 Natalia Cobian
Natalia Cobian Allison Garlet2
Allison Garlet2 Claudio Hidalgo-Cantabrana
Claudio Hidalgo-Cantabrana Rodolphe Barrangou
Rodolphe Barrangou- 1Department of Food, Bioprocessing and Nutrition Sciences, North Carolina State University, Raleigh, NC, United States
- 2BASF Corporation, Tarrytown, NY, United States
Cutibacterium acnes is an important member of the human skin microbiome and plays a critical role in skin health and disease. C. acnes encompasses different phylotypes that have been found to be associated with different skin phenotypes, suggesting a genetic basis for their impact on skin health. Here, we present a comprehensive comparative analysis of 255 C. acnes genomes to provide insights into the species genetic diversity and identify unique features that define various phylotypes. Results revealed a relatively small and open pan genome (6,240 genes) with a large core genome (1,194 genes), and three distinct phylogenetic clades, with multiple robust sub-clades. Furthermore, we identified several unique gene families driving differences between distinct C. acnes clades. Carbohydrate transporters, stress response mechanisms and potential virulence factors, potentially involved in competitive growth and host colonization, were detected in type I strains, which are presumably responsible for acne. Diverse type I-E CRISPR-Cas systems and prophage sequences were detected in select clades, providing insights into strain divergence and adaptive differentiation. Collectively, these results enable to elucidate the fundamental differences among C. acnes phylotypes, characterize genetic elements that potentially contribute to type I-associated dominance and disease, and other key factors that drive the differentiation among clades and sub-clades. These results enable the use of comparative genomics analyses as a robust method to differentiate among the C. acnes genotypes present in the skin microbiome, opening new avenues for the development of biotherapeutics to manipulate the skin microbiota.
Introduction
The Gram-positive anaerobic Cutibacterium (formerly Propionibacterium) acnes is one of the most dominant species in the pilosebaceous follicle and a key member of the human skin microbiome (Dreno et al., 2018). Originally classified and named as Propionibacterium acnes, it was recently reclassified as Cutibacterium acnes (Scholz and Kilian, 2016), a nomenclature broadly adopted despite discordance in the scientific community (Alexeyev et al., 2018; Dekio et al., 2019). Recent skin microbiome studies have attempted to decipher differences in microbial populations associated with healthy skin vs. disease states, and potential associations with gut microbiome composition and host immune health (Lee et al., 2019). Several studies have established that Cutibacterium, Corynebacterium, Staphylococcus, and Streptococcus are important members of the skin microbiota (Christensen et al., 2016; Barnard and Li, 2017) and observed a reduction in Cutibacterium relative abundance in certain skin diseases such as psoriasis, atopic dermatitis and rosacea (Kong et al., 2012; Chang et al., 2018; Woo et al., 2020). Due to the clinical importance of acne vulgaris, there is increasing interest in studying the skin microbiome and deciphering differences between healthy and disease populations regarding microbial, immunological and hormonal factors (Lee et al., 2019; McLaughlin et al., 2019; Ramasamy et al., 2019; Szegedi et al., 2019), as well as in other skin disease like rosacea (Thompson et al., 2020; Woo et al., 2020).
Cutibacterium acnes has several distinct genotypes within this species which can co-exist in healthy skin but lead to acne in cases where relative abundance shifts (McLaughlin et al., 2019). Three phylogroups or clades have been described, type I, II and III (McDowell et al., 2008), with several sub-clades described within type I (McLaughlin et al., 2019). While the type I clade is widely present in healthy individuals it is also enriched in acne vulgaris, with significantly higher relative abundance of sub-clades IA1 and IC. In contrast, sub-clades IA2, IB, and type II are more prevalent on healthy skin (Fitz-Gibbon et al., 2013; McLaughlin et al., 2019). Noteworthy, while type I and type II clades are present in both healthy and acne skin, albeit in different proportions. The type III clade is exclusively present in healthy skin and absent from acne-associated samples (McLaughlin et al., 2019), though it can be present in other skin diseases such as progressive macular hypomelanosis (Barnard et al., 2016; Petersen et al., 2017). Overall, a healthy skin typically presents a more balanced distribution of diverse C. acnes phylotypes compared to acnes disease, where lower diversity is often associated with type IA1 enrichment. As a result of the genetic diversity of Cutibacterium, recent efforts have focused on taxonomy and genotyping to compare strains associated with acne vulgaris. Originally, methods were based on PCR amplification and sequencing of variable sequences such as 16S rRNA, RecA, and tly, as well as multilocus sequence typing (MLST) encompassing seven to nine genes (McDowell et al., 2011, 2012, 2013; Dreno et al., 2018; Bangayan et al., 2020). More recently, the advent of next-generation sequencing (NGS) and bioinformatic technologies has enabled the determination of the genomes of numerous isolates. However, no extensive comparative genomic analysis has been performed to date in C. acnes, nor have their CRISPR-Cas systems been characterized and used for genotyping purposes. Unraveling the unique genetic determinants associated with each clade and sub-clade of this particular species will provide new insights into the skin microbiome, the virulence or pathogenicity of specific sub-clades and the skin virome based on the CRISPR spacers content.
Here, we carried out a comparative genomic analyses of C. acnes to investigate the phylogenetic relationship among 255 C. acnes strains, characterized CRISPR-Cas loci and provide insights into key genetic determinants that account for specific clades and sub-clades.
Materials and Methods
Genome Annotation
The 255 chromosomal sequences of Cutibacterium acnes available at NCBI on October 2019 were retrieved (Supplementary File 1). To ensure consistency in open reading frame (ORFs) identification and annotation, all genomes were reannotated with Prokka v1.13.3 with standard options (Seemann, 2014), using Prodigal (Hyatt et al., 2010) for gene prediction.
Core and Pan Genome Analyses
The core and pan genome analyses were carried out using Roary v3.12.0 (Page et al., 2015) with the flags -env -i 95 -cd 100, for a minimum percentage identity of 95% for BLASTp and strict threshold for the consideration of core genes, only if they occur in 100% of the genomes. Briefly, the predicted ORFs of each genome were used to perform the pan genome analyses to identify the total genes present in the pan genome, core genes, unique genes and new genes. The pan genome and new genes graph were depicted in RStudio v1.1.463 (R Core Team, 2015) using the create_pan_genome_plots script1. The openness of the pan genome was calculated according to Heap’s law (Tettelin et al., 2008). The functional COGs were assigned to pan-, core- and unique genes using EggNOG 5.02 (Huerta-Cepas et al., 2019) and then depicted in RStudio using ggplot2 (Wickham, 2009).
The heatmap containing the gene presence/absence results of the pangenome was generated using roary_plots.py script3. A customized version was generated in RStudio using heatmap.24 performing hierarchical clustering for both, genomes (rows) and genes (columns). The unique genes existing in each clade were identified using the query_pan_genome option implemented in Roary v3.12.0 (Page et al., 2015) with the flags: -a difference -g clustered_proteins -c 100. Similarly, the common genes shared among two groups were identified using the query_pan_genome with the flags: -a intersection -g clustered_proteins -c 100. The Venn Diagram was depicted in RStudio using the VennDiagram package.
Phylogenomic Analyses
The 1,194 core genes shared among the 255 C. acnes strains were aligned using the PRANK algorithm implemented in Roary v3.12.0 (Page et al., 2015) while performing core- pan genome analysis (see previous section). The resulting multi-FASTA alignment file was used as input in FastTree5 to infer the phylogenomic tree with generalized time reversible model (GTR), using standard flags -nt -gtr. The resulting output newick format file was used to depict the final tree using FigTree v1.4.46.
Identification of Virulence Factors
Virulence factors within C. acnes genomes were investigated using BLAST +. Briefly, a local blastx was performed creating a database with the amino acid sequence of previously identified virulent factors on C. acnes KPA171202 (McLaughlin et al., 2019; Supplementary Table 3), a type IB strain, to search among the other 254 genomes used as query. The results were hand-curated and only proteins with 70% identity across the full alignment were considered further. Finally, the blastx results for each virulent protein were represented as a heatmap displaying the percentage of identity with a blue gradient using the Heatmap package7 in RStudio (R Core Team, 2015), also representing the absence of each protein in white when applicable.
CRISPR-Cas System Identification and Characterization
CRISPR-Cas systems were identified and subtyped using CRISPRdisco providing the .fna and .faa files of each of the 255 C. acnes annotated genomes as input (Crawley et al., 2018). After identification and manual curation of each CRISPR-Cas locus, the downstream and upstream regions were analyzed to identify the overall chromosomal location and nucleotide conservation of cas genes and flanking sequences. Then, specific strains were selected as representatives for each CRISPR subtype to manually depict CRISPR loci of interest.
The Cas proteins alignment among C. acnes strains and with E. coli K12 type I-E was performed using ClustalW to identify the amino acid identity similarity. Then, the concatenated Cas proteins tree was generated using the genetic distance model Jukes-Cantor and the Neighbor-Joining tree build method. The final tree was depicted using FigTree v1.4.4 (see text footnote 6).
The CRISPR array, including the CRISPR repeats and CRISPR spacers, were identified, automatically extracted, visualized and aligned using CRISPRviz (Nethery and Barrangou, 2019a). Then, spacers for each strain were concatenated using the append_spacers.sh script implemented in CRISPRutils (Nethery and Barrangou, 2019b)8. The target (protospacer) identity from putative invasive nucleic acids was analyzed using BLAST (Camacho et al., 2009), with the flags: blastn -evalue 1e-3 -remote -db nt -outfmt 5. The results were trimmed to discard matches not related to phage, plasmid or prophages using blast_parser.py with the flags -ct implemented in CRISPRutils. The trimmed output file was used to depict a heatmap in RStudio, reflecting the number of positive hits in different targets. Finally, the alignment of the spacer-protospacer match also containing the flanking regions was extracted using blast_parser.py with the flag -p as implemented in CRISPRutils. The flank alignment file was used to represent the protospacer adjacent motif (PAM) sequence in WebLogo server (Crooks et al., 2004) based on a frequency chart where the height of each nucleotide represents the conservation of that nucleotide at each position.
The CRISPR repeat sequence was subjected to RNA folding analyses using NUPACK9 (Zadeh et al., 2011) to predict the hairpin structure of the mature CRISPR RNA (crRNA) and depicted by hand.
Statistical Analyses
Data distribution was analyzed using the Shapiro–Wilk test for genome size, %GC content, and number of predicted genes, under the hypothesis that each variable follows a Gaussian distribution. The resulting p-values < 0.05 displayed no normal distribution and therefore, non-parametric tests were used for subsequent analysis. The Kruskal–Wallis test was used to compare the number of genes between strain types to assess differences among groups, under the hypothesis of equal means among groups. Comparisons with a p-value < 0.05 were considered statistically significant. The statistical analyses were performed in R studio v1.1.463.
