
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 11 October 2021
Sec. Evolutionary and Genomic Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.763359
 Yang Liu1
Yang Liu1 Tao Pei1
Tao Pei1 Shuoxing Yi1
Shuoxing Yi1 Juan Du1
Juan Du1 Xianjiao Zhang1
Xianjiao Zhang1 Xiaoqin Deng1
Xiaoqin Deng1 Qing Yao2
Qing Yao2 Ming-Rong Deng1
Ming-Rong Deng1 Honghui Zhu1*
Honghui Zhu1*Rapid and accurate strain identification of the most closely related genera Myxococcus, Corallococcus, and Pyxidicoccus can enhance the efficiency of the mining of novel secondary metabolites through dereplication. However, the commonly used 16S rRNA gene sequencing cannot accurately differentiate members of the three genera above, and the whole-genome sequencing is unable to rapidly and inexpensively provide species assignation toward a large number of isolates. To overcome the limitations, the gyrB gene was investigated as a candidate genetic marker for exploring the phylogenetic relationships of bacteria within the three genera and for developing the gyrB-based typing method. Here, the bacterial phylogeny and species affiliations of the three genera were determined based on the phylogenomic reconstruction and the analysis of digital DNA–DNA hybridization values among 90 genomes, further confirming nine novel taxa and assigning over one-third of genomes to defined species. The phylogenetic relationships of these strains based on the gyrB gene sequences were congruent with those based on their genome sequences, allowing the use of the gyrB gene as a molecular marker. The gyrB gene-specific primers for the PCR-amplification and sequencing of bacteria within the three genera were designed and validated for 31 isolates from our group collection. The gyrB-based taxonomic tool proved to be able to differentiate closely related isolates at the species level. Based on the newly proposed 98.6% identity threshold for the 966-bp gyrB gene and the phylogenetic inference, these isolates were assigned into two known species and eight additional putative new species. In summary, this report demonstrated that the gyrB gene is a powerful phylogenetic marker for taxonomy and phylogeny of bacteria within the closely related genera Myxococcus, Corallococcus, and Pyxidicoccus, particularly in the case of hundreds or thousands of isolates in environmental studies.
The family Myxococcaceae belongs to the order Myxococcales and currently comprises five genera validly described as Aggregicoccus, Corallococcus, Myxococcus, Pyxidicoccus, and Simulacricoccus1. Just recently, the other four distinct genera Archangium, Cystobacter, Hyalangium, and Stigmatella have been proposed for reclassifying them into the family Myxococcaceae (Waite et al., 2020). Members of this family are widely distributed in terrestrial and marine environments (Liu et al., 2019), displaying great environmental adaptability. Among these genera above, the Myxococcus, Corallococcus, and Pyxidicoccus seem to be one of the most frequently isolated myxobacterial genera from almost all samples, especially from the soil (Dawid, 2000; Zhang et al., 2013; Wang et al., 2020) and cover the best studied and explored myxobacteria with a wide scope of research. For example, Myxococcus xanthus is most widely used as a model microorganism for studying bacterial social behaviors such as predation, swarming, fruiting-body formation, and sporulation (Zusman et al., 2007). The further fascinating feature of myxobacteria is that they can produce a large and variety of bioactive secondary metabolites acting as antimicrobials, antiparasitics, antivirals, cytotoxins, and anti-blood coagulants (Herrmann et al., 2017; Gregory et al., 2019). Many myxobacteria can prey on bacteria (Livingstone et al., 2017) and fungi (Li et al., 2019) by the secretion of antibiotic metabolites and hydrolytic enzymes, showing great application potential in plant disease control (Bull et al., 2002). Consequently, it is of great interest to be able to identify myxobacterial isolates, in a fast, reliable, and low-cost way, to determine their taxonomic affiliation, to monitor their ecological distribution and diversity in various environments.
The three genera Myxococcus, Corallococcus, and Pyxidicoccus are the most closely related among the family Myxococcaceae. From the early days to now, the morphology of cells and colonies and 16S rRNA gene analysis have been always used for the bacterial identification of the three genera above (Garcia et al., 2010; Reichenbach, 2015). However, the morphological characteristics such as typical shapes of vegetative cells and myxospores, the structure and color of fruiting bodies may vary under different culture conditions and media constituents and therefore are difficult to provide accurate myxobacterial taxonomic assignation (Reichenbach, 2015). These graphical and descriptive morphological features are also inconvenient for a data-based comparison among different research teams. Furthermore, the most frequently used 16S rRNA gene sequencing can only identify closely related strains at the genus level but not at the species level (Stackebrandt and Päuker, 2005; Stackebrandt et al., 2007). The multiple copies of the 16S rRNA gene in their genome may make the community abundance data distorted in microbiome surveys. With the advance of next-generation sequencing technologies, comparative genomics analysis based on the concatenated single-copy core genes has been successfully applied for identifying and classifying isolates of the three genera above (Chambers et al., 2020; Livingstone et al., 2020). For example, eight novel Corallococcus species, three novel Myxococcus species, and two novel Pyxidicoccus species have been recently proposed based on the analyses of comparative genomics and pan-genomics (Chambers et al., 2020; Livingstone et al., 2020), resulting in the number of described species to more than double. Although the genome-based analysis can produce reproducible and reliable phylogenetic relationships of bacteria within the three genera, it remains challenging according to the time-consuming and costly features, particularly in the case of hundreds or thousands of isolates. With the decrease in the sequencing cost and the advance in massive data analysis, it is reasonable to expect that the whole genome sequence-based comparison and characterization of bacteria within the three genera will become more feasible and practical than now. However, in the current case at least, the fast and accurate identification of bacteria belonged to the closely related genera Myxococcus, Corallococcus, and Pyxidicoccus is still a challenge whether using the 16S rRNA gene sequencing or whole-genome sequence comparison.
The conserved single-copy protein-coding genes, also known as housekeeping genes, have been proposed as alternative molecular markers for the study of microbial taxonomic relationships and diversity (Santos and Ochman, 2004; Vos et al., 2012; Ogier et al., 2019). Like the 16S rRNA gene, housekeeping genes are required for the maintenance of basic cellular functions and thus are essential and universally present in the bacterial kingdom. In contrast to the 16S rRNA gene, housekeeping genes are supposed to evolve at a much more rapid but constant rate and are therefore endowed with a better resolution power for differentiating different lineages that have recently diverged (Poirier et al., 2018). Moreover, single-copy housekeeping genes in bacterial genomes can avoid the overestimation of bacterial richness and abundance in ecological surveys based on 16S rRNA amplicon sequencing. As of now, some housekeeping genes have been used either to distinguish the closely related strains at the species level or to decipher microbial diversity (Poirier et al., 2018; Ogier et al., 2019). In the study of the heterogeneity of Corallococcus coralloides strains isolated from geographically diverse locations, the gyrB gene encoding DNA gyrase subunit B showed a higher resolution power against the 16S rRNA gene (Stackebrandt and Päuker, 2005). Soon after, the three housekeeping genes csgA, fibA, and pilA were used to investigate the small-scale genetic population structure of the soil bacterium M. xanthus (Vos and Velicer, 2006). The additional four taxonomic markers lepA, fusA, and rpoB were used to determine whether the genetic diversity of the three species within the genus Corallococcus is matched with their phenotypic properties (Stackebrandt et al., 2007). However, these molecular markers are too short to have enough genetic information to clearly distinguish the close relatives, and the use of these markers only focuses on a few species. More importantly, the classification criteria for these genes have not been established, to some extent due to the lack of myxobacterial genomic sequence at that time.
