 Min Wang
Min Wang Yangyi Zhang
Yangyi Zhang Cheng Huang3
Cheng Huang3 Genming Zhao
Genming Zhao Yuan Jiang
Yuan Jiang- 1Division of TB and HIV/AIDS Prevention, Shanghai Municipal Center for Disease Control and Prevention, Shanghai, China
- 2Department of Epidemiology, School of Public Health and Key Laboratory of Public Health Safety, Fudan University, Shanghai, China
- 3Department of Tuberculosis Control, Chongming District Center for Disease Control and Prevention, Shanghai, China
Background: Tuberculosis (TB) has remained a tough problem in China. This study aims to identify the risk of tuberculosis transmission and to assess its characteristics.
Methods: We performed a molecular epidemiological study for patients with culture-positive Mycobacterium tuberculosis (M. tuberculosis) in Shanghai, from 2009 to 2018. Demographic information was obtained from the Tuberculosis Information Management System. Whole-genome sequencing (WGS) was conducted with a threshold of 12 single-nucleotide polymorphisms (SNPs) to distinguish the genomic cluster. To analyze the characteristics of TB transmission, the contact investigation for clustered cases was performed.
Results: In total, 94 (27.25%) of the 345 enrolled patients were grouped into 42 genomic clusters, indicating local transmission of M. tuberculosis strains. Compared to a health system delay <14 days, patients with a health system delay ≥14 days [adjusted odds ratios (AOR) = 2.57, 95% confidence interval (CI): 1.34–4.95] were more likely to be clustered. Patients under 65 years old (AOR = 3.11, 95% CI: 1.76–5.49), residents (AOR = 2.43, 95% CI: 1.18–4.99), and Beijing genotype strains (AOR = 3.35, 95% CI: 1.32–8.53) were associated with increased risk of clustering. Interestingly, patients with resistance to isoniazid (AOR = 2.36, 95% CI: 1.15–4.88) had a higher risk of transmission. Sixteen confirmed/probable epidemiological links were identified. Local transmission of imported cases and household transmission were prominent.
Conclusion: Health system delay is a crucial factor for TB transmission. Patients with resistance to isoniazid should be priority targets for contact investigation to reduce transmission.
Introduction
Tuberculosis (TB) is a chronic communicable disease and remains a serious public threat worldwide. Despite great efforts at curbing the spreading of Mycobacterium tuberculosis (M. tuberculosis), the WHO has estimated that, of the 10.0 million (range, 8.9–11.0 million) people who fell ill with TB, an estimated 1.2 million TB-related deaths were among HIV-negative people in 2019 (World Health Organization [WHO], 2020).
Standard TB infection control focuses on patients already on treatment, but the spread of TB should not be ignored under frequent social activities. Transmission leads to new infections and cases. Next-generation sequencing technologies have facilitated the development of molecular epidemiology, and we can now study the transmission of TB at a high resolution (Gagneux, 2017). Although molecular approaches can help differentiate between reactivation or reinfection among TB cases, whole-genome sequencing (WGS) has been proven invaluable to illustrate the network and direction of transmission in recent years (Walker et al., 2014; Comin et al., 2020).
Identifying genomic clusters and understanding the transmission dynamics may be helpful to interpret the networks of TB spreading (Anderson et al., 2014; Walker et al., 2014; Kruczak et al., 2019). However, most previous WGS-based studies have only focused on exploring the transmission of multidrug-resistant/drug-resistant tuberculosis (MDR/DR-TB) (Clark et al., 2013; Senghore et al., 2017; Yang et al., 2017; Bouzouita et al., 2019; Dixit et al., 2019; Jiang et al., 2019) or describing the dynamics of TB in metropolitan cities under mass migrations (Yang et al., 2018; Abascal et al., 2019; Jiang et al., 2019). To develop tailored control strategies, it is essential to evaluate the real situation of TB transmission in a specific context (Auld et al., 2017). Few studies fully delineate the TB transmission in a relatively stable population and exclude the impact of population migration.
Factors that contribute to TB transmission could increase the risk of the population acquiring TB, which remains a threat to end TB. Identifying the risk factors and taking target measures may be helpful to reduce the morbidity of TB. Also, despite the advance in knowing the importance of early case finding and treatment, millions of TB cases are being missed due to health system delay, which results in sustained transmission (Floyd et al., 2018). Health system delay is the time interval between patients’ first seeking medical care and the date of diagnosis (Getnet et al., 2017), which has been reported to be longer in smear-negative or extrapulmonary TB (Broderick et al., 2018; Datiko et al., 2020). However, the role of the health system delay contributing to the dissemination of the disease is not known yet.
Chongming is a suburban island in Shanghai with a unique geographical location and limited population movement. About 0.678 million people live in 18 townships, and Shanghai is promoting this district as a world-class ecological island. Although several control measures have been implemented, the morbidity of TB declined slowly. To delineate the risk and characteristics of TB transmission in a relatively insular population, we conducted a WGS-based epidemiological study for all M. tuberculosis isolates in this district over a 10-year period.
Materials and Methods
Sample Source and Study Population
In China, all suspected pulmonary TB patients are referred to local TB-designated hospitals for diagnosis, treatment, and registration management. Through a routine surveillance system, the Shanghai Municipal Centre for Disease Control and Prevention (CDC) collected and preserved all culture-positive M. tuberculosis isolates from local TB-designated hospitals. However, the isolates of patients who were first diagnosed in the Shanghai Pulmonary Hospital were not sent to the Shanghai CDC. Our study obtained all clinical isolates of TB patients in the Chongming district, which were collected from the Shanghai CDC.
This study included patients aged 15 years or older with culture-confirmed pulmonary TB reported in Shanghai, between January 1, 2009 and December 31, 2018. Demographic information of TB patients was collected from the Tuberculosis Information Management System, which has updated small modules according to the work needs during the 10 years. Despite this, the main data collected in our study were not affected, such as age, sex, residential address, diagnostic time, occupation, and census register (residents refer to local patients with census registration in the Chongming district rather than migrants from other provinces in China).
