 Yang Yu
Yang Yu Hao Tan
Hao Tan Tianhai Liu1,2
Tianhai Liu1,2- 1National-Local Joint Engineering Laboratory of Breeding and Cultivation of Edible and Medicinal Fungi, Institute of Agricultural Resources and Environment, Sichuan Academy of Agricultural Sciences, Chengdu, China
- 2National Observing and Experimental Station of Agricultural Microbiology, Ministry of Agriculture and Rural Affairs, Chengdu, China
- 3School of Bioengineering, Jiangnan University, Wuxi, China
Morels (Morchella spp.) are economically important mushrooms cultivated in many countries. However, their production and quality are hindered by white mold disease because of Paecilomyces penicillatus infection. In this study, we aimed to understand the genetic mechanisms of interactions between P. penicillatus and Morchella. M. sextelata, the most prevalent species of Morchella in China, was inoculated with P. penicillatus; then, the expression profiles of both fungi were determined simultaneously at 3 and 6 days post-inoculation (dpi) using a dual RNA-Seq approach. A total of 460 and 313 differentially expressed genes (DEGs) were identified in P. penicillatus and M. sextelata, respectively. The CAZymes of β-glucanases and mannanases, as well as subtilase family, were upregulated in P. penicillatus, which might be involved in the degradation of M. sextelata cell walls. Chitin recognition protein, caffeine-induced death protein, and putative apoptosis-inducing protein were upregulated, while cyclin was downregulated in infected M. sextelata. This indicates that P. penicillatus could trigger programmed cell death in M. sextelata after infection. Laccase-2, tyrosinases, and cytochrome P450s were also upregulated in M. sextelata. The increased expression levels of these genes suggest that M. sextelata could detoxify the P. penicillatus toxins and also form a melanin barrier against P. penicillatus invasion. The potential pathogenic mechanisms of P. penicillatus on M. sextelata and the defense mechanisms of M. sextelata against P. penicillatus were well described.
Introduction
Morels (Morchella spp.), members of the Ascomycetes, Pezizomycetes, Pezizales, Morchellaceae, and Morchella, are commercially important edible mushrooms that are consumed throughout the world (Du et al., 2012). After more than 100years of trials, the commercial cultivation of Morchella in the field was successful (Ower et al., 1986; Dahlstrom et al., 2000; Masaphy, 2010). Since 2012, the industry that cultivates Morchella in China, with Sichuan as a typical example, developed rapidly (Peng et al., 2016). The cultivation area of morels in China has expanded from 200ha in 2012 to 10,000ha in 2020 (Zhao, 2021). The considerable economic benefits of morel cultivation have attracted many farmers and have become their main source of income. The Morchella species currently cultivated in China include M. importuna, M. sextelata, and M. septimelata (Liu et al., 2018). Among them, M. sextelata comprises >90% of the total area of Morchella cultivation (Deng et al., 2021). However, the white mold disease (WMD) caused by Paecilomyces penicillatus (Ascomycetes) seriously harms Morchella (He et al., 2017). WMD outbreak spreads quickly, infecting a large area. In severe cases, 60–80% of Morchella are infected in China, resulting in a decline in their production and commodity value (Chen et al., 2017; He et al., 2017). In addition, a high proportion of P. penicillatus in soil microbial community could inhibit the formation of f Morchella fruiting bodies (Tan et al., 2021a). A large number of farmers suffer financial hardship owing to WMD. Therefore, it is extremely urgent to prevent and control WMD.
Edible mushrooms are primarily cultivated and managed indoors. Therefore, the prevention and control of edible mushroom diseases rely on environmental controls during cultivation, including the control of temperature, light, water, and air, as well as the use of physical and chemical sterilization methods (Gea et al., 2021). Morchella is artificially cultivated in the field (Liu et al., 2018). Hence, the common strategies to prevent and control diseases of edible mushrooms are not effective for the control of Morchella diseases, such as WMD. Similar to other major crops, breeding resistant varieties are an effective way to control mushroom diseases (Kage et al., 2016). Several studies have addressed the immune response of mushrooms to mycoparasites, although this area of research is in its infancy. The activity of laccase, particularly encoded by lcc2 gene, was found to contribute to the metabolism of toxin production in Trichoderma aggressivum and enhanced the resistance to green mold disease in Agaricus bisporus (Sjaarda et al., 2015). A total of 17 simple sequence repeat markers associated with resistance to Mycogone perniciosa in A. bisporus have been established (Fu et al., 2016). A quantitative trait loci analysis was used to identify the locations, numbers, and effects of genomic regions associated with resistance to Lecanicillium fungicola in A. bisporus, and four traits related to resistance were analyzed (Foulongne-Oriol et al., 2012). Further research and data mining will help design classical breeding and genetic modification schemes, thus, producing resistant varieties.
Recently, the genome-wide profiles of several species of Morchella, including M. importuna, M. sextelata, and M. septimelata, as well as that of the pathogen P. penicillatus, were reported (Wingfield et al., 2018; Mei et al., 2019; Tan et al., 2019; Wang et al., 2020). P. penicillatus was found to harbor many CAZymes, particularly clusters of chitinase genes, which could be related to its pathogenicity (Wang et al., 2020). Cheng et al. (2021) reported the regulatory mechanism of interaction between P. penicillatus and M. importuna. However, the lack of a control group of M. importuna at the same developmental stage complicated the conclusions from that study. To date, the genetic and pathogenic mechanisms that underlie the interaction between P. penicillatus and Morchella remain unclear, which makes it difficult to efficiently breed resistant varieties and develop specific biological or chemical methods to control WMD in Morchella. In this study, M. sextelata, the most prevalent species of Morchella in China, was selected as the research object and artificially inoculated with P. penicillatus to develop WMD in the field. The transcriptional changes of P. penicillatus in different infection stages were analyzed. The differences in transcriptional changes were compared between different growth stages of infected and healthy M. sextelata. We aimed to profile the infection mechanism of P. penicillatus, the response mechanism of M. sextelata to P. penicillatus infection, and the candidate defense genes of M. sextelata that protect against P. penicillatus. This will provide theoretical support for the effective control of WMD and reduce the economic risk caused by WMD in the cultivation of Morchella.
Materials and Methods
Fungal Strains and Experimental Treatment
Cultivated strains of M. sextelata were purchased from Jindi Tianlingjian Biotechnology Co., Ltd., in Chengdu, Sichuan, China. P. penicillatus was isolated in our previous study (He et al., 2017) and was deposited in the Culture Collection Center of the Soil and Fertilizer Institute, Sichuan Academy of Agricultural Sciences, Chengdu, China. The strains of M. sextelata used in this study were cultivated on a farm in Xindu, Chengdu, China (30.8°N, 104.2E). The cultivation, growth, and management of morels were done according to standard practices (Tan et al., 2021b). P. penicillatus was cultured on potato dextrose agar medium for 5 d at 25°C before inoculation. When the fruiting bodies of M. sextelata reached 7cm high, they were inoculated with P. penicillatus mycelia. Needle tip-sized mycelia were picked up with sterilized toothpicks and inoculated on Morchella fruiting bodies. A total of 120 M. sextelata fruiting bodies were used for the field experiment. Half of them were inoculated with P. penicillatus as explained above, while the remaining half were not inoculated. According to the phenotypic changes of the lesion, samples were collected at 3 and 6 days post-inoculation (dpi). One cm2 fruiting body centered on the lesion was collected for each inoculated morel, while healthy tissue from the same site was collected from the non-inoculated morels. The samples were divided into four groups, including non-inoculated groups on 3 and 6 dpi and inoculated groups on 3 and 6 dpi. Each group contained three biological replicates, and groups of six fruiting bodies were pulled into one biological replicate. All the samples were stored at −80°C for subsequent transcriptome analysis.
RNA Extraction and qRT-PCR
Total RNA was isolated and purified from healthy and inoculated morels using the TRIzol reagent (Invitrogen, Carlsbad, CA, United States) following the manufacturer’s instructions. The purity, concentration, and integrity of each sample were determined using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, MA, United States), a Qubit RNA Kit (Life Technologies, Carlsbad, CA, United States), and a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, United States), respectively.
