 Michael Gorka1
Michael Gorka1 Amgalanbaatar Baldansuren2
Amgalanbaatar Baldansuren2 Amanda Malnati2
Amanda Malnati2 Elijah Gruszecki2
Elijah Gruszecki2 John H. Golbeck1,3*
John H. Golbeck1,3* K. V. Lakshmi2*
K. V. Lakshmi2*- 1Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA, United States
- 2Department of Chemistry and Chemical Biology and The Baruch ’60 Center for Biochemical Solar Energy Research, Rensselaer Polytechnic Institute, Troy, NY, United States
- 3Department of Chemistry, The Pennsylvania State University, University Park, PA, United States
Chlorophylls (Chl)s exist in a variety of flavors and are ubiquitous in both the energy and electron transfer processes of photosynthesis. The functions they perform often occur on the ultrafast (fs–ns) time scale and until recently, these have been difficult to measure in real time. Further, the complexity of the binding pockets and the resulting protein-matrix effects that alter the respective electronic properties have rendered theoretical modeling of these states difficult. Recent advances in experimental methodology, computational modeling, and emergence of new reaction center (RC) structures have renewed interest in these processes and allowed researchers to elucidate previously ambiguous functions of Chls and related pheophytins. This is complemented by a wealth of experimental data obtained from decades of prior research. Studying the electronic properties of Chl molecules has advanced our understanding of both the nature of the primary charge separation and subsequent electron transfer processes of RCs. In this review, we examine the structures of primary electron donors in Type I and Type II RCs in relation to the vast body of spectroscopic research that has been performed on them to date. Further, we present density functional theory calculations on each oxidized primary donor to study both their electronic properties and our ability to model experimental spectroscopic data. This allows us to directly compare the electronic properties of hetero- and homodimeric RCs.
Introduction
Photosynthesis is perhaps one of the most important processes in nature. The ability of oxygenic photosynthesis to utilize the virtually inexhaustible supply of solar energy has powered the planet for billions of years. Some of the earliest signs of the presence of anoxygenic phototrophs, found in South Africa, date to more than 3.4 Ga (Tice and Lowe, 2004) and are believed to have used hydrogen (H2) and/or iron (Fe) as the source reducing equivalents for carbon fixation (Widdel et al., 1993; Fischer et al., 2016). In the modern era, evolutionary history is marked by the advent of cyanobacteria [∼ 2.4 Ga (Hofmann, 1976; Bekker et al., 2004; Rasmussen et al., 2008)] that utilizes the free energy provided by the sun to generate a highly oxidizing species that splits water, producing reducing equivalents that are ultimately stored as NADPH or ‘biohydrogen’ for use in atmospheric CO2 fixation. This process has evolved to be incredibly efficient, with a quantum efficiency of 0.97 for energy capture (Şener et al., 2011). Furthermore, the energy loss in these systems is mitigated by electron-transfer processes where approx. 60% of the energy of a red photon is conserved (Ke et al., 1973).
Light-driven electron-transfer in photosynthesis originates in a structure known as the reaction center (RC). Reaction centers are large, multi-subunit pigment-protein complexes that harvest light energy through a network of internal or external chlorophyll (Chl) or bacteriochlorophyll (BChl) molecules and store the energy through charge separation and mobilization (Golbeck, 2006; Vinyard et al., 2013). Photoexcitation of the RC leads to rapid charge separation between two (or more) (B)Chl molecules bound in the polypeptide core of the RC. The initial charge separation is subsequently stabilized by electron transfer through a series of cofactors, which extends the lifetime of charge separation from the picosecond to the physiologically relevant millisecond time scale. There are two types of RCs in nature, Type I and Type II, that can be differentiated in several ways. The most obvious difference is the identity of the terminal electron acceptors of the RCs. The electron transfer pathway(s) in Type I RCs utilize iron-sulfur ([4Fe–4S]) clusters, whereas, Type II RCs use quinone molecules as terminal acceptors. Moreover, the identity and orientation of the primary electron acceptor(s) are different in Type I and II RCs (Figure 1). The primary acceptor of Type II RCs is a (bacterio)pheophytin [(B)Pheo], lacking a central metal ion, whereas the primary donor of the RC [with the exception of the RC from Acaryochloris marina (Chen et al., 2005; Tomo et al., 2007)] is a (B)Pheo derivative, (B)Chl, of the same type. In contrast, Type I RCs always contain (B)Chl a (or related derivative) as the primary acceptor regardless of the identity of the primary donor (Orf et al., 2018).
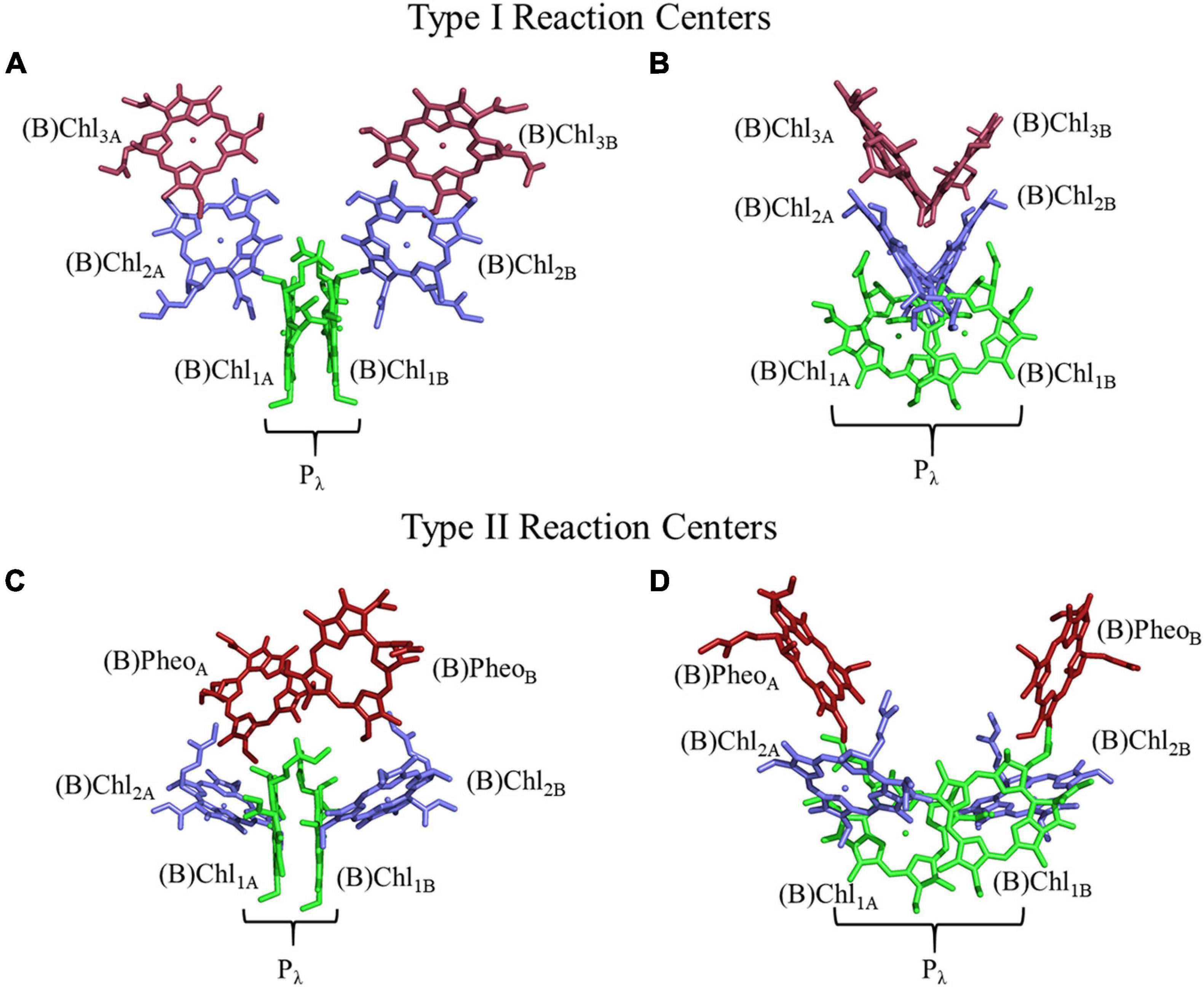
Figure 1. Structure and orientation of early (B)Chl acceptors in Type I (top) and Type II (bottom) RCs viewed from different angles. (A,C) A parallel orientation of the primary donor macrocycle planes, and (B,D) a perpendicular orientation of the primary donor macrocycle planes. The (B)Chl molecules of the primary donor, (B)Chl1, are shown in green, (B)Chl2 in blue, and (B)Chl3/(B)Pheo in red. The two (pseudo-)symmetric branches of cofactors are denoted as ‘A’ and ‘B,’ respectively.
Reaction centers can be further categorized as homodimers and heterodimers. The core of homodimeric RCs is comprised of a dimer of polypeptide subunits that are encoded by a single gene, e.g., the RC of Heliobacterium modesticaldum. Homodimeric RCs tend to be simpler in function, containing fewer polypeptide subunits and electron-transfer cofactors (Gisriel et al., 2017). While there are two branches of symmetric cofactors available for electron transfer in homodimeric RCs, it is predicted that since there is no means of differentiating between the two branches, it is axiomatic that both are employed equally. In contrast, heterodimeric RCs contain two distinct polypeptide subunits with reasonably high sequence homology. These RCs also contain two branches of cofactors for electron transfer, which appear symmetric. However, the heterogeneity of the protein-matrix leads to significant differences in the usage and time scales of electron transfer. For example, in the heterodimeric Type I RC of cyanobacteria, photosystem I (PS I), electron transfer in the A-branch is preferred over the B-branch by a factor of ∼ 2 (Agalarov and Brettel, 2003; Poluektov et al., 2005). This situation is pushed to the extreme in heterodimeric Type II RCs, such as, photosystem II (PS II) and the bacterial RC (bRC) from Rhodobacter (Rba.) sphaeroides and Rhodopseudomonas (Rps.) viridis, where the A-branch is exclusively used for electron transfer (Mimuro et al., 1995; Wakeham and Jones, 2005; Williams and Allen, 2009; Vinyard et al., 2013). Although the protein matrix effects responsible for the free energy differences that lead to branch specificity in RCs are yet to be fully elucidated (Wakeham and Jones, 2005), the study of site-specific genetic variants has yielded partial insight on the factors that influence directionality in heterodimeric RCs (Kirmaier et al., 1991, 1999; de Boer et al., 2002).
(Bacterio)chlorophyll molecules are amongst the most ubiquitous cofactors in RCs, with BChls commonly found in anoxygenic phototrophic bacteria, such as, the green sulfur bacterium, Chlorobaculum tepidum, and purple bacterium, Rba. sphaeroides, while Chls are found in oxygenic phototrophs, such as, higher plants and cyanobacteria (Oren, 2011). Both Chl and BChl molecules are tetrapyrrole macrocycles that bind a central metal ion, which is usually Mg2+ (but also Zn2+ in rare cases) (Tomi et al., 2007; He et al., 2019; Charles et al., 2020). The (B)Chl molecules can be distinguished by the extent of delocalization of the π-system across the tetrapyrrole macrocycle, which leads to a shift in the absorbance maximum, with the BChls absorbing farther to the red (Strain and Svec, 1966). The (B)Chls can be further divided into different ‘flavors,’ differentiated by the substituents on the macrocycle and are often denoted by a letter, such as, Chl a and Chl b. There are six Chl molecules, Chl a, 81-hydroxy-Chl a, divinyl-Chl a/b, Chl b, Chl d, and Chl f, and six BChl molecules, BChl a, BChl b, BChl c, BChl d, BChl e and BChl g, currently known (Chew and Bryant, 2007). Divinyl (DV)Chls are a variation of Chls, where the ethyl group of the pyrrole ring (II) is changed to a vinyl group (Steglich et al., 2003; Ito and Tanaka, 2011; Barrera-Rojas et al., 2018). This relatively minor change allows for absorption of blue light with virtually no impact on the lifetime of the excited state (see below) or their fluorescence quantum yield (Steglich et al., 2003). They are predominately found in Prochlorococcus, a deep-sea growing marine cyanobacterium that uses DVChls to absorb the plentiful blue light found deeper in the water column (Ralf and Repeta, 1992; Moore et al., 1995; Partensky et al., 1999). In general, the various substituents on the (B)Chl macrocycles lead to changes of the extended π-system, which results in shifts of the absorbance bands. The structures of three of the most common (B)Chl molecules are shown in Figure 2. In addition to a functionalized tetrapyrrole macrocycle, (B)Chls contain a long hydrophobic tail that is often a phytol group (Chew and Bryant, 2007), but can also be other substituents, such as, farnesol in heliobacteria (Hb) (Kobayashi et al., 1991) and green sulfur bacteria (Gsb) (Frigaard et al., 2006). While the function of the phytol tail is likely to serve as a lipophilic anchor within the hydrophobic trans-membrane region(s) of proteins, it has been suggested that steric effects can impact the electronic properties of the macrocycle, as well as control the chelation properties of the central metal ion (Fiedor et al., 2008).
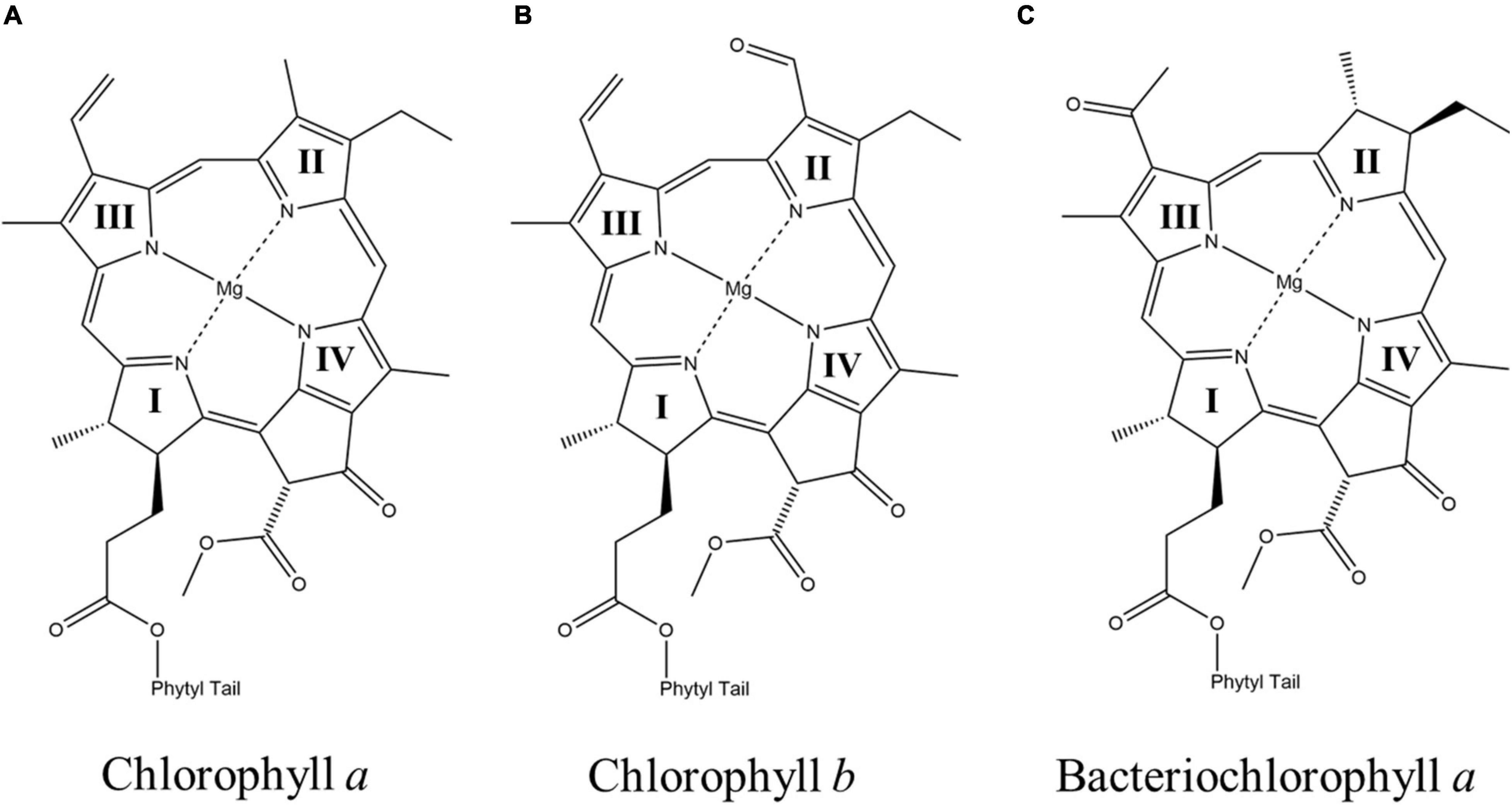
Figure 2. The chemical structures of (A) chlorophyll a (Chl a) (B) chlorophyll b (Chl b), and (C) bacteriochlorophyll a (BChl a). The nitrogen atoms of the four pyrrole rings in each macrocycle are labeled as I, II, III, and IV, respectively.
The arrangement of (B)Chl molecules in Type I and Type II RCs are presented in Figure 1. For the purposes of this review article, we will follow a common nomenclature for the (B)Chl cofactors across a variety of hetero- and homodimeric RCs. The two branches of cofactors will be denoted as ‘A’ and ‘B,’ as is common for Type I RCs. It should be noted that for Type II RCs, ‘A’ corresponds to the ‘L’ and ‘D1’ branches of the bRC and PS II, respectively. Similarly, ‘B’ refers to the ‘M’ and ‘D2’ branches of the bRC and PS II, respectively. The two closely spaced (B)Chl molecules near the luminal side of each RC are termed Chl1X, where X is the associated A- or B-branch (Figure 1). These dimeric (B)Chl1A/1B molecules comprise the primary donor, Pλ, where λ is the wavelength of maximal absorbance. As both the chemical identity of the pair of (B)Chls in the primary donor, and the surrounding electrostatic environment vary between species, the value of λ is unique for each RC. We term the second pair of (B)Chl molecules of the RC as ‘(B)Chl2A/2B.’ The (B)Chl in this position is also commonly referred to as the ‘accessory (B)Chl,’ as well as BA/B and ChlA/B in the bRC and PS II, respectively. The orientation of (B)Chl2A/2B in Type I RCs is nearly perpendicular to the membrane plane, and parallel to the plane of (B)Chl3A/3B, while Type II RCs display a roughly parallel orientation of (B)Chl2A/2B with the membrane plane and are almost perpendicular to the neighboring (B)Pheo. We label the final pair of (B)Chl molecules in the RC as (B)Chl3A/3B, but we refer to these cofactors as (B)PheoA/B in Type II RCs (this in keeping with the nomenclature used for Type II RCs). The (B)Chl3A/3B molecules are also commonly referred to as HA/B and A0 in the bRC and PS I, respectively.
There are several reasons that (B)Chl molecules are ubiquitous in photosynthesis [reviewed in Mauzerall et al. (1976) and Björn et al. (2009)], and in the interest of brevity we will only address a few reasons here. First, Chl molecules have an extended and modifiable π-system, drastically reducing the energy gap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), which allows for absorption of photons in the visible region of the electromagnetic spectrum. As described below, slight modification of the π-system of Chl derivatives allows for the absorption of different wavelengths of light. This, in turn, allows organisms to exist in a variety of different ecological niches, or to switch their relative Chl abundance in response to changes in the environment (Humbeck et al., 1984). Second, the extended lifetime of Chl excitation allows for efficient photochemistry. It should be noted that the time scale of Chl de-excitation in vivo is ∼ 0.3–2.3 ns (Brody and Rabinowitch, 1957; Mar et al., 1972; Morales et al., 2001) which is marginally faster than in vitro [5.1 and 3.9 ns for Chl a and Chl b, respectively (Brody and Rabinowitch, 1957; Brody, 2002)]. The enhanced time scale of de-excitation of Chl molecules in vivo is due to the interaction with the surrounding protein matrix. Regardless, these values are significantly slower than charge separation events, which occur with lifetimes of ∼ 100 fs (Shelaev et al., 2010) therefore providing ample time for photochemistry to occur.
Another important factor that dictates the use of (B)Chls is the tunability of the electronic properties through interactions with the surrounding protein matrix. Figure 3 showcases the redox potentials (both in vitro and in vivo) for a variety of monomeric (B)Chl molecules and primary donors of RCs. While smart protein-matrix interactions are not unique to (B)Chls, there are a multitude of methods by which the protein matrix can affect the redox properties of a Chl monomer or multimer (Srinivasan and Golbeck, 2009; Olson et al., 2013; Allen and Williams, 2014). One method for shifting a (B)Chl toward either a more positive or more negative redox potential is through the addition or elimination of charged residues within a distance of ∼10 Å of the cofactor (Williams et al., 2001; Johnson and Parson, 2002; Johnson et al., 2002; Ishikita et al., 2006). This method has immense flexibility in altering the potential depending on the charge of the added residue, with the impact being roughly controlled by the distance to that residue. The amplitude of this shift has been observed to vary for different cofactors, with a ∼50–60 mV shift (in either direction) for P865 (Allen and Williams, 2014) from the bRC, and up to +144 mV for the phylloquinones and iron-sulfur cluster, FX, of PS I (Karyagina et al., 2007; Srinivasan and Golbeck, 2009). Hydrogen bonding provides yet another means of tuning the redox properties of cofactors, with the added benefit of serving as a structural component. The redox potential of P865 has been shown to increase by 60–125 mV when a hydrogen bond is added, while removal of a preexisting bond decreases the potential by a similar amount (Allen and Williams, 2014). This effect is mirrored in the quinone acceptors of PS I (Srinivasan and Golbeck, 2009) and the bRC (Taguchi et al., 2013; Vermaas et al., 2015), to such an extent that the Gly225L residue that is hydrogen bonded to the secondary quinone acceptor, QB, of the bRC is thought to be primarily responsible for the difference of the redox potential between the primary and secondary quinone, QA, and QB (Wraight, 1979; Kleinfeld et al., 1984a; Taguchi et al., 2013). Indeed, the addition of a hydrogen bond to the phylloquinone acceptor, A1, in PS I (by altering the axial ligand of the Chl3 molecule) shifts its redox potential such that it is too positive to participate in forward electron transfer (Sun et al., 2014; Gorka et al., 2021a).
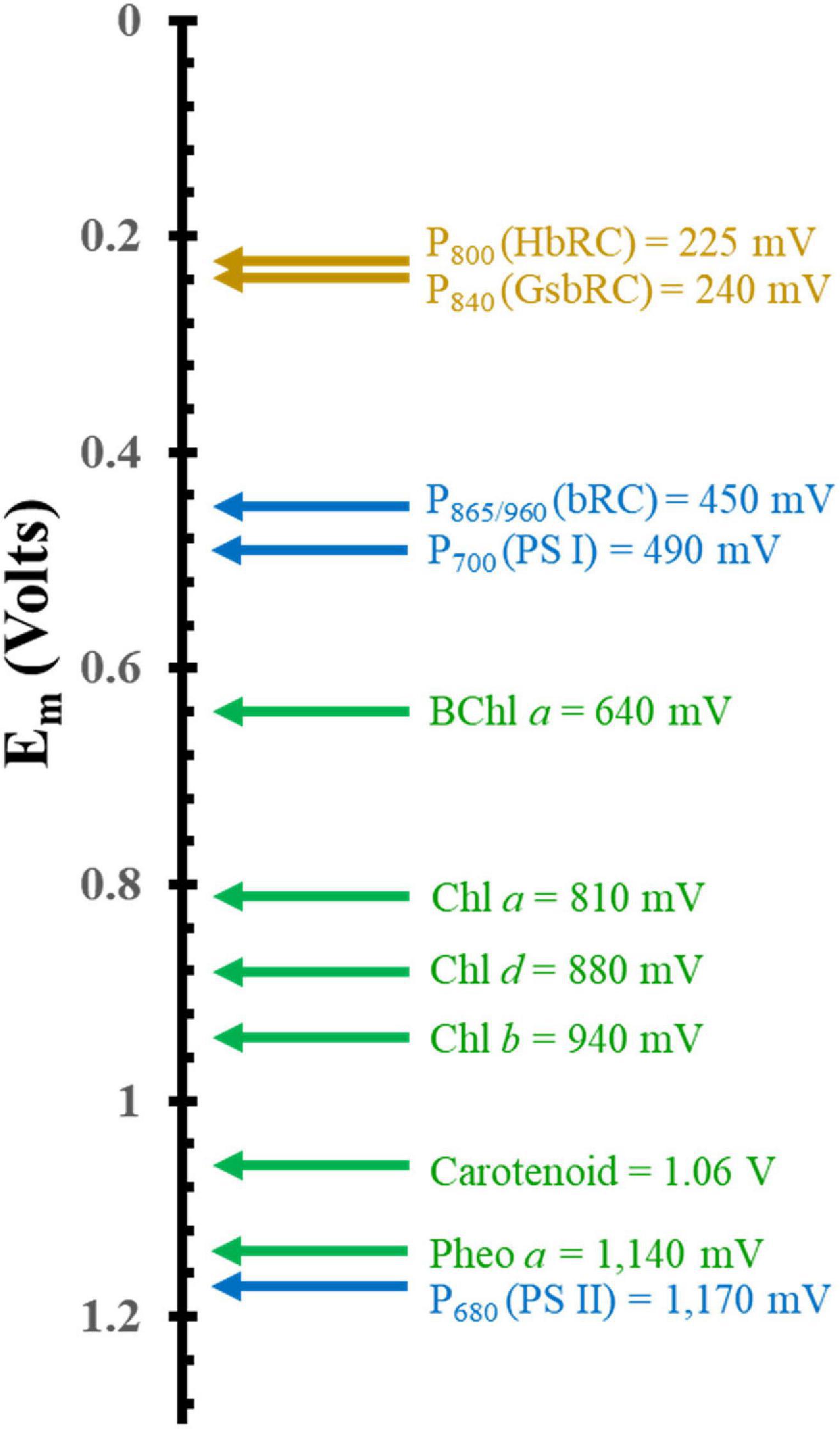
Figure 3. Comparison of the mid-point potential of a variety of (B)Chls in vitro and primary donors in vivo. Isolated in vitro values are depicted in green and hetero and homodimeric primary donors are in blue and dark yellow, respectively.