Phenotypic drug susceptibility testing (DST) against first-line anti-tuberculosis drugs was performed using the proportion method on Lowenstein–Jensen medium at the following concentrations: rifampicin 40.0 μg/ml, isoniazid 0.2 μg/ml, streptomycin 4.0 μg/ml, and ethambutol 2.0 μg/ml, respectively (World Health Organization [WHO], 2018).
Definitions
Genomic Cluster
Mycobacterium tuberculosis isolates with a genomic difference (s) ≤ 12 single-nucleotide polymorphisms (SNPs) were defined as a genomic cluster (Yang et al., 2017).
Health System Delay
The time interval between patients’ first consultation with TB-designated hospitals and the date of diagnosis (Getnet et al., 2017).
A Bacteriologically Confirmed Tuberculosis Case
One from whom a biological specimen is positive by smear microscopy, culture, or WHO-approved rapid diagnostics (such as Xpert MTB/RIF).
Tuberculosis-Designated Hospital
Refers to the medical institution designated by the administrative department of health to engage in the diagnosis, treatment, report registration, follow-up inspection, and management of tuberculosis during treatment in China (Beijing Municipal Health Commission [BMHC], 2021).
Confirmed Epidemiological Links
Patients knew each other and have confirmed relationships such as family, workmates, schoolmates, friends, and so on (Yang et al., 2017).
Probable Epidemiological Links
Patients did not know each other but shared locations where TB transmission probably occurred, including in a neighborhood complex or lived in the same town (Yang et al., 2017).
No Epidemiological Links
Patients did know each other, did not live in the same town, or lacked a common neighborhood or setting.
Whole-Genome Sequencing and Bioinformatics Analysis
Extraction and purification of genomic DNA were carried out following QIAamp DNA Mini Kit (Qiagen) protocols. Libraries were constructed by Nextera XT library prep (Illumina), and paired-end 150-bp DNA sequencing was performed on a Hiseq 2500 platform (Illumina) with an expected coverage of 100×. The bioinformatics analysis was performed following a previously validated pipeline (Yang et al., 2018). M. tuberculosis H37Rv strain (GenBank accession number NC_000962.3) was used as the reference template. Sequencing reads were mapped to the reference genome using Bowtie2 (v2.3.1). SAMtools (v1.6) was used for SNP calling with mapping quality greater than 30. Fixed mutations (frequency ≥ 75%) were identified using VarScan (v2.3.6) with at least 10 reads supporting and the strand bias filter option on. We excluded all SNPs annotated in regions, such as PPE/PE-PGRS family genes, phage sequences, insertion, or mobile genetic elements (Casali et al., 2016).
To assess the genetic drug resistance, the data of SNPs were compared with a list of the mutations related to anti-TB drug resistance (Coll et al., 2015). Excluding mutations in drug-resistance-associated genes, all SNP locations were combined into a non-redundant consensus list and were recalled with the mpileup2cns function of VarScan to build a phylogenetic tree. Nucleotide positions with missing calls present on more than 5% of the isolates were removed (Liu et al., 2018; Liu et al., 2020). A phylogenetic tree was constructed with MEGA X (version 10.1) using the maximum-likelihood method and a general time-reversible model of nucleotide substitution, with a gamma distribution (GTRGAMMA) and 500 bootstraps. Visualization of the bacteriological information was performed at Interactive Tree of Life.1 We adapted a recently described hierarchical nomenclature to define nodes and subclades within the tree in the definition of sub-lineages (Coll et al., 2014).
Epidemiological Investigation
Clustered cases were invited to participate in a contact investigation. After acquiring the informed consent, we have conducted a questionnaire in person or telephone interviews. To understand the social relationship between clustered cases, we collected data on close contacts, social contacts, and places they frequented before their diagnosis of TB, including the information of medical records, workplace, and residential address.
Statistical Analysis
We compared the characteristics between “clustered” and “non-clustered” using the chi-square test. Multivariate logistic regression was conducted to identify the risk factors of TB transmission, such as demographic, bacteriological, phenotypic resistance, and clinical characteristics. Factors with a P-value less than 0.05 in the final model were independently associated with genomic clusters. The adjusted odds ratios (AORs) and 95% confidence intervals (95% CIs) were calculated. Our statistical analysis was conducted with SAS (version 9.4). The clustering rate was calculated as the percentage of clustered patients among the number of total patients (number of clustered patients/number of total patients).
Ethics Approval and Institutional Safety Procedures
This study protocol was approved by the Ethical Review Committee at the Shanghai CDC (No. 2020-14). All participants provided written informed consent after completing the description of the study.
All biological experimentations in our study were performed after acquiring approval from the Biosafety Commission, Shanghai CDC. Our study abides by National Rules on Biosafety Management of Pathogenic Microorganism Laboratory (State Council of the People’s Republic of China [SCPRC], 2018) to conduct all specific procedures, such as microscopy, culture, DST, and molecular testing. A set of essential biosafety measures was enacted to minimize risks in our TB laboratory, including risk assessment, plans for emergency preparedness and response, codes of practice, personal protective equipment and clothing, as well as disposal procedures for contaminated materials.
Genomic Data Availability
Sequence data associated with this study were deposited in the Sequence Read Archive (SRA) of NCBI under project accession PRJNA760838.
Results
Basic Information
As shown in Figure 1, 1,708 TB cases were reported in the Chongming district between 2009 and 2018, including 685 bacteriologically confirmed TB cases. There were 525 culture-positive isolates and 160 smear-positive but culture-negative isolates. Of those, 370 patients (70.48%, 370/525) with culture-positive M. tuberculosis isolates were enrolled in this study. The rest of patients (155 culture-positive isolates) who were first diagnosed in the Shanghai Pulmonary Hospital were not included. In total, 16 isolates were excluded due to DNA extraction failure, contamination, or insufficient quality of WGS data. Seven patients had a relapse of pulmonary TB and one patient relapsed twice, so we excluded the nine isolates of relapsed patients. For the final analysis, 345 M. tuberculosis isolates were included.
