-Pardo, L., Pelechano, V., Coloma, M. D., and Tordera, V. (2014). Dynamic remodeling of histone modifications in response to osmotic stress in Saccharomyces cerevisiae. BMC Genomics 15, 1–16. doi: 10.1186/1471-2164-15-247
Masumoto, H., Hawke, D., Kobayashi, R., and Verreault, A. (2005). A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature 436, 294–298. doi: 10.1038/nature03714
Miyamoto, Y., Ishii, Y., Honda, A., Masunaka, A., Tsuge, T., Yamamoto, M., et al. (2009). Function of genes encoding acyl-CoA synthetase and enoyl-CoA hydratase for host-selective ACT-toxin biosynthesis in the tangerine pathotype of Alternaria alternata. Phytopathology 99, 369–377. doi: 10.1094/PHYTO-99-4-0369
Nagarajan, P., Ge, Z., Sirbu, B., Doughty, C., Garcia, P. A. A., Schlederer, M., et al. (2013). Histone acetyl transferase 1 is essential for mammalian development, genome stability, and the processing of newly synthesized histones H3 and H4. PLoS Genet 9:e1003518. doi: 10.1371/journal.pgen.1003518
Nguyen, T. V., Schäfer, W., and Bormann, J. (2012). The stress-activated protein kinase FgOS-2 is a key regulator in the life cycle of the cereal pathogen Fusarium graminearum. Mol. Plant Microbe Interact. 25, 1142–1156. doi: 10.1094/MPMI-02-12-0047-R
Nugent, R. L., Johnsson, A., Fleharty, B., Gogol, M., Xue-Franzén, Y., Seidel, C., et al. (2010). Expression profiling of S. pombe acetyltransferase mutants identifies redundant pathways of gene regulation. BMC Genomics 11:59. doi: 10.1186/1471-2164-11-59
Nützmann, H.-W., Reyes-Dominguez, Y., Scherlach, K., Schroeckh, V., Horn, F., Gacek, A., et al. (2011). Bacteria-induced natural product formation in the fungus Aspergillus nidulans requires Saga/Ada-mediated histone acetylation. Proc. Natl. Acad. Sci. U.S.A. 108, 14282–14287. doi: 10.1073/pnas.1103523108
Oezcan, S., Vallier, L. G., Flick, J. S., Carlson, M., and Johnston, M. (1997). Expression of the SUC2 gene of Saccharomyces cerevisiae is induced by low levels of glucose. Yeast 13, 127–137. doi: 10.1002/(SICI)1097-0061(199702)13:23.0.CO;2-#
Ortega, P., Gómez-González, B., and Aguilera, A. (2019). Rpd3L and Hda1 histone deacetylases facilitate repair of broken forks by promoting sister chromatid cohesion. Nat. Commun. 10, 1–15. doi: 10.1038/s41467-019-13210-5
Ozdemir, A., Spicuglia, S., Lasonder, E., Vermeulen, M., Campsteijn, C., Stunnenberg, H. G., et al. (2005). Characterization of lysine 56 of histone H3 as an acetylation site in Saccharomyces cerevisiae. J. Biol. Chem. 280, 25949–25952. doi: 10.1074/jbc.c500181200
Park, H.-S., and Yu, J.-H. (2012). Genetic control of asexual sporulation in filamentous fungi. Curr. Opin. Microbiol. 15, 669–677. doi: 10.1016/j.mib.2012.09.006
Pijnappel, W. P., Schaft, D., Roguev, A., Shevchenko, A., Tekotte, H., Wilm, M., et al. (2001). The S. cerevisiae SET3 complex includes two histone deacetylases. Hos2 and Hst1, and is a meiotic-specific repressor of the sporulation gene program. Genes Dev. 15, 2991–3004. doi: 10.1101/gad.207401
Polevoda, B., and Sherman, F. (2003). Composition and function of the eukaryotic N-terminal acetyltransferase subunits. Biochem. Biophys. Res. Commun. 308, 1–11. doi: 10.1016/S0006-291X(03)01316-0
Sun, X., Wang, J., Feng, D., Ma, Z., and Li, H. (2011). PdCYP51B, a new putative sterol 14α-demethylase gene of Penicillium digitatum involved in resistance to imazalil and other fungicides inhibiting ergosterol synthesis. Appl. Microbiol. Biotechnol. 91:1107. doi: 10.1007/s00253-011-3355-7
Tkach, J. M., Yimit, A., Lee, A. Y., Riffle, M., Costanzo, M., Jaschob, D., et al. (2012). Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat. Cell Biol. 14, 966–976. doi: 10.1038/ncb2549
Torres-Machorro, A. L., Clark, L. G., Chang, C. S., and Pillus, L. (2015). The set3 complex antagonizes the MYST acetyltransferase Esa1 in the DNA damage response. Mol. Cell. Biol. 35, 3714–3725. doi: 10.1128/MCB.00298-15
Tsai, H.-C., Yang, S. L., and Chung, K.-R. (2013). Cyclic AMP-dependent protein kinase a negatively regulates conidia formation by the tangerine pathotype of Alternaria alternata. World J. Microbiol. Biotechnol. 29, 289–300. doi: 10.1007/s11274-012-1182-3
Vidal, M., and Gaber, R. F. (1991). RPD3 encodes a second factor required to achieve maximum positive and negative transcriptional states in Saccharomyces cerevisiae. Mol. Cell. Biol. 11, 6317–6327. doi: 10.1128/MCB.11.12.6317
Vogelauer, M., Rubbi, L., Lucas, I., Brewer, B. J., and Grunstein, M. (2002). Histone acetylation regulates the time of replication origin firing. Mol. Cell 10, 1223–1233. doi: 10.1016/S1097-2765(02)00702-5