In recent years, with the rapid increase of myxobacterial isolates, it is urgent to establish an efficient method for the rapid and accurate identification of these bacteria. Many genome sequences belonged to the genera Myxococcus, Corallococcus, and Pyxidicoccus available in public databases provide an ideal opportunity for determining the threshold of a single molecular marker for species delineation. In comparison with other genetic markers, the gyrB gene with the much higher resolution has been fairly frequently used for determining phylogenetic relationships of closely related strains at the species level (Poirier et al., 2018; Martínez-Hidalgo et al., 2020; Klemetsen et al., 2021) and has been produced numerous reference sequences in public databases. Inspired by these results, we thus choose the gyrB gene as the candidate housekeeping gene. The present study aimed to determine the potential of the gyrB gene in the identification, genotyping, and phylogenetics of strains within the closely related three genera.
Genome sequences of bacteria within the three genera were retrieved from the NCBI database. The genomic quality was evaluated using the software CheckM version 1.1.2 (Parks et al., 2015). The general genomic characteristics of strains including genomic sizes and DNA G + C contents were conducted using the software QUAST version 5.0.2 (Gurevich et al., 2013). For consistency, all genome sequences were re-annotated using the software Prokka version 1.13 (Seemann, 2014). The 16S rRNA and gyrB gene sequences of each strain were extracted using a local BLAST search with default parameters. The digital DNA–DNA hybridization (dDDH) values were estimated using the genome-to-genome distance calculator (GGDC) version 2.1 online service with the recommended formula 2 (Auch et al., 2010). The dDDH values were visualized using the “HeatMap” tool of the software TBtools version 1.0981 (Chen et al., 2020). The pairwise identities of the 16S rRNA and gyrB gene sequences were conducted using the software DNAMAN version 8 (Lynnon Biosoft2) with default parameters after the multiple sequence alignment (MSA). The intraspecies and interspecies identities of the 16S rRNA and gyrB gene sequences were compared by the Student’s t-test using Microsoft Excel 2019. Correlation analyses between dDDH values, gyrB, and 16S rRNA gene sequences identities were undertaken by using the “basicTrendline” library in R3.
To obtain core genes of genome sequences used in this study, the pan-genome analysis was performed using the software Bacterial Pan Genomes Analysis Pipeline (BPGA) version 1.3 with default parameters. The maximum-likelihood (ML) tree of the core genomes were reconstructed using the software IQ-TREE version 2.1.2 (Minh et al., 2020) based on the LG + F + R4 model of amino acid substitution with default parameters (Stamatakis, 2014), which was selected by the ModelFinder (Kalyaanamoorthy et al., 2017) according to the Bayesian information criterion (default) (Schwarz, 1978). For the gyrB and 16S rRNA gene sequences, the MSA was performed using the software MAFFT version 6.240 with the FFT-NS-2 algorithm (Katoh and Standley, 2013). And then, the ML trees based on the 16S rRNA and gyrB gene sequences were reconstructed by using the IQ-TREE, respectively, under the TIM2 + F + R2 and TIM + F + I + G4 nucleotide substitution models. Support for the two single-gene phylogenetic trees was inferred by ultrafast bootstrapping with 10,000 replicates (Hoang et al., 2018). The visualization and annotation of the resulting phylogenetic trees were performed using the software MEGA version X (Kumar et al., 2018). Strain Aggregicoccus sp. 17bor-14 was used as an outgroup in all phylogenetic analyses.
To design primers of the gyrB gene for the amplification and sequencing of bacteria within the three genera, the complete gyrB gene sequences from genome sequences were aligned using the DNAMAN. A specific primer set of the gyrB gene was designed using the software Primer Premier version 5.0 (PREMIER Biosoft International, CA, United States). The 31 isolates that belonged to the above three genera based on the preliminary identification of the 16S rRNA gene sequencing (unpublished data) from our group were used for verifying the primer of the gyrB gene. All tested isolates were grown in the VY/2 medium (Reichenbach, 2015) at 28°C for a week. The genomic DNA of each isolate was extracted from fresh cells using the HiPure Bacterial DNA Kit (Magen Biotech Co., Ltd., Guangzhou, China) following the manufacturer’s instructions. The gyrB gene fragment was amplified in a 25 μL PCR mixture composed of 12.5 μL 2 × PCR Master Mix with 3 mmol/L MgCl2 (G-Clone Biotech Co., Ltd.), 0.5 μL each primer (10 mmol/L), 0.5 μL template DNA (ca. 50 ng/μL), and 11 μL deionized water. Gradient PCRs were performed to determine the optimal annealing temperature for the primers pair of the gyrB gene. The PCR reaction was done in a T100TM Thermal Cycler (Bio-Rad, CA, United States) with the following thermal PCR profile: initial denaturation at 94°C for 5 min, 30 cycles of denaturation at 95°C for 30 s, annealing at 63°C for 30 s, and extension at 72°C for 70 s, followed by a final extension at 72°C for 10 min. PCR products were screened by electrophoresis on a 1% agarose gel and sequenced by Suzhou Genewiz Biotechnology Co., Ltd. (Suzhou, China). In this study, sequencing primers were the same as amplification primers. The genomic sequencing, de novo assembly, and quality assessment of representative isolates were determined based on the previously described methods (Liu et al., 2021). The 966-bp gyrB gene sequences identities and dDDH values between representative isolates and their closely related relatives were conducted by the DNAMAN and GGDC, respectively, as mentioned above.
The sequence data generated in this study were deposited in the GenBank database under accession numbers provided in Supplementary Tables 1, 8.
A total of 106 genomes designated by the GenBank database as the genera Myxococcus, Corallococcus, and Pyxidicoccus were obtained in this study, also including that of strain Aggregicoccus sp. 17bor-14 as an outgroup. To ensure the reliability of the subsequent analysis, the quality of all genomes was assessed using the CheckM. Based on the estimation from the CheckM, 94 genomes were to be considered high quality based on more than 95% of completeness and less than 5% of contamination. Meanwhile, for multiple genomes of the same strain with different numbers, only one genome with the highest quality was selected for analysis. Therefore, there were 14 complete and 77 draft genomes used in this study (Table 1). The general features of genomes are summarized in Table 1. The genomic sizes ranged from 8.80 Mbp (Myxococcus sp. AM009) to 13.53 Mbp (Pyxidicoccus fallax DSM 14698T) with an average of 10.10 ± 1.07 Mbp. The genomic DNA G + C contents ranged from 68.74% (“Myxococcus llanfairensis” AM401T, this name originated from “Myxococcus llanfairpwllgwyngyllgogerychwyrndrobwllllantysiliogogogochensis” and was abbreviated to “M. llanfairensis”, the same below) to 70.74% (Corallococcus sp. Z5C101001) with a mean of 69.57 ± 0.59%. These results indicated that bacteria of the three genera above had two distinctive features with large genomes and high genomic DNA G + C contents relative to most bacterial taxa (Figure 1A) (Whitworth and Zwarycz, 2020).