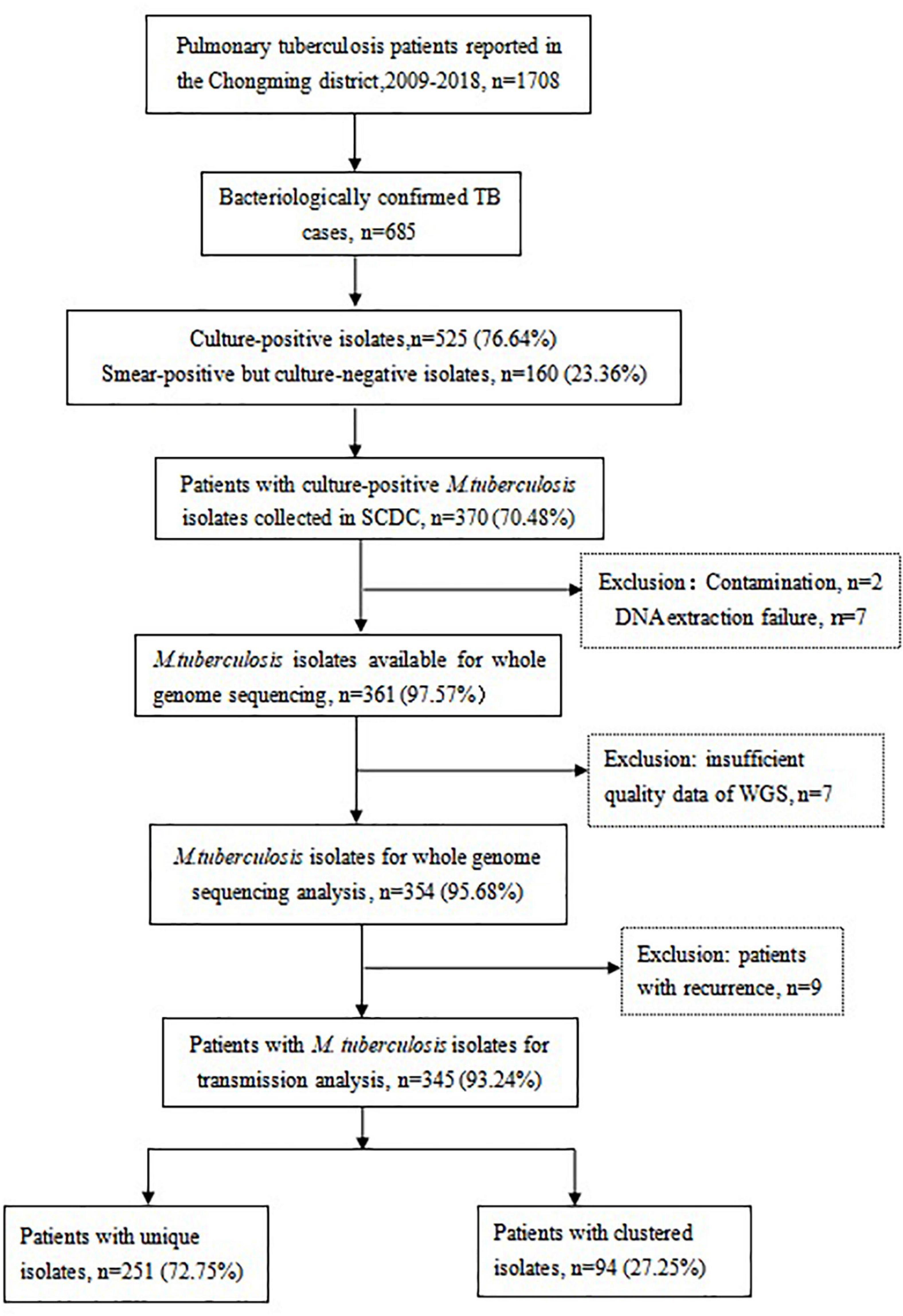
Figure 1. Flow chart of study population and selection.
The median age of the TB patients was 60 years (range 38–74). A total of 277 patients (80.29%, 277/345) were residents with census register in the Chongming district. About 86.09% of the patients (297/345) were new TB cases. The percentage of patients with resistance to isoniazid (INH) was 12.46% (43/345). The median time of health system delay was 3 days (range 2–9 days).
Most of the M. tuberculosis isolates belonged to the lineage 2 (n = 298, 86.38%) followed by lineage 4 (n = 46, 13.33%), and lineage 3 (n = 1, 0.29%). Sub-lineage 2.2.1 (n = 289, 83.76%) was the main sub-lineage. The phylogenetic tree revealed that three lineages of M. tuberculosis isolate circulated in the Chongming district (Figure 2). The tree also displays the drug resistance profile for 12 anti-TB drugs based on the presence of validated resistance-conferring mutations. The mutations in drug resistance genes mainly occurred in rpsL, katG, and rpoB.