While the addition of charged residues and hydrogen bonds constitute common strategies for controlling the redox properties of cofactors in proteins, there are aspects that are unique to (B)Chls in RCs. First, a typical strategy involves π-stacking of a nearby amino acid residue or neighboring cofactor. This has an ancillary benefit of facilitating the binding of cofactors (Mao et al., 2003), however, it is limited in that π-stacking only alters the potential in one direction, i.e., toward a more negative potential. This strategy has been used to alter the potential of the phylloquinone cofactors, A1A/1B, of PS I leading to a decrease in the potential from approx. 27 mV (Srinivasan and Golbeck, 2009) to 150 mV (Kaupp, 2002) by the presence of a π-stacked Trp679A/Trp677B residue. To date, there have been few studies of this phenomenon in (B)Chl molecules, but given the extended π-system of the (B)Chls, and the recent discovery of a π-stacked Phe residue in the green-sulfur bacterial (Gsb)RC (Chen et al., 2020), this appears to be a feasible approach. Additionally, the wide variety of available axial ligands that are employed in the binding of (B)Chls in RCs can have a sizeable impact on the redox potential (Heimdal et al., 2007). While most Chl molecules in the photosynthetic antenna complexes are bound by the imidazole of a His residue, as expected by hard-soft acid-base theory (Pearson, 1963), other groups are frequently employed as axial ligands as well. For example, a Met residue is used to bind the Chl molecules, Chl3A/3B, involved in electron transfer in PS I (Jordan et al., 2001), and changes to this axial ligand have been demonstrated to significantly impact the electronic (Gorka et al., 2021a) and electron-transfer (Cohen et al., 2004; Dashdorj et al., 2005; van der Est et al., 2009; Santabarbara et al., 2010) properties of Chl3A/3B. Finally, planar ring distortions can lead to changes in the absorptive features of (B)Chl molecules (Zucchelli et al., 2007). These protein matrix effects work in tandem to help tune the spectral, electronic, and redox properties of (B)Chl molecules so as to serve multiple functions, even within the same RC.
(B)Chls are critical to photosynthesis as they are not only used to harvest solar energy but these are the only pigments used to generate a charge-separated state, and play an integral role in photosynthetic electron transfer. Light harvesting in photosynthesis is often performed through a pool of densely packed (B)Chl molecules that allow energy transfer to neighboring pigments over very short time scales using fluorescence resonance energy transfer (FRET) (Oppenheimer and Schwinger, 1941; Şener et al., 2011). In the case of PS I, the antenna pool is contained within the RC itself (Jordan et al., 2001; Şener et al., 2011) and is comprised of nearly 100 Chl molecules, although additional antenna complexes are also formed under differing physiological conditions (Bibby et al., 2001). For example, the bacterial reaction center (bRC) and PS II contain light-harvesting complexes (LHCs), termed LHC-I and LHC-II, which aid in light harvesting and energy transfer through closely packed (B)Chl molecules (Deisenhofer et al., 1985; McDermott et al., 1995; Koepke et al., 1996; Papiz et al., 2003; Roszak et al., 2003; Şener et al., 2011; van Amerongen and Croce, 2013). This process can also occur externally through energy transfer from the chlorosome to the RC via the Fenna-Matthews-Olson (FMO) protein (Olson, 2004), as is the case for green sulfur bacteria (Olson, 2013). However, energy transfer to the RC in green sulfur bacteria is unusually inefficient (Blankenship et al., 1995; Francke et al., 1996; Neerken et al., 1998; Oh-oka et al., 1998; Olson, 1998), with an estimated value of only ∼ 30%. The energy of an absorbed photon migrates across this network of (B)Chl molecules until the energy finds its way to a thermodynamic well in the RC, known as the trap. Excitation of the trap leads to the charge-separation reaction in the RC. This process occurs on ultrafast time scales, from fs (Shelaev et al., 2010; Song et al., 2021) to ps (Holzapfel et al., 1989; Peloquin et al., 1994; Holzwarth and Müller, 1996), and has thus been difficult to study. Recent advances in theoretical and experimental techniques have led to several proposed models for charge separation in RCs. While a detailed analysis of every model is outside the scope of this review article, we will provide brief descriptions of the types that have been put forth. It should also be noted that given the variations in cofactor identity, structure, and organization in the different RCs, the exact mechanism of charge separation may vary between them. Therefore, the models for each RC are described in greater detail in the respective sections [for a comprehensive review, please see Savikhin and Jankowiak (2014)].
In general, three categories of models for charge-separation have been proposed in the literature, where the process originates either at the: (i) primary donor, (ii) accessory Chl, or (ii) a larger group of highly coupled cofactors. Shown in Scheme 1 is a simple representation of the models.
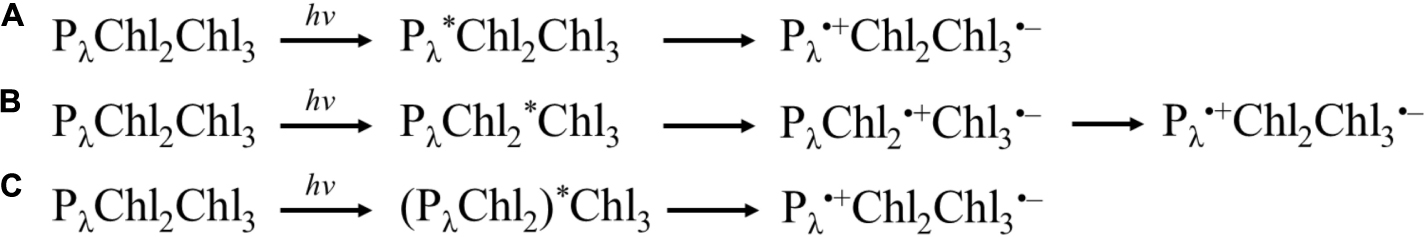
Scheme 1. Simple archetypes of charge separation models. (A) Excitation of primary donor, Pλ, leading to charge separation between Pλ and Chl3. (B) Excitation of Chl2, leading to charge separation between Chl2 and Chl3, followed by the hole on Chl2 being filled by Pλ. (C) Excitation of highly coupled Pλ, Chl2 (and potentially Chl3). Please note that Chl3 is a Pheo in Type II RCs.
For many years, the primary donor (P) had been thought of as the initial site for charge separation (White et al., 1996; Savikhin et al., 2000; Gibasiewicz et al., 2001; Gobets and van Grondelle, 2001; Gobets et al., 2001; Melkozernov, 2001; Savikhin and Jankowiak, 2014), whereby this electronically coupled special pair of (B)Chl molecules serves as an energy sink for the antenna complexes. While it was unclear whether the act of charge separation or energy transfer to the trap was the limiting factor, all of the models assumed that the low-potential reductant emerged from the primary donor, initially reducing (B)Chl2. Advances in ultrafast optical spectroscopy have made it possible to investigate the femto- and picosecond transient states of the RCs, which led to various proposed models for the charge separation reaction. A thorough review of ultrafast optical spectroscopy methods can be found in Berera et al. (2009) and Schlau-Cohen et al. (2012). Ultrafast spectroscopy has been employed for the study of the bRC (Van Brederode et al., 1997; van Brederode et al., 1998, 1999; van Stokkum et al., 1997; Vos et al., 1997; van Brederode and van Grondelle, 1999; Konar et al., 2018; Ma et al., 2019), PS II (Schatz et al., 1987; Peterman et al., 1998; Novoderezhkin et al., 2007; Myers et al., 2010), PS I (Müller et al., 2003, 2010; Holzwarth et al., 2006a; Cherepanov et al., 2017, 2020, 2021), and the HbRC (Kojima et al., 2020; Song et al., 2021). Early research on the bRC demonstrated that charge separation with the successful formation of the reduced QA⋅– state still occurs in site-directed genetic variants with significantly slower energy transfer from BChl2A∗ to P865 (Van Brederode et al., 1997; van Brederode et al., 1998). These results, coupled with the observation that direct excitation of BChl2A resulted in the formation of P865∗, led to a model in which BChl2A was the genesis of the charge-separated state. Other studies around the same time period suggested instead that the early BChls should be considered a hexamer, where both P865 and BChl2 can be coherently excited (Vos et al., 1997). Much later, multiple two-dimensional electronic spectroscopy with better time resolution showed that when vibrational and electronic factors are considered, it is more reasonable to conclude that P865 initiates charge separation (Niedringhaus et al., 2018; Ma et al., 2019) and that P865 and BChl2A/2B are electronically coupled, allowing for excited state migration (Konar et al., 2018). In principle, this could lead to the presence of multiple redundant mechanisms of charge separation in the system.
Analogous models for charge separation were also proposed for PS I using ultrafast spectroscopy methods. Ultrafast difference spectroscopy of wild-type (WT) PS I showed a mixture of the excited P700∗ state and the radical states associated with the charge-separated state, P700⋅+Chl2A/B⋅–. Moreover, perturbation of the protein environment surrounding the Chl2A/2B cofactors through site-directed genetic variants appeared to slow the formation of the charge-separated state in the symmetric branch, e.g., changes to Chl2B suppressed the formation of P700⋅+Chl2A⋅– state. This suggested the presence of a highly coupled system of Chl molecules in PS I. Most recently, ultrafast spectroscopy of both WT PS I and genetic variants of the protein environment in the vicinity of the Chl2 cofactor have suggested the formation of an initial excited state (Chl2AP700Chl2B)∗ (Cherepanov et al., 2020, 2021), which rapidly leads to the formation of the first charge-separated state, P700⋅+Chl2⋅–.
In contrast, Müller and coworkers have proposed a mechanism of charge separation in PS I that originates at Chl2 (Müller et al., 2003; Holzwarth et al., 2006a), where the first charge-separated state is Chl2⋅+Chl3⋅–. Here, the hole on Chl2 was suggested to be reduced by P700 (Chl1), forming the first stable charge-separated state, P700⋅+Chl3⋅–. Theoretical studies of PS II have found that this is a promising model, as Chl2 was observed to have the lowest site energy of the initial Chl/Pheo molecules in the core (Sirohiwal et al., 2020). Recent findings have indicated common ground with both of the models described above, suggesting that the initial four (or six) Chl molecules of PS I are highly electronically coupled, and participate in charge separation to various extents. Similar models have also been proposed for the homodimeric RC from H. modesticaldum, invoking both the BChl2/BChl3 pair (Song et al., 2021) and/or all six core BChl molecules (Kojima et al., 2020; Song et al., 2021) in the excited state. It should be noted that these models are not necessarily mutually exclusive as different models likely take priority under different excitation conditions and may vary between RCs.
With the recent availability of the X-ray crystal and cryo-electron microscopy structures of the HbRC and GsbRC (Gisriel et al., 2017; Chen et al., 2020), respectively, we have the unique opportunity to compare the electronic structure of the primary donors in a variety of RCs. The goal of this article is to review research on the geometric and electronic structure of a variety of primary donors of hetero and homodimeric RCs, with an emphasis on pulsed electron paramagnetic resonance (EPR) and density functional theory (DFT) methods. We will focus on six RCs: (i) The heterodimers, P700 of Photosystem I (PS I), P680 of Photosystem II (PS II), P865 and P960 of the bRCs from Rba. sphaeroides and Rps. viridis, respectively, and (ii) homodimers, P800 of H. modesticaldum and P840 of the green-sulfur bacterium, C. tepidum.
Geometric Structures
Geometric Structure of the Primary Donor of Heterodimeric Reaction Centers
The Primary Donor, P700, of Photosystem I
We begin by describing the extensively studied primary donor, P700, of PS I. Cyanobacterial PS I [reviewed in Golbeck (2006)] is a composed of 12 or 13 polypeptide subunits that bind the light-harvesting and electron-transfer cofactors. There are 90 Chl a molecules that function as the light-harvesting antenna while the electron-transfer cofactors are comprised of two pseudo-symmetric branches of six Chl a molecules (Chl1A/1B, Chl2A/2B, and Chl3A/3B), two phylloquinones (A1A/1B), and three [4Fe–4S] clusters, FX, FA and FB (Figures 4A,B). The electron-transfer cofactors are largely bound in the transmembrane PsaA and PsaB polypeptide subunits, with the terminal [4Fe–4S] clusters, FA and FB, bound by PsaC on the stromal side of the RC (Figure 4A).
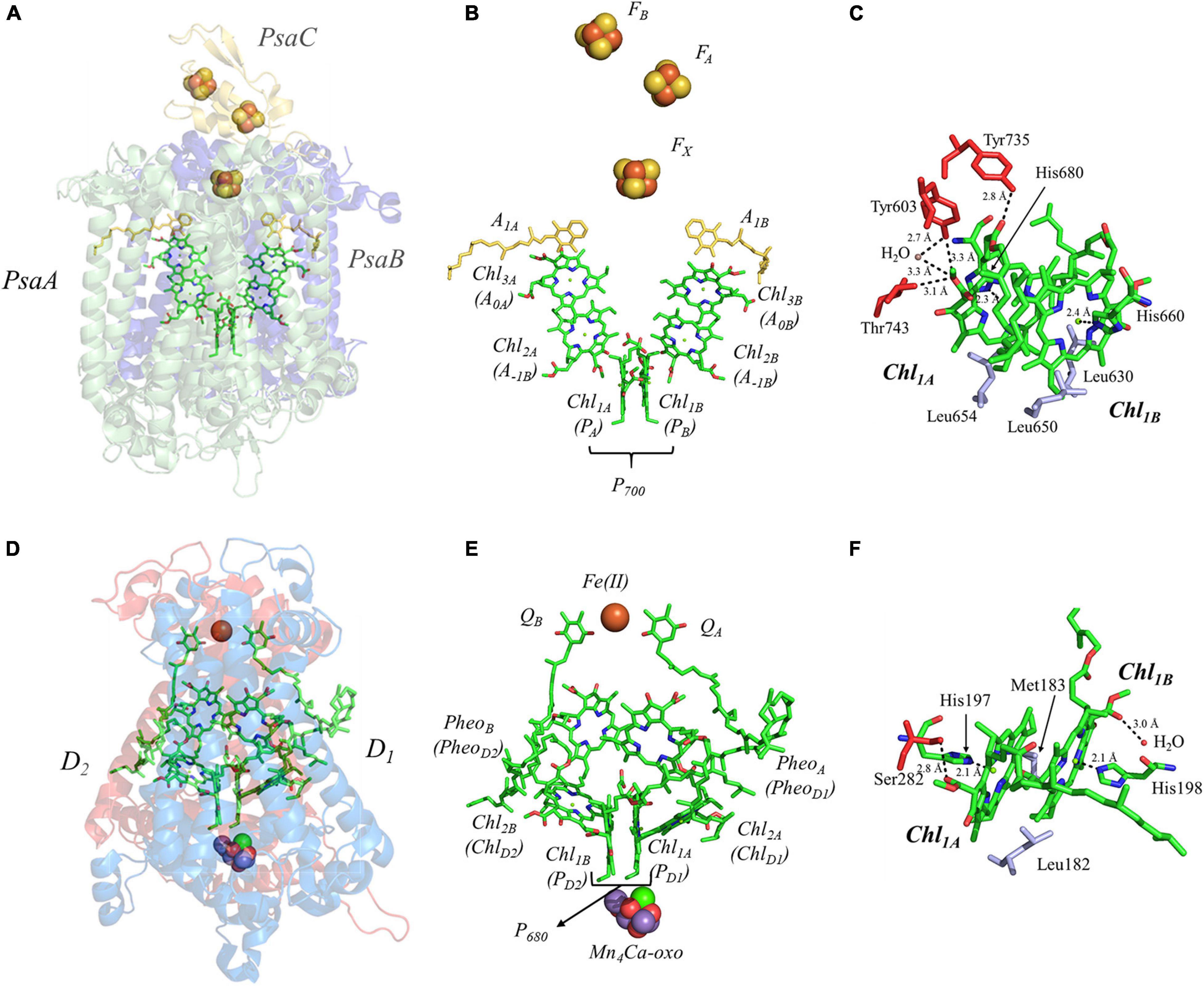
Figure 4. (A) The core subunits (PsaA, green; PsaB, blue; and PsaC, yellow) and cofactors of PS I as observed by X-ray crystallography (PDB ID: 1jb0), (B) the cofactors that participate in the primary electron transfer pathway of PS I and (C) the binding pocket of the primary donor, P700. (D) The core subunits (D1, red; D2, blue) and cofactors of PS II as observed by X-ray crystallography (PDB ID: 3wu2), (E) the cofactors that participate in the primary electron transfer pathway of PS II and (F) the binding pocket of the primary donor, P680. Commonly used labels are in parentheses. Part (C,F) emphasize the prominent protein-matrix effects of the primary donor of PS I and PS II, respectively, where the residues that are hydrogen bonded to the primary donors are shown in red, and nearby non-polar or π-stacked residues are colored in gray.
There is currently a lack of consensus on the mechanism of charge separation in PS I. Early models had suggested that P700 functioned as the source of the electron (White et al., 1996; Savikhin et al., 2000; Gibasiewicz et al., 2001; Gobets and van Grondelle, 2001; Gobets et al., 2001; Melkozernov, 2001; Savikhin and Jankowiak, 2014), whereby the dimerization of Chl1A/Chl1B created a lower energy site that traps the excitation energy. Subsequently, it was proposed that Chl2, instead of acting as the first electron acceptor, served to trap the energy and was the source of the charge-separated state (Müller et al., 2003; Holzwarth et al., 2006a). It was proposed that Chl2 would reduce Chl3 and the hole that was generated would migrate to P700, ultimately creating the charge-separated P700⋅+Chl3⋅– state. However, theoretical studies have suggested that the redox potentials of the Chl2/Chl3 pair make this mechanism thermodynamically unlikely (Ptushenko et al., 2008). More recently, advances in theoretical and experimental methods have led to a highly electronically coupled model, wherein (P700Chl2AChl2B)∗ form an exciplex, with the negative charge localized primarily on Chl2 (Cherepanov et al., 2020, 2021). While there are differences that remain in the field, and the mechanism may vary based on experimental conditions and across organisms, it should be noted that similar models have been proposed in other Type I RCs (Kojima et al., 2020; Song et al., 2021).
While the overall mechanism remains under study, the lifetime of the various electron-transfer steps of PS I are reasonably well understood. Exciton migration from the antenna system to the trap appears to be the limiting step in charge separation, occurring with a lifetime of ∼ 5 ps (Wasielewski et al., 1987; Shelaev et al., 2010). However, charge separation is known to occur within 100 fs of photoexcitation of the trap (Shelaev et al., 2010). It is upon charge separation that the electron-transfer pathway bifurcates between the A- and B-branches, forming the P700⋅+A0A⋅– or P700⋅+A0B⋅– state. Unlike Type II RCs, each branch is utilized, albeit unequally, with the A-branch being favored in cyanobacteria by a factor of two in comparison with the B-branch (Agalarov and Brettel, 2003; Poluektov et al., 2005). Electron transfer then proceeds to the A1A and A1B cofactors within ∼ 30–50 ps (Brettel, 1997; Itoh et al., 2001), and then within 20 or 200 ns, respectively (Agalarov and Brettel, 2003; Kurashov et al., 2018) to the inter-polypeptide [4Fe–4S] cluster, FX, where the A- and B-branches are known to converge (Figure 4B). Subsequently, electron transfer occurs linearly though the FA and then FB clusters (Díaz-Quintana et al., 1998) with a lifetime of ∼ 200 ns, after which the electron is transferred to a soluble electron acceptor, ferredoxin (Mondal and Bruce, 2018) or flavodoxin (Pierella Karlusich and Carrillo, 2017), for downstream processes. For a detailed analysis of the electron-transfer and charge-recombination lifetimes in PS I, please see (Kurashov et al., 2018).
It is important to understand the structural and electronic factors that influence the redox and kinetic properties of the primary donor, P700, as it is critical for light-driven electron transfer in PS I. P700 is a dimer of a Chl a’ and Chl a molecules, Chl1A and Chl1B, where Chl a’ is the 132 epimer of Chl a (Kobayashi, 1996; Webber and Lubitz, 2001; Figure 4C). Of all of the primary donors that are discussed in this review, P700 appears to have the most extensive and asymmetric protein-cofactor interactions based on the X-ray crystal structure (Jordan et al., 2001). Perhaps an obvious indication is the striking asymmetry of the hydrogen (H)-bonding environment of the two Chl molecules, Chl1A and Chl1B, that comprise the primary donor. As can be seen in Figure 4C, Chl1A has three hydrogen bonds, provided by the Tyr735PsaA, Tyr603PsaA, and Thr743PsaA residues, with a hydrogen H-bonding distance of 2.8, 3.3, and 3.1 Å, respectively. Additionally, there is a water molecule within 3.3 Å of Chl1A, which is not present near Chl1B. While it is possible that the hydrogen bonds may facilitate selective binding of the Chl a’ epimer, these are also likely to serve as a means to alter the redox potential of Chl1A relative to Chl1B, providing strict control over the distribution of electron density. The hydrogen bonds do not appear to be the only significant deviation in the protein matrix that could influence asymmetry between Chl1A and Chl1B, as three Leu residues (Leu630PsaB, Leu650PsaA, and Leu654PsaA) preferentially interact with Chl1B, although Leu654Psa and Leu630PsaB are found in the non-overlapping regions of the rings. Beyond this, both contain axially ligated His residues, which should not contribute to any asymmetrical effects.
Alterations to the primary donor, P700, of PS I from Chlamydomonas reinhardtii with the axial ligands, His656PsaB and His676PsaA, have demonstrated profound effects on its properties. Initial studies, where the His656PsaB residue was replaced by Asn or Ser, displayed a shift in the mid-point potential of P700 by +40 mV, with a corresponding alteration in its spectral features. While such changes are to be expected, electron-nuclear double resonance (ENDOR) spectroscopy of P700⋅+ in the genetic variants revealed an increase in electron spin density at the methyl group in position 12 by ∼ 20%, suggesting a significant effect on the electron delocalization over the two halves of the dimer (Webber et al., 1996). Subsequently, additional variants were studied that included changes to the axial ligand of Chl1A on the A-side, His676PsaA, and non-polar, acidic, basic, and uncharged residues in the vicinity. Changes to the redox properties of P700 displayed an expected pattern, where soft base ligands, such as, the S atom of a Cys residue, had the most profound effect on the redox potential, shifting the potential by up to ∼ + 140 mV. The effects on the electronic properties of P700⋅+ were subtle and highly asymmetric.
Shown in Figure 5A are the inter-cofactor distances and orientation of the Chl1A/1B molecules of P700. P700 has perhaps the most archetypal parallel orientation of the two Chl macrocycles of Chl1A and Chl1B in the dimer, with the distance between the macrocycles varying from 3.5–3.6 Å. There is significant overlap of the ring planes of the Chl1A and Chl1B macrocycles, as the center-to-center distance between the two Mg2+ ions is 6.3 Å. Much like every other primary donor, the most prominent overlap occurs over the pyrrole group of the N3 atom, with a small overlap of the pyrrole of N2. The space-filling model in Figure 6A shows the extent of the overlap between the two macrocycles. Because of the significant overlap, the nearest ring nitrogen atoms are amongst the closest amongst all of the RCs discussed here. The most relevant are the N3A–N3B, N2A–N2B, and N3A–N2B distances of 3.7, 4.4, and 4.9 Å, respectively. These values are shown in Table 1 for comparison with analogous distances in other RCs. Figure 6A also highlights the typical orientation of the phytol tail of P700 in comparison with that of other Type I RCs, i.e., a configuration that is tight to the central rings of the macrocycles with a small bend influenced by the presence of a water molecule.