Results
Cutibacterium acnes Core and Pan Genome Determination
The initial genome screening of the 255 genomes of C. acnes publicly available at NCBI (October 2019) (Supplementary File 1) did not reveal significant overall differences in genome size (2.51 Mbp ± 0.04) (Mean ± SD), or GC content (60.1% ± 0.08). The number of putative protein-coding sequences in genomes varied between 2,214 and 2,567, with an average of 2,331 ± 40.31. However, there was a significantly (p-value < 0.005) higher number of genes detected in type III strains (2,469 ± 38.82) (Figure 1A). Type III strain had a larger genome size (65 Kb) than the average type I and type II strains (2.57 Mbp ± 0.04 vs. 2.51 Mbp ± 0.036, respectively), although differences in available genomes for each strain type (203 type I, 47 type II, 5 type III) may have influenced this observation. No correlation was observed between the number of genes predicted and the number of contigs of the genome, suggesting that sequencing depth and genome assembly quality was not an influencing factor for subsequent analyses (Figure 1B). In fact, 80.78% (206/255) of the genomes have less than 20 contigs. Overall, the “typical” C. acnes genome is similar to C. avidum (2.5 Mbp), but slightly bigger than other Cutibacterium species like C. granulosum (2.18 Mbp). Noteworthy, the Propionibacterium genus, where C. acnes was previously classified (Scholz and Kilian, 2016), contains species which genome size ranging between 2.6 and 5 Mbp, with a 67% GC content, relatively higher than the 60% displayed by C. acnes.
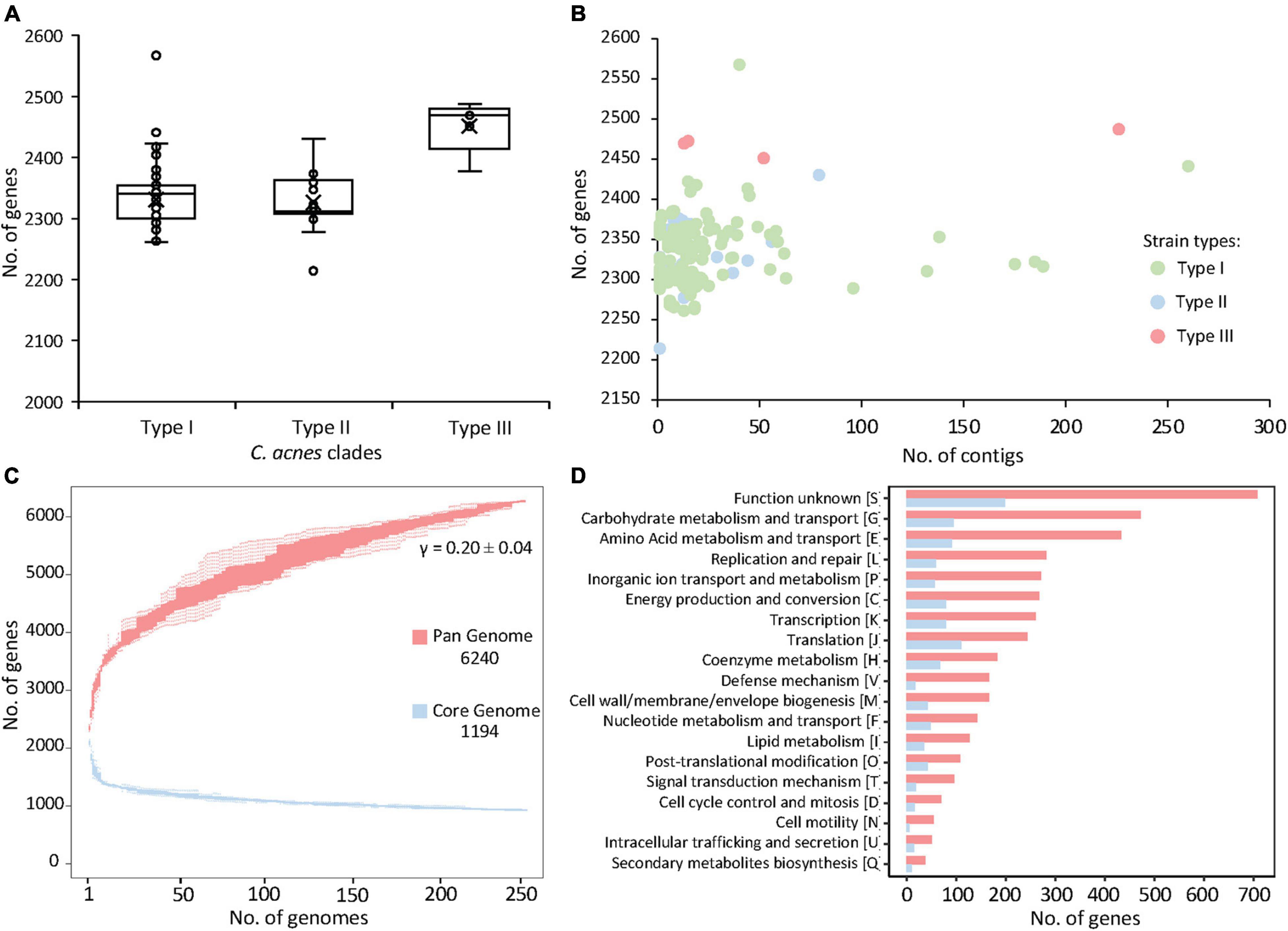
Figure 1. Pan and core genome of the 255 Cutibacterium acnes. (A) Distribution of the number of genes per genome with C. acnes strains organized by clades. (B) Number of genes plotted against the number of contigs of the same genome. (C) The number of genes increases in the pan genome (red) and decreases in the core genome (blue) with the addition of more genomes. (D) Number of genes in the pan genome (red) and core genome (blue) corresponding to each functional category.
To assess genomic conservation across the 255 strains, the total coding sequences were used to determine the pan genome using Roary (Page et al., 2015). This analysis revealed a total of 6,240 genes representing the pan genome of 255 C. acnes strains (Figure 1C). Remarkably, the pan genome did not reach a plateau, as the addition of each new genome still increased the number of genes in the pan genome (Figure 1C). The Heap’s law calculation (Tettelin et al., 2008) corroborated the openness (γ > 0) of this pan genome with γ = 0.20 ± 0.04 and K = 3.32 ± 0.10. Of the 6,240 identified coding sequences, 1,194 represent the core genes, shared across all strains. There are 5,046 accessory genes that are present in the pan genome outside the core genes, which contributes to major genetic and presumably phenotypic differences among strains.
The functional categories for the core and pan genes were annotated through EggNOG (Huerta-Cepas et al., 2019) and the number of genes for each functional category was represented with bar graphs (Figure 1D). The majority of the core genes were related to basic biological functions such as carbohydrate and amino acid metabolism, energy production, transcription and translation, and coenzyme metabolism; besides the genes of unknown function. Similar trends were observed with the predicted functional categories of the coding sequences of the pan genome (Figure 1D).
The overall visualization of 6,240 genes occurrence across 255 genomes (1.6 M data points) using a heatmap revealed several distinct groups and subgroups of strains based on the gene presence/absence (Figure 2). For instance, the dendrogram of the hierarchical clustering displayed that two major clades exist, with the bottom branch divided in two sub-clades. These three clades correspond to the three phylogenetic groups previously described in C. acnes and named as type I, II and III strains clades (McLaughlin et al., 2019). The major type I clade (green), containing the majority of genomes, is distinct and separate from C. acnes type II clade (blue) and C. acnes type III clade (red). Noteworthy, the type I clade is further segregated in several sub-clades, that correlate with some of the previously described sub-clades IA, IB and IC (Lomholt and Kilian, 2010; Bruggemann et al., 2012; McLaughlin et al., 2019). The current nomenclature distinguishes only two sub-groups within the sub-clade IA (IA1 and IA2) (Figure 2, column i), however, no consistency in the IA1-IA2 sub-grouping was identified in our analysis (Figure 2, column ii), where several sub-groups exist (see Figure 3). Regarding the type II clade, we identified two main sub-clades (IIA and IIB), that were not previously established. Overall, these results confirm the presence of three main types and several sub-types within the type I and type II clades.
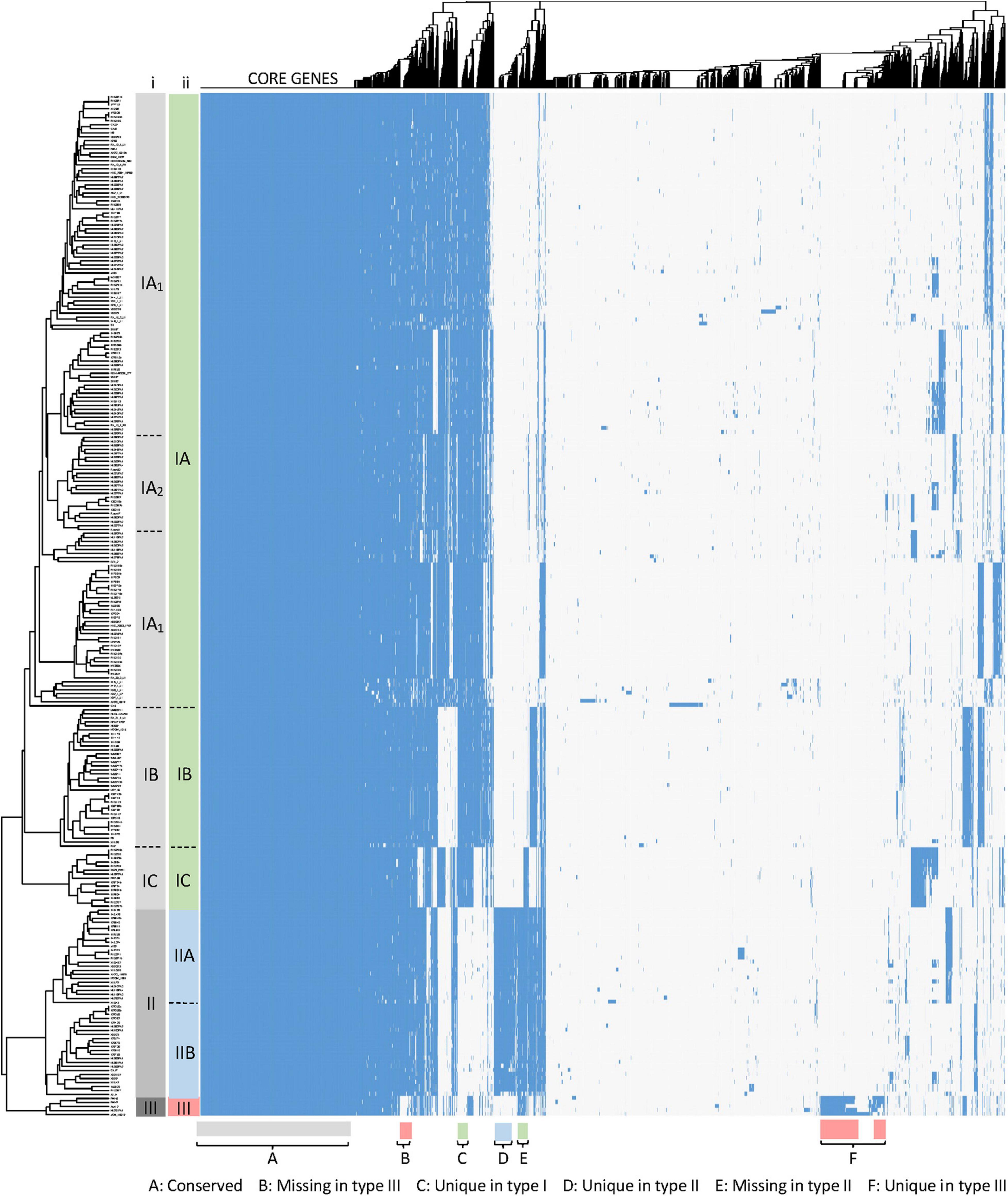
Figure 2. Gene cluster matrix of presence/absence (blue)/(white) of the 6,240 genes (columns) that constitute the pan genome of the 255 C. acnes strains (rows). Hierarchical clustering was performed for both rows and columns and dendrograms were depicted. The main clades and sub-clades of strains are labeled (i) in gray according to the current nomenclature and (ii) color coded for type I, type II and type III, according to our newly proposed classification (see Figure 3). Signature gene blocks of each clade are indicated in the bottom.
