 Huawei Yi1†
Huawei Yi1† Qin Wang
Qin Wang Xiaowei Gao
Xiaowei Gao- 1Clinical Laboratory, The First Affiliated Hospital of Yangtze University, Jingzhou, China
- 2School of Pharmacy, Southwest Medical University, Luzhou, China
- 3Department of Chemistry, Zhejiang University, Hangzhou, China
Incorporation of noncanonical amino acids (ncAAs) into proteins has been proven to be a powerful tool to manipulate protein structure and function, and to investigate many biological processes. Improving the yields of ncAA-containing proteins is of great significance in industrial-scale applications. Escherichia coli C321.ΔA was generated by the replacement of all known amber codons and the deletion of RF1 in the genome and has been proven to be an ideal host for ncAA-containing protein expression using genetic code expansion. In this study, we investigated the transcriptome and proteome profiles of this first codon reassignment strain and found that some functions and metabolic pathways were differentially expressed when compared with those of its parent strain. Genes involved in carbohydrate and energy metabolism were remarkably downregulated. Our results may provide important clues about the growth defects in E. coli C321.ΔA. Furthermore, we improved the yields of ncAA-containing proteins in E. coli C321.ΔA by integrating the T7 RNA polymerase system.
Introduction
DNA and RNA are genetic information carriers that determine the lengths and sequences of proteins (Crick, 1970). As the main gene products, proteins participate in nearly every cellular process and have a wide range of functions, including signal transduction, transcriptional regulation, catalytic reaction, and cytoskeleton formation. Despite the diverse functional roles of proteins, nearly all of them are composed of 20 canonical amino acid building blocks (Polycarpo et al., 2004). In nature, posttranslational modifications of proteins, such as methylation, phosphorylation, and glycosylation at specific residues, usually occur. These modifications can augment new chemistries in proteins and make the functions of proteins more versatile (Seo and Lee, 2004). It has also been proven that that site-specific incorporation of noncanonical amino acids (ncAAs) with different functional structures into proteins has the potential to enhance the physical, chemical, and biological properties of proteins, and even generate new functions (Chin, 2014; Xiao and Schultz, 2016).
Genetic code expansion has been proven to be a powerful tool to site-specifically incorporate ncAAs into proteins in vivo. More than 200 ncAAs with different functional groups have been genetically encoded in both prokaryotic and eukaryotic organisms with high fidelity and efficiency (Xiao and Schultz, 2016). A heterogeneous and orthogonal tRNA/synthetase pair is needed to insert ncAAs of interest in response to a nonsense or frameshift codon. Although genetic code expansion has been successfully applied in yeast, mammalian cells, and even animals, Escherichia coli remains an attractive expression host because of its robustness, low cost, facile manipulation, and high levels of protein expression (Chin, 2014; Wals and Ovaa, 2014). Several orthogonal tRNA/synthetase pairs from Methanocaldococcus jannaschii, Methanosarcina barkeri, Methanosarcina mazei, Saccharomyces cerevisiae, and Pyrococcus horikoshii have been successfully engineered in E. coli to genetically encode a large number of ncAAs with novel functions, including unique chemical reactivities, metal binding, photo-crosslinking, photocaging, and fluorescence (Liu and Schultz, 2010).
To date, the amber codon UAG is the most frequently used codon for ncAA incorporation in genetic code expansion. Naturally, UAG is recognized by release factor 1 (RF1) and mediates peptide chain synthesis termination during translation in E. coli (Ryden et al., 1986). Due to the competition of RF1, the recognition and binding efficiency of UAG by the engineered orthogonal UAG-decoding tRNA-synthetase pair is not sufficient, and the suppression rate for a single amber codon is limited to 10–20% (Young et al., 2010). To address this issue, considerable effort has been made with different strategies, including increasing the copy number of orthogonal synthetase/tRNA genes, modification of Ef-tu (elongation factor thermo-unstable) to accommodate ncAAs-tRNA substrates, engineering of orthogonal ribosomes, and deletion of RF1 in E. coli (Wang et al., 2007; Young et al., 2010; Johnson et al., 2011; Park et al., 2011; Chatterjee et al., 2013).
Previously, it was reported that RF1 is essential for E. coli to decode UAG and terminate translation, and knockout of the prfA gene, encoding RF1, from the E. coli genome is lethal (Ryden and Isaksson, 1984). However, Mukai et al. (2010) found that eliminating RF1 could be achieved after engineering seven essential genes: coaD, hda, hemA, mreC, murF, lolA, and lpxK, of E. coli to end with ochre codon UAA. Johnson et al. (2011) found that RF1 could also be removed by deactivation of the in-frame UAG autoregulation element and mutation of Thr246 to alanine in RF2. Both studies demonstrated that the yields of the ncAA-containing proteins were increased, and the efficiency of incorporation of ncAAs at multiple sites in a single polypeptide was also improved when these RF1 deleted E. coli strains were used as expression hosts. However, the growth rates of these E. coli strains decreased remarkably.
Deletion of RF1 in E. coli would result in C-terminal extensions of many proteins unexpectedly due to the read-through of amber stop codons, which might cause cellular growth defects (Johnson et al., 2011). By using multiplex automated genome engineering and conjugative assembly genome engineering, Lajoie et al. (2013) replaced all known amber codons UAG in E. coli MG1655 with synonymous ochre codons UAA and deleted RF1 to generate the strain E. coli C321.ΔA. In E. coli C321.ΔA, UAG is no longer a stop codon, but a blank codon that can be used to assign unambiguously to a natural amino acid or ncAA. The yields of full-length proteins bearing one or multiple ncAAs were remarkably increased and comparable to those of wild-type proteins when using E. coli C321.ΔA as an expression host. Up to 30 ncAAs can be incorporated into a single polypeptide with high yields (~50mg/L) and accuracy (>95%; Amiram et al., 2015). Thus, E. coli C321.ΔA is an ideal expression host for genetic code expansion and an ideal chassis cell to reassign other synonymous codon to generate more blank codons.
To de novo design and synthesis of non-canonical polymers in living cells, chassis cell with robust ncAAs containing protein expression ability is needed to perform further genomic, metabolic, and protein engineering. It would be helpful to obtain more information about the physiological and biochemical characteristics and reveal the potential effects of codon reassignment on E. coli. Considered that codon reassignment in the whole genome may affect gene transcription and translation, we comparatively analyzed the transcriptome and proteome of the first codon reassignment strain E. coli C321.ΔA in this study. Because T7 RNA polymerase elongates peptide chains approximately five times faster than the E. coli RNA polymerase, the T7 RNA polymerase system has become one of the most widely used expression systems in E. coli (Balzer et al., 2013). The pET-derived plasmids based on the T7 RNA polymerase system have become the preferred expression systems in industry due to their high expression capabilities. To be able to use the T7 RNA polymerase system in E. coli C321.ΔA, we integrated the T7 RNA polymerase gene generated from E. coli BL21(DE3) into the genome of E. coli C321.ΔA.exp. We comparatively tested the ability of this strain to express ncAA-containing proteins with pET-derived plasmids and found that the yields of the target proteins were remarkably increased. Thus, the strains E. coli C321.ΔA exp T7 obtained in this study could facilitate and further improve the yields of ncAA-containing proteins when used as expression hosts with pET-derived plasmids.
Materials and Methods
Materials
FastDigest restriction enzymes were purchased from Fermentas (Burlington, Canada). The EasyGeno Assembly Cloning kit was purchased from Tiangen Biotech (Beijing, China). The 2× Taq PCR Master Mix was purchased from Beijing Solarbio Science & Technology Co., Ltd. (Beijing, China). KOD-Plus-Neo DNA polymerase was purchased from Toyobo Co., Ltd. (Osaka, Japan). Oligonucleotide primers and genes were synthesized by GENEWIZ Bio Inc. (Suzhou, China). All other chemicals and reagents used were of analytical grade.
Bacteria Strains and Growth Conditions
Detailed information on the strains used in this study is presented in Table 1. Escherichia coli EcNR2, E. coli C321.ΔA, and E. coli C321.ΔA.exp were obtained from Addgene (Watertown, MA, United States). Escherichia coli DH10B and E. coli BL21(DE3) were purchased from Thermo Fisher Scientific (Rockford, IL, United States) and Novagen (Darmstadt, Germany), respectively. Unless otherwise indicated, bacteria were grown in LB medium at 37 or 30°C (for E. coli EcNR2 and E. coli C321.ΔA only) supplemented with kanamycin (30μg/ml), spectinomycin (50μg/ml), chloramphenicol (34μg/ml), and/or ampicillin (50μg/ml) when needed.
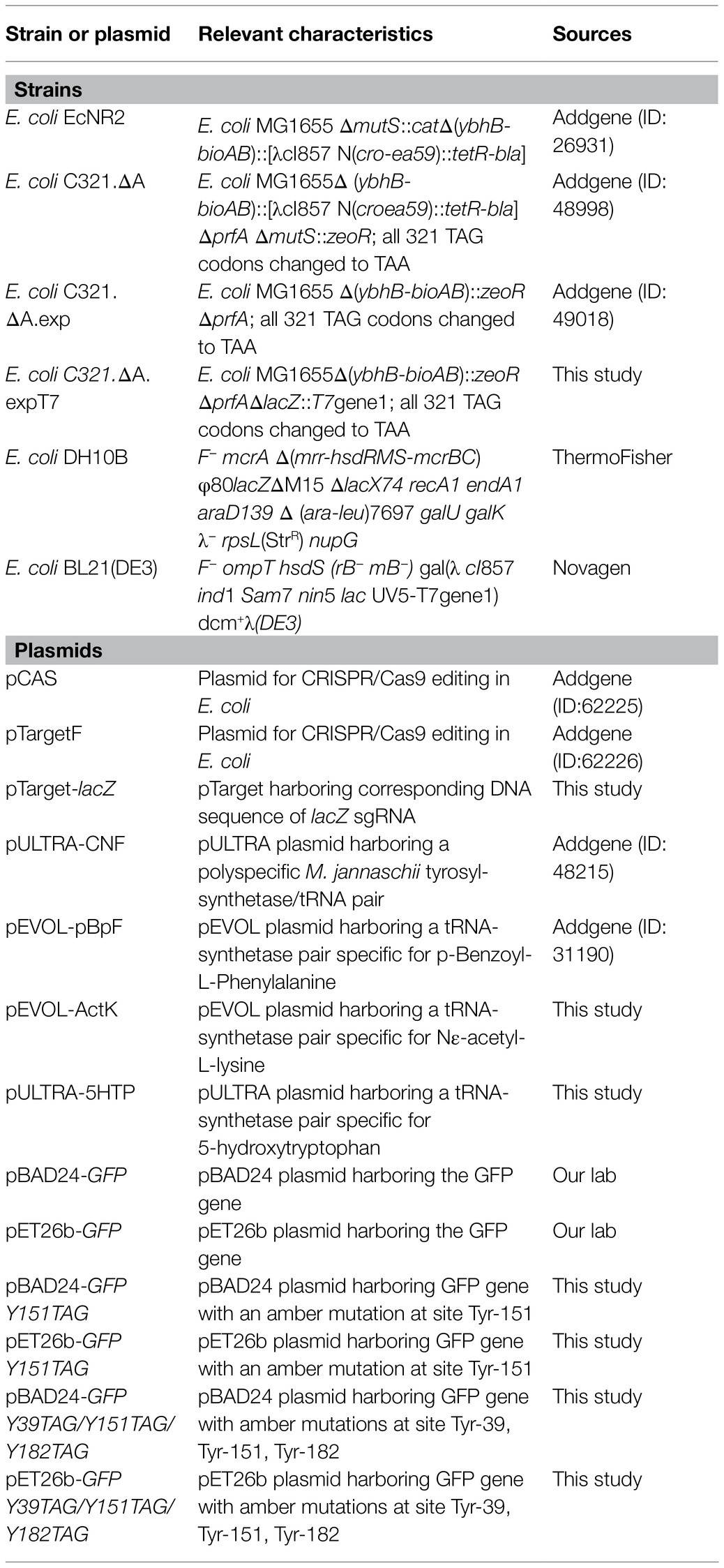
Table 1. Bacterial strains and plasmids used in this study.
Transcriptomic Analyses of E. coli EcNR2 and E. coli C321.ΔA
After the OD600 values of the culture reached 0.6~0.8, the cells were harvested by centrifugation and washed three times with sterile phosphate buffer at 4°C. Total RNA was extracted from the pellet cells using the RNeasy Plant Mini Kit (Qiagen, CA, United States) according to the manufacturer’s protocol. The purity and concentration of the resulting RNA were determined using a NanoDrop instrument (Thermo Scientific, Wilmington, DE, United States), and the integrity was analyzed using 1% agarose gel electrophoresis. RNA integrity number values were calculated using the Agilent Bioanalyzer 2,100 system (Agilent Technologies, Santa Clara, United States). Ribosomal RNA (rRNA) was depleted from the samples using the Illumina Ribo-Zero rRNA removal kit, and the resulting mRNA was fragmented with an average insert length of approximately 200bp. After construction of the cDNA library using the TruSeq Stranded Total RNA Library Prep Kit (Illumina, San Diego, CA, United States), the sequences were determined using the Illumina HiSeq platform at Majorbio Bio-Pharm Technology Co., Ltd., Shanghai, China. To gain statistical confidence, RNA-seq experiments were performed in three independent biological replicates. The datasets generated for this study can be found in the NCBI with an accession number of PRJNA748036.
The quality of the raw reads was filtered and controlled using SeqPrep1 and Sickle software (Version 1.33). The resulting valid sequences were then mapped and aligned to the reference genome (E. coli C321.ΔA)2 using BLAST+ (version 2.9.0) and Bowtie2 (version 2.3.5).3 The rRNA contamination assessment was performed by randomly selecting 10,000 raw reads in each sample and aligning them to Rfam database4 with blast method. Based on the annotation results, percentage of rRNA in each sample is calculated, which is estimated as rRNA contamination. The bioinformatics analyses were performed using the free online platform of Majorbio Cloud Platform5 from Shanghai Majorbio Bio-pharm Technology Co., Ltd. The gene expression level analyses were performed using RSEM tool.6 FPKM and TPM method are used to calculated expression level, FPKM represents fragments per kilobase of exon model per million mapped reads, and TPM represents transcript per million mapped reads. The FPKM and TPM are able to eliminate the influence of different gene length and sequencing discrepancy on the calculation of gene expression. Therefore, the calculated gene expression can be directly used for comparing the difference of gene expression among samples. Principal coordinate analysis (PCoA) was completed using edgeR.7 The differentially expressed genes (DEGs) and transcripts in each sample were determined using the Salmon (version 0.14.1) and DESeq2 (version 1.24.0)8 program with default settings. Significant differences in gene expression were evaluated using one-way ANOVA with a set of value of p<0.05, and fold-change ≥2. Goatools9 is exploited to identify statistically significantly enriched Gene Ontology (GO) term using Fisher’s exact test. The purpose of performing FDR correction is to reduce the Type-1 error by Bonferroni, Holm, BY, BH (multiple hypothesis test method). After multiple testing correction, GO terms with corrected value of p≤0.05 are significantly enriched in DEGs. KOBAS 2.010 is exploited to identify statistically significantly enriched pathway using Fisher’s exact test. The purpose of performing FDR correction is to reduce the Type-1 error by Bonferroni, Holm, BY, BH (multiple hypothesis test method). The calculating formula of value of p and corrected value of p is similar with that in GO analysis. After multiple testing correction, we choose pathways with value of p<0.05 that are significantly enriched in DEGs. Functional analyses of the DEGs were performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG), GO, Swiss-Prot (version 2019.7.1), and Pfam (version v32.0) databases.
Proteomics Analyses of E. coli EcNR2 and E. coli C321.ΔA
After washing three times with phosphate buffer at 4°C, the pelleted cells were resuspended in 2ml of lysis buffer (20mM HEPES, 10mM NaOH, pH 7.5) containing 150mM NaCl and 1% protease inhibitor cocktail (GE Healthcare, Pittsburgh, United States) and sonicated on ice. After centrifugation at 15,000×g for 20min to remove cell debris, the protein samples were precipitated using trichloroacetic acid at a final concentration of 20% (w/v) and washed with ice-cold acetone. After air drying, the precipitated proteins were stored at −80°C until use. Total proteins were analyzed by SDS-PAGE, and the concentrations were determined using the Bradford method with bovine serum albumin as a standard. Before mass spectrometric analysis, the protein samples were dissolved in 8M urea/100mM NH4HCO3 and subjected to reductive alkylation and trypsin hydrolysis. Three biological replicates were performed for each strain in the proteomic analyses.
The Mascot and Proteome Discoverer software was used to search the raw LC–MS/MS data against the UniProt E. coli database. Significant differences in protein expression were evaluated using one-way ANOVA with a set of value of p<0.05 and fold-change ≥2. Differentially expressed proteins were analyzed by KEGG pathway analysis and GO analysis using the Swissport database and the Pfam database to identify significantly enriched KEGG pathways and GO functions. Based on the GO and KEGG categories, proteins were classified according to cellular components, molecular functions, and biological processes.
Plasmid Construction and Mutagenesis
Detailed information on the plasmids used in this study is presented in Table 1. The plasmids pCAS, pTargetF, pULTRA-CNF, and pEVOL-pBpF were obtained from Addgene. The plasmids pBAD24-GFP and pET26b-GFP were previously used in our laboratory. The gene of a previously reported orthogonal tRNA-synthetase pair that incorporated 5-hydroxytryptophan (5-HTP) into proteins was chemically synthesized and cloned into a pULTRA plasmid to generate the plasmid pULTRA-5HTP. The primers used in this study are listed in Table 2. The primer pairs GFP-Y39TAG-F and GFP-Y39TAG-R, GFP-Y151TAG-F and GFP-Y151TAG-R, and GFP-Y182TAG-F and GFP-Y182TAG-R were used to construct amber mutations at the permissive sites of GFP using the QuikChange site-directed mutagenesis method (Papworth, 1996).
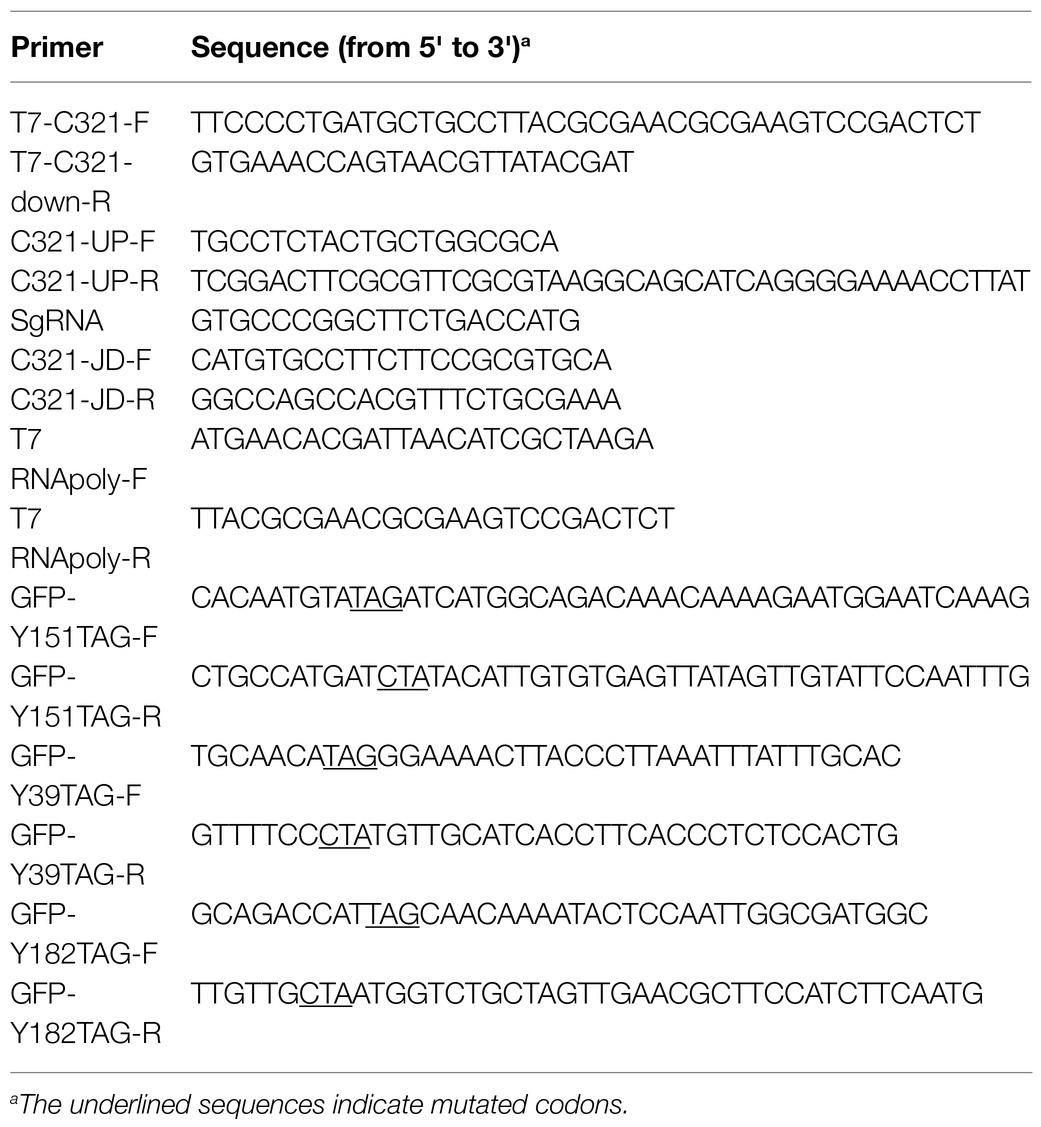
Table 2. Primers used in this study.
Integration of T7 RNA Polymerase Into the Genome of E. coli C321.ΔA.exp
T7 RNA polymerase was integrated into the genome of E. coli C321.ΔA.exp using the CRISPR-Cas9 system (Jiang et al., 2015). The T7 RNA polymerase gene was first amplified by PCR from the genome of E. coli BL21(DE3) using the primer T7-C321-F/T7-C321down-R pair, and the homology arms were amplified using the genome of E. coli C321.ΔA.exp as a template with the primer C321-UP-F/C321-UP-R pair. The two resulting PCR products were used to amplify the T7 RNA polymerase expression cassettes by overlapping PCR with the primers C321-UP-F/T7-C321down-R. Genome editing experiments were performed as described by Jiang et al. (2015). Briefly, E. coli C321.ΔA.exp was first transformed with pCas and induced with arabinose (10mM final concentration) to express recombination-related proteins. After induction, the harvested cells were used to generate competent cells. Then, 100ng of pTarget-lacZ plasmid and 400ng of T7 RNA polymerase expression cassette DNA were co-electroporated into competent cells. After recovery at 30°C for 1h, the cells were spread onto LB agar containing kanamycin and spectinomycin and incubated overnight at 30°C. Transformants were identified by colony PCR with the primer C321-JD-F/C321-JD-R pair, and DNA sequencing with the primer T7 RNApoly-F/T7 RNApoly-R pair.
To cure the gene editing-related plasmids, the verified edited colony was inoculated into 5ml of LB medium containing IPTG and kanamycin and cultured at 30°C for 12h. The culture was spread onto LB plates containing kanamycin and incubated at 30°C overnight. Then, the colonies were confirmed to be cured of plasmid pTarget-lacZ by determining their sensitivity to spectinomycin. The pCas plasmid was cured by incubating the colonies without plasmid pTarget-lacZ in LB medium overnight at 37°C. Finally, E. coli C321.ΔA.expT7 with a free plasmid was verified by colony PCR.
Growth Assay of Different E. coli Strains
A single colony of each E. coli strain was picked and grown in LB medium at 37°C overnight. The resulting cultures were normalized and diluted to an OD600 of 0.05, in 50ml of fresh LB medium. The strains were incubated at 37°C, and the OD600 values were measured every 30min for 12h.
In-Cell Fluorescence Assay
The E. coli strains harboring two plasmids, one containing different orthogonal tRNA-synthetase pairs and the other containing GFP variant genes, were grown at 37°C in 50ml of LB supplemented with antibiotics. After the OD600 values of the cultures reached 0.4, 1mM of ncAAs was added to the cultures and shaken for another 40min. Then, the inducer IPTG or/and arabinose with final concentrations of 0.4 and 10mM was added to the medium to induce protein expression. After induction at 30°C for 10h, 1ml of the culture was harvested by centrifugation at 8,000×g for 10min. The cell pellets were washed three times and suspended in 1ml of phosphate buffer. After normalization, the GFP fluorescence of the cells was measured using a plate reader (Ex/Em=488/513nm). Notably, the cells used to express 5HTP containing proteins were cultured in M9 medium.
Protein Expression and Purification
Recombinant GFP variants containing different ncAAs were produced in E. coli strains, as described above. The harvested cells were suspended in phosphate buffer and sonicated on ice. After centrifugation at 13,000×g for 10min at 4°C, the soluble fraction containing the recombinant proteins was recovered and purified using a Ni2+-charged chelating Sepharose Fast Flow column (GE Healthcare, Uppsala, Sweden). The purified proteins were analyzed by SDS-PAGE, and the concentrations were measured using the Bradford method with bovine serum albumin as a standard (Bradford, 1976).
Statistical Analysis
All data in this study were evaluated using one-way ANOVA in SPSS version 20.0. Statistical significance was set at p<0.05.
Results
Comparative Analyses of the Transcriptome Profiles of E. coli C321.ΔA
The strain E. coli EcNR2 is the parent strain of E. coli C321.ΔA (Lajoie et al., 2013). To reveal the potential effects of UAG codon reassignment on E. coli, the transcriptomic information of E. coli C321.ΔA and E. coli EcNR2 was identified and compared using transcript sequencing. After removing the low-quality reads, the number of clean reads obtained from each sample ranged from 31.25 to 35.48 million, with genome mapped ratios from 97.50 to 99.31%. PCA analysis and correlation coefficient analysis revealed that greater variability existed between the two E. coli strains than within each biological replicate (Supplementary Figures S1, S2). The results suggested that the replicates in each group had reproducible transcriptome profiles, and UAG codon reassignment could alter the transcriptomic pattern of E. coli. The total number of expressed genes in E. coli C321.ΔA and E. coli EcNR2 was 3,722 and 3,676, respectively (Figure 1A). A total of 132 genes were uniquely expressed in E. coli C321.ΔA, whereas 86 genes were uniquely expressed in E. coli EcNR2. The threshold for identification of significantly DEGs was set as at least twofold-change and p<0.05. Compared with E. coli EcNR2, 394 genes were upregulated, and 389 genes were downregulated in E. coli C321.ΔA (Figure 1B). Thus, DEGs accounted for approximately 21.04% of the total number of expressed genes. A scatter plot was constructed to represent the overall changes in the DEGs between E. coli C321.ΔA and E. coli EcNR2 (Figure 1C). Detailed information on the upregulated and downregulated genes, including gene names, fold changes, and function categories, is shown in Supplementary Tables S1 and S2.
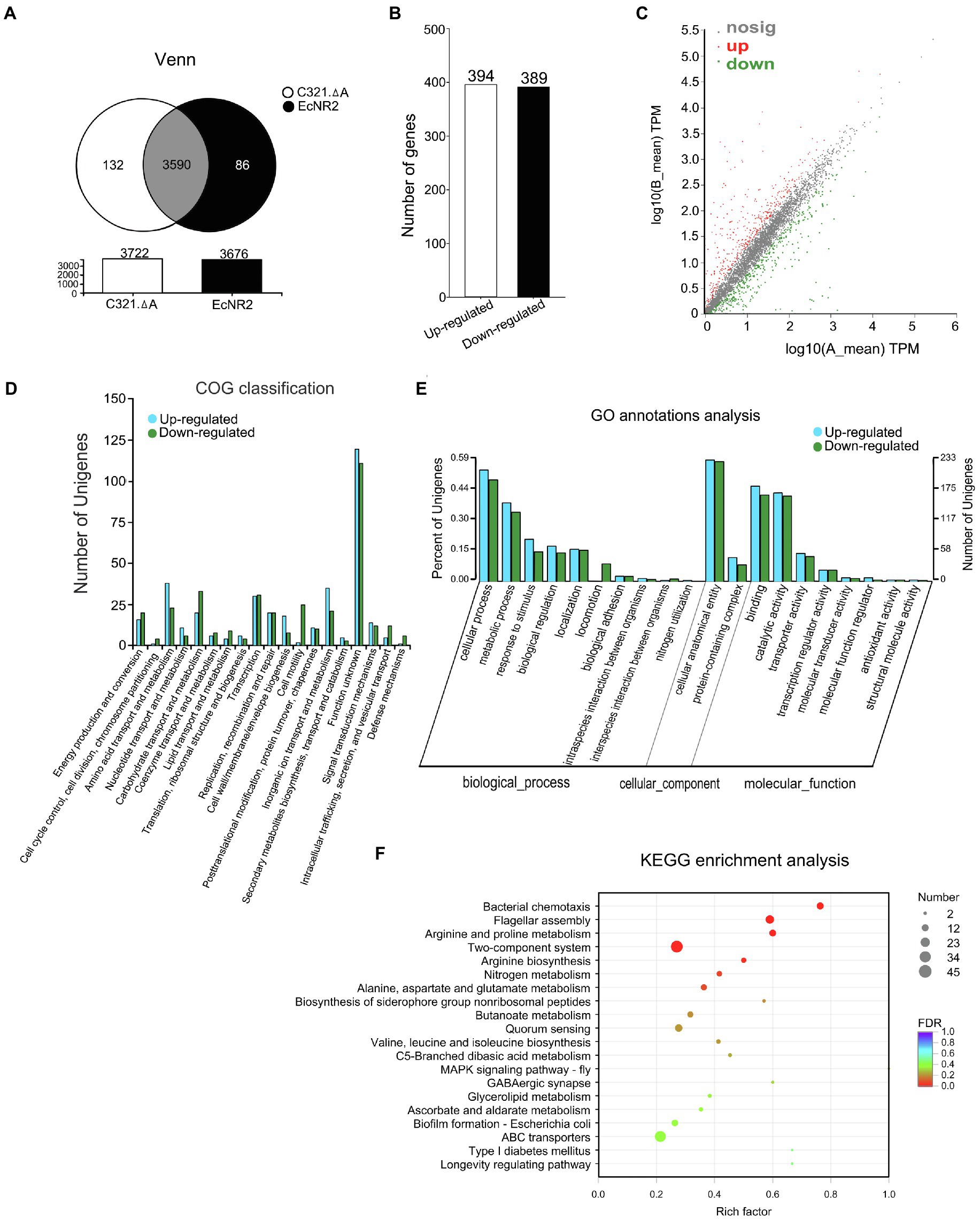
Figure 1. The transcriptome profiles of Escherichia coli C321.ΔA and its parent strain. (A) A venn diagram of shared and unique of DEGs between E. coli C321.ΔA and its parent strain. (B) Up- and down-regulated expressed gene count. (C) A scatter plot represents the overall changes of the transcriptomic profiles of E. coli C321.ΔA. The horizontal and vertical coordinates represent the expression of genes in the two strains. The values of the horizontal and vertical coordinates are logarithmic, with each point representing a specific gene. The red points in the graph indicate significantly up-regulated genes, the green points indicate significantly down-regulated genes, and the gray points are non-significantly different genes. When all genes are mapped, the closer the points are to zero, the lower the expression; those points that deviate more from the diagonal indicate that the gene is more differentially expressed between the two samples. (D) Functional classification of the DEGs according to COG. In the horizontal coordinates, the functional classification names were shown. (E) Gene Ontology (GO) annotations analyses of the DEGs. (F) Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of the DEGs. The vertical axis represents the pathway name, and the horizontal axis represents the Rich factor. The size of the dots indicates the number of genes in the pathway, while the color of the dots corresponds to the different FDR-value ranges.
A total of 783 DEGs identified from the transcripts were assigned to 19 clusters of orthologous groups (COG; Figure 1D). According to the COG classification analysis, 231 genes were classified into the category “unknown function, [S],” accounting for 29.50% of the total number of DEGs. The function class containing the relatively most significantly up-expressed genes was “Cell wall/membrane/envelope biogenesis, [M],” whereas the function class containing the relatively most significantly down-expressed genes was “Cell motility, [N].”
To further assess the effects of the transcript changes caused by UAG codon reassignment on E. coli, we performed GO annotation and KEGG pathway analysis to identify the functional classification of the DEGs. As shown in Figure 1E, GO analysis mapped all of the DEGs into biological processes, cellular components, and molecular functions via biological process functional categorization. Within the “biological process” function class, the largest numbers of DEGs were related to the “cellular process” subclass, whereas within the “cellular component” class, the “cellular anatomical entity” subclass contained the most DEGs. Within the “molecular function” function class, the “binding” and “catalytic activity” subclasses had the largest number of DEGs. The metabolic pathways affected by the DEGs were revealed by KEGG pathway analysis. The 20 most abundant metabolic pathways in the DEGs are shown in Figure 1F, and four significantly (p<0.05) enriched KEGG pathways were “Flagellar assembly,” “Bacterial chemotaxis,” “Arginine and proline metabolism,” and “Two-component system.” The metabolic pathway “Two-component system” contained the largest number of DEGs when compared with other enriched KEGG pathways.
Comparative Analyses of the Proteome Profiles of E. coli C321.ΔA
To determine the potential effects of UAG codon reassignment on the proteome of E. coli, the protein synthesis profiles of E. coli C321.ΔA and E. coli EcNR2 were identified and compared using label-free proteomic sequencing analysis. The whole cell proteins of E. coli C321.ΔA and E. coli EcNR2 were first analyzed by SDS-PAGE (Figure 2A). According to the identified proteomic information, the total number of expressed proteins in E. coli C321.ΔA and E. coli EcNR2 was 2,089 and 2,221, respectively (Supplementary Figure S3). The number of unique proteins identified in E. coli C321.ΔA and E. coli EcNR2 was 31 and 63, respectively. The threshold for identification of significantly differentially expressed proteins was set as at least twofold-change, and p<0.05. The overall significant changes in the proteome of E. coli C321.ΔA were represented by a differential volcano plot (Figure 2B). Compared with E. coli EcNR2, 256 proteins were downregulated, and 125 proteins were upregulated. Thus, the significantly differentially expressed proteins accounted for roughly 18.24% of the total expressed proteins in E. coli C321.ΔA. The locations of the differentially expressed proteins in E. coli are shown in Figure 2C, and 91.60% of the identified differentially expressed proteins, including 111 upregulated and 238 downregulated proteins, were cytoplasmic.
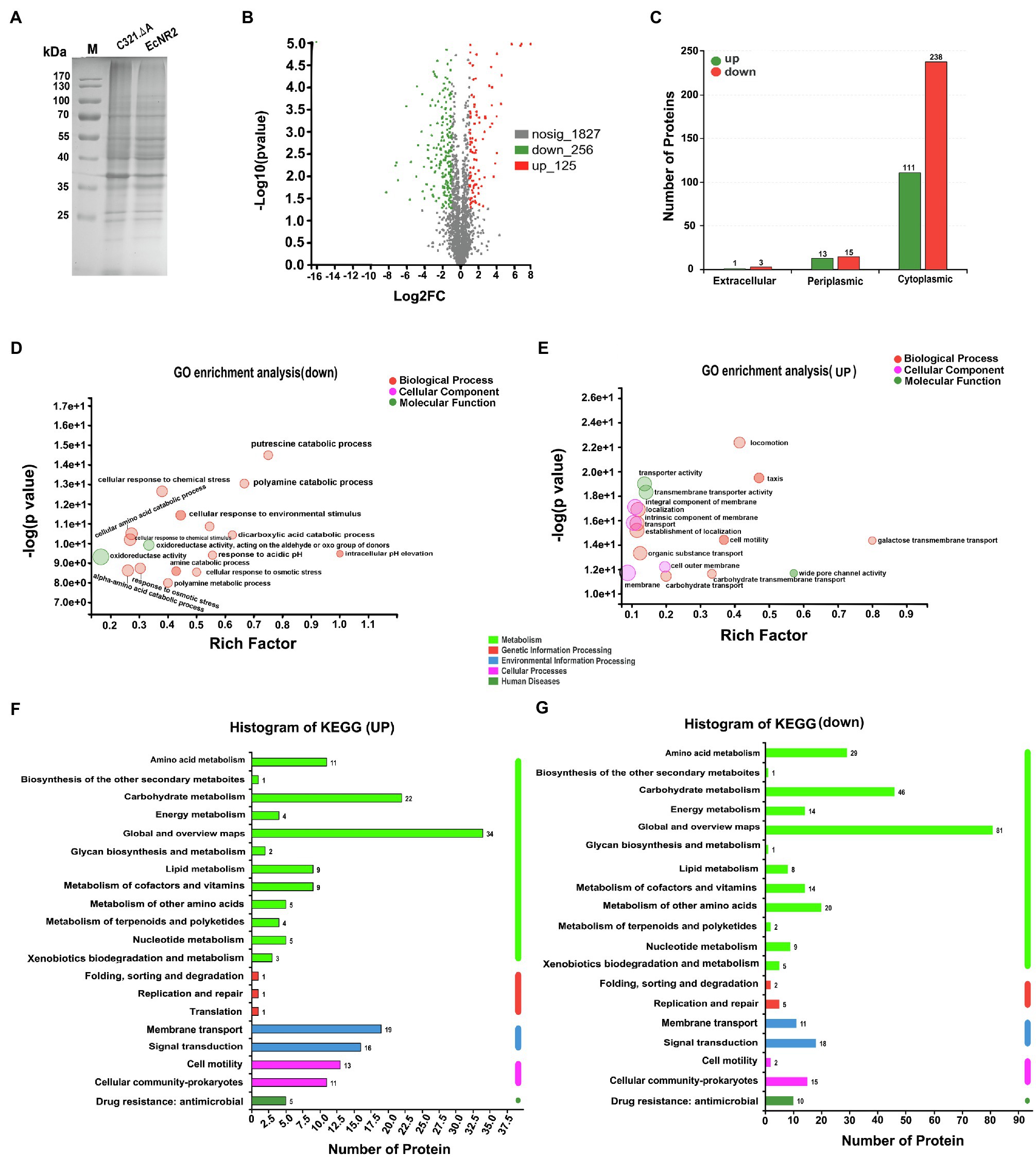
Figure 2. The proteome profiles of E. coli C321.ΔA and its parent strain. (A) SDS-PAGE analysis of the whole cell proteins of the strain E. coli C321.ΔA and its parent strain. (B) Differential volcano plot of different expressed proteins. The horizontal coordinate is the value of the fold change in protein expression between the two strains, and the vertical coordinate is the statistical test value of the difference in gene expression. Each point in the graph represents a specific protein, the further to the left and up the more significant the difference in expression. (C) Prediction analysis of subcellular localization of the significantly different expressed proteins. (D,E) GO enrichment analyses of the different expressed proteins. (F,G) KEGG pathways analysis of the different expressed proteins.
The significantly differentially expressed proteins were analyzed by GO enrichment and KEGG pathway analyses. According to GO categorization, the 10 most relatively abundant down-expressed groups were “intracellular pH elevation,” “pH elevation,” “putrescine catabolic process,” “glucarate catabolic process,” “glucarate metabolic process,” “D-glucarate catabolic process,” “D-glucarate metabolic process,” “glycine decarboxylation via glycine cleavage system,” “cellular response to acidic pH,” and “polyamine catabolic process,” whereas the top 10 relatively abundant up-expressed groups were “galactose transmembrane transport,” “methylgalactoside transport,” “glycoside transport,” “L-lyxose metabolic process,” “methyl accepting chemotaxis protein complex,” “transmembrane signaling receptor activity,” “porin activity,” “wide pore channel activity,” “hemotaxis,” and “taxis” (Figures 2D,E). As shown in Figure 2F, a large proportion of the upregulated proteins was enriched in seven KEGG pathways, including “Global and overview maps,” “Carbohydrate metabolism,” “Membrane transport,” “Signal transduction,” “Cell motility,” “Cellular community – prokaryotes,” and “Amino acid metabolism.” The down-expressed proteins were abundant in “Global and overview maps,” “Carbohydrate metabolism,” “Amino acid metabolism,” “Metabolism of other amino acids,” “Signal transduction,” “Cellular community – prokaryotes,” “Metabolism of cofactors and vitamins,” “Energy metabolism,” “Membrane transport,” and “Drug resistance: antimicrobial” (Figure 2G).
Integration of the T7 RNA Polymerase Gene Into the Genome of E. coli C321.ΔA. exp
Without the competition of RF1, the orthogonal tRNA-synthetase pair could efficiently decode the amber UAG codon and remarkably improve the yields of ncAA-containing proteins in E. coli C321.ΔA (Lajoie et al., 2013). However, the fitness of E. coli C321.ΔA is far lower than that of its non-recoded ancestor, which limits its application in many fields, including industrial-scale protein production, metabolic engineering, and live cell imaging (Wannier et al., 2018). Using multiplex genome engineering and predictive modeling, Kuznetsov et al. (2017) recovered the fitness deficit of E. coli C321.ΔA and generated the strain E. coli C321.ΔA. exp. Escherichia coli C321.ΔA. exp showed slightly enhanced ncAA-dependent protein expression. To facilitate the incorporation of ncAAs into proteins using the pET series plasmids in E. coli C321.ΔA. exp and further improve the yields of ncAA-containing proteins, we tried to integrate the T7 RNA polymerase gene into the genome using the CRISPR-Cas9 system in this study.
The T7 RNA polymerase expression cassette was amplified from the E. coli BL21(DE3) genome and inserted into the genome of E. coli C321.ΔA. exp to replace the lac Z gene via the CRISPR-Cas9 system, as described in the “Materials and Methods” section (Figure 3A). Genomic PCR screening and sequencing verified that T7 RNA polymerase was integrated into the genome of E. coli C321.ΔA. exp (Figure 3B). The T7 RNA polymerase inserted strain was named E. coli C321.ΔA. exp T7. We tested whether insertion of the T7 RNA polymerase gene affected the growth of E. coli C321.ΔA. exp. According to Figure 3C, the growth rate of E. coli C321.ΔA. exp T7 was similar to that of E. coli C321.ΔA. exp, indicating that integration of the T7 RNA polymerase gene had limited effects on the strain growth. Next, we investigated whether the inserted T7 RNA polymerase gene could work well in E. coli C321.ΔA. exp by performing recombinant protein expression using pET derivative plasmids. As shown in Figure 3D, both E. coli C321.ΔA. exp and E. coli C321.ΔA. exp T7 could detect strong GFP fluorescence when harboring the pBAD24-GFP plasmid after induction with arabinose, but only E. coli C321.ΔA.exp T7 showed remarkable GFP fluorescence when both strains harbored the pET26b-GFP plasmid after induction with IPTG. Thus, we experimentally verified that pET derivative plasmids could not be used in E. coli C321.ΔA.exp directly because of the lack of T7 RNA polymerase, but could work well in our strain E. coli C321.ΔA.exp T7.
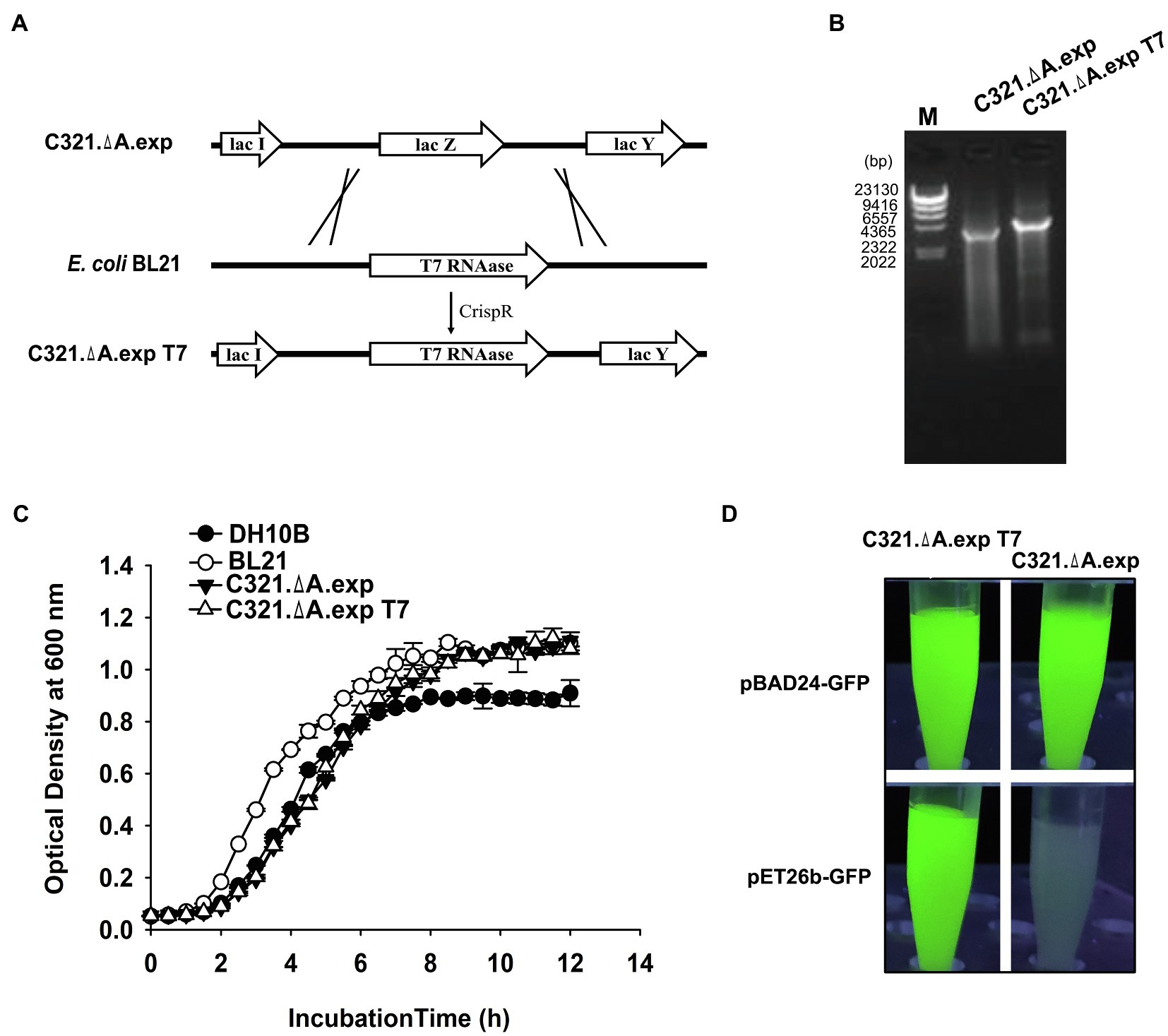
Figure 3. Integration of T7 RNA polymerase gene in E. coli C321.ΔA. exp and verification. (A) Display of the overall strategy used in this study to integrate T7 RNA polymerase into the genome of E. coli C321.ΔA.exp. The crossing lines mean that the LacZ gene in the strain C321. exp is replaced by the T7 RNAase gene amplified from the BL21 genome. (B) PCR verification of the successful integration of T7 RNA polymerase in the genome of E. coli C321.ΔA.exp. (C) Growth curves of the E. coli strains used for recombinant protein expression in this study. (D) Expression of GFP in E. coli C321.ΔA.exp and E. coli C321.ΔA.exp T7 with plasmids pBAD24-GFP and pET26b-GFP.
The Ability of Incorporation of Different ncAAs at Multiple UAG Sites in E. coli C321.ΔA.exp T7
To test the ability of incorporation of different ncAAs at multiple UAG sites in E. coli C321.ΔA.exp T7, we used two widely reported orthogonal plasmids (pEVOL and pUltra derivatives) to incorporate six ncAAs into GFP with amber mutations. For comparison, we also performed the same experiments in E. coli BL21(DE3), E. coli DH10B, and E. coli C321.ΔA.exp. The structures of ncAAs and the main features of the plasmids used in this experiment are shown in Figures 4A,B. According to the growth curves, E. coli BL21(DE3) grew faster in the exponential phase than E. coli C321.ΔA.exp and E. coli C321.ΔA.exp T7, but the same biomass accumulated in the stationary phase. Escherichia coli DH10B showed a similar growth rate to E. coli C321.ΔA.exp and E. coli C321.ΔA.exp T7, but had a slightly lower biomass in the stationary phase (Figure 3C).
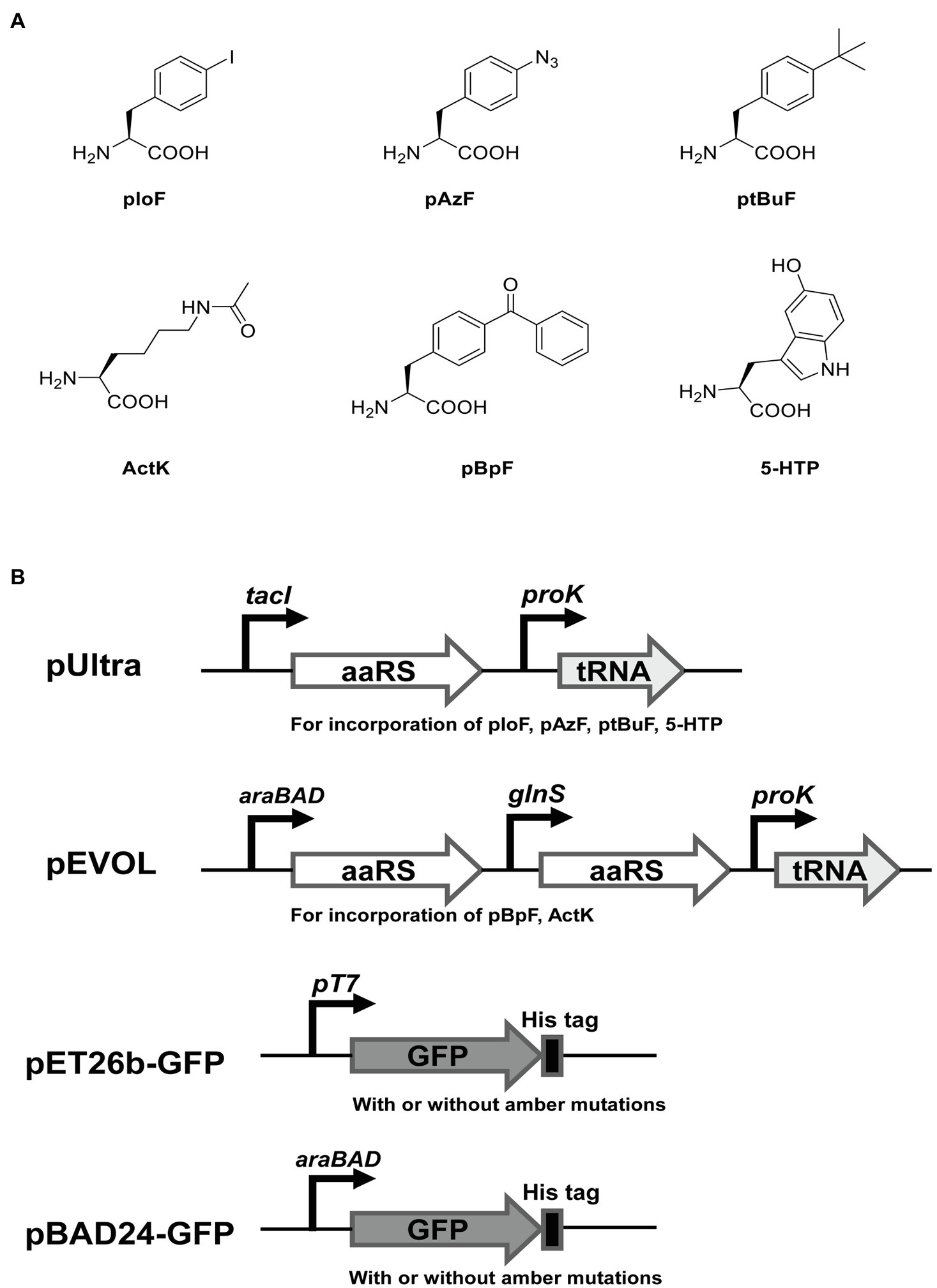
Figure 4. The noncanonical amino acids (ncAAs) and plasmids used in this study for recombinant GFP expression. (A) Structures of ncAAs. (B) The main features of plasmids.
We first compared the GFP expression ability of the four strains when using pIoF as a building block. As shown in Figure 5A, when using pET26b as a vector for expression of the GFP Y151TAG gene, only strains E. coli BL21(DE3) and E. coli C321.ΔA.exp T7 had strong in-cell GFP fluorescence when supplemented with 1mM pIoF. The strains E. coli DH10B and E. coli C321.ΔA.exp did not show any visible fluorescence due to the lack of T7 RNA polymerase and the inability to use the T7 promoter in the pET26 plasmid. In E. coli strain C321.ΔA.exp T7 harboring plasmid pET26b-GFPY151TAG, a relatively high GFP fluorescence was also detected in the absence of pIoF. We speculated that the reason for this phenomenon is that UAGs in the GFP gene could be misread by endogenous tRNAs after RF1 removal in E. coli C321.ΔA.exp T7, as described by Johnson et al. (2012). When using plasmid pBAD24 as a vector for expression of the GFP Y151TAG gene, all four strains showed remarkable in-cell GFP fluorescence in the presence of 1mM pIoF. Escherichia coli DH10B showed stronger fluorescence than E. coli BL21(DE3), which may be due to the inability of E. coli DH10B to catabolize the inducer L-arabinose and could express more intact pIoF-containing GFP variants. Escherichia coli BL21(DE3) showed weaker fluorescence than E. coli C321.ΔA.exp and E. coli C321.ΔA.exp T7 may be attributed to the competition between RF1 and premature translation termination in E. coli BL21(DE3). We also determined the ability of the four strains to incorporate pIoF at the three UAG sites in GFP. As shown in Figure 5B, E. coli C321.ΔA.exp T7 harboring plasmid pET26b-GFPY39TAG/Y151TAG/Y182TAG had stronger fluorescence than the other three strains, indicating that the yields of the pIoF-containing protein could be remarkably improved with the expression of proteins with multiple UAG sites in E. coli C321.ΔA.exp T7.
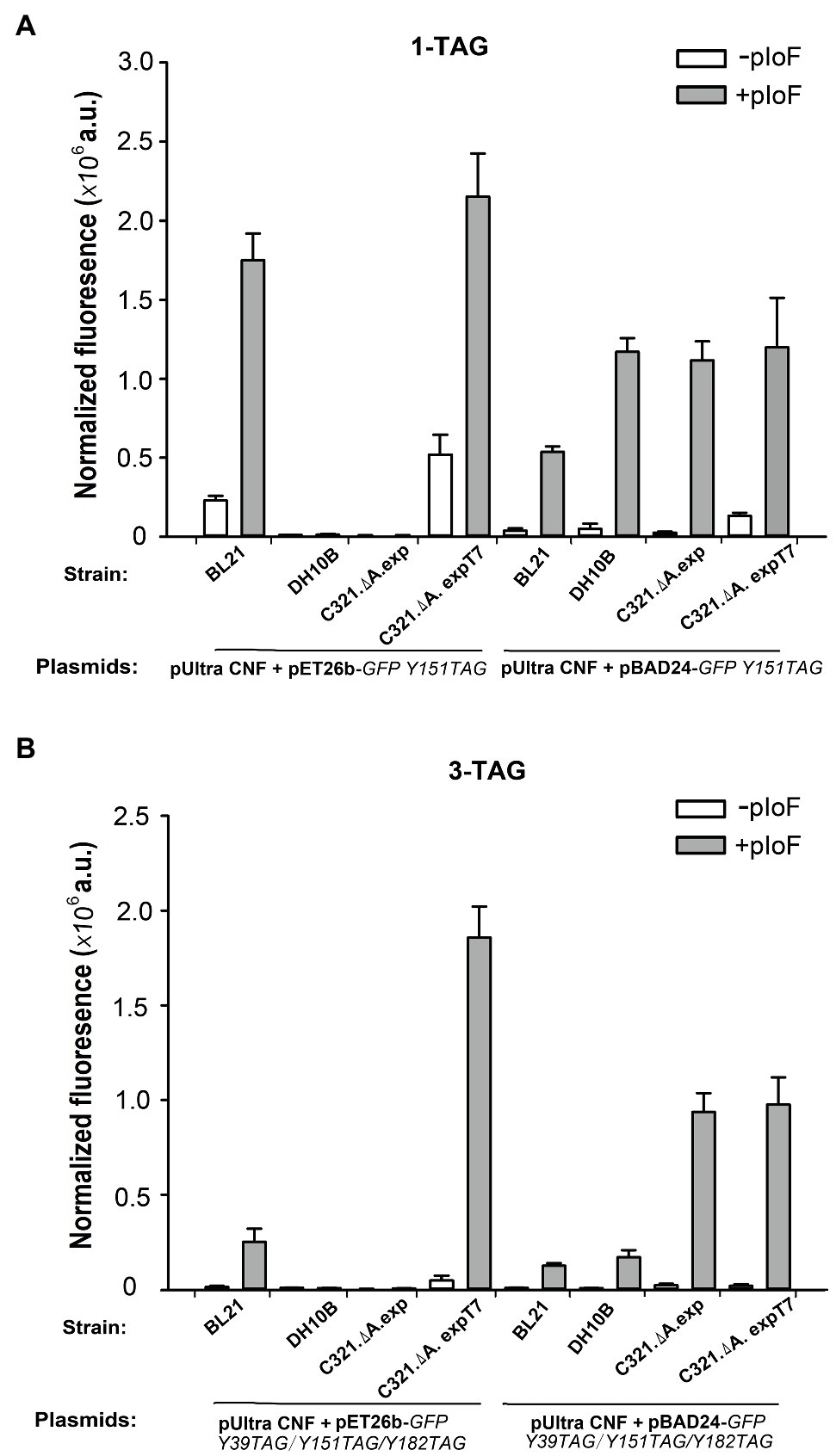
Figure 5. Incorporation of pIoF into GFP containing amber mutations in different strains. (A) In-cell GFP fluorescence intensity assay of different strains harboring pUltra-CNF, and pBAD24-GFPY151TAG or pET26b-GFPY151TAG in the presence or absence of 1mM pIoF in the medium. (B) In-cell GFP fluorescence intensity assay of different strains harboring pUltra-CNF, and pBAD24-GFPY39TAG/Y151TAG/Y182TAG or pET26b-GFPY39TAG/Y151TAG/Y182TAG in the presence or absence of 1mM pIoF in the medium. The values are expressed as means±SD from three independent experiments.
Next, we compared the ability of the four strains to incorporate five other ncAAs at three UAG sites in GFP. As shown in Figure 6, regardless of the ncAAs and orthogonal tRNA-synthetase pairs used, E. coli C321.ΔA.exp T7 harboring the plasmid pET26b-GFPY39TAG/Y151TAG/Y182TAG showed the highest fluorescence among the samples. This result demonstrates that pET derivative plasmids are better than pBAD derivative plasmids in improving the yields of ncAA-containing proteins in E. coli C321.ΔA derivative strains. The purified GFP with different ncAAs expressed in E. coli C321.ΔA.exp T7 harboring plasmid pET26b-GFPY39TAG/Y151TAG/Y182TAG is shown in Supplementary Figure S3. Escherichia coli C321.ΔA.exp and E. coli C321.ΔA.exp T7 harboring pBAD24-GFPY39TAG/Y151TAG/Y182TAG also showed relatively strong in-cell fluorescence, but was lower than that of E. coli C321.ΔA.exp T7 harboring the plasmid pET26b-GFPY39TAG/Y151TAG/Y182TAG. We speculate that this may be because E. coli C321.ΔA.exp and E. coli C321.ΔA.exp T7 degrade and reduce the concentration of the inducer L-arabinose during recombinant protein expression, thus, decreasing the yield of the protein products.
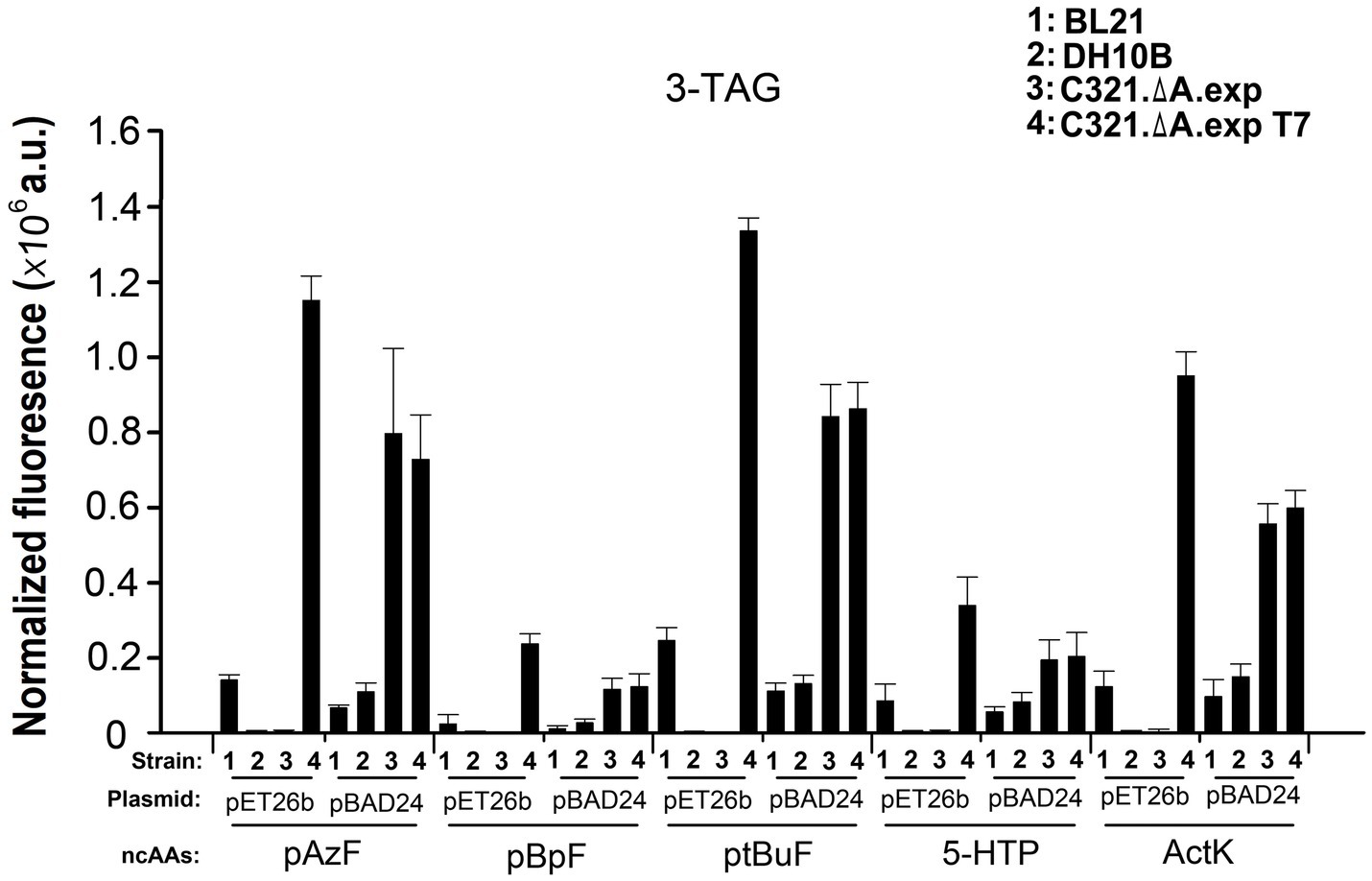
Figure 6. Incorporation of various ncAAs into GFP containing three amber mutations at different positions in different strains. In-cell GFP fluorescence intensity assay of different strains harboring pUltra-CNF, and pBAD24-GFP or pET26b-GFP containing 3-TAG mutations in the presence or absence of 1mM various ncAAs in the medium. The values are expressed as means±SD from three independent experiments.
Discussion
The incorporation of ncAAs with different functional groups into proteins has been used in many biological fields, including exploring biological processes, designing fluorescent protein probes, capturing transient protein–protein interactions, enhancing protein drugs, and enzyme-directed evolution (Xiao and Schultz, 2016). Thus, the production of ncAA-containing proteins on an industrial scale is of great significance. In the past few decades, researchers have developed many techniques to incorporate ncAAs into proteins. One such technique is solid-phase peptide synthesis (Kimmerlin and Seebach, 2005). Polypeptide chains no larger than 50 residues can be directly synthesized using canonical amino acids and ncAAs. This method cannot be used to synthesize large proteins (de Graaf et al., 2009). However, this drawback can be partially compromised by some intein-based peptide ligation reactions. Cell-free translation systems can also be used to express proteins with ncAAs (Noren et al., 1989). The key point of this method is the preparation of aminoacylated tRNAs with ncAAs, which can be used by ribosomes to synthesize ncAA-containing proteins. Some ncAAs (structural analogs of canonical amino acids) can be mischarged onto wild-type tRNAs by endogenous corresponding aminoacyl-tRNA synthetases in vivo (Link et al., 2003; Hendrickson et al., 2004). In a strain auxotrophic for a certain canonical amino acid, the corresponding ncAA-analog in the medium can be taken by the cells, replacing the canonical amino acid at all sites in the whole proteome of the strain (Ibba and Hennecke, 1995; Sharma et al., 2000). This method may have toxic effects on host cells and restrict the yield of the target protein. Although these methods are useful for incorporating ncAAs into target proteins, they have their own limitations that include not being site-specific, being unsustainable, and only being applicable for canonical amino acid analogs.
Genetic code expansion can overcome most of the aforementioned drawbacks. It can be used to incorporate various ncAAs site-specifically and at multiple sites into target proteins in all living organisms (Chin, 2014). However, due to the competition between RFs and restriction of the endogenous ribosome, the yields of ncAA-containing proteins are usually lower. It has been proven that E. coli C321.ΔA and its derivative strains used as expression hosts could significantly improve the yields of ncAA-containing proteins, especially with expressed proteins containing nsAAs at multiple sites (Amiram et al., 2015). To further understand the molecular biology of E. coli C321.ΔA, we analyzed the transcriptome and proteome of this strain by high-throughput sequencing and label-free proteomic sequencing, respectively. Our results revealed that both the transcriptome and proteome of E. coli C321.ΔA showed significant differences when compared with that of its parent strain E. coli EcNR2. Some differentially expressed functions and metabolic pathways were identified in this study. The genes with the function of “Carbohydrate transport and metabolism” and “Cell motility” were significantly downregulated at the transcriptomic level in E. coli C321.ΔA. More strikingly, the gene products with the function of “Carbohydrate metabolism,” “Energy metabolism,” and “Amino acid metabolism” were remarkably downregulated at the proteomic level in E. coli C321.ΔA. These results indicate that deletion of RF1 and/or UAG codon reassignment may weaken carbohydrate utilization and energy generation in E. coli, which may be one of the reasons why E. coli C321.ΔA shows a growth defect in the phenotype. However, more studies are needed to strengthen this conclusion, and the molecular mechanism underlying these changes awaits future experimental endeavor.
Although the deletion of RF1 and reassignment of the UAG codon could significantly enhance the production of ncAA-containing proteins, in many cases, the yields of the ncAA-containing proteins are lower than those of the wild-type (Amiram et al., 2015). This phenomenon may be attributed to several factors. The enzymatic activity of the orthogonal tRNA/synthetase pairs usually generated from other living kingdoms may be insufficient in E. coli. This problem has been partially overcome by altering the copy number of the orthogonal plasmids, the copy number of orthogonal synthetase/tRNA genes in the plasmids, and the expression strength of the promoters (Young et al., 2010; Chatterjee et al., 2013). Increasing the enzymatic activity of orthogonal translation systems by directed evolution has also been reported to remarkably improve the yields of proteins containing multiple nsAAs (Amiram et al., 2015). The Ef-tu may have suboptimal activity with amino-acylated ncAA-tRNA substrates when delivered into the ribosome, and engineering of Ef-tu to accommodate ncAAs–tRNA substrates could further improve the ncAA-containing protein yields (Park et al., 2011). The development of orthogonal ribosomes is another approach that has been successfully used to enhance the incorporation of unnatural amino acids in vivo (Wang et al., 2007). The orthogonal ribosome is generated by directed evolution and can function in parallel with the natural ribosome. This was achieved by designing mutations in 16S rRNA. The mutated 16S rRNA could direct the orthogonal ribosome to the orthogonal messenger RNA containing corresponding complementary mutations in the Shine–Dalgarno sequence. Orthogonal ribosomes can be used to selectively translate orthogonal message RNAs that are not substrates for natural ribosomes.
In genetic code expansion, supplementing ncAAs into the medium is essential to express ncAA-containing proteins during culture fermentation. This would undoubtedly increase the production cost and limit the application of ncAA-containing proteins on an industrial scale, especially when ncAAs are expensive. Autonomous biosynthesis of ncAAs in cells by enzyme evolution and metabolic pathway engineering, and coupling it with genetic code expansion in situ could avoid supplementing ncAAs exogenously and significantly save costs. Much progress has been made in this field. By introducing a synthetic pathway from Streptomyces venezuelae, p-amino-phenylalanine (pAF) has been synthesized in E. coli and used as building blocks to produce pAF-containing proteins by genetic code expansion (Chen et al., 2018). 5-HTP has been successfully synthesized in E. coli harboring a phenylalanine 4-hydroxylase from Xanthomonas campestris and an artificial cofactor regeneration pathway (Chen et al., 2020). The biosynthesized 5-HTP could be genetically incorporated into proteins with high fidelity by genetic code expansion. L-phosphothreonine has been biosynthesized by Salmonella enterica kinase in E. coli and incorporated into proteins site-specially using a phosphoseryl-tRNA synthetase/tRNA pair (Zhang et al., 2017a). By exogenous feeding of ammonia, catechol, and pyruvate, L-dihydroxyphenylalanine was biosynthesized by a tyrosine phenol-lyase and directly incorporated into proteins by genetic code expansion in E. coli (Kim et al., 2018). Similarly, by feeding allyl mercaptan, S-allyl-L-cysteine is biosynthesized from O-acetylserine by PLP-dependent acetylserine sulfhydrylase isozymes of E. coli and subsequently incorporated into proteins by an evolved S-allylcysteinyl-tRNA synthetase/tRNA pair (Exner et al., 2017).
Several major challenges need to be addressed to synthesize non-canonical polymers in living cells. First, more aminoacyl-tRNA synthetase tRNA pairs that are orthogonal to not only the natural host synthetases and tRNAs but also to each other need to be developed. Early experiments have demonstrated that the MjTyrRS/tRNA pair and the P. horikoshii lysyl-tRNA synthetase/tRNA pair or PylRS/tRNA pair are mutually orthogonal and can be used to incorporate two different ncAAs into a single polypeptide (Wei et al., 2010). Efforts in de novo design and generation of multiple mutually orthogonal aaRS/tRNAs have also been reported (Neumann et al., 2010). Italia et al. (2019) developed a set of triply orthogonal pyrrolysyl–tRNA synthetase/tRNA pairs that can be used to decode three distinct non-canonical amino acids in a single polypeptide. Second, new blank codons are required to encode the incorporation of distinct ncAAs into proteins. Since the 61 triplet codons are used in the genome of most organisms for encoding natural amino acids into proteins, stop codons and special quadruplet codons have been used to encode the incorporation of unnatural amino acids before (Chatterjee et al., 2014; Tharp et al., 2021). More blank codons are required to encode non-canonical polymers in living cells. The UAG codon in E. coli C321.ΔA was the first blank codon to be assigned for ncAA incorporation. Since then, efforts for synonymous codon replacement, compression, and reassignment in the whole genome have been carried out (Wang et al., 2016). Up to seven codons have been replaced with synonymous alternatives in E. coli, and the engineered cells have been used to encode the translation of diverse noncanonical heteropolymers and macrocycles (Ostrov et al., 2016; Robertson et al., 2021). Recently, expanding the genetic alphabet with unnatural base pairs to generate new codons in E. coli has been made big breakthroughs by Malyshev et al. (2014). The unnatural codons can be decoded in vivo and incorporated ncAAs into proteins (Zhang et al., 2017b). With continuous progress in biochemistry, molecular biology, and chemical biology as described above, we believe that de novo design and synthesis of non-canonical polymers in living cells could be achieved in the future.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA748036.
Author Contributions
XGa conceived and supervised the experiments and wrote the initial draft of the manuscript. HY, JZ, FK, XGu, JY, and PX performed the experiments. XGa analyzed the data. LL and QW revised the initial draft of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the China Postdoctoral Science Foundation (2020M683364), the Department of Science and Technology of Sichuan Province (2020YJ0129), the Collaborative Fund of the Science and Technology Agency of the Luzhou Government and Southwest Medical University (2020LZXNYDJ29), and the Science Fund Project of the Southwest Medical University (2019ZQN024 and 2020ZRQNA007).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.744284/full#supplementary-material
Footnotes
1. ^https://github.com/jstjohn/SeqPrep
2. ^https://www.ncbi.nlm.nih.gov/nuccore/CP006698.1
3. ^http://bowtie-bio.sourceforge.net/bowtie2/index.shtml
6. ^http://deweylab.github.io/RSEM/
7. ^http://www.bioconductor.org/packages/2.12/bioc/html/edgeR.html
8. ^http://bioconductor.org/packages/release/bioc/html/DESeq2.html
References
Amiram, M., Haimovich, A. D., Fan, C., Wang, Y. S., Aerni, H. R., Ntai, I., et al. (2015). Evolution of translation machinery in recoded bacteria enables multi-site incorporation of nonstandard amino acids. Nat. Biotechnol. 33, 1272–1279. doi: 10.1038/nbt.3372
Balzer, S., Kucharova, V., Megerle, J., Lale, R., Brautaset, T., and Valla, S. (2013). A comparative analysis of the properties of regulated promoter systems commonly used for recombinant gene expression in Escherichia coli. Microb. Cell Factories 12:26. doi: 10.1186/1475-2859-12-26
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. doi: 10.1016/0003-2697(76)90527-3
Chatterjee, A., Lajoie, M. J., Xiao, H., Church, G. M., and Schultz, P. G. (2014). A bacterial strain with a unique quadruplet codon specifying non-native amino acids. Chembiochem 15, 1782–1786. doi: 10.1002/cbic.201402104
Chatterjee, A., Sun, S. B., Furman, J. L., Xiao, H., and Schultz, P. G. (2013). A versatile platform for single-and multiple-unnatural amino acid mutagenesis in Escherichia coli. Biochemistry 52, 1828–1837. doi: 10.1021/bi4000244
Chen, Y., Loredo, A., Gordon, A., Tang, J., Yu, C., Ordonez, J., et al. (2018). A noncanonical amino acid-based relay system for site-specific protein labeling. Chem. Commun. 54, 7187–7190. doi: 10.1039/C8CC03819H
Chen, Y., Tang, J., Wang, L., Tian, Z., Cardenas, A., Fang, X., et al. (2020). Creation of bacterial cells with 5-hydroxytryptophan as a 21st amino acid building block. Chem 6, 2717–2727. doi: 10.1016/j.chempr.2020.07.013
Chin, J. W. (2014). Expanding and reprogramming the genetic code of cells and animals. Annu. Rev. Biochem. 83, 379–408. doi: 10.1146/annurev-biochem-060713-035737
de Graaf, A. J. D., Kooijman, M., Hennink, W. E., and Mastrobattista, E. (2009). Nonnatural amino acids for site-specific protein conjugation. Bioconjug. Chem. 20, 1281–1295. doi: 10.1021/bc800294a
Exner, M. P., Kuenzl, T., To, T. M. T., Ouyang, Z., Schwagerus, S., Hoesl, M. G., et al. (2017). Design of S-allylcysteine in situ production and incorporation based on a novel pyrrolysyl-tRNA synthetase variant. Chembiochem 18, 85–90. doi: 10.1002/cbic.201600537
Hendrickson, T. L., De Crécy-Lagard, V., and Schimmel, P. (2004). Incorporation of nonnatural amino acids into proteins. Annu. Rev. Biochem. 73, 147–176. doi: 10.1146/annurev.biochem.73.012803.092429
Ibba, M., and Hennecke, H. (1995). Relaxing the substrate specificity of an aminoacyl-tRNA synthetase allows in vitro and in vivo synthesis of proteins containing unnatural amino acids. FEBS Lett. 364, 272–275. doi: 10.1016/0014-5793(95)00408-2
Italia, J. S., Addy, P. S., Erickson, S. B., Peeler, J. C., Weerapana, E., and Chatterjee, A. (2019). Mutually orthogonal nonsense-suppression systems and conjugation chemistries for precise protein labeling at up to three distinct sites. J. Am. Chem. Soc. 141, 6204–6212. doi: 10.1021/jacs.8b12954
Jiang, Y., Chen, B., Duan, C., Sun, B., Yang, J., and Yang, S. (2015). Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 81, 2506–2514. doi: 10.1128/AEM.04023-14
Johnson, D. B., Wang, C., Xu, J., Schultz, M. D., Schmitz, R. J., Ecker, J. R., et al. (2012). Release factor one is nonessential in Escherichia coli. ACS Chem. Biol. 7, 1337–1344. doi: 10.1021/cb300229q
Johnson, D. B., Xu, J., Shen, Z., Takimoto, J. K., Schultz, M. D., Schmitz, R. J., et al. (2011). RF1 knockout allows ribosomal incorporation of unnatural amino acids at multiple sites. Nat. Chem. Biol. 7, 779–786. doi: 10.1038/nchembio.657
Kim, S., Sung, B. H., Kim, S. C., and Lee, H. S. (2018). Genetic incorporation of L-dihydroxyphenylalanine (DOPA) biosynthesized by a tyrosine phenol-lyase. Chem. Commun. 54, 3002–3005. doi: 10.1039/C8CC00281A
Kimmerlin, T., and Seebach, D. (2005). “100 years of peptide synthesis”: ligation methods for peptide and protein synthesis with applications to beta-peptide assemblies. J. Pept. Res. 65, 229–260. doi: 10.1111/j.1399-3011.2005.00214.x
Kuznetsov, G., Goodman, D. B., Filsinger, G. T., Landon, M., Rohland, N., Aach, J., et al. (2017). Optimizing complex phenotypes through model-guided multiplex genome engineering. Genome Biol. 18:100. doi: 10.1186/s13059-017-1217-z
Lajoie, M. J., Rovner, A. J., Goodman, D. B., Aerni, H. R., Haimovich, A. D., Kuznetsov, G., et al. (2013). Genomically recoded organisms expand biological functions. Science 342, 357–360. doi: 10.1126/science.1241459
Link, A. J., Mock, M. L., and Tirrell, D. A. (2003). Non-canonical amino acids in protein engineering. Curr. Opin. Biotechnol. 14, 603–609. doi: 10.1016/j.copbio.2003.10.011
Liu, C. C., and Schultz, P. G. (2010). Adding new chemistries to the genetic code. Annu. Rev. Biochem. 79, 413–444. doi: 10.1146/annurev.biochem.052308.105824
Malyshev, D. A., Dhami, K., Lavergne, T., Chen, T., Dai, N., Foster, J. M., et al. (2014). A semi-synthetic organism with an expanded genetic alphabet. Nature 509, 385–388. doi: 10.1038/nature13314
Mukai, T., Hayashi, A., Iraha, F., Sato, A., Ohtake, K., Yokoyama, S., et al. (2010). Codon reassignment in the Escherichia coli genetic code. Nucleic Acids Res. 38, 8188–8195. doi: 10.1093/nar/gkq707
Neumann, H., Slusarczyk, A. L., and Chin, J. W. (2010). De novo generation of mutually orthogonal aminoacyl-tRNA synthetase/tRNA pairs. J. Am. Chem. Soc. 132, 2142–2144. doi: 10.1021/ja9068722
Noren, C. J., Anthony-Cahill, S. J., Griffith, M. C., and Schultz, P. G. (1989). A general method for site-specific incorporation of unnatural amino acids into proteins. Science 244, 182–188. doi: 10.1126/science.2649980
Ostrov, N., Landon, M., Guell, M., Kuznetsov, G., Teramoto, J., Cervantes, N., et al. (2016). Design, synthesis, and testing toward a 57-codon genome. Science 353, 819–822. doi: 10.1126/science.aaf3639
Papworth, C. (1996). Site-directed mutagenesis in one day with> 80% efficiency. Strategies 8, 3–4. doi: 10.1080/08924562.1996.11000299
Park, H. S., Hohn, M. J., Umehara, T., Guo, L. T., Osborne, E. M., Benner, J., et al. (2011). Expanding the genetic code of Escherichia coli with phosphoserine. Science 333, 1151–1154. doi: 10.1126/science.1207203
Polycarpo, C., Ambrogelly, A., Bérubé, A., Winbush, S. M., McCloskey, J. A., Crain, P. F., et al. (2004). An aminoacyl-tRNA synthetase that specifically activates pyrrolysine. Proc. Natl. Acad. Sci. U. S. A. 101, 12450–12454. doi: 10.1073/pnas.0405362101
Robertson, W. E., Funke, L. F. H., de la Torre, D., Fredens, J., Elliott, T. S., Spinck, M., et al. (2021). Sense codon reassignment enables viral resistance and encoded polymer synthesis. Science 372, 1057–1062. doi: 10.1126/science.abg3029
Ryden, S., and Isaksson, L. (1984). A temperature-sensitive mutant of Escherichia coli that shows enhanced misreading of UAG/A and increased efficiency for tRNA nonsense suppressors. Mol. Gen. Genet. MGG 193, 38–45. doi: 10.1007/BF00327411
Ryden, M., Murphy, J., Martin, R., Isaksson, L., and Gallant, J. (1986). Mapping and complementation studies of the gene for release factor 1. J. Bacteriol. 168, 1066–1069. doi: 10.1128/jb.168.3.1066-1069.1986
Seo, J. W., and Lee, K. J. (2004). Post-translational modifications and their biological functions: proteomic analysis and systematic approaches. BMB Rep. 37, 35–44. doi: 10.5483/BMBRep.2004.37.1.035
Sharma, N., Furter, R., Kast, P., and Tirrell, D. A. (2000). Efficient introduction of aryl bromide functionality into proteins in vivo. FEBS Lett. 467, 37–40. doi: 10.1016/S0014-5793(00)01120-0
Tharp, J. M., Vargas-Rodriguez, O., Schepartz, A., and Söll, D. (2021). Genetic encoding of three distinct noncanonical amino acids using reprogrammed initiator and nonsense codons. ACS Chem. Biol. 16, 766–774. doi: 10.1021/acschembio.1c00120
Wals, K., and Ovaa, H. (2014). Unnatural amino acid incorporation in E. coli: current and future applications in the design of therapeutic proteins. Front. Chem. 2:15. doi: 10.3389/fchem.2014.00015
Wang, K., Fredens, J., Brunner, S. F., Kim, S. H., Chia, T., and Chin, J. W. (2016). Defining synonymous codon compression schemes by genome recoding. Nature 539, 59–64. doi: 10.1038/nature20124
Wang, K., Neumann, H., Peak-Chew, S. Y., and Chin, J. W. (2007). Evolved orthogonal ribosomes enhance the efficiency of synthetic genetic code expansion. Nat. Biotechnol. 25, 770–777. doi: 10.1038/nbt1314
Wannier, T. M., Kunjapur, A. M., Rice, D. P., McDonald, M. J., Desai, M. M., and Church, G. M. (2018). Adaptive evolution of genomically recoded Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 115, 3090–3095. doi: 10.1073/pnas.1715530115
Wei, W., Ying, H., Wang, Z., Russell, W., Pai, P. J., Russell, D., et al. (2010). A facile system for genetic incorporation of two different noncanonical amino acids into one protein in Eescherichia coli. Angew. Chem. 122, 3279–3282. doi: 10.1002/anie.201000465
Xiao, H., and Schultz, P. G. (2016). At the interface of chemical and biological synthesis: an expanded genetic code. Cold Spring Harb. Perspect. Biol. 8:a023945. doi: 10.1101/cshperspect.a023945
Young, T. S., Ahmad, I., Yin, J. A., and Schultz, P. G. (2010). An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 395, 361–374. doi: 10.1016/j.jmb.2009.10.030
Zhang, M. S., Brunner, S. F., Huguenin-Dezot, N., Liang, A. D., Schmied, W. H., Rogerson, D. T., et al. (2017a). Biosynthesis and genetic encoding of phosphothreonine through parallel selection and deep sequencing. Nat. Methods 14, 729–736. doi: 10.1038/nmeth.4302
Keywords: genetic code expansion, noncanonical amino acids, transcriptome, proteome, Escherichia coli , T7 RNA polymerase
Citation: Yi H, Zhang J, Ke F, Guo X, Yang J, Xie P, Liu L, Wang Q and Gao X (2021) Comparative Analyses of the Transcriptome and Proteome of Escherichia coli C321.△A and Further Improving Its Noncanonical Amino Acids Containing Protein Expression Ability by Integration of T7 RNA Polymerase. Front. Microbiol. 12:744284. doi: 10.3389/fmicb.2021.744284
Edited by:
Richard Allen White III, University of North Carolina at Charlotte, United StatesReviewed by:
Jeffery M. Tharp, Yale University, United StatesDenis Jacob Machado, University of North Carolina at Charlotte, United States
Copyright © 2021 Yi, Zhang, Ke, Guo, Yang, Xie, Liu, Wang and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaowei Gao, eGlhb3dlaWdhb0Bzd211LmVkdS5jbg==
†These authors have contributed equally to this work