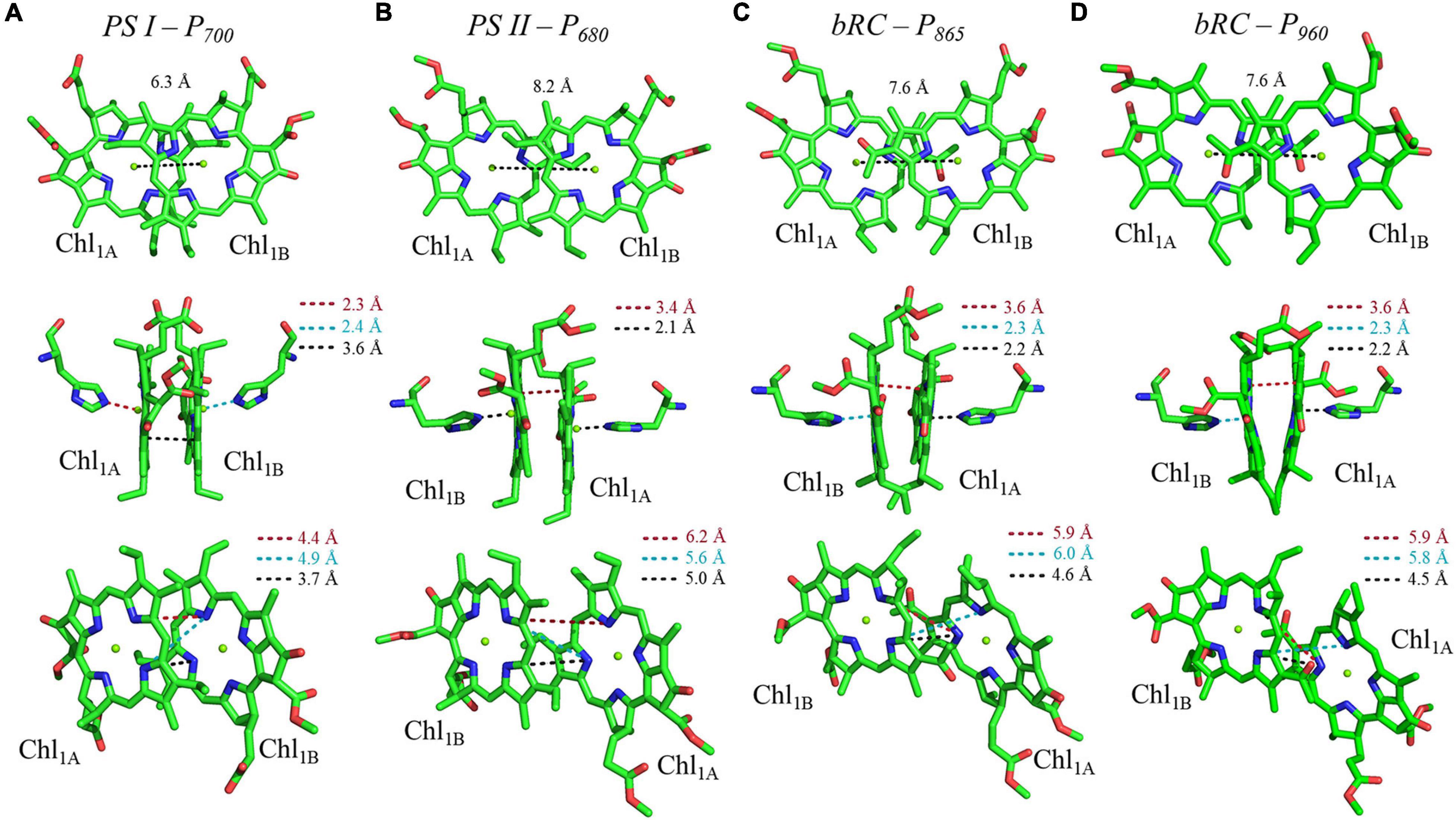
Figure 5. Analysis of inter-cofactor distances and relative orientation of primary donors from the heterodimeric RCs: (A) Photosystem I, (B) Photosystem II, and the bacterial RC from (C) Rba. sphaeroides and (D) Rps. viridis.
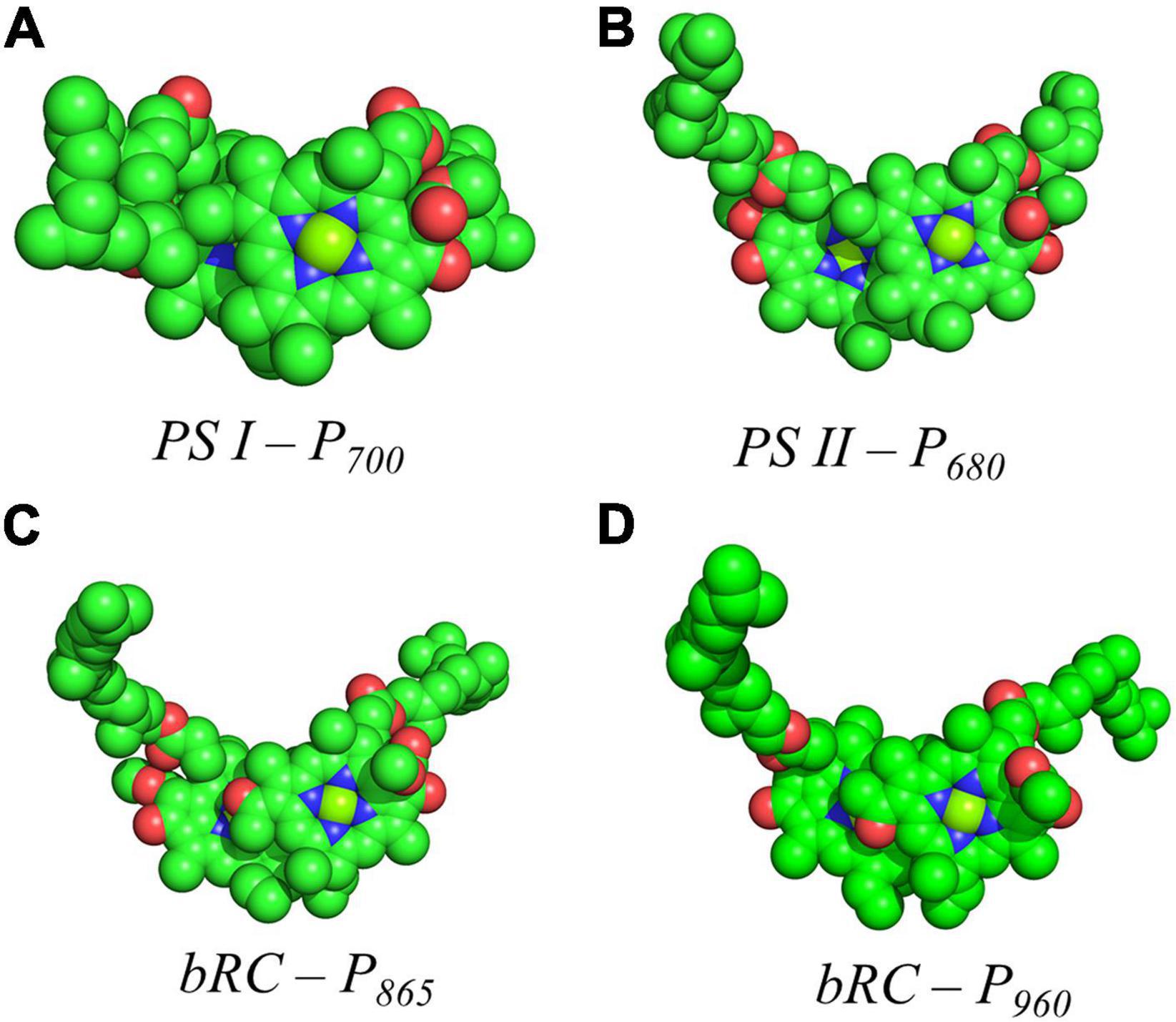
Figure 6. Space filling models of (A) P700 from PS I, (B) P680 from PS II, (C) P865 from the bRC of Rba. Sphaeroides and (D) P960 from the bRC of Rps. Viridis.
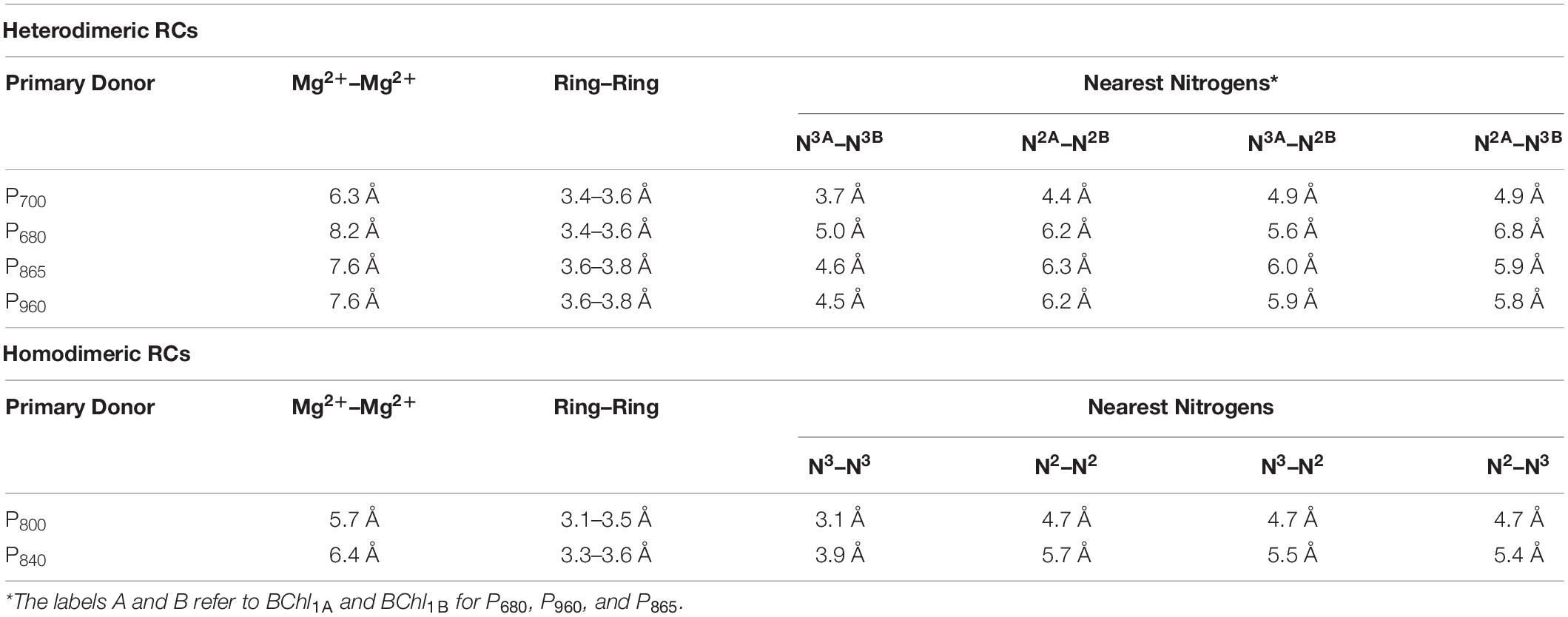
Table 1. Distance parameters of the primary donors of heterodimeric and homodimeric RCs.
The Primary Donor, P680, of Photosystem II
This section is focused on the Type II heterodimeric RC, photosystem II (PS II) [reviewed in Vinyard et al. (2013) and Lakshmi et al. (2014)]. The primary electron-transfer cofactors of PS II are present in the core polypeptide subunits, D1 and D2, and encompassed by ∼ 22–23 smaller polypeptides, whose identity varies among differing organisms. The primary electron-transfer pathway of PS II is composed of the following cofactors that are common to heterodimeric Type II RCs: four pseudo-symmetric Chl a molecules, two pheophytins (PheoA and PheoB) and two quinones (QA and QB) (Figures 4D,E). In contrast to other Type II RCs, the electron-transfer pathways also contain two redox-active tyrosine residues, YD and YZ, and a unique tetranuclear manganese calcium-oxo (Mn4Ca-oxo) cluster, which is the well-known water-splitting catalytic moiety of PS II. The redox properties of the primary donor, P680, further differentiate PS II from other Type I and Type II RCs. The primary donor, P680, of PS II is by far the most oxidizing of any RC discovered thus far, with an Em of ∼ 1,200 mV (Rappaport et al., 2002; Ishikita et al., 2005, 2006; Figure 3). In fact, it is one of the most oxidizing species that is found in nature. The evolutionary reason for this is clear, as P680 needs to be a strong oxidant in order to allow for successive oxidations of the Mn4Ca-oxo cluster that leads to the catalytic conversion of two substrate water molecules into dioxygen. The extremely high oxidizing potential of P680 showcases the amazing ability of nature in modulating the redox potential of identical or highly similar cofactors for different purposes (Ishikita et al., 2006).
Once again, there are various proposals in literature on the location of the primary charge separation of PS II, but recent findings suggest that charge separation originates at the ‘accessory’ Chl, termed Chl2A (Groot et al., 2005; Holzwarth et al., 2006b; Romero et al., 2010; Duan et al., 2017; Sirohiwal et al., 2020; Tamura et al., 2020). Theoretical modeling of site energies has indicated that contrary to previous assumptions, Chl2A has the lowest energy of all of the initial Chls. The reason for this deviation from other Type II heterodimeric RCs (see below) is attributed to the need to incorporate the Mn4Ca-oxo cluster used in water splitting (Tamura et al., 2020). In other studies, Stark spectroscopy was used to observe multiple mixed exciton charge-transfer states: (Chl1Bδ+Chl1Aδ–Chl2A)∗, (Chl2Aδ+PheoDδ–)∗, and (Chl1B+Chl1A–)δ*, suggesting a highly electronically coupled model. Interestingly, multiple empirical methods have found two events associated with charge separation, at 100–400 fs and ∼1.8 ps (Romero et al., 2010; Duan et al., 2017). Regardless of the mechanism, within ∼ 1.5 ps of photoexcitation of the trap, an electron is transferred to PheoA, forming the charge-separated [P680⋅+PheoA⋅–] state. Unlike PS I, electron transfer within PS II is branch specific, only occurring through the A-side involving the cofactors associated with the D1 polypeptide subunit (with the exception of the primary quinone, QA, that is bound in the D2 polypeptide). The stabilization of the relatively short-lived P680⋅+PheoA⋅– state is achieved through rapid forward electron transfer to the primary quinone acceptor, QA, within 200 ps and then to the secondary quinone, QB, within ∼ 200–400 μs. Much like the bRC, QB is doubly reduced and doubly protonated after two turnovers of the RC, forming the labile quinol, QBH2, with the second electron transfer step occurring notably slower than the first one (∼500–800 μs) (Bowes et al., 1980; Robinson and Crofts, 1983; de Wijn and van Gorkom, 2001; Sauer and Yachandra, 2004). As previously mentioned, the terminal quinone acceptors are a hallmark of Type II RCs as are the [4Fe–4S] clusters of the Type I RC, PS I. Meanwhile, on the acceptor side, the hole on P680⋅+ is reduced by the redox-active tyrosine, YZ, which, in turn, is re-reduced by the Mn4Ca-oxo cluster. Two turnovers of PS II are required for the complete reduction of QB and a total of four turnovers results in the oxidation of two molecules of substrate water to dioxygen at the catalytic Mn4Ca-oxo cluster (Vinyard et al., 2013).
The primary donor of PS II, P680, is a dimer of Chl a molecules, Chl1A and Chl1B. In contrast with P700, which displays an asymmetry of interactions with the protein matrix, P680 of shows less asymmetry in the protein matrix for each Chl a molecule. P680 contains one hydrogen bond at each Chl a provided by the Ser282D2 residue and a water molecule at Chl1A and Chl1B, respectively (Figure 4F). Moreover, the axial ligand to each Chl a is a His residue, His197D1 and His198D2, where the His197D1 residue also serves to bind a water molecule in proximity of the Chl1A cofactor. It should be noted that the importance of the His198D2 residue is in question and may well be species-dependent. While the alteration of the His198D2 residue in PS II from Synechocystis sp. PCC 6803 displayed changes in the spectral profile of P680 with a corresponding drop in the redox potential of ∼ 80 mV (Diner et al., 2001), analogous genetic variants of His198D2 in PS II from Thermosynechococcus elongatus displayed no discernable effects on the spectral, redox, or kinetic properties of P680 (Sugiura et al., 2008). Aside from the H-bonding interactions, the only other deviation between the environment of Chl1A and Chl1B of P680 are the residues Leu182D2 (near Chl1B) and Met183D1 (near Chl1B), in the non-overlapping region of the Chl a macrocycles.
The primary donor, P680, further differentiates itself from P700 in terms of the inter-cofactor distances and relative orientation of the Chl a molecules. While the macrocycles adopt a parallel orientation with a similar distance between the ring planes of Chl1A and Chl1B of 3.4–3.6 Å, the distance between the Mg2+ ions increases to 8.2 Å in P680. This results in minimal overlap of Chl1A and Chl1B, which is evident only in part of the N3 pyrrole group (Figure 5B, Top Panel). The limited overlap of the ring planes is further manifest as an increased distance between the nearest nitrogen atoms of Chl1A and Chl1B. The distance for the three nearest nitrogen atoms are: N3D1–N3D2 (5.0 Å), N3D1–N2D2 (5.6 Å), and N2D1–N2D2 (6.2 Å) (Figure 5B, bottom panel). This reveals that not only is there an increase in the distance between proximal nitrogen atoms of Chl1A and Chl1B, but the longest distance of the three nitrogen atoms is observed in N2D1–N2D2 (as opposed to N3A–N2B for P700). This suggests that the macrocycles exhibit a small degree of ‘outward’ rotation relative to each other. However, the orientation of the tail of P680 is typical of other Type II RCs, where it extends outward toward the stromal side of the protein. The space-filling model shown in Figure 6B provides a visual representation of the change in ring overlap, slight outward rotation, and the change of the tail orientation of P680.
The Primary Donor, P865 and P960, of the Bacterial Reaction Centers
The final set of heterodimeric primary donors is that of the bRCs from Rba. sphaeroides and Rps. viridis (more recently known as Bl. viridis) [reviewed in Leonova et al. (2011) and Savikhin and Jankowiak (2014)]. The determination of the X-ray crystal structure of these RCs dates back to several decades. Indeed, the first X-ray crystal structure of a membrane protein ever obtained was from Rps. viridis, for which Hartmut Michel, Robert Huber, and Johann Deisenhofer were awarded the Nobel Prize in chemistry in 1988 (Deisenhofer et al., 1985). In contrast with the other heterodimeric RCs, the bRC is relatively simple, being composed of only three polypeptide subunits, L, M, and H. An additional subunit containing four cytochrome c polypeptides is present in Rps. viridis, but the electron-transfer cofactors are entirely contained within the L and M subunits of the bRC. Similar to PS II, the electron transfer chain of the bRC is comprised of four bacteriochlorophylls (BChl), two bacteriopheophytins (BPheoA and BPheoB) and two quinones (QA and QB) (Figures 7A,B,D,E).
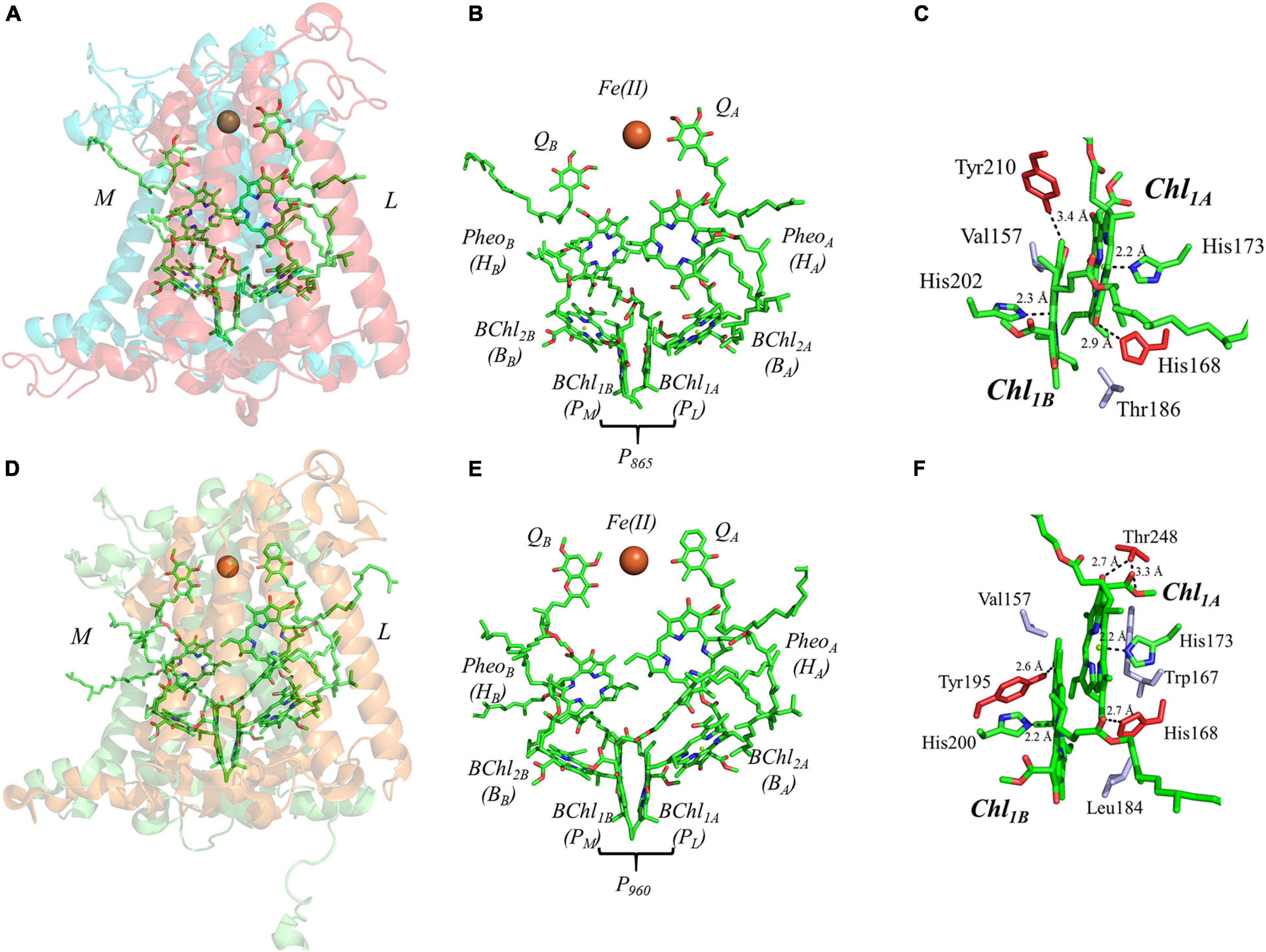
Figure 7. (A) The core subunits (M, blue; and L, red) and cofactors of the bRC from Rba. sphaeroides as observed by X-ray crystallography (PDB ID: 1aij), (B) the cofactors that participate in the primary electron transfer pathway of the bRC from Rba. sphaeroides and (C) the binding pocket of the primary donor, P865. (D) The core subunits (M, blue; and L, red) and cofactors of the bRC from Rps. viridis as observed by X-ray crystallography (PDB ID: 2jbl), (E) the cofactors that participate in the primary electron transfer pathway of the bRC from Rps. viridis and (F) the binding pocket of the primary donor, P960. Commonly used labels are in parentheses. Part (C,F) emphasize the prominent protein-matrix effects of the primary donor of the bRC from Rba. sphaeroides and Rps. viridis, respectively, where the residues that are hydrogen bonded to the primary donors are shown in red, and nearby non-polar or π-stacked residues are colored in gray.
The predominant model of charge separation in the bRC includes the initiation of electron transfer at the primary donor (Niedringhaus et al., 2018), P865 and P960, in Rba. sphaeroides and Rps. viridis, respectively. Theoretical studies have even suggested that charge separation can occur in the primary donor between BChl1A and BChl1B itself (Xu et al., 2002). However, Huang and coworkers used polarization selective spectroscopic methods to demonstrate that the excitation of BChl2 can lead to two sub-populations, wherein there is rapid formation of BChl2A⋅+PheoA⋅– in approximately half of the RCs, and the excitation migrates to P865 in the remaining RCs (Huang et al., 2012). The photoexcitation of the primary donor results in charge separation within 3–5 ps in Rba. sphaeroides (Martin et al., 1986; Holzapfel et al., 1989; Lauterwasser et al., 1991; Peloquin et al., 1994; Holzwarth and Müller, 1996) and a slightly faster rate of ∼ 2 ps in Rps. viridis (Wasielewski and Tiede, 1986; Huppman et al., 2002). The rapid formation of the PheoA⋅– state is unexpected given its long distance from the primary donor. Hence, it was proposed that the accessory BChl, BChl2, may play a role in charge separation. Through a combination of transient absorption spectroscopy (Arlt et al., 1993; van Stokkum et al., 1997), ultrafast mid-IR spectroscopy (Pawlowicz et al., 2008), mutagenesis (Shkuropatov and Shuvalov, 1993; Heller et al., 1996; Kennis et al., 1997; Roberts et al., 2001), and cofactor replacement experiments (Kirmaier et al., 1995a,b), the presence of BChl2⋅– was indeed detected and it was found to play an integral role in electron transfer. However, the first stable electron transfer intermediate is largely considered to be PheoA⋅–, after which the electron is transferred to the primary quinone, QA, in ∼ 200 ps. The reduction of QA, and the subsequent change in the presence of an electric field causes a structural perturbation to the solvation (and ultimately the protein) structure (Arata and Parson, 1981; Kleinfeld et al., 1984b; McMahon et al., 1998), which allows for the final step of electron transfer and the reduction of the secondary quinone, QB. Similar to PS II, two turnovers of the bRC are required for the complete reduction of QB, which functions as a two-electron/two-proton acceptor, forming the quinol, QBH2 (Wraight, 2004).