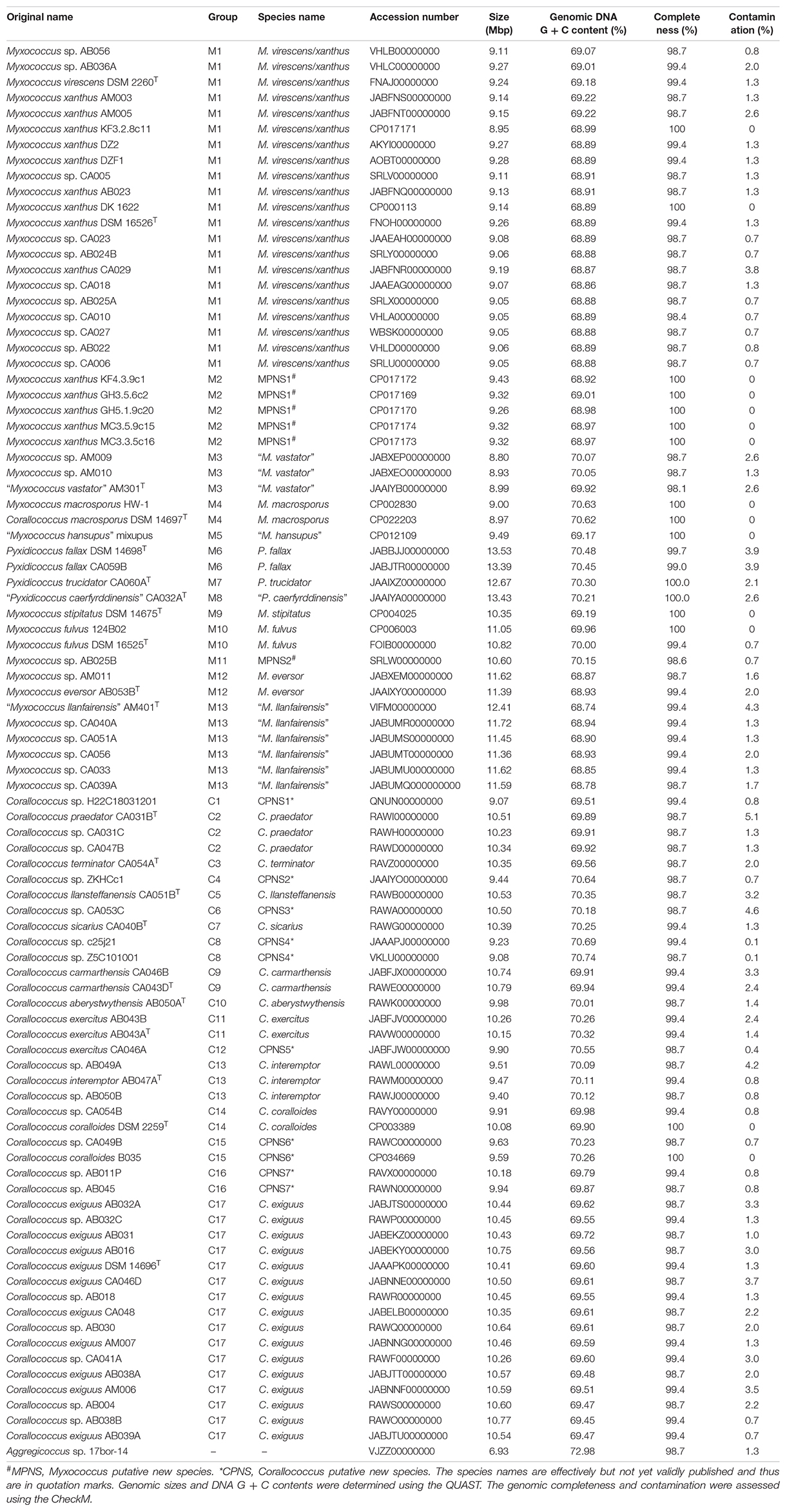
Table 1. The detailed information on genomes used in this study.
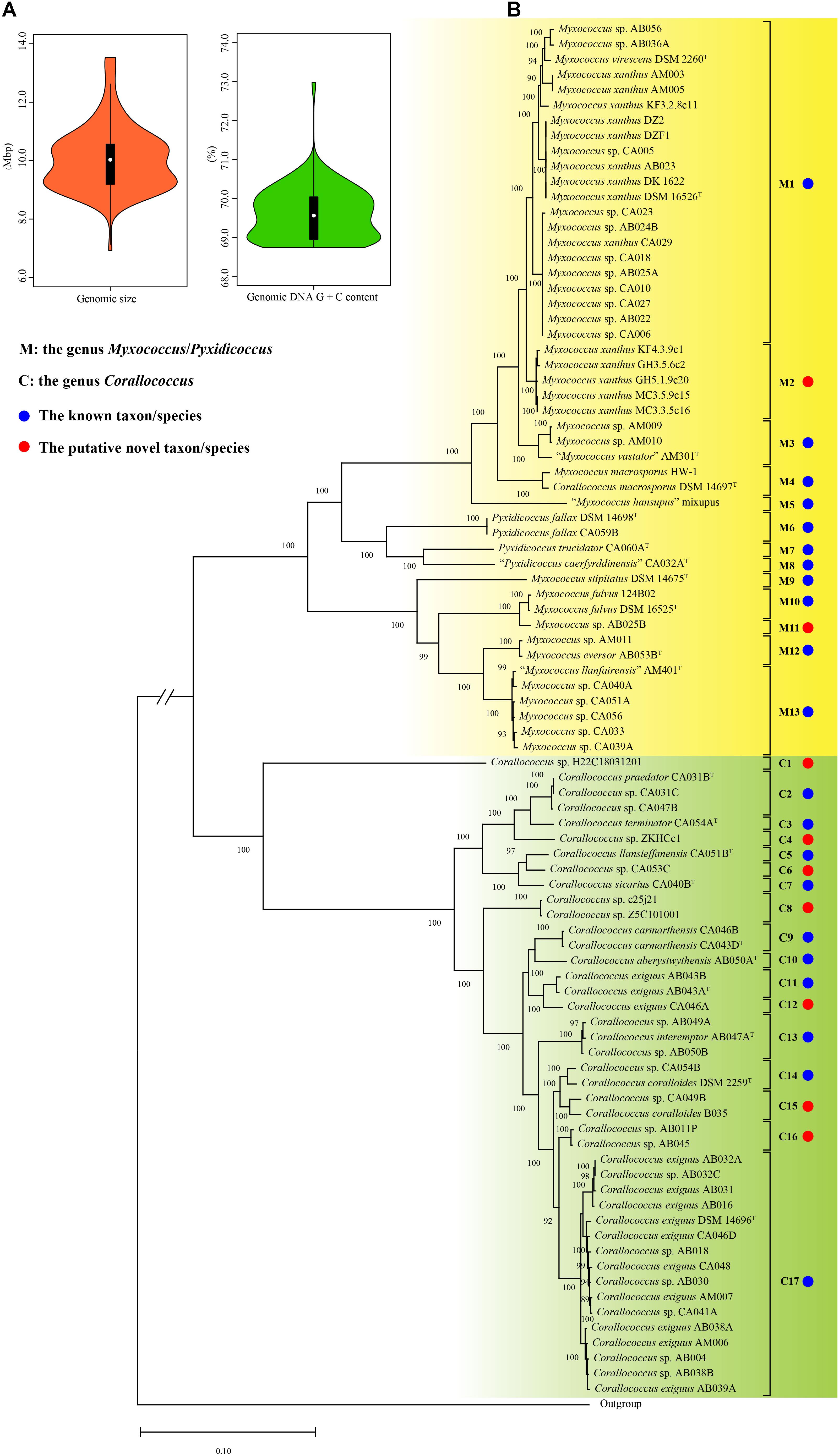
Figure 1. The violin plots of genomic sizes and DNA G + C contents (A), the maximum-likelihood (ML) phylogenetic tree of 90 strains based on 1,887 orthologous protein sequences of 91 genome sequences (B). The ML tree was reconstructed using the IQ-TREE with the LG + F + R4 model. The species names are effectively but not yet validly published and thus are in quotation marks. Strain Aggregicoccus sp. 17bor-14 was used as an outgroup. Bootstrap values great than 80% were shown at branch points. Bar: 0.1 represents the number of substitutions per site. The branch length of the outgroup was cut down to fit the image size and indicated by parallel oblique lines “//”.