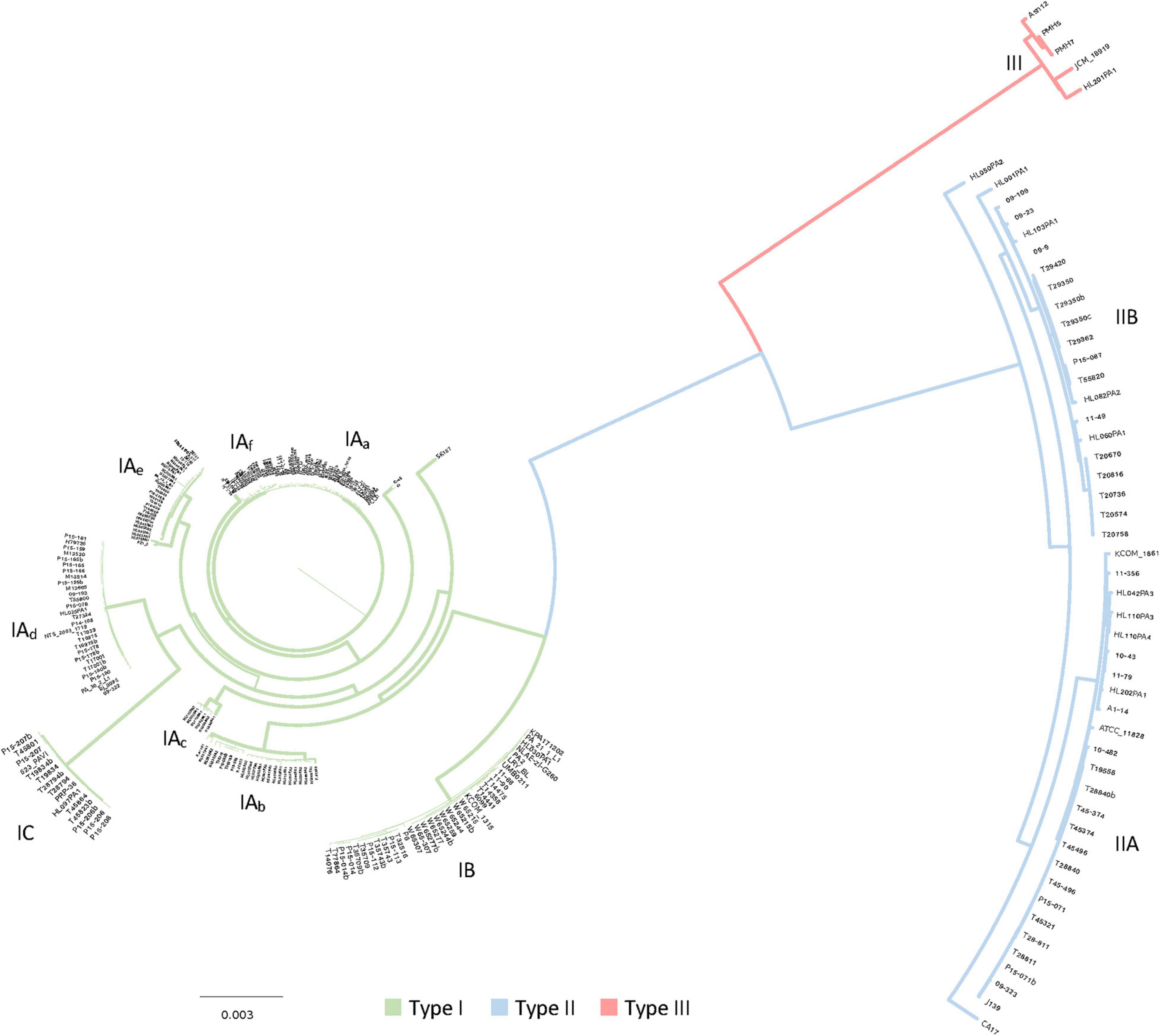
Figure 3. Core genome based phylogenomic tree with the 1,194 core genes detected in the 255 C. acnes strains. The main clades of strains were identified, and color coded in the branches for type I, type II and type III, with green, blue and red respectively. Sub-clades within the type I (IA, IB, and IC) and type II clades (IIA, IIB) were also identified, together with six sub-groups within the sub-clade IA, labeled as IAa to IAf.
Besides the strain clustering, it is important to visualize the gene occurrence patterns (Figure 2), revealing the relative size of the core genome, and variability of the genes representing the pan genome, with groups of genes uniquely present/absent in the three clades (Figure 2, bottom part), and clusters of genes accounting for the various type I subtypes and type II subtypes.
Phylogenomics of Cutibacterium acnes
To further decipher the phylogenetic relationship among the strains we generated a phylogenomic tree based on the alignment of the 1,194 core genes previously detected (see section above). The results of this analysis are consistent with the heatmap of the pan genome, displaying a clear segregation of the three major clades, and the presence of multiple type I sub-clades and type II sub-clades (Figure 3). Within the sub-clade IA, six different subgroups were identified, potentially corresponding to six different genotypes, labeled as IAa to IAf according to the clustering reflected in this phylogenomic tree based on 1,194 core genes (Figure 3). Likewise, the newly proposed sub-clades IIA and IIB, identified in the pan genome results (Figure 2), were also identified here in the phylogenomic tree using the core genes, highlighting the consistency of the strain and genome clustering results. The overall distribution of the 255 genomes among clades and sub-clades, after the phylogenomic analyses was as follow: 153 type IA, 35 type IB, 15 type IC, 24 type IIA, 23 type IIB and 5 type III.
Unique Genes in Various Cutibacterium acnes Clades
To investigate the genetic determinants that drive the differentiation between different strain types, we identified unique genes that occurred in each C. acnes clade, using the query_pan_genome option implemented in Roary (Page et al., 2015). Interestingly, 2,371 genes occurred uniquely in type I strains (in at least one strain) whereas 410 unique genes were identified in type II clade including cas genes from CRISPR-Cas systems (see the corresponding section), and 546 in type III (Figure 4A). These numbers drastically decrease to 51, 1, and 178 unique genes for each clade (I, II, III, respectively) when only the unique genes present in all the strains of each clade were considered. These results suggest that most of the unique genes of each clade are only present in some strains (strain genetic variability) with few of them driving the uniqueness and differentiation of that clade.
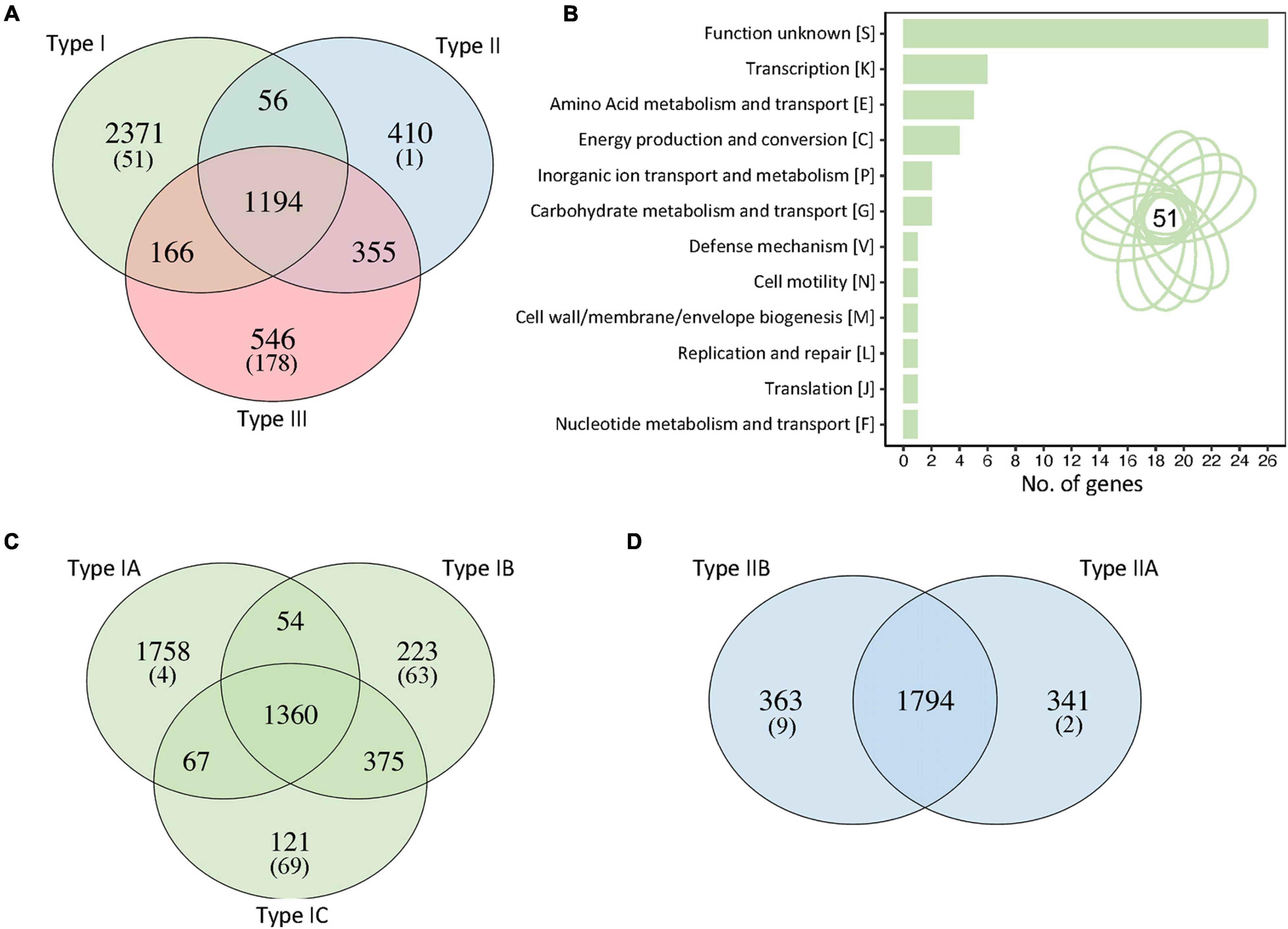
Figure 4. Unique genes in each C. acnes clade. (A) Identification of the number of unique genes for each clade, genes shared by two clades and core genes. The number between brackets represent the number of unique genes present in all the strains of that particular clade. (B) Functional categories of the 51 unique genes present in all type I strains. Manual curation and annotation of these 51 unique genes allowed to elucidate their functionality (see Supplementary Tables 1, 2). (C) Unique genes and shared genes within the sub-clades of type I strains. (D) Unique genes and shared genes within the sub-clades of type II strains.