The primary donor of the bRC is a dimer of two BChl molecules that are bound by the L and M polypeptide subunits. It is interesting that the identity of the BChls, and thus the spectral features of the primary donors, P865 and P960, of the bRC from Rba. sphaeroides and Rps. viridis are different. In Rba. sphaeroides, the primary donor, P865, is a dimer of BChl a molecules, while P960 of Rps. viridis is a dimer of BChl b molecules. In this section, we will focus on both bRCs as the differences between the two are minor. The binding site of P865 and P960 suggests a moderately asymmetric H-bonding environment that could influence the relative redox potential of each primary donor. As shown in Figures 7C,F, there is a hydrogen bond to the BChl molecules of P865 and P960 from the Tyr210L and Tyr195L residue that are within 3.4 and 2.6 Å, of BChl1B, respectively, and the His168L that is within 2.9 and 2.7 Å of BChl1A in P865 and P960, respectively. There is an additional residue that provides a hydrogen bond to BChl1A in P960, Thr248L, with the hydroxyl and carboxyl group at a distance of 2.7 and 3.3 Å, respectively. Additional differences between the two bRCs include the residues that are located in the non-overlapping region of the BChl rings. In P960, there are two non-polar residues, Val157L and Leu184M, whereas, in P865 there is a non-polar, Val157L, and an unusual polar residue, Thr186M. The axial ligands of all of the BChl molecules, however, remain similar to all of the other primary donors, with His202M (P865)/His200M (P960) and His173L ligating BChl1B and BChl1A, respectively.
Further analysis of the inter-cofactor distances and relative orientation of the BChl macrocycles of P865 of the bRC from Rba. sphaeroides reveals a structure that is similar to P680 of PS II, albeit with unusual features. A view of the macrocycle plane (Figure 5C, Top Panel) indicates an overlapping structure that is similar to PS II, whereby the overlap of the two BChl rings is nearly exclusively on the pyrrole associated with N3. The Mg–Mg distance of P865 is 7.6 Å, which is in between PS II (8.2 Å) and PS I (6.3 Å); however, there is significantly more overlap than exists in P680. But perhaps the most defining feature of the BChl dimer of P865 is in how it deviates from a parallel orientation of the ring planes. The macrocycles form a ‘V’ shape when viewed parallel to the ring plane, pointing toward the luminal side of the protein. While most prominently seen in Rps. viridis, it is also observed in Rba. sphaeroides (Figures 5C,D). As expected, this results in a change of the distance between the nearest nitrogen atoms of the macrocycles. While the N3 nitrogen atoms remain the closest at 4.6 Å and the N3M–N2L distance is next at 6.0 and 5.9 Å in P865 and P960, respectively, N2M is located at a distance of 5.9 and 5.8 Å from N3L in P865 and P960, respectively, which is different from the pattern that is observed in other RCs. The space-filling model P865 and P960 shown in Figures 6C,D displays the macrocycle overlap and tail orientation that is common to other Type II RCs.
Geometric Structure of the Primary Donor of Homodimeric Reaction Centers
The Primary Donor, P800, of Heliobacterium modesticaldum
The homodimeric RC from H. modesticaldum, HbRC, is a simplified complex which is often considered an exemplar of ancestral Type I RCs (Ferlez et al., 2016; Orf et al., 2018). It has long been known that the HbRC is composed of a dimer of PshA polypeptide subunits, but it was not until the availability of the X-ray crystal structure that another identical pair of polypeptide subunits, PshX, were found on the periphery of each PshA polypeptide subunit (Gisriel et al., 2017; Figure 8A). The electron-transfer cofactors are completely housed in the PshA polypeptide core, and are comprised of four symmetric BChl g molecules, two 81-OH Chl a molecules, and a single inter-polypeptide [4Fe–4S] cluster, FX (Figure 8B). Note the lack of the two quinones and two additional [4Fe–4S] clusters bound by a small soluble ferredoxin-like protein present in the other Type I RC, PS I, discussed above (Romberger and Golbeck, 2012).
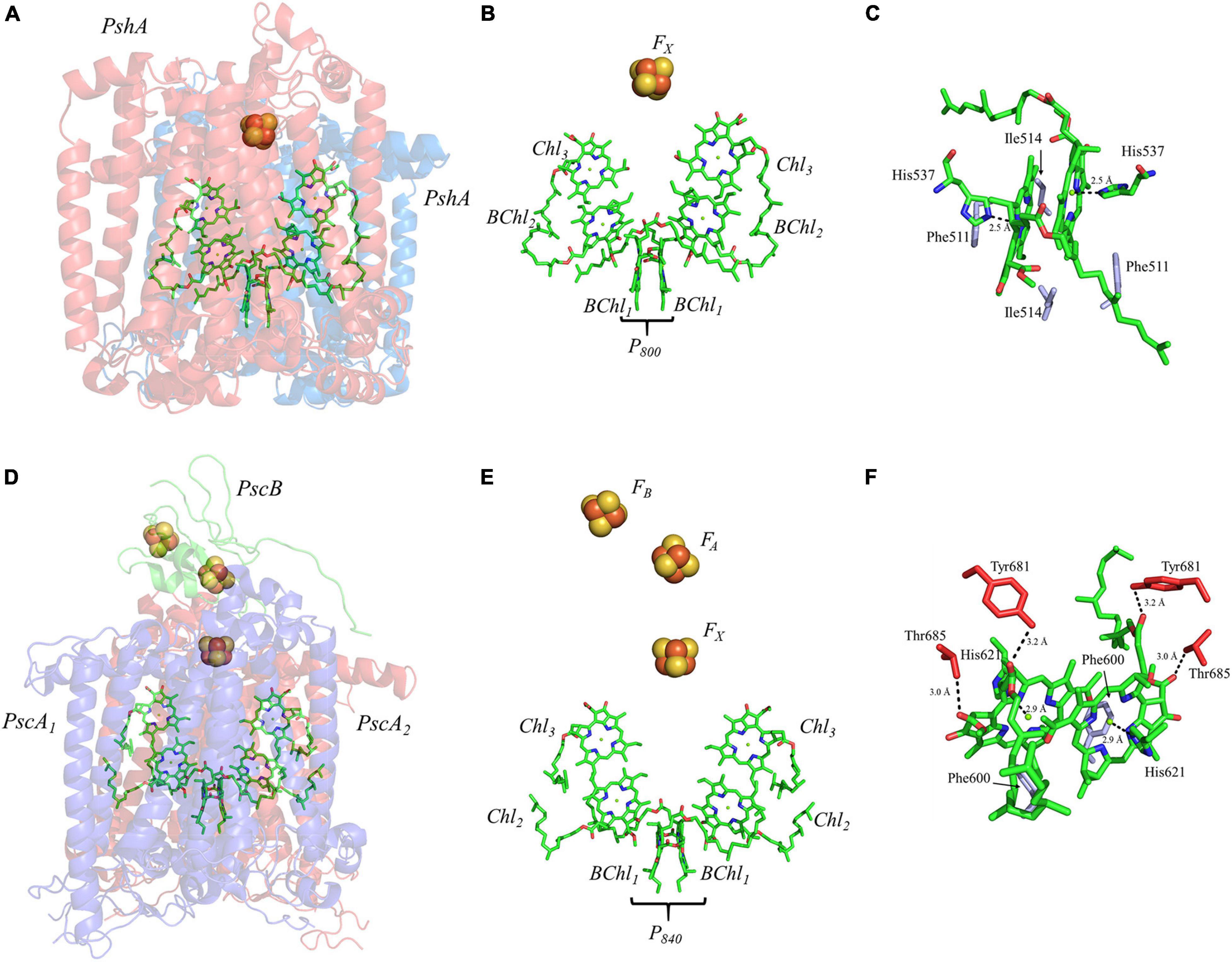
Figure 8. (A) The core subunits (PshA, Blue and Red) and cofactors of the HbRC from H. modesticaldum as observed by X-ray crystallography (PDB ID: 5v8k), (B) the cofactors that participate in the primary electron transfer pathway of the HbRC and (C) the binding pocket of the primary donor, P800. (D) The core subunits (PscA1, blue; PscA2, red; and PscB, green) and cofactors GsbRC from C. tepidum as observed by cryo-electron microscopy (PDB ID: 6m32), (E) the cofactors that participate in the primary electron transfer pathway of the GsbRC and (F) the binding pocket of the primary donor, P840. The residues in part (C,F) that are providing hydrogen bonds are shown in red, and nearby non-polar or π-stacked residues are colored in gray.
The mechanism(s) of primary charge separation in the HbRC has been the focus of several recent studies. The unusual pigment composition of the HbRC, where multiple types of (B)Chl molecules are involved in electron transfer, lends itself particularly well to the direct observation of the transient excited states using optical spectroscopic methods. Ultrafast pump-probe spectroscopy was initially employed to study energy transfer amongst the various pools of BChl molecules as well as the initial states of charge separation, suggesting a highly coupled system of early BChl molecules (Kojima et al., 2020). Subsequently, global analysis of two-dimensional electronic spectra suggested multiple pathways of charge separation. The direct photoexcitation of the trap was suggested to generate the excited state [BChl2/Chl3]∗, which then extended to [P800/BChl2/Chl3]∗ in ∼ 90 fs and led to the formation of the first charge-separated state, [P800/BChl2]⋅–Chl3⋅+, within 900 fs (Song et al., 2021). Alternately, the excitation of the antenna system led to the formation of the initial charge-separated state, [P800/BChl2]⋅–Chl3⋅+, within 2.2 ps. Regardless of which mechanism is involved, the final charge separated state, P800⋅+A0⋅–, was formed within 20–25 ps, which is in agreement with previous observations (Chauvet et al., 2013; Kojima et al., 2020). Once the stable P800⋅+A0⋅– state is formed in the HbRC, the electron is transferred to the FX cluster in 600–800 ps (Nuijs et al., 1985a; Chauvet et al., 2013).
Much like PS I, the primary donor of the HbRC, P800, is composed of an epimer of the primary pigment of the RC, which is BChl g’ (Kobayashi et al., 1991). The P800 dimer may be the most interesting of all the cases discussed here, not because it contains extensive protein-matrix effects like those observed for P700, but rather due to the lack of apparent protein matrix effects from the surrounding environment. While FTIR spectroscopy had previously suggested that a cysteine residue, either Cys469 or Cys601, may be hydrogen bonded to the primary donor itself (Noguchi et al., 1997), the high-resolution X-ray crystal structure of the HbRC has revealed that this is not the case (Gisriel et al., 2017). Instead, the differential signal at 2550/2560 cm–1 that was observed in the FTIR spectra was likely due to Cys601, a residue that is in close proximity to the axial His ligands, His537, of P800. Indeed, there are no apparent hydrogen bonds that have been observed in either of the BChl g’ molecules of the HbRC (Figure 8C). Also lacking are any effects from neighboring water molecules. The only observable residues are Ile514 that resides in the non-overlapping region of the BChl g’ macrocycles and a nearby Phe511 residue within 4.4 Å that is oriented in a pseudo π-stacked fashion.
Several facets of the inter-cofactor distances and relative macrocycle orientation further showcase the unique nature of the BChl g’ dimer of P800. The Mg–Mg distance is the shortest of all known RCs, at 5.7 Å, which results in a significant amount overlap of the BChl g’ macrocycles (Figure 9A). Moreover, while the macrocycles maintain a roughly parallel orientation, there is a minor bend in each BChl, which causes the distance between the rings to vary from 3.1 to ∼ 3.5 Å, the former being the shortest distance between ring planes seen thus far. An interesting artifact of the macrocycle structure is reflected in the distance between the nearest nitrogen atoms of the rings. Not only is N3–N3 distance of 3.1 Å representative of the closest point between the two macrocycles, the distance between N2–N2 and N3–N2 for each ring is identical at 4.7 Å (the distance parameters for homodimeric RCs are summarized in Table 1). This suggests a highly coupled dimer resulting in nearly symmetric electron density on each macrocycle. The extensive overlap of the two macrocycles of P800 is evident in the space-filling model shown in Figure 10A, with a tail orientation typical of Type I RCs.
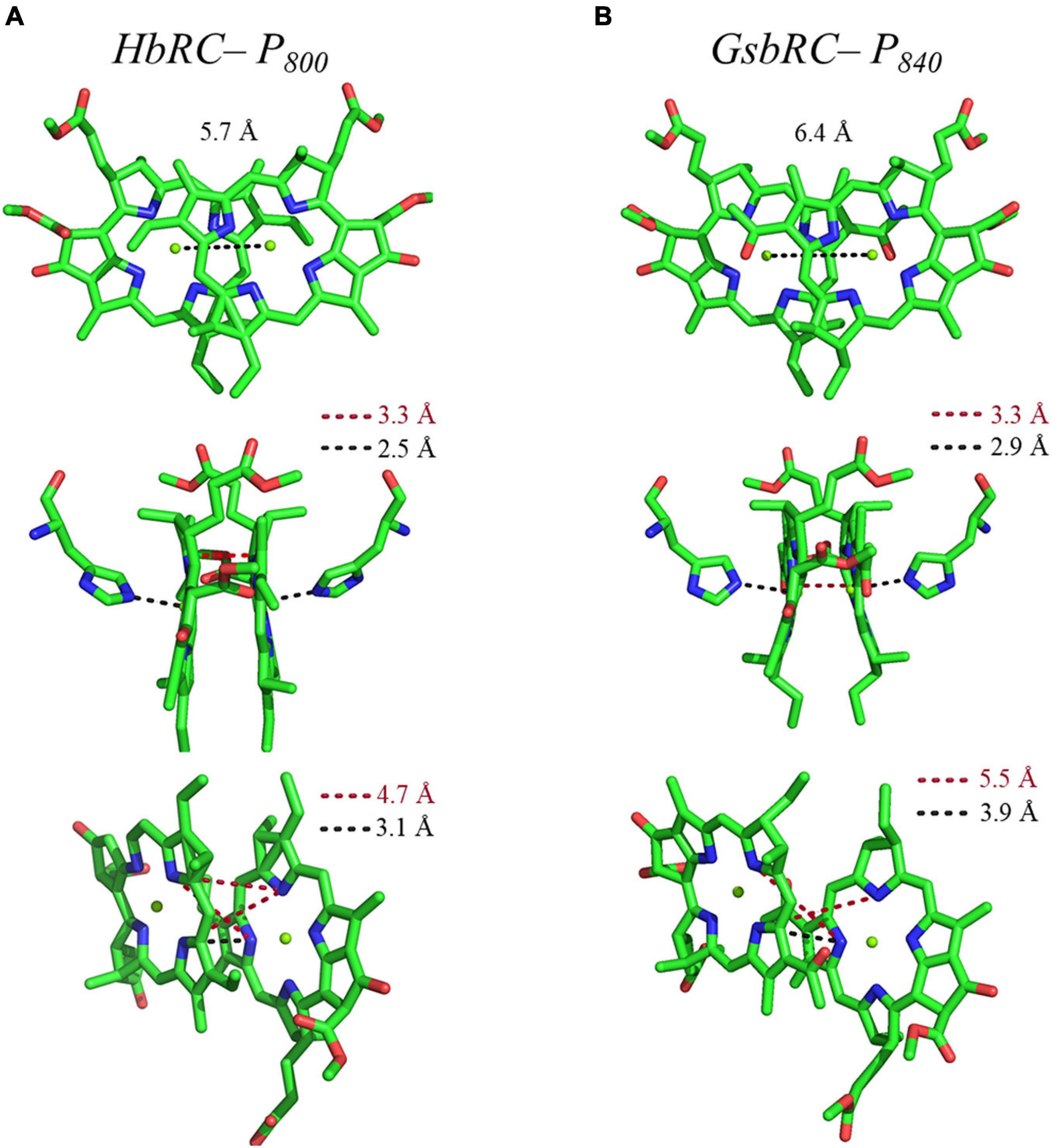
Figure 9. Analysis of inter-cofactor distances and relative orientation of primary donors from the homodimeric RCs: (A) P800 from the HbRC, and (B) P840 from the GsbRC.
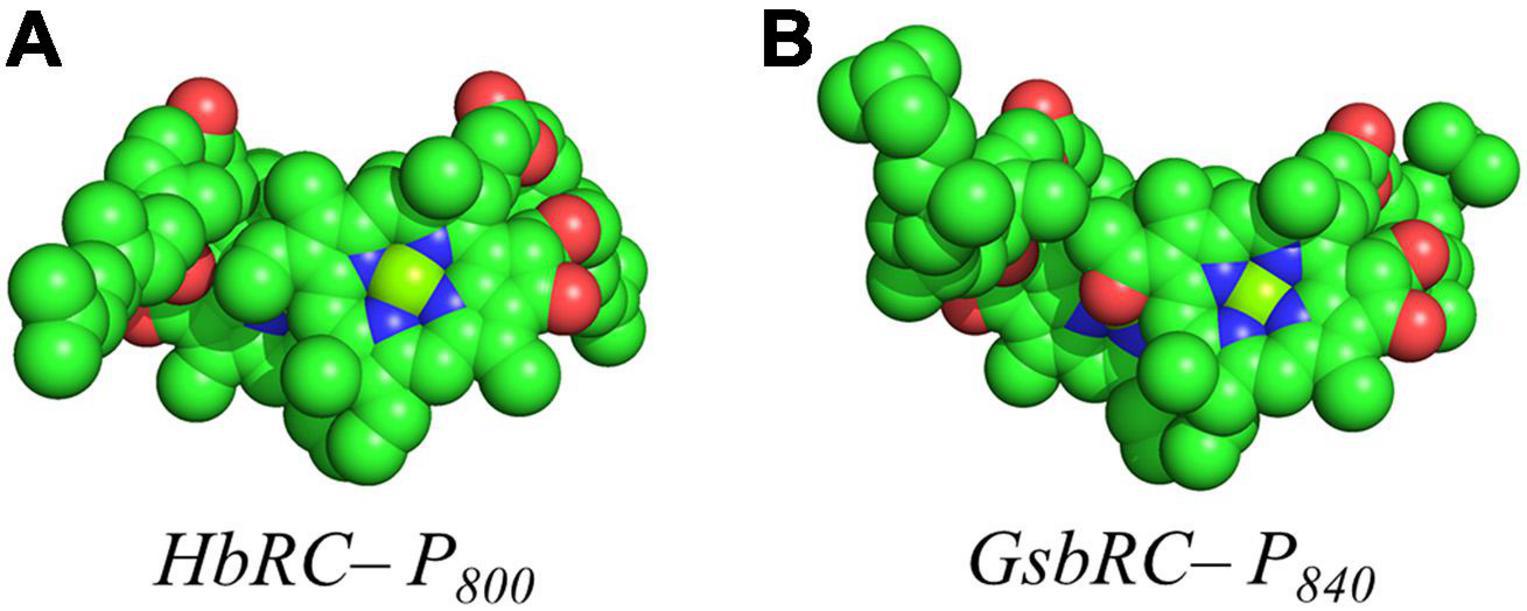
Figure 10. Space filling models of (A) P800 from the HbRC and (B) P840 from the GsbRC.
The Primary Donor, P840, of Chlorobaculum tepidum
The RC from the green sulfur bacterium C. tepidum, GsbRC, is a homodimer of the membrane-bound PscA polypeptide subunit (Figures 8D,E), which is encoded by a single gene, pscA (Hager-Braun et al., 1999; Hauska et al., 2001). The GsbRC core also includes PscB, a polypeptide analogous to PsaC in PS I, however, this polypeptide subunit is less tightly bound than PsaC and can be removed with detergent treatment (Miller et al., 1992; Schmidt et al., 2000) or at relatively low salt concentration (Miller et al., 1992; Jagannathan and Golbeck, 2008). While the polypeptide core and accompanying cofactors of the GsbRC should be considered a homodimer, the symmetry is broken by the PscB polypeptide, whose arbitrary orientation (in conjunction with PscD) directs the binding of the FMO-1, -2, and -3 proteins on the cytoplasmic side of the membrane (Chen et al., 2020). The electron-transfer cofactors reside within the homodimeric core and are comprised of two BChl molecules that constitute the primary donor, P840, four Chl a molecules, and three [4Fe–4S] clusters, FX, FA, and FB. Similar to the HbRC, note the absence of any quinones between Chl3 and the [4Fe–4S] clusters. For a detailed review of the GsbRC, please see (Hauska et al., 2001).
Charge separation in the GsbRC appears to follow a pattern similar to that of other Type I RCs. Time-resolved transient absorption spectroscopy at various temperatures has suggested that a highly electronically coupled system of (B)Chls are responsible for energy trapping and primary charge separation (Neerken et al., 1998, 1999). It should be noted that similar data is not available for the homodimeric HbRC, hence, a detailed comparison of the mechanism is not possible here. Regardless, photoexcitation of the trap appears to be complete within 2 ps, allowing charge separation to occur within 25 ps (Francke et al., 1996; Neerken et al., 1998, 1999), a lifetime that is nearly identical to the HbRC. Optical spectroscopy on the picosecond time scale showed that subsequent electron transfer from A0⋅– to FX occurs within 600 ps (Nuijs et al., 1985b; Shuvalov et al., 1986), notably slower than A0⋅– oxidation in PS I. Electron transfer then proceeds linearly through the two [4Fe–4S] clusters, FA and FB, that are bound by the PscB subunit in <5 μs (Hauska et al., 2001).
Similar to PS I and HbRC, the primary donor of the GsbRC is comprised of a 132-epimer, BChl a’ (Kobayashi et al., 2000). Examination of the primary donor of the GsbRC in Figure 8F reveals that the protein-matrix effects are much more pronounced than those of the analogous homodimeric RC, the HbRC, from H. modesticaldum. As discussed recently, the markedly lower resolution of the cryo-electron microscopy structure (as compared to other X-ray structures) suggests that assignments of protein matrix interactions should be considered tentative (Gisriel et al., 2021). There exists a total of four hydrogen bonds, two on each BChl macrocycle, provided by the Tyr681 and Thr685 residues (Figure 8F). However, as this is a homodimeric RC, it is expected that the effects of these interactions will impact the redox properties of each BChl a molecule equally and only serve to alter the redox potential of the primary donor, P840, as opposed to influencing the localization of the charge density. Another interesting feature of the protein matrix is the identity of the non-polar residues that fill the non-overlapping region of the macrocycles. While these tend to be amino acid residues with a short aliphatic side chain, such as Val and Ile in both heterodimeric and homodimeric RCs, there are aromatic residues in the GsbRC, e.g., Phe600. The side chain of the Phe residues are orthogonal to the BChl a macrocycles, suggesting that there are no π-stacking interactions present. However, the aromatic residues are within a distance of 3.1 Å of the BChl a macrocycles and are likely to impact the redox properties of the P840 dimer.
The inter-cofactor distances and relative orientation of the BChl a macrocycles of the primary donor, P840, appear to be similar to P700 and P800 of PS I and the HbRC, respectively. The Mg–Mg distance of 6.4 Å for P840 (Figure 9B, Top Panel) is close to that of the P700 donor of PS I (6.3 Å), resulting in significant overlap of the two macrocycles, including both the pyrrole rings of N3 and to a lesser extent the pyrrole ring associated with N2. However, when viewed along the ring planes, it is clear that the macrocycles remain in a roughly parallel orientation, but adopt a non-planar or domed shape. While analogous to the HbRC, the deviation from planarity is much more extensive in P840. This bending results in a slight deviation in the identity of the nearest nitrogens of the macrocycles, where N3s remain proximal to each other at 3.9 Å, and both N2s are proximal to the opposing N3, at 5.4 and 5.5 Å. The space-filling model shows overlapping rings and a tail orientation that shares a high similarity to that of P700 (Figure 10B).