A total of 1887 orthologous protein sequences (Supplementary Table 2) from the core genome by the BPGA were used to infer the phylogeny of 90 bacteria within the three genera. As shown in Figure 1B, the ML phylogenomic tree was characterized by high bootstrap values, indicating that the protein sequences selected were reflective of a robust evolutionary relatedness between bacteria. The ML tree showed that bacteria of the three genera were grouped into two distinct groups. Group M was composed of 48 strains, including eleven type strains and 37 non-type strains. In Group M, the bacteria from the two genera Myxococcus and Pyxidicoccus were mixed. Similar to the study by Chambers et al. (2020), the two genera were therefore proposed as a single genus, referring to it hereafter as the genus Myxococcus/Pyxidicoccus. Group C was found to be comprised of ten type strains and 32 non-type strains and matched exactly to the genus Corallococcus. In the phylogenomic tree, the type strain Corallococcus macrosporus DSM 14697T was located in Group M and shared a close relationship with strain Myxococcus macrosporus HW-1. Actually, these studies have proved that strain C. macrosporus DSM 14697T was more closely related to members of the genus Myxococcus than to those of the genus Corallococcus (Lang and Stackebrandt, 2009). But, M. macrosporus is still shown to be a homotypic synonym of C. macrosporus in the LPSN4. From a taxonomic standpoint, it is necessary to clarify in the following study.
The DNA–DNA hybridization (DDH) values have been used continuously for over half a century as the gold standard for prokaryotic species circumscription at the genomic level (Chun et al., 2018) and can be obtained from the GGDC. In contrast with DDH values, various algorithms available for calculating the average nucleotide identity could produce different values and thus did not provide consistent results regarding the conspecificity of isolates (Palmer et al., 2020). In view of this situation, in the current study, the species assignation for all strains was determined only based on the dDDH and phylogenomic analyses and was also used as a criterion in the following analyses of the gyrB and 16S rRNA genes. Based on the 70% DDH threshold, all strains were divided into 30 species (Supplementary Figure 1 and Supplementary Table 3). Group M included 13 species (marked with M1 to M13) corresponding to 11 well-defined species indicated by blue solid circles (the same below) and two putative novel species indicated by red solid circles (the same below). Group C contained 17 species (marked with C1–C17) corresponding to 10 known species and seven putative novel species. Based on the new species assignation, more than one-third of genomes with unresolved or incorrect specific epithets were reclassified into defined species in this study (Table 1). Unexpectedly, the two type strains Myxococcus virescens DSM 2260T and M. xanthus DSM 16526T both belonged to the M1 taxon and shared a 73.1% dDDH above the threshold for bacterial species delineation, indicating that they should be conspecific. In this case, the M1 taxon was preliminarily designated as the species M. virescens/xanthus in this study, as shown in Table 1. As a result, combining with the phylogenomic and dDDH values analyses, a robust phylogeny of the genera Myxococcus, Corallococcus, and Pyxidicoccus and reliable species assignation of their bacteria were conducted and would provide a solid foundation for establishing a new single housekeeping gene-based identification of bacteria within the three genera.
Inspired by previous studies (Stackebrandt and Päuker, 2005; Stackebrandt et al., 2007), the gyrB gene was used as a candidate marker to infer the bacterial phylogeny of the genera Myxococcus, Corallococcus, and Pyxidicoccus in this study. For all 90 strains analyzed, only one copy of the gyrB gene was identified in each genome sequence. The complete gyrB gene sequences varied in size between 2,448 and 2,466 bp. As shown in Figure 2A, similar to the genome-based tree, all strains in the gyrB gene-based tree were also clustered into two distinct groups. At the species level, all strains in the gyrB gene-based tree were further divided into 30 subgroups, matching exactly 30 species from the phylogenomic and dDDH values analyses. Some small differences in tree topologies of the genome- and gyrB-based trees were observed. For example, the two species C9 and C10 were found to be sister taxa in the genome-based tree, while the species C9 was the outer taxon against the species C10 in the gyrB gene-based tree. A similar situation was found for the two species C11 and C12. In more detail, the intraspecific phylogenetic relationships of some strains in the gyrB gene-based tree were different from those in the genome-based tree (Figures 1B, 2A). The intraspecies identities of the gyrB gene sequences ranged from 97.3 to 100% with a mean value of 98.8%, and the interspecies identities ranged from 85.7 to 98.5% with a mean value of 89.5% (Supplementary Figure 2 and Supplementary Table 4). The intraspecies identities of the complete gyrB gene sequences were significantly higher than and interspecies identities (t-test, p < 0.001, Supplementary Figure 2). These results indicated that the gyrB gene can provide reliable evolutionary relationships and species assignation of the three genera and thus seems to be a better alternative as a powerful molecular marker to infer their phylogeny.
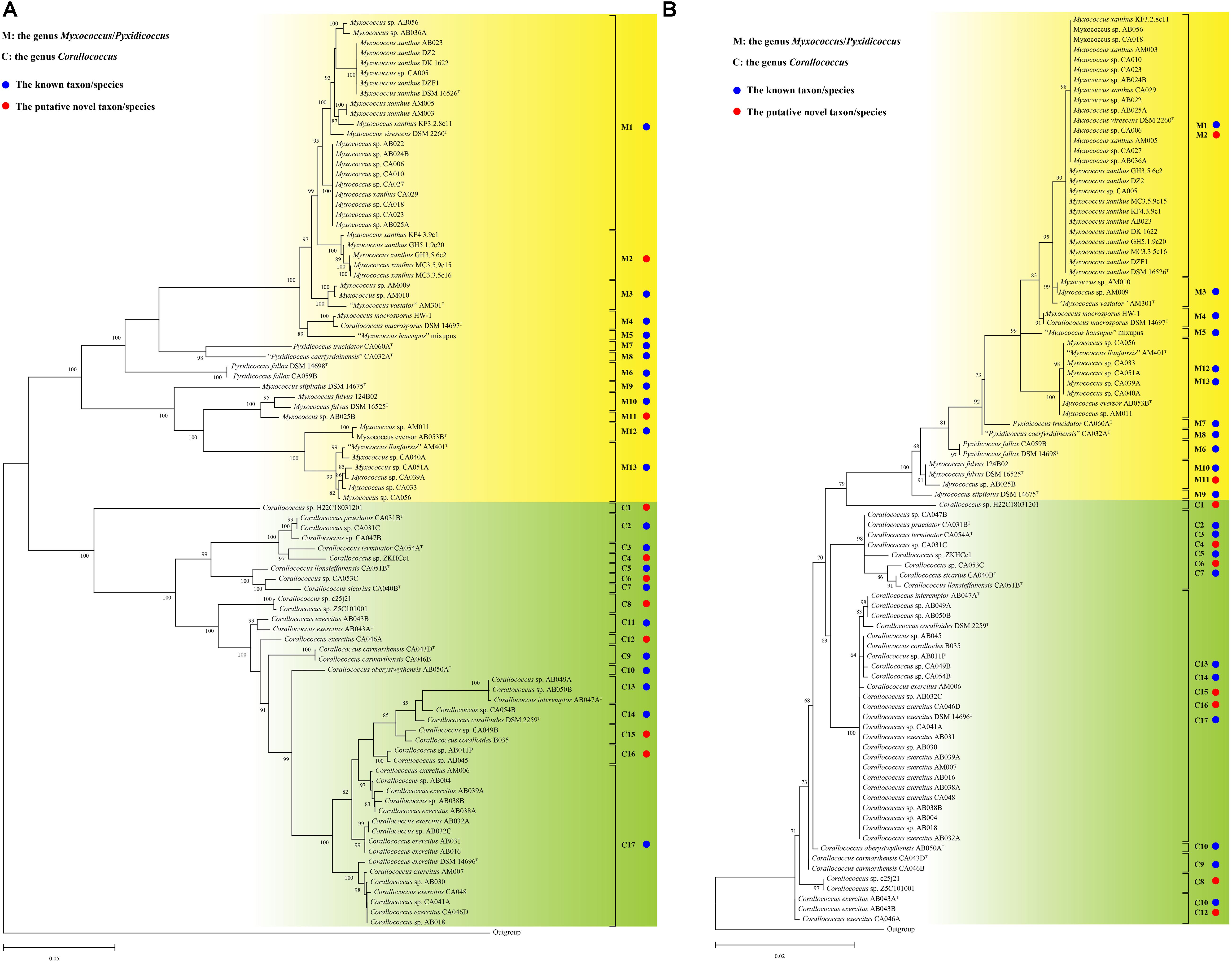
Figure 2. The ML phylogenetic trees based on complete gyrB (A) and 16S rRNA gene (B) sequences. The ML trees of the gyrB and 16S rRNA gene were reconstructed using the IQ-TREE with the TIM + F + I + G4 and TIM2 + F + R2 models, respectively. The species names are effectively but not yet validly published and thus are in quotation marks. Strain Aggregicoccus sp. 17bor-14 was used as an outgroup. Bootstrap values great than 80% in gyrB-based tree and 50% in 16S rRNA-based tree were shown at branch points. Bar: 0.05/0.02 represents the number of substitutions per site.