About 2μg of total RNA from each sample was reverse-transcribed into cDNA using a RevertAid First Strand cDNA Synthesis Kit (THERMO, United Kingdom). The profiles of expression of the genes expression profiles were determined using SYBR Green Premix (TaKaRa Code RR820A; TaKaRa, Shiga, Japan) on a Roche LightCycle480 system (Roche Applied Science, Rotkreuz, Switzerland). All the PCR reactions were conducted using 40cycles at 98°C for 10s, 60°C for 10s, and 72°C for 10s, in a 20μl reaction mixture that contained 10pmol of each primer and 2μl of cDNA as a template. All the reactions were performed in triplicate, and 5.8S rRNA was used as the internal control for normalization. Supplementary Table S1 lists the primers used for quantitative real-time reverse transcriptase–PCR (qRT-PCR).
cDNA Library Construction and Sequencing
cDNA library preparation and sequencing were conducted by the Personal Biotechnology Co., Ltd., in Shanghai, China. All the libraries were sequenced using an Illumina Nova-Seq platform (Illumina, San Diego, CA, United States). The FASTX toolkit was used to filter the raw data, short-fragment reads, and sequencing adapters, while other low-quality reads were filtered to obtain the clean reads. After preprocessing the RNA-Seq data, the reads were mapped to the P. penicillatus and M. sextelata reference genomes using HISAT2 software.1 The RNA reads were classified as M. sextelata or P. penicillatus based on their similarity to the corresponding genome sequence. Fragments per kilobase of transcript per million mapped fragments were used to normalize the abundance of transcripts (Trapnell et al., 2012).
Differential Expression Analysis
Differential expression analysis was conducted using DESeq2 (Love et al., 2014). The resulting value of p were adjusted using Benjamini and Hochberg’s approach to control the false discovery rate. Genes with an adjusted p <0.05 and log2 (fold change) >1 that were detected by DESeq were designated as differentially expressed.
TopGO was used for the Gene Ontology (GO) enrichment analysis, and the calculated value of p was then obtained using the Wallenius non-central hypergeometric distribution method to identify significantly enriched GO terms (p <0.05) from all of the DEGs (Ashburner et al., 2000). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed on the DEGs using clusterProfiler. The value of p was calculated using the hypergeometric distribution method (Kanehisa et al., 2004).
Data Availability
The Illumina Nova-Seq sequencing data from this study were deposited in the NCBI Sequence Read Archive.2 The reference genome of P. penicillatus can be downloaded from https://www.ncbi.nlm.nih.gov/genome/79861?genome_assembly_id=1473685, and the reference genome of M. sextelata can be downloaded from https://www.ncbi.nlm.nih.gov/genome/86229?genome_assembly_id=748597.
Results
Overview of the RNA-Seq Data
White spots were observed on the M. sextelata fruiting bodies at 3days after inoculation with P. penicillatus, indicating that P. penicillatus was successfully germinated and began to infect Morchella. The typical symptom of white mold disease started to appear at 6 dpi (Figure 1). Based on the observed phenotypic change, we sampled tissues at 3 and 6 dpi, respectively. The non-inoculated tissues at 3 and 6 dpi were used as the control groups for the subsequent dual RNA-Seq analysis.

Figure 1. Field phenotype of the white mold disease (WMD) in Morchella sextelata. (A,C) represent the non-inoculated morels at 3 and 6days, respectively. (B,D) represent the morels at 3 and 6days post-Paecilomyces penicillatus inoculation, respectively. The red arrow shows the WMD symptom.
An average of 75,728,447 raw reads was generated for each sample with a Q20 and Q30 of 97.81 and 94.11%, respectively (Supplementary Table S2). After quality control, an average of 68,239,578 clean reads was generated. All the clean reads were mapped to a corresponding reference genome. An average of 92.87% clean reads of the non-inoculated samples was mapped to the M. sextelata genome, while none (< 0.05%) was mapped to the P. penicillatus genome. A total of 90.56 and 84.72% clean reads of the inoculated samples were mapped to the genome of M. sextelata at the early and late stages of infection, respectively. However, 2.01 and 6.85% clean reads from the infected sites were mapped to the P. penicillatus genome at the early and late stages of infection, respectively (Supplementary Table S3).
Quantitative RT-PCR Validation
We selected 19 genes, including 11 (laccase-2, ABCB1, tyrosinase, primary amine oxidase, pckA, probable acetate kinase, aconitate hydratase, alcohol dehydrogenase 6, indoleamine 2,3-dioxygenase, pyruvate decarboxylase, and sophorolipid transporter) from M. sextelata and eight (chitinase 1, CMB1, GH20, 3-isopropylmalate dehydratase, phosphotransferase, glucosamine-6-phosphate deaminase, leuB, and serine dehydratases) from P. penicillatus, to validate the RNA-Seq data. qRT-PCR assays were conducted to test their patterns of expression at 3 and 6 dpi. As shown in Figure 2, the qPCR assay identified nine upregulated genes in M. sextelata and two were downregulated. Among them, laccase-2 was highly upregulated at both infection stages by 10.9- (3 dpi) and 21.4-fold (6 dpi). In P. penicillatus, three were upregulated, and two were downregulated. The patterns of expression obtained from the qRT-PCR were consistent with the RNA-Seq results, indicating that the RNA-Seq data were well suited to analyze the interaction between P. penicillatus and M. sextelata.
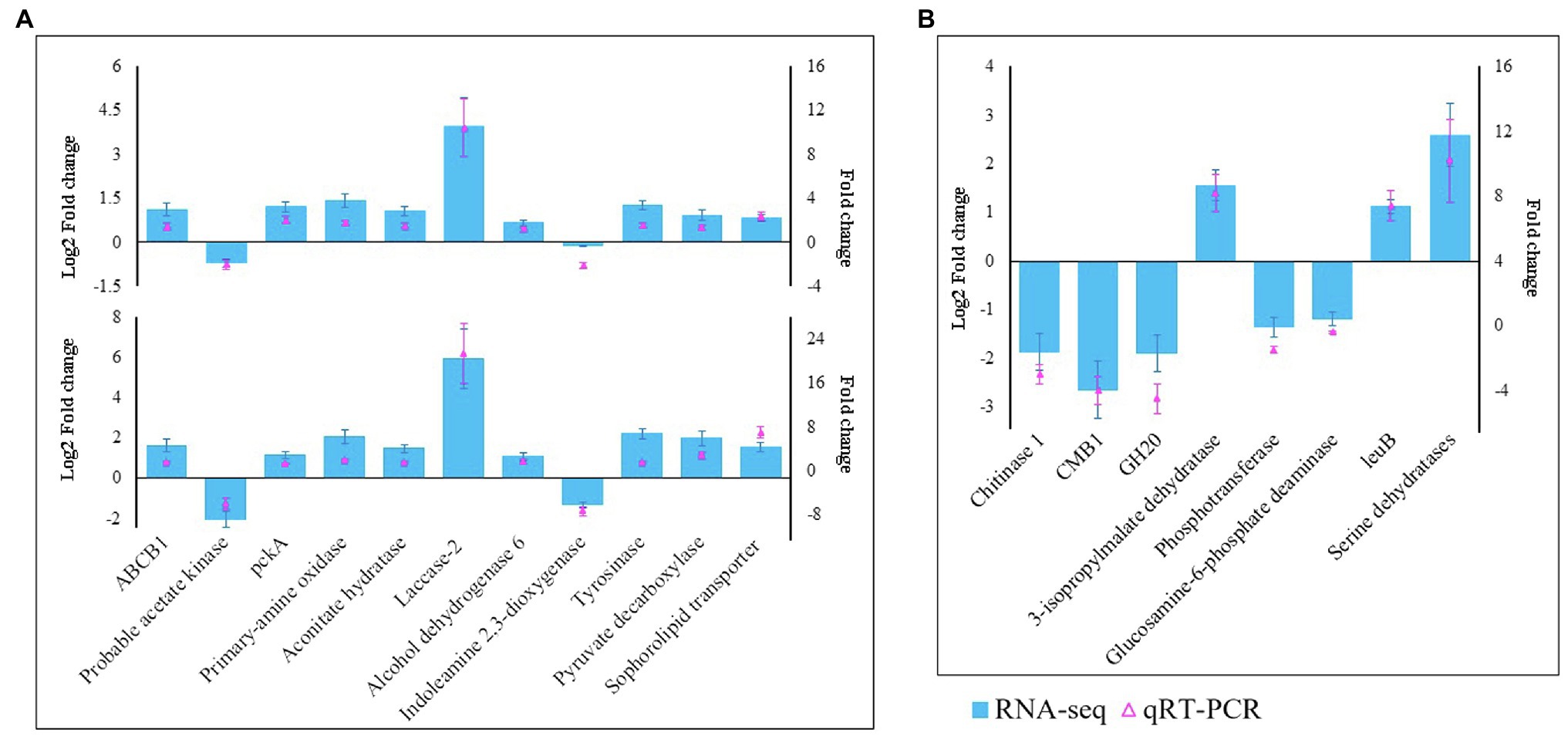
Figure 2. Validation of the RNA-Seq data by qRT-PCR. (A) 11 DEGs from M. sextelata were selected for validation, and the y-axis shows the fold change expression at 3 and 6 days post-inoculation (dpi) compared with the non-inoculated samples. (B) Eight DEGs from P. penicillatus were selected, and the y-axis shows the fold change expression at 6 dpi compared with 3 dpi. Each data point was obtained from three biological replicates. DEGs, differentially expressed genes; qRT-PCR, quantitative real-time reverse transcriptase–PCR.
Differentially Expressed Genes in P. penicillatus
The transcriptional expression of P. penicillatus on 3 and 6 dpi was analyzed. A total of 460 DEGs in P. penicillatus were identified by comparing the abundance of transcripts on 6 dpi with those on 3 dpi. A total of 336 were upregulated, and 124 were downregulated. Two proteases, including subtilase family protease and serine proteinase, were the most highly expressed among these DEGs (Figure 3A; Supplementary Table S4). The expressions of CAZymes in P. penicillatus, which play an important role in mycoparasitic fungi (Wang et al., 2020), were analyzed. A total of 45 CAZymes were identified from all the DEGs in P. penicillatus (Figure 3B), including 25 glycosyl hydrolases (GHs), seven glycosyltransferases (GTs), seven auxiliary activities (AAs) enzymes, four carbohydrate-binding modules, and one carbohydrate esterase (CE). Among them, GH 17 (glucan endo-1,3-β-glucosidase), AA6 (1,4-benzoquinone reductase), and GT2 (chitin synthase 1) were the top three genes that were expressed, and they were all upregulated during the late stage of P. penicillatus infection (Supplementary Table S4), indicating their importance in the pathogenicity of P. penicillatus. Out of the 25 GHs, six β-glucanases, three α-mannosidases, and two α-1,6-mannanases were upregulated, while two chitinases (GH18) were downregulated (Figure 3B). This information indicated that the main cell wall-degrading enzymes (CWDEs) were glucanases and mannanases rather than chitinase when P. penicillatus infected M. sextelata.
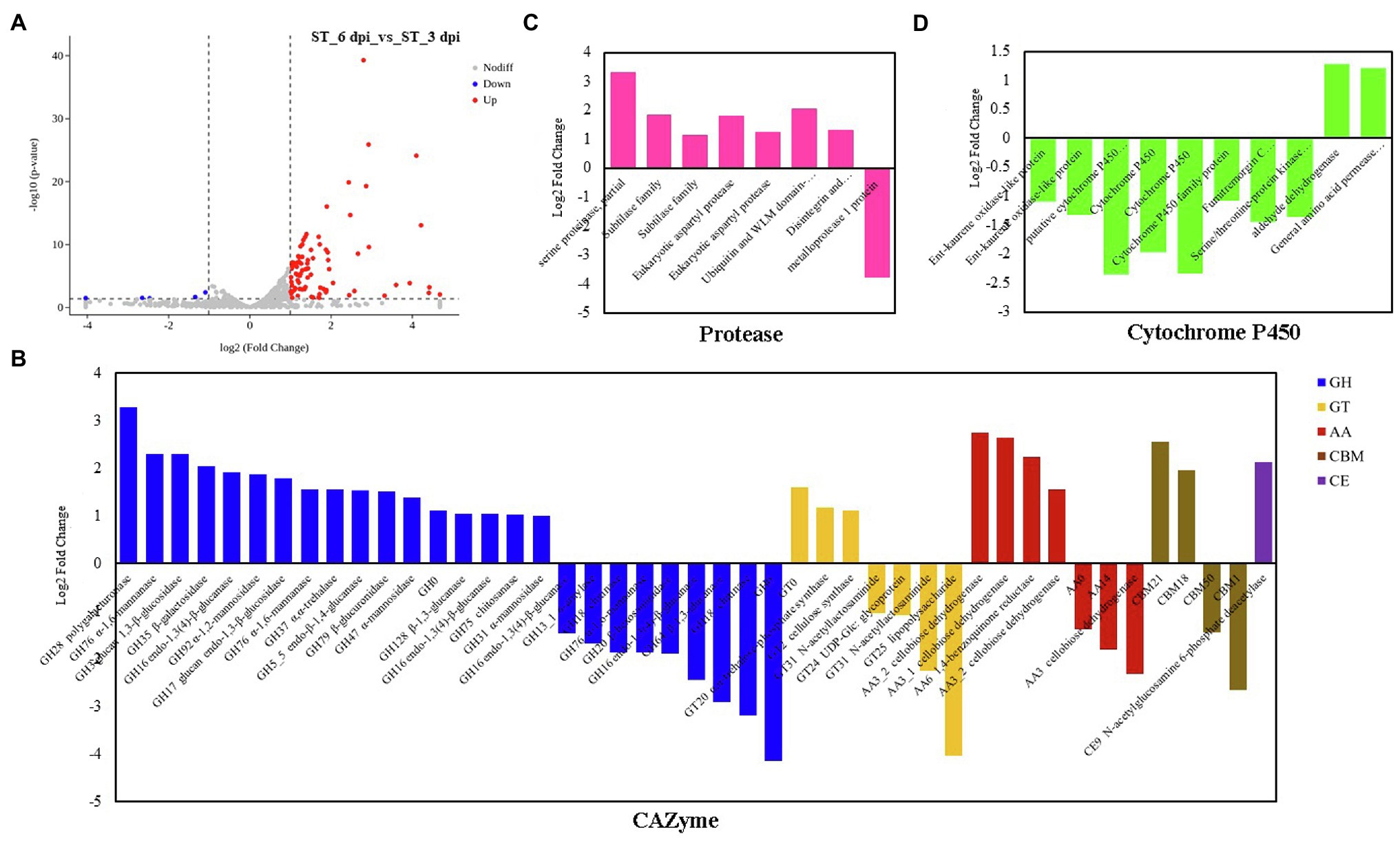
Figure 3. DEGs at 6 days post-inoculation (dpi) compared with 3 dpi in Paecilomyces penicillatus. (A) Volcano plot of the DEGs in P. penicillatus. Differentially expressed CAZymes (B), proteases (C) and cytochrome P450s (D) in P. penicillatus during the infection process. ST, inoculated samples; 3 and 6 dpi, 3 and 6 days post-inoculation; DEGs, differentially expressed genes.
Functional Analysis of Differentially Expressed Genes in P. penicillatus
GO category enrichment analysis was utilized to elucidate the functional enrichment of the DEGs in P. penicillatus. A total of 73 GO terms were enriched (p<0.05) in P. penicillatus (Supplementary Table S5). Four “hydrolase activity (GO:0004553, GO:0016813, GO:0016810, and GO:0016798),” three “oxidoreductase activity (GO:0016614, GO:0016616, and GO:0016491),” one “NAD binding (GO:0008172)” in the category of molecular function, one “carbohydrate metabolic process (GO:0005975)” in the biological processes category, and three “membrane (GO:0016020, GO:0016021, and GO:0031224)” in the cellular component category were among the top 20 GO terms (Figure 4). The DEGs in the “carbohydrate metabolic process” were analyzed. Among the 34 DEGs, 16 of them were CAZymes and also belonged to the GO term of “hydrolase activity, hydrolyzing O-glycosyl compounds” (Supplementary Table S5). Eight proteases were identified from “hydrolase activity,” including three metalloprotease proteins, two proteinase T-like proteins, two eukaryotic aspartyl proteases, and one serine proteinase (Figure 3C), which could act as CWDEs when P. penicillatus infected the morels. Ten cytochrome P450 (CYP) superfamily proteins were identified in the GO term of oxidoreductase activity, and eight of them were downregulated (Figure 3D).
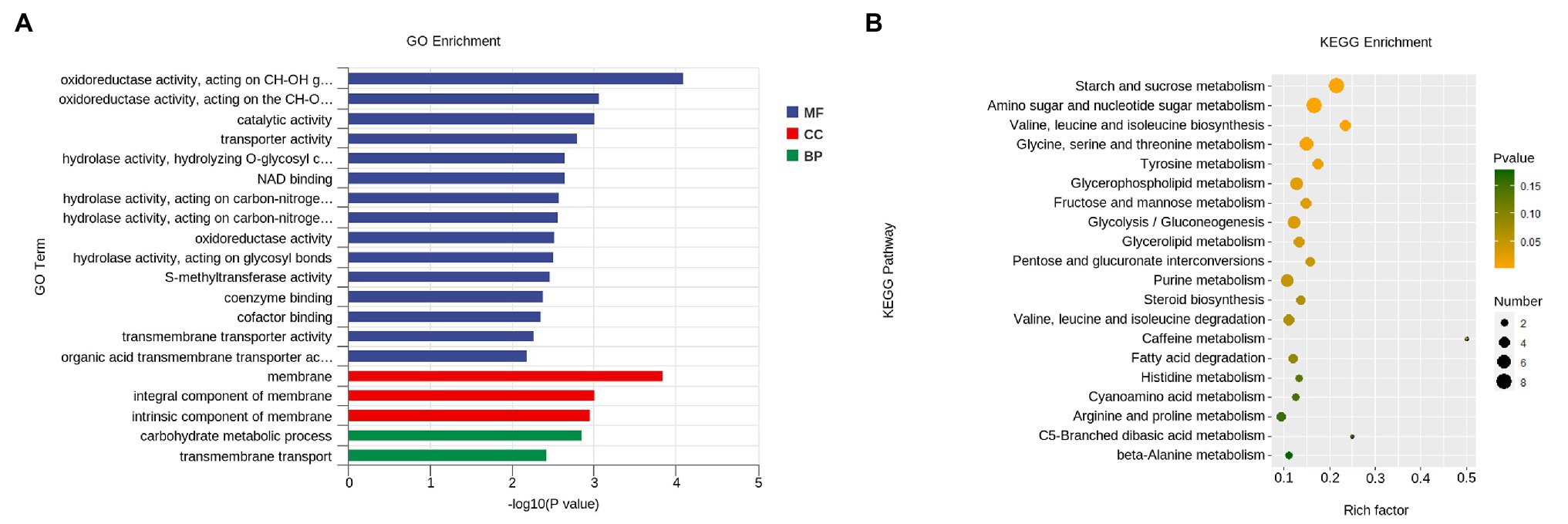
Figure 4. Functional enrichment analyses for DEGs in P. penicillatus during the infection process. (A) GO enrichment analyses. GO term enrichment values of p are indicated on the x-axis. BP, biological process; MF, molecular function; CC, cellular component. (B) KEGG enrichment analyses. The x-axis represents the rich factor. The size of the dot indicates the number of DEGs involved in the pathway. Color bars on the right represent the value of p of the KEGG pathway. DEGs, differentially expressed genes; GO, gene ontology; KEGG, kyoto encyclopedia of genes and genomes.
The KEGG pathway analysis showed that 11 pathways were significantly enriched (p<0.05) in P. penicillatus, including five “carbohydrate metabolism,” three “amino acid metabolism,” two “lipid metabolism,” and one “nucleotide metabolism.” Among them, “starch and sucrose metabolism (ko00500)” and “amino sugar and nucleotide sugar metabolism (ko00520)” were the two most significantly enriched pathways (Figure 4; Supplementary Table S5). These results indicate that carbohydrate metabolism plays an important role in the pathogenicity of P. penicillatus on M. sextelata, which was similar to the results of GO category enrichment analysis.
Differentially Expressed Genes in M. sextelata
We also analyzed the transcriptional expression of M. sextelata after infection with P. penicillatus. A total of 313 DEGs were identified in M. sextelata by comparing the abundance of transcripts of the inoculated morels with those of the non-inoculated morels, and 90 and 186 DEGs were identified on 3 and 6 dpi, respectively. Among them, 63 genes were common DEGs in the two infection stages (Figure 5; Supplementary Table S6). Notably, all the 63 common DEGs were upregulated during the two infection stages. Among them, a laccase-2 gene was the most highly upregulated (log2fc=5.9; Supplementary Table S6). Interestingly, chitin recognition protein, cyclin-dependent protein kinase, caffeine-induced death protein 2, and allergen Asp f 15 precursor involved in immune responses were identified in these common DEGs (Supplementary Table S6). A total of 85 upregulated and five downregulated genes were found to be the specific DEGs on 3 dpi in M. sextelata, while 164 upregulated and 22 downregulated genes were specifically identified on 6 dpi. A cyclin was downregulated on 3 dpi and expressed at normal levels on 6 dpi (Supplementary Table S6).
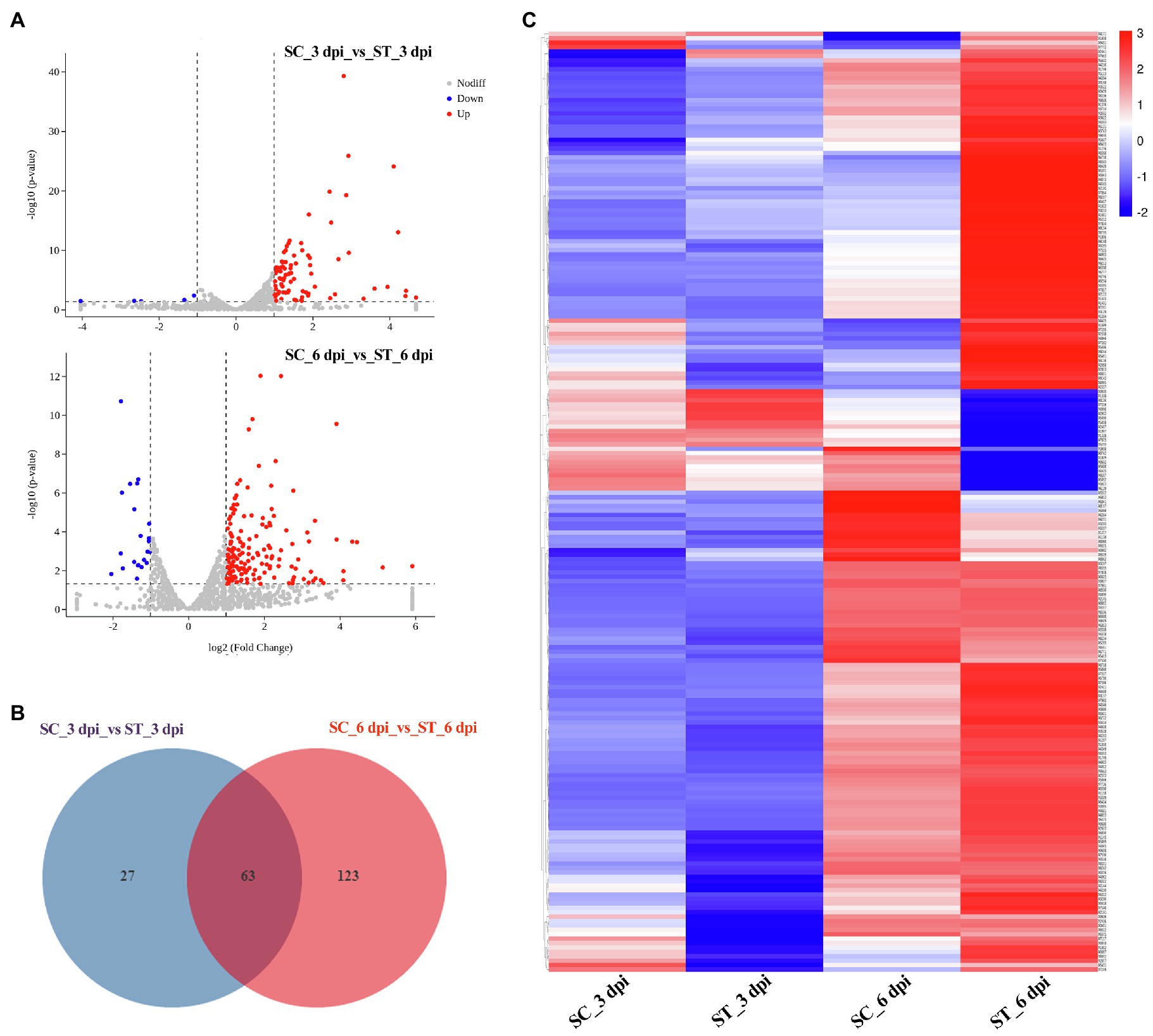
Figure 5. DEGs in M. sextelata at 3 and 6 days post-inoculation. Volcano plot (A), Venn diagrams (B) and heat maps (C) of the DEGs modulated by P. penicillatus infection in M. sextelata. Blue indicates downregulated DEGs, while red indicates upregulated DEGs in heat maps. ST and SC represent the inoculated and non-inoculated samples, respectively. DEGs, differentially expressed genes; dpi, days post-inoculation.
Functional Analysis of Differentially Expressed Genes in M. sextelata
GO category enrichment analysis was conducted in more detail, and 77 and 71 GO terms were identified (p<0.05) on 3 and 6 dpi, respectively (Supplementary Table S7). “Oxidoreductase activity (GO:0016491),” “oxidation-–reduction process (GO:0055114),” “carboxy-lyase activity (GO:0016831),” and several pathways associated with these GO terms were commonly enriched in both infection stages (Figure 6; Supplementary Table S7). DEGs in the oxidation-reduction process were screened, and sets of genes associated with immunity were identified, including five CYPs (benzoate 4-monooxygenase CYP-like protein, NADPH-P450 reductase, CYP52A4, CYP52A4, and similar to isotrichodermin C-15 hydroxylase), four tyrosinases, three flavin oxidoreductases, two multicopper oxidases (laccase-2 and bilirubin oxidase), one copper amine oxidase, and one cytochrome c peroxidase (Figure 7; Supplementary Table S8), were all upregulated after infection with P. penicillatus and could be related to the detoxification of P. penicillatus toxins. In addition, a putative apoptosis-inducing protein was upregulated (Figure 7; Supplementary Table S8). GO terms as serine-type peptidase activity (GO:0008236), hormone activity (GO:0005179), pantothenate metabolic process (GO:0015939), and FMN binding (GO:0010181) were specifically enriched on 6 dpi (Supplementary Table S7), indicating that M. sextelata could distinguish between the early and late stages of infection with P. penicillatus.