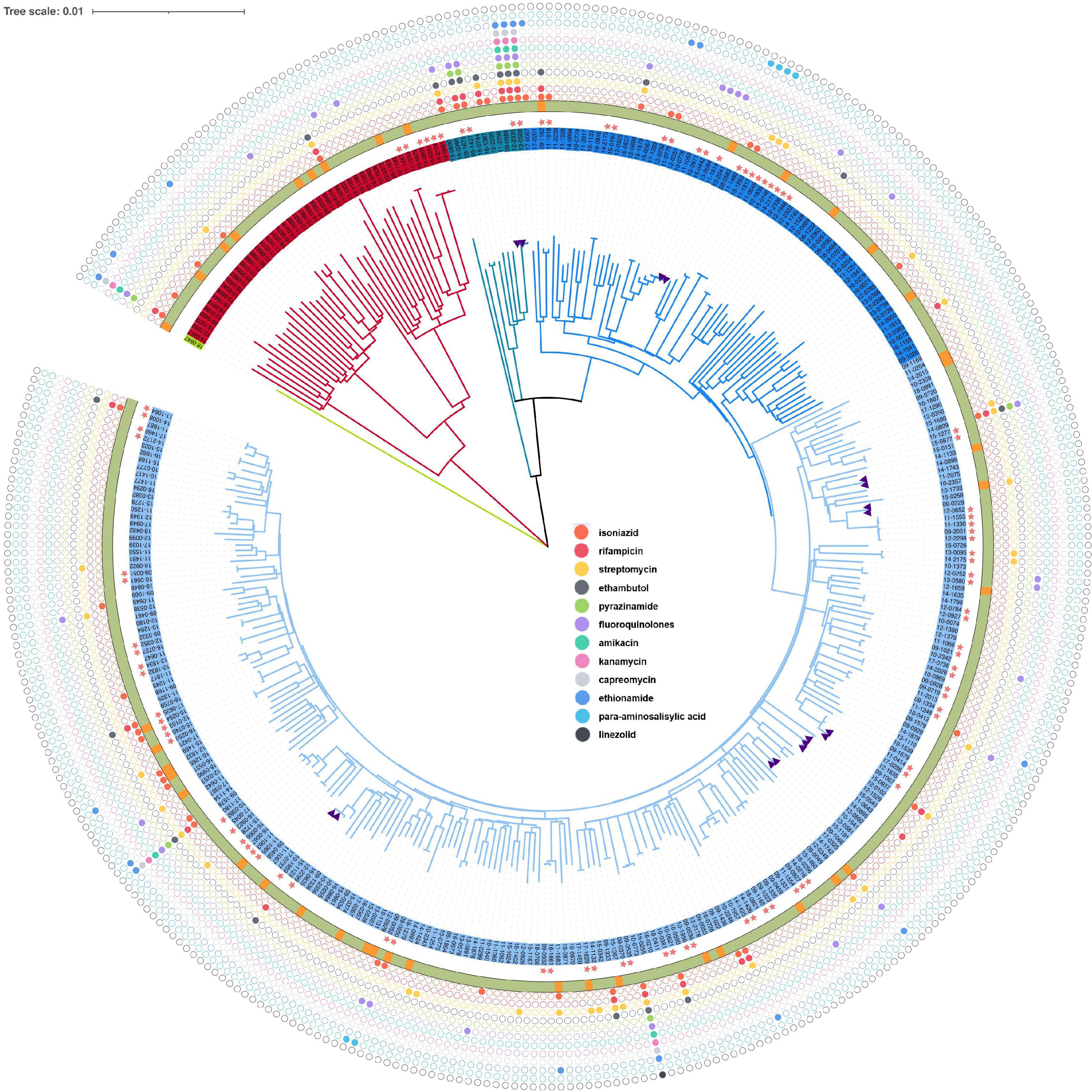
Figure 2. Phylogenetic tree for the 354 Mycobacterium tuberculosis isolates from Shanghai, China, 2009–2018. The purple triangles represent isolates from relapsed cases (n = 9). The pink five-pointed stars indicate the clustered isolates, whose genetic distance is ≤12 single-nucleotide polymorphisms (SNPs). The outer ring shows the census register of patients, of which the green represents resident and the orange represents migrant. From inside to outside, the solid colorful circles represent drug resistance mutations for isoniazid, rifampicin, streptomycin, pyrazinamide, amikacin, kanamycin, capreomycin, ethionamide, para-aminosalicylic acid, and linezolid.
The Clustering Rate
Among 345 TB cases, 94 were grouped into 42 genomic clusters. The clustering rate was 27.25%, which indicated TB transmission in this district. The genomic clusters consisted of two to five strains, and the majority of the clusters had two strains, accounting for 74.47% (Table 1) of the clustered cases (70/94).

Table 1. The cluster size and the number of genomic clusters of 94 Mycobacterium tuberculosis isolates.
Risk Factors Associated With Genomic Clusters