The lengths of complete 16S rRNA gene sequences from the respective genome ranged from 1,536 to 1,538 bp. Among all genomes, each complete genome contained the same three or four copies of the 16S rRNA gene and each high-quality draft genome contained only one copy (data not shown). Therefore, a complete 16S rRNA gene sequence in each genome was obtained for the phylogenetic analysis. In the phylogenetic tree based on 16S rRNA gene sequences, many species that were well differentiated based on genome and gyrB gene sequences were clustered together (Figure 2B). For example, the two species M1 and M2 were on the same branch. Similar situations were observed for several other species, such as species M10–M11, M12–M13, C2–C7, and C13–C17. The phylogenetic relationships of 90 strains in the 16S rRNA gene-based tree were significantly different from those in the genome- and gyrB gene-based trees. One of the striking differences was that the five species C13–C17 clustered together in 16S rRNA gene tree, but they formed five different clusters/species in the genome- and gyrB gene-based trees. More obviously, the phylogenetic tree of 16S rRNA gene sequences presented much shorter branch lengths and lower bootstrap values than those of genome- and gyrB-based trees. As shown in Supplementary Figure 2, the intraspecies identities of 16S rRNA gene sequences ranged from 99.7 to 100% with a mean value of 99.97%, and the interspecies identities ranged from 97.2 to 100% with a mean value of 98.4% (Supplementary Table 5). The intraspecies identities of the 16S rRNA gene sequences were significantly higher than the interspecies identities (t-test, p < 0.001, Supplementary Figure 2). The same significance levels of identities of the gyrB and 16S rRNA gene sequences did not match their distinct phylogenetics. The inconsistency was mainly due to over-representative identities between strains of some subgroups capable of being distinguished by the 16S rRNA gene. In short, the 16S rRNA gene was inappropriate for accurate identification of bacteria within of the closely related three genera due to its poor resolution.
The above-described in silico analyses demonstrated that the gyrB gene is a powerful phylogenetic marker to differentiate bacteria of the closely related three genera at the species level. Therefore, a PCR-based method that could be widely used by researchers working with the three genera would be extremely valuable as a simple tool in the classification of isolates.
To establish the gyrB gene-based PCR method, amplification primers of this gene were first designed based on 90 complete gyrB gene sequences from their genomes. The pair of primers covering a 1,079 bp fragment without any insertions and deletions (corresponding to the position 348–1,426 within the complete gyrB gene sequence from type strain Myxococcus fulvus DSM 16525T with the locus_tag = “SAMN05443572_101133”) was proposed: Myxoco_gyrBF (5′-AGCAAGTTCGGCAACG G-3′) and Myxoco_gyrBR (5′-AGCATCTTCTCGAAGCG-3′). Temperature gradients from 56 to 66°C were performed for confirming an optimum annealing temperature in PCR. The optimal annealing temperature was chosen at 63°C based on the detection of the brightness and uniqueness of PCR products/brands in agarose gel electrophoresis (Supplementary Figure 3). Logically, strains isolated from different habitats usually have more genotypes, and the use of more genotypic myxobacteria is conducive to better validate this method. Therefore, in this study, 31 bacteria isolated from multiple sources (Supplementary Table 1) by our group were related to the three genera Myxococcus, Corallococcus, and Pyxidicoccus based on the preliminary 16S rRNA gene sequencing analysis (unpublished data) and were used as target strains. All tested strains produced the expected PCR fragments of the gyrB gene and PCR products were sequenced using the amplification primers. After the MSA of the gyrB gene, all ambiguous bases at both ends were trimmed and the final sequence length for comparison was 966 bp. Phylogenetic tree based on 966-bp gyrB gene sequences from 31 tested isolates and 90 strains were reconstructed, as shown in Supplementary Figure 4. Among all tested isolates, 16 were distributed between the above four subgroups M1, M11, C8, and C9; 15 formed eight independent subgroups different from the above 30 ones obtained by the genome analysis. The comparison of the 966-bp gyrB gene sequences from 90 genomes showed that the intraspecies identities ranged from 97.6 to 100% with a mean value of 98.5%, and the interspecies identities ranged from 88.5 to 99.1% with a mean value of 90.7% (Supplementary Figure 2 and Supplementary Table 6). And apparently, an overlapped area between intraspecies and interspecies identities was from 97.6 to 99.1% (Supplementary Figure 2). Interestingly, almost all identities in the overlapping area came from the two subgroups M1 and C17 (Supplementary Tables 6, 7). But, as shown in Supplementary Figure 4, the accurate identification of these strains that were in the identity overlapped area was resolved using the phylogenetic analysis of the 966-bp gyrB gene. For example, strains AB036A and DZF1 shared a 97.7% identity in the overlapping area, but they could cluster into the subgroup M1. The identities of the complete gyrB gene sequences and the 966-bp ones were highly linear (R2 = 0.9998, Figure 3A), suggesting that their ability to infer phylogenetic relationships of bacteria within the three genera was almost equal. In addition, the intraspecies identities of the 966-bp gyrB gene sequences were statistically significantly higher than the interspecies identities (t-test, p < 0.001, Supplementary Figure 2). These results indicated that the PCR primers designed in this study were suitable for amplifying and sequencing the target region of the gyrB gene for bacteria of the three genera.