Electronic Structure of the Primary Donor of Heterodimeric and Homodimeric Reaction Centers
Continuous-Wave and Pulsed Electron Paramagnetic Resonance Spectroscopy Measurements
Continuous-wave (CW) and pulsed electron paramagnetic resonance (EPR) spectroscopy has been used for the study of the electronic structure and function of (B)Chl cofactors of RCs (Commoner et al., 1956; Käβ et al., 1995, 1996; Käβ and Lubitz, 1996). As described above, developments in protein crystallization and X-ray crystallography led to outstanding high-resolution structures of PS I, PS II, the bRC and HbRC from cyanobacteria, purple bacteria and heliobacteria, respectively (Stowell et al., 1997; Jordan et al., 2001; Umena et al., 2011; Gisriel et al., 2017). These advances have been complemented by other techniques, such as, cryo-electron microscopy, that provided the structure of the GsbRC from green sulfur bacteria (Chen et al., 2020). While these structures have delivered insight on the geometry and relative topology of the (B)Chl dimers in the neutral ground state, spectroscopic measurements have been instrumental in determining the electronic properties of the individual primary donors. CW and pulsed EPR methods have been especially well suited to probe the structure of the oxidized primary donors as the high sensitivity and specificity of the detection of unpaired electron spin(s) has helped overcome limitations of the large size of the RCs, and the use of powder samples has largely eliminated the need for soluble or crystalline material.
Photooxidation of the primary donor, P, generates a paramagnetic center with an unpaired electron spin, S, of 1/2 which makes it possible to probe the P⋅+ state by EPR spectroscopy. The EPR signal of P⋅+ is known to display a relatively small g-anisotropy, with a featureless resonance at X-band (9.6 GHz) frequency (Commoner et al., 1956; Norris et al., 1971). One of the earliest applications of CW EPR spectroscopy demonstrated that steady-state photoaccumulation of the primary donor cation, P700⋅+, of PS I (Commoner et al., 1956), resulted in a signal at a g value of 2.0025. Subsequently, Norris and coworkers showed that the EPR signal of P700⋅+ arises from a strongly excitonically coupled Chl a dimer (Norris et al., 1971; Davis et al., 1993; Mac et al., 1998; Käβ et al., 2001) with a line width that is narrower than that of monomeric Chl a⋅+ in vitro (Norris et al., 1971). This observation provided initial evidence for the delocalization of the unpaired electron spin across the dimeric Chl1A/1B cofactors of P700⋅+ (Norris et al., 1971). The line width of the excitonically coupled dimeric Chl1A/1B molecules of P700⋅+ is related to the width of a monomeric cation signal through the relationship, ΔHN = 1/√N⋅ΔHM (where ΔHN and ΔHM are the line width of the multimeric chlorophyll cation with spin delocalization, ChlN⋅+, and a monomeric Chl⋅+ cation, respectively). The presence of a narrower line width, and hence the possible delocalization of the unpaired electron spin, was also observed in the primary donor, P680⋅+ and P865⋅+, of PS II and the bRC from Rba. sphaeroides, respectively (van Gorkom et al., 1974; Davis et al., 1979).
Although the g-anisotropy of an oxidized primary donor, P⋅+, is not resolved at X-band (9.6 GHz) frequency, it is possible to observe the anisotropy at higher EPR frequencies (Webber and Lubitz, 2001). One of us has previously demonstrated that the g-anisotropy of P700⋅+ can be resolved at D-band (130 GHz) EPR frequency using perdeuterated PS I from Synechococcus lividus (Poluektov et al., 2002). These studies suggested that the g-anisotropy of P700⋅+ is smaller than that of monomeric Chl a⋅+ in vitro, which was explained by the delocalized electronic character of P700⋅+ or a heteromeric model of the primary donor. Using higher EPR frequencies, Angerhofer, Redding and coworkers determined the g-anisotropy of P700⋅+ of PS I from Chlamydomonas reinhardtii (Petrenko et al., 2004) and higher plants (Bratt et al., 1997, 2000), which suggested that the electron spin distribution of P700⋅+ may be more monomeric than dimeric. A similar comparison of the g-anisotropy of P865⋅+ of the bRC from Rba. sphaeroides with monomeric BChl a⋅+ in vitro also revealed little difference in the respective line widths (Burghaus et al., 1993). However, these results were in contrast with previous experimental and computational studies. Thus, it appeared that the magnitude of g-anisotropy of the primary donor cation, P⋅+, alone was not sufficient to provide definitive evidence on the extent of the delocalization of the electron spin over the (B)Chl molecules.
In principle, the direct measurement of magnetic hyperfine couplings between the unpaired electron spin and NMR-active nuclei could reveal the distribution of the electron spin density and hence, the electronic structure of the Chl1A/1B dimer in the oxidized primary donor, P⋅+. However, it is not possible to measure the electron-nuclear hyperfine interactions using CW EPR spectroscopy due to the inhomogeneous broadening of the peaks in the spectrum. Therefore, hyperfine spectroscopy methods, such as ENDOR, electron-spin-echo envelope modulation (ESEEM) and two-dimensional (2D) hyperfine sub-level correlation (HYSCORE) spectroscopy, in conjunction with computational modeling, have been used to obtain information on the electronic structure of the oxidized primary donors (Britt, 1993; Deligiannakis et al., 2000; Lakshmi and Brudvig, 2000, 2001; Prisner et al., 2001; Harmer et al., 2009). The electron-nuclear hyperfine interactions determined by both experimental and computational methods have been used to identify the CW EPR signals, characterize the surrounding environment and coordination geometry, and quantitatively determine the hyperfine couplings and electron spin density distribution of the oxidized primary donors, P⋅+, in RCs. These measurements have included hyperfine interactions of the unpaired electron spin of the oxidized primary donor, P⋅+, with NMR-active nuclei, such as 1H, 13C, 15N and 14N of the (B)Chl molecules.
Electron-nuclear double resonance spectroscopy is a double resonance technique with reasonably high spectral resolution that has typically allowed for the measurement of small hyperfine couplings. Initial ENDOR spectroscopy studies of P⋅+ utilized CW radio-frequency irradiation, although this was replaced by pulsed methods, such as, Mims and Davies ENDOR, which have the advantage of more selective measurement of the nuclear hyperfine couplings (Hoffman, 2003; Kulik and Lubitz, 2009). More recently, ESEEM spectroscopy has been applied to the study of the oxidized primary donor, P⋅+, as it overcomes the inhomogeneous broadening of EPR resonances and provides access to unresolved electron-nuclear hyperfine couplings (Deligiannakis et al., 2000). The hyperfine couplings that are measured by ENDOR and ESEEM spectroscopy are orientation dependent, which means that information on the hyperfine anisotropy is lost in powder EPR samples with random orientations. Therefore, the hyperfine measurements of P⋅+ have also been performed on single-crystals of RCs or at high EPR frequencies, where the g-anisotropy of P⋅+ is better resolved making it possible to select for specific orientations of the molecules with respect to the applied magnetic field.
The nuclear transitions of multiple abundant spins, such as, 1H or 14N, in powder samples or frozen solutions have often been difficult to resolve by one-dimensional ENDOR and ESEEM spectroscopy due to spectral overcrowding of signals. Hence, we and others have been employing HYSCORE spectroscopy, which is a two-dimensional version of ESEEM, to obtain correlations between nuclear transitions to facilitate the detection and assignment of multiple hyperfine-coupled proton and nitrogen atoms of the oxidized primary donors, P⋅+, in two-dimensional frequency space (Höfer et al., 1986). All three of the hyperfine methods, ENDOR, ESEEM and HYSCORE, are highly sensitive as the electron spin-coupled nuclear transitions are monitored through the observation of a paramagnetic electron spin (Webber and Lubitz, 2001). Additionally, these methods are versatile as they can detect weak hyperfine couplings to less sensitive nuclei with smaller magnetic moments, such as, 14N and 15N atoms.
The hyperfine interactions of protons (1H), carbon (13C) and nitrogen (both 14N and isotope-labeled 15N) atoms of the oxidized primary donor, P700⋅+, P865⋅+, P960⋅+, P680⋅+, and P840⋅+ of the heterodimeric RCs, PS I, the bRC from Rba. sphaeroides and Rps. viridis, PS II and the homodimeric RCs, GsbRC from Chlorobium limicola and Chloroacidobacterium thermophilum (Cab. RC) (Charles et al., 2020), respectively, have been determined by pulsed EPR spectroscopy methods. As shown in Figures 4C,F, 7C,F, 8F, the primary donors of PS I, PS II, the bRCs and GbRC are comprised of a dimer of (B)Chl1A and (B)Chl1B molecules (Stowell et al., 1997; Jordan et al., 2001; Umena et al., 2011). Earliest estimates of the hyperfine coupling parameters of P700⋅+ of PS I were obtained by Dikanov, Astashkin and coworkers using ENDOR and ESEEM spectroscopy of the nitrogen atoms (both 14N and 15N) of the Chl a and Chl a’ macrocycles interacting with the unpaired electron spin (Dikanov et al., 1983; Astashkin et al., 1987). It was observed that the unpaired electron spin of P700⋅+ displayed magnetic interactions with four nitrogen atoms, where two of the nitrogens were strongly hyperfine coupled with an isotropic hyperfine coupling, Aiso, of ∼ 2 MHz, and two other nitrogens were weakly coupled with a much smaller Aiso of ∼ 0.2 MHz. While the early results were semi-qualitative, they established the feasibility of hyperfine measurements on P700⋅+ and highlighted the need for measurements that would yield higher spectral resolution.
Subsequently, the unpaired electron spin density distribution of the oxidized primary donors was determined by quantitative measurements of the hyperfine interactions of the protons and nitrogen atoms, which were compared with those of monomeric Chl a⋅+ and BChl a⋅+ in vitro (Astashkin et al., 1988). There is broad consensus in the field that the oxidized heterodimeric primary donors are comprised of a dimer of coupled (B)Chl molecules, albeit with small differences on the extent of asymmetry of the electron spin delocalization across the dimer. The asymmetric spin density distribution (ratio 3:1 to 5:1) of P700⋅+ has been observed by 1H, 14N and 15N ENDOR, HYSCORE and single crystal EPR and ENDOR measurements (Käβ et al., 1995; Käβ and Lubitz, 1996; Deligiannakis and Rutherford, 2001; Käβ et al., 2001; Webber and Lubitz, 2001; Chestnut et al., 2021). Recently, using 14N HYSCORE spectroscopy we conclusively established that there are at least four distinct 14N atoms (and likely more than four nitrogen atoms if we consider the possibility of overlapping cross-peaks in the spectrum) that are interacting with the unpaired electron spin of P700⋅+ (Gorka et al., 2021b). The isotropic hyperfine couplings, Aiso, range from 1.4–2.8 MHz, indicating that the electron spin is distributed on at least four nitrogen atoms. In conjunction with previous and current findings, this indicates that P700⋅+ is comprised of a strongly electronically coupled Chl a dimer, where the unpaired electron spin density distribution is asymmetric over the two Chl a molecules (Käβ et al., 1995; Käβ and Lubitz, 1996; Deligiannakis and Rutherford, 2001; Käβ et al., 2001; Webber and Lubitz, 2001; Chestnut et al., 2021). A summary of the experimental hyperfine and quadrupolar couplings of the nitrogen atoms of P700⋅+ is presented in Table 2.
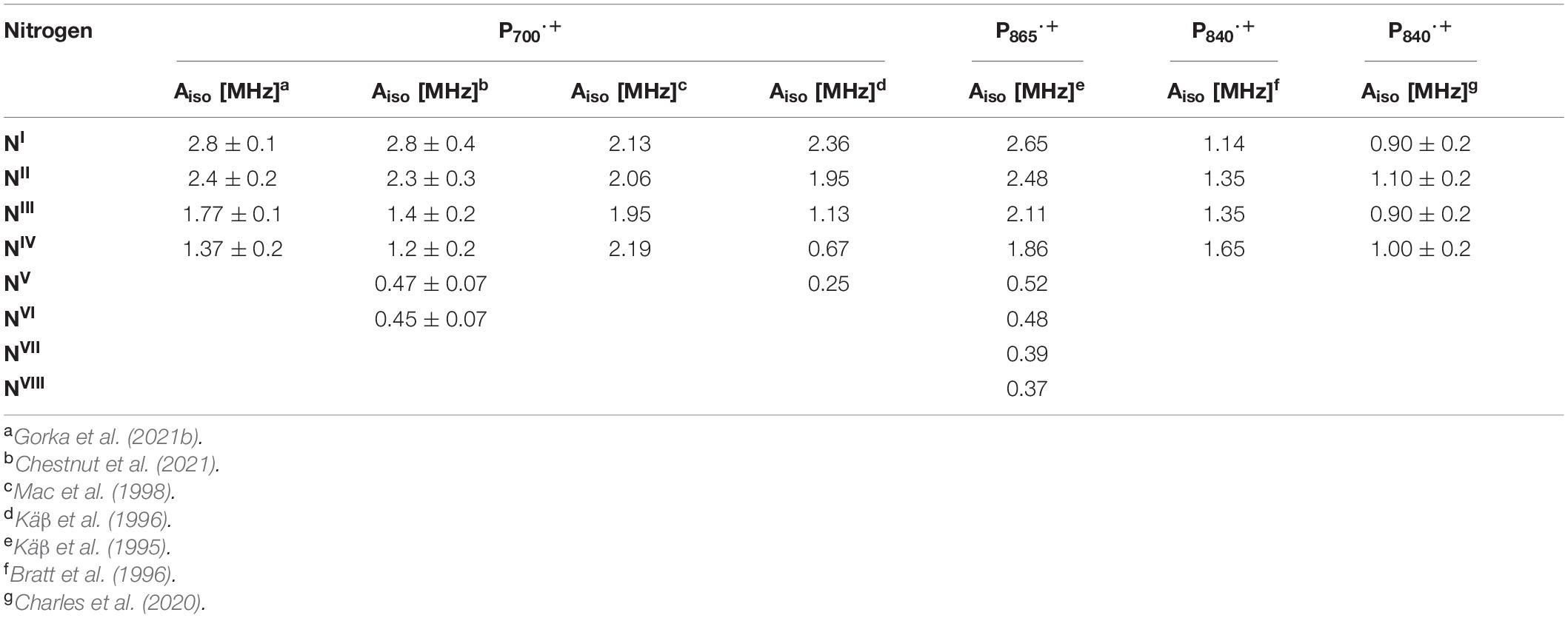
Table 2. Experimental 14N isotropic hyperfine couplings of the nitrogen atoms of P700⋅+, P865⋅+, and P840⋅+ of the GsbRC and Cab. RC.
Early experimental measurements of P865⋅+ of the bRC from Rba. sphaeroides had determined two or four nitrogen hyperfine coupling tensors, which were on average smaller than those of the monomeric BChl a⋅+ in vitro by a factor of 2 (Lubitz et al., 1984). At the time, this seemed adequate as only four reduced hyperfine couplings were expected for the nitrogen atoms in a symmetric BChl dimer model of P865⋅+. Consequently, the first ENDOR and ESEEM spectroscopy studies were interpreted on the basis of an essentially symmetric spin density distribution across BChl1A and BChl1B of P865⋅+ (Lubitz et al., 1984; Lin et al., 1986; Astashkin et al., 1988). A detailed 1H ENDOR study performed on single crystals of the bRC from Rba. sphaeroides near room temperature showed that the 3:1 electron spin density distribution in favor of the Chl1A (Lendzian et al., 1993), which was also observed for P960⋅+ of the bRC from Rps. viridis (Lendzian et al., 1988). This observation was further supported by the determination of 15N hyperfine couplings under similar conditions using 15N-labeled single crystals of the bRC (Lendzian et al., 1992). However, the complete hyperfine tensors were not determined in these experiments and hence, the assignment of the hyperfine couplings was only possible by comparison with molecular orbital calculations. These results were further confirmed by ESEEM measurements of P865⋅+ and P960⋅+ of the bRC from Rba. sphaeroides and Rps. viridis (Davis et al., 1993). Subsequently, a combined ESEEM and HYSCORE spectroscopy study of monomeric 15N-labeled BChl a⋅+ in solution and P865⋅+ in I5N-labeled bRC from Rba. sphaeroides yielded a much higher asymmetry of the spin density ratio of approximately 5:1 (Käβ et al., 1995; Table 2).
Proton ENDOR and HYSCORE spectroscopy have also been employed for the study of the primary donor cation, P680⋅+, in core preparations of PS II containing the D1, D2 and cytochrome b559 polypeptides (Rigby et al., 1994a). The hyperfine parameters of the protons of methyl groups obtained from 1H ENDOR spectroscopy was used to calculate the electron spin density distribution of P680⋅+. Comparison of the hyperfine parameters and electron spin density distribution of P680⋅+ with the Chl a⋅+ monomer in vitro indicated an apparent reduction in the unpaired electron spin density for P680⋅+. Similar to P700⋅+ and P865⋅+, these studies suggested that P680⋅+ was a weakly coupled Chl1A/1B dimer with 82% of the unpaired electron spin located on one of the chlorophyll molecules. Based on the experimental hyperfine parameters of P700⋅+, P865⋅+, P960⋅+, and P680⋅+, it is evident that the asymmetric spin density distribution that is observed does not appear to be caused by the structural difference of the (B)Chl molecules of P⋅+ in heterodimeric RCs, but by the interaction of the cofactors with the protein environment.
There have been far fewer reports on the primary donors of the homodimeric RC, HbRC and GsbRC. The oxidized primer donor, P840⋅+, of the GsbRC in Chlorobium limicola membranes was studied by 1H ENDOR and Triple electron-nuclear-nuclear spectroscopy (Rigby et al., 1994b). These studies showed that P840⋅+ is comprised of a BChl a dimer with a highly symmetrical distribution of electron spin density between BChl1A and BChl1B. Moreover, Triple spectroscopy resolved the separate contributions of the two halves of the dimeric primary donor and revealed small deviations from a 1:1 electron spin density distribution. Subsequently, 14N ESEEM spectroscopy of the GsbRC in Chlorobium limicola membranes (Table 2) confirmed that the electron spin density distribution of P840⋅+ is shared equally between the two BChl a molecules (Bratt et al., 1996). In these studies, P840⋅+ was found to be closest yet to the symmetrical ‘dimer’ that was originally thought to exist in bRCs.
Most recently, we probed the oxidized primary donor, P840⋅+, of Chloroacidobacterium (Cab.) thermophilum with 14N and 67Zn HYSCORE spectroscopy (Charles et al., 2020). Cab. thermophilum is a microaerophilic, chlorophototrophic species in the phylum Acidobacteria that employs a homodimeric RC with BChl molecules. The Cab. RC is highly unusual, as pigment analyses have shown the presence of three (B)Chl molecules, BChl aP, Chl aPD, and Zn2+-BChl aP′, in the ratio 7.1:5.4:1 (He et al., 2019). While Chl aPD was shown to be the primary electron acceptor, we demonstrated that the primary electron donor, P840, contains a dimer of Zn2+-BChl aP′ molecules. The 14N and 67Zn hyperfine couplings (Table 2) and DFT calculations have indicated that the electron spin density is distributed nearly symmetrically over the two Zn2+-(B)Chl aP′ molecules of P840⋅+ (Charles et al., 2020), as expected in a homodimeric RC. To our knowledge, this is the only example of a photochemical RC in which the (B)Chl molecules of the primary donor are metalated differently from those of the antenna.
Computational Studies of the Spin Density Distribution
The electronic structure of the oxidized primary donors, P700⋅+, P680⋅+, P865⋅+, and P960⋅+, of the heterodimeric RC, PS I, PS II and the bRC, have been probed by semi-empirical molecular orbital, quantum mechanics/molecular modeling (QM/MM) and density functional theory (DFT) methods. Early RHF-INDO/SP calculations by Lubitz and coworkers suggested that P700⋅+ of PS I is formed by a dimer of Chl a and Chl a’ molecules, Chl1A and Chl1B, respectively, with an asymmetric charge and electron spin density distribution in favor of the Chl a half of the dimer (Käβ et al., 1995; Webber and Lubitz, 2001; Plato et al., 2003). The predicted asymmetry of the charge and spin density distribution across Chl1A and Chl1B was in agreement with previously reported EPR and ENDOR spectroscopy studies (Käβ et al., 2001). Interestingly, the stepwise inclusion of the electrostatic interactions of the Chl1A and Chl1B molecules of P700⋅+ with the neighboring amino acid residues, such as, Thr743PsaA, which forms a putative hydrogen bond with the keto group of Chl a’, and His680PsaA and His660PsaB, serving as axial ligands to the Mg atoms of Chl1A and Chl1B, respectively, led to systematic enhancement of the electronic asymmetry that yielded a spin density ratio of almost 5:1. Molecular orbital calculations indicated that hydrogen bonding specifically stabilized the Chl1B molecule of the dimer, which suggested that the unpaired electron of P700⋅+ would predominantly reside at this site. This was corroborated by the DFT calculations performed by Sun and coworkers, who found that the asymmetry of the spin density of P700⋅+ was mainly due to the hydrogen bond to the 131-keto-O group of Chl1A (Sun et al., 2004). More recently, using QM/MM methods Saito and Ishikita (Saito and Ishikita, 2011) estimated the asymmetry of spin density of P700⋅+ as 22.4:77.6 in favor of the Chl1B molecule. This ratio was in good agreement with the experimental value of 25:75–20:80 that was obtained for the spin density distribution for P700⋅+ of PS I from spinach (Davis et al., 1993) and 15:85 for P700⋅+ of PS I from Thermosynechococcus elongatus (Käβ et al., 2001). The general consensus has been that there are three factors that significantly contribute to a larger spin population of Chl1B relative to that of Chl1A, namely, (i) the presence of the Thr743PsaA residue that forms a hydrogen bond with the 131-keto-O group of Chl1A, which is absent in Chl1B, (ii) the identity of Chl1A as a Chl a epimer, which leads the methyl ester group of Chl1A and Chl1B to be oppositely oriented with respect to the chlorin plane and (iii) the conserved pair of Arg750PsaA and Ser734PsaB residues that interact with Chl1A and Chl1B.
The charge and spin density distribution of P680⋅+ of PS II has also been studied by DFT and QM/MM methods. The charge distribution determined by natural population analysis indicated that the positive charge of P680⋅+ was significantly delocalized over the two Chl a molecules, Chl1A and Chl1B, with a slight bias in favor of the Chl1B molecule (ratio of 0.46:0.54) (Takahashi et al., 2008). However, the charge delocalization, and similar spin density distribution, on Chl1A and Chl1B of P680⋅+ in this study (Takahashi et al., 2008) was in contrast with experimental observations that the charge is mostly localized on one of the Chl a molecules, Chl1A, of P680⋅+ (Rigby et al., 1994b). More recently, the delocalization of the charge and spin density across the Chl1A and Chl1B dimer of P680⋅+ was studied by Saito and coworkers using QM/MM methods (Saito and Ishikita, 2011). The Chl1A:Chl1B charge and spin density distribution was found to be 76.9:23.1 and 80.6:19.4, respectively, based on the complete structure of PS II that was obtained from the 1.9 Å X-ray crystal structure (Umena et al., 2011). The calculated spin density distribution was more asymmetric than the charge delocalization in the previous QM/MM study, which was also observed in computational studies of the other oxidized heterodimeric primary donors. The ratio of the spin density distribution on the Chl1A and Chl1B dimer of 80.6:19.4 was in agreement with the ratio of 82:18 that was obtained from experimental 1H ENDOR spectroscopy of P680⋅+ of PS II from spinach (Rigby et al., 1994a) and the ratio of 80:20 from flash-induced spectroscopic studies of PS II from Synechocystis sp. PCC 6803 (Diner et al., 2001). This suggested a preferential localization of the cationic state on Chl1A over Chl1B irrespective of the homology of the protein sequences between the D1 and D2 polypeptide subunits of PS II. Interestingly, the removal of the protein subunits of PS II in the QM/MM calculations yielded an isolated Chl1A:Chl1B ratio of 57.5:42.5 in vacuum. The significantly lower ratio of Chl1A:Chl1B in the absence of the polypeptide subunits, in comparison with that obtained in their presence, suggested that the remarkable asymmetric distribution of the cationic state among Chl1A and Chl1B of P680⋅+ was not due to the geometry of the Chl molecules but due to the asymmetric protein environment provided by PS II. Based on this observation, it was concluded that the Chl1A:Chl1B ratio of ∼ 80:20 was mainly due to the difference in the amino acid residues that were interacting with Chl1A and Chl1B of P680.
Initial semi-empirical RHF-INDO/SP calculations of the 15N hyperfine couplings of P865⋅+ of the bRC had yielded a spin density ratio of 1.8:1 across the BChl1A and BChl1B dimer, which appeared to be in agreement with the early 1H ENDOR data (Lendzian et al., 1992). However, later ENDOR, ESEEM and HYSCORE spectroscopy measurements of both single-crystal and powder samples of the bRC from Rba. sphaeroides and Rps. viridis indicated that the ratio of the electron spin density distribution is 3:1 or 5:1 in favor of BChl1A (Lendzian et al., 1988, 1992, 1993). Subsequent semi-empirical molecular orbital calculations by Lubitz and coworkers on the oxidized primary donor, P865⋅+, using RHF-INDO/SP methods (Käβ et al., 1995) reproduced the experimental isotropic hyperfine couplings to within 10% error, which facilitated the assignment of the hyperfine couplings of all of the nitrogen atoms of the BChl1A and BChl1B dimer and a ratio of the electron spin density distribution of 5:1 was deduced from the measured and assigned hyperfine couplings of the 15N atoms of P865⋅+. Most recently, the asymmetry of the spin density distribution and electronic coupling of the (B)Chl molecules of the oxidized primary donor, P865⋅+, from Rba. sphaeroides, as well as P700⋅+ and P680⋅+, of PS I and PS II from Synechococcus elongatus, and Thermosynechococcus vulcanus, respectively, was estimated by frozen-density embedding diabatic (FDE-diab) methodology (Artiukhin and Neugebauer, 2018). The calculated ratio of spin densities was in agreement with previous experimental results for PS II and the bRC, where 82% and 66% of the spin density of P680⋅+ and P865⋅+ was found to be located on the Chl1A molecule.