Beijing genotype strains were more frequent in clustered cases than in non-clustered cases (P < 0.05). Resistance to isoniazid was associated with genomic clusters (P < 0.05). Additionally, positive sputum smears and cavitation on chest radiography did not differ between clustered cases and non-clustered cases (P > 0.05) (Table 2).
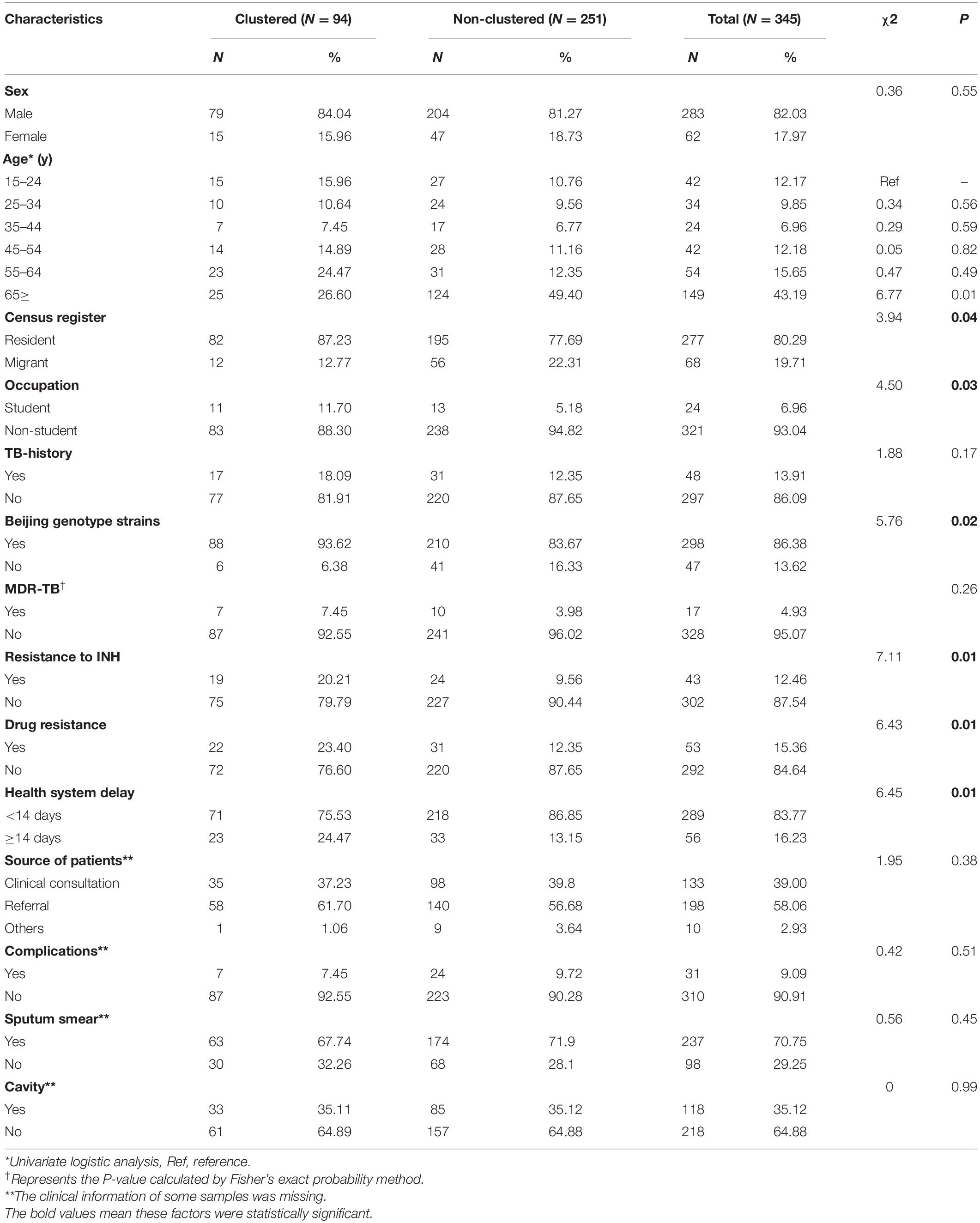
Table 2. Demographic, bacteriological, phenotypic resistance, and clinical characteristics of genomic clustered and non-clustered cases in Shanghai, China, 2009–2018.
In the multivariable logistic analysis, factors independently associated with genomic cluster included age, census register, health system delay, genotype, and resistance to isoniazid. As shown in Table 3, patients under 65 years old were more likely to be clustered than patients aged 65 years or older (AOR = 3.11, 95% CI: 1.76–5.49). Residents (AOR = 2.43, 95% CI: 1.18–4.99) and Beijing genotype strains (AOR = 3.35, 95% CI: 1.32–8.53) had a higher risk of clustering. Interestingly, patients with resistance to isoniazid were more likely to be clustered than others (AOR = 2.36, 95% CI: 1.15–4.88). Compared to a health system delay < 14 days, a health system delay ≥ 14 days increases by 2.57 (95% CI: 1.34–4.95) times the risk to form genomic clusters.
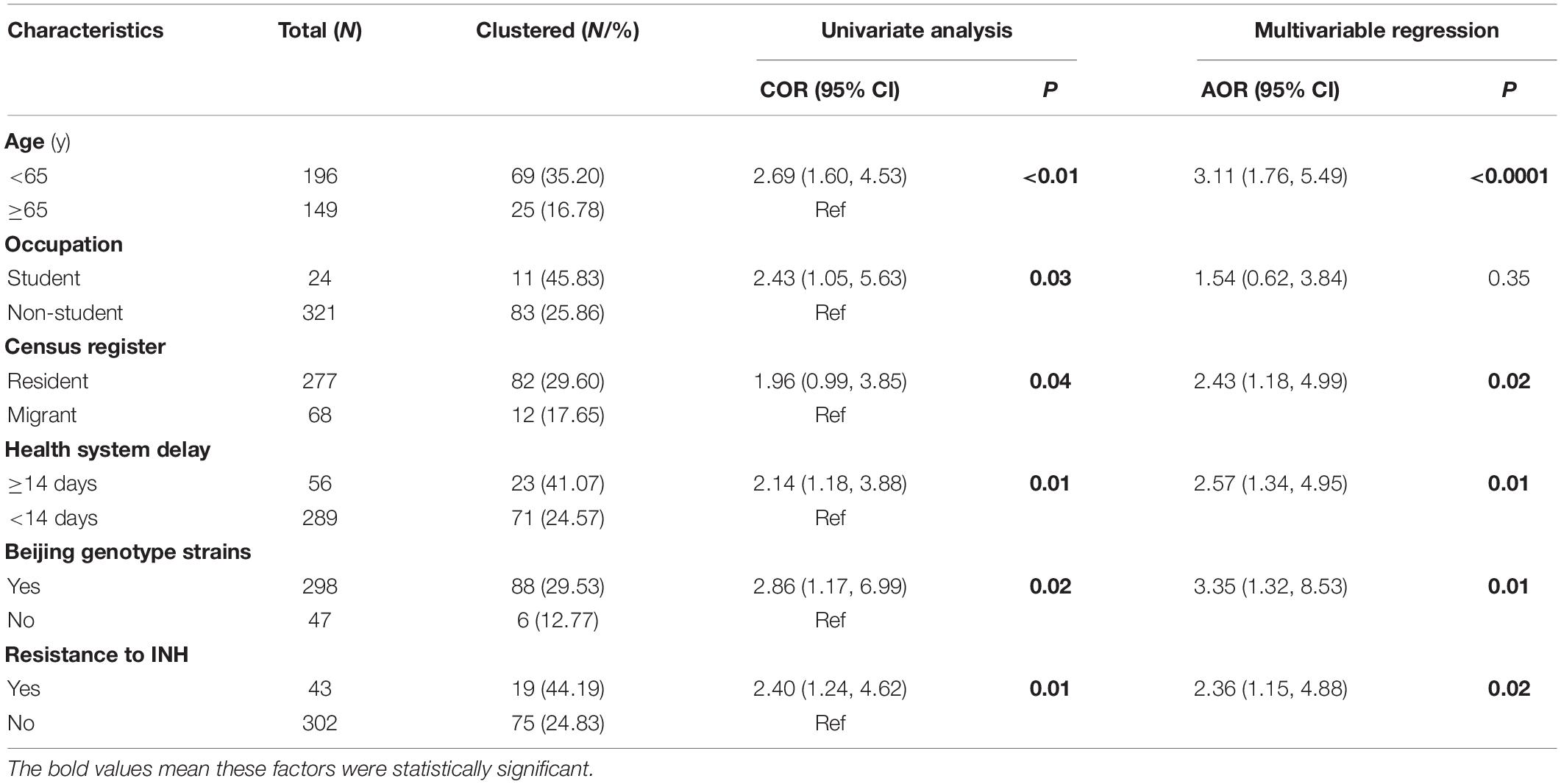
Table 3. Univariate and multivariable regression analysis on the risk factors of genomic clustering.