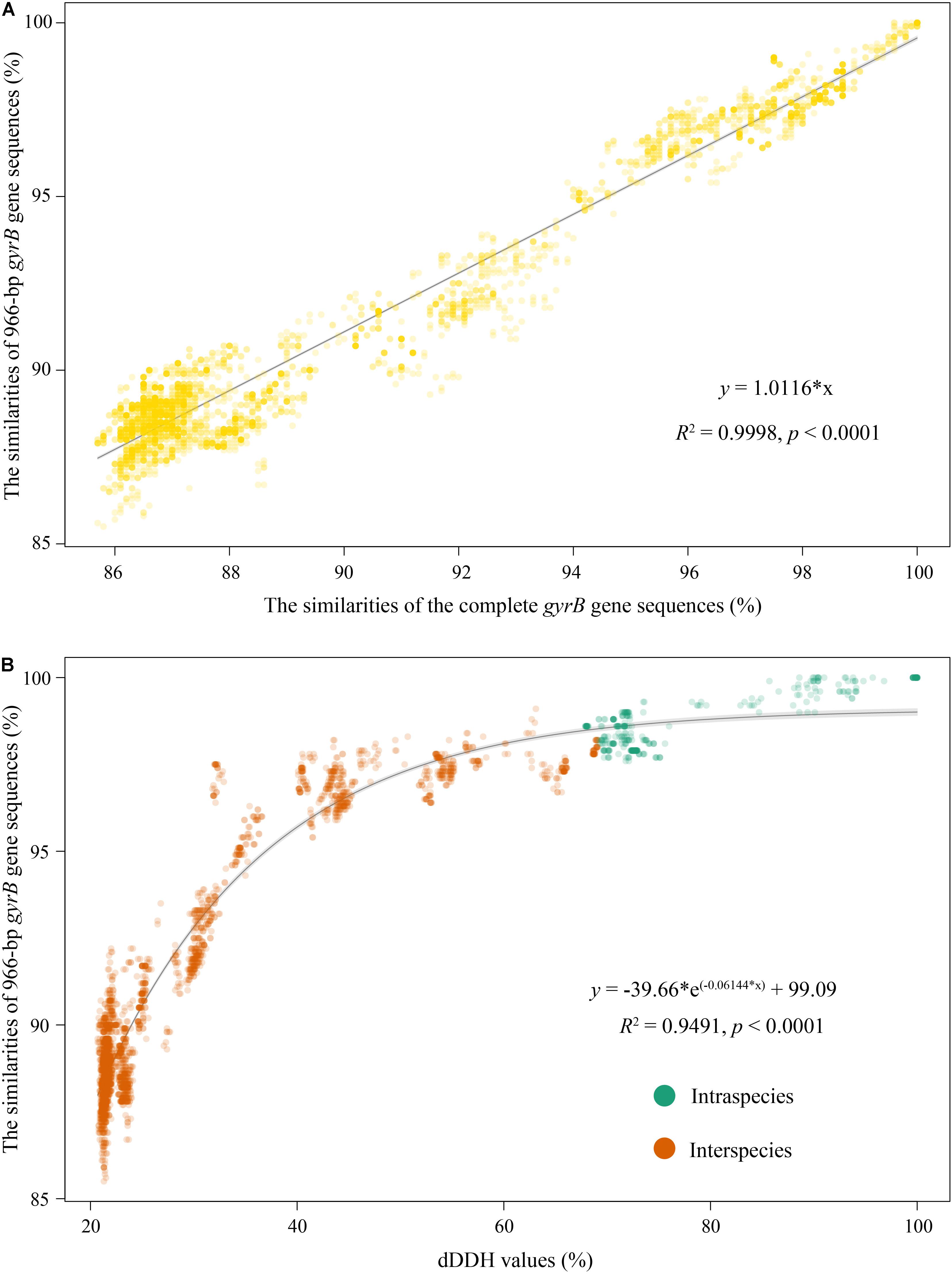
Figure 3. Correlation analyses of identities between the complete gyrB gene sequences and 966-bp gyrB ones (A), and the dDDH values and the 966-bp gyrB gene sequence identities (B). Correlation analyses of the former and latter were, respectively, simulated using the Excel linear regression and “exp3P” model of the “basicTrendline” library in R.
The threshold of the gyrB gene for species delineation is an important parameter in the identification of strains with the genera Myxococcus, Corallococcus, and Pyxidicoccus. The correlation between dDDH values and marker gene sequence identities shows the accuracy with which a marker gene reflects the genome variation rate and indicates how precisely a marker gene predicts the phylogenetic relationship of genome sequences between strains. Consequently, we attempted to determine the identity threshold of the gyrB gene for species delineation using the correlation analysis. The dDDH values were highly correlated with identities of the complete gyrB gene sequences (R2 = 0.9760 and p < 0.0001, Supplementary Figure 5) and the 966-bp ones (R2 = 0.9491 and p < 0.0001, Figure 3B). This indicated that the change rate in nucleotide sequences of the complete gyrB gene was consistent with that of the 966-bp ones for bacteria of the three genera. Based on respective regression equations, 70% dDDH was equivalent to 98.4% of the complete gyrB gene or 98.6% of the 966-bp gyrB gene, both of which were used as thresholds for species delineation of bacteria within the three genera.
Applying the 98.6% threshold of the 966-bp gyrB gene, 16 isolates were assigned into the four known species M1, M11, C8, and C9, while the other 15 isolates were divided into the eight putative novel species. To further confirm these new species, the genomic sequences of the five representative isolates were determined in this study. Based on the estimation from the CheckM, the five genome sequences were also of high quality by comparing the standard above (Supplementary Table 8). The gyrB gene sequences of the five isolates extracted from each genome using a local BLAST search were the same as those obtained by the PCR, further confirming the authenticity of the final genome assembly. As shown in Supplementary Table 9, the dDDH values between strains AB025B, XM-1-1-1, and AS-1-15 were above the 70% threshold for bacterial species delineation, while those between each strain from the species M11 and other strains were below this threshold, indicating that M11 should represent a novel genospecies. The dDDH value between isolates AS-1-6 and AS-1-12 was 90.2%, demonstrating that they should belong to the same genospecies. The dDDH values between each isolate (AS-1-6 and AS-1-12) and other strains, RHSTA-1-4 and other strains were below the 70% threshold. The dDDH values analysis of these five representative isolates strengthened the accuracy of species assignation based on the 966-bp gyrB gene analysis. As a result, the 98.6% identity of the 966-bp gyrB gene can be used as a reliable threshold for rapidly and accurately identifying isolates of the above three most closely related genera to the species level in practice. In addition, for these potential new taxa, we will endeavor to determine phenotypic and genotypic characteristics to describe them as novel species in the future.
The current phylogenomic study demonstrated that the gyrB gene sequencing is a powerful molecular tool for identifying and classifying bacteria of the closely related genera Myxococcus, Corallococcus, and Pyxidicoccus at the species level. The analyses of the complete and 966-bp gyrB gene sequences showed that this marker provides accurate phylogenetic relationships of these bacteria consistent with genomic analysis and has greater discriminatory power than the widely used 16S rRNA gene, in particular when analyzing closely related strains.
The 16S rRNA gene has been a mainstay for the classification and identification of members of myxobacteria for decades (Shimkets and Woese, 1992; Garcia et al., 2010; Mohr, 2018). However, the 16S rRNA gene (which represents only 0.03% of an average 5-Mbp prokaryotic genome) shows a limited resolution for highly related strains at the interspecies and even intergeneric levels, making reliable species and genus level identifications not possible. In the last decade, with the advance of whole-genome sequencing and bioinformatic tool development, many genome-based methods have been developed and applied for microbial taxonomy, for example, the pan-genome analysis (Chambers et al., 2020; Livingstone et al., 2020) and the dDDH analysis (Auch et al., 2010), both of which have also been used to distinguish closely related bacteria in this study. However, considering that the whole-genome sequencing and analysis for a large number of isolates was a costly, time- and computer resource-consuming process and required bioinformatic skills for data processing and the effective interpretation of results, comparison of the DNA sequences of protein-encoding genes is an alternative approach to the analysis of whole-genome relatedness. Therefore, multiple housekeeping genes have been used as group-level taxonomic markers for some myxobacterial taxa (Stackebrandt and Päuker, 2005; Stackebrandt et al., 2007). But the use of these marker genes was only used for one or several species of the genera Myxococcus and Corallococcus, and was also a lack of group- or species-specific primers, resulting in negative amplification reactions for some isolates. In this study, a simple and accurate gyrB gene-based PCR method extending to the three genera and with specific primers was proposed and can bridge the gap between 16S rRNA gene sequence and genome analyses for taxonomic affiliation and phylogenetic relationships of bacteria within the three closely related genera above.