Density functional theory and QM/MM methods are powerful tools for the investigation of the electronic structure of the oxidized primary donors. As described above, there are several reports in literature on computational studies of P700⋅+, P680⋅+, P865⋅+, and P960⋅+ of PS I, PS II and the bRC from Rba. sphaeroides and Rps. viridis, respectively. However, to our best knowledge quantum-mechanical calculations to determine the hyperfine and quadrupolar parameters of the 14N atoms of the primary donors of the homodimeric RCs, with the exception of the Cab. RC (Charles et al., 2020), are lacking at this time. This may be because the high-resolution structures of homodimeric RCs have been determined only very recently. In order to facilitate a comparison of the oxidized primary donor of hetero- and homodimeric RCs, we performed DFT calculations on computational models of P800⋅+ and P840⋅+ of the HbRC and GsbRC from heliobacteria and green sulfur bacteria, respectively. The goal is to obtain a better understanding of the effect of the relative geometry, symmetry and protein matrix effects on the electronic structure of the dimeric BChl molecules in the oxidized primary donor of homodimeric RCs.
In order to assess the computational methods that were employed in this study, we performed DFT calculations on models of P700⋅+, P680⋅+ P865⋅+, and P960⋅+ that were derived from the X-ray crystal structure of PS I (Jordan et al., 2001) (PDB ID: 1jb0), PS II (Umena et al., 2011) (PDB ID: 3wu2) and the bRC from Rba. sphaeroides (PDB ID: 1pcr) (Ermler et al., 1994) and Rps. viridis (PDB ID: 1prc) (Deisenhofer et al., 1995). The computational model of each primary donor included the dimeric (B)Chl molecules, (B)Chl1A and (B)Chl1B, the axial histidine ligands of the (B)Chls and proximal hydrogen-bonding and hydrophobic residues in the protein matrix as observed in the respective structures (Figures 4C,F, 7C,F). The dimeric models contained the complete (B)Chl molecules with the exception that the phytol tail was truncated by a methyl group after 23 carbon atoms. Please note that there may be an effect of the accessory (B)Chl molecules, (B)Chl2A and (B)Chl2B, on the spin density distribution of the oxidized primary donors, which was not investigated in this study.
The single-point energy of each model was calculated employing the hybrid-generalized gradient approximation (hybrid-GGA) B3LYP functional (Dunham et al., 1971; Becke, 1988; Lee et al., 1988) along with the special EPR-optimized EPR-II (Barone, 1995) basis set for the lighter atoms and 6-31G(d) for magnesium, respectively, for most calculations, and the valence polarization basis sets (SVP and TZVP) (Schäfer et al., 1992; Weigend and Ahlrichs, 2005; Weigend, 2006) with the decontracted auxiliary basis sets (i.e., the coulomb fitting def2/J) (Weigend, 2006) when necessary. The calculations were performed in the spin-unrestricted mode for nuclear quadrupole couplings and isotropic hyperfine interactions of pyrrole nitrogen atoms. To account for the influence of solvent effects, a model of uniform dielectric constant of solvents using the conductor-like polarizable continuum model (CPCM) was used in the calculations (Klamt and Schüürmann, 1993; Cossi et al., 2003). The CPCM used a dielectric constant, ε, of 4.0 for incorporating the effects of the protein environment in all of the DFT calculations. All of the calculations included dispersion correction using a DFT-D3 approach with Becke-Johnson damping (D3BJ). In order to estimate the effect of the exchange functional, the hyperfine tensor calculations were also performed with the hybrid-meta-GGA TPSSh (Tao et al., 2003) functional along with the chain of spheres (RIJCOSX) (Staroverov et al., 2003; Neese et al., 2009) approximation and an EPR-II (Barone, 1995) and 6-31G(d) basis set for the lighter atoms and magnesium, that were previously used in the single-point energy calculations. The DFT calculations of P700⋅+, P680⋅+, P865⋅+, and P960⋅+ were validated by comparison of the calculated and previously published experimental hyperfine coupling constants of the 14N atoms. Although the absorbance features of chromophores have been reasonably estimated through quantum mechanical calculations [e.g., the primary donor of Rba. sphaeroides (Eccles et al., 1988)], we focused on the prediction of hyperfine coupling constants for the purpose of this review article.
As a starting point for the DFT calculations, we selected a simple computational model for P700⋅+ that was comprised only of the Chl1A and Chl1B molecules. In this model, the electron spin density across the singly occupied molecular orbital (SOMO) of isolated Chl1A/1B⋅+ was distributed predominantly on the Chl1A molecule. This was in contrast with experimental findings where the electron spin was predominantly located on Chl1B (see above). To determine the effects of protein-cofactor interactions, we systematically expanded the computational model of P700⋅+ to include the axial ligands, His680PsaA and His660PsaB, putative hydrogen-bonding residues, Thr743PsaA, Tyr735PsaA, and Tyr603PsaA and the hydrophobic residues, Leu630PsaB and Leu650PsaA. As observed in Figure 4C, the protein matrix effects appear to be completely asymmetric across the Chl1A/1B dimer of P700 as the hydrogen bonding and electrostatic interactions are limited to Chl1A, while the hydrophobic effects are localized in the vicinity of Chl1B. We observed that despite the presence of the axial His ligands and putative hydrogen bonding residues, the asymmetric electron spin density distribution in the SOMO that was observed in the DFT calculations was still in favor of Chl1A, albeit there was a slight change in the distribution of the spin density within the Chl1A molecule. Interestingly, it was the introduction of the hydrophobic effects from the addition of the residues, Leu630PsaB and Leu650PsaA, which resulted in a shift of the spin density distribution in favor of the Chl1B half of the dimer of P700⋅+.
For the DFT calculations of P865⋅+ of the bRC from Rba. sphaeroides, the computational model included the Tyr210L, Val157M, His202L, Thr186L, His168M, and His173M residues that are proximal to the BChl1A and BChl1B molecules in the X-ray crystal structure (Figure 7C; Ermler et al., 1994). Similarly, the computational model of P960⋅+ of the bRC from Rps. viridis contained the Val157M, Tyr195L, His200L, Leu184L, His168M, Trp167M, His173M, and Thr248M residues (Figure 7F). We observed that the calculated electron spin density distribution in the SOMO of P865⋅+ and P960⋅+ was in favor of the BChl1A molecule, both in the absence and presence of the hydrogen-bonding and hydrophobic amino acid residues. We observed a similar trend in the DFT calculation of P680⋅+ of PS II with a model comprised of the Chl1A and Chl1B dimer with the Ser282D1, Leu182D2, Met183D1, His197D1, and His198D2 residues (Figure 4F), where the spin density in the SOMO was located on the Chl1A half of the dimer. The localization of the electron spin density on the (B)Chl1A molecule of P865⋅+, P960⋅+, and P680⋅+ was in contrast with that of P700⋅+, where the asymmetric distribution of the electron spin density was in favor of the Chl1B molecule.
The hyperfine coupling constants of the 14N atoms of the oxidized primary donors, (B)Chl1A and (B)Chl1B, of P700⋅+, P680⋅+, P865⋅+, and P960⋅+ that were obtained from the DFT calculations are presented in Table 3. In the case of P700⋅+ and P865⋅+ of PS I and the bRC from Rba. sphaeroides, respectively, a comparison of the experimental (Table 2) and calculated (Table 3) 14N hyperfine couplings indicates that the calculations corroborate the experimental values obtained by ENDOR, ESEEM and HYSCORE spectroscopy. Further, the ratio of the asymmetric spin density distribution across the (B)Chl1A and (B)Chl1B dimer of P700⋅+ and P865⋅+ that was estimated from the spin populations was 5.7:1 and 5.6:1, respectively, which was in good agreement with the approximate estimate of Chl1B:Chl1A:5:1 and BChl1A:BChl1B:5:1 that was obtained from spectroscopic measurements of P700⋅+ and P865⋅+ (Käβ et al., 1995; Käβ et al., 1995; Käβ and Lubitz, 1996; Deligiannakis and Rutherford, 2001; Käβ et al., 2001; Chestnut et al., 2021). To our best knowledge, the 14N hyperfine coupling constants of P680⋅+ are not available in literature. However, the asymmetric spin density distribution that was determined in the DFT calculations of P680⋅+ of Chl1A:Chl1B:6:1 was roughly in agreement with the ratio of Chl1A:Chl1B:80:20 in literature. The broad agreement between the calculated and experimental hyperfine coupling constants and the high asymmetry of the electron spin density distribution of the oxidized heterodimeric primary donors indicates that the DFT methods used in this study are comparable with the experimental data in literature. However, it is important to note that the Kohn-Sham DFT methods employed may have resulted in slight over delocalization of the electron spin density across the oxidized primary donors (Artiukhin and Neugebauer, 2018). Further studies are in progress to address this possibility.
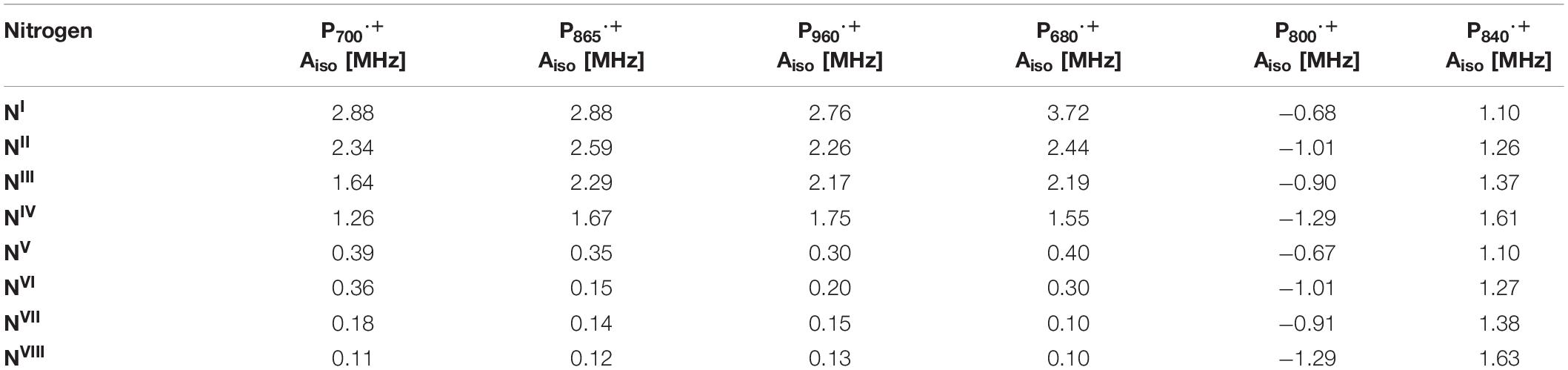
Table 3. Calculated 14N isotropic hyperfine coupling parameters of the nitrogen atoms of P700⋅+, P865⋅+, P960⋅+, P680⋅+, P800⋅+, P840⋅+ (of the GsbRC).
The DFT calculations on the oxidized heterodimeric primary donors of PS I, PS II and the bRC were consistent the magnetic parameters and asymmetric electron spin distribution over the (B)Chl dimer of P700⋅+, P680⋅+, P865⋅+, and P960⋅+. Following which, we performed DFT calculations to understand better the effects of the electronic structure and local symmetry on the hyperfine and quadrupolar parameters of the 14N atoms in the oxidized primary donors, P800⋅+ and P840⋅+, of the RC from the HbRC and GsbRC, respectively. The coordinates for the atoms in the computational models of P800 and P840 were derived from the structures of the HbRC and GsbRC from H. modesticaldum (PDB ID: 5v8k) (Gisriel et al., 2017) and C. tepidum (PDB ID: 6m32) (Chen et al., 2020), respectively. Similar to the computational models of the heterodimeric primary donors, the models for the homodimeric ones included the dimeric BChl molecules, BChl1A and BChl1B, and the axial ligands to each BChl monomer (Figures 8C,F). Once again, the models contained the complete BChl molecules with the exception that the farnesol tail was truncated by a methyl group after 23 carbon atoms. Both the HbRC and GsbRC are comprised of a homodimeric polypeptide subunit core encoded by the single genes, pshA and pscA, respectively. However, the symmetry of the homodimeric core of the GsbRC is broken by the presence of an additional PscB polypeptide (Chen et al., 2020). Additionally, while the primary donor, P840, of the GsbRC contains a symmetric BChl a’ dimer with four hydrogen-bonding residues (Figure 8F), two on each BChl molecule, P800 of the HbRC is comprised of a dimer of BChl g′ molecules that lack hydrogen-bonding interactions (Figure 8C). However, it is expected that the effects of the hydrogen-bonding interactions of P840 of the GsbRC will impact the BChl molecules equally, which will not influence the localization of the charge or spin density distribution. Since there are no residues that appear to be participating in hydrogen bonds with P800 of the HbRC, the spin density is also expected to be completely symmetric.
The DFT calculations of the oxidized primary donor models, P800⋅+ and P840⋅+, of the HbRC and GsbRC were also performed with B3LYP level of theory using an EPR-II and 6-31G(d) basis set for the lighter atoms and Mg, respectively. The calculated 14N hyperfine coupling constants (Table 3) and the spin populations of BChl1A and BChl1B that were obtained from the DFT calculations indicated that the ratio of the electron spin density distribution across the dimer of P800⋅+ and P840⋅+ of the HbRC and GsbRC, respectively, are completely symmetric. The unpaired electron spin density distribution in the singly occupied molecular orbital (SOMO) of P800⋅+ and P840⋅+ was also symmetric (Figures 11A,B). The calculated hyperfine couplings of the nitrogen atoms of P800⋅+ of the HbRC and P840⋅+ of the GsbRC (Table 3) were in agreement with the experimental couplings obtained from 14N ESEEM and HYSCORE spectroscopy measurements of P840⋅+ from Chlorobium limicola and Cab. thermophilum membranes, respectively (Table 2), respectively. This is indeed interesting as P800 of the HbRC is comprised of a BChl g’ dimer, while P840 of Cab. thermophilum has been shown to a dimer of Zn2+-BChl a′ molecules. This suggests that the protein environment surrounding P⋅+ in the HbRC and GsbRC is likely similar to that of the Cab. thermophilum RC.
Figure 11. Electron spin density distribution on the Chl1A macrocycles of the homodimeric oxidized primary donor (A) P800⋅+ of the HbRC from Heliobacterium modesticaldum (PDB ID: 5v8k) (Gisriel et al., 2017) and (B) P840⋅+ of the GsbRC from Chlorobaculum tepidum (PDB ID: 6m32) (Chen et al., 2020) in the singly occupied molecular orbital (SOMO) as determined through DFT calculations.
Although the symmetry of the homodimeric core in the structure of the GsbRC is broken due to the presence of the additional PscB polypeptide, it appears that this did not impact the magnetic parameters of the BChl dimer of P840⋅+. This finding is in agreement with the 1H ENDOR and HYSCORE spectroscopy studies that demonstrated that the electron distribution of P840⋅+ of the GsbRC from Chlorobium limicola was symmetrical in nature (Rigby et al., 1994b). However, there is the possibility that the small asymmetric effects from the extended protein environment, leading to minor deviations from total symmetry were not observed in the DFT calculations. This would most likely be due to the limited computational model of P840⋅+ that was adopted in this study.
The spatial arrangements of (B)Chl molecules in the primary donor of PS I, PS II, and the bRC are similar, where there are small differences in the inter-cofactor distance and relative orientation of the (B)Chl molecules. However, the protein matrix effects on the two halves of the (B)Chl dimer are highly asymmetric and this results in the localization of the spin density on a single (B)Chl molecule. We observed that the asymmetry of the spin density distribution is not solely caused by the structural differences of the (B)Chl macrocycles, but by the interaction of the cofactors with the protein environment. In contrast, these asymmetric effects are not present in homodimeric RCs, resulting in nearly equal sharing of spin density between the two BChl molecules (Figures 11A,B). The absence of asymmetric electrostatic and/or hydrophobic effects from the surrounding protein environment leads to a symmetric distribution of the charge and spin density in the homodimeric primary donors, P800⋅+ and P840⋅+. This effect is also mirrored in the distance between the macrocycles to ensure that spin density is not localized on one molecule. A direct consequence of the symmetric electronic structure of P800 and P840 is that it allows for electron transfer through both branches of cofactors with equal efficiency, which is not the case with the heterodimers examined in this study.
Comparison of the Primary Donors in Hetero and Homodimeric Reaction Centers
Overall, the DFT calculations of the hetero- and homodimeric oxidized primary donors indicated that the presence of the surrounding protein matrix has profound effects on the distribution of the electron spin density across the (B)Chl molecules. Although the spatial arrangement of (B)Chl molecules in the primary donor of PS I, PS II, and the bRC are similar, there are small differences in the inter-cofactor distance and relative orientation of the (B)Chl molecules. As expected, at larger inter-molecular separations the spin density distribution becomes more localized on a single (B)Chl molecule. The increased localization of spin density in the cationic state is caused by the absolute magnitudes of the electronic coupling that decreases exponentially with the inter-molecular separation. It appears that this asymmetry of the spin density distribution is not solely caused by the structural differences of the (B)Chl macrocycles, but by the interaction of the cofactors with the protein environment. This is perhaps most apparent in P700, where the protein environment is highly asymmetric with respect to hydrogen bonds and non-polar residues around the two Chls. Interestingly, it is the combination of both hydrogen bonds and non-polar residues that provide the preferential localization on Chl1B, since, in the absence of the Leu residues, localization is expected to be completely inverted in favor of Chl1A. This observation may merit further detailed investigation. In contrast, these asymmetric effects are not present in homodimeric RCs, resulting in nearly equal sharing of spin density between the two BChl molecules.
The protein environment and its effect on the neutral P700 state was recently probed by Mitsuhashi et al. using a combination of computational methods (QM/MM/PCM and time-dependent DFT). They found that this asymmetric environment led to a high coupling for the HOMO of the neutral Chl1A/Chl1B pair (85 meV), but a low coupling of the LUMO (15 meV) (Mitsuhashi et al., 2021). They concluded that this caused a preferential (but not exclusive) localization of the excited state on Chl1A, resulting the strong preference for A-branch electron transfer. Interestingly, this does not extend to the bRC, where the neutral HOMO and LUMO couplings were found to be high (115 and 134 meV, respectively), implying that the excited state is shared nearly equally among the two BChl molecules. The deciding factor in branch specificity instead occurs at the BChl2A level, in line with previous mutagenesis experiments (Kirmaier et al., 2001). It appears that manipulations to the electronic structure of the primary donor alone are not enough to confer the quantitative branch specificity required in Type II RCs, but are sufficient to lend a slight preference to branch usage, as observed in PS I. This may explain why Type II RCs display less asymmetry of protein matrix effects while attaining high asymmetry in the branch usage.
In addition to affecting the electronic coupling between the two (B)Chl monomers, the protein matrix also influences the dimer as a whole. As highlighted in Figure 3, the monomeric (B)Chls as well as the primary donors exist on a wide distribution of possible redox potentials. From most oxidizing species, P680 of PS II with a potential of ∼ 1,200 mV, to the least oxidizing, P800 of the HbRC at ∼ 225 mV, the Em values span a range of nearly 1,000 mV, thus showcasing both the versatility and profound impact that the protein matrix effects can have in altering the electronic properties of (B)Chl molecules. An interesting example is the comparison of P700 of PS I to the P865 and P960 of the bRCs. There are significant differences in redox properties of BChl a [Em = + 640 mV (Fajer et al., 1975)], Chl a [Em = + 800 mV (Fajer et al., 1982; Maggiora et al., 1985)], and Chl b [Em = + 940 mV (Kobayashi et al., 2007)], and yet the primary donors of P700 (composed of Chl a), P865 (composed of BChl a), and P865 (composed of BChl b) that have nearly the same redox potential of ∼ 500 mV (Moss et al., 1991; Williams et al., 1992; Nagarajan et al., 1993; Lin et al., 1994; Brettel, 1997; Alric et al., 2004). It should be noted that the redox potential for BChl b is unknown, but it is likely more oxidizing than BChl a and close to that of Chl a. Regardless, through a combination of (B)Chl dimerization and implementation of extensive protein-matrix effects, heterodimeric RCs can modulate the redox potential of the primary donors by at least 300 mV as compared to the respective monomers. Perhaps even more impressive is the remarkably precise control that proteins have to tailor their interactions with chemically dissimilar molecules to achieve a highly similar mid-point potential.
Not only are RCs capable of tuning the mid-point potentials of disparate Chls to similar values, but the reverse is also observed, where proteins are able to modify chemically identical primary donors to achieve vastly different redox potentials. Nowhere is this effect more pronounced than in the comparison of P680 and P700 of PS II and PS I, respectively. Even though P680 and P700 are both composed of Chl a molecules, their redox potentials vary by over 600 mV, with the Em of P680 and P700 ∼ 1,200 mV (Rappaport et al., 2002; Ishikita et al., 2005) and ∼ 500 mV, respectively. Indeed, nature is able to take a modestly oxidizing species and form one of the most oxidizing cofactors in biology. Please note that the value of the redox potential of P700 is approximate, as it has been shown to vary in different organisms, with P700 of spinach PS I reporting a value of +470 and ∼ +400 mV for the cyanobacterium Gloeobacter violaceus (Nakamura et al., 2011). Regardless, the difference of ∼700 mV for a dimer of chemically identical Chl molecules is noteworthy. While this difference is staggering, it must be acknowledged that these RCs ultimately are primed to serve vastly different functions. PS I must generate a low potential reductant sufficient to reduce a soluble ferredoxin, while PS II needs to generate a strong oxidant for water splitting at the Mn4Ca-oxo cluster.
In contrast to heterodimers, the primary donors of homodimeric reaction centers exhibit remarkably similar midpoint potentials. Redox titrations on the primary donor, P840, of the GsbRC yielded a reduction potential of +217 (Azai et al., 2016)–+240 mV (Fowler et al., 1971; Prince and Olson, 1976; Hauska et al., 2001). Related experiments on P800 from the HbRC yielded a surprisingly similar Em of +225 mV (Prince et al., 1985). This is notable given the differences in the surrounding protein environment and chemical identity of the BChl molecules. While they share the same axial ligands, P800 lacks any hydrogen bonds while P840 has two putative hydrogen bonds to each BChl in the dimer and two unique aromatic residues in close proximity. There are currently no empirical measurements in the literature for the Em of BChl g, so it is difficult to assess the degree to which the redox potential of P800 must be altered to achieve a value of +225 mV. However, the fact that the HbRC, containing BChl g, can achieve the same redox potential as GsbRC, containing BChl a, without the extensive protein-matrix effects suggests a more oxidizing potential of BChl g. The reason for the Em of each RC to shift by ∼ 250 mV in comparison to the heterodimeric Type I RC, PS I, is not well understood, as both RCs are capable of reducing soluble ferredoxins (Seo et al., 2001; Romberger and Golbeck, 2012). It should be noted that the more simplified RCs have fewer electron transfer cofactors and therefore do not need to account for the loss of ΔG. However, that the GsbRC is associated with a cytochrome c551 that is only 53 mV more negative than P840 (Kusumoto et al., 1999) may be a contributing factor. Further insight can be gained from the HbRC, as the BChl g pigments that compose the primary donor are subject to relatively controlled oxidation to Chl aox in the presence of light and O2. Interestingly, partial oxidation of the pair, resulting in BChl g/Chl aox remains functionally active and able to generate a charge-separated state (Ferlez et al., 2015). However, full oxidation of both BChl g molecules results in a loss of activity. Whether this loss of activity arises from a decoupling of the dimer, a shifting of its redox potential, or another substantial change to its electronic properties remains an open question.
Alterations to the redox and electronic properties of primary donors and its effects on the efficiency of charge separation have been studied in literature. Interestingly, small changes to the local protein environment can substantially impact the redox potential of the primary donor. Allen and Williams (Allen and Williams, 1995) investigated a series of site-specific genetic variants that either altered, added, or deleted hydrogen bonds to P865. Removing all of the hydrogen bonds by replacing a His residue by a Phe decreased the redox potential of P865 by 95 mV, while the addition of three His residues resulting in a total of four hydrogen bonds increased the potential by 260 mV. Concomitant with increase of the redox potential was a decrease of the recombination rate of the P865⋅+QA⋅– state by 60% and an increase in the lifetime of the excited state decay by ∼ 15 times. Similar genetic variants of other bRCs have displayed similar effects (Jia et al., 1993; Wachtveitl et al., 1993a,b). For example, a change in the electrostatic environment when the Tyr210L residue in the bRC from Rps. viridis was replaced by Phe, Ile, or Trp, each resulted in a redox potential that was sequentially more positive (Alden et al., 1996). A better understanding of the factors that influence the redox properties of primary donors has allowed for exciting progress in re-directing electron transfer within the bRCs (Wakeham and Jones, 2005; Williams and Allen, 2009).