Voichek, Y., Bar-Ziv, R., and Barkai, N. (2016). Expression homeostasis during DNA replication. Science 351, 1087–1090. doi: 10.1126/science.aad1162
Voorrips, R. E. (2002). MapChart: software for the graphical presentation of linkage maps and QTLs. J. Hered. 93, 77–78. doi: 10.1093/jhered/93.1.77
Wang, J.-J., Cai, Q., Qiu, L., Ying, S.-H., and Feng, M.-G. (2018). The histone acetyltransferase Mst2 sustains the biological control potential of a fungal insect pathogen through transcriptional regulation. Appl. Microbiol. Biotechnol. 102, 1343–1355. doi: 10.1007/s00253-017-8703-9
Wang, M., and Kaufman, R. J. (2016). Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529, 326–335. doi: 10.1038/nature17041
Wang, M., Fu, H., Shen, X. X., Ruan, R., Rokas, A., and Li, H. (2019). Genomic features and evolution of the conditionally dispensable chromosome in the tangerine pathotype of Alternaria alternata. Mol. Plant Pathol. 20, 1425–1438. doi: 10.1111/mpp.12848
Wang, M., Sun, X., Yu, D., Xu, J., Chung, K., and Li, H. (2016). Genomic and transcriptomic analyses of the tangerine pathotype of Alternaria alternata in response to oxidative stress. Sci. Rep. 6:32437. doi: 10.1038/srep32437
Wang, N.-Y., Lin, C.-H., and Chung, K.-R. (2010). A Gα subunit gene is essential for conidiation and potassium efflux but dispensable for pathogenicity of Alternaria alternata on citrus. Curr. Genet. 56, 43–51. doi: 10.1007/s00294-009-0278-2
Wang, P.-H., Wu, P.-C., Huang, R., and Chung, K.-R. (2020). The role of a nascent polypeptide-associated complex subunit alpha in siderophore biosynthesis, oxidative stress response, and virulence in Alternaria alternata. Mol. Plant Microbe Interact. 33, 668–679. doi: 10.1094/MPMI-11-19-0315-R
Wieser, J., Lee, B. N., Fondon, J. W., and Adams, T. H. (1994). Genetic requirements for initiating asexual development in Aspergillus nidulans. Curr. Genet. 27, 62–69. doi: 10.1007/BF00326580
Wirén, M., Silverstein, R. A., Sinha, I., Walfridsson, J., Lee, H. M., Laurenson, P., et al. (2005). Genome wide analysis of nucleosome density histone acetylation and HDAC function in fission yeast. EMBO J. 24, 2906–2918. doi: 10.1038/sj.emboj.7600758
Xin, Q., Gong, Y., Lv, X., Chen, G., and Liu, W. (2013). Trichoderma reesei histone acetyltransferase Gcn5 regulates fungal growth, conidiation, and cellulase gene expression. Curr. Microbiol. 67, 580–589. doi: 10.1007/s00284-013-0396-4
Yago, J. I., Lin, C. H., and Chung, K. R. (2011). The SLT2 mitogen-activated protein kinase-mediated signalling pathway governs conidiation, morphogenesis, fungal virulence and production of toxin and melanin in the tangerine pathotype of Alternaria alternata. Mol. Plant Pathol. 12, 653–665. doi: 10.1111/j.1364-3703.2010.00701.x
Yang, S. L., and Chung, K. R. (2012). The NADPH oxidase-mediated production of hydrogen peroxide (H2O2) and resistance to oxidative stress in the necrotrophic pathogen Alternaria alternata of citrus. Mol. Plant Pathol. 13, 900–914. doi: 10.1111/j.1364-3703.2012.00799.x
Yang, S. L., and Chung, K. R. (2013). Similar and distinct roles of NADPH oxidase components in the tangerine pathotype of Alternaria alternata. Mol. Plant Pathol. 14, 543–556. doi: 10.1111/mpp.12026
Yang, S. L., Yu, P. L., and Chung, K. R. (2016). The glutathione peroxidase-mediated reactive oxygen species resistance, fungicide sensitivity and cell wall construction in the citrus fungal pathogen Alternaria alternata. Environ. Microbiol. 18, 923–935. doi: 10.1111/1462-2920.13125
Yang, X.-J., and Seto, E. (2008). The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 9, 206–218. doi: 10.1038/nrm2346
Yu, G., Wang, L.-G., Han, Y., and He, Q.-Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. doi: 10.1089/omi.2011.0118
Keywords: histone acetylation, transcriptional regulation, DNA damage repair, pathogenicity, Alternaria alternata
Citation: Ma H, Li L, Gai Y, Zhang X, Chen Y, Zhuo X, Cao Y, Jiao C, Gmitter FG Jr and Li H (2021) Histone Acetyltransferases and Deacetylases Are Required for Virulence, Conidiation, DNA Damage Repair, and Multiple Stresses Resistance of Alternaria alternata. Front. Microbiol. 12:783633. doi: 10.3389/fmicb.2021.783633
Received: 26 September 2021; Accepted: 02 November 2021;
Published: 22 November 2021.
Edited by:
Xiuling Yang, Institute of Plant Protection, Chinese Academy of Agricultural Sciences (CAAS), ChinaReviewed by:
Huiquan Liu, Northwest A&F University, ChinaCopyright © 2021 Ma, Li, Gai, Zhang, Chen, Zhuo, Cao, Jiao, Gmitter and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fred G. Gmitter Jr., ZmdtaXR0ZXJAdWZsLmVkdQ==; Hongye Li, aHlsaUB6anUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.