Epidemiological Investigation and Pathway of Tuberculosis Transmission
Thirty-three clustered cases were investigated, with a response rate of 35.11% (33/94), and those cases covered 52.38% of the genomic clusters (22/42). We found four clusters with confirmed epidemiological links, in which one cluster included two prisoners from the same prison and three clusters included six family members. Based on the residential address, we found that several patients in the same clusters lived in the same towns. Thus, probable epidemiological links could be established for 28 patients from 12 clusters. No epidemiological links were found in eight genomic clusters.
Many clustered patients reported a history of exposure to TB with family members (C11, C12, and C25) or classmates (C02, C15, C16, and C26). There were five patients of four clusters (C02, C15, C24, and C29) that worked in other places before they were diagnosed. The WGS data indicated that they were in the same clusters with other residents. Several clustered cases were from the same sex, shared the same occupation, and had similar age. The young people often went to internet cafes and school, while the older group often went to markets, chess, and card rooms. It means that casual contact transmission could occur in those same groups due to their lifestyles.
Discussion
Summary of the Principal Findings
This genomic epidemiological study systematically delineates the relative contribution of TB transmission and identifies principal risk factors that may be responsible for TB transmission in Shanghai, from 2009 to 2018. Our study concludes that health system delay and resistance to isoniazid are principal contributing factors for the dissemination of the disease. A total of 27.25% (94/345) clustered cases were identified, implying that local transmission is a threat to end TB. WGS was performed for all available M. tuberculosis isolates in a 10-year period in the Chongming district, Shanghai, which could accurately reflect the level of TB transmission.
Our study shows the capability of WGS to identify exact clusters. The epidemiological investigation has further validated that confirmed or probable epidemiological links exist in some genomic clusters. We have found 16 clusters with confirmed/probable epidemiological links by contact investigation. Based on the medical records, WGS data, and epidemiological information, the transmission relationships of clustered patients were explored, and the characteristics of TB transmission were summarized. Household transmission is an important characteristic in this area. Several patients in the same cluster share family relationships, and many clustered patients have a history of family contact with TB.
Significance of the Study
Our study suggested that patients under 65 years old or residents had a higher risk for transmission. To halt the spread of TB, active case finding should be focused on these high-risk groups. A study in the United Kingdom suggests that most patients who migrate from high-incidence countries do not lead to local transmission (Walker et al., 2014). However, our results are distinct from a study in the Songjiang district, Shanghai, which inferred that TB transmission from residents to migrants occurs as commonly as transmission from migrants to residents (Yang et al., 2018). One possible explanation is that older residents in the Chongming district have lower awareness of TB and have more frequent social contact with others, which increases the chance to be infected and transmit to others. Also, most migrants in the Songjiang district could be younger groups with stronger bodies.
This study noted that Beijing genotype strains had a higher risk of clustering. This finding is consistent with previous studies (Yang et al., 2015; Jiang et al., 2019), indicating that the Beijing genotype may have increased transmissibility. However, it contrasts with a study (Lalor et al., 2017) conducted in England, which noted that the Beijing genotype strains are not associated with transmissibility among household contacts.
We detected a significant association between resistance to isoniazid and genomic clusters. A previous study (Dheda et al., 2017) has showed that close contacts of drug-resistant TB cases are more likely to be infected with a more infectious M. tuberculosis strain. A recent study (Theron et al., 2020) reported similar results, showing that drug-resistant TB patients have higher numbers of culturable M. tuberculosis in an aerosol. Our results also showed that 20.21% (19/94) of clustered isolates were resistant to isoniazid. A systematic review reported that treatment of isoniazid-resistant TB with the standardized first-line regimen led to treatment failure in 11% of the cases, compared with 2% among drug-susceptible TB patients (Gegia et al., 2017).
In our study, patients with a health system delay ≥ 14 days have a higher risk to transmit the disease. Chongming district is at the mouth of the Yangtze River, with relatively inconvenient traffic and a low economic level. This delay may be due to the unavailability of rapid and accurate diagnostic technologies required to confirm TB. A study (Segagni Lusignani et al., 2013) indicated that living in rural areas is associated with prolonged health system delay. It is worth noting that the diagnostic delay could prolong the infectious period. A meta-analysis (Bao et al., 2019) revealed that the case finding delay is a major contributing factor to the spread of TB in outbreaks. Also, the diagnostic delay might result in worse treatment outcome and increased risk of transmission (Cheng et al., 2013; Shiferaw and Zegeye, 2019). However, the main diagnostic tests for TB are rapid molecular assays, smear microscopy, and culture-based methods. Culture-based methods are the reference standard, which require a higher level of TB laboratory and can take up to 8 weeks to get results (World Health Organization [WHO], 2020). To achieve early diagnosis and promote early treatment, health system delay should be reduced by introducing rapid and accurate diagnostic tools such as sequencing technologies.
Strengths and Limitations of the Study
Our study has several limitations and strengths. First, the level of TB transmission may be underestimated. About 30% (155/525) of culture-positive isolates were not collected in our study. Also, it is unable to analyze the TB transmission of patients without clinical strains. Second, we obtained limited information about clustered cases due to the retrospective study design. Therefore, epidemiological links between clustered cases were difficult to interpret clearly. However, this was the first population-based study using WGS to explore the transmission of TB in the Chongming district, Shanghai. Also, the long-span study timeframe facilitates the capture of all TB cases and provides insights into TB transmission dynamics. We have traced the infection source of the population studied at the molecular level, and the transmission chains of TB patients have been detected.