Nature has many robust tools for the tuning and control of photosynthetic electron transfer cofactors that are tailored to its various needs, especially the primary donors of photosynthetic RCs. It has demonstrated the ability to not only align the redox potential of differing cofactors, but also push identical molecules to extreme oxidative potentials. Protein-matrix effects have a profound impact on the electronic properties of both the neutral and oxidized states of the primary donor, which we are just beginning to understand. As shown in this review, modern computational methods can be employed to model these states and accurately re-create the electronic properties that are observed experimentally. As we are able to understand better the influence of hydrogen bonds, charged and/or hydrophobic residues, the geometry and relative orientation of the (B)Chls, and protein motion (Gorka et al., 2020) on the highly efficient processes of light harvesting and electron transfer, it delivers insight on the evolutionary photosynthetic origins in our past and provides a blueprint for the design of artificial photosynthetic systems in the future.
Author Contributions
MG, KVL, and JHG were responsible for the writing of the manuscript. MG and KVL were responsible for the geometric analysis of reaction centers, while AB, AM, EG, and KVL performed the computational analysis. All authors contributed to the article and approved the submitted version.
Funding
This study is supported by the Photosynthetic Systems Program, Office of Basic Energy Sciences of the United States Department of Energy under the contracts DE-FG02-07ER15903 (KVL) and DE-FG-05-05-ER46222 (JHG).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors acknowledge support from the NSF REU Program in Physics (Grant no. 1560266) for EG and AM and thank the Center for Computational Innovations at RPI for computational resources.
References
Agalarov, R., and Brettel, K. (2003). Temperature dependence of biphasic forward electron transfer from the phylloquinone(s) A1 in photosystem I: only the slower phase is activated. Biochim. Biophys. Acta 1604, 7–12. doi: 10.1016/s0005-2728(03)00024-0
Alden, R. G., Parson, W. W., Chu, Z. T., and Warshel, A. (1996). Orientation of the OH dipole of tyrosine (M)210 and its effect on electrostatic energies in photosynthetic bacterial reaction centers. J. Phys. Chem. 100, 16761– 16770. doi: 10.1021/jp961271s
Allen, J. P., and Williams, J. C. (1995). Relationship between the oxidation potential of the bacteriochlorophyll dimer and electron transfer in photosynthetic reaction centers. J. Bioenerg. Biomembr. 27, 275–283. doi: 10.1007/BF02110097
Allen, J. P., and Williams, J. C. (2014). “Energetics of cofactors in photosynthetic complexes: relationship between protein–cofactor interactions and midpoint potentials,” in The Biophysics of Photosynthesis, eds J. Golbeck and A. van der Est (New York City, NY: Springer), 275–295.
Alric, J., Cuni, A., Maki, H., Nagashima, K. V. P., Verméglio, A., and Rappaport, F. (2004). Electrostatic interaction between redox cofactors in photosynthetic reaction centers. J. Biol. Chem. 279, 47849–47855. doi: 10.1074/jbc.M408888200
Arata, H., and Parson, W. W. (1981). Enthalpy and volume changes accompanying electron transfer from P870 to quinones in Rhodopseudomonas sphaeroides reaction centers. Biochim. Biophys. Acta 636, 70–81. doi: 10.1016/0005-2728(81)90077-3
Arlt, T., Schmidt, S., Kaiser, W., Lauterwasser, C., Meyer, M., Scheer, H., et al. (1993). The accessory bacteriochlorophyll: a real electron carrier in primary photosynthesis. Proc. Natl. Acad. Sci. U S A 90, 11757–11761. doi: 10.1073/pnas.90.24.11757
Artiukhin, D. G., and Neugebauer, J. (2018). Frozen-density embedding as a quasi-diabatization tool: charge-localized states for spin-density calculations. J. Chem. Phys. 148:214104. doi: 10.1063/1.5023290
Astashkin, A. V., Dikanov, S. A., and Tsvetkov, Y. D. (1987). Spectrometer dead time: effect on electron spin echo modulation spectra in disordered systems. Chem. Phys. Lett. 136, 204–208. doi: 10.1016/0009-2614(87)80442-6
Astashkin, A. V., Dikanov, S. A., and Tsvetkov, Y. D. (1988). Hyperfine interactions of deuterium nuclei in the nearest surroundings of trapped electrons in alkaline glass. Chem. Phys. Lett. 144, 258–264. doi: 10.1016/0009-2614(88)87150-1
Azai, C., Sano, Y., Kato, Y., Noguchi, T., and Oh-oka, H. (2016). Mutation-induced perturbation of the special pair P840 in the homodimeric reaction center in green sulfur bacteria. Sci. Rep. 6:19878. doi: 10.1038/srep19878
Barone, V. (1995). “Structure, magnetic properties and reactivities of open-shell species from density functional and self-consistent hybrid methods,” in Recent Advances in Density Functional Methods, ed. D. P. Chong (Singapore: World Scientific), 287–334.
Barrera-Rojas, J., de la Vara, L. G., Ríos-Castro, E., Leyva-Castillo, L. E., and Gómez-Lojero, C. (2018). The distribution of divinyl chlorophylls a and b and the presence of ferredoxin-NADP+ reductase in Prochlorococcus marinus MIT9313 thylakoid membranes. Heliyon 4, e01100. doi: 10.1016/j.heliyon.2018.e01100
Becke, A. D. (1988). Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098–3100. doi: 10.1103/PhysRevA.38.3098
Bekker, A., Holland, H. D., Wang, P. L., Rumble, D., Stein, H. J., Hannah, J. L., et al. (2004). Dating the rise of atmospheric oxygen. Nature 427, 117–120. doi: 10.1038/nature02260
Berera, R., van Grondelle, R., and Kennis, J. T. M. (2009). Ultrafast transient absorption spectroscopy: principles and application to photosynthetic systems. Photosynth. Res. 101, 105–118. doi: 10.1007/s11120-009-9454-y
Bibby, T. S., Nield, J., and Barber, J. (2001). Iron deficiency induces the formation of an antenna ring around trimeric photosystem I in cyanobacteria. Nature 412, 743–745. doi: 10.1038/35089098
Björn, L. O., Papageorgiou, G. C., Blankenship, R. E., and Govindjee. (2009). A viewpoint: why chlorophyll a? Photosynth. Res. 99, 85–98. doi: 10.1007/s11120-008-9395-x
Blankenship, R. E., Olson, J. M., and Miller, M. (1995). “Antenna complexes from green photosynthetic bacteria,” in Anoxygenic Photosynthetic Bacteria, eds R. E. Blankenship, M. T. Madigan, and C. E. Bauer (Dordrecht, Netherland: Springer), 399–435.
Bowes, J., Crofts, A. R., and Arntzen, C. J. (1980). Redox reactions on the reducing side of photosystem II in chloroplasts with altered herbicide binding properties. Arch. Biochem. Biophys. 200, 303–308. doi: 10.1016/0003-9861(80)90359-8
Bratt, P. J., Muhiuddin, I. P., Evans, M. C. W., and Heathcote, P. (1996). 14N electron spin echo envelope modulation (ESEEM) spectroscopy of the cation radical P840+, the primary electron donor of the Chlorobium limicola reaction center. Photochem. Photobiol. 64, 20–25. doi: 10.1111/j.1751-1097.1996.tb02416.x
Bratt, P. J., Poluektov, O. G., Thurnauer, M. C., Krzystek, J., Brunel, L.-C., Schrier, J., et al. (2000). The g-factor anisotropy of plant chlorophyll a⋅+. J. Phys. Chem. B 104, 6973–6977.
Bratt, P. J., Rohrer, M., Krzystek, J., Evans, M. C. W., Brunel, L.-C., and Angerhofer, A. (1997). Submillimeter high-feld EPR studies of the primary donor in plant photosystem I P700⋅+. J. Phys. Chem. B 101, 9686–9689. doi: 10.1021/jp9725238
Brettel, K. (1997). Electron transfer and arrangement of the redox cofactors in photosystem I. Biochim. Biophys. Acta Bioenerg. 1318, 322–373. doi: 10.1016/S0005-2728(96)00112-0
Britt, R. D. (1993). Time-domain electron paramagnetic resonance spectroscopy. Curr. Opin. Struct. Biol. 3, 774–779. doi: 10.1016/0959-440X(93)90063-Q
Brody, S. S. (2002). Fluorescence lifetime, yield, energy transfer and spectrum in photosynthesis, 1950–1960∗. Photosynth. Res. 73:127. doi: 10.1023/A:1020405921105
Brody, S. S., and Rabinowitch, E. (1957). Excitation lifetime of photosynthetic pigments in vitro and in vivo. Science 125:555. doi: 10.1126/science.125.3247.555
Burghaus, O., Plato, M., Rohrer, M., Moebius, K., MacMillan, F., and Lubitz, W. (1993). 3-mm High-field EPR on semiquinone radical anions Q– related to photosynthesis and on the primary donor P+ and acceptor QA– in reaction centers of Rhodobacter sphaeroides R-26. J. Phys. Chem. B 97, 7639–7647.
Charles, P., Kalendra, V., He, Z., Khatami, M. H., Golbeck, J. H., van der Est, A., et al. (2020). Two-dimensional 67Zn HYSCORE spectroscopy reveals that a Zn-bacteriochlorophyll aP’ dimer is the primary donor (P840) in the type-1 reaction centers of Chloracidobacterium thermophilum. Phys. Chem. Chem. Phys. 22, 6457–6467. doi: 10.1039/c9cp06556c
Chauvet, A., Sarrou, J., Lin, S., Romberger, S. P., Golbeck, J. H., Savikhin, S., et al. (2013). Temporal and spectral characterization of the photosynthetic reaction center from Heliobacterium modesticaldum. Photosynth. Res. 116, 1–9. doi: 10.1007/s11120-013-9871-9
Chen, J.-H., Wu, H., Xu, C., Liu, X.-C., Huang, Z., Chang, S., et al. (2020). Architecture of the photosynthetic complex from a green sulfur bacterium. Science 370:eabb6350. doi: 10.1126/science.abb6350
Chen, M., Telfer, A., Lin, S., Pascal, A., Larkum, A. W. D., Barber, J., et al. (2005). The nature of the photosystem II reaction centre in the chlorophyll d-containing prokaryote, Acaryochloris marina. Photochem. Photobiol. Sci. 4, 1060–1064. doi: 10.1039/b507057k
Cherepanov, D. A., Shelaev, I. V., Gostev, F. E., Aybush, A. V., Mamedov, M. D., Shuvalov, V. A., et al. (2020). Generation of ion-radical chlorophyll states in the light-harvesting antenna and the reaction center of cyanobacterial photosystem I. Photosynth. Res. 146, 55–73. doi: 10.1007/s11120-020-00731-0
Cherepanov, D. A., Shelaev, I. V., Gostev, F. E., Mamedov, M. D., Petrova, A. A., Aybush, A. V., et al. (2017). Mechanism of adiabatic primary electron transfer in photosystem I: femtosecond spectroscopy upon excitation of reaction center in the far-red edge of the QY band. Biochim. Biophys. Acta Bioenerg. 1858, 895–905. doi: 10.1016/j.bbabio.2017.08.008
Cherepanov, D. A., Shelaev, I. V., Gostev, F. E., Petrova, A., Aybush, A. V., Nadtochenko, V. A., et al. (2021). Primary charge separation within the structurally symmetric tetrameric Chl2APAPBChl2B chlorophyll exciplex in photosystem I. J. Photochem. Photobiol. B 217:112154. doi: 10.1016/j.jphotobiol.2021.112154
Chestnut, M. M., Milikisiyants, S., Chatterjee, R., Kern, J., and Smirnov, A. I. (2021). Electronic structure of the primary electron donor P700⋅+ in photosystem I studied by multifrequency HYSCORE spectroscopy at X- and Q-Band. J. Phys. Chem. B 125, 36–48. doi: 10.1021/acs.jpcb.0c09000
Chew, A. G. M., and Bryant, D. A. (2007). Chlorophyll biosynthesis in bacteria: the origins of structural and functional diversity. Annu. Rev. Microbiol. 61, 113–129. doi: 10.1146/annurev.micro.61.080706.093242
Cohen, R. O., Shen, G., Golbeck, J. H., Xu, W., Chitnis, P. R., Valieva, A. I., et al. (2004). Evidence for asymmetric electron transfer in cyanobacterial photosystem I: analysis of a methionine-to-leucine mutation of the ligand to the primary electron acceptor A0. Biochemistry 43, 4741–4754. doi: 10.1021/bi035633f
Commoner, B., Heise, J. J., and Townsend, J. (1956). Light-induced paramagnetism in chloroplasts. Proc. Natl. Acad. Sci. U S A 42, 710–718. doi: 10.1073/pnas.42.10.710
Cossi, M., Rega, N., Scalmani, G., and Barone, V. (2003). Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 24, 669–681. doi: 10.1002/jcc.10189
Dashdorj, N., Xu, W., Cohen, R. O., Golbeck, J. H., and Savikhin, S. (2005). Asymmetric electron transfer in cyanobacterial photosystem I: charge separation and secondary electron transfer dynamics of mutations near the primary electron acceptor A0. Biophys. J. 88, 1238–1249. doi: 10.1529/biophysj.104.050963
Davis, I. H., Heathcote, P., MacLachlan, D. J., and Evans, M. C. W. (1993). Modulation analysis of the electron spin echo signals of in vivo oxidised primary donor 14N chlorophyll centres in bacterial, P870 and P960, and plant photosystem I, P700, reaction centres. Biochim. Biophys. Acta Bioenerg. 1143, 183–189. doi: 10.1016/0005-2728(93)90141-2
Davis, M. S., Forman, A., and Fajer, J. (1979). Ligated chlorophyll cation radicals: their function in photosystem II of plant photosynthesis. Proc. Natl. Acad. Sci. USA 76, 4170–4174. doi: 10.1073/pnas.76.9.4170
de Boer, A. L., Neerken, S., de Wijn, R., Permentier, H. P., Gast, P., Vijgenboom, E., et al. (2002). B-branch electron transfer in reaction centers of Rhodobacter sphaeroides assessed with site-directed mutagenesis. Photosynth. Res. 71, 221–239. doi: 10.1023/A:1015533126685
de Wijn, R., and van Gorkom, H. J. (2001). Kinetics of electron transfer from QA to QB in photosystem II. Biochemistry 40, 11912–11922. doi: 10.1021/bi010852r
Deisenhofer, J., Epp, O., Miki, K., Huber, R., and Michel, H. (1985). Structure of the protein subunits in the photosynthetic reaction centre of Rhodopseudomonas viridis at 3Å resolution. Nature 318, 618–624. doi: 10.1038/318618a0
Deisenhofer, J., Epp, O., Sinning, I., and Michel, H. (1995). Crystallographic refinement at 2.3 Å resolution and refined model of the photosynthetic reaction centre from Rhodopseudomonas viridis. J. Mol. Biol. 246, 429–457. doi: 10.1006/jmbi.1994.0097
Deligiannakis, Y., Louloudi, M., and Hadjiliadis, N. (2000). Electron Spin Echo Envelope Modulation (ESEEM) spectroscopy as a tool to investigate the coordination environment of metal centers. J. Coord. Chem. 204, 1–112. doi: 10.1016/S0010-8545(99)00218-0
Deligiannakis, Y., and Rutherford, A. W. (2001). Electron spin echo envelope modulation spectroscopy in photosystem I. Biochim. Biophys. Acta Bioenerg. 1507, 226–246. doi: 10.1016/S0005-2728(01)00201-8
Díaz-Quintana, A., Leibl, W., Bottin, H., and Sétif, P. (1998). Electron transfer in photosystem I reaction centers follows a linear pathway in which iron-sulfur cluster FB is the immediate electron donor to soluble ferredoxin. Biochemistry 37, 3429–3439. doi: 10.1021/bi972469l
Dikanov, S. A., Astashkin, A. V., Tsvetkov, Y. D., and Goldfeld, M. G. (1983). Comparative modulation analysis of electron spin echo signals from oxidized chlorophyll a in vitro and P700 centres in chloroplasts. Chem. Phys. Lett. 101, 206–210. doi: 10.1016/0009-2614(83)87372-2
Diner, B. A., Schlodder, E., Nixon, P. J., Coleman, W. J., Rappaport, F., Lavergne, J., et al. (2001). Site-directed mutations at D1-His198 and D2-His197 of Photosystem II in Synechocystis PCC 6803: sites of primary charge separation and cation and triplet stabilization. Biochemistry 40, 9265–9281. doi: 10.1021/bi010121r
Duan, H.-G., Prokhorenko, V. I., Wientjes, E., Croce, R., Thorwart, M., and Miller, R. J. D. (2017). Primary charge separation in the photosystem II reaction center revealed by a global analysis of the two-dimensional electronic spectra. Sci. Rep. 7:12347. doi: 10.1038/s41598-017-12564-4
Dunham, W. R., Bearden, A. J., Salmeen, I. T., Palmer, G., Sands, R. H., Orme-Johnson, W. H., et al. (1971). The two-iron ferredoxins in spinach, parsley, pig adrenal cortex, Azotobacter vinelandii, and Clostridium pasteurianum: studies by magnetic field Mössbauer spectroscopy. Biochim. Biophys. Acta Bioenerg. 253, 134–152. doi: 10.1016/0005-2728(71)90240-4
Eccles, J., Honig, B., and Schulten, K. (1988). Spectroscopic determinants in the reaction center of Rhodopseudomonas viridis. Biophys. J. 53, 137–144. doi: 10.1016/S0006-3495(88)83075-3
Ermler, U., Fritzsch, G., Buchanan, S. K., and Michel, H. (1994). Structure of the photosynthetic reaction centre from Rhodobacter sphaeroides at 2.65 Å resolution: cofactors and protein-cofactor interactions. Structure 2, 925–936. doi: 10.1016/s0969-2126(94)00094-8
Fajer, J., Brune, D. C., Davis, M. S., Forman, A., and Spaulding, L. D. (1975). Primary charge separation in bacterial photosynthesis: oxidized chlorophylls and reduced pheophytin. Proc. Natl. Acad. Sci. U S A 72, 4956–4960. doi: 10.1073/pnas.72.12.4956
Fajer, J., Fujita, I., Davis, M. S., Forman, A., Hanson, L. K., Smith, K. M., et al. (1982). “Electrochemical and spectrochemical studies of biological redox components,” in ACS division of analytical chemistry, ed. K. M. Kadish (Atlanta, GA: ACS Publication), 489–513.
Ferlez, B., Cowgill, J., Dong, W., Gisriel, C., Lin, S., Flores, M., et al. (2016). Thermodynamics of the electron acceptors in Heliobacterium modesticaldum: an exemplar of an early homodimeric Type I photosynthetic reaction center. Biochemistry 55, 2358–2370. doi: 10.1021/acs.biochem.5b01320
Ferlez, B., Dong, W., Siavashi, R., Redding, K., Hou, H. J. M., Golbeck, J. H., et al. (2015). The effect of bacteriochlorophyll g oxidation on energy and electron transfer in reaction centers from Heliobacterium modesticaldum. J. Phys. Chem. B 119, 13714–13725. doi: 10.1021/acs.jpcb.5b03339
Fiedor, L., Kania, A., Myśliwa-Kurdziel, B., and Orzeł, Ł, and Stochel, G. (2008). Understanding chlorophylls: central magnesium ion and phytyl as structural determinants. Biochim. Biophys. Acta Bioenerg. 1777, 1491–1500. doi: 10.1016/j.bbabio.2008.09.005
Fischer, W. W., Hemp, J., and Johnson, J. E. (2016). Evolution of oxygenic photosynthesis. Annu. Rev. Earth Planet Sci. 44, 647–683. doi: 10.1146/annurev-earth-060313-054810
Fowler, C. F., Nugent, N. A., and Fuller, R. C. (1971). The isolation and characterization of a photochemically active complex from Chloropseudomonas ethylica. Proc. Natl. Acad. Sci. USA 68, 2278–2282. doi: 10.1073/pnas.68.9.2278
Francke, C., Otte, S. C. M., Miller, M., Amesz, J., and Olson, J. M. (1996). Energy transfer from carotenoid and FMO-protein in subcellular preparations from green sulfur bacteria. Spectroscopic characterization of an FMO-reaction center core complex at low temperature. Photosynth. Res. 50, 71–77. doi: 10.1007/BF00018222
Frigaard, N.-U., Maqueo Chew, A. G., Maresca, J. A., and Bryant, D. A. (2006). “Bacteriochlorophyll biosynthesis in green bacteria,” in Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions and Applications, eds B. Grimm, R. J. Porra, W. Rüdiger, and H. Scheer (Netherlands: Springer), 201–221.
Gibasiewicz, K., Ramesh, V. M., Melkozernov, A. N., Lin, S., Woodbury, N. W., Blankenship, R. E., et al. (2001). Excitation dynamics in the core antenna of PS I from Chlamydomonas reinhardtii CC 2696 at room temperature. J. Phys. Chem. B 105, 11498–11506. doi: 10.1021/jp012089g
Gisriel, C., Sarrou, I., Ferlez, B., Golbeck, J. H., Redding, K. E., and Fromme, R. (2017). Structure of a symmetric photosynthetic reaction center-photosystem. Science 357, 1021–1025. doi: 10.1126/science.aan5611
Gisriel, C. J., Azai, C., and Cardona, T. (2021). Recent advances in the structural diversity of reaction centers. Photosynth. Res. doi: 10.1007/s11120-021-00857-9
Gobets, B., and van Grondelle, R. (2001). Energy transfer and trapping in photosystem I. Biochim. Biophys. Acta Bioenerg. 1507, 80–99. doi: 10.1016/s0005-2728(01)00203-1
Gobets, B., van Stokkum, I. H., Rögner, M., Kruip, J., Schlodder, E., Karapetyan, N. V., et al. (2001). Time-resolved fluorescence emission measurements of photosystem I particles of various cyanobacteria: a unified compartmental model. Biophys. J. 81, 407–424. doi: 10.1016/S0006-3495(01)75709-8
Golbeck, J. H. (2006). Photosystem I: The Light-Driven Plastocyanin: Ferredoxin Oxidoreductase. Dordrecht: Springer.
Gorka, M., Charles, P., Kalendra, V., Baldansuren, A., Lakshmi, K. V., and Golbeck, J. H. (2021a). A dimeric chlorophyll electron acceptor differentiates type I from type II photosynthetic reaction centers. iScience 24:102719. doi: 10.1016/j.isci.2021.102719
Gorka, M., Cherepanov, D. A., Semenov, A. Y., and Golbeck, J. H. (2020). Control of electron transfer by protein dynamics in photosynthetic reaction centers. Crit. Rev. Biochem. Mol. Biol. 55, 425–468. doi: 10.1080/10409238.2020.1810623
Gorka, M., Gruszecki, E., Charles, P., Kalendra, V., Lakshmi, K. V., and Golbeck, J. H. (2021b). Two-dimensional HYSCORE spectroscopy reveals a histidine imidazole as the axial ligand to Chl3A in the M688HPsaA genetic variant of Photosystem I. Biochim. Biophys. Acta Bioenerg. 1862:148424. doi: 10.1016/j.bbabio.2021.148424
Groot, M. L., Pawlowicz, N. P., van Wilderen, L. J. G. W., Breton, J., van Stokkum, I. H. M., and van Grondelle, R. (2005). Initial electron donor and acceptor in isolated photosystem II reaction centers identified with femtosecond mid-IR spectroscopy. Proc. Natl. Acad. Sci. USA 102, 13087–13092. doi: 10.1073/pnas.0503483102
Hager-Braun, C., Zimmermann, R., and Hauska, G. (1999). “The homodimeric reaction center of chlorobium,” in The Phototrophic Prokaryotes, eds G. A. Peschek, W. Löffelhardt, and G. Schmetterer (Boston, MA: Springer), 169–181.