Species are regarded as the fundamental units in the taxonomy of bacteria and archaea, and the standard of species demarcation is an important parameter for taxonomic studies (Rosselló-Móra and Amann, 2015). For the myxobacteria, the assignation of strains to known or novel species is often performed using the morphology of vegetative cells, swarming colonies, fruiting bodies, and myxospores (Garcia and Müller, 2014). But, due to the lack of clear and quantifiable standards for species definition and over-reliance on personal experience and skills, these morphological characteristics seem somewhat subjective and unsuitable as a practical taxonomic criterion. Therefore, single-locus nucleotide-based and genome-based approaches have been applied for the myxobacterial taxonomy, such as the 16S rRNA gene (Chambers et al., 2020), a single housekeeping gene (Stackebrandt et al., 2007), and core genes (Livingstone et al., 2020). Among these approaches, the traditional 97% (Stackebrandt and Goebel, 1994) or updated 98.65% (Kim et al., 2014) of 16S rRNA gene sequence identities are not suitable as the thresholds for species delineation of highly related strains such as those from the genera Myxococcus, Corallococcus, and Pyxidicoccus, and previous studies did not provide thresholds of these molecular markers for species delineation (Stackebrandt and Päuker, 2005; Stackebrandt et al., 2007). To address the issue, the threshold for species delineation of the three genera in the current study was proposed through phylogenomics and the correlation analysis of dDDH values and the gyrB gene identities, and will greatly improve its practicability and comparability. The use of the gyrB gene threshold for the accurate and rapid identification of large batches of environmental isolates at the species level is advantageous in terms of resolution but also from a practical and financial point of view. Moreover, with the dramatic increase of the gyrB gene sequences in public databases, we anticipate that the gyrB gene will also become an interesting target to characterize the in situ diversity and abundance of the three genera and to guide further isolation endeavors from environmental samples.
Myxobacteria represents a highly diverse and ubiquitous group. As the most frequently isolated myxobacteria, the accurate, affordable, and fast identification of bacteria within the three genera Myxococcus, Corallococcus, and Pyxidicoccus is a major challenge owing to the limited discriminative power of the 16S rRNA gene. The current study has proposed a useful molecular identification tool using the gyrB gene sequencing as an alternative to the traditional 16S rRNA gene. Following this study, some potential important topics, but not limited to these, will be interesting to be determined by us and/or other researchers: (i) improving and then extending the gyrB-based method for strain identification to other taxa or even the whole myxobacteria, rather than just concentrating on the genera Myxococcus, Corallococcus, and Pyxidicoccus; (ii) establishing a curated DNA sequence database and a web-based tool for gyrB-based identification of myxobacterial isolates for much broader and easier use of this genetic marker in worldwide researchers; (iii) developing a gyrB amplicon sequencing protocol compatible with the high-throughput sequencing platform for profiling in situ myxobacterial community structure, species diversity, and temporal distribution from environmental samples; (iv) clarifying the taxonomic status of the genus Pyxidicoccus and the species C. macrosporus and identifying the potential new taxa obtained from this study using the polyphasic taxonomic approach in the subsequent study.
In this report, we have contributed the framework of phylogenetic relationships of bacteria within the genera Myxococcus, Corallococcus, and Pyxidicoccus, and determines the well-suited gyrB gene for species assignation. We believe that the discovery of the phylogenetic power of the gyrB gene and the establishment of a PCR method that can be used in amplification and sequencing of the gene is of general interest, whether for use alone or together with the genome-based analysis. This research also provides a paradigm for selecting and determining a single molecular marker for the simple and reliable species delineation and phylogenetic inference of other bacterial taxa.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
YL and HZ: conceptualization and project administration. YL: data curation, software, visualization, and writing – original draft. YL and TP: formal Analysis and methodology. XZ, XD, M-RD, and HZ: funding acquisition. YL, TP, SY, JD, XZ, and XD: investigation. HZ: resources and supervision. YL, TP, and QY: validation. YL, QY, and HZ: writing – review and editing. All authors contributed to the article and approved the submitted version.
This work was jointly supported by the Key-Area Research and Development Program of Guangdong Province (2018B020205003), the National Natural Science Fund of China (32000007), the GDAS’ Project of Science and Technology Development (2021GDASYL-20210103020), and the Science and Technology Plan Project of Guangdong Province (2019B030316017).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.763359/full#supplementary-material
Auch, A. F., von Jan, M., Klenk, H.-P., and Göker, M. (2010). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Bull, C. T., Shetty, K. G., and Subbarao, K. V. (2002). Interactions between myxobacteria, plant pathogenic fungi, and biocontrol agents. Plant Dis. 86, 889–896. doi: 10.1094/pdis.2002.86.8.889
Chambers, J., Sparks, N., Sydney, N., Livingstone, P. G., Cookson, A. R., and Whitworth, D. E. (2020). Comparative genomics and pan-genomics of the Myxococcaceae, including a description of five novel species: Myxococcus eversor sp. nov., Myxococcus llanfairpwllgwyngyllgogerychwyrndrobwllllantysiliogogogochensis sp. nov., Myxococcus vastator sp. nov., Pyxidicoccus caerfyrddinensis sp. nov., and Pyxidicoccus trucidator sp. nov. Genome Biol. Evol. 12, 2289–2302. doi: 10.1093/gbe/evaa212
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chun, J., Oren, A., Ventosa, A., Christensen, H., Arahal, D. R., da Costa, M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. doi: 10.1099/ijsem.0.002516
Dawid, W. (2000). Biology and global distribution of myxobacteria in soils. FEMS Microbiol. Rev. 24, 403–427. doi: 10.1111/j.1574-6976.2000.tb00548.x
Garcia, R., and Müller, R. (2014). “The Family Myxococcaceae” in The Prokaryotes: Deltaproteobacteria and Epsilonproteobacteria. (eds) E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson. (Berlin: Springer). 191–212.