Implications for Practice or Policy
Health system delay is a crucial factor for the transmission of TB in rural areas. The public health strategies should be redirected to reduce health system delay by introducing rapid and accurate diagnostic tools. Additionally, it could be feasible to take targeted interventions on patients with resistance to isoniazid.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA760838.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethical Review Committee of Shanghai Municipal Center for Disease Control and Prevention. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
MW, YZ, and CH performed the material preparation, data collection, and data analysis. MW wrote the first draft of the manuscript. YZ, JL, and XS helped to proofread the manuscript. QP, GZ, and YJ supervised the study and made a critical revision to the manuscript. All authors contributed to the study conception and design, and approved the submitted version.
Funding
This study was supported by the National Key Research and Development Program of China (No. 2017YFD0500301), the National Natural Science Foundation of China (81872679), the Shanghai Municipal Health Commission (No. 20194Y0169), the Three-Year Action Plan of Shanghai Public Health System Construction—Key Discipline Construction (2020–2022) (No. GWV-10.1-XK03), and the Shanghai Municipal Project for Academic Leaders in Public Health (No. GWV-10.2-XD23). The funding body had no role in the study design, collection, analysis, interpretation of data, and in writing the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all the doctors, laboratory technicians, and project administrators who made contributions to the databases. We are grateful to Biao Xu from Fudan University for the critical reading and helpful discussions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.768659/full#supplementary-material
Footnotes
References
Abascal, E., Pérez-Lago, L., and García-de-Viedma, D. (2019). Whole genome sequencing-based analysis of tuberculosis (TB) in migrants: rapid tools for cross-border surveillance and to distinguish between recent transmission in the host country and new importations. Euro Surveill. 24:1800005. doi: 10.2807/1560-7917.ES.2019.24.4.1800005
Anderson, L. F. D., Tamne, S. R., and Abubakar, I. P. (2014). Transmission of multidrug-resistant tuberculosis in the UK: a cross-sectional molecular and epidemiological study of clustering and contact tracing. Lancet Infect. Dis. 14, 406–415. doi: 10.1016/S1473-3099(14)70022-2
Auld, S. C., Kasmar, A. G., and Shah, N. S. (2017). Research Roadmap for Tuberculosis Transmission Science: where Do We Go From Here and How Will We Know When We’re There? J. Infect. Dis. 216, S662–S668. doi: 10.1093/infdis/jix353
Bao, H., Liu, K., and Jiang, J. (2019). Tuberculosis outbreaks among students in mainland China: a systematic review and meta-analysis. BMC Infect. Dis. 19:972. doi: 10.1186/s12879-019-4573-3
Beijing Municipal Health Commission [BMHC] (2021). Notice the Issuance of Standards for the Establishment of Designated Tuberculosis Medical Institutions at the District Level in Beijing. Available online at: http://wjw.beijing.gov.cn/zwgk_20040/zxgk/202107/t20210714_2435813.html [Accessed December 10, 2021]
Bouzouita, I., Cabibbe, A. M., and Saidi, L. S. (2019). Whole-Genome Sequencing of Drug-Resistant Mycobacterium tuberculosis Strains, Tunisia, 2012–2016. Emerg. Infect. Dis. 25, 538–546. doi: 10.3201/eid2503.181370
Broderick, C., Hopkins, S., Mack, D. J. F., Aston, W., Pollock, R., Skinner, J. A., et al. (2018). Delays in the diagnosis and treatment of bone and joint tuberculosis in the United Kingdom. Bone Joint J. 100–B, 119–124. doi: 10.1302/0301-620X.100B1.BJJ-2017-0357.R1
Casali, N., Broda, A., and Drobniewski, F. (2016). Whole Genome Sequence Analysis of a Large Isoniazid-Resistant Tuberculosis Outbreak in London: a Retrospective Observational Study. PLoS Med. 13:e1002137. doi: 10.1371/journal.pmed.1002137
Cheng, S., Chen, W., and Jia, Z. (2013). Effect of Diagnostic and Treatment Delay on the Risk of Tuberculosis Transmission in Shenzhen, China: an Observational Cohort Study, 1993-2010. PLoS One 8:e67516. doi: 10.1371/journal.pone.0067516
Clark, T. G., Mallard, K., and McNerney, R. (2013). Elucidating emergence and transmission of multidrug-resistant tuberculosis in treatment experienced patients by whole genome sequencing. PLoS One 8:e83012. doi: 10.1371/journal.pone.0083012
Coll, F., McNerney, R., and Clark, T. G. (2014). A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat. Commun. 5:4812. doi: 10.1038/ncomms5812
Coll, F., McNerney, R., and Clark, T. G. (2015). Rapid determination of anti-tuberculosis drug resistance from whole-genome sequences. Genome Med. 7:51. doi: 10.1186/s13073-015-0164-0
Comin, J., Chaure, A., and Samper, S. (2020). Investigation of a rapidly spreading tuberculosis outbreak using whole-genome sequencing. Infect. Genet. Evol. 81:104184. doi: 10.1016/j.meegid.2020.104184
Datiko, D. G., Jerene, D., and Suarez, P. (2020). Patient and health system delay among TB patients in Ethiopia: nationwide mixed method cross-sectional study. BMC Public Health 20:1126. doi: 10.1186/s12889-020-08967-0
Dheda, K., Gumbo, T., and Warren, R. M. (2017). The epidemiology, pathogenesis, transmission, diagnosis, and management of multidrug-resistant, extensively drug-resistant, and incurable tuberculosis. Lancet Respir Med. S2213–2600, 30079–6. doi: 10.1016/S2213-2600(17)30079-6
Dixit, A., Freschi, L., and Farhat, M. R. (2019). Whole genome sequencing identifies bacterial factors affecting transmission of multidrug-resistant tuberculosis in a high-prevalence setting. Sci. Rep. 9:5602. doi: 10.1038/s41598-019-41967-8
Floyd, K., Glaziou, P., and Raviglione, M. (2018). The global tuberculosis epidemic and progress in care, prevention, and research: an overview in year 3 of the End TB era. Lancet Respir Med. 6, 299–314. doi: 10.1016/S2213-2600(18)30057-2
Gagneux, S. (2017). Strain Variation in the Mycobacterium tuberculosis Complex: its Role in Biology, Epidemiology and Control. Germany: Springer.
