
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 21 October 2021
Sec. Microbe and Virus Interactions with Plants
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.744897
 Jianbo Xie1,2†
Jianbo Xie1,2† Ghada E. Dawwam1,2,3†
Ghada E. Dawwam1,2,3† Amira E. Sehim1,2,3†
Amira E. Sehim1,2,3† Xian Li1,2
Xian Li1,2 Jiadong Wu1,2
Jiadong Wu1,2 Sisi Chen1,2
Sisi Chen1,2 Deqiang Zhang1,2*
Deqiang Zhang1,2*Drought is a major threat to crop productivity and causes decreased plant growth, poor yields, and crop failure. Nevertheless, the frequency of droughts is expected to increase in the coming decades. The microbial communities associated with crop plants can influence how plants respond to various stresses; hence, microbiome manipulation is fast becoming an effective strategy for improving the stress tolerance of plants. The effect of drought stress on the root microbiome of perennial woody plants is currently poorly understood. Using Populus trees as a model ecosystem, we found that the diversity of the root microbial community decreased during drought treatment and that compositional shifts in microbes during drought stress were driven by the relative abundances of a large number of dominant phyla, including Actinobacteria, Firmicutes, and Proteobacteria. A subset of microbes, including Streptomyces rochei, Bacillus arbutinivorans, B. endophyticus, B. megaterium, Aspergillus terreus, Penicillium raperi, Trichoderma ghanense, Gongronella butleri, and Rhizopus stolonifer, was isolated from the drought-treated poplar rhizosphere soils, which have potentially beneficial to plant fitness. Further controlled inoculation experiments showed that the isolated bacterial and fungal isolates positively impacted plant growth and drought tolerance. Collectively, our results demonstrate the impact of drought on root microbiome structure and provide a novel example of manipulating root microbiomes to improve plant tolerance.
Drought stress is a serious and increasing problem in agriculture, as it negatively affects plant growth and development (Abid et al., 2019). Plant species are strongly associated with diverse archaeal, bacterial, and fungal communities (Veach et al., 2020). Microbiome communities are highly associated with plant health, productivity, and environmental adaptation (Berendsen et al., 2012) and have the potential to improve sustainable agricultural practices. Increasing evidence suggests that certain plant-microbe interactions can enhance plant stress tolerance, which has prompted researchers to manipulate microbial communities to promote plant growth during periods of environmental stress (Armada et al., 2018). The composition of plant-associated microbiomes is influenced by several host-associated and environmental factors (Baetz and Martinoia, 2013; Xu et al., 2018). Plants intimately interact with communities of microorganisms that share their environment (Naylor et al., 2017; Hu et al., 2018). Perturbations in plant metabolism, such as those triggered by abiotic stresses, can alter their microbiome with potential consequences for host fitness (Franzosa et al., 2019). Changes in soil moisture availability can significantly impact plant-associated microbiomes (Baetz and Martinoia, 2013; Santosmedellin et al., 2017; Xu et al., 2018). As drought stress represents a significant and increasing threat to crop productivity (Schwalm et al., 2017), current efforts are focused on improving the drought tolerance of various plant species (Rolli et al., 2015).
Plant species are highly associated with the microbial communities that inhabit rhizosphere (Berendsen et al., 2012; Cregger et al., 2018; Veach et al., 2019; Cregger et al., 2021). Variation in edaphic conditions selects for specific microbial communities. Increasing evidence demonstrates the importance of the microbiome, which consists of the entire complex of rhizosphere-associated microbes, their genetic elements and their interactions, in determining plant health (Berendsen et al., 2012). Several studies have shown that bacterial communities are dynamically shaped by several factors such as root exudates, soil, plant genotype, and chemotype (Veach et al., 2019). The complexity of the belowground plant microbiome enables its rapid adaption to changing soil conditions, which in turn can improve host adaptation (Timm et al., 2018; Veach et al., 2020). Therefore, environmental stressors are essential factors that could influence plant hosts or their microbiomes. Recent studies have demonstrated that drought has a strong impact on the structure of root microbiome communities and is associated with enrichment of specific bacterial lineages (Naylor et al., 2017; Santosmedellin et al., 2017; Edwards et al., 2018). Furthermore, plant root microbial ecosystems can act as “resource islands” for cultivating microorganisms in the surrounding soil (Marasco et al., 2012; Simmons et al., 2020). However, the structural and functional dynamics of microbial communities in perennial plant root systems during periods of abiotic stress are currently unknown. Alongside traditional breeding and genetic approaches, the manipulation of plant root microbiomes can potentially confer benefits capable of reducing the impact of abiotic stress on crops (Xu et al., 2018), thus enabling greater crop stability for feeding populations in drought-prone agroecosystems (Niu et al., 2017).
Poplars (Populus spp.) are model perennial systems for studying angiosperm tree physiology and genetics; they can also be employed to study the microbial composition of root rhizospheres and their responses to environmental changes (Munné-Bosch, 2007; Deyou et al., 2019; Wang et al., 2020a). Elucidation of these interactions is critical, as trees are economically essential to industry (Cregger et al., 2018). A variety of studies has examined total microbial communities associated with the roots of different species of Populus (Gottel et al., 2011; Armada et al., 2018; Veach et al., 2019, 2020; Tian et al., 2020). In nature, Populus species form mutualistic associations with specialized rhizosphere microbiomes that may affect nutrient uptake or water-absorbing efficiency from the soil under extreme conditions (Bissonnette et al., 2010; Timm et al., 2018; Wang et al., 2020b; Liu et al., 2021). Microbial community isolates from Populus have been shown to enhance the health, growth, and development of their plant hosts (Taghavi et al., 2009; Henning et al., 2016; Timm et al., 2016). Furthermore, the long-lived and clonal nature of Populus spp. could lead to functional relationships with specific microbes that may be underdeveloped or are absent in many annual model plant species that have typically served as functional organisms. Using Populus as a model system, this study seeks to characterize how the microbiome communities vary during drought stress treatment. We also studied the functional dynamics of the rhizosphere microbial communities using high-resolution metagenomic analyses. To determine whether some rhizosphere microbiomes contribute to changes in host phenotype under drought conditions, a series of functional bacterial and fungal strains were isolated and inoculated for host drought stress tests. We found that drought stress significantly shifted the composition of the Populus root microbiome. The drought-responsive microbes could potentially benefit their host as they may contribute to drought stress tolerance. These analyses revealed the dynamic interactions of microbial communities within the rhizosphere and changes in the functional gene pool under drought stress.
Silt-clay loam soil was collected from 20 forest locations (at a depth of 30–75cm in 20 tree root balls), in Guan Xian County, Shandong Province, China (36°3'N, 115°47'E), using root cuttings from the entire natural distribution of Populus tomentosa (30–40°N, 105–125°E; Denyer et al., 2019). Then, the soils were sieved to <4mm, thoroughly homogenized and scooped into pots (24cm inner diameter; 28cm outer diameter; 26cm height). Soil pH was measured in a soil water (1:2.5 w/v) suspension using a pH meter. The elemental composition [concentrations of total nitrogen (N), phosphorus (P), and potassium (K)] of the soil was determined by a previously described method (Liu et al., 2021). Prior to seedling transplantation, sufficient water was added to submerge the soils. Physicochemical characteristics of the soil used for the growth of the plants were as follows: total N (mgkg−1), 842.5±9.3; available P (mgkg−1), 48.7±2.4; available K (mgkg−1), 354.7±2.4; and organic matter content (gkg−1), 9.2±9.1. The pH value was 0.75±0.06.
The shoot tips collected from actively growing poplar were sterilized by washing in 10% bleach, 70% ethanol, and three times in deionized water. Tips were rooted in tissue culture medium (1× Murashige and Skoog basal salt mixture, 0.5mg/L indole-3-butyric acid) to produce rooted cuttings. Initial rooted cuttings were serially cultured in the same medium to generate germ-free experimental poplar seedlings. For this experiment, P. tomentosa rooted cuttings were transplanted into a growth chamber with sterilized soil for 7days, and healthy plants were selected for subsequent experiment.
The germ-free healthy plants were transplanted into individual pots with collected nature soils for 6months in a greenhouse at Beijing Forestry University, Beijing, China [16-h light/8-h dark; 20–24°C; 40°000'N, 116°200'E]. Nutrient water was supplied every 7days before drought treatment. Subsequently, soils were allowed to dry. Percent depletion of relative soil moisture contents (RSMCs) shown represents the proportion of potentially plant-available stored soil water in the ~5cm zones of the soil profile (the same zone in which root-associated microbiomes were sampled) estimated. Poplars were harvested at different time points: sample DA: RSMC 70–75% soil water content, 16/04/2019; sample DB: RSMC 50–55% soil water content, 27/04/2019; sample DC: RSMC 10–15% soil water content, 16/05/2019; and sample DD: rewatered for 2days (RSMC 30–35%), 06/06/2019. Poplars grown under standard watering conditions were included as a negative control and sampled at the same time points (CA; CB; CC; CD; RSMC 70–75%). Gravimetric water content of the soil was used to determine volumetric soil water percentage. Three biological replicates were used for each treatment.
Plant samples were collected by manually extracting whole plants with their root systems. We collected rhizosphere samples (soil tightly adhering to the root surface) by collecting and pooling roots severed with sterile blades at each time point. Roots were vortexed in epiphyte removal buffer (0.75% KH2PO4, 0.95% K2HPO4, and 1% Triton X-100 in ddH2O; filter-sterilized at 0.2μM) for 5min and then centrifuged at 3,500rpm for 5min after removal of the root tissue to pellet the resulting rhizosphere soil. Bulk soil samples were collected ~5cm below the soil surface at distance from the sample plants. DNA extractions were performed immediately after sampling, following the DNeasy PowerSoil Kit (Qiagen, Hilden, Germany) protocol.
Aliquots of each DNA sample were mechanically sheared, and products were size-selected to 350bp and gel purified. Sequencing was performed using the Illumina HiSeq platform 2×150-bp sequencing technology. Quality control of raw reads was carried out as follows: removal of reads containing more than 40bp of low-quality nucleotides (sQ≤38); removal of reads containing more than 10bp 3' discarding 5'adapter and adapter-adapter contaminants. The qualified reads that passed the filters above were then assembled using MEGAHIT (Dinesh et al., 2017) with the parameter --presets meta-large. Clean data from each sample were realigned to scaftigs using Bowtie21 to get unaligned paired-end reads with parameters --end-to-end, --sensitive, -I 200, and -X 400. All unaligned reads were used to assemble scaftigs. Gene prediction was conducted using MetaGeneMark,2 and gene redundancies were removed using CD-HIT3 with parameters -c 0.95, -G 0, -aS 0.9, -g 1, and -d 0. Gene catalogues with less than two read alignments were removed from the gene set. Species annotation was performed by searching the bacterial, fungal, archaeal, and viral sequences in the NR database using DIAMOND (Buchfink et al., 2015) with the parameter blastp e-value ≤1e−5. The assembled reads in our data set were 89.22% bacterial, 1.71% archaeal, 0.04% viral, and 8.9% unclassified.
Bulk soil DNA samples were amplified using a dual-indexed 16S rRNA Illumina iTags primer set specific to the V5–V7 region: 799F (5'-AACMGGATTAGATACCCKG-3') and 1193R (5'-ACGTCATCCCCACCTTCC-3'). For fungal libraries, the ITS1 region was amplified using universal primers. All polymerase chain reaction (PCR) reactions were carried out with 15μl of Phusion High-Fidelity PCR Master Mix (New England Biolabs), 0.2μM of forward and reverse primers, and ~10ng of template DNA. Thermal cycling consisted of initial denaturation at 98°C for 1min; followed by 30cycles of denaturation at 98°C for 10s, annealing at 50°C for 30s, and elongation at 72°C for 30s; and a final elongation at 72°C for 5min.
For PCR product quantification and qualification, the same volume of 1× loading buffer (containing SYBR green) was mixed with the PCR products and electrophoresed on a 2% agarose gel for detection. PCR products were mixed in equal ratios. The mixture of PCR products was purified with a Qiagen Gel Extraction Kit (Qiagen). For library preparation, sequencing libraries were generated using the TruSeq DNA PCR-Free Sample Preparation Kit (Illumina, San Diego, CA, United States) following the manufacturer’s recommendations. The library was sequenced on the Illumina NovaSeq platform, and 250-bp paired-end reads were generated.
Paired-end reads were merged using FLASH (V1.2.7).4 Unjoined reads were discarded, and assembled reads were assigned to samples from barcodes by using split libraries. High-quality clean tags were subjected to the QIIME pipeline (V1.9.1).5 Tags were compared with the reference database (Silva database)6 using the UCHIME algorithm7 to obtain effective tags, and chimeric, mitochondria, and chloroplasts sequences were removed. Sequences with ≥97% similarity were assigned to the same operational taxonomic unit (OTU). Representative sequences for each OTU were screened for taxonomic annotation based on the Mothur algorithm. After taxonomies were assigned to each OUT, we discarded (1) all OTUs that were not assigned a Kingdom level RDP classification score of at least 0.5 and (2) all OTUs that were not assigned to Kingdom Bacteria. To remove low abundance OTUs that are in many cases artifacts generated through the sequencing process, we removed OTUs without at least five reads in at least three samples. These thresholds were found to be suitable using technical replicates in a data set published previously (Xu et al., 2018).
All analyses were conducted in the R Environment version 3.2.3. For principal coordinate analysis (PCoA), OTU or unigene counts were normalized using the variance-stabilizing transformation implemented in DESeq2. Weighted UniFrac distances were then calculated with phyloseq. Unconstrained PCoA was performed with the PCoA function in the ape package. Shannon diversity analysis was performed based on the openxlsx, reshape2, and vegan packages in R. Group difference analyses were performed using Metastats. The significance of different factors on community dissimilarity was tested with PERMANOVA or nested PERMANOVA using the “adonis” function of the VEGAN package in R based on weighted UniFrac distances.
The increased abundance of Actinobacteria and Firmicutes under drought conditions could be due to their high tolerance for stress, although direct selection from root exudates might have occurred. To investigate the potential contributions of drought-enriched microbes and their interactions with plants, we performed controlled inoculation experiments using seedlings grown in sterile soil. Bacterial and fungal species isolated from drought-stressed poplar roots were applied individually or in consortia to pre-sterilized soil. We collected rhizosphere samples (soil tightly adhering to the poplar root surface) of poplar seedlings under all drought treatment and pooling all root samples. Roots were washed with sterile water, ground in 10mM MgSO4 with a mortar and pestle and then centrifuged at 10,000×g for 10min to pellet the root-associated microbes. For bacterial isolation, 1ml of the suspension was plated onto King’s B medium and nutrient agar medium (24). For the isolation of fungal strains, 1ml of the suspension was plated onto potato dextrose agar (PDA) and malt extract agar (MEA) media supplemented with 50ppm of chloramphenicol to prevent bacterial growth and 35ppm of Rose Bengal to inhibit fast-growing fungi (Numponsak et al., 2018). Plates were then sealed with parafilm and incubated at 28°C for 7days. Representative colonies were picked and transferred to fresh nutrient agar medium for further studies.
To investigate the effects of water stress on the growth of the bacterial isolates, nutrient broth supplemented with varying concentrations of PEG-6000 (0, 15, 25, and 35%) was inoculated with 1% bacterial culture overnight. Cultures were incubated at 28°C with 120rpm of shaking. Growth was estimated by measuring the optical density (OD) of the cultures at 600nm using a spectrophotometer. The influence of water stress on fungal biomass was investigated by cultivating isolates in modified Melin-Norkrans medium supplemented with varying concentrations of PEG-6000 (0, 15, 25, and 35%). The cultures were incubated at 25°C in the dark for 7days. Mycelial mats were removed from the liquid medium, rinsed three times with distilled water, dried overnight at 80°C, and weighed (Zhang et al., 2011). The experiment was carried out using three repeats. Four bacterial and five fungal drought-tolerant strains were identified and selected for subsequent analyses.
For bacterial strain identification, the 16S rRNA of the drought-tolerant bacterial strains was amplified via PCR using the following primers: 16S-F (forward, 5'-AGAGTTTGATCCTGGCTCAGAACGAACGCT-3') and 16S-R (reverse, 5'-TACGGCTACCTTGTTACGACTTCACCCC-3'). Each 20-μl PCR mixture contained 1μl of forward primer, 1μl of reverse primer, and 10μl of 2× Es Taq Master Mix. The PCR was programmed as follows: initial denaturation for 5min at 95°C, 36cycles of denaturation (0.5min at 95°C), annealing (0.5min at 55°C), and extension (1.5min at 72°C), and final extension for 5min at 72°C. PCR products were purified using a TaKaRa MiniBEST Agarose Gel DNA Extraction kit and analyzed via horizontal electrophoresis in 1% agarose gel. Similarities of the 16S rRNA sequences were examined using the National Center for Biotechnology Information BLAST program.
For fungal strain identification, genomic DNA was extracted using an EZgene TM Fungal DNA Miniprep kit according to the manufacturer’s instructions. Amplification of the fungal ITS region was performed via PCR using a forward primer (ITS1: 5'-TCCGTAGGTGAACCTGCGG-3') and a reverse primer (ITS4: 5'-TCCTCCGCTTATTGATATGC-3'; Wendrich et al., 2020). The thermocycling conditions were as follows: initial denaturation for 5min at 94°C, 35cycles of denaturation (1min at 94°C), annealing (40s at 55°C), and extension (1min at 72°C), and final extension for 5min at 72°C. The PCR products were analyzed using 1% agarose gel and sequenced using an ABI3730XL DNA Analyzer (Beijing Ribio Biotech). Similarities in the ITS sequences were examined using BLAST.
Antagonism between selected fungal pairs was examined in vitro via the dual culture technique using PDA medium (Abdallah et al., 2015). A mycelial disc of one selected fungus (diameter 5mm) was deposited on one side of a Petri dish, and another fungal disc was deposited equidistantly on the other side of the same plate. After 7days of incubation at 28°C, antagonism was detected if zones of inhibited growth were present. For bacteria, strains were inoculated in lines on the surface of nutrient agar media. Other bacterial strains were inoculated in lines perpendicular to the initial inoculations. The plates were incubated for 24h at 37°C. Antagonistic activity was estimated by identifying zones of inhibited growth (Seyfferth et al., 2021).
To measure the phosphate solubilization activity of the microbial isolates, both bacterial and fungal isolates were inoculated separately in Pikovskaya’s solid medium containing insoluble tri-calcium phosphate (0.5%). The solubilization zones and colony diameters were measured after 120h of incubation at 30°C. The results were expressed as solubilization efficiency (SE; Liu et al., 2020):
Indole-3-acetic acid (IAA) is a primary plant hormone that regulates plant growth and development. To measure microbial IAA production, test strains were grown in MAZ medium (per liter: K2HPO4·3H2O, 6.0g; KH2PO4, 4.0g; MgSO4·7H2O, 0.2g; NaCl, 0.1g; CaCl2, 0.02g; FeCl3·6 H2O, 0.01g; H3BO3, 2.8mg; MnSO4·H2O, 2.1mg; NaMoO4·2 H2O, 2mg; ZnSO4·7H2O, 0.24mg; CuSO4·5H2O, 0.016mg) supplemented with 34mM malate (carbon source), 100μgml−1 tryptophan (IAA precursor), and 10mM NH4Cl (nitrogen source). Bacterial strains were inoculated into LB broth, while fungal strains were grown in Czapek’s medium amended with 1,000μgml−1 Trp (IAA precursor). The ability of isolates to produce IAA was determined using Salkowski’s reagent via a previously described colorimetric method (Rich-Griffin et al., 2020).
For the inoculation experiment, P. tomentosa LM50 tissue culture seedlings were transplanted into growth chamber with sterilized soil for 7days, and then, healthy seedlings were inoculated with bacterial and fungal strains. For bacterial inoculation, we inoculated 10ml of LB medium with a fresh colony and incubated the culture at 37°C overnight with 250rpm of shaking. The following day, 300μl of the culture medium was transferred to 10ml of LB medium and incubated for 6h, or until an OD (at 600nm) of 0.6 was reached. Then, 5ml of each culture was pelleted via centrifugation (2,000×g, 10min), and the pellet was resuspended in 1ml of sterilized water. For fungal inoculation, fungal isolates were cultured on PDA slants for 1week at 25°C. To prepare conidial suspensions for inoculation, approximately 9ml of sterile distilled water was added onto the surface of each fungal slant. The conidial suspensions were spread with a sterile glass rod and then filtered to remove mycelia. The spore suspension density was adjusted to 1×105ml−1. The suspension was mixed with autoclaved water to a total volume of 400ml and added to the growth chamber. Plants were grown under a 16-h light/8-h dark photoperiod in a greenhouse at Beijing Forestry University, Beijing, China (40°00'N, 116°200'E). The suspension was added every 7days for the treatment group plants. After 30days, all plants except control samples were subjected to severe drought stress by suspending watering for 18days. Plants of the control group were watered with 400ml sterilized water every 7days.
Seven traits were measured after inoculation, and three biological replicates were used for each trait. Plant samples were collected by manually extracting whole plants with root systems. Plant height was measured from the apex to the base; root length was measured from the base to the longest root apex. Mature leaves from the same position were harvested and used to measure growth and physiological characteristics. The RWC (relative water content) of leaves was calculated using the following equation: RWC=(FW−DW)/(TW−DW)×100%; where FW=leaf fresh weight, TW=leaf turgid weight (leaves were placed in water for ~24h in darkness before weighing), and DW=leaf dry weight (leaves were dried for 24h at 90°C before weighing). Total protein concentration was measured by Bradford’s method (Gururani and Upadhyaya, 2013) using bovine serum albumin as the standard. The ascorbate peroxidase (APX), peroxidase (POD), and superoxide dismutase (SOD) activity levels were measured with customized kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s instruction. Malondialdehyde (MDA) content was determined according to a previously described method (Beckers et al., 2017). The total sugar content was measured by anthrone colorimetry, as described previously (Munné-Bosch and Alegre, 2002). The proline content was measured by ninhydrin colorimetry using a previously described method (Dubrovina and Kiselev, 2016).
To investigate the effects of drought stress on microbial recruitment in the rhizosphere, Populus seedlings were subjected to soil water deficiency at four RSMCs, selected according to previous research (Niu et al., 2017). Shotgun metagenomics was applied to examine community composition throughout the drought treatment. After quality control, a total of 155.98G 150-bp Illumina sequences were obtained (Supplementary Table S1). Assembly of the reads produced more than 642,520 scaffolds, ranging from 628.57 to 998.68bp in length (Supplementary Table S2). On average, 981,950 open reading frames were predicted (Supplementary Table S3). To explore the impact of RSMC on microbial community composition, unconstrained PCoAs were performed using Bray-Curtis distances. Samples taken from the drought treatments clustered into distinct groups according to treatment, and samples taken at the same timepoint approximately clustered into the same group. DA and CA samples were also grouped together (Figure 1A). This indicated that the microbial communities were strongly influenced by drought treatment and RSMC (Figure 1A). A comparison of drought and control treatments revealed that Shannon’s diversity level was significant difference between drought-treated samples and the control samples (Figure 1B; p<0.05, Tukey’s test). Shannon’s diversity level in the rhizosphere at the start was similar between treatment and control samples (Figure 1B); by contrast, levels of Shannon’s diversity in the soil remained largely unchanged (Supplementary Figure S1). We next examined the composition of the rhizosphere microbiome. At the phylum level, each community was dominated by Proteobacteria, Actinobacteria, Verrucomicrobia, and Firmicutes (Figures 2A,B). These bacterial phyla are relatively abundant and ubiquitous in soil (Figures 2C,D). Additional phyla, including Acidobacteria, Chloroflexi, and Bacteroidetes, were also found in nearly all samples, but their relative abundances were variable and typically represented less than 5% of the total reads (Supplementary Table S4).
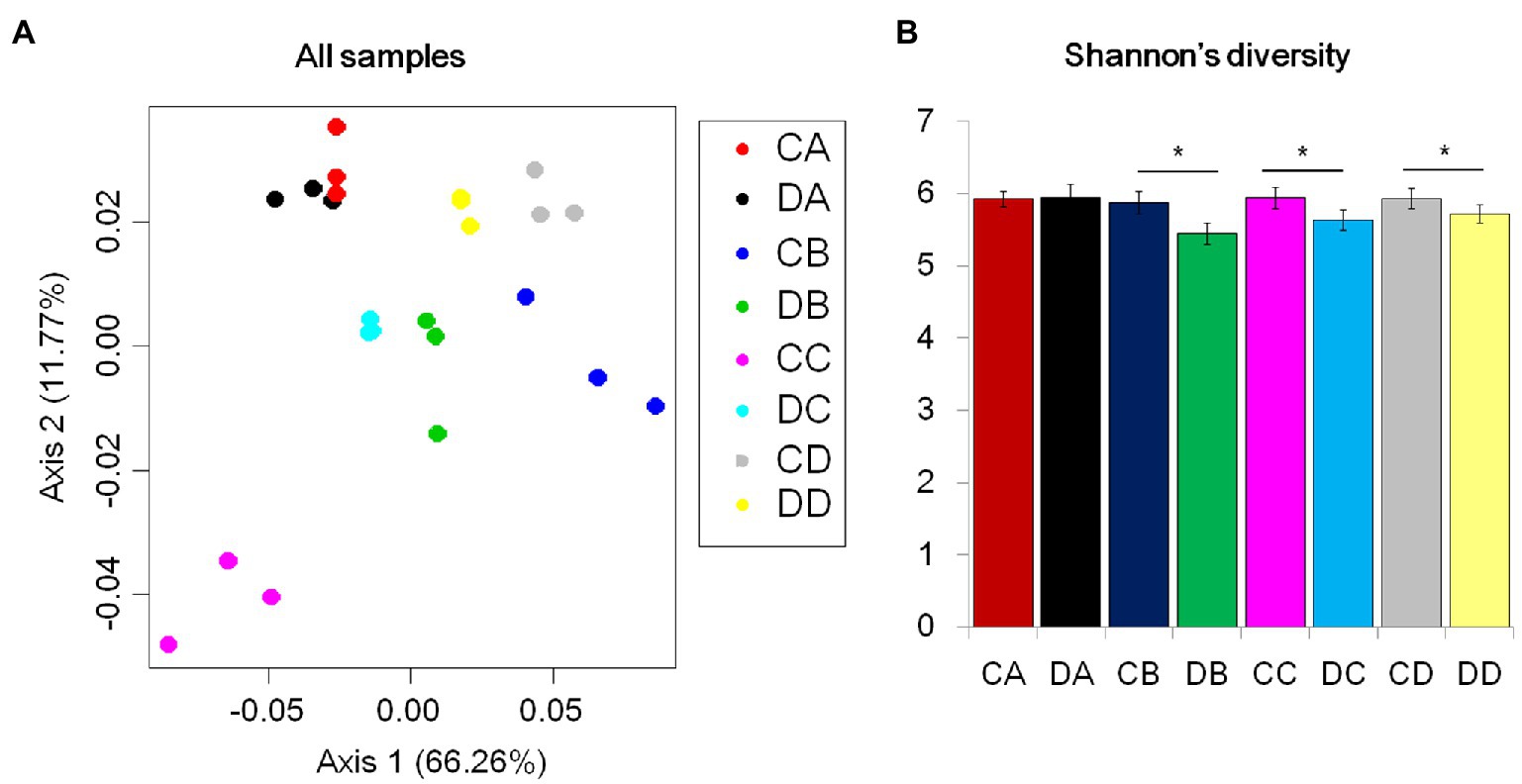
Figure 1. Drought impacts on root and soil microbiome development. (A) Principal coordinate analysis (PCoA) of Bray-Curtis distances for all control and drought samples. The color of each point indicates the control or drought treatment. (B) Mean Shannon’s diversity across the rhizosphere. (*p<0.05; *Tukey’s test).
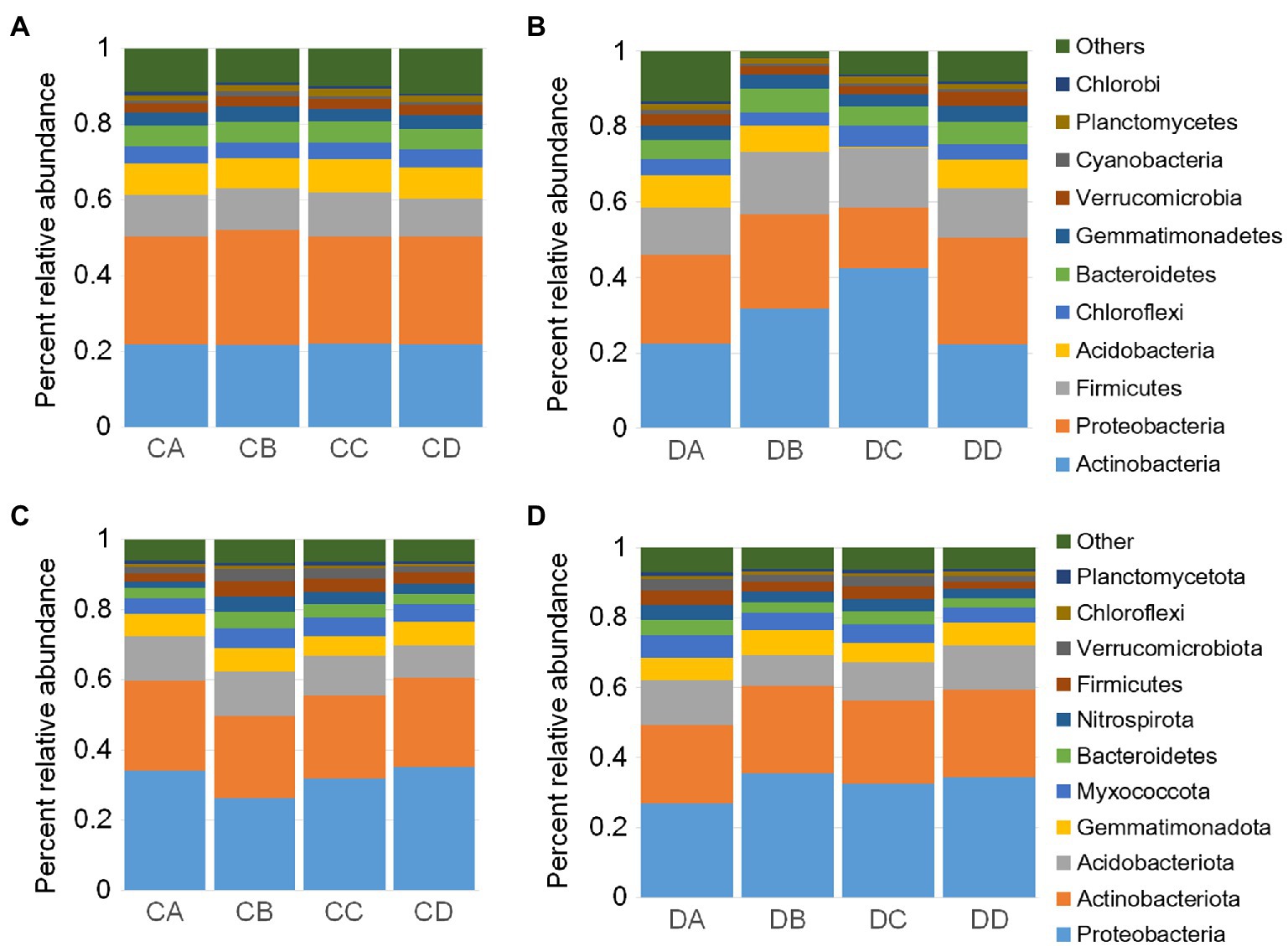
Figure 2. Drought impacts root microbiome development. Percent relative abundances of the top 11 most abundant phyla for rhizosphere samples of the control (A) and drought treatment (B). Percent relative abundances of the top 11 most abundant phyla for bulk soil samples of the control (C) and drought treatment (D). DA, drought sample A; DB, drought sample B; DC, drought sample C; DD, drought sample D; CA, control sample A; CB, control sample B; CC, control sample C; CD, control sample D.
We then investigated temporal dynamics in the root microbiome under drought treatment by analyzing the relative abundance of each phylum in the microbial communities. The rhizosphere communities exhibited dynamic shifts during the drought treatment. Similar shifts were not observed for the control group, which indicated that this short period of plant development did not significantly influence the community composition (Figure 2A). To explore how bacterial community composition varied across both growth and drought treatment, NMDS ordinations and nested PERMANOVA analyses were performed (Supplementary Table S5). The analyses revealed significant differences in composition across the data set are driven primarily by drought treatment for rhizosphere (F statistic=6.216, p<0.001) and for bulk samples (F statistic=7.523, p<0.001). The compositional changes in the rhizosphere were largely due to dominant phyla such as Proteobacteria, Actinobacteria, Firmicutes, and Acidobacteria (Figure 2A). The compositional shifts in the microbial communities during the drought treatment were driven by the relative abundances of a large number of dominant phyla, including Actinobacteria, Firmicutes, and Proteobacteria (Figure 2A). At the class level, most of the bacteria belonged to Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria, Deltaproteobacteria, and Actinobacteria (Supplementary Figure S1A). Nitrosomonadaceae, Bacillaceae, Streptomycetaceae, and Xanthobacteraceae were the abundant families (Supplementary Figure S1B). Pseudolabrys, Streptomyces, Bacillus, and Gaiella were the abundant genera (Supplementary Figure S1C). Notably, we observed significant enrichment of Actinobacteria and Firmicutes, which was consistent with previous studies (Naylor et al., 2017; Santosmedellin et al., 2017; Cregger et al., 2018) and a likely contributing factor for the decrease in microbial diversity within the rhizosphere. Strikingly, after re-watering, the Actinobacteria and Firmicutes lineages in drought-treated plants decreased rapidly, by 12.3 and 10.6%, respectively (Figures 2A,B; p<0.01; one-sided Student’s t test).
At higher taxonomic resolution, we further identified specific bacteria that exhibit relative abundance patterns that differ between samples (Supplementary Data S1). Four genera were found enriched or depleted over the course of drought treatment, including Bacillus, Actinotignum, Aneurinibacillus, and Streptomyces. Strikingly, the most significantly enriched genera in drought treatment samples were Streptomyces and Bacillus, with 1.6- to10.4-fold higher abundance in drought-treated rhizosphere samples compared to control (Supplementary Data S1; p<0.001; Fisher’s exact test).
Samples of bulk soil were collected to determine the influence of drought on soil microbial communities, which were investigated using Illumina MiSeq sequencing of the V5–V7 region of the 16S rRNA gene. Proteobacteria, Actinobacteria, Acidobacteria, Gemmatimonadetes, Myxococcota, Bacteroidetes, Nitrospirae, Firmicutes, Verrucomicrobia, Chloroflexi, and Planctomyces were dominant phyla in soil samples (Figures 2C,D). Cyanobacteria were not dominant in soil samples (Figures 2C,D). We further observed that Proteobacteria was the most abundant phylum in soil, while Actinobacteria was most abundant in the rhizosphere (Figures 2A,B).
To determine whether the drought-induced rhizosphere shifts correlated with changes in microbial function, we performed functional analyses on all samples. First, we investigated changes in rhizosphere community function during drought treatment. We found a significant increase in genes associated with signal transduction, membrane transport, and transport and catabolism. By contrast, pathways related to the metabolism of terpenoids and polyketides, as well as cell motility, exhibited a relative decrease in all three drought-treated samples (Figure 3A; p<0.01). The abundance of genes associated with the hydrolysis of carbohydrate esters, non-hydrolytic cleavage of glycosidic bonds, formation of glycosidic bonds, and hydrolysis and/or rearrangement of glycosidic bonds was significantly lower in drought-treated samples than in control samples (Fisher’s exact test: p<0.039; Figure 3B). A high-resolution EggNOG analysis of the functional categories influenced by drought in the rhizosphere revealed that the sub-functions of a large number of genes were related to resource transport, including phosphonate ABC transport, sulfate ABC transport, and putrescine ABC transport (Supplementary Data S2).
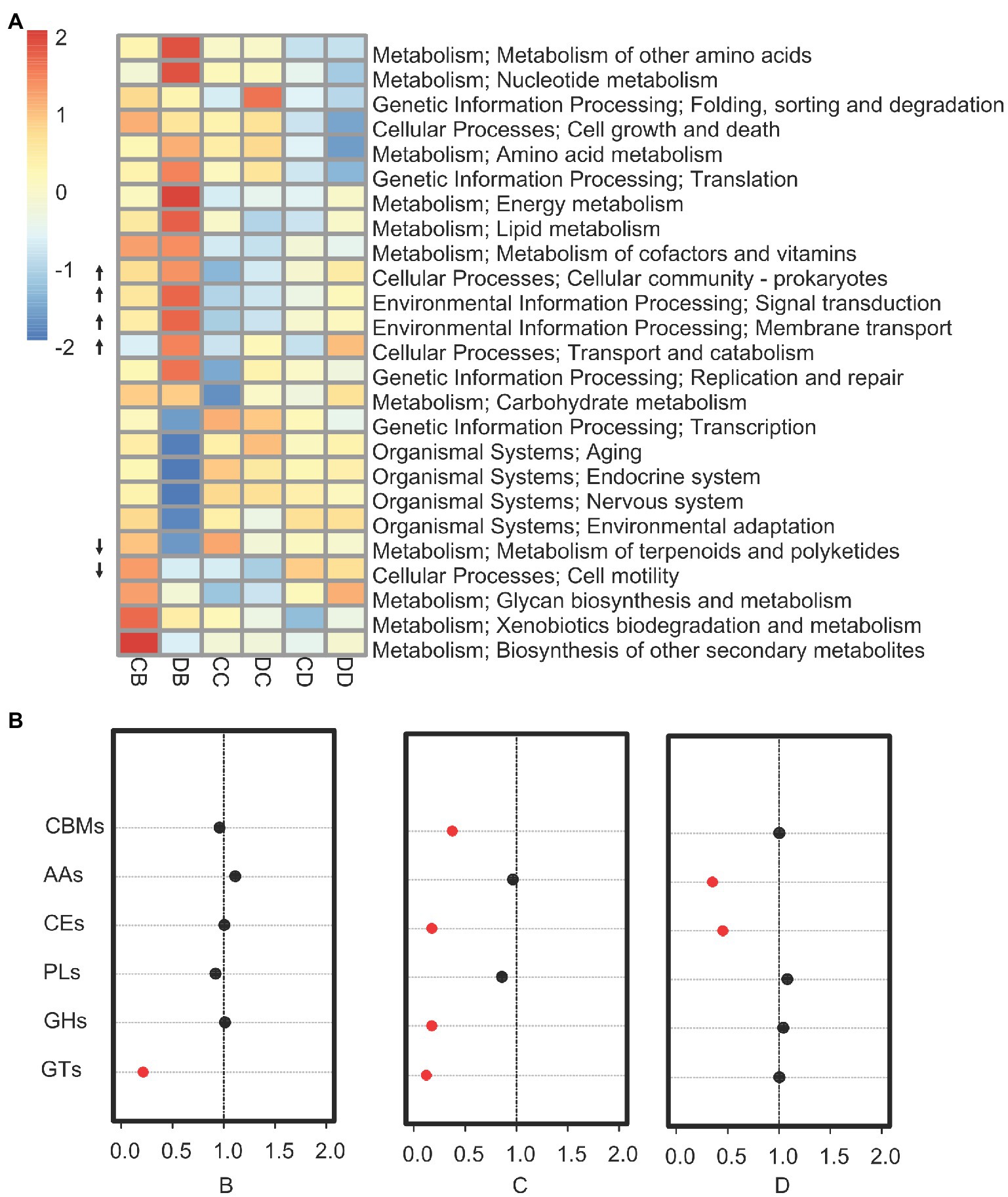
Figure 3. Drought impacts on the rhizosphere microbiome gene pool. (A) Kyoto Encyclopedia of Genes and Genomes enrichment analysis for all genes showing enrichment under drought for rhizosphere microbiomes. Categories significantly enriched are indicated (p≤0.05; Fisher’s exact test). The up (down) arrow indicates categories for which there are more (less) genes in the drought samples than in the control. (B) Enrichment analysis for all enzymes that degrade, modify, or create glycosidic bonds annotated in the CAZy database. The red circles indicate categories for which the enrichment had a value of p<0.05 in a Fisher’s exact test. The values on the x-axis indicate the fold enrichment ratio of annotated CAZy genes from samples B–D (drought treatment/control). CBMs, carbohydrate-binding modules; AAs, auxiliary activities; CEs, carbohydrate esterases; PLs, polysaccharide lyases; GHs, glycoside hydrolases; and GTs, glycosyltransferases.
The bacteria chosen include three strains from Firmicutes (Bacillus megaterium bc32, Bacillus endophyticus bc39, and Bacillus arbutinivorans bc49) and one Actinobacteria strain (Streptomyces rochei bc68); the fungal strains comprised Aspergillus terreus fg7, Penicillium raperi fg11, Trichoderma ghanense fg18, Gongronella butleri fg16, and Rhizopus stolonifer fg20 (Supplementary Figure S2; Supplementary Tables S6 and S7). No obvious antagonism was detected among the bacterial isolates (Supplementary Figure S3), whereas antagonism was present among all fungal isolates, with the exception of strains fg18 and fg20. Antagonism was also detected between some bacterial and fungal isolates. Thus, individual strains and representative groups of strains were chosen for this experiment.
To examine the potential growth-promoting effects of the selected microbial strains on plant development, levels of the plant growth hormone IAA were measured for each test strain using a colorimetric assay. IAA production was detected in all strains, with the highest levels detected in B. endophyticus bc39 (78.82μgml−1) and the lowest in P. raperi fg11 (13.42μgml−1). Six of the strains, B. megaterium bc32, B. endophyticus bc39, B. arbutinivorans bc49, A. terreus fg7, P. raperi fg11, and T. ghanense fg18, were able to solubilize phosphate. Bacillus megaterium bc32 and A. terreus fg7 were the most effective phosphorus solubilizers, exhibiting phosphorus solubilization indices of 165.57 and 150.36%, respectively (Supplementary Figure S4).
Significant improvement in poplar growth parameters (plant height and root length) was observed following inoculation with several of the microbial species compared to the sterile control (Figure 4). We observed that the average root length of plants that were inoculated with all three bacterial strains simultaneously was 49.2% longer compared to the control (p<0.01; one-sided Student’s t test). Average root length was also 29.6% longer than the control in plants inoculated with consortia containing fg18 and fg20 (Figure 4; p<0.01; one-sided Student’s t test). Furthermore, six physiological characteristics, including leaf total proline, MDA content, APX activity, POD activity, sugar content, and SOD activity, were examined for each group (Supplementary Figure S4). All six characteristics differed significantly between inoculated plants and control plants under the drought treatment (p<0.05; one-sided Student’s t test). For example, the total proline content was significantly different in the inoculated group under drought treatment compared to the control drought treatment group (Supplementary Figure S5; p<0.05; one-sided Student’s t test).
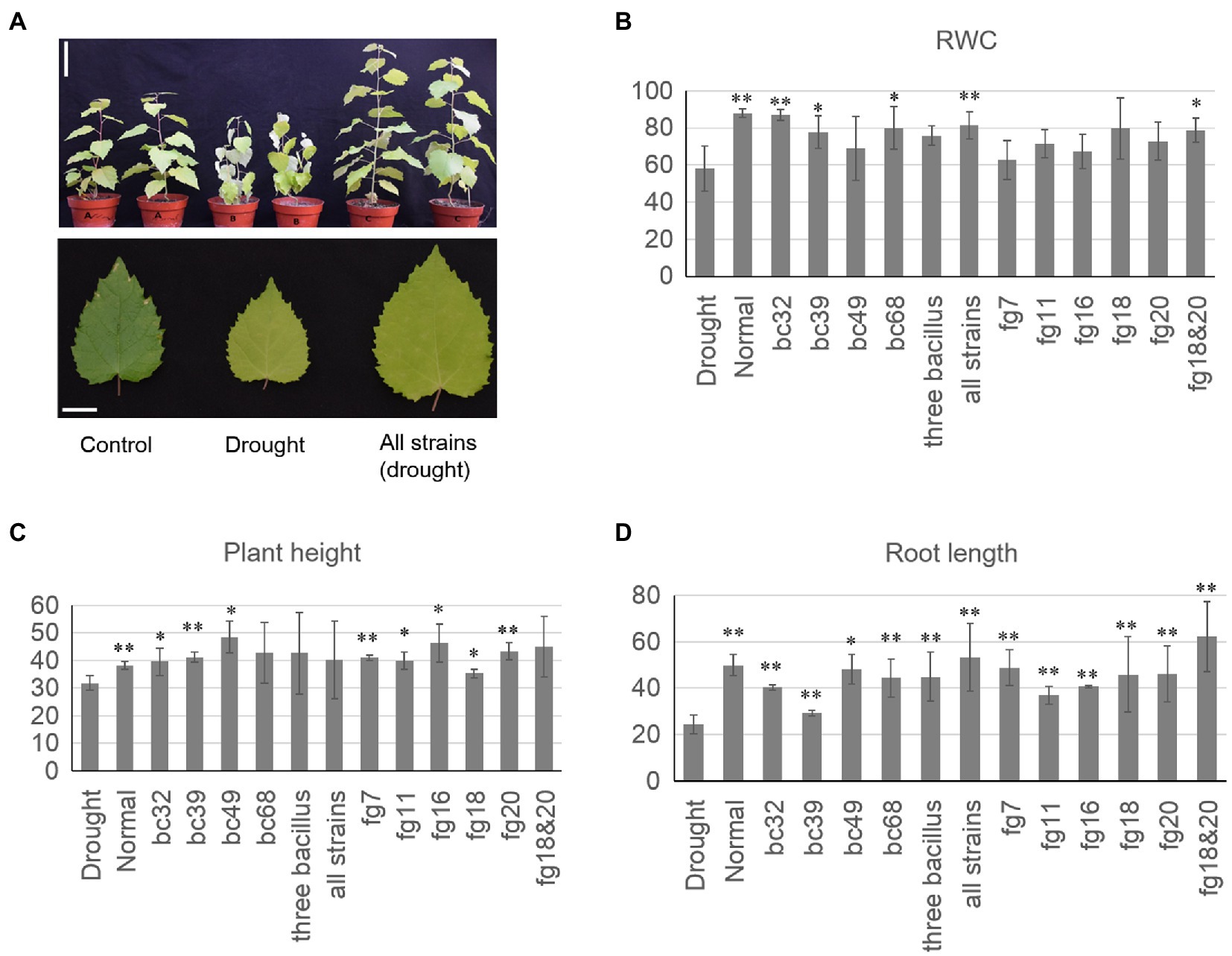
Figure 4. Isolated strains enhance the drought tolerance of poplar. (A) Inoculation of all mixed bacteria strains enhanced the drought tolerance of poplar. Bars (up)=10cm. Bars (down)=2cm. (B) The relative water content (RWC) of different inoculation groups. (C) The plant height of different inoculation groups. (D) The root length of different inoculation groups. RWC=[(fresh weight of leaves−dry weight of leaves)/(Turgid weight of leaves−dry weight of leaves)]×100. All strains (Bacillus megaterium bc32, Bacillus endophyticus bc39, Bacillus arbutinivorans bc49, and Streptomyces rochei bc68); three Bacillus (Bacillus megaterium bc32, Bacillus endophyticus bc39, and Bacillus arbutinivorans bc49; *p<0.05; **p<0.01).
In this work, we characterized the rhizosphere communities associated with a perennial plant under drought stress using high-throughput metagenomics. We found that the microbial community within the rhizosphere changed significantly during drought treatment, highlighting the dynamic nature of microbial communities associated with abiotic environments. Moreover, we isolated a series of bacterial and fungal strains from poplar roots that can directly or indirectly benefit plant growth. Our results suggest that the structure of the rhizosphere microbiome is relatively stable, while drought stress induces highly dynamic changes for some microbial lineages. In general, taxonomic classification represents the closest ancestor or relative in many cases. Despite the limitations, we identified similarities of rhizosphere microbial communities both in Populus and other genera, with strong preferential enrichment for Actinobacteria, Proteobacteria, Acidobacteria, and Chloroflexi (Zelicourt et al., 2013; Santosmedellin et al., 2017; Cregger et al., 2018; Xu et al., 2018). As a result, taxa belonging to these phyla were opportunistic during the drought stress. Firmicutes taxa were also comparatively abundant during the drought treatment. These taxonomic trends were observed across all soil types despite their intrinsic compositional differences, indicating that soil serves as a seed bank for rhizosphere recruitment. The result also indicates that the differences in the microbiome composition of the rhizosphere are not due to unwanted heterogeneity within the soil inoculum.
In our study, the drought treatment significantly influenced microbial community composition, which might have been due to the dramatic shifts in soil moisture. The coexistence of microbial species typically depends on metabolic trade-offs, where each species has fitness advantages under particular abiotic and biotic conditions (Xu et al., 2018). The enrichment of particular microbial strains may arise due to the differing life strategies of distinct lineages. For example, the spore-forming ability of Firmicutes and Actinobacteria enables them to enter a stable and quiescent state during periods of environmental stress; this strategy likely enabled them to persist under drought conditions, whereas less fit bacterial lineages decreased in abundance (Vanessa et al., 2013; Naylor et al., 2017). These bacterial communities likely carry a competitive advantage that allows them to respond more rapidly to environmental conditions (Wang et al., 2020b; Zhang et al., 2021). These may have the potential to confer benefits to a wide range of hosts as drought events become more prevalent globally (Lobell et al., 2011). Additionally, several members of Actinobacteria are characterized by their filamentous growth habit and their ability to produce stress-resistant spores (Hacquard et al., 2015; Santosmedellin et al., 2017), two traits that could help this group resist drought stress. Several other mechanisms, such as biofilm formation, and osmoprotectant production may be applied by the set of microorganisms enriched under drought conditions (Hacquard et al., 2015). The dynamic shifts in distinct lineages, including Actinobacteria and Firmicutes, could be host-specific and a result of differing drought-response mechanisms. Some microorganisms, such as Rhizophagus, Mesorhizobium, and Streptomyces, are reported to promote drought tolerance (Berendsen et al., 2012). One possible explanation of Actinobacteria and Firmicutes enriched under drought that is related to fast stress response of these lineages such as Bacillus. The two bacterial lineages composed almost entirely of monoderms, which have thick cell walls, and producing a large set of general stress proteins (Xu et al., 2018). Environmental changes shift the gene expressions of the plant host toward the production of cell wall components, plant hormones, and metabolic pathways of plant secondary metabolism (Satterlee et al., 2020; Shaw et al., 2021), which presumably has subsequent downstream effects on the plant-associated microbiome communities (Timm et al., 2018).
Metagenomics is powerful tool for understanding the functions of the rhizosphere (Nayfach and Pollard, 2015). In this study, we explored the functional diversity of root microbial communities during drought treatment. Most of the gene sequences were related to carbohydrate metabolism, including those encoding carbohydrate esterases, polysaccharide lyases, glycoside hydrolases, and glycosyltransferases. Carbohydrates, which participate in a multitude of biological processes, can combine to form an enormous number of compounds through the activity of carbohydrate-active enzymes (Lombard et al., 2014). Soil microbial communities play an important role in the carbon cycle, from serving as a carbon reserve to producing structural molecules (Lombard et al., 2014). Our results suggest that the drought treatment inhibited the gene pool associated with carbohydrate-active enzymes, implying that microbial metabolism and vital processes were less active in the drought-treated samples. Recent evidence has shown that environmental stress drives the changes in exudation of specific plant metabolites (Badri et al., 2013), further influencing the composition of the plant-associated microbiome. Pathways associated with transport and catabolism in the rhizosphere exhibited increased activity during the drought treatment, whereas signal transduction and metabolic pathways were reduced in drought-treated soil. Evidence showed that catabolic enzymes and catabolite transporters could be regulated by environmental changes (Maurer et al., 2005), which might contribute to tolerance to drought and other abiotic stresses in specific taxa. These functional groups might have been related to the stress responses of the microbial community in the drought-treated soil groups. Taken together, these data suggest that drought stress had a significant effect on the gene pool of rhizosphere-associated microbiomes and that rhizosphere genes associated with carbohydrate and amino acid metabolism and transport increased under drought.
Following the isolation of individual root microbial species, we investigated potential plant-microbiota interactions, as well as the effects of individual and multi-strain inoculation on plant growth during drought stress. We inoculated plants with four bacterial and five fungal strains that were abundant in the rhizosphere both individually and in combinations and then quantified the height, root length, and RWC of poplar seedlings. Following inoculation, the plants exhibited increased growth under drought treatment compared to the well-watered groups. It is possible that these strains promoted growth under drought stress through the production of specific plant hormones. Our analyses revealed that several strains were able to produce auxins and solubilize phosphate, two features that can enhance plant growth. The microbial production of other growth regulators, such as jasmonic and abscisic acids, can also improve plant performance under stressful soil conditions (Forchetti et al., 2010). In addition, extensive crosstalk exists among different signaling pathways (Selvakumar et al., 2014), leading to the possibility that specific signaling components may integrate inputs from multiple hormones to enhance plant tolerance. Several plant-promoting characteristics, such as ACC deaminase activity and siderophore-mediated iron assimilation, are widely distributed in bacteria and fungi. Examples include Streptomyces and Amycolatopsis species, which have been shown to enhance plant growth (Forchetti et al., 2010). Consistent with this, a recent study on the effect of drought on the microbiome observed a significant correlation between the relative abundance of the genus Streptomyces in plant roots and host drought tolerance (Xu et al., 2018). However, few studies exist in which bacteria and fungi were isolated from stressful environments and screened for their potential to produce ACC deaminase and siderophores. Our results suggest that inoculation of plant soil with specific strains or strain combinations can potentially benefit plant fitness, within the context of a controlled laboratory setting and given the absence of other microbes. Further investigation with a wide range of monoderm lineages will be necessary to understand the mechanisms driving microbe-associated drought tolerance.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
AS and GD performed the experiment. JX wrote the manuscript. DZ designed the experiment, obtained funding, and responsible for this article. XL and JW provided valuable suggestions to the manuscript. SC and DZ revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by funding from the Project of the National Natural Science Foundation of China (Nos. 31872671, 31972954, and 32022057), Young Elite Scientists Sponsorship Program by CAST (2018QNRC001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.744897/full#supplementary-material
1. ^http://bowtie-bio.sourceforge.net/bowtie2/index.shtml
2. ^http://exon.gatech.edu/GeneMark/meta_gmhmmp.cgi
3. ^http://www.bioinformatics.org/cd-hit/
4. ^http://ccb.jhu.edu/software/FLASH
Abdallah, R. A. B., Jabnoun-Khiareddine, H., Mejdoub-Trabelsi, B., and Daami-Remadi, M. (2015). Soil-borne and compost-borne Aspergillus species for biologically controlling post-harvest diseases of potatoes incited by Fusarium sambucinum and Phytophthora erythroseptica. J. Plant Pathol. Microbiol. 6, 1–9. doi: 10.4172/2157-7471.1000313
Abid, U., Adnan, A., Luo, Q., Hamid, K. A., Hakim, M., Muhammad, S., et al. (2019). Microbiome diversity in cotton rhizosphere under normal and drought conditions. Microb. Ecol. 77, 429–439. doi: 10.1007/s00248-018-1260-7
Armada, E., Leite, M. F. A., Medina, A., Azcón, R., and Kuramae, E. E. (2018). Native bacteria promote plant growth under drought stress condition without impacting the rhizomicrobiome. FEMS Microbiol. Ecol. 94, 1–13. doi: 10.1093/femsec/fiy092
Badri, D. V., Chaparro, J. M., Zhang, R., Shen, Q., and Vivanco, J. M. (2013). Application of natural blends of phytochemicals derived from the root exudates of Arabidopsis to the soil reveal that phenolic-related compounds predominantly modulate the soil microbiome. J. Biol. Chem. 288, 4502–4512. doi: 10.1074/jbc.M112.433300
Baetz, U., and Martinoia, E. (2013). Root exudates, the hidden part of plant defense. Trends Plant Sci. 19, 90–98. doi: 10.1016/j.tplants.2013.11.006
Beckers, B., Beeck, M., Weyens, N., Boerjan, W., and Vangronsveld, J. (2017). Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. Microbiome 5:25. doi: 10.1186/s40168-017-0241-2
Berendsen, R. L., Pieterse, C., and Bakker, P. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Bissonnette, L., St-Arnaud, M., and Labrecque, M. (2010). Phytoextraction of heavy metals by two Salicaceae clones in symbiosis with arbuscular mycorrhizal fungi during the second year of a field trial. Plant Soil 332, 55–67. doi: 10.1007/s11104-009-0273-x
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12:59. doi: 10.1038/nmeth.3176
Cregger, M., Carper, D. L., Christel, S., Doktycz, M., and Schadt, C. (2021). Plant-microbe interactions, from genes to ecosystems using Populus as a model system. Phytobiomes J. 5, 1–42. doi: 10.1094/PBIOMES-01-20-0009-FI
Cregger, M. A., Veach, A. M., Yang, Z. K., Crouch, M. J., Vilgalys, R., Tuskan, G. A., et al. (2018). The Populus holobiont: dissecting the effects of plant niches and genotype on the microbiome. Microbiome 6:31. doi: 10.1186/s40168-018-0413-8
Denyer, T., Ma, X., Klesen, S., Scacchi, E., Nieselt, K., and Timmermans, M. (2019). Spatiotemporal developmental trajectories in the Arabidopsis root revealed using high-throughput single-cell RNA sequencing. Dev. Cell 48, 840.e845–852.e845. doi: 10.1016/j.devcel.2019.02.022
Deyou, Q., Shenglong, B., Jianchao, M., Lisha, Z., Fenjuan, S., Kaikai, Z., et al. (2019). The genome of Populus alba x Populus tremula var. glandulosa clone 84K. DNA Res. 5, 423–431. doi: 10.1093/dnares/dsz020
Dinesh, R., Srinivasan, V., Sheeja, T. E., Anandaraj, M., and Srambikkal, H. (2017). Endophytic actinobacteria, diversity, secondary metabolism and mechanisms to unsilence biosynthetic gene clusters. Crit. Rev. Microbiol. 43, 546–566. doi: 10.1080/1040841X.2016.1270895
Dubrovina, A. S., and Kiselev, K. V. (2016). Age-associated alterations in the somatic mutation and DNA methylation levels in plants. Plant Biol. 18, 185–196. doi: 10.1111/plb.12375
Edwards, J. A., Santos-Medellin, C. M., Liechty, Z. S., Nguyen, B., Lurie, E., Eason, S., et al. (2018). Compositional shifts in root-associated bacterial and archaeal microbiota track the plant life cycle in field-grown rice. PLoS Biol. 16:e2003862. doi: 10.1371/journal.pbio.2003862
Forchetti, G., Masciarelli, O., Izaguirre, M. J., Alemano, S., Alvarez, D., and Abdala, G. (2010). Endophytic bacteria improve seedling growth of sunflower under water stress, produce salicylic acid, and inhibit growth of pathogenic fungi. Curr. Microbiol. 61, 485–493. doi: 10.1007/s00284-010-9642-1
Franzosa, E. A., Sirota-Madi, A., Avila-Pacheco, J., Fornelos, N., Haiser, H. J., Reinker, S., et al. (2019). Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 4, 293–305. doi: 10.1038/s41564-018-0306-4
Gottel, N. R., Castro, H. F., Kerley, M., Yang, Z., Pelletier, D. A., Podar, M., et al. (2011). Distinct microbial communities within the endosphere and rhizosphere of Populus deltoides roots across contrasting soil types. Appl. Environ. Microbiol. 77, 5934–5944. doi: 10.1128/AEM.05255-11
Gururani, M. A., and Upadhyaya, C. (2013). Plant growth-promoting rhizobacteria enhance abiotic stress tolerance in Solanum tuberosum through inducing changes in the expression of ROS-scavenging enzymes and improved photosynthetic performance. J. Plant Growth Regul. 32, 245–258. doi: 10.1007/s00344-012-9292-6
Hacquard, S., Garrido-Oter, R., González, A., Spaepen, S., Ackermann, G., Lebeis, S., et al. (2015). Microbiota and host nutrition across plant and animal kingdoms. Cell Host Microbe 17, 603–616. doi: 10.1016/j.chom.2015.04.009
Henning, J. A., Weston, D. J., Pelletier, D. A., Timm, C. M., Jawdy, S. S., and Classen, A. T. (2016). Root bacterial endophytes alter plant phenotype, but not physiology. PeerJ. 4:e2606. doi: 10.7717/peerj.2606
Hu, L., Robert, C., Selma, C., Zhang, X., Meng, Y., Li, B., et al. (2018). Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat. Commun. 9:2738. doi: 10.1038/s41467-018-05122-7
Liu, Q., Liang, Z., Feng, D., Jiang, S., Wang, Y., Du, Z., et al. (2021). Transcriptional landscape of rice roots at the single-cell resolution. Mol. Plant 14, 384–394. doi: 10.1016/j.molp.2020.12.014
Liu, Z., Zhou, Y., Guo, J., Li, J., and Sun, X. (2020). Global dynamic molecular profiles of stomatal lineage cell development by single-cell RNA sequencing. Mol. Plant 13, 1178–1193. doi: 10.1016/j.molp.2020.06.010
Lobell, D. B., Schlenker, W., and Costa-Roberts, J. (2011). Climate trends and global crop production since 1980. Science 333, 616–620. doi: 10.1126/science.1204531
Lombard, V., Ramulu, H. G., Drula, E., Coutinho, P. M., and Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. doi: 10.1093/nar/gkt1178
Marasco, R., Rolli, E., Ettoumi, B., Vigani, G., Mapelli, F., Borin, S., et al. (2012). A drought resistance-promoting microbiome is selected by root system under desert farming. PLoS One 7:e48479. doi: 10.1371/journal.pone.0048479
Maurer, L. M., Yohannes, E., Bondurant, S. S., Radmacher, M., and Slonczewski, J. L. (2005). pH regulates genes for flagellar motility, catabolism, and oxidative stress in Escherichia coli K-12. J. Bacteriol. 187, 304–319. doi: 10.1128/JB.187.1.304-319.2005
Munné-Bosch, S. (2007). Aging in perennials. Crit. Rev. Plant Sci. 26, 123–138. doi: 10.1080/07352680701402487
Munné-Bosch, S., and Alegre, L. (2002). Plant aging increases oxidative stress in chloroplasts. Planta 214, 608–615. doi: 10.1007/s004250100646
Nayfach, S., and Pollard, K. S. (2015). Average genome size estimation improves comparative metagenomics and sheds light on the functional ecology of the human microbiome. Genome Biol. 16:51. doi: 10.1186/s13059-015-0611-7
Naylor, D., Degraaf, S., Purdom, E., and Coleman-Derr, D. (2017). Drought and host selection influence bacterial community dynamics in the grass root microbiome. ISME J. 11, 2691–2704. doi: 10.1038/ismej.2017.118
Niu, X., Song, L., Xiao, Y., and Ge, W. (2017). Drought-tolerant plant growth-promoting rhizobacteria associated with foxtail millet in a semi-arid agroecosytem and their potential in alleviating drought stress. Front. Microbiol. 8:2580. doi: 10.3389/fmicb.2017.02580
Numponsak, T., Kumla, J., Suwannarach, N., Matsui, K., Lumyong, S., and Sarrocco, S. (2018). Biosynthetic pathway and optimal conditions for the production of indole-3-acetic acid by an endophytic fungus, Colletotrichum fructicola CMU-A109. PLoS One 13:e0205070. doi: 10.1371/journal.pone.0205070
Rich-Griffin, C., Stechemesser, A., Finch, J., Lucas, E., Ott, S., and Schafer, P. (2020). Single-cell transcriptomics: a high-resolution avenue for plant functional genomics. Trends Plant Sci. 25, 186–197. doi: 10.1016/j.tplants.2019.10.008
Rolli, E., Marasco, R., Vigani, G., Ettoumi, B., Mapelli, F., Deangelis, M. L., et al. (2015). Improved plant resistance to drought is promoted by the root-associated microbiome as a water stress-dependent trait. Environ. Microbiol. 17, 316–331. doi: 10.1111/1462-2920.12439
Santosmedellin, C., Edwards, J., Liechty, Z., Nguyen, B., and Sundaresan, V. (2017). Drought stress results in a compartment-specific restructuring of the rice root-associated microbiomes. MBio 8:e00764-17. doi: 10.1128/mBio.00764-17
Satterlee, J. W., Strable, J., and Scanlon, M. J. (2020). Plant stem-cell organization and differentiation at single-cell resolution. Proc. Natl. Acad. Sci. U. S. A. 117, 33689–33699. doi: 10.1073/pnas.2018788117
Schwalm, C. R., Anderegg, W. R. L., Michalak, A. M., Fisher, J. B., Biondi, F., Koch, G., et al. (2017). Global patterns of drought recovery. Nature 548, 202–205. doi: 10.1038/nature23021
Selvakumar, G., Panneerselvam, P., and Ganeshamurthy, A. N. (2014). “Diversity utility and potential of actinobacteria in the agro-ecosystem,” in Bacterial Diversity in Sustainable Agriculture. Sustainable Development and Biodiversity. Vol 1. ed. D. Maheshwari (Cham: Springer International Publishing), 23–40.
Seyfferth, C., Renema, J., Wendrich, J. R., Eekhout, T., Seurinck, R., Vandamme, N., et al. (2021). Advances and opportunities of single-cell transcriptomics for plant research. Annu. Rev. Plant Biol. 72, 847–866. doi: 10.1146/annurev-arplant-081720-010120
Shaw, R., Tian, X., and Xu, J. (2021). Single-cell transcriptome analysis in plants: advances and challenges. Mol. Plant 14, 115–126. doi: 10.1016/j.molp.2020.10.012
Simmons, T., Styer, A. B., Pierroz, G., Gonçalves, A. P., and Coleman-Derr, D. (2020). Drought drives spatial variation in the millet root microbiome. Front. Plant Sci. 11:599. doi: 10.3389/fpls.2020.00599
Taghavi, S., Garafola, C., Monchy, S., Newman, L., Hoffman, A., Weyens, N., et al. (2009). Genome survey and characterization of endophytic bacteria exhibiting a beneficial effect on growth and development of poplar trees. Appl. Environ. Microbiol. 75, 748–757. doi: 10.1128/AEM.02239-08
Tian, C., Du, Q., Xu, M., Du, F., and Jiao, Y. (2020). Single-nucleus RNA-seq resolves spatiotemporal developmental trajectories in the tomato shoot apex. bioRxiv 20200920305029. doi: 10.1101/20200920305029
Timm, C. M., Carter, B. K. R., Carrell, C. A. A., Jun, S. R., and Jawdy, D. S. S. (2018). Abiotic stresses shift belowground Populus-associated bacteria toward a core stress microbiome. mSystems 3:e00070-17. doi: 10.1128/mSystems.00070-17
Timm, C. M., Pelletier, D. A., Jawdy, S. S., Gunter, L. E., Henning, J. A., Nancy, E., et al. (2016). Two poplar-associated bacterial isolates induce additive favorable responses in a constructed plant-microbiome system. Front. Plant Sci. 7:497. doi: 10.3389/fpls.2016.00497
Vanessa, N. K., Gouvêa, T. R., Duarte, L. O. M., Dini, A. F., Rodrigo, M., Itamar, S. D. M., et al. (2013). Water regime influences bulk soil and rhizosphere of Cereus jamacaru bacterial communities in the Brazilian Caatinga biome. PLoS One 8:e73606. doi: 10.1371/journal.pone.0073606
Veach, A. M., Chen, H. H., Yang, Z. K., Labbe, A. D., Engle, N. L., Tschaplinski, T. J., et al. (2020). Plant hosts modify belowground microbial community response to extreme drought. mSystems 5:e00092-20. doi: 10.1128/mSystems.00092-20
Veach, A. M., Morris, R., Yip, D. Z., Yang, Z. K., Engle, N. L., Cregger, M. A., et al. (2019). Rhizosphere microbiomes diverge among Populus trichocarpa plant-host genotypes and chemotypes, but it depends on soil origin. Microbiome 7:76. doi: 10.1186/s40168-019-0668-8
Wang, L., Cui, J., Jin, B., Zhao, J., and Lin, J. (2020a). Multifeature analyses of vascular cambial cells reveal longevity mechanisms in old Ginkgo biloba trees. Proc. Natl. Acad. Sci. U. S. A. 117, 2201–2210. doi: 10.1073/pnas.1916548117
Wang, Y., Huan, Q., Chu, X., Li, K., and Qian, W. (2020b). Single-cell transcriptome analyses recapitulate the cellular and developmental responses to abiotic stresses in rice. bioRxiv 20200130926329 doi: 10.1101/20200130926329
Wendrich, J. R., Yang, B., Vandamme, N., Verstaen, K., Smet, W., Van de Velde, C., et al. (2020). Vascular transcription factors guide plant epidermal responses to limiting phosphate conditions. Science 370:eaay4970. doi: 10.1126/science.aay4970
Xu, L., Naylor, D., Dong, Z., Simmons, T., Pierroz, G., Hixson, K. K., et al. (2018). Drought delays development of the sorghum root microbiome and enriches for monoderm bacteria. Proc. Natl. Acad. Sci. U. S. A. 115, E4284–E4293. doi: 10.1073/pnas.1717308115
Zelicourt, D., Al-Yousif, M., and Hirt, H. (2013). Rhizosphere microbes as essential partners for plant stress tolerance. Mol. Plant 6, 242–245. doi: 10.1093/mp/sst028
Zhang, T. Q., Chen, Y., and Wang, J. W. (2021). A single-cell analysis of the Arabidopsis vegetative shoot apex. Dev. Cell 56, 1056–1107. doi: 10.1016/j.devcel.2021.02.021
Keywords: drought, microbiome, poplar, inoculation, interplay between drought stress and plant microbiome
Citation: Xie J, Dawwam GE, Sehim AE, Li X, Wu J, Chen S and Zhang D (2021) Drought Stress Triggers Shifts in the Root Microbial Community and Alters Functional Categories in the Microbial Gene Pool. Front. Microbiol. 12:744897. doi: 10.3389/fmicb.2021.744897
Edited by:
Marco Scortichini, Council for Agricultural and Economics Research (CREA), ItalyReviewed by:
Olivera Topalovic, Aarhus University, DenmarkCopyright © 2021 Xie, Dawwam, Sehim, Li, Wu, Chen and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deqiang Zhang, ZGVxaWFuZ3poYW5nQGJqZnUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.