Garcia, R., Gerth, K., Stadler, M., Dogma, I. J. Jr., and Müller, R. (2010). Expanded phylogeny of myxobacteria and evidence for cultivation of the ‘unculturables’. Mol. Phylogenet. Evol. 57, 878–887. doi: 10.1016/j.ympev.2010.08.028
Gregory, K., Salvador, L. A., Akbar, S., Adaikpoh, B. I., and Stevens, D. C. (2019). Survey of biosynthetic gene clusters from sequenced myxobacteria reveals unexplored biosynthetic potential. Microorganisms 7:181. doi: 10.3390/microorganisms7060181
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Herrmann, J., Fayad, A. A., and Müller, R. (2017). Natural products from myxobacteria: novel metabolites and bioactivities. Nat. Prod. Rep. 34, 135–160. doi: 10.1039/c6np00106h
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q., and Vinh, L. S. (2018). UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, M., Oh, H.-S., Park, S.-C., and Chun, J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351. doi: 10.1099/ijs.0.059774-0
Klemetsen, T., Karlsen, C. R., and Willassen, N. P. (2021). Phylogenetic revision of the genus Aliivibrio: intra- and inter-species variance among clusters suggest a wider diversity of species. Front. Microbiol. 12:626759. doi: 10.3389/fmicb.2021.626759
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lang, E., and Stackebrandt, E. (2009). Emended descriptions of the genera Myxococcus and Corallococcus, typification of the species Myxococcus stipitatus and Myxococcus macrosporus and a proposal that they be represented by neotype strains. Request for an Opinion. Int. J. Syst. Evol. Microbiol. 59, 2122–2128. doi: 10.1099/ijs.0.003566-0
Li, Z., Ye, X., Liu, M., Xia, C., Zhang, L., Luo, X., et al. (2019). A novel outer membrane beta-1,6-glucanase is deployed in the predation of fungi by myxobacteria. ISME J. 3, 2223–2235. doi: 10.1038/s41396-019-0424-x
Liu, Y., Pei, T., Du, J., Huang, H., Deng, M.-R., and Zhu, H. (2021). Comparative genomic analysis of the genus Novosphingobium and the description of two novel species Novosphingobium aerophilum sp. nov. and Novosphingobium jiangmenense sp. nov. Syst. Appl. Microbiol. 44:126202. doi: 10.1016/j.syapm.2021.126202
Liu, Y., Yao, Q., and Zhu, H. (2019). Meta-16S rRNA gene phylogenetic reconstruction reveals the astonishing diversity of cosmopolitan myxobacteria. Microorganisms 7:551. doi: 10.3390/microorganisms7110551
Livingstone, P. G., Ingleby, O., Girdwood, S., Cookson, A. R., Morphew, R. M., and Whitworth, D. E. (2020). Predatory organisms with untapped biosynthetic potential: descriptions of novel Corallococcus species C. aberystwythensis sp. nov., C. carmarthensis sp. nov., C. exercitus sp. nov., C. interemptor sp. nov., C. llansteffanensis sp. nov., C. praedator sp. nov., C. sicarius sp. nov., and C. terminator sp. nov. Appl. Environ. Microbiol. 86, e1931–1919. doi: 10.1128/AEM.01931-19
Livingstone, P. G., Morphew, R. M., and Whitworth, D. E. (2017). Myxobacteria are able to prey broadly upon clinically-relevant pathogens, exhibiting a prey range which cannot be explained by phylogeny. Front. Microbiol. 8:1593. doi: 10.3389/fmicb.2017.01593
Martínez-Hidalgo, P., Flores-Félix, J. D., Velázquez, E., Brau, L., Trujillo, M. E., and Martínez-Molina, E. (2020). High taxonomic diversity of Micromonospora strains isolated from Medicago sativa nodules in Western Spain and Australia. Syst. Appl. Microbiol. 43:126043. doi: 10.1016/j.syapm.2019.126043
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Mohr, K. I. (2018). Diversity of myxobacteria-we only see the tip of the iceberg. Microorganisms 6:84. doi: 10.3390/microorganisms6030084
Ogier, J. C., Pages, S., Galan, M., Barret, M., and Gaudriault, S. (2019). rpoB, a promising marker for analyzing the diversity of bacterial communities by amplicon sequencing. BMC Microbiol. 19:171. doi: 10.1186/s12866-019-1546-z
Palmer, M., Steenkamp, E. T., Blom, J., Hedlund, B. P., and Venter, S. N. (2020). All ANIs are not created equal: implications for prokaryotic species boundaries and integration of ANIs into polyphasic taxonomy. Int. J. Syst. Evol. Microbiol. 70, 2937–2948. doi: 10.1099/ijsem.0.004124
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Poirier, S., Rue, O., Peguilhan, R., Coeuret, G., Zagorec, M., Champomier-Verges, M. C., et al. (2018). Deciphering intra-species bacterial diversity of meat and seafood spoilage microbiota using gyrB amplicon sequencing: A comparative analysis with 16S rDNA V3-V4 amplicon sequencing. PLoS One 13:e0204629. doi: 10.1371/journal.pone.0204629
Rosselló-Móra, R., and Amann, R. (2015). Past and future species definitions for Bacteria and Archaea. Syst. Appl. Microbiol. 38, 209–216. doi: 10.1016/j.syapm.2015.02.001
Santos, S. R., and Ochman, H. (2004). Identification and phylogenetic sorting of bacterial lineages with universally conserved genes and proteins. Environ. Microbiol. 6, 754–759. doi: 10.1111/j.1462-2920.2004.00617.x
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shimkets, L., and Woese, C. R. (1992). A phylogenetic analysis of the myxobacteria: basis for their classification. Proc. Natl. Acad. Sci. U. S. A. 89, 9459–9463. doi: 10.1073/pnas.89.20.9459
Stackebrandt, E., and Goebel, B. M. (1994). Taxonomic note: a place for DNA-DNA reassociation and 16s rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 44, 846–849. doi: 10.1099/00207713-44-4-846
Stackebrandt, E., and Päuker, O. (2005). Gene sequence heterogeneity of Corallococcus coralloides strains isolated from geographically diverse locations. Environ. Microbiol. 7, 1017–1023. doi: 10.1111/j.1462-2920.2005.00773.x
Stackebrandt, E., Päuker, O., Steiner, U., Schumann, P., Sträubler, B., Heibei, S., et al. (2007). Taxonomic characterization of members of the genus Corallococcus: molecular divergence versus phenotypic coherency. Syst. Appl. Microbiol. 30, 109–118. doi: 10.1016/j.syapm.2006.03.002
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Vos, M., and Velicer, G. J. (2006). Genetic population structure of the soil bacterium Myxococcus xanthus at the centimeter scale. Appl. Environ. Microbiol. 72, 3615–3625. doi: 10.1128/AEM.72.5.3615-3625.2006
Vos, M., Quince, C., Pijl, A. S., de Hollander, M., and Kowalchuk, G. A. (2012). A comparison of rpoB and 16S rRNA as markers in pyrosequencing studies of bacterial diversity. PLoS One 7:e30600. doi: 10.1371/journal.pone.0030600
Waite, D. W., Chuvochina, M., Pelikan, C., Parks, D. H., Yilmaz, P., Wagner, M., et al. (2020). Proposal to reclassify the proteobacterial classes Deltaproteobacteria and Oligoflexia, and the phylum Thermodesulfobacteria into four phyla reflecting major functional capabilities. Int. J. Syst. Evol. Microbiol. 70, 5972–6016. doi: 10.1099/ijsem.0.004213
Wang, C., Lv, Y., Li, A., Yao, Q., Feng, G., and Zhu, H. (2020). Culture-dependent and -independent methods revealed an abundant myxobacterial community shaped by other bacteria and pH in Dinghushan acidic soils. PLoS One 15:e0238769. doi: 10.1371/journal.pone.0238769
Whitworth, D. E., and Zwarycz, A. (2020). A genomic survey of signalling in the Myxococcaceae. Microorganisms 8:1739. doi: 10.3390/microorganisms8111739
Zhang, X., Yao, Q., Cai, Z., Xie, X., and Zhu, H. (2013). Isolation and identification of myxobacteria from saline-alkaline soils in Xinjiang. China. PLoS One 8:e70466. doi: 10.1371/journal.pone.0070466
Keywords: phylogenomic analysis, novel marker gyrB gene, species identification, differentiate, the most closely related genera
Citation: Liu Y, Pei T, Yi S, Du J, Zhang X, Deng X, Yao Q, Deng M-R and Zhu H (2021) Phylogenomic Analysis Substantiates the gyrB Gene as a Powerful Molecular Marker to Efficiently Differentiate the Most Closely Related Genera Myxococcus, Corallococcus, and Pyxidicoccus. Front. Microbiol. 12:763359. doi: 10.3389/fmicb.2021.763359
Received: 23 August 2021; Accepted: 22 September 2021;
Published: 11 October 2021.
Edited by:
Iain Sutcliffe, Northumbria University, United KingdomReviewed by:
David Cole Stevens, University of Mississippi, United StatesCopyright © 2021 Liu, Pei, Yi, Du, Zhang, Deng, Yao, Deng and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Honghui Zhu, emh1aGhAZ2RpbS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.