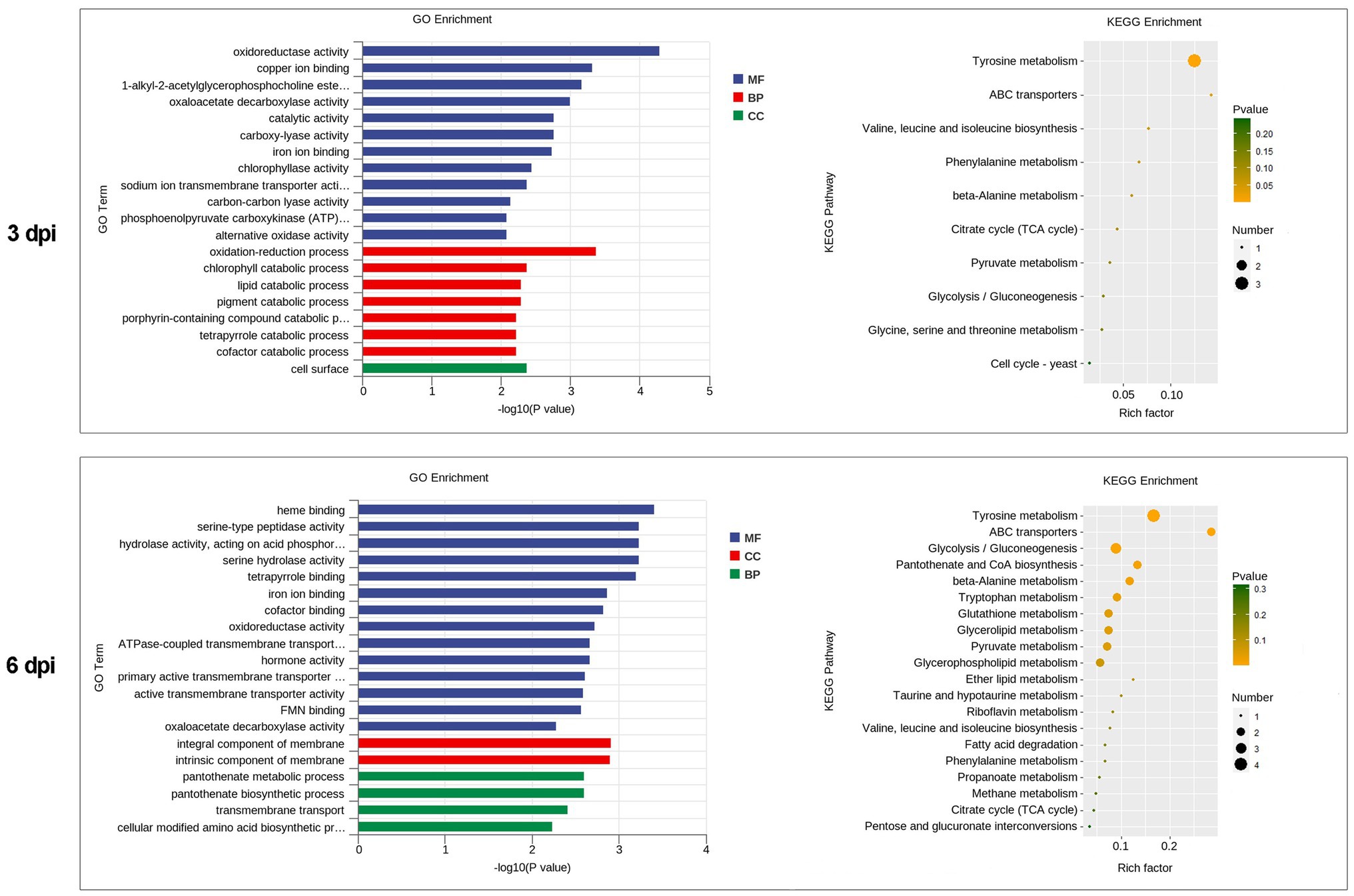
Figure 6. Functional enrichment analyses for DEGs in M. sextelata at 3 and 6 days post-inoculation. GO enrichment analyses, GO term enrichment values of p are indicated on the x-axis. BP, biological process; MF, molecular function; CC, cellular component. KEGG enrichment analyses, x-axis represents the rich factor; the dot size indicates the number of DEGs involved in the pathway; Color bars on the right represent the values of p of the KEGG pathway. 3 and 6 dpi represent 3 and 6 days post-inoculation. DEGs, differentially expressed genes; GO, gene ontology; KEGG, kyoto encyclopedia of genes and genomes.
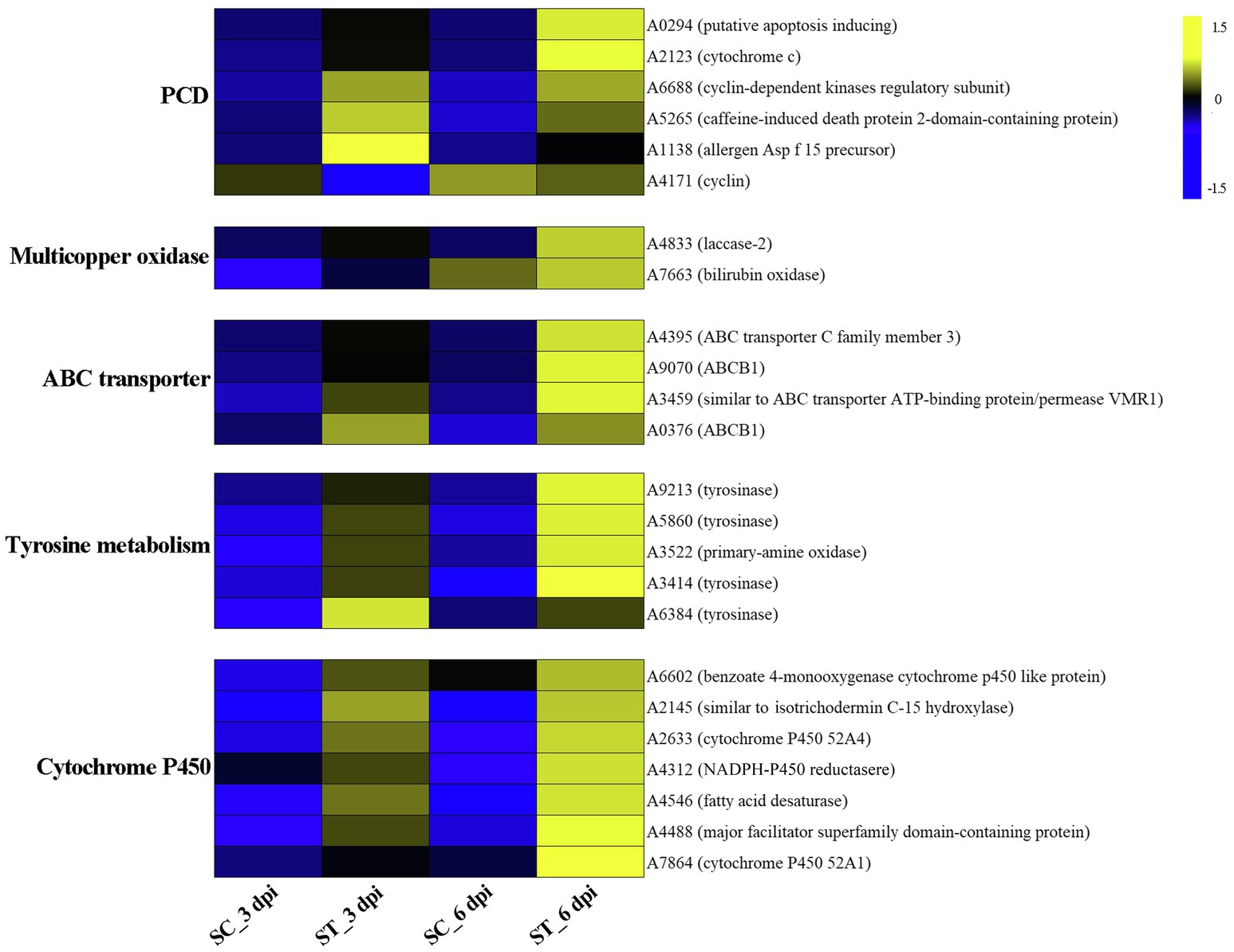
Figure 7. Heat map of DEGs that responded to P. penicillatus infection in M. sextelata. PCD, programmed cell death; ST and SC represent the inoculated and non-inoculated samples, respectively; dpi, days post-inoculation; blue indicates downregulated DEGs, while yellow indicates upregulated DEGs. KEGG, kyoto encyclopedia of genes and genomes.
KEGG pathway analysis identified two and nine significantly enriched pathways on 3 and 6 dpi, respectively. Among them, “tyrosine metabolism (ko00350)” and “ABC transporters (ko02010)” were the common enriched pathways in both infection stages (Supplementary Table S7). The DEGs in these two pathways were screened. Five tyrosinases and two ABC transporter family proteins were found to be upregulated in the infected morel, confirming their important roles in the response of M. sextelata to P. penicillatus (Figure 7; Supplementary Table S8). “Glycolysis/Gluconeogenesis (ko00010),” “pantothenate and CoA biosynthesis (ko00770),” and several other pathways were specifically enriched on 6 dpi (Supplementary Table S7), which is similar to the results of GO category enrichment analysis.
Discussion
Owing to the lack of resistant varieties and the prevalence of P. penicillatus in many regions, WMD poses a serious threat to the cultivation of morels in China. This study identified a number of genes involved in the regulation of the pathogenicity of P. penicillatus to M. sextelata and the response of M. sextelata to infection with P. penicillatus. We defined how P. penicillatus successfully parasitizes M. sextelata and how M. sextelata encodes its immune response to invasion by P. penicillatus.
Mycoparasitic fungi usually produce various fungal CWDEs to lyse the host cell wall, including glucanases, chitinase, and proteases (Gruber and Seidl-Seiboth, 2012). A previous study found that P. penicillatus encodes approximately 300 CAZymes, which could be involved in the degradation of fungal cell walls (Wang et al., 2020). P. penicillatus activated six β-glucanases, three α-mannosidases, and two α-1,6-mannanases when it infected M. sextelata (Figure 3B), indicating that glucanases and mannanases were the primary CAZymes of P. penicillatus that are involved in the degradation of M. sextelata cell walls. Only two chitinases (GH18) were identified in P. penicillatus, even though they were the most annotated CAZyme family in the P. penicillatus genome (Wang et al., 2020). They were all unexpectedly downregulated (Figure 3B). Previous studies have shown that mycoparasitic species of Trichoderma activated chitinase when interacting with A. bisporus (Guthrie and Castle, 2006; Ihrmark et al., 2010), in contrast to the findings of this study. However, the chitinases of T. gamsii were all downregulated when interacting with Fusarium graminearum (Zapparata et al., 2021), which was consistent with the data of this study. This suggests that P. penicillatus could change the expression of chitinases based on demands of specific situations when infecting other Ascomycetes fungi. In this study, chitin synthase 1 was upregulated in P. penicillatus. One possible explanation for this upregulation is that these chitinases were involved in the cell wall remodeling during P. penicillatus growth. Their downregulation could result in strengthened cell walls during interaction. Another possible explanation is that when in contact with M. sextelata, P. penicillatus suppresses its secretion of fungal chitinases to avoid recognition and the induction of defense reactions in M. sextelata. Several proteases, including subtilase family protease, metalloprotease, and serine proteinase, were activated during the infection process of P. penicillatus (Figure 3C). These genes were reported to be involved in the regulation of cell wall degradation in plants and microorganisms (Feng et al., 2014; Schaller et al., 2018). In particular, the subtilase family proteases are good candidates for this function since they were reported to be induced during mycoparasitism before and during contact with the host in different Trichoderma species (Suárez et al., 2007; Seidl et al., 2009). This indicated that these CWDEs play an important role in the parasitism of P. penicillatus on morels.
M. sextelata activated a series of mechanisms to respond once it sensed the invasion of P. penicillatus. A chitin recognition protein and an allergen Asp f 15 precursor in M. sextelata were upregulated during both infection stages (Figure 7; Supplementary Table S8), which could activate the downstream immune response (Sanchez-Vallet et al., 2015). M. sextelata upregulated a caffeine-induced death protein 2-domain-containing protein and a putative apoptosis-inducing protein, as well as downregulating a cyclin (Figure 7; Supplementary Table S8); these genes are widely reported to promote immunity by initiating programmed cell death (Saiki et al., 2011; Qi and Zhang, 2019). We hypothesize that M. sextelata activates an apoptotic response to escape the proliferation of the pathogen when P. penicillatus invades.
A change in the reduction-oxidation status is one of the earliest responses detected when cells are attacked (Frederickson Matika and Loake, 2014). The reduction-oxidation process plays a key role in both the host and pathogen during the interaction of P. penicillatus and M. sextelata, while the regulatory mechanisms differ. The cytochrome P450 family genes in M. sextelata and P. penicillatus showed an opposite trend of expression. Six of the seven cytochrome P450 in P. penicillatus were downregulated, while all five in M. sextelata were upregulated (Figures 3D, 7). A previous study reported that ent-kaurene-derived diterpenoids act as a virulence factor interacting with the bacterium Xanthomonas oryzae pv. oryzicola and rice (Lu et al., 2015) and inhibited the growth of Staphylococcus aureus and Bacillus subtilis (Fatope et al., 2010). Two ent-kaurene oxidase-like proteins that are cytochrome P450s were downregulated in P. penicillatus, which could lead to increased synthesis of diterpenoid cytotoxins in P. penicillatus (Wang et al., 2012). Once the diterpenoid cytotoxins were secreted into M. sextelata cells, the pathogenicity of P. penicillatus to M. sextelata might be enhanced. Interestingly, one isotrichodermin C-15 hydroxylase gene was downregulated in P. penicillatus, while another was upregulated in M. sextelata. Previous study reported that blocking an isotrichodermin C-15 hydroxylase in Fusarium sporotrichioides resulted in the accumulation of trichothecenes, a virulence factor of Fusarium. spp. (McCormick and Hohn, 1997; Ward et al., 2002). This study also observed increased expression of CYPs superfamily genes in M. sextelata after infection with P. penicillatus, including benzoate 4-monooxygenase CYP-like protein and NADPH-P450 reductase. Benzoate 4-monooxygenase of ascomycete Cochliobolus lunatus was reported to be involved in detoxification of benzoic acid (BA), a key intermediate in metabolism of aromatic compounds in fungi, as well as the detoxification of phenolic compounds (Podobnik et al., 2008; Berne et al., 2012). NADPH-P450 reductase was reported to plays a central role in chemical detoxification and insecticide resistance in insects and fungi (Lian et al., 2011; Xu et al., 2021). This information suggests that these CYPs in M. sextelata might participated in the detoxification of toxic metabolites produced by P. penicillatus. We also identified two ABCB1 proteins that were upregulated in M. sextelata following infection with P. penicillatus (Figure 7). We hypothesize that M. sextelata could transport harmful substances secreted by P. penicillatus out of its cells by activating the expression of ABCB1. This has been confirmed by previous studies that ABCB1, a member of the ABC transporter family, plays an important physiological role in protecting the tissues from xenobiotics and endogenous metabolites (Roy et al., 2021).
In fungi, tyrosinases are generally associated with the formation and stability of spores, defense and virulence, and melanin production (Halaouli et al., 2006). Once the cells have been damaged, melanin produced by the reaction of tyrosinase with its substrate forms a melanin barrier to protect against the invasion of pathogenic bacteria or their toxins (Janusz et al., 2020). In this study, tyrosinase metabolism was the most enriched KEGG pathway in the infected M. sextelata (Figure 6). Five tyrosinase genes in M. sextelata were upregulated after infection by P. penicillatus (Figure 7). This indicated their importance in M. sextelata against infection with P. penicillatus.
Fungal laccase, a multicopper oxidase, is usually induced by a variety of phenolic compounds (Piscitelli et al., 2011) and was reported to have a wide range of functions, including defense against stressful conditions (Lakshmanan and Sadasivan, 2016; Chakraborty et al., 2020). One 1,4-benzoquinone reductase was upregulated in P. penicillatus (Figure 3B; Supplementary Table S4) and could be involved in the downstream synthesis of phenolic compounds. Interestingly, laccase-2 was the most upregulated gene in infected M. sextelata (Figure 7; Supplementary Table S6). A previous study found that the activity of laccase-2 enhanced the resistance of A. bisporus to the T. aggressivum toxin (Sjaarda et al., 2015), which is 3,4-dihydro-8-hydroxy-3-methyl isocoumarin that contains a phenolic hydroxyl and lactone ring (Krupke et al., 2003). This suggests that P. penicillatus could synthesize phenolic toxins and secrete them into M. sextelata, and M. sextelata could detoxify these phenolic toxins by activating laccase expression. However, further experiments are needed to verify this hypothesis.
Conclusion
During P. penicillatus infection of M. sextelata, cell wall-degrading enzymes, such as glucanases, mannanases, and proteases were probably secreted by P. penicillatus to degrade the cell wall of M. sextelata. M. sextelata triggered programmed cell death to prevent the excessive proliferation of P. penicillatus and synthesize cytochrome P450 and laccase to detoxify the P. penicillatus toxins. The melanin barrier formed by tyrosinase was also a possible immune pathway in M. sextelata. Although these possible regulatory pathways proposed in this study still need further verification, they provided a theoretical basis for researchers to breed WMD-resistant varieties and develop prevention and control methods for WMD.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
YY: conceptualization, funding acquisition, validation, investigation, data curation, visualization, writing – original draft preparation, and writing – review and editing. HT: funding acquisition, validation, and investigation. TL: investigation and data curation. LL: resources and investigation. JT: resources. WP: funding acquisition and supervision. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This research was funded by National Science Foundation of China (NSFC31901119), Science and Technology Project of Sichuan Province (2021YFYZ0026), SAAS-International Cooperation Project (2021ZSSFGH04), Edible Fungus Innovation Team of Sichuan Province (SCCXTD-2021-7), and Special Fund for Talent Introduction and Training of SAAS (510000-01-114852).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.760444/full#supplementary-material
Footnotes
References
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., and Cherry, J. M. (2000). Gene ontology: tool for the unification of biology. Gene Ontol. Cons. Nature Gene. 25, 25–29. doi: 10.1038/75556
Berne, S., Podobnik, B., Zupanec, N., Novak, M., Krasevec, N., Turk, S., et al. (2012). Virtual screening yields inhibitors of novel antifungal drug target, benzoate 4-monooxygenase. J. Chem. Inf. Model. 52, 3053–3063. doi: 10.1021/ci3004418
Chakraborty, T., Toth, R., Nosanchuk, J. D., and Gacser, A. (2020). Multicopper oxidases in Saccharomyces cerevisiae and human pathogenic fungi. J. Fungi 6:56. doi: 10.3390/jof6020056
Chen, C., Li, Q., Huang, W., Wang, J., Fu, R., Luo, X., et al. (2017). Effects of Morchella white mold disease on soil fungal community structure. Microbiology 44, 2652–2659. doi: 10.13344/j.microbiol.china.170030
Cheng, C. A., Rf, A., Jian, W. A., Xl, A., Xc, A., Qiang, L. B., et al. (2021). Genome sequence and transcriptome profiles of pathogenic fungus Paecilomyces penicillatus reveal its interactions with edible fungus Morchella importuna. Computational and structural. Biotechnol. J. 19, 2607–2617. doi: 10.1016/j.csbj.2021.04.065
Dahlstrom, J. L., Smith, J. E., and Weber, N. S. (2000). Mycorrhiza-like interaction by Morchella with species of the Pinaceae in pure culture synthesis. Mycorrhiza 9, 279–285. doi: 10.1007/PL00009992
Deng, K., Lan, X., Fang, Q., Li, M., Xie, G., and Xie, L. (2021). Untargeted metabolomics reveals alterations in the primary metabolites and potential pathways in the vegetative growth of Morchella sextelata. Front. Mol. Biosci. 8:632341. doi: 10.3389/fmolb.2021.632341
Du, X. H., Zhao, Q., O'Donnell, K., Rooney, A. P., and Yang, Z. L. (2012). Multigene molecular phylogenetics reveals true morels (Morchella) are especially species-rich in China. Fungal Genet. Biol. 49, 455–469. doi: 10.1016/j.fgb.2012.03.006
Fatope, M. O., Varma, G. B., Alzri, N. M., Marwah, R. G., and Nair, R. S. (2010). Ent-Kaurene diterpenoids from Blepharispermum hirtum. Chem. Biodivers. 7, 1862–1870. doi: 10.1002/cbdv.200900389
Feng, T., Nyffenegger, C., Hojrup, P., Vidal-Melgosa, S., Yan, K. P., Fangel, J. U., et al. (2014). Characterization of an extensin-modifying metalloprotease: N-terminal processing and substrate cleavage pattern of Pectobacterium carotovorum Prt1. Appl. Microbiol. Biotechnol. 98, 10077–10089. doi: 10.1007/s00253-014-5877-2
Foulongne-Oriol, M., Rodier, A., and Savoie, J. M. (2012). Relationship between yield components and partial resistance to Lecanicillium fungicola in the button mushroom, Agaricus bisporus, assessed by quantitative trait locus mapping. Appl. Environ. Microbiol. 78, 2435–2442. doi: 10.1128/AEM.07554-11
Frederickson Matika, D. E., and Loake, G. J. (2014). Redox regulation in plant immune function. Antioxid. Redox Signal. 21, 1373–1388. doi: 10.1089/ars.2013.5679
Fu, Y., Wang, X., Li, D., Liu, Y., Song, B., Zhang, C., et al. (2016). Identification of resistance to wet bubble disease and genetic diversity in wild and cultivated strains of Agaricus bisporus. Int. J. Mol. Sci. 17:1568. doi: 10.3390/ijms17101568
Gea, F. J., Navarro, M. J., Santos, M., Dianez, F., and Carrasco, J. (2021). Control of fungal diseases in mushroom crops while dealing with fungicide resistance. Rev. Micro. 9:585. doi: 10.3390/microorganisms9030585
Gruber, S., and Seidl-Seiboth, V. (2012). Self versus non-self: fungal cell wall degradation in Trichoderma. Microbiology 158, 26–34. doi: 10.1099/mic.0.052613-0
Guthrie, J. L., and Castle, A. J. (2006). Chitinase production during interaction of Trichoderma aggressivum and Agaricus bisporus. Can. J. Microbiol. 52, 961–967. doi: 10.1139/w06-054
Halaouli, S., Asther, M., Sigoillot, J. C., Hamdi, M., and Lomascolo, A. (2006). Fungal tyrosinases: new prospects in molecular characteristics, bioengineering and biotechnological applications. J. Appl. Microbiol. 100, 219–232. doi: 10.1111/j.1365-2672.2006.02866.x
He, X. L., Peng, W. H., Miao, R. Y., Tang, J., Chen, Y., Liu, L. X., et al. (2017). White mold on cultivated morels caused by Paecilomyces penicillatus. FEMS Microbiol. Lett. 364:364. doi: 10.1093/femsle/fnx037
Ihrmark, K., Asmail, N., Ubhayasekera, W., Melin, P., Stenlid, J., and Karlsson, M. (2010). Comparative molecular evolution of trichoderma chitinases in response to mycoparasitic interactions. Evol. Bioinformatics Online 6, 1–26. doi: 10.4137/ebo.s4198
Janusz, G., Pawlik, A., Swiderska-Burek, U., Polak, J., Sulej, J., Jarosz-Wilkolazka, A., et al. (2020). Laccase properties, physiological functions, and evolution. Int. J. Mol. Sci. 21:966. doi: 10.3390/ijms21030966
Kage, U., Kumar, A., Dhokane, D., Karre, S., and Kushalappa, A. C. (2016). Functional molecular markers for crop improvement. Crit. Rev. Biotechnol. 36, 917–930. doi: 10.3109/07388551.2015.1062743
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y., and Hattori, M. (2004). The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, D277–D280. doi: 10.1093/nar/gkh063
Krupke, O. A., Castle, A. J., and Rinker, D. L. (2003). The north American mushroom competitor, Trichoderma aggressivum F. aggressivum, produces antifungal compounds in mushroom compost that inhibit mycelial growth of the commercial mushroom Agaricus bisporus. Mycol. Res. 107, 1467–1475. doi: 10.1017/s0953756203008621
Lakshmanan, D., and Sadasivan, C. (2016). Trichoderma viride laccase plays a crucial role in defense mechanism against antagonistic organisms. Front. Microbiol. 7:741. doi: 10.3389/fmicb.2016.00741
Lian, L. Y., Widdowson, P., McLaughlin, L. A., and Paine, M. J. (2011). Biochemical comparison of Anopheles gambiae and human NADPH P450 reductases reveals different 2'-5'-ADP and FMN binding traits. PLoS One 6:e20574. doi: 10.1371/journal.pone.0020574
Liu, Q., Ma, H., Zhang, Y., and Dong, C. (2018). Artificial cultivation of true morels: current state, issues and perspectives. Crit. Rev. Biotechnol. 38, 259–271. doi: 10.1080/07388551.2017.1333082
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lu, X., Hershey, D. M., Wang, L., Bogdanove, A. J., and Peters, R. J. (2015). An ent-kaurene-derived diterpenoid virulence factor from Xanthomonas oryzae pv. Oryzicola. New Phytol. 206, 295–302. doi: 10.1111/nph.13187
Masaphy, S. (2010). Biotechnology of morel mushrooms: successful fruiting body formation and development in a soilless system. Biotechnol. Lett. 32, 1523–1527. doi: 10.1007/s10529-010-0328-3
McCormick, S. P., and Hohn, T. M. (1997). Accumulation of Trichothecenes in liquid cultures of a Fusarium sporotrichioides mutant lacking a functional Trichothecene C-15 hydroxylase. Appl. Environ. Microbiol. 63, 1685–1688. doi: 10.1128/aem.63.5.1685-1688.1997
Mei, H., Qingshan, W., and Baiyintala, and Wuhanqimuge,. (2019). The whole-genome sequence analysis of Morchella sextelata. Sci. Rep. 9:15376. doi: 10.1038/s41598-019-51831-4
Ower, R. D., Mills, G. L., and Malachowski, J. A. (1986). Cultivation of morchella. United States patent US4,866,878. 19 Sep 1989.
Peng, W., Tang, J., He, X., Chen, Y., and Tan, H. (2016). Status analysis of morel artificial cultivation in Sichuan. Edible Med. Mushrooms 24, 145–150.
Piscitelli, A., Giardina, P., Lettera, V., Pezzella, C., Sannia, G., and Faraco, V. (2011). Induction and transcriptional regulation of laccases in fungi. Curr. Genomics 12, 104–112. doi: 10.2174/138920211795564331
Podobnik, B., Stojan, J., Lah, L., Krasevec, N., Seliskar, M., Rizner, T. L., et al. (2008). CYP53A15 of Cochliobolus lunatus, a target for natural antifungal compounds. J. Med. Chem. 51, 3480–3486. doi: 10.1021/jm800030e
Qi, F., and Zhang, F. (2019). Cell cycle regulation in the plant response to stress. Front. Plant Sci. 10:1765. doi: 10.3389/fpls.2019.01765
Roy, S., Breakspear, A., Cousins, D., Torres-Jerez, I., Jackson, K. J., Kumar, A., et al. (2021). Three common symbiotic ABC-B transporters in Medicago truncatula are regulated by a NIN-independent branch of the symbiosis signalling pathway. Mol. Plant-Microbe Interact. doi: 10.1094/MPMI-02-21-0036-R [Epub ahead of print]
Saiki, S., Sasazawa, Y., Imamichi, Y., Kawajiri, S., Fujimaki, T., Tanida, I., et al. (2011). Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy 7, 176–187. doi: 10.4161/auto.7.2.14074
Sanchez-Vallet, A., Mesters, J. R., and Thomma, B. P. (2015). The battle for chitin recognition in plant-microbe interactions. FEMS Microbiol. Rev. 39, 171–183. doi: 10.1093/femsre/fuu003
Schaller, A., Stintzi, A., Rivas, S., Serrano, I., Chichkova, N. V., Vartapetian, A. B., et al. (2018). From structure to function - a family portrait of plant subtilases. New Phytol. 218, 901–915. doi: 10.1111/nph.14582
Seidl, V., Song, L., Lindquist, E., Gruber, S., Koptchinskiy, A., Zeilinger, S., et al. (2009). Transcriptomic response of the mycoparasitic fungus Trichoderma atroviride to the presence of a fungal prey. BMC Genomics 10:567. doi: 10.1186/1471-2164-10-567
Sjaarda, C. P., Abubaker, K. S., and Castle, A. J. (2015). Induction of lcc2 expression and activity by Agaricus bisporus provides defence against Trichoderma aggressivum toxic extracts. Microb. Biotechnol. 8, 918–929. doi: 10.1111/1751-7915.12277
Suárez, M., Vizcaíno, J., Llobell, A., and Monte, E. (2007). Characterization of genes encoding novel peptidases in the biocontrol fungus Trichoderma harzianum CECT 2413 using the TrichoEST functional genomics approach. Curr. Genet. 51, 331–342. doi: 10.1007/s00294-007-0130-5
Tan, H., Kohler, A., Miao, R., Liu, T., Zhang, Q., Zhang, B., et al. (2019). Multi-omic analyses of exogenous nutrient bag decomposition by the black morel Morchella importuna reveal sustained carbon acquisition and transferring. Environ. Microbiol. 21, 3909–3926. doi: 10.1111/1462-2920.14741
Tan, H., Liu, T., Yu, Y., Tang, J., Jiang, L., Martin, F. M., et al. (2021a). Morel production related to soil microbial diversity and evenness. Microbiol. Spectr. 9:e00229-21. doi: 10.1128/Spectrum.00229-21
Tan, H., Yu, Y., Tang, J., Liu, T., Miao, R., Huang, Z., et al. (2021b). Build your own mushroom soil: Microbiota succession and nutritional accumulation in semi-synthetic substratum drive the fructification of a soil-saprotrophic morel. Front. Microbiol. 12:656656. doi: 10.3389/fmicb.2021.656656
Trapnell, C. R. A., Goff, L., Pertea, G., Kim, D., and Kelley, D. R. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Wang, Q., Hillwig, M. L., Wu, Y., and Peters, R. J. (2012). CYP701A8: a rice ent-kaurene oxidase paralog diverted to more specialized diterpenoid metabolism. Plant Physiol. 158, 1418–1425. doi: 10.1104/pp.111.187518
Wang, X., Peng, J., Sun, L., Bonito, G., Guo, Y., Li, Y., et al. (2020). Genome sequencing of Paecilomyces Penicillatus provides insights into its phylogenetic placement and Mycoparasitism mechanisms on Morel mushrooms. Pathogens 9:834. doi: 10.3390/pathogens9100834
Ward, T. J., Bielawski, J. P., Kistler, H. C., Sullivan, E., and O'Donnell, K. (2002). Ancestral polymorphism and adaptive evolution in the trichothecene mycotoxin gene cluster of phytopathogenic Fusarium. Proc. Natl. Acad. Sci. 99, 9278–9283. doi: 10.1073/pnas.142307199
Wingfield, B. D., Bills, G. F., Dong, Y., Huang, W., Nel, W. J., Swalarsk-Parry, B. S., et al. (2018). IMA genome-F 9: draft genome sequence of Annulohypoxylon stygium, aspergillus mulundensis, Berkeleyomyces basicola (syn. Thielaviopsis basicola), Ceratocystis smalleyi, two Cercospora beticola strains, Coleophoma cylindrospora, Fusarium fracticaudum, Phialophora cf. hyalina, and Morchella septimelata. IMA Fungus 9, 199–223. doi: 10.5598/imafungus.2018.09.01.13
Xu, J. W., Liao, C. C., Hung, K. C., Wang, Z. Y., Tung, Y. T., and Wu, J. H. (2021). Proteomics reveals Octyl Gallate as an environmentally friendly wood preservative leading to reactive oxygen species-driven metabolic inflexibility and growth inhibition in white-rot fungi (Lenzites betulina and Trametes versicolor). J. Fungi. 7:145. doi: 10.3390/jof7020145
Zapparata, A., Baroncelli, R., Brandstrom Durling, M., Kubicek, C. P., Karlsson, M., Vannacci, G., et al. (2021). Fungal cross-talk: an integrated approach to study distance communication. Fungal Genet. Biol. 148:103518. doi: 10.1016/j.fgb.2021.103518
Zhao, Q. (2021). The 6th National Morchella Conference. Available at: https://www.emushroom.net/news/show-33505.html (Accessed April 28, 2021).
Keywords: Morchella sextelata, Paecilomyces penicillatus, pathogenic factors, response, transcriptomics, CAZymes, laccase, tyrosinase
Citation: Yu Y, Tan H, Liu T, Liu L, Tang J and Peng W (2021) Dual RNA-Seq Analysis of the Interaction Between Edible Fungus Morchella sextelata and Its Pathogenic Fungus Paecilomyces penicillatus Uncovers the Candidate Defense and Pathogenic Factors. Front. Microbiol. 12:760444. doi: 10.3389/fmicb.2021.760444
Edited by:
Anhuai Lu, Peking University, ChinaReviewed by:
José Ascención Martínez-Álvarez, University of Guanajuato, MexicoChenyang Huang, Institute of Agricultural Resources and Regional Planning, Chinese Academy of Agricultural Sciences (CAAS), China
Copyright © 2021 Yu, Tan, Liu, Liu, Tang and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Yu, eWFuZ3l1MDIyMUAxMzkuY29t; Weihong Peng, cHdoNDI0QDE2My5jb20=