Gegia, M., Winters, N., and Menzies, D. (2017). Treatment of isoniazid-resistant tuberculosis with first-line drugs: a systematic review and meta-analysis. Lancet Infect. Dis. 17, 223–234. doi: 10.1016/S1473-3099(16)30407-8
Getnet, F., Demissie, M., and Worku, A. (2017). Delay in diagnosis of pulmonary tuberculosis in low-and middle-income settings: systematic review and meta-analysis. BMC Pulm. Med. 17:202. doi: 10.1186/s12890-017-0551-y
Jiang, Q., Liu, Q., and Gao, Q. (2019). Citywide Transmission of Multidrug-resistant Tuberculosis Under China’s Rapid Urbanization: a Retrospective Population-based Genomic Spatial Epidemiological Study. Clin. Infect. Dis. 24, 142–151. doi: 10.1093/cid/ciz790
Kruczak, K., Augustynowicz-Kopeć, E., and Sładek, K. (2019). Tuberculosis transmission in the population of patients from the Krakow Region (Poland) based on the epidemiological and molecular methods. Int. J. Mycobacteriol. 8:60. doi: 10.4103/ijmy.ijmy_11_19
Lalor, M. K., Anderson, L. F., and Thomas, H. L. (2017). Recent household transmission of tuberculosis in England, 2010–2012: retrospective national cohort study combining epidemiological and molecular strain typing data. BMC Med. 15:105. doi: 10.1186/s12916-017-0864-y
Liu, Q., Ma, A., and Gao, Q. (2018). China’s tuberculosis epidemic stems from historical expansion of four strains of Mycobacterium tuberculosis. Nat. Ecol. Evol. 2, 1982–1992. doi: 10.1038/s41559-018-0680-6
Liu, Q., Wei, J., Li, Y., and Gao, Q. (2020). Mycobacterium tuberculosis clinical isolates carry mutational signatures of host immune environments. Sci. Adv. 6:eaba4901. doi: 10.1126/sciadv.aba4901
Segagni Lusignani, L., Quaglio, G., and Manenti, F. (2013). Factors associated with patient and health care system delay in diagnosis for tuberculosis in the province of Luanda, Angola. BMC Infect. Dis. 13:168–168. doi: 10.1186/1471-2334-13-168
Senghore, M., Otu, J., and Antonio, M. (2017). Whole-genome sequencing illuminates the evolution and spread of multidrug-resistant tuberculosis in Southwest Nigeria. PLoS One 12:e0184510. doi: 10.1371/journal.pone.0184510
Shiferaw, M. B., and Zegeye, A. M. (2019). Delay in tuberculosis diagnosis and treatment in Amhara state, Ethiopia. BMC Health Serv. Res. 19:232. doi: 10.1186/s12913-019-4056-7
State Council of the People’s Republic of China [SCPRC] (2018). National Rules on Biosafety Management of Pathogenic Microorganism Laboratory. Available online at: http://www.gov.cn/zhengce/2020-12/27/content_5574545.htm. [Accessed December 12, 2021]
Theron, G., Limberis, J., Venter, R., Smith, L., Pietersen, E., Esmail, A., et al. (2020). Bacterial and host determinants of cough aerosol culture positivity in patients with drug-resistant versus drug-susceptible tuberculosis. Nat. Med. 26, 1435–1443. doi: 10.1038/s41591-020-0940-2
Walker, T. M., Lalor, M. K., and Conlon, C. P. (2014). Assessment of Mycobacterium tuberculosis transmission in Oxfordshire, UK, 2007–12, with whole pathogen genome sequences: an observational study. Lancet Respirat. Med. 2, 285–292. doi: 10.1016/S2213-2600(14)70027-X
World Health Organization [WHO] (2018). Technical Manual for Drug Susceptibility Testing of Medicines Used in the Treatment of Tuberculosis. Available online at: https://apps.who.int/iris/handle/10665/275469 [Accessed 28 December 2021.
World Health Organization [WHO] (2020). Global Tuberculosis Report. Available online at: https://www.who.int/teams/global-tuberculosis-programme/tb-reports. [Access on 18 June 2021.]
Yang, C., Lu, L., and Cohen, T. (2018). Internal migration and transmission dynamics of tuberculosis in Shanghai, China: an epidemiological, spatial, genomic analysis. Lancet Infect. Dis. 18, 788–795. doi: 10.1016/S1473-3099(18)30218-4
Yang, C., Luo, T., and Gao, Q. (2017). Transmission of multidrug-resistant Mycobacterium tuberculosis in Shanghai, China: a retrospective observational study using whole-genome sequencing and epidemiological investigation. Lancet Infect. Dis. 17, 275–284. doi: 10.1016/S1473-3099(16)30418-2
Keywords: tuberculosis, transmission, whole-genome sequencing, diagnostic delay, genomic cluster, epidemiological investigation
Citation: Wang M, Zhang Y, Huang C, Li J, Shen X, Zhao G, Jiang Y and Pan Q (2022) A Whole-Genome Sequencing-Based Study to Delineate the Risk and Characteristics of Tuberculosis Transmission in an Insular Population Over 10 Years in Shanghai. Front. Microbiol. 12:768659. doi: 10.3389/fmicb.2021.768659
Received: 01 September 2021; Accepted: 31 December 2021;
Published: 16 February 2022.
Edited by:
Maurizio Sanguinetti, Catholic University of the Sacred Heart, ItalyReviewed by:
Abdolrazagh Hashemi Shahraki, University of Florida, United StatesDiana Machado, New University of Lisbon, Portugal
Copyright © 2022 Wang, Zhang, Huang, Li, Shen, Zhao, Jiang and Pan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuan Jiang, amlhbmd5dWFuQHNjZGMuc2guY24=; Qichao Pan, cGFucWljaGFvQHNjZGMuc2guY24=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship