 Chonghui Xu
Chonghui Xu Jizheng Chen
Jizheng Chen Xinwen Chen
Xinwen Chen- 1State Key Laboratory of Virology, Wuhan Institute of Virology, Center for Biosafety Mega-Science, Chinese Academy of Sciences, Wuhan, China
- 2University of Chinese Academy of Sciences, Beijing, China
- 3Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou, China
Hepatitis viruses are primary causative agents of hepatitis and represent a major source of public health problems in the world. The host innate immune system forms the first line of defense against hepatitis viruses. Hepatitis viruses are sensed by specific pathogen recognition receptors (PRRs) that subsequently trigger the innate immune response and interferon (IFN) production. However, hepatitis viruses evade host immune surveillance via multiple strategies, which help compromise the innate immune response and create a favorable environment for viral replication. Therefore, this article reviews published findings regarding host innate immune sensing and response against hepatitis viruses. Furthermore, we also focus on how hepatitis viruses abrogate the antiviral effects of the host innate immune system.
Introduction
Hepatitis is an inflammation of the liver that gives rise to a wide spectrum of systemic manifestations and can cause death. It continues to be a global human health threat and economic burden. According to the World Health Organization (WHO), about 325 million people suffer from chronic hepatitis infection. There are five main strains of the hepatitis virus [type A, B, C, D, and E] (Carman and Thomas, 1990; Purcell, 1993)—all of which cause liver disease, but differ in some aspects, such as mode of transmission, severity of illness, geographical distribution, and prevention methods (Acorn et al., 1995; Rasche et al., 2019). Hepatitis A virus (HAV) and hepatitis E virus (HEV) are responsible for a large proportion of cases of acute hepatitis, while hepatitis B virus (HBV) and hepatitis C virus (HCV) are the most common causes of chronic viral hepatitis. Hepatitis D virus (HDV) is a satellite virus that requires the envelope proteins of HBV for cell release and uptake (Hughes et al., 2011; Tu et al., 2017; Smith and Simmonds, 2018).
The host innate immune response is the first line of defense against virus invasion. During virus infection, the pathogen-associated molecular patterns (PAMPs) of the virus are recognized by host pathogen recognition receptors (PRRs), which is the first step of antiviral innate immunity activation (Lester and Li, 2014; Streicher and Jouvenet, 2019). Viral nucleic acid, which represents a major PAMP, can be recognized by various PRRs, including retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs), Toll-like receptors (TLRs), and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), as well as cytosolic DNA sensors, such as cyclic GMP-AMP (cGAMP) synthase (cGAS), interferon (IFN) regulatory factors (DAIs), and interferon gamma inducible protein 16 (IFI16). PRRs then recruit downstream adaptor proteins, which typically include mitochondrial antiviral-signaling protein (MAVS), TIR-domain-containing adaptor-inducing IFN-β (TRIF), myeloid differentiation primary response gene 88 (MyD88), and intracellular stimulator of IFN genes (STING). These adaptor proteins ultimately lead to the activation of the transcription factor nuclear factor-kappa B (NF-κB) and interferon regulatory factor 3 (IRF3), subsequently inducing the expression of type I IFNs and pro-inflammatory cytokines, which contribute to the establishment of the “antiviral state” (Koyama et al., 2008; Wan et al., 2020; Carty et al., 2021).
As a key, frontline immune tissue, the liver is able to mount a rapid and robust immune response under appropriate conditions. It is enriched with innate immune cells, including Kupffer cells (KCs), natural killer (NK) cells, natural killer T (NKT) cells, and hepatic dendritic cells [DCs] (Gao, 2016; Ringelhan et al., 2018). Evidence suggests that hepatocytes are responsible for the biosynthesis of 80–90% of innate immune proteins, including complement components and secreted PRRs, and the liver can express membrane-bound PRRs, such as TLRs (Gao et al., 2008, 2011). However, viruses have also evolved a variety of strategies to evade the innate immune response in order to optimize their replication capacity. Research regarding hepatitis viruses, in particular HBV and HCV, has identified several molecular mechanisms hepatitis viruses employ to evade the host innate immune system (Foy et al., 2003; Johnson et al., 2007; Pugnale et al., 2009; Dong et al., 2012; Hong et al., 2015; Liu et al., 2015; Yi et al., 2015; Feng and Lemon, 2019; Wang et al., 2019). Here, we provide an overview of how hepatitis viruses trigger and evade the host antiviral innate immune response.
Hepatitis A Virus
Hepatitis A virus is a small, hepatotropic, positive-strand RNA virus transmitted by the fecal–oral route. In contrast to HBV and HCV, which cause chronic hepatitis, HAV generally causes only acute disease, and patients may develop multiple symptoms of acute hepatitis, such as acute liver failure [ALF] (Shin and Jeong, 2018). To date, the innate immune response to acute HAV infection has been reported, but not carefully examined. Previous studies have shown that TLR3, RIG-I, and melanoma differentiation-associated gene-5 (MDA5) are the main PRRs involved in HAV recognition, which leads to the induction of an antiviral state (Fensterl et al., 2005; Yang et al., 2007; Qu et al., 2011). Compared with acute resolving HCV infection, there is a very limited induction of type I IFN-stimulated genes in HAV-infected animals (Lanford et al., 2011). This similar result is also found in the study by Brack et al. (2002). They demonstrated that HAV can inhibit double-stranded RNA (dsRNA)-induced IFN-β gene expression (Brack et al., 2002). These results indicated that HAV is stealthier than HCV in acute resolving infection. Consistent with the conclusions of that study, some studies have shown that HAV can actively employ strategies to evade the innate immune response. These strategies, to some degree, contribute to the stealth of HAV. MAVS is a central adaptor protein that plays a critical role in RIG-I- and MDA5-mediated signaling. The activation of MAVS leads to the induction of type I IFNs through the activation of transcription NF-κB and IRF3. Yang et al. (2007) showed that downregulation of MAVS by HAV 3ABC disrupts the RIG-I and MDA5 pathways, thereby inhibiting the type I IFN response (Figure 1). They also found that HAV disrupts TLR3 signaling by targeting the essential adaptor protein, TRIF. Moreover, the degradation of TRIF was shown to be caused by HAV 3CD (Qu et al., 2011). HAV can disrupt RIG-I, MDA5, and TLR3 signaling pathways through cleavage of essential adaptor proteins by two distinct protease precursors derived from the common 3ABCD polyprotein processing intermediate (Figure 1). TNF receptor-associated factor 6 (TRAF6) is also a key adaptor protein in RIG-I- and MDA5-mediated type I IFN signaling pathways. A recent study by Mo et al. (2021) demonstrated that, aside from MAVS and TRIF, HAV also disrupts IFN-β signaling, in part, through the cleavage of TRAF6 mRNA via microRNAs (miRNAs). They found HAV can increase the expression of hsa-miR-146a-5p, thus leading to the cleavage of TRAF6 (Figure 1; Mo et al., 2021). Furthermore, HAV 3Cpro cleaves nuclear factor-κB essential modulator (NEMO), the regulatory subunit for IκB kinase (IKK) that controls the NF-κB signaling pathway, at the Q304 residue, which negates the signaling adaptor function of NEMO, thereby abrogating RIG-I, MDA5, and TLR3 pathways (Figure 1; Wang et al., 2014). In addition to HAV 3ABC, another HAV protein, 2B, also has an inhibitory effect on MAVS-mediated IRF3 activation. HAV 2B interferes with the activity of MAVS and affects the function of IRF3 kinases, TANK-binding kinase 1 (TBK1), and the inhibitor of NF-κB kinase ε [IKKε] (Paulmann et al., 2008). Furthermore, experiments in a mouse model of HAV infection have also indicated that HAV can also evade the MAVS-mediated type I IFN response (Hirai-Yuki et al., 2016). According to the above results and existing literature, cleavage of important adaptor proteins is the primary way for HAV to antagonize the innate immune response. In addition, studies have also shown that plasmacytoid dendritic cells (pDCs) can rapidly produce a large amount of type I IFNs in response to foreign nucleic acids, and this function of pDCs is connected with TLR7 and TLR9 (Gilliet et al., 2008; Reizis et al., 2011). As for HAV, pDCs produce large amounts of IFN-α via TLR7 signaling when co-cultured with HAV-infected cells. Interestingly, while host membrane envelopment has been shown to protect HAV from host neutralizing antibodies, it also contributes to the early detection of HAV by pDCs (Feng et al., 2015).
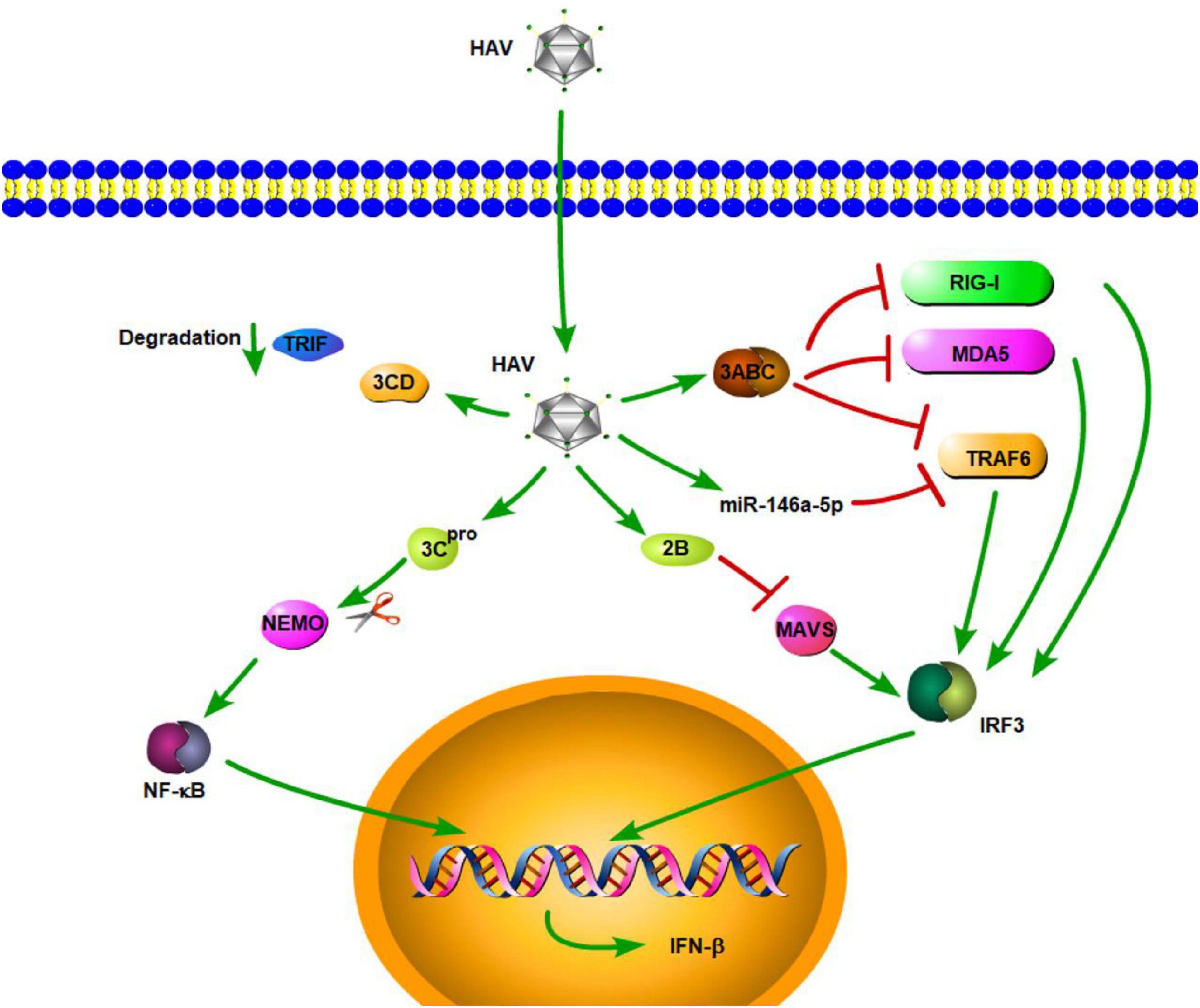
Figure 1. Hepatitis A virus suppression of type I interferon (IFN) response (green arrows indicate the related signaling pathways and T-arrows in red indicate blocking the targets). HAV 3ABC downregulates MAVS to disrupt the RIG-I and MDA5 pathways, thereby inhibiting the type I IFN response. HAV increased the expression of hsa-miR-146a-5p, thus leading to the cleavage of TRAF6.HAV 3CD can induce TRIF degradation. HAV 3Cpro cleaves nuclear factor-κB essential modulator (NEMO), the regulatory subunit for IκB kinase (IKK) that controls the NF-κB signaling pathway. HAV 2B interferes with the activity of MAVS and affects the function of IRF3 kinases.
Hepatitis B Virus
Hepatitis B virus is a partially double-stranded DNA virus belonging to the Hepadnaviridae family. HBV infection remains the most common chronic viral infection in the world. If left untreated, chronic HBV infection progress to end-stage liver disease, such as liver cirrhosis and hepatocellular carcinoma [HCC] (Li H. et al., 2020). During infection, HBV has the ability to stimulate the innate immune response. According to published studies, HBV triggers the innate immune system, but it also has the capability of using a variety of powerful strategies to evade the innate immune response and induce immunosuppression.
Toll-Like Receptors
Toll-like receptors play significant roles against HBV infection. TLRs, including TLR3, TLR4, TLR5, TLR7, and TLR9, can inhibit HBV replication in an IFN-α/β-dependent manner in HBV transgenic mice within 24 h (Isogawa et al., 2005). HBV RNA is produced in infected cells, and dsRNA is the main target for TLR3. Chen et al. (2015) reported that the expression of TLR3 in human HBV-associated HCC tissues is related to HBV infection. The upregulation of the synthesis of dsRNA in HCC with HBV infection can activate TLR3, which in turn, promotes interstitial immunoreactive cells and induces production of inflammatory cytokines (Chen et al., 2015). As for TLR4, a previous study showed that TLR4 activation represses HBV infection through the NF-κB pathway (Das et al., 2016; Jing et al., 2018). Another study also revealed that peripheral blood mononuclear cells (PBMCs) isolated from chronic HBV (CHB)-infected patients have decreased expression of TRIF, which may attenuate TLR3 and TLR4 signaling subsequent to HBV infection (Ayoobi et al., 2013) and might be one of the reasons why HBV infection is stable in CHB patients. Numerous studies have reported various TLR7 agonists, which have been used as adjuvants to induce immune responses, to have diverse effects against HBV (Sepehri et al., 2016), including R-848 (a ligand for TLR7), which promotes an immune response against HBsAg (Ma et al., 2007). Xu et al. reported that PBMCs from CHB patients showed reduced production of IFN-α in response to Loxoribine [a ligand for TLR7] (Xu et al., 2012). These results suggested that TLR7 plays a key role in the induction of an immune response against HBV. Another study also revealed that TLR9 can recognize HBV DNA, resulting in activation of immune cells (Sajadi et al., 2013; Shahrakyvahed et al., 2014). Furthermore, a recent study by Zhang et al. (2020) found that TLR2 is involved in the recognition of HBV during the early infection process and initiates innate immune signaling in primary human hepatocytes (PHHs), demonstrating that TLR2 is an important TLR in the fight against HBV. Additionally, several TLRs, including TLR3 (An et al., 2007; Li et al., 2009), TLR4 (Chen et al., 2008), TLR7 (Xu et al., 2012; Lin et al., 2013), and TLR9 (Vincent et al., 2011; Sajadi et al., 2013; Shahrakyvahed et al., 2014), are downregulated in CHB patients. Thus, downregulating TLRs might be a strategy for HBV to evade host TLR-activated innate immune responses.
Moreover, studies have shown that HBV surface antigen (HBsAg), HBV e antigen (HBeAg), and even HBV virions can suppress TLR-mediated innate immunity and IFN-β induction in liver cells (Figure 2; Wu et al., 2009). A previous study demonstrated that HBsAg inhibits TLR9-mediated IRF7 expression and nuclear translocation, which are important for induction of IFN-α gene transcription (Figure 2; Xu et al., 2009). Furthermore, HBeAg can associate with NEMO and inhibit TRAF6-mediated K63-linked ubiquitination of NEMO (Figure 2). The decreased ubiquitination of NEMO contributes to the downregulation of NF-κB activity, thus promoting the replication of HBV (Wang et al., 2019). Besides regulating the modification of NEMO, HBeAg protein also can modulate the NF-κB pathway in hepatoma cells via suppressing IL-18 and IL-1β-mediated NF-κB signaling (Wilson et al., 2011; Jegaskanda et al., 2014). Lang et al. provided a molecular mechanism describing HBeAg immunomodulation of the innate immune signal transduction pathways. They found that HBeAg, but not the HBV core antigen, co-localizes with Toll/IL-1receptor (TIR)-containing proteins TRAM and Mal at the plasma membrane. HBeAg can then suppress TIR-mediated activation of the inflammatory transcription factor NF-κB and IFN-β promoter activity and disrupt homotypic TIR:TIR interaction, which is critical for TLR-mediated signaling (Lang et al., 2011). Additionally, a previous study demonstrated that after culturing PBMCs with various concentrations of HBeAg for 72 h, HBeAg can inhibit proliferation of lymphocytes, increase TLR3, TLR4, and PD-1 expression, and decrease IFN-γ production (Chen et al., 2017). The inhibition of IFN-γ expression is also found in HBeAg-positive rather than HBeAg-negative CHB patients (Jegaskanda et al., 2014). MyD88 is induced by IFN-α and acts as a critical component in the signaling cascade through TLRs. Xiong et al. (2004) found that the levels of HBV proteins and viral replicative intermediates are effectively reduced in MyD88-expressing cells, indicating that MyD88 has antiviral activity against HBV replication. Based on this, Wu et al. (2007) reported that HBV polymerase can inhibit expression of the IFN-α-inducible MyD88 by inhibiting the activity of the MyD88 promoter, possibly by blocking the nuclear translocation of activator of transcription 1 [STAT1] (Figure 2). In addition, transcriptional activity of the IFN-stimulated response element (ISRE) promoter and expression of interferon-stimulated genes (ISGs) are also downregulated by HBV polymerase (Figure 2; Wu et al., 2007). In a certain respect, these results indicate that HBV viral proteins play important roles against TLR-mediated innate immune responses.
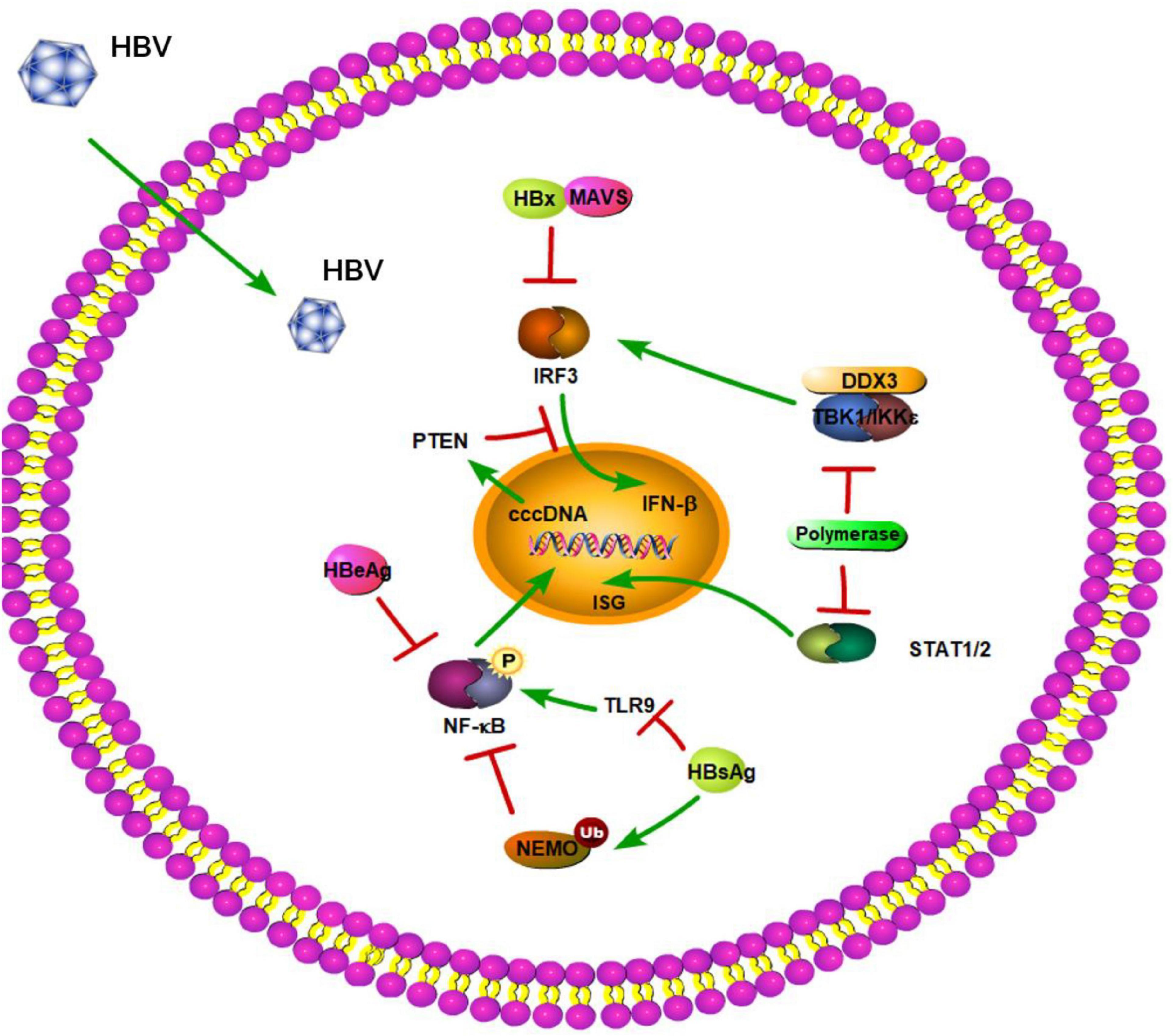
Figure 2. Hepatitis B virus suppression of type I interferon (IFN) response (green arrows indicate the related signaling pathways and T-arrows in red indicate blocking the targets). HBsAg inhibits TLR9 and mediates the ubiquitination of NEMO to suppress the NF-kB pathway. HBeAg can inhibit phosphorylation of NF-kB. HBV polymerase can block the nuclear translocation of activator of transcription 1 (STAT1). HBV polymerase is able to disrupt the interaction between TBK1/IKKε and DEAD box protein 3 (DDX3) to play a novel role in HBV counteraction of IFN-β production. HBx interacts with MAVS to act as a suppressor of virus-triggered induction of type I IFNs. HBV infection can also reduce PTEN protein expression to inhibit IFN signaling pathway.
RIG-I-Like Receptors
RIG-I-like receptors are key PRRs that detect virus-derived RNAs. A recent clinical study not only showed that RLRs have an effect on HBV clearance in the Chinese Han population, but also identified three single-nucleotide polymorphisms (SNPs; DDX58 rs3824456, DHX58 s2074160, and IFIH1 rs2111485) in RLRs that may be associated with spontaneous clearance of HBV (Yao et al., 2021). Lee et al. (2021) also revealed that activation of the RIG-I pathway offers therapeutic benefits toward elimination of HBV covalently closed circular DNA [cccDNA]. Different HBV genotypes can be recognized by diverse RLRs. In a previous study, Sato et al. (2015) reported that RIG-I can sense HBV genotype A, B, and C and recognize the 5′-ε region of HBV pgRNA. Furthermore, RIG-I dampens HBV replication through counteracting the interaction between HBV polymerase and pgRNA as well (Sato et al., 2015). In turn, HBV polymerase was shown to be able to disrupt the interaction between TBK1/IKKε and DEAD box protein 3 (DDX3) to play a novel role in HBV counteraction of IFN-β production (Yu et al., 2010). Recently, a further study by Zhou et al. also revealed that HBV impedes RIG-I-induced IFN production through sequestering MAVS from RIG-I by forming a ternary complex including hexokinase (HK). This study provided a novel mechanism of HBV immune escape in which HBV promotes glucose metabolism and suppresses RIG-I-MAVS signaling (Zhou et al., 2021). Another RLR, MDA5, can sense the genotype D of HBV and associate with HBV DNA (Lu and Liao, 2013). However, it is unknown whether the association of MDA5 with DNA is required for MDA5-mediated signaling. Interestingly, across-sectional study showed that mRNA levels of MDA5 and RIG-1 are significantly decreased and increased, respectively, in CHB patients when compared to healthy controls (Ebrahim et al., 2015). It is easy to understand that decreased expression of MDA5 results in impaired HBV recognition and consequently disrupts the immune response in CHB patients. However, it is confusing that RIG-I plays an opposing role in CHB patients. Subsequently, Zeng et al. (2019) revealed that the levels of RIG-I are significantly decreased in CHB patients in the active phase when compared to healthy controls, which is contradictory to the results of Ebrahim et al. (2015). This suggested that the expression level of RIG-I might be related to the progression of CHB infection. MAVS is a key adaptor protein in RIG-I- and MDA5-mediated signaling. Downregulation of MAVS-induced signaling by HBV-encoded X protein (HBx) has already been proposed (Figure 2). A study by Kumar et al. (2011) identified MAVS as one of the novel cytoplasmic binding partners of HBx, which specifically inhibits the double-stranded DNA (dsDNA) pathway of IFN-β activation in a dose-dependent manner. Wang et al. (2010) also found HBx interacts with MAVS and acts as a suppressor of virus-triggered induction of type I IFNs by disrupting the association of MAVS with its upstream and downstream components (Figure 2). Moreover, HBx promotes the degradation of MAVS, thereby attenuating the antiviral response of the innate immune system (Figure 2). Further analysis of clinical samples also revealed that MAVS protein is downregulated in HBV-induced HCC (Wei et al., 2010). These results provide possible explanations into how HBV is able to avoid activating MAVS-induced innate immune responses during virus infection. Apart from affecting key innate immune adaptor proteins, the roles of m6A in RNAs are emerging in viral immune regulation. Kim et al. (2020) demonstrated that m6A modification of HBV pgRNA and HCV genomic RNA promotes their interactions with YTHDF proteins, and thereby, disrupts RIG-I sensing activity. Recently, they also revealed that HBV can affect tumor suppression, including phosphatase and tensin homolog (PTEN), which activates IFN signaling pathway by dephosphorylation of IRF3 at Ser97 (Figure 2). HBV infection can also reduce PTEN protein expression by increasing the m6A methylation of PTEN mRNA (Kim et al., 2021). The elimination of PTEN results in the inhibition of p-IRF3 nuclear import. Kim et al. (2020, 2021) provided new insights into the mechanism of immune evasion via m6A modification of RNAs.
NOD-Like Receptors and STING-Dependent DNA Sensor
Currently, the NLR family pyrin domain-containing 3 (NLRP3) inflammasome is the most studied inflammasome. Yu et al. (2017) found that HBeAg, but not HBsAg, suppresses LPS-induced NLRP3 inflammasome activation and IL-1β production via inhibiting NF-κB phosphorylation and ROS production. This inhibitory effect of HBeAg has been confirmed in CHB patients and might contribute to the HBV persistence and immune tolerance (Yu et al., 2017).
STING-dependent DNA sensors include cGAS, DAIs, and IFI16. These DNA sensors play important roles in the defense against invasion of many DNA viruses, including HBV. A previous study showed that HBV genome-derived dsDNA induces the innate immune response through the cGAS-STING pathway and suppresses HBV assembly and replication (Dansako et al., 2016; He J. et al., 2016). Moreover, Liu et al. found MITA-related protein (MRP), a spliced isoform of MITA/STING, significantly inhibits HBV replication through the NF-κB pathway in vitro (Liu et al., 2017). Chen et al. also demonstrated that DAIs can inhibit HBV replication via activation of NF-κB but not IRF3 signaling in Huh7 cells (Chen et al., 2012). Recently, a study by Yang et al. (2020) showed that IFI16 serves as a unique nuclear sensor for HBV cccDNA and suppresses cccDNA function by integrating innate immune activation and epigenetic regulation by targeting the ISRE of cccDNA. However, higher HBsAg levels are associated with a stronger downregulation of innate immunity pathway regulators/effectors, including IFI16 (Lebossé et al., 2017). The DNA sensors mentioned above and their downstream adaptor protein, STING, have critical roles in the recognition and defense against HBV infection. However, CHB patients are unable to express appropriate levels of STING in PBMCs, which might result in impairing HBV DNA recognition, and subsequently, disruption of innate immune responses (Karimi-Googheri et al., 2015). This result is consistent with Kasap et al. (2020). Another study demonstrated that human and murine hepatocytes have limited expression of STING (Thomsen et al., 2016). The lack of a functional innate DNA-sensing pathway creates a suitable environment for HBV replication and contributes to the weak capacity to clear HBV infection. Likewise, a recent study by Lauterbach-Rivière et al. (2020) revealed that PHHs express reduced levels of DNA sensors as compared to myeloid cells, and the HBV DNA in the capsids is less likely to trigger the low functionality of the cGAS-STING pathway. These conditions likely allow HBV to escape the STING-dependent innate immune response.
Hepatitis C Virus
Hepatitis C virus is a positive-strand RNA virus of the Hepacivirus genus, which belongs to the Flaviviridae family. HCV infection ultimately results in liver cirrhosis, hepatic failure, or HCC, all of which can cause death (Chevaliez and Pawlotsky, 2021). Numerous studies have shown that HCV is mainly sensed by RLRs, TLRs, and NLRs and has evolved routes to inactivate innate immune signaling.
RIG-I-Like Receptors
RIG-I-like receptors are pathogen receptors that regulate cellular permissiveness to HCV replication and play key roles against HCV infection. A previous study demonstrated that RIG-I can recognize the polyuridine motif of the HCV genome 3′ non-translated region (NTR) as a PAMP. The 3′ NTR of HCV stimulates RIG-I-dependent signaling to induce a hepatic innate immune response in vivo and triggers IFN and ISG expression to suppress HCV infection in vitro (Saito et al., 2008). Wu et al. also found two SNPs in RIG-I (DDX58 rs10813831 and rs10738889) that are associated with spontaneous clearance of HCV in HCV-persistent infectors of the Chinese Han population. The two SNPs of RIG-I might be identified as a predictive marker in local HCV patients (Wu et al., 2019). Similar to HAV 3ABC, HCV NS3/4A can cleave MAVS at C508, releasing MAVS from the mitochondrial membrane and resulting in the subcellular redistribution of MAVS and loss of interaction with RIG-I (Figure 3; Loo et al., 2006). Moreover, HCV NS3/4A can also inactivate MAVS, which results in the downregulation of the RIG-I-dependent antiviral response, thereby allowing HCV to avoid triggering both IRF3 and NF-κB activation during infection (Foy et al., 2005; Meylan et al., 2005). A previous study revealed HCV NS3/4A negatively regulates other steps of the antiviral response as well, such as targeting the E3 ubiquitin ligase Riplet, which is known to activate RIG-I by triggering K63-linked polyubiquitination (Oshiumi et al., 2013). A further study by Vazquez et al. identified a Tyr-16-Phe (Y16F) change in the NS4A transmembrane domain that can prevent NS3-NS4A targeting of Riplet, but not MAVS. This result demonstrated that the NS4A Y16 residue regulates a non-canonical Riplet-TBK1-IRF3-dependent, but RIG-I-MAVS-independent signaling pathway, which limits HCV infection (Figure 3; Vazquez et al., 2019). In addition, Otsuka et al. (2005) found that HCV NS3 protein suppresses IRF-3 activation through disrupting the association between TBK1 and IRF3 (Figure 3). Like NS3/4A, the NS4B protein also blocks IFN signaling mediated by RIG-I. NS4B suppresses RIG-I-mediated IFN-β production signaling through a direct protein interaction with STING, and the disruption of this interaction might restore the cellular antiviral response (Figure 3; Nitta et al., 2013). Another mechanistic study indicated that NS4B can disrupt the interaction between STING and TBK1 to allow HCV to evade the host innate immune system (Ding et al., 2013). Another HCV protein, NS5A, in addition to its role in viral replication and assembly, is also able to bind with LRPPRC and exploit the ability of LRPPRC to inhibit MAVS-regulated antiviral signaling (Refolo et al., 2019). MDA5 is another RLR and plays an important role in sensing HCV infection to trigger IFN response (Israelow et al., 2014). Cao et al. (2015) demonstrated that MDA5 can recognize HCV to initiate the host innate immune response during HCV infection. They also considered that HCV infection is capable of inducing IFN production, which is mainly dependent upon MDA5 rather than RIG-I (Cao et al., 2015). At the same time, however, a study by Hiet et al. (2015) reported that HCV can activate both RIG-I- and MDA5-mediated signaling, and it is probably in a sequential manner, with MDA5-mediated signaling likely taking place during the later stages of the viral replication cycle as compared with RIG-I-mediated signaling. This suggested that RIG-I- and MDA5-mediated innate immune response may be connected with the progression of HCV infection. Such a complementary role of RIG-I and MDA5 has also been previously reported for another Flaviviridae family member, West Nile Virus (Fredericksen et al., 2008; Errett et al., 2013).
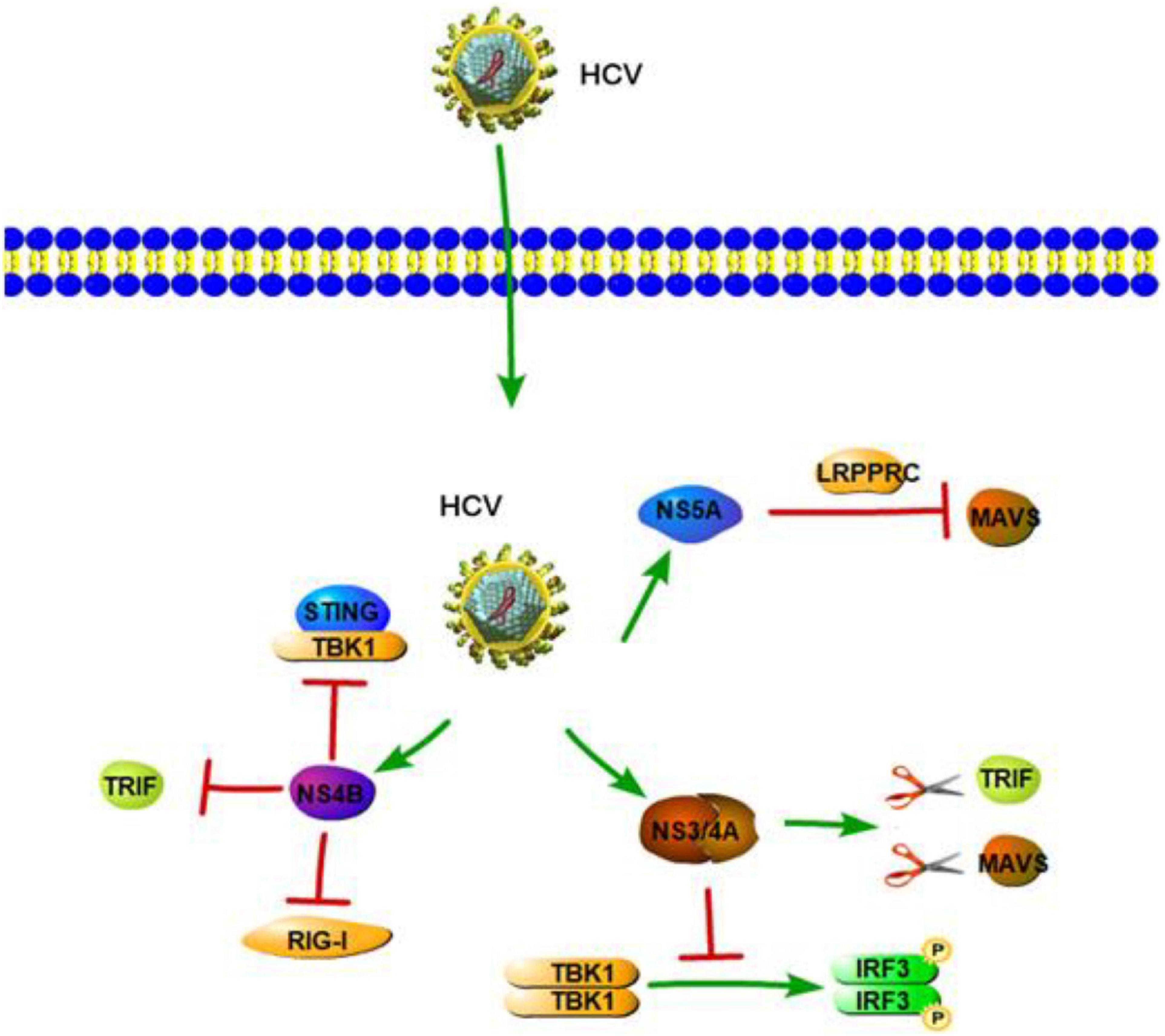
Figure 3. Hepatitis C virus suppression of type I interferon (IFN) response (green arrows indicate the related signaling pathways and T-arrows in red indicate blocking the targets). HCV NS3/4A serine protease cleaves TRIF and MAVS to inhibit IFN-b pathway. HCV NS3 protein suppresses IRF-3 activation through disrupting the association between TBK1 and IRF3. NS4B suppresses RIG-I-mediated IFN-β production signaling through a direct protein interaction with STING or disrupting the interaction between STING and TBK1.NS4B also could inhibit TLR3-mediated IFN signaling by downregulating TRIF protein levels via caspase 8. NS5A is able to bind with LRPPRC and exploit the ability of LRPPRC to inhibit MAVS-regulated antiviral signaling.
Toll-Like Receptors and NOD-Like Receptors
As for TLRs and NLRs, a previous study revealed IL-1β production and hepatic inflammation in HCV infection are linked and driven via virus-induced TLR7 and NLRP3 inflammasome signaling in liver macrophages (Negash et al., 2013). Zhang et al. (2009) provided direct evidence that HCV RNA components can enhance immune activation via TLR7 in multiple types of cells, including PBMCs, pDCs, THP-1, and Huh7. Similar with HAV, Takahashi et al. (2010) showed that pDCs produce type I IFN by direct sensing of HCV-infected cells. Importantly, active HCV replication and TLR7 signaling are also required in this process (Takahashi et al., 2010). Although viral RNA is not a typical substrate of TLR9, it has been proposed that TLR9, a DNA-sensing TLR, takes part in HCV antiviral effects, such as activation of pDCs via TLR9 through the HCV RNA polyuridine tail (Howell et al., 2013). Zhou et al. (2009) demonstrated that HCV infection leads to a decreased expression of TLR9 mRNA and protein, which are negatively correlated with serum viral copies. Further, a clinical study showed that the TLR9 promoter SNPs are associated with the natural course of HCV infection and have higher transcriptional activities in women, suggesting that the DNA sensor TLR9 has an effect in HCV infection (Fischer et al., 2017). Moreover, TLR3-mediated chemokine and inflammatory cytokine response also plays a significant role in the host immune response to HCV. TLR3 can sense HCV dsRNA and induce NF-κB activation with the production of numerous chemokines and inflammatory cytokines (Li et al., 2012). The studies that HCV NS3/4A serine protease cleaves TRIF and also MAVS (Li et al., 2005a,b). NS3/4A-mediated cleavage of TRIF can inhibit the immune response induced by TLR3 (Li et al., 2005a). Moreover, NS4B, another HCV-encoded protein, also inhibits TLR3-mediated IFN signaling by downregulating TRIF protein levels via caspase 8 (Figure 3; Liang et al., 2018). Another clinical study showed that, as compared to healthy controls, the expression of TRIF, TLR3, and MAVS is downregulated in chronic hepatitis C (CHC) patients (Firdaus et al., 2014; Kar et al., 2017). According to the studies mentioned above, the ability of HCV NS3/4A/4B proteins to inhibit the innate immune occurs mainly via two ways. One is to downregulate crucial innate immune signaling pathway molecules, and the other is to inhibit the association between upstream and downstream signaling molecules. These two strategies prevent downstream activation of the immune response and help HCV to evade the host innate immune system.
Aside from inhibiting the innate immune response, viral proteins can also induce and maintain the innate immune response. HCV, through the action of its NS5A protein, induces expression of TLR4 in B cells and hepatocytes to improve the TLR4-mediated inflammatory response (Machida et al., 2006). The HCV core and NS3 proteins also trigger TLR2-specific cellular activation and induce inflammatory pathways in a TLR2-dependent manner, suggesting that the HCV core and NS3 protein are likely to contribute to inflammatory activation during HCV infection (Dolganiuc et al., 2004). Further study is needed to show that immune recognition mediated by TLR2 involves TLR1 and TLR6 (Loo et al., 2006).
Hepatitis D Virus
Hepatitis D virus is the smallest known human virus with a circular RNA genome. As a satellite RNA virus, HDV does not encode its own envelope proteins for packaging of its ribonucleoprotein (RNP) and typically depends on the surface glycoproteins (GPs) of HBV for virion assembly, envelopment, and transmission (Hughes et al., 2011; Rizzetto, 2016). According to Giersch et al. (2015) significant induction of hISGs (hISG15, hSTATs, and hHLA-E) and human-specific cytokines (hIP10, hTGF-β, hIFN-β, and hIFN-γ) are detected in during HBV and HDV co-infection as compared with HBV mono-infection in humanized uPA/SCID/beige (USB) mice. Previous studies have also demonstrated that HDV can induce IFN-β and IFN-γ in hepatocytes and mouse models (Giersch et al., 2015; He et al., 2015; Alfaiate et al., 2016).
Although numerous molecules have been reported to participate in detecting intracellular viral RNA and triggering innate immune responses, to date it is not well-known which PRR(s) is/are involved in HDV infection and if this defense affects HDV replication. TLR3 uses TRIF as the main intermediate signaling molecule to induce IFN-β, while MDA5 and RIG-I use MAVS. In order to elucidate if one or both of the pathways is involved in the detection of HDV-RNA and IFN-β induction, Suárez-Amarán et al. (2017) established a new mouse model by using adeno-associated viruses (AAV; AAV-HDV and AAV-HBV) to deliver HDV- and HBV-replication competent genomes. This new HDV mouse model reproduces several important characteristics of human HDV infection and disease. The authors used TRIF- and MAVS-deficient mice to establish the HDV model. The results showed that in TRIF-deficient mice, IFN-β mRNA levels are equivalent to those observed in wild-type mice, while they are obviously reduced but not completely abolished in MAVS-deficient mice. A strong reduction in the expression level of all of the ISGs, except IP-10, was also observed in MAVS-deficient mice (Suárez-Amarán et al., 2017). This finding suggested that MAVS is the key adaptor protein and MDA5 or RIG-I, but not TLR3, might play a dominant part in the detection of cytoplasmic HDV dsRNA. Subsequently, a further study by Zhang et al. showed that HDV can induce IFN levels remained unaltered upon RIG-I or TLR3 knockdown, but are almost completely abolished upon MDA5 depletion. Although it has a minimal effect on HDV replication, MDA5 depletion greatly suppresses HDV-induced IFN response. These results revealed that MDA5 is the key sensor for the recognition of HDV-replicative intermediates rather than RIG-I and TLR3. According to Zhang et al. (2018) the precise mechanism of how HDV RNA is recognized by MDA5 still needs further research. In addition to this, a previous study by Pugnale et al. (2009) revealed that HDV impairs the IFN-α-stimulated JAK-STAT signaling pathway by inhibiting tyrosine phosphorylation of STAT1, STAT2, and receptor associated kinase Tyk2 (Figure 4). This result revealed a countermeasure for HDV to evade the IFN response by targeting the JAK-STAT transduction cascade. In general, the role of other PRRs and HDV viral factors in the innate immune system initiation and evasion still need further research. In order to achieve this, a more suitable HBV and HDV co-infection system should be established as soon as possible.
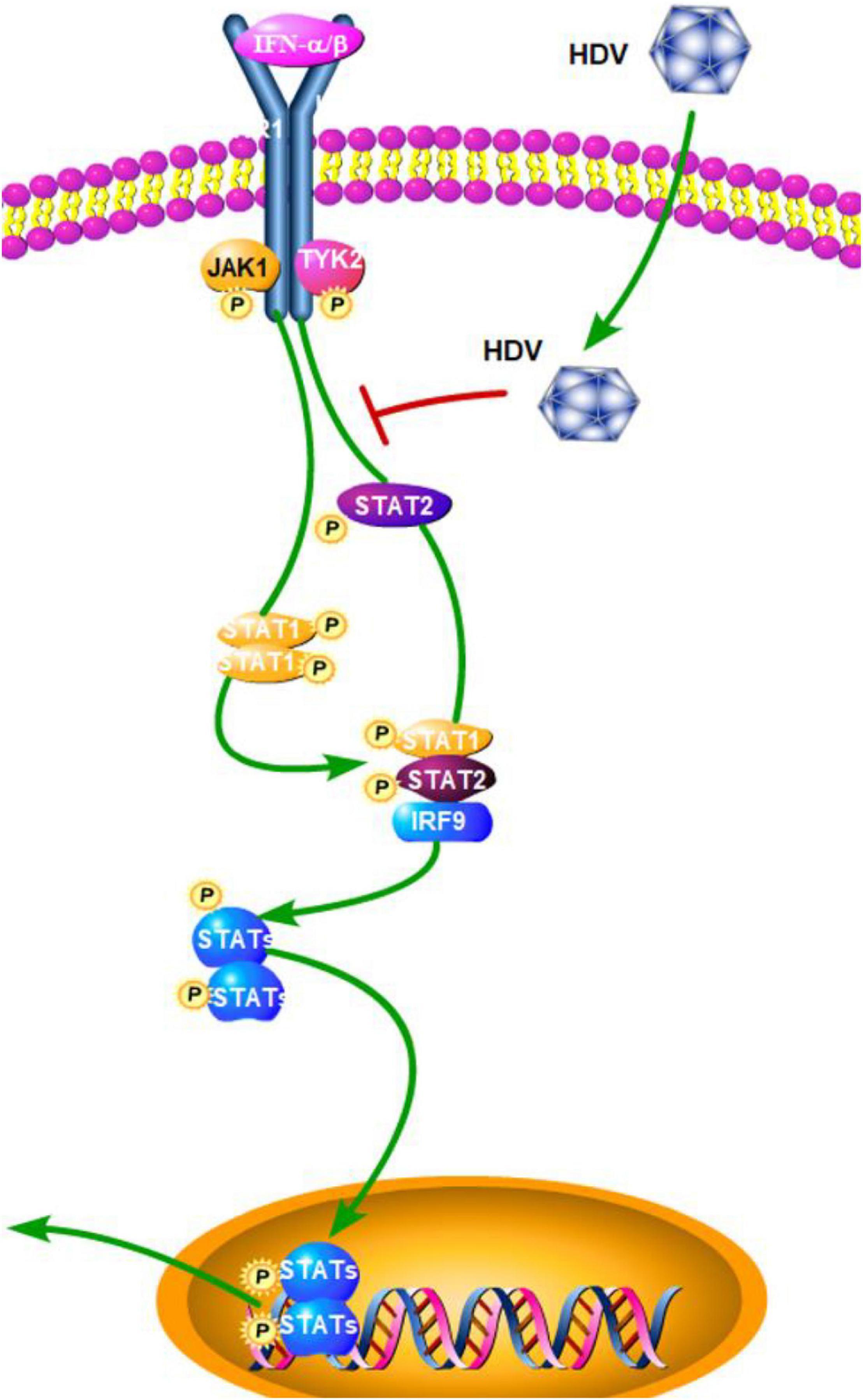
Figure 4. Hepatitis D virus suppression of type I interferon (IFN) response (green arrows indicate the related signaling pathways and T-arrows in red indicate blocking the targets). HDV impairs the IFN-α-stimulated JAK-STAT signaling pathway by inhibiting tyrosine phosphorylation of STAT1, STAT2, and receptor associated kinase Tyk2.
Hepatitis E Virus
The Recognition of Hepatitis E Virus
Aside from HAV, HEV is also a common cause of acute viral hepatitis in human. HEV is a non-enveloped virus with a positive-sense, single-stranded RNA genome that belongs to the family of Hepeviridae (Wang et al., 2016; Sander et al., 2018). During HEV infection, RLRs and TLRs mainly recognize HEV and trigger innate immune responses against HEV. A previous study revealed that a U-rich region in the 3′ UTR of the HEV genome mainly acts as a potent RIG-I PAMP, which induces an IFN response that is specific to the infected host cell type (Sooryanarain et al., 2020). Ectopic overexpression of RIG-I activates the transcription of many antiviral ISGs, partly through the activation of the JAK-STAT cascade without IFN production. In turn, the transcription of these antiviral ISGs helps establish an anti-HEV status (Xu et al., 2017). Unlike HAV and HCV, HEV does not target MAVS and can induce a sustained type III IFN response, which is dependent on both RIG-I and MDA5. However, the IFN level is insufficient to eliminate the virus (Yin et al., 2017). Li Y. et al. (2020) revealed that MDA5 can potently inhibit the replication of HEV without IFN production through the stimulation of a non-canonical IFN-like response, which is partially dependent on the JAK-STAT cascade.
Hepatitis E Virus Proteins
Similar HEV sequences have been isolated from many different animals, and the HEV mainly contains open-reading frames (ORFs) 1, 2, and 3. According to previous studies, these ORFs encode HEV proteins, which both activate and suppress the innate immune response during HEV infection. HEV ORF1 encodes non-structural polyproteins, and some of them emerge as potential IFN antagonists. HEV methyltransferase (MeT), a non-structural protein of HEV, significantly inhibits RIG-I/MDA5-mediated IFN-β signaling and NF-κB activation in a dose-dependent manner, leading to attenuation of IFN-β production (Figure 5; Kang et al., 2018; Min and Min, 2019). This suppression of IFN-β signaling is mainly caused by inhibiting the phosphorylation and activation of IRF3 and the p65 subunit of NF-κB, thus blocking efficient activation of the IFN-β promoter (Myoung et al., 2019). Aside from Met, papain-like cysteine protease domain (PCP) and X domain, other products of HEV ORF1, have also been demonstrated to act as IFN antagonists, which can inhibit RIG-I-mediated signaling in different steps (Figure 5). PCP mediates the deubiquitination of RIG-I and TBK-1, while the X domain inhibits IRF-3 phosphorylation (Figure 5; Nan et al., 2014b). These are potential molecular mechanisms of HEV ORF1-mediated antagonism of IFN pathways.
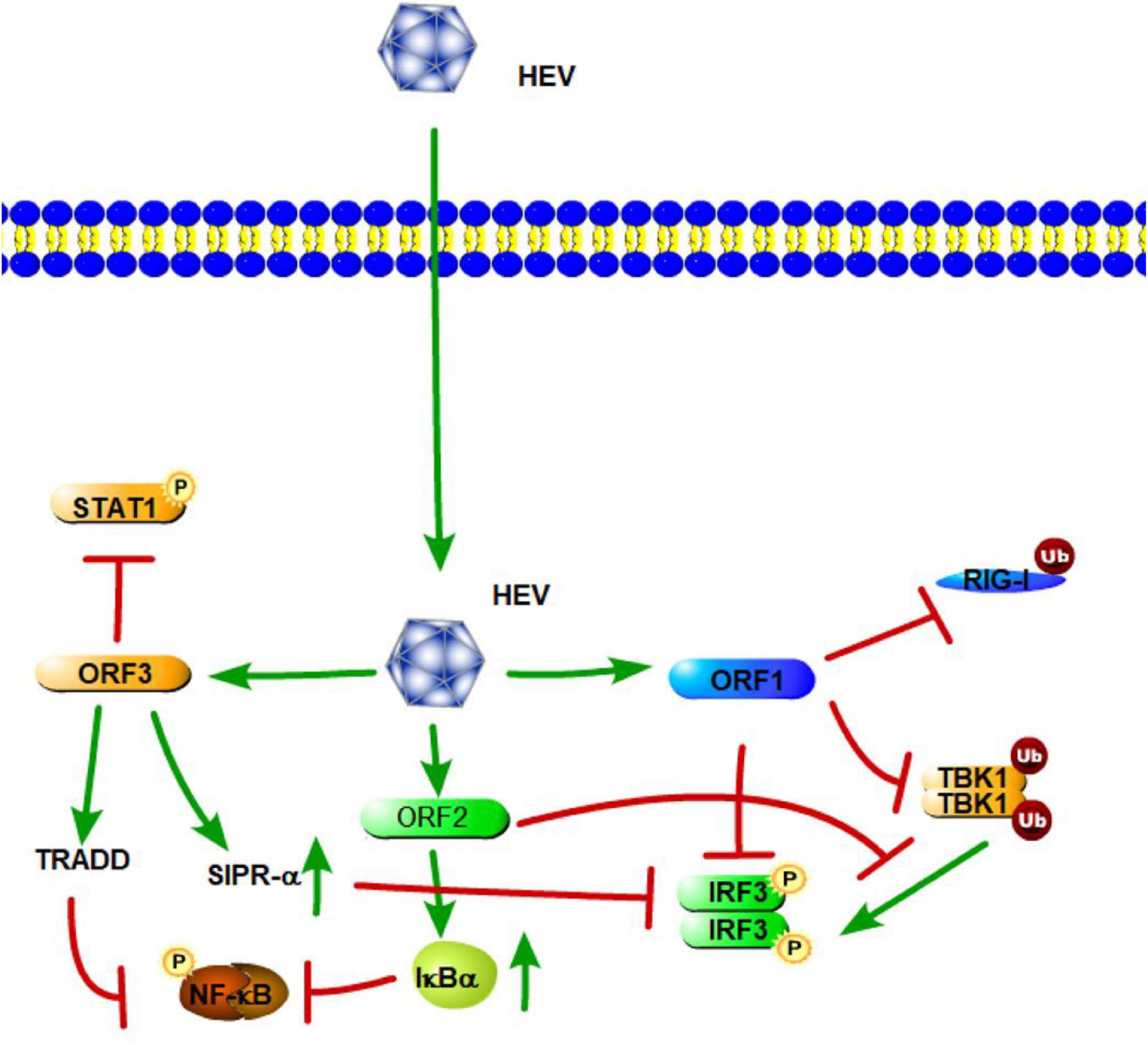
Figure 5. Hepatitis E virus suppression of type I interferon (IFN) response (green arrows indicate the related signaling pathways and T-arrows in red indicate blocking the targets). HEV ORF1 mediates the deubiquitination of RIG-I and TBK-1 and inhibits IRF-3 phosphorylation. The ORF2 protein affects RIG-I and TLR signaling pathways to block the phosphorylation of IRF3 by TBK1. The ORF2 protein also can stabilize inhibitor kappa B alpha (IκBα), thereby inhibiting NF-κB activity. HEV ORF3 contributes to the activation of SIRP-α to downregulate phosphorylation of IRF3. ORF3 of HEV suppresses TLR3-mediated NF-κB signaling via tumor necrosis factor receptor 1-associated death domain protein (TRADD). HEV ORF3 protein has the ability to inhibit IFN-α signaling through the regulation STAT1 phosphorylation.
ORF2 encodes the capsid protein of HEV. A previous study has shown that the capsid protein interacts with the MAVS-TBK1-IRF3 complex to block the phosphorylation of IRF3 (Figure 5). The arginine-rich motif of the N-terminal domain of the capsid protein has been found to be indispensable for inhibiting IRF3 activation (Nan et al., 2014a). Hingane et al. (2020) also indicated that the HEV ORF2 protein affects both the IFN-β and NF-κB axes of the RIG-I and TLR signaling pathways via targeting innate immune signaling downstream of adapter proteins and upstream of IRF3. Moreover, the ORF2 protein of HEV can stabilize inhibitor kappa B alpha (IκBα), thereby inhibiting NF-κB activity in human hepatoma cells (Figure 5; Surjit et al., 2012). Thus, HEV ORF2 protein most likely leads to a general suppression of the innate immune response.
HEV open-reading frame 3 (ORF3) encodes a 13-kDa multifunctional protein (vp13). A previous study showed that HEV vp13 can enhance poly (I:C)-induced IFN expression by increasing the RIG-I protein level. Additionally, vp13 interacts with the N-terminal domain of RIG-I and can enhance its K63-linked ubiquitination, which is essential for RIG-I activation (Figure 5; Nan et al., 2014a). Xu et al. (2014) also revealed that the ORF3 of HEV transiently activates NF-κB through an unfolded protein response (UPR) during the early stage of infection; but in the late phase, HEV ORF3 inhibits NF-κB signaling to create a favorable environment for HEV replication. During HEV infection, the D1 domain of HEV ORF3 contributes to the activation of SIRP-α, and thus, downregulates phosphorylation of IRF3, which results in the suppression of IFN-β (Huang et al., 2016). With regard to the TLRs involved in HEV, a study by Majumdar et al. (2015) revealed that TLR3 plays an important role in HEV disease pathogenesis. Patients expressing high levels of TLR3 are able to limit the disease and recover uneventfully (Majumdar et al., 2015). He M. et al. (2016) also demonstrated that the ORF3 of HEV downregulated TLR3-mediated NF-κB signaling via tumor necrosis factor receptor 1-associated death domain protein (TRADD) and receptor-interacting protein kinase 1 (RIP1) (Figure 5). Furthermore, Lei et al. (2018) found that HEV ORF3 protein significantly inhibits type I IFN induction by suppressing expression of TLR3/TLR7 and impairing multiple signaling pathways, including NF-κB, JAK/STAT, and JNK/MAPK. Aside from IFN-β signaling, a study by Dong et al. (2012) found that the HEV ORF3 protein has the ability to inhibit IFN-α signaling through the regulation STAT1 phosphorylation and reduced IFN-α-stimulated gene expression. Overall, these results showed how HEV triggers innate immunity through the direct or indirect inhibition of TLR/RLR-mediated activation of host innate immune responses.
Conclusion
Many studies have documented how hepatitis viruses trigger and evade the host innate immune response. From these studies, it could conclude that host recognition of the various hepatitis viruses via PRRs is the first step to initiate the host innate immune response. TLRs and RLRs are the main PRRs that recognize hepatitis viruses. In addition, HBV dsDNA also can be recognized by DNA sensors. During infection, hepatitis viruses have evolved the ability to compromise the innate immune system. There are some similarities in immune evasion techniques between the different types of hepatitis viruses. Key signaling molecules, including PRRs and central adaptor proteins, play important roles in innate immune signaling pathways. Decreased expression of these key signaling molecules is an efficient and common strategy that hepatitis viruses use to escape immune supervision. The expression of several TLRs, RLRs, and adaptor proteins, such as TLR3, RIG-I, MAVS, and TRIF, decreased in some types of hepatitis virus infection. Hepatitis virus proteins or factors also have the capacity of interfering with the innate immune response, which aids in the evasion of the innate immune response. Some hepatitis viral proteins are involved in the cleavage and degradation of key signaling molecules, such as HAV 3ABC and HCV NS3/4A, which can cleave MAVS to disrupt host innate immune signaling pathways. Other hepatitis viral proteins directly or indirectly interfere with the function of key signaling molecules. For instance, HEV ORF3 can suppress innate immune signaling directly through TRADD and RIP1. It also activates SIRP-α and downregulates phosphorylation of IRF3 in an indirect manner. Moreover, some limiting factors still prevent the further research of hepatitis viruses. One limiting factor is the lack of suitable and well-defined small animal models, as it is important to validate the relationships between hepatitis viruses and host immunity under physiological conditions. With suitable animal models and systems construction, the relationship between hepatitis viruses and host innate immunity will be better understood, which will provide insights into anti-hepatitis virus therapy.
Author Contributions
CX and JC wrote the manuscript. XC and JC revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the National Natural Sciences Foundation of China (Grant Number 32070176) and the Youth Innovation Promotion Association, Chinese Academy of Sciences.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript. We would also like to thank Jielin Tang and Qi Yang from the Wuhan Institute of Virology for useful help in literature collection.
References
Acorn, C. J., Millar, C. C., Hoffman, S. K., and Lyle, W. M. (1995). Viral hepatitis: a review. Optom. Vis. Sci. 72, 763–769. doi: 10.1097/00006324-199510000-00010
Alfaiate, D., Lucifora, J., Abeywickrama-Samarakoon, N., Michelet, M., Testoni, B., Cortay, J.-C., et al. (2016). HDV RNA replication is associated with HBV repression and interferon-stimulated genes induction in super-infected hepatocytes. Antiviral Res. 136, 19–31. doi: 10.1016/j.antiviral.2016.10.006
An, B., Xie, Q., Lin, L., Shen, H., Jia, N., Wang, H., et al. (2007). [Expression of Toll-like receptor 3 on peripheral blood dendritic cells in HBeAg positive patients with chronic hepatitis B]. Zhonghua Gan Zang Bing Za Zhi 15, 729–733.
Ayoobi, F., Hassanshahi, G., Zainodini, N., Khorramdelazad, H., Arababadi, M. K., and Kennedy, D. (2013). Reduced expression of TRIF in chronic HBV infected Iranian patients. Clin. Res. Hepatol. Gastroenterol. 37, 491–495. doi: 10.1016/j.clinre.2012.11.005
Brack, K., Berk, I., Magulski, T., Lederer, J., Dotzauer, A., and Vallbracht, A. (2002). Hepatitis A virus inhibits cellular antiviral defense mechanisms induced by double-stranded RNA. J. Virol. 76, 11920–11930. doi: 10.1128/jvi.76.23.11920-11930.2002
Cao, X., Ding, Q., Lu, J., Tao, W., Huang, B., Zhao, Y., et al. (2015). MDA5 plays a critical role in interferon response during hepatitis C virus infection. J. Hepatol. 62, 771–778. doi: 10.1016/j.jhep.2014.11.007
Carman, W. F., and Thomas, H. C. (1990). Hepatitis viruses. Baillieres Clin. Gastroenterol. 4, 201–232. doi: 10.1016/0950-3528(90)90047-k
Carty, M., Guy, C., and Bowie, A. G. (2021). Detection of viral infections by innate immunity. Biochem. Pharmacol. 183:114316. doi: 10.1016/j.bcp.2020.114316
Chen, L.-M., Fan, X.-G., Ma, J., He, B., and Jiang, Y.-F. (2017). Molecular mechanisms of HBeAg in persistent HBV infection. Hepatol. Int. 11, 79–86. doi: 10.1007/s12072-016-9734-5
Chen, Q.-Y., Liu, Y.-H., Li, J.-H., Wang, Z.-K., Liu, J.-X., and Yuan, Z.-H. (2012). DNA-dependent activator of interferon-regulatory factors inhibits hepatitis B virus replication. World J. Gastroenterol. 18, 2850–2858. doi: 10.3748/wjg.v18.i22.2850
Chen, X.-L., Xu, Y.-Y., Chen, L., Wang, G.-L., and Shen, Y. (2015). TLR3 Plays Significant Roles against HBV-Associated HCC. Gastroenterol. Res. Pract. 2015:572171. doi: 10.1155/2015/572171
Chen, Z., Cheng, Y., Xu, Y., Liao, J., Zhang, X., Hu, Y., et al. (2008). Expression profiles and function of Toll-like receptors 2 and 4 in peripheral blood mononuclear cells of chronic hepatitis B patients. Clin. Immunol. 128, 400–408. doi: 10.1016/j.clim.2008.04.006
Chevaliez, S., and Pawlotsky, J.-M. (2021). “HCV genome and life cycle,” in Hepatitis C Viruses: Genomes and Molecular Biology, ed. S.-L. Tan (Norfolk: Horizon Bioscience).
Dansako, H., Ueda, Y., Okumura, N., Satoh, S., Sugiyama, M., Mizokami, M., et al. (2016). The cyclic GMP-AMP synthetase-STING signaling pathway is required for both the innate immune response against HBV and the suppression of HBV assembly. FEBS J. 283, 144–156. doi: 10.1111/febs.13563
Das, D., Sarkar, N., Sengupta, I., Pal, A., Saha, D., Bandopadhyay, M., et al. (2016). Anti-viral role of toll like receptor 4 in hepatitis B virus infection: an in vitro study. World J. Gastroenterol. 22, 10341–10352. doi: 10.3748/wjg.v22.i47.10341
Ding, Q., Cao, X., Lu, J., Huang, B., Liu, Y.-J., Kato, N., et al. (2013). Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J. Hepatol. 59, 52–58. doi: 10.1016/j.jhep.2013.03.019
Dolganiuc, A., Oak, S., Kodys, K., Golenbock, D. T., Finberg, R. W., Kurt-Jones, E., et al. (2004). Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 127, 1513–1524. doi: 10.1053/j.gastro.2004.08.067
Dong, C., Zafrullah, M., Mixson-Hayden, T., Dai, X., Liang, J., Meng, J., et al. (2012). Suppression of interferon-α signaling by hepatitis E virus. Hepatology 55, 1324–1332. doi: 10.1002/hep.25530
Ebrahim, M., Mirzaei, V., Bidaki, R., Shabani, Z., Daneshvar, H., Karimi-Googheri, M., et al. (2015). Are RIG-1 and MDA5 Expressions Associated with Chronic HBV Infection? Viral Immunol. 28, 504–508. doi: 10.1089/vim.2015.0056
Errett, J. S., Suthar, M. S., McMillan, A., Diamond, M. S., and Gale, M. (2013). The essential, nonredundant roles of RIG-I and MDA5 in detecting and controlling West Nile virus infection. J. Virol. 87, 11416–11425. doi: 10.1128/JVI.01488-13
Feng, Z., and Lemon, S. M. (2019). Innate immunity to enteric hepatitis viruses. Cold Spring Harb. Perspect. Med. 9:a033464. doi: 10.1101/cshperspect.a033464
Feng, Z., Li, Y., McKnight, K. L., Hensley, L., Lanford, R. E., Walker, C. M., et al. (2015). Human pDCs preferentially sense enveloped hepatitis A virions. J. Clin. Invest. 125, 169–176. doi: 10.1172/JCI77527
Fensterl, V., Grotheer, D., Berk, I., Schlemminger, S., Vallbracht, A., and Dotzauer, A. (2005). Hepatitis A virus suppresses RIG-I-mediated IRF-3 activation to block induction of beta interferon. J. Virol. 79, 10968–10977. doi: 10.1128/JVI.79.17.10968-10977.2005
Firdaus, R., Biswas, A., Saha, K., Mukherjee, A., Pal, F., Chaudhuri, S., et al. (2014). Modulation of TLR 3, 7 and 8 expressions in HCV genotype 3 infected individuals: potential correlations of pathogenesis and spontaneous clearance. Biomed. Res. Int. 2014:491064. doi: 10.1155/2014/491064
Fischer, J., Weber, A. N. R., Böhm, S., Dickhöfer, S., El Maadidi, S., Deichsel, D., et al. (2017). Sex-specific effects of TLR9 promoter variants on spontaneous clearance of HCV infection. Gut 66, 1829–1837. doi: 10.1136/gutjnl-2015-310239
Foy, E., Li, K., Sumpter, R., Loo, Y.-M., Johnson, C. L., Wang, C., et al. (2005). Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl. Acad. Sci. U.S.A. 102, 2986–2991. doi: 10.1073/pnas.0408707102
Foy, E., Li, K., Wang, C., Sumpter, R., Ikeda, M., Lemon, S. M., et al. (2003). Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science 300, 1145–1148. doi: 10.1126/science.1082604
Fredericksen, B. L., Keller, B. C., Fornek, J., Katze, M. G., and Gale, M. (2008). Establishment and maintenance of the innate antiviral response to West nile virus involves both RIG-I and MDA5 signaling through IPS-1. J. Virol. 82, 609–616. doi: 10.1128/JVI.01305-07
Gao, B., Jeong, W.-I., and Tian, Z. (2008). Liver: an organ with predominant innate immunity. Hepatology 47, 729–736. doi: 10.1002/hep.22034
Gao, B., Seki, E., Brenner, D. A., Friedman, S., Cohen, J. I., Nagy, L., et al. (2011). Innate immunity in alcoholic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G516–G525. doi: 10.1152/ajpgi.00537.2010
Giersch, K., Allweiss, L., Volz, T., Helbig, M., Bierwolf, J., Lohse, A. W., et al. (2015). Hepatitis Delta co-infection in humanized mice leads to pronounced induction of innate immune responses in comparison to HBV mono-infection. J. Hepatol. 63, 346–353. doi: 10.1016/j.jhep.2015.03.011
Gilliet, M., Cao, W., and Liu, Y.-J. (2008). Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 8, 594–606. doi: 10.1038/nri2358
He, J., Hao, R., Liu, D., Liu, X., Wu, S., Guo, S., et al. (2016). Inhibition of hepatitis B virus replication by activation of the cGAS-STING pathway. J. Gen. Virol. 97, 3368–3378. doi: 10.1099/jgv.0.000647
He, M., Wang, M., Huang, Y., Peng, W., Zheng, Z., Xia, N., et al. (2016). The ORF3 protein of genotype 1 hepatitis E virus suppresses TLR3-induced NF-κB signaling via TRADD and RIP1. Sci. Rep. 6:27597. doi: 10.1038/srep27597
He, W., Ren, B., Mao, F., Jing, Z., Li, Y., Liu, Y., et al. (2015). Hepatitis D virus infection of mice expressing human sodium taurocholate co-transporting polypeptide. PLoS Pathog. 11:e1004840. doi: 10.1371/journal.ppat.1004840
Hiet, M.-S., Bauhofer, O., Zayas, M., Roth, H., Tanaka, Y., Schirmacher, P., et al. (2015). Control of temporal activation of hepatitis C virus-induced interferon response by domain 2 of nonstructural protein 5A. J. Hepatol. 63, 829–837. doi: 10.1016/j.jhep.2015.04.015
Hingane, S., Joshi, N., Surjit, M., and Ranjith-Kumar, C. T. (2020). Hepatitis E virus ORF2 inhibits RIG-I mediated interferon response. Front. Microbiol. 11:656. doi: 10.3389/fmicb.2020.00656
Hirai-Yuki, A., Hensley, L., McGivern, D. R., González-López, O., Das, A., Feng, H., et al. (2016). MAVS-dependent host species range and pathogenicity of human hepatitis A virus. Science 353, 1541–1545. doi: 10.1126/science.aaf8325
Hong, Y., Zhou, L., Xie, H., and Zheng, S. (2015). Innate immune evasion by hepatitis B virus-mediated downregulation of TRIF. Biochem. Biophys. Res. Commun. 463, 719–725. doi: 10.1016/j.bbrc.2015.05.130
Howell, J., Angus, P., Gow, P., and Visvanathan, K. (2013). Toll-like receptors in hepatitis C infection: implications for pathogenesis and treatment. J. Gastroenterol. Hepatol. 28, 766–776. doi: 10.1111/jgh.12170
Huang, F., Yang, C., Yu, W., Bi, Y., Long, F., Wang, J., et al. (2016). Hepatitis E virus infection activates signal regulator protein α to down-regulate type I interferon. Immunol. Res. 64, 115–122. doi: 10.1007/s12026-015-8729-y
Hughes, S. A., Wedemeyer, H., and Harrison, P. M. (2011). Hepatitis delta virus. Lancet 378, 73–85. doi: 10.1016/S0140-6736(10)61931-9
Isogawa, M., Robek, M. D., Furuichi, Y., and Chisari, F. V. (2005). Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J. Virol. 79, 7269–7272. doi: 10.1128/JVI.79.11.7269-7272.2005
Israelow, B., Narbus, C. M., Sourisseau, M., and Evans, M. J. (2014). HepG2 cells mount an effective antiviral interferon-lambda based innate immune response to hepatitis C virus infection. Hepatology 60, 1170–1179. doi: 10.1002/hep.27227
Jegaskanda, S., Ahn, S. H., Skinner, N., Thompson, A. J., Ngyuen, T., Holmes, J., et al. (2014). Downregulation of interleukin-18-mediated cell signaling and interferon gamma expression by the hepatitis B virus e antigen. J. Virol. 88, 10412–10420. doi: 10.1128/JVI.00111-14
Jing, X., Tian, Z., Gao, P., Xiao, H., Qi, X., Yu, Y., et al. (2018). HBsAg/β2GPI activates the NF-κB pathway via the TLR4/MyD88/IκBα axis in hepatocellular carcinoma. Oncol. Rep. 40, 1035–1045. doi: 10.3892/or.2018.6507
Johnson, C. L., Owen, D. M., and Gale, M. (2007). Functional and therapeutic analysis of hepatitis C virus NS3.4A protease control of antiviral immune defense. J. Biol. Chem. 282, 10792–10803. doi: 10.1074/jbc.M610361200
Kang, S., Choi, C., Choi, I., Han, K.-N., Rho, S. W., Choi, J., et al. (2018). Hepatitis E virus methyltransferase inhibits type I interferon induction by targeting RIG-I. J. Microbiol. Biotechnol. 28, 1554–1562. doi: 10.4014/jmb.1808.08058
Kar, P., Kumar, D., Gumma, P. K., Chowdhury, S. J., and Karra, V. K. (2017). Down regulation of TRIF, TLR3, and MAVS in HCV infected liver correlates with the outcome of infection. J. Med. Virol. 89, 2165–2172. doi: 10.1002/jmv.24849
Karimi-Googheri, M., Daneshvar, H., Khaleghinia, M., Bidaki, R., and Kazemi Arababadi, M. (2015). Decreased expressions of STING but not IRF3 molecules in chronic HBV infected patients. Arch. Iran Med. 18, 351–354.
Kasap, B., Buruk, C. K., Kaklıkkaya, N., Özkaya, E., Aydın Kasap, Z., and Aydın, F. (2020). [Comparison of mRNA levels of stimulator of interferon genes (STING) in individuals with natural immunity to Hepatitis B virus (HBV), and in those with chronic hepatitis B infection and without HBV]. Mikrobiyol. Bul. 54, 66–78. doi: 10.5578/mb.68787
Kim, G.-W., Imam, H., Khan, M., Mir, S. A., Kim, S.-J., Yoon, S. K., et al. (2021). Induced increased N6 methyladenosine modification of PTEN RNA affects innate immunity and contributes to HCC. Hepatology 73, 533–547. doi: 10.1002/hep.31313
Kim, G.-W., Imam, H., Khan, M., and Siddiqui, A. (2020). N6-Methyladenosine modification of hepatitis B and C viral RNAs attenuates host innate immunity via RIG-I signaling. J. Biol. Chem. 295, 13123–13133. doi: 10.1074/jbc.RA120.014260
Koyama, S., Ishii, K. J., Coban, C., and Akira, S. (2008). Innate immune response to viral infection. Cytokine 43, 336–341. doi: 10.1016/j.cyto.2008.07.009
Kumar, M., Jung, S. Y., Hodgson, A. J., Madden, C. R., Qin, J., and Slagle, B. L. (2011). Hepatitis B virus regulatory HBx protein binds to adaptor protein IPS-1 and inhibits the activation of beta interferon. J. Virol. 85, 987–995. doi: 10.1128/JVI.01825-10
Lanford, R. E., Feng, Z., Chavez, D., Guerra, B., Brasky, K. M., Zhou, Y., et al. (2011). Acute hepatitis A virus infection is associated with a limited type I interferon response and persistence of intrahepatic viral RNA. Proc. Natl. Acad. Sci. U.S.A. 108, 11223–11228. doi: 10.1073/pnas.1101939108
Lang, T., Lo, C., Skinner, N., Locarnini, S., Visvanathan, K., and Mansell, A. (2011). The Hepatitis B e antigen (HBeAg) targets and suppresses activation of the Toll-like receptor signaling pathway. J. Hepatol. 55, 762–769. doi: 10.1016/j.jhep.2010.12.042
Lauterbach-Rivière, L., Bergez, M., Mönch, S., Qu, B., Riess, M., Vondran, F. W. R., et al. (2020). Hepatitis B Virus DNA is a Substrate for the cGAS/STING Pathway but is not Sensed in Infected Hepatocytes. Viruses 12:592. doi: 10.3390/v12060592
Lebossé, F., Testoni, B., Fresquet, J., Facchetti, F., Galmozzi, E., Fournier, M., et al. (2017). Intrahepatic innate immune response pathways are downregulated in untreated chronic hepatitis B. J. Hepatol. 66, 897–909. doi: 10.1016/j.jhep.2016.12.024
Lee, S., Goyal, A., Perelson, A. S., Ishida, Y., Saito, T., and Gale, M. (2021). Suppression of hepatitis B virus through therapeutic activation of RIG-I and IRF3 signaling in hepatocytes. iScience 24, 101969. doi: 10.1016/j.isci.2020.101969
Lei, Q., Li, L., Zhang, S., Li, T., Zhang, X., Ding, X., et al. (2018). ORF3 downregulates TLR7 to inhibit the generation of type I interferon via impairment of multiple signaling pathways. Sci. Rep. 8:8585. doi: 10.1038/s41598-018-26975-4
Lester, S. N., and Li, K. (2014). Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 426, 1246–1264. doi: 10.1016/j.jmb.2013.11.024
Li, H., Yan, L., Shi, Y., Lv, D., Shang, J., Bai, L., et al. (2020). Hepatitis B virus infection: overview. Adv. Exp. Med. Biol. 1179, 1–16. doi: 10.1007/978-981-13-9151-4_1
Li, K., Foy, E., Ferreon, J. C., Nakamura, M., Ferreon, A. C. M., Ikeda, M., et al. (2005a). Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. U.S.A. 102, 2992–2997. doi: 10.1073/pnas.0408824102
Li, X.-D., Sun, L., Seth, R. B., Pineda, G., and Chen, Z. J. (2005b). Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. U.S.A 102, 17717–17722. doi: 10.1073/pnas.0508531102
Li, K., Li, N. L., Wei, D., Pfeffer, S. R., Fan, M., and Pfeffer, L. M. (2012). Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on Toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology 55, 666–675. doi: 10.1002/hep.24763
Li, N., Li, Q., Qian, Z., Zhang, Y., Chen, M., and Shi, G. (2009). Impaired TLR3/IFN-beta signaling in monocyte-derived dendritic cells from patients with acute-on-chronic hepatitis B liver failure: relevance to the severity of liver damage. Biochem. Biophys. Res. Commun. 390, 630–635. doi: 10.1016/j.bbrc.2009.10.018
Li, Y., Yu, P., Qu, C., Li, P., Li, Y., Ma, Z., et al. (2020). MDA5 against enteric viruses through induction of interferon-like response partially via the JAK-STAT cascade. Antiviral Res. 176:104743. doi: 10.1016/j.antiviral.2020.104743
Liang, Y., Cao, X., Ding, Q., Zhao, Y., He, Z., and Zhong, J. (2018). Hepatitis C virus NS4B induces the degradation of TRIF to inhibit TLR3-mediated interferon signaling pathway. PLoS Pathog. 14:e1007075. doi: 10.1371/journal.ppat.1007075
Lin, K.-J., Lin, T.-M., Wang, C.-H., Liu, H.-C., Lin, Y.-L., and Eng, H.-L. (2013). Down-regulation of Toll-like receptor 7 expression in hepatitis-virus–related human hepatocellular carcinoma. Hum. Pathol. 44, 534–541. doi: 10.1016/j.humpath.2012.06.015
Liu, S., Zhao, K., Su, X., Lu, L., Zhao, H., Zhang, X., et al. (2017). MITA/STING and its alternative splicing isoform MRP restrict hepatitis B virus replication. PLoS One 12:e0169701. doi: 10.1371/journal.pone.0169701
Liu, Y., Li, J., Chen, J., Li, Y., Wang, W., Du, X., et al. (2015). Hepatitis B virus polymerase disrupts K63-linked ubiquitination of STING to block innate cytosolic DNA-sensing pathways. J. Virol. 89, 2287–2300. doi: 10.1128/JVI.02760-14
Loo, Y.-M., Owen, D. M., Li, K., Erickson, A. K., Johnson, C. L., Fish, P. M., et al. (2006). Viral and therapeutic control of IFN-β promoter stimulator 1 during hepatitis C virus infection. Proc. Natl. Acad. Sci. U.S.A. 103, 6001–6006. doi: 10.1073/pnas.0601523103
Lu, H.-L., and Liao, F. (2013). Melanoma differentiation-associated gene 5 senses hepatitis B virus and activates innate immune signaling to suppress virus replication. J. Immunol. 191, 3264–3276. doi: 10.4049/jimmunol.1300512
Ma, R., Du, J., Huang, J., and Wu, C. (2007). Additive effects of CpG ODN and R-848 as adjuvants on augmenting immune responses to HBsAg vaccination. Biochem. Biophys. Res. Commun. 361, 537–542. doi: 10.1016/j.bbrc.2007.07.028
Machida, K., Cheng, K. T. H., Sung, V. M.-H., Levine, A. M., Foung, S., and Lai, M. M. C. (2006). Hepatitis C Virus Induces Toll-Like Receptor 4 expression, leading to enhanced production of beta interferon and interleukin-6. J. Virol. 80, 866–874. doi: 10.1128/JVI.80.2.866-874.2006
Majumdar, M., Ratho, R. K., Chawla, Y., and Singh, M. P. (2015). Role of TLR gene expression and cytokine profiling in the immunopathogenesis of viral hepatitis E. J. Clin. Virol. 73, 8–13. doi: 10.1016/j.jcv.2015.09.011
Meylan, E., Curran, J., Hofmann, K., Moradpour, D., Binder, M., Bartenschlager, R., et al. (2005). Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437, 1167–1172. doi: 10.1038/nature04193
Min, J. M., and Min, K. (2019). Dose-Dependent Inhibition of Melanoma Differentiation-Associated Gene 5-Mediated Activation of Type I Interferon Responses by Methyltransferase of Hepatitis E Virus. J. Microbiol. Biotechnol. 29, 1137–1143. doi: 10.4014/jmb.1905.05040
Mo, L., Zeng, Z., Deng, R., Li, Z., Sun, J., Hu, N., et al. (2021). Hepatitis A virus-induced hsa-miR-146a-5p attenuates IFN-β signaling by targeting adaptor protein TRAF6. Arch. Virol. 166, 789–799. doi: 10.1007/s00705-021-04952-z
Myoung, J., Lee, J. Y., and Min, K. S. (2019). Methyltransferase of a cell culture-adapted hepatitis E inhibits the MDA5 receptor signaling pathway. J. Microbiol. 57, 1126–1131. doi: 10.1007/s12275-019-9478-8
Nan, Y., Yu, Y., Ma, Z., Khattar, S. K., Fredericksen, B., and Zhang, Y.-J. (2014b). Hepatitis E virus inhibits type I interferon induction by ORF1 products. J. Virol. 88, 11924–11932. doi: 10.1128/JVI.01935-14
Nan, Y., Ma, Z., Wang, R., Yu, Y., Kannan, H., Fredericksen, B., et al. (2014a). Enhancement of interferon induction by ORF3 product of hepatitis E virus. J. Virol. 88, 8696–8705. doi: 10.1128/JVI.01228-14
Negash, A. A., Ramos, H. J., Crochet, N., Lau, D. T. Y., Doehle, B., Papic, N., et al. (2013). IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 9:e1003330. doi: 10.1371/journal.ppat.1003330
Nitta, S., Sakamoto, N., Nakagawa, M., Kakinuma, S., Mishima, K., Kusano-Kitazume, A., et al. (2013). Hepatitis C virus NS4B protein targets STING and abrogates RIG-I-mediated type I interferon-dependent innate immunity. Hepatology 57, 46–58. doi: 10.1002/hep.26017
Oshiumi, H., Miyashita, M., Matsumoto, M., and Seya, T. (2013). A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog. 9:e1003533. doi: 10.1371/journal.ppat.1003533
Otsuka, M., Kato, N., Moriyama, M., Taniguchi, H., Wang, Y., Dharel, N., et al. (2005). Interaction between the HCV NS3 protein and the host TBK1 protein leads to inhibition of cellular antiviral responses. Hepatology 41, 1004–1012. doi: 10.1002/hep.20666
Paulmann, D., Magulski, T., Schwarz, R., Heitmann, L., Flehmig, B., Vallbracht, A., et al. (2008). Hepatitis A virus protein 2B suppresses beta interferon (IFN) gene transcription by interfering with IFN regulatory factor 3 activation. J. Gen. Virol. 89, 1593–1604. doi: 10.1099/vir.0.83521-0
Pugnale, P., Pazienza, V., Guilloux, K., and Negro, F. (2009). Hepatitis delta virus inhibits alpha interferon signaling. Hepatology 49, 398–406. doi: 10.1002/hep.22654
Purcell, R. H. (1993). The discovery of the hepatitis viruses. Gastroenterology 104, 955–963. doi: 10.1016/0016-5085(93)90261-A
Qu, L., Feng, Z., Yamane, D., Liang, Y., Lanford, R. E., Li, K., et al. (2011). Disruption of TLR3 signaling due to cleavage of TRIF by the hepatitis A virus protease-polymerase processing intermediate, 3CD. PLoS Pathog. 7:e1002169. doi: 10.1371/journal.ppat.1002169
Rasche, A., Sander, A.-L., Corman, V. M., and Drexler, J. F. (2019). Evolutionary biology of human hepatitis viruses. J. Hepatol. 70, 501–520. doi: 10.1016/j.jhep.2018.11.010
Refolo, G., Ciccosanti, F., Di Rienzo, M., Basulto Perdomo, A., Romani, M., Alonzi, T., et al. (2019). Negative regulation of mitochondrial antiviral signaling protein-mediated antiviral signaling by the mitochondrial protein LRPPRC during hepatitis C virus infection. Hepatology 69, 34–50. doi: 10.1002/hep.30149
Reizis, B., Bunin, A., Ghosh, H. S., Lewis, K. L., and Sisirak, V. (2011). Plasmacytoid dendritic cells: recent progress and open questions. Annu. Rev. Immunol. 29, 163–183. doi: 10.1146/annurev-immunol-031210-101345
Ringelhan, M., Pfister, D., O’Connor, T., Pikarsky, E., and Heikenwalder, M. (2018). The immunology of hepatocellular carcinoma. Nat. Immunol. 19, 222–232. doi: 10.1038/s41590-018-0044-z
Rizzetto, M. (2016). The adventure of delta. Liver Int. 36(Suppl. 1), 135–140. doi: 10.1111/liv.13018
Saito, T., Owen, D. M., Jiang, F., Marcotrigiano, J., and Gale, M. (2008). Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454, 523–527. doi: 10.1038/nature07106
Sajadi, S. M. A., Mirzaei, V., Hassanshahi, G., Khorramdelazad, H., Daredor, H. Y., Hosseini, S. M. H., et al. (2013). Decreased expressions of Toll-like receptor 9 and its signaling molecules in chronic hepatitis B virus-infected patients. Arch. Pathol. Lab. Med. 137, 1674–1679. doi: 10.5858/arpa.2012-0415-OA
Sander, A.-L., Corman, V. M., Lukashev, A. N., and Drexler, J. F. (2018). Evolutionary origins of enteric hepatitis viruses. Cold Spring Harb. Perspect. Med. 8:a031690. doi: 10.1101/cshperspect.a031690
Sato, S., Li, K., Kameyama, T., Hayashi, T., Ishida, Y., Murakami, S., et al. (2015). The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 42, 123–132. doi: 10.1016/j.immuni.2014.12.016
Sepehri, Z., Kiani, Z., Alavian, S. M., Arababadi, M. K., and Kennedy, D. (2016). The link between TLR7 signaling and hepatitis B virus infection. Life Sci. 158, 63–69. doi: 10.1016/j.lfs.2016.06.026
Shahrakyvahed, A., Sanchooli, J., Sanadgol, N., Arababadi, M. K., and Kennedy, D. (2014). TLR9: an important molecule in the fight against hepatitis B virus. Postgrad. Med. J. 90, 396–401. doi: 10.1136/postgradmedj-2013-132309
Shin, E.-C., and Jeong, S.-H. (2018). Natural history, clinical manifestations, and pathogenesis of hepatitis A. Cold Spring Harb. Perspect. Med. 8:a031708. doi: 10.1101/cshperspect.a031708
Smith, D. B., and Simmonds, P. (2018). Classification and genomic diversity of enterically transmitted hepatitis viruses. Cold Spring Harb. Perspect. Med. 8:a031880. doi: 10.1101/cshperspect.a031880
Sooryanarain, H., Heffron, C. L., and Meng, X.-J. (2020). The U-rich untranslated region of the hepatitis E virus induces differential type I and type III interferon responses in a host cell-dependent manner. mBio 11:e03103-19. doi: 10.1128/mBio.03103-19
Streicher, F., and Jouvenet, N. (2019). Stimulation of innate immunity by host and viral RNAs. Trends Immunol. 40, 1134–1148. doi: 10.1016/j.it.2019.10.009
Suárez-Amarán, L., Usai, C., Di Scala, M., Godoy, C., Ni, Y., Hommel, M., et al. (2017). A new HDV mouse model identifies mitochondrial antiviral signaling protein (MAVS) as a key player in IFN-β induction. J. Hepatol. 67, 669–679. doi: 10.1016/j.jhep.2017.05.010
Surjit, M., Varshney, B., and Lal, S. K. (2012). The ORF2 glycoprotein of hepatitis E virus inhibits cellular NF-κB activity by blocking ubiquitination mediated proteasomal degradation of IκBα in human hepatoma cells. BMC Biochem. 13:7. doi: 10.1186/1471-2091-13-7
Takahashi, K., Asabe, S., Wieland, S., Garaigorta, U., Gastaminza, P., Isogawa, M., et al. (2010). Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc. Natl. Acad. Sci. U.S.A. 107, 7431–7436. doi: 10.1073/pnas.1002301107
Thomsen, M. K., Nandakumar, R., Stadler, D., Malo, A., Valls, R. M., Wang, F., et al. (2016). Lack of immunological DNA sensing in hepatocytes facilitates hepatitis B virus infection. Hepatology 64, 746–759. doi: 10.1002/hep.28685
Tu, T., Bühler, S., and Bartenschlager, R. (2017). Chronic viral hepatitis and its association with liver cancer. Biol. Chem. 398, 817–837. doi: 10.1515/hsz-2017-0118
Vazquez, C., Tan, C. Y., and Horner, S. M. (2019). Hepatitis C virus infection is inhibited by a noncanonical antiviral signaling pathway targeted by NS3-NS4A. J. Virol. 93:e0725-19. doi: 10.1128/JVI.00725-19
Vincent, I. E., Zannetti, C., Lucifora, J., Norder, H., Protzer, U., Hainaut, P., et al. (2011). Hepatitis B virus impairs TLR9 expression and function in plasmacytoid dendritic cells. PLoS One 6:e26315. doi: 10.1371/journal.pone.0026315
Wan, D., Jiang, W., and Hao, J. (2020). Research advances in how the cGAS-STING pathway controls the cellular inflammatory response. Front. Immunol. 11:615. doi: 10.3389/fimmu.2020.00615
Wang, D., Fang, L., Wei, D., Zhang, H., Luo, R., Chen, H., et al. (2014). Hepatitis A virus 3C protease cleaves NEMO to impair induction of beta interferon. J. Virol. 88, 10252–10258. doi: 10.1128/JVI.00869-14
Wang, X., Li, Y., Mao, A., Li, C., Li, Y., and Tien, P. (2010). Hepatitis B virus X protein suppresses virus-triggered IRF3 activation and IFN-beta induction by disrupting the VISA-associated complex. Cell Mol. Immunol. 7, 341–348. doi: 10.1038/cmi.2010.36
Wang, Y., Cui, L., Yang, G., Zhan, J., Guo, L., Chen, Y., et al. (2019). Hepatitis B e antigen inhibits NF-κB activity by interrupting K63-linked ubiquitination of NEMO. J. Virol. 93:e0667-18. doi: 10.1128/JVI.00667-18
Wang, Y., Zhao, C., Qi, Y., and Geng, Y. (2016). Hepatitis E Virus. Adv. Exp. Med. Biol. 948, 1–16. doi: 10.1007/978-94-024-0942-0_1
Wei, C., Ni, C., Song, T., Liu, Y., Yang, X., Zheng, Z., et al. (2010). The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J. Immunol. 185, 1158–1168. doi: 10.4049/jimmunol.0903874
Wilson, R., Warner, N., Ryan, K., Selleck, L., Colledge, D., Rodgers, S., et al. (2011). The hepatitis B e antigen suppresses IL-1β-mediated NF-κB activation in hepatocytes. J. Viral Hepat. 18, e499–e507. doi: 10.1111/j.1365-2893.2011.01484.x
Wu, J., Meng, Z., Jiang, M., Pei, R., Trippler, M., Broering, R., et al. (2009). Hepatitis B virus suppresses toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology 49, 1132–1140. doi: 10.1002/hep.22751
Wu, M., Xu, Y., Lin, S., Zhang, X., Xiang, L., and Yuan, Z. (2007). Hepatitis B virus polymerase inhibits the interferon-inducible MyD88 promoter by blocking nuclear translocation of Stat1. J. Gen. Virol. 88, 3260–3269. doi: 10.1099/vir.0.82959-0
Wu, X., Zang, F., Liu, M., Zhuo, L., Wu, J., Xia, X., et al. (2019). Genetic variants in RIG-I-like receptor influences HCV clearance in Chinese Han population. Epidemiol. Infect. 147:e195. doi: 10.1017/S0950268819000827
Xiong, W., Wang, X., Liu, X., Xiang, L., Zheng, L., and Yuan, Z. (2004). Interferon-inducible MyD88 protein inhibits hepatitis B virus replication. Virology 319, 306–314. doi: 10.1016/j.virol.2003.11.005
Xu, J., Wu, F., Tian, D., Wang, J., Zheng, Z., and Xia, N. (2014). Open reading frame 3 of genotype 1 hepatitis E virus inhibits nuclear factor-κappa B signaling induced by tumor necrosis factor-α in human A549 lung epithelial cells. PLoS One 9:e100787. doi: 10.1371/journal.pone.0100787
Xu, L., Wang, W., Li, Y., Zhou, X., Yin, Y., Wang, Y., et al. (2017). RIG-I is a key antiviral interferon-stimulated gene against hepatitis E virus regardless of interferon production. Hepatology 65, 1823–1839. doi: 10.1002/hep.29105
Xu, N., Yao, H.-P., Lv, G.-C., and Chen, Z. (2012). Downregulation of TLR7/9 leads to deficient production of IFN-α from plasmacytoid dendritic cells in chronic hepatitis B. Inflamm. Res. 61, 997–1004. doi: 10.1007/s00011-012-0493-z
Xu, Y., Hu, Y., Shi, B., Zhang, X., Wang, J., Zhang, Z., et al. (2009). HBsAg inhibits TLR9-mediated activation and IFN-α production in plasmacytoid dendritic cells. Mol. Immunol. 46, 2640–2646. doi: 10.1016/j.molimm.2009.04.031
Yang, Y., Liang, Y., Qu, L., Chen, Z., Yi, M., Li, K., et al. (2007). Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl. Acad. Sci. U.S.A. 104, 7253–7258. doi: 10.1073/pnas.0611506104
Yang, Y., Zhao, X., Wang, Z., Shu, W., Li, L., Li, Y., et al. (2020). Nuclear sensor interferon-inducible protein 16 inhibits the function of hepatitis B virus covalently closed circular DNA by integrating innate immune activation and epigenetic suppression. Hepatology 71, 1154–1169. doi: 10.1002/hep.30897
Yao, Y., Shen, Y., Shao, H., Liu, Y., Ji, Y., Du, G., et al. (2021). Polymorphisms of RIG-I-like receptor influence HBV clearance in Chinese Han population. J. Med. Virol. 93, 4957–4965. doi: 10.1002/jmv.26969
Yi, Z., Chen, J., Kozlowski, M., and Yuan, Z. (2015). Innate detection of hepatitis B and C virus and viral inhibition of the response. Cell Microbiol. 17, 1295–1303. doi: 10.1111/cmi.12489
Yin, X., Li, X., Ambardekar, C., Hu, Z., Lhomme, S., and Feng, Z. (2017). Hepatitis E virus persists in the presence of a type III interferon response. PLoS Pathog. 13:e1006417. doi: 10.1371/journal.ppat.1006417
Yu, S., Chen, J., Wu, M., Chen, H., Kato, N., and Yuan, Z. (2010). Hepatitis B virus polymerase inhibits RIG-I- and Toll-like receptor 3-mediated beta interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKepsilon and DDX3. J. Gen. Virol. 91, 2080–2090. doi: 10.1099/vir.0.020552-0
Yu, X., Lan, P., Hou, X., Han, Q., Lu, N., Li, T., et al. (2017). HBV inhibits LPS-induced NLRP3 inflammasome activation and IL-1β production via suppressing the NF-κB pathway and ROS production. J. Hepatol. 66, 693–702. doi: 10.1016/j.jhep.2016.12.018
Zeng, Y., Wu, W., Fu, Y., Chen, S., Chen, T., Yang, B., et al. (2019). Toll-like receptors, long non-coding RNA NEAT1, and RIG-I expression are associated with HBeAg-positive chronic hepatitis B patients in the active phase. J. Clin. Lab. Anal. 33:e22886. doi: 10.1002/jcla.22886
Zhang, Y.-L., Guo, Y.-J., Bin, L., and Sun, S.-H. (2009). Hepatitis C virus single-stranded RNA induces innate immunity via Toll-like receptor 7. J. Hepatol. 51, 29–38. doi: 10.1016/j.jhep.2009.03.012
Zhang, Z., Filzmayer, C., Ni, Y., Sültmann, H., Mutz, P., Hiet, M.-S., et al. (2018). Hepatitis D virus replication is sensed by MDA5 and induces IFN-β/λ responses in hepatocytes. J. Hepatol. 69, 25–35. doi: 10.1016/j.jhep.2018.02.021
Zhang, Z., Trippler, M., Real, C. I., Werner, M., Luo, X., Schefczyk, S., et al. (2020). Hepatitis B virus particles activate toll-like receptor 2 signaling initially upon infection of primary human hepatocytes. Hepatology 72, 829–844. doi: 10.1002/hep.31112
Zhou, J., Huang, Y., Tian, D., Xu, D., Chen, M., and Wu, H. (2009). Expression of toll-like receptor 9 in peripheral blood mononuclear cells from patients with different hepatitis B and C viral loads. J. Huazhong Univ. Sci. Technol. Med. Sci. 29, 313–317. doi: 10.1007/s11596-009-0310-2
Keywords: hepatitis virus, viral infection, antiviral innate immunity, immune evasion, replication
Citation: Xu C, Chen J and Chen X (2021) Host Innate Immunity Against Hepatitis Viruses and Viral Immune Evasion. Front. Microbiol. 12:740464. doi: 10.3389/fmicb.2021.740464
Received: 13 July 2021; Accepted: 29 September 2021;
Published: 03 November 2021.
Edited by:
Xulin Chen, Jinan University, ChinaReviewed by:
Zongdi Feng, The Ohio State University, United StatesXiaoyong Zhang, Southern Medical University, China
Copyright © 2021 Xu, Chen and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jizheng Chen, Y2hlbmp6QHdoLmlvdi5jbg==; Xinwen Chen, Y2hlbnh3QHdoLmlvdi5jbg==