Then, we focused the downstream analyses on type I clade to understand the genetic determinants of this group related with acne vulgaris disease. From the 2,371 unique genes, only 51 are present in all the type I strains, perhaps representing the key genetic content that distinguishes type I strains from the other two clades (Figure 4B). Functional categories were analyzed and after annotation and manual curation of the 51 genes, 71% of them were assigned to specific functions, although 29% remain with unknown functions (Supplementary Table 1). Interestingly, several unique genes present in type I C. acnes strains are related to carbohydrate metabolism and sugar transporters, like glycosyl hydrolase, trehalose transporters sugA, PTS transporters and ABC transporters; and two of the unique genes displayed homology to the type II toxin-antitoxin system HicAB from E. coli and other pathogens (Supplementary Table 1). Other unique genes identified in type I strains are related to energy production, transport and metabolism pathways such as ATPase associated diverse cellular activities (AAA proteins), N-acetyltransferases, cyanate permease, metal-dependent amidase/carboxypeptidase, and few transcriptional regulators, among others (Supplementary Table 1).
To further investigate the sub-clade clustering in type I strains, the unique genes and core genes within type I sub-clades were identified (Figure 4C). The type IA sub-clade, containing the higher number of strains, also contains the higher number of unique genes (1,758) due to strain diversity, with only four of these unique genes shared among all type IA strains (Figure 4C). Interestingly, sub-clade IB and IC have a similar number of unique genes present in all strains of each sub-clade, 63 and 69 respectively, despite the number of genomes of each sub-clade (35 and 15 respectively), although the total number of unique genes are different (223 and 121). A higher number of genomes increases strain diversity genotype and therefore the number of unique genes. Noteworthy, sub-clades IB and IC shared a higher number of core genes than the other subclades (IA-IB or IA-IC), representing a close phylogenic relationship, as previously established by the pan genome clustering (Figure 2).
Regarding the type II clade, we described the existence of two clear sub-clades, IIA and IIB, based on our pan genome results and phylogenomic analyses with core genes (Figures 2, 3), that were not previously documented in the literature. The unique genes analyses displayed more than three hundred genes in each sub-clade accounting for this sub-aggrupation, but with only a handful of genes present in all the strains of each sub-group (Figure 4D). In this regard, the type IIA clade only displayed two unique genes shared across all twenty-four strains, and these two genes are gatC2 (part of a galactitol-specific PTS system) and NAD-dependent glycerol-3-phosphate dehydrogenase (Supplementary Table 2). For subclade IIB, nine unique genes are present in every single strain out of the twenty-three genomes within of this group, with certain genes related to maltose metabolism, ABC transporter and others (Supplementary Table 2).
Virulence Factors
We investigated the presence of thirty-three virulence factors across all studied genomes and specifically determined whether they could account for C. acnes clade or sub-clade differences in virulence, especially for type I strains in general, and IA strains in particular. Surprisingly, 27 out of 33 virulent proteins were detected in all of the strains, regardless of clade and sub-clade, albeit with slight amino acid sequences differences among strains (Supplementary Figure 1A). Three of the PTRs proteins (PTR-3, PTR-4, PTR-6) were missing in few strains but overall seemed widely distributed across groups. Noteworthy, the amino acid identity of these widely distributed genetic determinants account for the strain clustering in the previously described C. acnes clades (I, II, III) including the differentiation of sub-clades IIA-IIB.
Interestingly, the hyaluronate lyase (HYL), an enzyme involves in the degradation of the main polysaccharide component of the dermal and epidermal matrix (hyaluronan or hyaluronic acid), was detected in both type I and type II clades but completely absent in the type III clade, which correlates with its absence in acne-associated skin. More importantly, the PTR-1_PPA0180 was uniquely detected in type I strains and is very conserved across strains, with no differences in amino acid identity (Supplementary Figure 1A). On the contrary, DeoR_PPA0299 which is a repressor of porphyrin biosynthesis, was absent in some sub-clades of type I, but present in others, and also present in most of type II strains, and all type III genomes.
The Christie Atkins Munch-Petersen (CAMP) factors are membrane pore-forming toxins that act as host tissue degradation enzymes, with a hemolytic and cytotoxic effect involved in colonization and inflammation, and potentially implicated in acne development (Christie et al., 1944; Wang et al., 2018). However, no differences were detected across the five CAMP factors analyzed across strain types. Rather, these virulent proteins seem widespread among C. acnes and not specifically associated with any type. Slight differences in the amino acid identity of the CAMP1_ PPA1340 were detected between type I strains and the type II and type III subsets, ranging from 95.5–100%, with similar findings for CAMP2_PPA0687 (Supplementary Figure 1A).
Finally, the chromosomal location of these 33 virulent genes was investigated to assess the potential existence of genomic islands associated with pathogenicity. However, the virulent factors seem to be widespread across the genome and not located in particular islands, as illustrated using the genome of the type IB strain KPA171202 (Supplementary Figure 1B).
CRISPR-Cas System Identification and Characterization
CRISPR-Cas systems constitute the adaptive immune system of bacteria, against invasive genetic elements such as phages and plasmids (Barrangou et al., 2007). Here, we determined the occurrence and diversity of CRISPR-Cas systems across the 255 C. acnes strains, using previously developed pipelines (Crawley et al., 2018).
Overall, 45 CRISPR-Cas systems subtype I-E were identified (Figure 5A), based on the presence of the corresponding signature cas genes. Interestingly, these 45 type I-E CRISPR-Cas systems occurred exclusively in C. acnes type II strains, and not in type I nor type III strains, in concordance with previous results of a smaller dataset (Bruggemann et al., 2012). Moreover, the type I-E CRISPR-Cas systems seems to be widespread among type II strains with 95.74% strains (45/47) carrying a locus. The detailed analyses of these CRISPR-Cas loci revealed a canonical structure for this subtype I-E (Makarova et al., 2018), encompassing the signature nuclease cas3 involved in DNA cleavage, the associated cas genes that constitute the Cascade complex (Cas8-Cas11-Cas7-Cas5-Cas6) (Brouns et al., 2008), and the cas1 and cas2 genes, involved in new spacer acquisition (Figure 5B). The sequence comparison of the Cas proteins among the 45 C. acnes strains displayed high identity (99%) among strains, whereas the amino acid sequence and protein length was distant from the canonical E. coli I-E Cas proteins with less than 30% identity (Supplementary Figure 2).
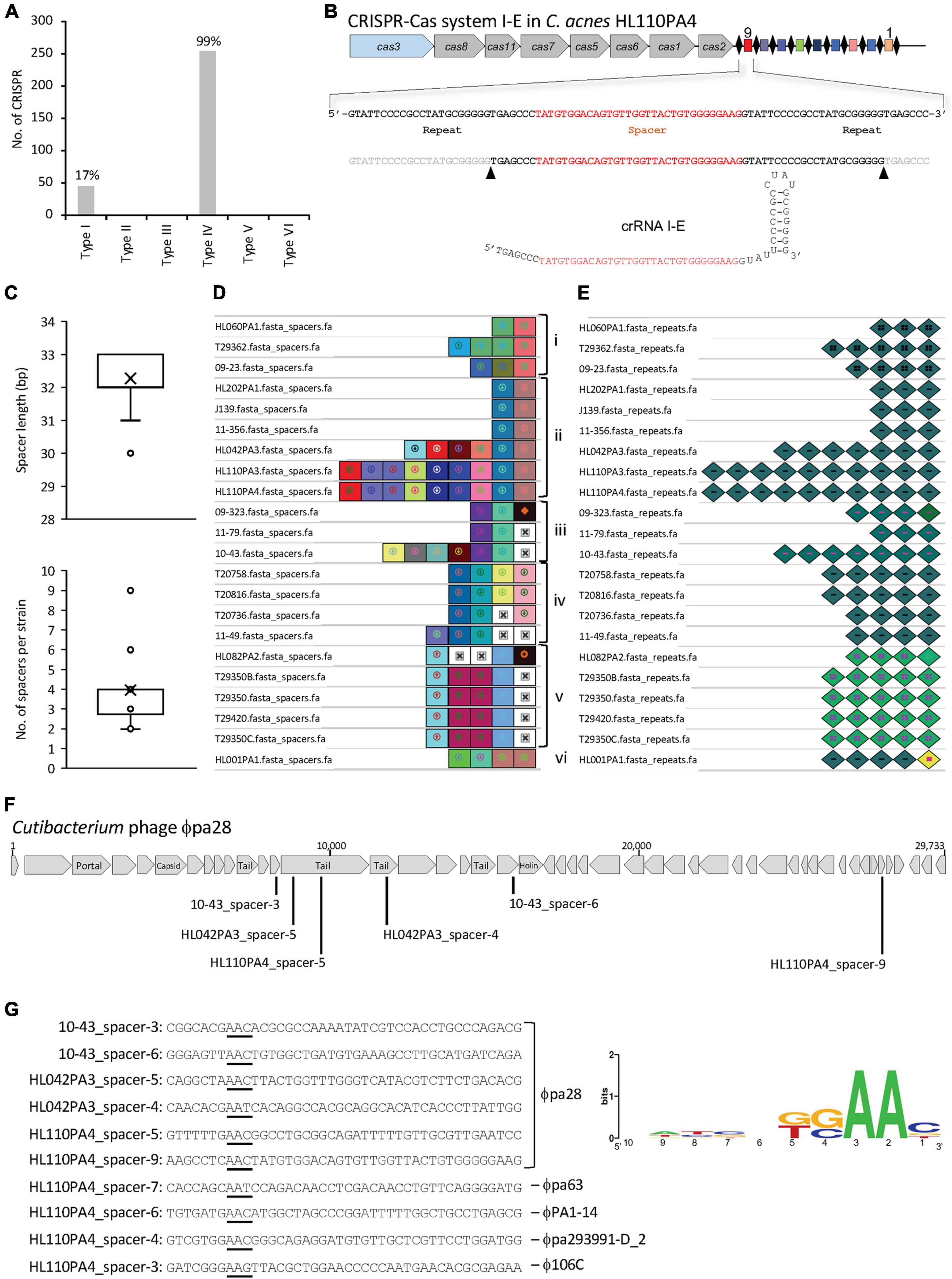
Figure 5. CRISPR-Cas systems in C. acnes. (A) Occurrence and diversity of CRISPR-Cas systems in 255 C. acnes strains. (B) CRISPR-Cas system I-E displaying a canonical profile with signature cas3 gene colored in blue, Cascade in gray, CRISPR array with the repeats as black diamonds and the spacers as colored squares; with the mature CRISPR RNA (crRNA) displaying the hairpin structure on the bottom. (C) Distribution of the spacer length and number of spacers per strain. (D) Visualization of the CRISPR spacers with unique colors and shapes for each unique spacer sequence, aligned based on nucleotide identity for genotyping purposes, with the oldest spacer (ancestor) on the right and the most recently acquire spacer on the left. Groups of strains based on CRISPR spacers-genotyping are labeled from i-vi. (E) Visualization of the CRISPR repeats with unique colors and shapes for each unique repeat sequence. Mutated terminal repeats (corresponding to the ancestor) were identified. (F) Schematic representation of the Cutibacterium phage ϕpa28 targeted by several CRISPR spacers of various C. acnes strains. (G) Protospacer sequences (targeted by CRISPR spacers) with the corresponding protospacer adjacent motif (PAM) on the 5′-end underlined, and representation of the PAM conservation (right panel).
Then, the CRISPR array, encompassing the repeat and spacer sequences, was identified for 22 of the 45 CRISPR-Cas systems previously mentioned, with the remaining 23 not encoding a CRISPR array. For these 22 strains, high cas sequence conservation was detected across strains (99.25% ± 0.44), and noteworthy, the chromosomal location and genetic context of each CRISPR-Cas locus in the genome was exactly the same in all strains, as determined by the analysis of the 10 kb flanking CRISPR loci (data not shown).
The CRISPR repeats are conserved sequences which nucleotide length and structure variable across CRISPR-Cas subtype and bacterial species (Crawley et al., 2018). All the identified loci have a repeat sequence length of 28–29 nucleotides (33.3 and 66% occurrence respectively), with a conserved sequence across strains, with slight variants. The presumed mature CRISPR RNA (crRNA), which comprises a spacer sequence flanked by portions of the CRISPR repeat (Hidalgo-Cantabrana and Barrangou, 2020), was determined using the RNA folding hairpin structure (Figure 5B).
Each CRISPR spacer represents an immunization event providing the bacterium the ability to confront any predator carrying a complementary sequence. The identified CRISPR spacers (n = 87) among the 22 C. acnes strains, displayed a variable length between 30–33 nucleotides, with 32–33 nt being the most prevalent (66.6 and 31% respectively) (Figure 5C, top panel). A range between 2 and 9 spacers were detected for all strains, with an average of four spacers per strain (Figure 5C, bottom panel). Based on the CRISPR-spacers content, six groups were identified among the 22 strains (Figure 5D, i-vi) when the spacers were aligned and visualized (Nethery and Barrangou, 2019a), with the ancestral spacer on the right, representing a common origin for the strains within each group. The strain HL001 was a unique strain that shared no common ancestral space with any other strain. The representation of the CRISPR repeats allowed to identify the mutated terminal repeat in the strains 09-323, HL082PA2 and HL001PA1 (Figure 5E).
The origin of the invasive nucleic acid corresponding to each spacer of the 22 strains was investigated with BLAST (Camacho et al., 2009). Across the 87 spacers, 47 spacers provided 769 hits with 97–100% identity between the spacer and the protospacer (counterpart of the spacer in the invasive nucleic acid). The C. acnes spacers were targeting Propionibacterium-Cutibacterium phages and plasmids, with 694 and 75 hits respectively. Interestingly, several strains contained spacers against the same phage, e.g., Cutibacterium phage ϕpa28, targeting close regions encompassing the tail protein, minor tail protein and other phage proteins (Figure 5F). The spacer-protospacer alignment and the flanking regions were extracted from CRISPRviz (Nethery and Barrangou, 2019b), to identify the existence of a flanking region carrying the protospacer adjacent motif (PAM) (Horvath et al., 2008; Mojica et al., 2009; Marraffini and Sontheimer, 2010). The PAM motif is a conserved sequence, located at the 5′-end of protospacers in type I CRISPR-Cas systems, which is required for DNA binding and cleavage and is also essential for new spacer acquisition. Ten protospacers, targeting different phages and corresponding to spacers from different strains, were considered for the prediction of the PAM 5′-AA-3′ (Figure 5G). These results, reflecting an AT rich PAM are in concordance with previously PAM sequences described for other type I CRISPR-Cas systems in different genera and species (Mulepati and Bailey, 2013; Leenay et al., 2016; Hidalgo-Cantabrana et al., 2017, 2019).
Discussion
We have performed comparative genomics analyses in 255 C. acnes genomes to elucidate the different genotypes existing in this commensal species related to skin health and disease, understanding the strain diversity and the key genetic features that drive the differentiation among groups. The pan genome analyses displayed an open pan genome which is double the size of the pan genome previously described for a smaller data set of 82 C. acnes genomes (Tomida et al., 2013). Although the results of both studies are consistent, previous studies describe a relatively low diversity within this species and an open pan genome. The pan genome representation of gene presence/absence displayed the accessory genes or groups of genes that are responsible for the C. acnes clades and sub-clade differentiation. Besides the well-known existence of three clades (Type I-II-III), we have been able to show for the first time the existence of two main sub-clades in clade II (Figure 2). Moreover, this aggrupation was also consistent on the phylogenomics analyses performed based on nucleotide similarity of the core genes, elucidating also several sub-groups for the sub-clade IA (Figure 3), the most prevalent in acnegenic skin.
Over the last decade, different approaches have been used to understand the genetic diversity and phylogenetic groups of C. acnes mainly based on single locus sequence analyses (16S rRNA, RecA, tly) (Scholz et al., 2014; McDowell, 2017; Yang et al., 2019), multilocus sequence typing (MLST) and more recently combined with whole genome sequence analyses (Lomholt and Kilian, 2010; McDowell et al., 2011, 2012, 2013; Bangayan et al., 2020). While some of those typing methods can identify clonal complexes (CC) and sequence types (STs), they cannot differentiate among the sub-clades of type I strains. In fact, the commonly used ribotypes (RTs) in C. acnes for strain clustering are based on nucleotide differences among the 16S rRNA sequence, with up to 532 ribotypes (Fitz-Gibbon et al., 2013), while the general clustering classification in clades I, II, III was based on RecA sequence, with further classification including the clades and sub-clades IA1, IA2, IB, IC, II and III, according to MLST (McDowell et al., 2005; Lomholt and Kilian, 2010; Fitz-Gibbon et al., 2013). Moreover, 16S ribotypes and specific SNPs variation has been used to trace C. acnes between owner and owner-possessions implementing machine learning models with high accuracy (Yang et al., 2019) but this model does not provide information on the overall strain genotype diversity and taxonomy. Alternatively, Scholz and collaborators defined a SLST typing based on 800 nt sequence specific of C. acnes that allows for precise strain classification (Scholz et al., 2014), however, not suitable for understanding the overall genomic content, strain diversity and the key genetic features of each clade and sub-clade.
Such phylogenomic and comparative genomic analyses have been previously used to study commensal and pathogenic bacteria, especially to understand the epidemiology of a rising pathogen and the genetic content that drives pathogenicity (Bhardwaj and Somvanshi, 2017; Laing et al., 2017; Knight et al., 2019; Nethery et al., 2019; Pan et al., 2020). Our results showed how both the pan genome and the phylogenomic analyses with core genes can be used to study C. acnes population genetics, understand strain diversity, and differentiate between clades, sub-clades and strains, enabling the identification of diverse genotypes in complex microbiotas with higher robustness than other methods. Moreover, the addition of new genomes to the pipeline will provide comprehensive results, allowing to compare among different studies and strains, while enhancing strain clustering without the need of different typing approaches. Thus, such genome analyses should be used for C. acnes typing and taxonomic assignation.
The analyses of the unique genes present in all type I strains allowed to understand the key genetic determinants of this particular clade, highly abundant in acne vulgaris disease. Interestingly, several are related to carbohydrate metabolism and sugar transporters, like glycosyl hydrolase, trehalose transporters sugA, PTS transporters and ABC transporters, which can confer an ecological advantage for growth and colonization with a broader metabolic repertoire to scavenge carbohydrates and energy sources (Supplementary Table 1). Trehalose sugA is part of the complex transporter system specific for the uptake and catabolism of trehalose typically encompasses a transcriptional regulator, a PTS transporter and a carbohydrate hydrolase (Duong et al., 2006). The presence of both the PTS transporter and the hydrolase enzyme are required for trehalose fermentation, as previously demonstrated in Lactobacillus acidophilus with the inactivated-gene mutants unable to grow in the presence of trehalose as carbohydrate source (Duong et al., 2006). Moreover, the ability to uptake this sugar, through translocation of the substrate across the membrane, conferring a metabolic advantage has been described as essential during some stage of infection in M. tuberculosis (Kalscheuer et al., 2010). Trehalose is not synthesized by mammalian cells but is produce by a wide range of organisms like bacteria, yeast and fungi (even insects and plants) and can be secreted to the outside of the cell (Ruhal et al., 2013), so the production of trehalose by other members of the skin microbiome could facilitate the overgrowth of type I C. acnes strains that has the ability to uptake and catabolize this sugar (trehalose catabolism by type I strains was confirmed in our laboratory, data not shown). Thus, it is important to decipher the interplay within the skin microbiome to understand health and disease in the context of a dynamic and complex microbial community.
Noteworthy, two of the unique genes displayed homology to the type II toxin-antitoxin system HicAB present in E. coli, Salmonella enterica, Citrobacter rodentium, and Klebsiella pneumoniae. HicAB consists of two adjacent genes that constitute an operon, where HicA is the toxin and HicB the antitoxin. HicA toxin activity is based on mRNA interference activity causing cleavage of mRNA, whereas HicB forms a stable complex with HicA to inhibit its function (Daimon et al., 2015; Winter et al., 2018). Toxin-antitoxin systems have been associated with the appearance of persister cells that can act as a reservoir for chronic infections and could presumably have a similar impact in acne proliferation. In this regard, HicA overexpression has been associated with cell growth arrest and an increasing number of persister cells tolerant to several antibiotics, in the opportunistic human pathogen Burkholderia pseudomallei and E. coli (Butt et al., 2014). In C. acnes type I strains, the HicA subunit is a small protein of 64 amino acids (aa), and the HicB subunit consists of 123 aa with a 79.7 and 97% identity to homologs in E. coli, respectively. This is relevant since antibiotic treatment has been related to alterations in skin microbial diversity modifying the composition, specifically affecting C. acnes and acne severity (Park et al., 2020). The HicAB system could thus constitute an ecological advantage in C. acnes type I strains that allows overgrowth under certain physiological conditions of the skin (hormonal changes or drug treatments), which has been described in acne, with a relative increase of type I strains overcoming other C. acnes clades and other members of the skin microbiome.
Since the very first C. acnes genome sequence was determined almost two decades ago (Bruggemann et al., 2004), several genetic features have been described as potentially implicated in inflammation or tissue damage induction in the host, like co-hemolytic Christie-Atkins-Munch-Peterson (CAMP) factors, hyaluronate lyase (HYL), lipases, and sialidases and endoglycoceramidases, among others, (Achermann et al., 2014; Dreno et al., 2018; McLaughlin et al., 2019). Likewise, cell-surface associated proteins with multiple proline-threonine repeats (PTRs) and inflammatory potential were previously detected.
We here detected the presence of two different genetic variants (alleles) of HYL with differences between a sub-group of type I and type II strains, and no presence in type III strains, in concordance with previously reported results in smaller datasets (Tyner and Patel, 2015; Scholz et al., 2016; Nazipi et al., 2017). These results reflects that the two HYL alleles are a sub-clade characteristic and they do not randomly occur. Moreover, Nazipi and collaborators demonstrated that the two HYL genetic variants has differential activity degrading hyaluronic acid, with the variant present in type IB and type II strains being highly active (dark blue in our Supplementary Figure 1A), and the variant present in type IA (light blue in our Supplementary Figure 1A) barely functional (Nazipi et al., 2017). Thus, the presence of HYL and its presumed effect on the host tissue, destroying certain components of the skin, could promote inflammation in acne vulgaris disease.
This virulent factor PTR-1_PPA0180, uniquely detected in type I strains (Supplementary Figure 1A), also detected within the 51 unique genes occurring in type I strains (Supplementary Table 1), is an ImmA/IrrE family metallo-endopeptidase involved in xenobiotic response, acting as a DNA binding transcriptional regulator under certain environmental conditions. Differential xenobiotic responses (transcriptional profile based on mRNA) of various members of the gut microbiome have been related to the presence of certain drugs, antibiotics as well as pH variations (Maurice et al., 2013). This metallo-endopeptidase has also been identified in other human opportunistic pathogens such as Mycobacterium tuberculosis, Klebsiella pneumoniae, Staphylococcus aureus, and Nocardia farcinica. Stress response mechanisms, or xenobiotic response to environmental changes, confer tremendous advantages for adaptation to variable environmental conditions, potentially conferring an ecological advantage compared to other species and strains that lack these systems, enabling colonization of and persistence on or in certain environments and human tissues. Other colonization factors like RoxP have been shown to be essential for adherence to and colonization of the skin, but they are ubiquitous in every C. acnes strain, and not associate with specific genotypes (Allhorn et al., 2016). This ImmA/IrrE family metallo-endopeptidase endopeptidase exclusively present in type I strains may be considered as a potential indicator of pathogenicity or virulence in C. acnes and can also be used as a genetic marker for strain genotyping and taxonomy.
Bacterial porphyrin production, which are pro-inflammatory and related to skin disease, is significantly higher in type I strains, compared to types II and III and also compared to other skin bacteria such as C. avidum and C. granulosum (Barnard et al., 2020). The absence of the repressor of porphyrin biosynthesis DeoR_PPA0299 has been implicated in higher production of porphyrins in type IA1 strains isolated from acnegenic skin (Johnson et al., 2016), and porphyrins production and secretion to the perifollicular area has been related to an inflammatory reaction of the follicle. Thus, our findings on the absence of this repressor in type IA1 strains (Supplementary Figure 1A) is consistency with previous literature and correlates with the higher production of porphyrins in the most virulent C. acnes genotype, which is predominant in acne vulgaris disease.
The CAMP factors seem widespread among C. acnes and not specifically associated with any type. Slight differences in the amino acid identity of the CAMP1_ PPA1340 were detected between type I strains and the type II and type III subsets, ranging from 95.5–100%, with similar findings for CAMP2_PPA0687 (Supplementary Figure 1A). The high distribution of CAMP factors across clades and sub-clades was previously described and transcription regulation may be involved, with CAMP-2 highly expressed in type IA and not in type II strains, according to immunoblotting quantification (Valanne et al., 2005). On the contrary, CAMP-1 is highly produced in type II strains and type IB, but not in type IA which are the majority of C. acnes strains.
Overall, previously documented virulent factors are surprisingly widespread among C. acnes strains and clades. This contrasts with previous literature reports, though we note differences in amino acid sequences that support consistent C. acnes clades clustering in the previously described clades. We report the uniquely occurrence of PTR-1_PPA0180 in type I strains, a genetic marker for this clade, and a potential contributor to pathogenicity. Moreover, we corroborated the existence of different alleles of HYL and the absence of deoR in the most virulent sub-clade IA1. Besides genomic content, transcriptomic and proteomics analyses of virulence factors should be considered to assess their relative activity across C. acnes strains and clades, as previously demonstrated for porphyrins, CAMP factors, dermatan-sulfate adhesins, lipase GehA and others (Valanne et al., 2005; Brzuszkiewicz et al., 2011; Johnson et al., 2016; Barnard et al., 2020).
CRISPR-Cas systems represent the adaptive immune system of bacteria and archaea (Barrangou et al., 2007). Understanding their occurrence and diversity provides insights into the predatory challenges that a bacterium is exposed to within a microbiome (Hidalgo-Cantabrana et al., 2018) while also providing a basis for the development of genome editing tools (Hidalgo-Cantabrana and Barrangou, 2020). We here performed a detailed characterization of CRISPR-Cas systems allowing us to elucidate all the necessary elements to repurpose these systems for downstream applications (Figure 5). Interestingly, CRISPR-Cas systems in C. acnes uniquely belong to the type I-E family, and they exclusively occur in type II strains. This association established these CRISPR loci as a specific trait that might be used for strain typing, phylogenetic analysis or taxonomy for clade designation. Moreover, investigating the CRISPR spacers allowed us to elucidate historical and evolutionary relationship among C. acnes strains, with six clear groups identified, based on spacers-genotyping (Figure 5D). These results illustrate the potential of CRISPR spacers for genotyping purposes, for identification and traceability of each strain, while reflecting the divergent evolution of C. acnes from different ancestors. CRISPR spacers have been used for genotyping of human pathogens previously, providing high-resolution phylogeny for pathogenic bacteria such as C. difficile and Salmonella (Bachmann et al., 2014; Shariat et al., 2015; Sola et al., 2015; Andersen et al., 2016; Almeida et al., 2017). Noteworthy, the spacer content displayed that the predatory attacks suffered by C. acnes are mainly related to phages present in the skin microbiome. Although less efforts have been performed understanding the skin virome, rather than the microbiome, certain metagenomically studies have elucidated that the majority of the skin virome is constituted by bacteriophages against Propionoibacterium (Cutibacterium), Staphylococcus, Corynebacterium and Enterococcus among other groups (Hannigan et al., 2015, 2017).
Thus, next generation biotherapeutics for skin health could include alternative technologies to modulate the skin microbiome toward a healthy balance to prevent the increase of type I strain population in acne vulgaris, using probiotics (Goodarzi et al., 2020; O’Neill et al., 2020), including specific strains of C. acnes (Karoglan et al., 2019) or even skin microbiota transplant (Paetzold et al., 2019). Moreover, naturally existing phages, engineered phages and/or heterologous CRISPR-Cas systems provide new opportunities to alter the skin microbiota composition toward a healthy skin with no impact on other members of the microbiome.
Conclusion
With diverse C. acnes strains coexisting within the skin microbiome, molecular typing methods and whole genome sequencing can provide valuable insights into the roles that different phylogenetic clades may play in health vs. disease. Comparative genomics analyses represent a robust approach enabling large genome data sets comparisons. Our results contribute to the overall understanding of the genetic diversity among the different C. acnes clades and sub-clades while elucidating the genetic features that are uniquely present in C. acnes type I clade, the most predominant in acne vulgaris disease. For instance, carbohydrate metabolic pathways, transcriptional regulators and stress response mechanisms are key elements that can potentially confer type I strains an ecological advantage for competitive growth and colonization, outcompeting others C. acnes clades. Elucidating the differential genetic content among C. acnes sub-clades open new avenues to study the mechanisms that drive virulence and pathogenicity, contributing to the development of skin microbiome therapies focused on the selective targeting or manipulation of specific genotypes in acne vulgaris disease.
Data Availability Statement
The 255 chromosomal genome sequences of the Cutibacterium acnes strains analyzed in this study were those publicly available in the NCBI database as of October 2019 (Supplementary File 1).
Author Contributions
NC and CH-C designed the study, performed bioinformatic analyses, and participated in the manuscript drafting. AG edited the manuscript. CH-C and RB coordinated and supervised the study and edited the manuscript. All authors approved the final manuscript.
Funding
This study received funding from NC State University and BASF Corporation.
Conflict of Interest
RB and CH-C are co-inventors on several patents related to CRISPR-Cas systems and their uses. RB is a shareholder of Caribou Biosciences, Intellia Therapeutics, Locus Biosciences, TreeCo, Inari Ag, and CRISPR Biotechnologies. CH-C is a shareholder of Microviable Therapeutics and CRISPR Biotechnologies. AG is an employee of BASF Corporation. The authors declare that this study received funding from BASF Corporation. The funder had the following involvement in the study: edited and approved the final manuscript.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank their colleagues, especially Meichen Pan for bioinformatic support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.758749/full#supplementary-material
Supplementary Figure 1 | Occurrence of virulent genes in C. acnes. (A) Heatmap of the presence/absence (blue/white) and percentage of identity (blue gradient) of 33 virulent genes (columns) across the 255 C. acnes strains used in this study. Hierarchical clustering was performed for both rows and columns and dendrograms were depicted. The main clades of strains were identified, and color coded for type I, type II and type III, with green, blue and red respectively. (B) Chromosomal location of the 33 virulent genes displayed in the strain C. acnes KPA171202 (subtype IB), with GC-AT content represented as blue-green lines.
Supplementary Figure 2 | (A) Cas proteins alignment of the type I-E CRISPR-Cas system in C. acnes HL110PA4 (as representative) and E. coli K12 the displaying the identity (%) at amino acid level. (B) Cas proteins alignment of the 45 C. acnes strains and E. coli K12, and (C) corresponding phylogenetic tree of the concatenated Cas proteins.
Supplementary File 1 | Cutibacterium acnes strains and genome accession numbers.
Footnotes
- ^ https://github.com/sanger-pathogens/Roary/blob/master/bin/create_pan_genome_plots.R
- ^ http://eggnog5.embl.de/#/app/home
- ^ https://github.com/sanger-pathogens/Roary/tree/master/contrib/roary_plots
- ^ https://www.rdocumentation.org/packages/gplots/versions/3.0.3/topics/heatmap.2
- ^ http://www.microbesonline.org/fasttree/
- ^ http://tree.bio.ed.ac.uk/software/figtree/
- ^ https://www.r-graph-gallery.com/heatmap
- ^ https://github.com/CRISPRlab/CRISPRutils
- ^ http://nupack.org/
References
Achermann, Y., Goldstein, E. J., Coenye, T., and Shirtliff, M. E. (2014). Propionibacterium acnes: from commensal to opportunistic biofilm-associated implant pathogen. Clin. Microbiol. Rev. 27, 419–440. doi: 10.1128/CMR.00092-13
Alexeyev, O. A., Dekio, I., Layton, A. M., Li, H., Hughes, H., Morris, T., et al. (2018). Why we continue to use the name Propionibacterium acnes. Br. J. Dermatol. 179:1227. doi: 10.1111/bjd.17085
Allhorn, M., Arve, S., Bruggemann, H., and Lood, R. (2016). A novel enzyme with antioxidant capacity produced by the ubiquitous skin colonizer Propionibacterium acnes. Sci. Rep. 6:36412. doi: 10.1038/srep36412
Almeida, F., Medeiros, M. I., Rodrigues, D. D., Allard, M. W., and Falcao, J. P. (2017). Molecular characterization of Salmonella Typhimurium isolated in Brazil by CRISPR-MVLST. J. Microbiol. Methods 133, 55–61. doi: 10.1016/j.mimet.2016.12.020
Andersen, J. M., Shoup, M., Robinson, C., Britton, R., Olsen, K. E., and Barrangou, R. (2016). CRISPR Diversity and Microevolution in Clostridium difficile. Genome Biol. Evol. 8, 2841–2855. doi: 10.1093/gbe/evw203
Bachmann, N. L., Petty, N. K., Ben Zakour, N. L., Szubert, J. M., Savill, J., and Beatson, S. A. (2014). Genome analysis and CRISPR typing of Salmonella enterica serovar Virchow. BMC Genomics 15:389. doi: 10.1186/1471-2164-15-389
Bangayan, N. J., Shi, B., Trinh, J., Barnard, E., Kasimatis, G., Curd, E., et al. (2020). MG-MLST: Characterizing the Microbiome at the Strain Level in Metagenomic Data. Microorganisms 8:684. doi: 10.3390/microorganisms8050684
Barnard, E., and Li, H. (2017). Shaping of cutaneous function by encounters with commensals. J. Physiol. 595, 437–450. doi: 10.1113/JP271638
Barnard, E., Johnson, T., Ngo, T., Arora, U., Leuterio, G., McDowell, A., et al. (2020). Porphyrin Production and Regulation in Cutaneous Propionibacteria. mSphere 5, e793–e719. doi: 10.1128/mSphere.00793-19
Barnard, E., Liu, J., Yankova, E., Cavalcanti, S. M., Magalhaes, M., Li, H., et al. (2016). Strains of the Propionibacterium acnes type III lineage are associated with the skin condition progressive macular hypomelanosis. Sci. Rep. 6:31968. doi: 10.1038/srep31968
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712.
Bhardwaj, T., and Somvanshi, P. (2017). Pan-genome analysis of Clostridium botulinum reveals unique targets for drug development. Gene 623, 48–62. doi: 10.1016/j.gene.2017.04.019
Brouns, S. J., Jore, M. M., Lundgren, M., Westra, E. R., Slijkhuis, R. J., Snijders, A. P., et al. (2008). Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964. doi: 10.1126/science.1159689
Bruggemann, H., Henne, A., Hoster, F., Liesegang, H., Wiezer, A., Strittmatter, A., et al. (2004). The complete genome sequence of Propionibacterium acnes, a commensal of human skin. Science 305, 671–673. doi: 10.1126/science.1100330
Bruggemann, H., Lomholt, H. B., Tettelin, H., and Kilian, M. (2012). CRISPR/cas loci of type II Propionibacterium acnes confer immunity against acquisition of mobile elements present in type I P. acnes. PLoS One 7:e34171. doi: 10.1371/journal.pone.0034171
Brzuszkiewicz, E., Weiner, J., Wollherr, A., Thurmer, A., Hupeden, J., Lomholt, H. B., et al. (2011). Comparative genomics and transcriptomics of Propionibacterium acnes. PLoS One 6:e21581. doi: 10.1371/journal.pone.0021581
Butt, A., Higman, V. A., Williams, C., Crump, M. P., Hemsley, C. M., Harmer, N., et al. (2014). The HicA toxin from Burkholderia pseudomallei has a role in persister cell formation. Biochem. J. 459, 333–344. doi: 10.1042/BJ20140073
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Chang, H. W., Yan, D., Singh, R., Liu, J., Lu, X., Ucmak, D., et al. (2018). Alteration of the cutaneous microbiome in psoriasis and potential role in Th17 polarization. Microbiome 6:154.
Christensen, G. J., Scholz, C. F., Enghild, J., Rohde, H., Kilian, M., Thurmer, A., et al. (2016). Antagonism between Staphylococcus epidermidis and Propionibacterium acnes and its genomic basis. BMC Genomics 17:152. doi: 10.1186/s12864-016-2489-5
Christie, R., Atkins, N. E., and Munch-Petersen, E. (1944). A note on a lytic phenomenon shown by group B streptococci. Aust. J. Exp. Biol. 22, 197–200. doi: 10.1038/icb.1945.30
Crawley, A. B., Henriksen, J. R., and Barrangou, R. (2018). CRISPRdisco: An Automated Pipeline for the Discovery and Analysis of CRISPR-Cas Systems. CRISPR J. 2018, 171–181. doi: 10.1089/crispr.2017.0022
Crooks, G. E., Hon, G., Chandonia, J. M., and Brenner, S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. doi: 10.1101/gr.849004
Daimon, Y., Narita, S., and Akiyama, Y. (2015). Activation of Toxin-Antitoxin System Toxins Suppresses Lethality Caused by the Loss of sigmaE in Escherichia coli. J. Bacteriol. 197, 2316–2324. doi: 10.1128/JB.00079-15
Dekio, I., McDowell, A., Sakamoto, M., Tomida, S., and Ohkuma, M. (2019). Proposal of new combination, Cutibacterium acnes subsp. elongatum comb. nov., and emended descriptions of the genus Cutibacterium, Cutibacterium acnes subsp. acnes and Cutibacterium acnes subsp. defendens. Int. J. Syst. Evol. Microbiol. 69, 1087–1092. doi: 10.1099/ijsem.0.003274
Dreno, B., Pecastaings, S., Corvec, S., Veraldi, S., Khammari, A., and Roques, C. (2018). Cutibacterium acnes (Propionibacterium acnes) and acne vulgaris: a brief look at the latest updates. J. Eur. Acad. Dermatol. Venereol. 32, (Suppl. 2), 5–14. doi: 10.1111/jdv.15043
Duong, T., Barrangou, R., Russell, W. M., and Klaenhammer, T. R. (2006). Characterization of the tre locus and analysis of trehalose cryoprotection in Lactobacillus acidophilus NCFM. Appl. Environ. Microbiol. 72, 1218–1225. doi: 10.1128/AEM.72.2.1218-1225.2006
Fitz-Gibbon, S., Tomida, S., Chiu, B. H., Nguyen, L., Du, C., Liu, M., et al. (2013). Propionibacterium acnes strain populations in the human skin microbiome associated with acne. J. Invest. Dermatol. 133, 2152–2160. doi: 10.1038/jid.2013.21
Goodarzi, A., Mozafarpoor, S., Bodaghabadi, M., and Mohamadi, M. (2020). The potential of probiotics for treating acne vulgaris: A review of literature on acne and microbiota. Dermatol. Ther. 2020:e13279.
Hannigan, G. D., Meisel, J. S., Tyldsley, A. S., Zheng, Q., Hodkinson, B. P., SanMiguel, A. J., et al. (2015). The human skin double-stranded DNA virome: topographical and temporal diversity, genetic enrichment, and dynamic associations with the host microbiome. mBio 6, e1578–e1515. doi: 10.1128/mBio.01578-15
Hannigan, G. D., Zheng, Q., Meisel, J. S., Minot, S. S., Bushman, F. D., and Grice, E. A. (2017). Evolutionary and functional implications of hypervariable loci within the skin virome. PeerJ. 5:e2959. doi: 10.7717/peerj.2959
Hidalgo-Cantabrana, C., and Barrangou, R. (2020). Characterization and applications of Type I CRISPR-Cas systems. Biochem. Soc. Trans. 48, 15–23. doi: 10.1042/bst20190119
Hidalgo-Cantabrana, C., Crawley, A. B., Sanchez, B., and Barrangou, R. (2017). Characterization and Exploitation of CRISPR Loci in Bifidobacterium longum. Front. Microbiol. 8:1851. doi: 10.3389/fmicb.2017.01851
Hidalgo-Cantabrana, C., Goh, Y. J., Pan, M., Sanozky-Dawes, R., and Barrangou, R. (2019). Genome editing using the endogenous type I CRISPR-Cas system in Lactobacillus crispatus. Proc. Natl. Acad. Sci. U S A. 116, 15774–15783. doi: 10.1073/pnas.1905421116
Hidalgo-Cantabrana, C., Sanozky-Dawes, R., and Barrangou, R. (2018). Insights into the Human Virome Using CRISPR Spacers from Microbiomes. Viruses 10:479. doi: 10.3390/v10090479
Horvath, P., Romero, D. A., Coute-Monvoisin, A. C., Richards, M., Deveau, H., Moineau, S., et al. (2008). Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J. Bacteriol. 190, 1401–1412. doi: 10.1128/JB.01415-07
Huerta-Cepas, J., Szklarczyk, D., Heller, D., Hernandez-Plaza, A., Forslund, S. K., Cook, H., et al. (2019). eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314. doi: 10.1093/nar/gky1085
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 11:119. doi: 10.1186/1471-2105-11-119
Johnson, T., Kang, D., Barnard, E., and Li, H. (2016). Strain-Level Differences in Porphyrin Production and Regulation in Propionibacterium acnes Elucidate Disease Associations. mSphere 1, e23–e15. doi: 10.1128/mSphere.00023-15
Kalscheuer, R., Weinrick, B., Veeraraghavan, U., Besra, G. S., and Jacobs, W. R. Jr. (2010). Trehalose-recycling ABC transporter LpqY-SugA-SugB-SugC is essential for virulence of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U S A. 107, 21761–21766. doi: 10.1073/pnas.1014642108
Karoglan, A., Paetzold, B., Pereira, de Lima, J., Bruggemann, H., Tuting, T., et al. (2019). Safety and Efficacy of Topically Applied Selected Cutibacterium acnes Strains over Five Weeks in Patients with Acne Vulgaris: An Open-label, Pilot Study. Acta Derm. Venereol. 99, 1253–1257. doi: 10.2340/00015555-3323
Knight, D. R., Kullin, B., Androga, G. O., Barbut, F., Eckert, C., Johnson, S., et al. (2019). Evolutionary and Genomic Insights into Clostridioides difficile Sequence Type 11: a Diverse Zoonotic and Antimicrobial-Resistant Lineage of Global One Health Importance. MBio 10, e446–e419. doi: 10.1128/mBio.00446-19
Kong, H. H., Oh, J., Deming, C., Conlan, S., Grice, E. A., Beatson, M. A., et al. (2012). Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 22, 850–859. doi: 10.1101/gr.131029.111
Laing, C. R., Whiteside, M. D., and Gannon, V. P. J. (2017). Pan-genome Analyses of the Species Salmonella enterica, and Identification of Genomic Markers Predictive for Species, Subspecies, and Serovar. Front. Microbiol. 8:1345. doi: 10.3389/fmicb.2017.01345
Lee, Y. B., Byun, E. J., and Kim, H. S. (2019). Potential Role of the Microbiome in Acne: A Comprehensive Review. J. Clin. Med. 8:987. doi: 10.3390/jcm8070987
Leenay, R. T., Maksimchuk, K. R., Slotkowski, R. A., Agrawal, R. N., Gomaa, A. A., Briner, A. E., et al. (2016). Identifying and Visualizing Functional PAM Diversity across CRISPR-Cas Systems. Mol. Cell. 62, 137–147. doi: 10.1016/j.molcel.2016.02.031
Lomholt, H. B., and Kilian, M. (2010). Population genetic analysis of Propionibacterium acnes identifies a subpopulation and epidemic clones associated with acne. PLoS One 5:e12277. doi: 10.1371/journal.pone.0012277
Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2018). Classification and Nomenclature of CRISPR-Cas Systems: Where from Here? CRISPR J. 1, 325–336. doi: 10.1089/crispr.2018.0033
Marraffini, L. A., and Sontheimer, E. J. (2010). Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463, 568–571. doi: 10.1038/nature08703
Maurice, C. F., Haiser, H. J., and Turnbaugh, P. J. (2013). Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 152, 39–50. doi: 10.1016/j.cell.2012.10.052
McDowell, A. (2017). Over a Decade of recA and tly Gene Sequence Typing of the Skin Bacterium Propionibacterium acnes: What Have We Learnt? Microorganisms 6:6010001. doi: 10.3390/microorganisms6010001
McDowell, A., Barnard, E., Nagy, I., Gao, A., Tomida, S., Li, H., et al. (2012). An expanded multilocus sequence typing scheme for Propionibacterium acnes: investigation of ‘pathogenic’, ‘commensal’ and antibiotic resistant strains. PLoS One 7:e41480. doi: 10.1371/journal.pone.0041480
McDowell, A., Gao, A., Barnard, E., Fink, C., Murray, P. I., Dowson, C. G., et al. (2011). A novel multilocus sequence typing scheme for the opportunistic pathogen Propionibacterium acnes and characterization of type I cell surface-associated antigens. Microbiology 157, 1990–2003. doi: 10.1099/mic.0.049676-0
McDowell, A., Nagy, I., Magyari, M., Barnard, E., and Patrick, S. (2013). The opportunistic pathogen Propionibacterium acnes: insights into typing, human disease, clonal diversification and CAMP factor evolution. PLoS One 8:e70897. doi: 10.1371/journal.pone.0070897
McDowell, A., Perry, A. L., Lambert, P. A., and Patrick, S. (2008). A new phylogenetic group of Propionibacterium acnes. J. Med. Microbiol. 57, 218–224. doi: 10.1099/jmm.0.47489-0
McDowell, A., Valanne, S., Ramage, G., Tunney, M. M., Glenn, J. V., McLorinan, G. C., et al. (2005). Propionibacterium acnes types I and II represent phylogenetically distinct groups. J. Clin. Microbiol. 43, 326–334. doi: 10.1128/JCM.43.1.326-334.2005
McLaughlin, J., Watterson, S., Layton, A. M., Bjourson, A. J., Barnard, E., and McDowell, A. (2019). Propionibacterium acnes and Acne Vulgaris: New Insights from the Integration of Population Genetic, Multi-Omic, Biochemical and Host-Microbe Studies. Microorganisms 7:128. doi: 10.3390/microorganisms7050128
Mojica, F. J., Diez-Villasenor, C., Garcia-Martinez, J., and Almendros, C. (2009). Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155, 733–740. doi: 10.1099/mic.0.023960-0
Mulepati, S., and Bailey, S. (2013). In vitro reconstitution of an Escherichia coli RNA-guided immune system reveals unidirectional, ATP-dependent degradation of DNA target. J. Biol. Chem. 288, 22184–22192. doi: 10.1074/jbc.M113.472233
Nazipi, S., Stodkilde-Jorgensen, K., Scavenius, C., and Bruggemann, H. (2017). The Skin Bacterium Propionibacterium acnes Employs Two Variants of Hyaluronate Lyase with Distinct Properties. Microorganisms 5:57. doi: 10.3390/microorganisms5030057
Nethery, M. A., and Barrangou, R. (2019a). CRISPR Visualizer: rapid identification and visualization of CRISPR loci via an automated high-throughput processing pipeline. RNA Biol. 16, 577–584. doi: 10.1080/15476286.2018.1493332
Nethery, M. A., and Barrangou, R. (2019b). Predicting and visualizing features of CRISPR-Cas systems. Methods Enzymol. 616, 1–25. doi: 10.1016/bs.mie.2018.10.016
Nethery, M. A., Henriksen, Daughtry, K. V., Johanningsmeier, S. D., and Barrangou, R. (2019). Comparative genomics of eight Lactobacillus buchneri strains isolated from food spoilage. BMC Genomics 20:902. doi: 10.1186/s12864-019-6274-0
O’Neill, A. M., Nakatsuji, T., Hayachi, A., Williams, M. R., Mills, R. H., Gonzalez, D. J., et al. (2020). Identification of a Human Skin Commensal Bacterium that Selectively Kills Cutibacterium acnes. J. Invest. Dermatol. 140, 1619.e–1628.e. doi: 10.1016/j.jid.2019.12.026
Paetzold, B., Willis, J. R., Pereira, de Lima, J., Knodlseder, N., Bruggemann, H., et al. (2019). Skin microbiome modulation induced by probiotic solutions. Microbiome 7:95. doi: 10.1186/s40168-019-0709-3
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Pan, M., Hidalgo-Cantabrana, C., and Barrangou, R. (2020). Host and body site-specific adaptation of Lactobacillus crispatus genomes. NAR Genomics Bioinformat. 2:lqaa001. doi: 10.1093/nargab/lqaa001
Park, S. Y., Kim, H. S., Lee, S. H., and Kim, S. (2020). Characterization and Analysis of the Skin Microbiota in Acne: Impact of Systemic Antibiotics. J. Clin. Med. 9:168. doi: 10.3390/jcm9010168
Petersen, R. L., Scholz, C. F., Jensen, A., Bruggemann, H., and Lomholt, H. B. (2017). Propionibacterium Acnes Phylogenetic Type III is Associated with Progressive Macular Hypomelanosis. Eur. J. Microbiol. Immunol. 7, 37–45. doi: 10.1556/1886.2016.00040
Ramasamy, S., Barnard, E., Dawson, T. L. Jr., and Li, H. (2019). The role of the skin microbiota in acne pathophysiology. Br. J. Dermatol. 181, 691–699. doi: 10.1111/bjd.18230
Ruhal, R., Kataria, R., and Choudhury, B. (2013). Trends in bacterial trehalose metabolism and significant nodes of metabolic pathway in the direction of trehalose accumulation. Microb. Biotechnol. 6, 493–502. doi: 10.1111/1751-7915.12029
Scholz, C. F. P., and Kilian, M. (2016). The natural history of cutaneous propionibacteria, and reclassification of selected species within the genus Propionibacterium to the proposed novel genera Acidipropionibacterium gen. nov., Cutibacterium gen. nov. and Pseudopropionibacterium gen. nov. Int. J. Syst. Evol. Microbiol. 66, 4422–4432. doi: 10.1099/ijsem.0.001367
Scholz, C. F., Bruggemann, H., Lomholt, H. B., Tettelin, H., and Kilian, M. (2016). Genome stability of Propionibacterium acnes: a comprehensive study of indels and homopolymeric tracts. Sci. Rep. 6, 20662. doi: 10.1038/srep20662
Scholz, C. F., Jensen, A., Lomholt, H. B., Bruggemann, H., and Kilian, M. (2014). A novel high-resolution single locus sequence typing scheme for mixed populations of Propionibacterium acnes in vivo. PLoS One 9:e104199. doi: 10.1371/journal.pone.0104199
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shariat, N., Timme, R. E., Pettengill, J. B., Barrangou, R., and Dudley, E. G. (2015). Characterization and evolution of Salmonella CRISPR-Cas systems. Microbiology 161, 374–386. doi: 10.1099/mic.0.000005
Sola, C., Abadia, E., Le Hello, S., and Weill, F. X. (2015). High-Throughput CRISPR Typing of Mycobacterium tuberculosis Complex and Salmonella enterica Serotype Typhimurium. Methods Mol. Biol. 1311, 91–109. doi: 10.1007/978-1-4939-2687-9_6
Szegedi, A., Dajnoki, Z., Biro, T., Kemeny, L., and Torocsik, D. (2019). Acne: Transient Arrest in the Homeostatic Host-Microbiota Dialog? Trends Immunol. 40, 873–876. doi: 10.1016/j.it.2019.08.006
Tettelin, H., Riley, D., Cattuto, C., and Medini, D. (2008). Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 12, 472–477. doi: 10.1016/j.mib.2008.09.006
Thompson, K. G., Rainer, B. M., Antonescu, C., Florea, L., Mongodin, E. F., Kang, S., et al. (2020). Comparison of the skin microbiota in acne and rosacea. Exp. Dermatol. 30, 1375–1380. doi: 10.1111/exd.14098
Tomida, S., Nguyen, L., Chiu, B. H., Liu, J., Sodergren, E., Weinstock, G. M., et al. (2013). Pan-genome and comparative genome analyses of Propionibacterium acnes reveal its genomic diversity in the healthy and diseased human skin microbiome. mBio 4, e00003–e00013. doi: 10.1128/mBio.00003-13
Tyner, H., and Patel, R. (2015). Hyaluronidase in Clinical Isolates of Propionibacterium acnes. Int. J. Bacteriol. 2015:218918.
Valanne, S., McDowell, A., Ramage, G., Tunney, M. M., Einarsson, G. G., O’Hagan, S., et al. (2005). CAMP factor homologues in Propionibacterium acnes: a new protein family differentially expressed by types I and II. Microbiology 151, 1369–1379. doi: 10.1099/mic.0.27788-0
Wang, Y., Hata, T. R., Tong, Y. L., Kao, M. S., Zouboulis, C. C., Gallo, R. L., et al. (2018). The Anti-Inflammatory Activities of Propionibacterium acnes CAMP Factor-Targeted Acne Vaccines. J. Invest. Dermatol. 138, 2355–2364. doi: 10.1016/j.jid.2018.05.032
Wickham, H. (2009). ggplot2: Elegant Graphics for Data Analysis. New York, NY: Springer-Verlag New York.
Winter, A. J., Williams, C., Isupov, M. N., Crocker, H., Gromova, M., Marsh, P., et al. (2018). The molecular basis of protein toxin HicA-dependent binding of the protein antitoxin HicB to DNA. J. Biol. Chem. 293, 19429–19440. doi: 10.1074/jbc.RA118.005173
Woo, Y. R., Lee, S. H., Cho, S. H., Lee, J. D., and Kim, H. S. (2020). Characterization and Analysis of the Skin Microbiota in Rosacea: Impact of Systemic Antibiotics. J. Clin. Med. 9:185. doi: 10.3390/jcm9010185
Yang, J., Tsukimi, T., Yoshikawa, M., Suzuki, K., Takeda, T., Tomita, M., et al. (2019). Cutibacterium acnes (Propionibacterium acnes) 16S rRNA Genotyping of Microbial Samples from Possessions Contributes to Owner Identification. mSystems 4, e594–e519. doi: 10.1128/mSystems.00594-19
Keywords: CRISPR, genomics, Cas, genotyping, phylogeny
Citation: Cobian N, Garlet A, Hidalgo-Cantabrana C and Barrangou R (2021) Comparative Genomic Analyses and CRISPR-Cas Characterization of Cutibacterium acnes Provide Insights Into Genetic Diversity and Typing Applications. Front. Microbiol. 12:758749. doi: 10.3389/fmicb.2021.758749
Received: 14 August 2021; Accepted: 15 October 2021;
Published: 03 November 2021.
Edited by:
Guangcai Duan, Zhengzhou University, ChinaReviewed by:
Pedro H. Oliveira, Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), FranceYingjun Li, Huazhong Agricultural University, China
Copyright © 2021 Cobian, Garlet, Hidalgo-Cantabrana and Barrangou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudio Hidalgo-Cantabrana, Y2hpZGFsZ0BuY3N1LmVkdQ==; Rodolphe Barrangou, cmJhcnJhbkBuY3N1LmVkdQ==