Harmer, J., Mitrikas, G., and Schweiger, A. (2009). “Advanced pulse EPR methods for the characterization of metalloproteins,” in High Resolution EPR: Applications to Metalloenzymes and Metals in Medicine, eds L. Berliner and G. Hanson (New York City, NY: Springer), 13–61. doi: 10.1039/b701568b
Hauska, G., Schoedl, T., Remigy, H., and Tsiotis, G. (2001). The reaction center of green sulfur bacteria. Biochim. Biophys. Acta 1507, 260–277. doi: 10.1016/S0005-2728(01)00200-6
He, Z., Ferlez, B., Kurashov, V., Tank, M., Golbeck, J. H., and Bryant, D. A. (2019). Reaction centers of the thermophilic microaerophile, Chloracidobacterium thermophilum (Acidobacteria) I: biochemical and biophysical characterization. Photosynth. Res. 142, 87–103. doi: 10.1007/s11120-019-00650-9
Heimdal, J., Jensen, K. P., Devarajan, A., and Ryde, U. (2007). The role of axial ligands for the structure and function of chlorophylls. J. Biol. Inorg. Chem. 12, 49–61. doi: 10.1007/s00775-006-0164-z
Heller, B. A., Holten, D., and Kirmaier, C. (1996). Effects of Asp residues near the L-side pigments in bacterial reaction centers. Biochemistry 35, 15418–15427. doi: 10.1021/bi961362f
Höfer, P., Grupp, A., Nebenführ, H., and Mehring, M. (1986). Hyperfine sublevel correlation (HYSCORE) spectroscopy: a 2D ESR investigation of the squaric acid radical. Chem. Phys. Lett. 132, 279–282. doi: 10.1016/0009-2614(86)80124-5
Hoffman, B. M. (2003). Electron-nuclear double resonance spectroscopy (and electron spin-echo envelope modulation spectroscopy) in bioinorganic chemistry. Proc. Natl. Acad. Sci. USA 100, 3575–3578. doi: 10.1073/pnas.0636464100
Hofmann, H. J. (1976). Precambrian microflora, Belcher Islands, Canada: significance and systematics. J. Paleontol. 50, 1040–1073. doi: 10.2307/1303547
Holzapfel, W., Finkele, U., Kaiser, W., Oesterhelt, D., Scheer, H., Stilz, H. U., et al. (1989). Observation of a bacteriochlorophyll anion radical during the primary charge separation in a reaction center. Chem. Phys. Lett. 160, 1–7. doi: 10.1016/0009-2614(89)87543-8
Holzwarth, A. R., and Müller, M. G. (1996). Energetics and kinetics of radical pairs in reaction centers from Rhodobacter sphaeroides. A femtosecond transient absorption study. Biochemistry 35, 11820–11831. doi: 10.1021/bi9607012
Holzwarth, A. R., Müller, M. G., Niklas, J., and Lubitz, W. (2006a). Ultrafast transient absorption studies on photosystem I reaction centers from Chlamydomonas reinhardtii. 2: mutations near the P700 reaction center chlorophylls provide new insight into the nature of the primary electron donor. Biophys. J. 90, 552–565. doi: 10.1529/biophysj.105.059824
Holzwarth, A. R., Müller, M. G., Reus, M., Nowaczyk, M., Sander, J., and Rögner, M. (2006b). Kinetics and mechanism of electron transfer in intact photosystem II and in the isolated reaction center: pheophytin is the primary electron acceptor. Proc. Natl. Acad. Sci. USA 103, 6895–6900. doi: 10.1073/pnas.0505371103
Huang, L., Ponomarenko, N., Wiederrecht, G. P., and Tiede, D. M. (2012). Cofactor-specific photochemical function resolved by ultrafast spectroscopy in photosynthetic reaction center crystals. Proc. Natl. Acad. Sci. USA 109, 4851–4856. doi: 10.1073/pnas.1116862109
Humbeck, K., Schumann, R., and Senger, H. (1984). “The influence of blue light on the formation of chlorophyll-protein complexes in Scenedesmus,” in Blue Light Effects in Biological Systems, ed. H. Senger (Berlin, Heidelberg: Springer), 359–365.
Huppman, P., Arlt, T., Penzkofer, H., Schmidt, S., Bibikova, M., Dohse, B., et al. (2002). Kinetics, energetics, and electronic coupling of the primary electron transfer reactions in mutated reaction centers of Blastochloris viridis. Biophys. J. 82, 3186–3197. doi: 10.1016/S0006-3495(02)75661-0
Ishikita, H., Loll, B., Biesiadka, J., Saenger, W., and Knapp, E.-W. (2005). Redox potentials of chlorophylls in the photosystem II reaction center. Biochemistry 44, 4118–4124. doi: 10.1021/bi047922p
Ishikita, H., Saenger, W., Biesiadka, J., Loll, B., and Knapp, E.-W. (2006). How photosynthetic reaction centers control oxidation power in chlorophyll pairs P680, P700, and P870. Proc. Natl. Acad. Sci. USA 103, 9855–9860. doi: 10.1073/pnas.0601446103
Ito, H., and Tanaka, A. (2011). Evolution of a divinyl chlorophyll-based photosystem in Prochlorococcus. Proc. Natl. Acad. Sci. USA 108, 18014–18019. doi: 10.1073/pnas.1107590108
Itoh, S., Iwaki, M., and Ikegami, I. (2001). Modification of photosystem I reaction center by the extraction and exchange of chlorophylls and quinones. Biochim. Biophys. Acta Bioenerg. 1507, 115–138. doi: 10.1016/s0005-2728(01)00199-2
Jagannathan, B., and Golbeck, J. H. (2008). Unifying principles in homodimeric type I photosynthetic reaction centers: properties of PscB and the FA, FB and FX iron–sulfur clusters in green sulfur bacteria. Biochim. Biophys. Acta Bioenerg. 1777, 1535–1544. doi: 10.1016/j.bbabio.2008.09.001
Jia, Y., DiMagno, T. J., Chan, C. K., Wang, Z., Popov, M. S., Du, M., et al. (1993). Primary charge separation in mutant reaction centers of Rhodobacter capsulatus. J. Phys. Chem. 97, 13180–13191. doi: 10.1021/j100152a024
Johnson, E. T., Müh, F., Nabedryk, E., Williams, J. C., Allen, J. P., Lubitz, W., et al. (2002). Electronic and vibronic coupling of the special pair of bacteriochlorophylls in photosynthetic reaction centers from wild-type and mutant strains of Rhodobacter sphaeroides. J. Phys. Chem. B 106, 11859– 11869. doi: 10.1021/jp021024q
Johnson, E. T., and Parson, W. W. (2002). Electrostatic interactions in an integral membrane protein. Biochemistry 41, 6483–6494. doi: 10.1021/bi012131y
Jordan, P., Fromme, P., Witt, H. T., Klukas, O., Saenger, W., and Krauss, N. (2001). Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution. Nature 411, 909–917. doi: 10.1038/35082000
Käβ, H., Rautter, J., Boenigk, B., Hoefer, P., and Lubitz, W. (1995). 2D ESEEM of the 15N-labeled radical cations of bacteriochlorophyll a and of the primary donor in reaction centers of Rhodobacter sphaeroides. J. Phys. Chem. 99, 436–448. doi: 10.1021/j100001a065
Karyagina, I., Pushkar, Y., Stehlik, D., van der Est, A., Ishikita, H., Knapp, E.-W., et al. (2007). Contributions of the protein environment to the midpoint potentials of the A1 phylloquinones and the FX iron-sulfur cluster in photosystem I. Biochemistry 46, 10804–10816. doi: 10.1021/bi700846z
Käβ, H., Fromme, P., Witt, H. T., and Lubitz, W. (2001). Orientation and electronic structure of the primary donor radical cation in photosystem I: a single crystal EPR and ENDOR Study. J. Phys. Chem. B 105, 1225–1239. doi: 10.1021/jp0032311
Kaupp, M. (2002). The function of photosystem I. quantum chemical insight into the role of tryptophan-quinone interactions. Biochemistry 41, 2895–2900. doi: 10.1021/bi0159783
Käβ, H., Bittersmann-Weidlich, E., Andréasson, L. E., Bönigk, B., and Lubitz, W. (1995). ENDOR and ESEEM of the 15N labelled radical cations of chlorophyll a and the primary donor P700 in photosystem I. Chem. Phys. 194, 419–432. doi: 10.1016/0301-0104(95)00021-F
Käβ, H., Fromme, P., and Lubitz, W. (1996). Quadrupole parameters of nitrogen nuclei in the cation radical P700.+ determined by ESEEM of single crystals of photosystem I. Chem. Phys. Lett. 257, 197–206. doi: 10.1016/0009-2614(96)00533-7
Käβ, H., and Lubitz, W. (1996). Evaluation of 2D ESEEM data of 15N-labeled radical cations of the primary donor P700 in photosystem I and chlorophyll a. Chem. Phys. Lett. 251, 193–203. doi: 10.1016/0009-2614(96)00129-7
Ke, B., Hansen, R. E., and Beinert, H. (1973). Oxidation-reduction potentials of bound iron-sulfur proteins of photosystem I. Proc. Natl. Acad. Sci. USA 70, 2941–2945. doi: 10.1073/pnas.70.10.2941
Kennis, J. T. M., Shkuropatov, A. Y., van Stokkum, I. H. M., Gast, P., Hoff, A. J., Shuvalov, V. A., et al. (1997). Formation of a long-lived P+BA– state in plant pheophytin-exchanged reaction centers of Rhodobacter sphaeroides R26 at low temperature. Biochemistry 36, 16231–16238. doi: 10.1021/bi9712605
Kirmaier, C., Gaul, D., DeBey, R., Holten, D., and Schenck, C. (1991). Charge separation in a reaction center incorporating bacteriochlorophyll for photoactive bacteriopheophytin. Science 251, 922–927. doi: 10.1126/science.2000491
Kirmaier, C., He, C., and Holten, D. (2001). Manipulating the direction of electron transfer in the bacterial reaction center by swapping Phe for Tyr near BChlM (L181) and Tyr for Phe near BChlL (M208). Biochemistry 40, 12132–12139. doi: 10.1021/bi0113263
Kirmaier, C., Laporte, L., Schenck, C. C., and Holten, D. (1995a). The nature and dynamics of the charge-separated intermediate in reaction centers in which bacteriochlorophyll replaces the photoactive bacteriopheophytin. 1. spectral characterization of the transient state. J. Phys. Chem. 99, 8903–8909. doi: 10.1021/j100021a067
Kirmaier, C., Laporte, L., Schenck, C. C., and Holten, D. (1995b). The nature and dynamics of the charge-separated intermediate in reaction centers in which bacteriochlorophyll replaces the photoactive bacteriopheophytin. 2. the rates and yields of charge separation and recombination. J. Phys. Chem. 99, 8910–8917. doi: 10.1021/j100021a068
Kirmaier, C., Weems, D., and Holten, D. (1999). M-side electron transfer in reaction center mutants with a lysine near the nonphotoactive bacteriochlorophyll. Biochemistry 38, 11516–11530. doi: 10.1021/bi9908585
Klamt, A., and Schüürmann, G. (1993). COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2, 799–805. doi: 10.1039/P29930000799
Kleinfeld, D., Okamura, M. Y., and Feher, G. (1984a). Electron transfer in reaction centers of Rhodopseudomonas sphaeroides. I. determination of the charge recombination pathway of D+QAQ-B and free energy and kinetic relations between Q–AQB and QAQB–. Biochim. Biophys. Acta Bioenerg. 766, 126–140. doi: 10.1016/0005-2728(84)90224-x
Kleinfeld, D., Okamura, M. Y., and Feher, G. (1984b). Electron-transfer kinetics in photosynthetic reaction centers cooled to cryogenic temperatures in the charge-separated state: evidence for light-induced structural changes. Biochemistry 23, 5780–5786. doi: 10.1021/bi00319a017
Kobayashi, M. (1996). Study of precise pigment composition of photosystem 1-type reaction centers by means of normal-phase HPLC. J. Plant Res. 109, 223–230.
Kobayashi, M., Ohashi, S., Iwamoto, K., Shiraiwa, Y., Kato, Y., and Watanabe, T. (2007). Redox potential of chlorophyll d in vitro. Biochim. Biophys. Acta Bioenerg. 1767, 596–602. doi: 10.1016/j.bbabio.2007.02.015
Kobayashi, M., Oh-oka, H., Akutsu, S., Akiyama, M., Tominaga, K., Kise, H., et al. (2000). The primary electron acceptor of green sulfur bacteria, bacteriochlorophyll 663, is chlorophyll a esterified with Δ 2,6-phytadienol. Photosynth. Res. 63, 269–280. doi: 10.1023/A:1006480629059
Kobayashi, M., van de Meent, E. J., Erkelens, C., Amesz, J., Ikegami, I., and Watanabe, T. (1991). Bacteriochlorophyll g epimer as a possible reaction center component of heliobacteria. Biochim. Biophys. Acta Bioenerg. 1057, 89–96. doi: 10.1016/S0005-2728(05)80087-8
Koepke, J., Hu, X., Muenke, C., Schulten, K., and Michel, H. (1996). The crystal structure of the light-harvesting complex II (B800-850) from Rhodospirillum molischianum. Structure 4, 581–597. doi: 10.1016/s0969-2126(96)00063-9
Kojima, R., Yamamoto, H., Azai, C., Uragami, C., Hashimoto, H., Kosumi, D., et al. (2020). Energy transfer and primary charge separation upon selective femtosecond excitation at 810 nm in the reaction center complex from Heliobacterium modesticaldum. J. Photochem. Photobiol. A 401:112758. doi: 10.1016/j.jphotochem.2020.112758
Konar, A., Sechrist, R., Song, Y., Policht, V. R., Laible, P. D., Bocian, D. F., et al. (2018). Electronic interactions in the bacterial reaction center revealed by two-color 2D electronic spectroscopy. J. Phys. Chem. Lett. 9, 5219–5225. doi: 10.1021/acs.jpclett.8b02394
Kulik, L., and Lubitz, W. (2009). Electron-nuclear double resonance. Photosynth. Res. 102, 391–401. doi: 10.1007/s11120-009-9401-y
Kurashov, V., Gorka, M., Milanovsky, G. E., Johnson, T. W., Cherepanov, D. A., Semenov, A. Y., et al. (2018). Critical evaluation of electron transfer kinetics in P700-FA/FB, P700-FX, and P700-A1 photosystem I core complexes in liquid and in trehalose glass. Biochim. Biophys. Acta Bioenerg. 1859, 1288–1301. doi: 10.1016/j.bbabio.2018.09.367
Kusumoto, N., Sétif, P., Brettel, K., Seo, D., and Sakurai, H. (1999). Electron transfer kinetics in purified reaction centers from the green sulfur bacterium Chlorobium tepidum studied by multiple-flash excitation. Biochemistry 38, 12124–12137. doi: 10.1021/bi990452s
Lakshmi, K. V., and Brudvig, G. W. (2000). “Electron paramagnetic resonance distance measurements in photosystems,” in Distance Measurements in Biological Systems by EPR, eds L. J. Berliner, G. R. Eaton, and S. S. Eaton (Boston, MA: Springer), 513–567.
Lakshmi, K. V., and Brudvig, G. W. (2001). Pulsed electron paramagnetic resonance methods for macromolecular structure determination. Curr. Opin. Struct. Biol. 11, 523–531. doi: 10.1016/s0959-440x(00)00242-6
Lakshmi, K. V., Coates, C. S., Smith, S., and Chatterjee, R. (2014). “The radical intermediates of photosystem II,” in The Biophysics of Photosynthesis, eds J. Golbeck and A. van der Est (New York City, NY: Springer), 299–320.
Lauterwasser, C., Finkele, U., Scheer, H., and Zinth, W. (1991). Temperature dependence of the primary electron transfer in photosynthetic reaction centers from Rhodobacter sphaeroides. Chem. Phys. Lett. 183, 471–477. doi: 10.1016/0009-2614(91)80161-P
Lee, C., Yang, W., and Parr, R. G. (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789. doi: 10.1103/physrevb.37.785
Lendzian, F., Geβern, C., Bönigk, B., Plato, M., Möbius, K., and Lubitz, W. (1992). “15N- and 1H-ENDOR/TRIPLE resonance of the primary donor cation radical D+⋅ in isotopically labelled reaction centers of Rhodobacter sphaeroides,” in Research in Photosynthesis, Proceedings of the IXth International Congress on Photosynthesis, Nagoya, Japan, ed. N. Murata (Dordrecht, Netherland: Springer), 433–436.
Lendzian, F., Huber, M., Isaacson, R. A., Endeward, B., Plato, M., Bönigk, B., et al. (1993). The electronic structure of the primary donor cation radical in Rhodobacter sphaeroides R-26: ENDOR and TRIPLE resonance studies in single crystals of reaction centers. Biochim. Biophys. Acta Bioenerg. 1183, 139–160. doi: 10.1016/0005-2728(93)90013-6
Lendzian, F., Lubitz, W., Scheer, H., Hoff, A. J., Plato, M., Tränkle, E., et al. (1988). ESR, ENDOR and TRIPLE resonance studies of the primary donor radical cation P960+⋅ in the photosynthetic bacterium Rhodopseudomonas viridis. Chem. Phys. Lett. 148, 377–385. doi: 10.1016/0009-2614(88)87191-4
Leonova, M. M., Fufina, T. Y., Vasilieva, L. G., and Shuvalov, V. A. (2011). Structure-function investigations of bacterial photosynthetic reaction centers. Biochemistry (Mosc) 76, 1465–1483. doi: 10.1134/S0006297911130074
Lin, C. P., Bowman, M. K., and Norris, J. R. (1986). Analysis of electron spin echo modulation in randomly oriented solids: 15N modulation of radical cations of bacteriochlorophyll a and the primary donor of photosynthetic bacterium Rhodospirillum rubrum. J. Chem. Phys. 85, 56–62. doi: 10.1063/1.451582
Lin, X., Murchison, H. A., Nagarajan, V., Parson, W. W., Allen, J. P., and Williams, J. C. (1994). Specific alteration of the oxidation potential of the electron donor in reaction centers from Rhodobacter sphaeroides. Proc. Natl. Acad. Sci. U S A. 91, 10265–10269. doi: 10.1073/pnas.91.22.10265
Lubitz, W., Isaacson, R. A., Abresch, E. C., and Feher, G. (1984). 15N electron nuclear double resonance of the primary donor cation radical P865+⋅ in reaction centers of Rhodopseudomonas sphaeroides: additional evidence for the dimer model. Proc. Natl. Acad. Sci. USA 81, 7792–7796. doi: 10.1073/pnas.81.24.7792
Ma, F., Romero, E., Jones, M. R., Novoderezhkin, V. I., and van Grondelle, R. (2019). Both electronic and vibrational coherences are involved in primary electron transfer in bacterial reaction center. Nat. Commun. 10:933. doi: 10.1038/s41467-019-08751-8
Mac, M., Bowlby, N. R., Babcock, G. T., and McCracken, J. (1998). Monomeric spin density distribution in the primary donor of photosystem I as determined by electron magnetic resonance: functional and thermodynamic implications. J. Am. Chem. Soc. 120, 13215–13223.
Maggiora, L. L., Petke, J. D., Gopal, D., Iwamoto, R. T., and Maggiora, G. M. (1985). Experimental and theoretical studies of Schiff base chlorophylls. Photochem. Photobiol. 42, 69–75. doi: 10.1021/bi00236a011
Mao, L., Wang, Y., and Hu, X. (2003). π−π stacking interactions in the peridinin-chlorophyll-protein of Amphidinium carterae. J. Phys. Chem. B 107, 3963–3971. doi: 10.1021/jp0276496
Mar, T., Govindjee, Singhal, G. S., and Merkelo, H. (1972). Lifetime of the excited state in vivo. I. chlorophyll a in algae, at room and at liquid nitrogen temperatures; rate constants of radiationless deactivation and trapping. Biophys. J. 12, 797–808. doi: 10.1016/S0006-3495(72)86123-X
Martin, J.-L., Breton, J., Hoff, A. J., Migus, A., and Antonetti, A. (1986). Femtosecond spectroscopy of electron transfer in the reaction center of the photosynthetic bacterium Rhodopseudomonas sphaeroides R-26: direct electron transfer from the dimeric bacteriochlorophyll primary donor to the bacteriopheophytin acceptor with a time constant of 2.8 ± 0.2 psec. Proc. Natl. Acad. Sci. USA 83, 957–961. doi: 10.1073/pnas.83.4.957
Mauzerall, D., Neuberger, A., and Kenner, G. W. (1976). Chlorophyll and photosynthesis. Philos. Trans. R Soc. B 273, 287–294. doi: 10.1098/rstb.1976.0014
McDermott, G., Prince, S. M., Freer, A. A., Hawthornthwaite-Lawless, A. M., Papiz, M. Z., Cogdell, R. J., et al. (1995). Crystal structure of an integral membrane light-harvesting complex from photosynthetic bacteria. Nature 374, 517–521. doi: 10.1038/374517a0
McMahon, B. H., Muller, J. D., Wraight, C. A., and Nienhaus, G. U. (1998). Electron transfer and protein dynamics in the photosynthetic reaction center. Biophys. J. 74, 2567–2587. doi: 10.1016/S0006-3495(98)77964-0
Melkozernov, A. N. (2001). Excitation energy transfer in Photosystem I from oxygenic organisms. Photosynth. Res. 70, 129–153. doi: 10.1023/A:1017909325669
Miller, M., Liu, X., Snyder, S. W., Thurnauer, M. C., and Biggins, J. (1992). Photosynthetic electron-transfer reactions in the green sulfur bacterium Chlorobium vibrioforme: evidence for the functional involvement of iron-sulfur redox centers on the acceptor side of the reaction center. Biochemistry 31, 4354–4363. doi: 10.1021/bi00132a028
Mimuro, M., Tomo, T., Nishimura, Y., Yamazaki, I., and Satoh, K. (1995). Identification of a photochemically inactive pheophytin molecule in the spinach D1-D2-cyt b559 complex. Biochim. Biophys. Acta Bioenerg. 1232, 81–88. doi: 10.1016/0005-2728(95)00116-2
Mitsuhashi, K., Tamura, H., Saito, K., and Ishikita, H. (2021). Nature of asymmetric electron transfer in the symmetric pathways of photosystem I. J. Phys. Chem. B 125, 2879–2885. doi: 10.1021/acs.jpcb.0c10885
Mondal, J., and Bruce, B. D. (2018). Ferredoxin: the central hub connecting photosystem I to cellular metabolism. Photosynthetica 56, 279–293. doi: 10.1007/s11099-018-0793-9
Moore, L. R., Goericke, R., and Chisholm, S. W. (1995). Comparative physiology of Synechococcus and Prochlorococcus: influence of light and temperature on growth, pigments, fluorescence and absorptive properties. Mar. Ecol. Prog. Ser. 116, 259–275.
Morales, F., Moise, N., Quílez, R., Abadía, A., Abadía, J., and Moya, I. (2001). Iron deficiency interrupts energy transfer from a disconnected part of the antenna to the rest of photosystem II. Photosynth. Res. 70, 207–220. doi: 10.1023/A:1017965229788
Moss, D. A., Leonhard, M., Bauscher, M., and Mäntele, W. (1991). Electrochemical redox titration of cofactors in the reaction center from Rhodobacter sphaeroides. FEBS Lett. 283, 33–36. doi: 10.1016/0014-5793(91)80547-g
Müller, M. G., Niklas, J., Lubitz, W., and Holzwarth, A. R. (2003). Ultrafast transient absorption studies on photosystem I reaction centers from Chlamydomonas reinhardtii. 1. a new interpretation of the energy trapping and early electron transfer steps in photosystem I. Biophys. J. 85, 3899–3922. doi: 10.1016/S0006-3495(03)74804-8
Müller, M. G., Slavov, C., Luthra, R., Redding, K. E., and Holzwarth, A. R. (2010). Independent initiation of primary electron transfer in the two branches of the photosystem I reaction center. Proc. Natl. Acad. Sci. U S A 107, 4123–4128. doi: 10.1073/pnas.0905407107
Myers, J. A., Lewis, K. L. M., Fuller, F. D., Tekavec, P. F., Yocum, C. F., and Ogilvie, J. P. (2010). Two-dimensional electronic spectroscopy of the D1-D2-cyt b559 photosystem II reaction center complex. J. Phys. Chem. 1, 2774–2780. doi: 10.1021/jz100972z
Nagarajan, V., Parson, W. W., Davis, D., and Schenck, C. C. (1993). Kinetics and free energy gaps of electron-transfer reactions in Rhodobacter sphaeroides reaction centers. Biochemistry 32, 12324–12336. doi: 10.1021/bi00097a008
Nakamura, A., Suzawa, T., Kato, Y., and Watanabe, T. (2011). Species dependence of the redox potential of the primary electron donor P700 in photosystem I of oxygenic photosynthetic organisms revealed by spectroelectrochemistry. Plant Cell. Physiol. 52, 815–823. doi: 10.1093/pcp/pcr034
Neerken, S., Permentier, H. P., Francke, C., Aartsma, T. J., and Amesz, J. (1998). Excited states and trapping in reaction center complexes of the green sulfur bacterium Prosthecochloris aestuarii. Biochemistry 37, 10792–10797. doi: 10.1021/bi9806899
Neerken, S., Schmidt, K. A., Aartsma, T. J., and Amesz, J. (1999). Dynamics of energy conversion in reaction center core complexes of the green sulfur bacterium Prosthecochloris aestuarii at low temperature. Biochemistry 38, 13216–13222. doi: 10.1021/bi9910981
Neese, F., Wennmohs, F., Hansen, A., and Becker, U. (2009). Efficient, approximate and parallel Hartree–Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree–Fock exchange. Chem. Phys. 356, 98–109. doi: 10.1016/j.chemphys.2008.10.036
Niedringhaus, A., Policht, V. R., Sechrist, R., Konar, A., Laible, P. D., Bocian, D. F., et al. (2018). Primary processes in the bacterial reaction center probed by two-dimensional electronic spectroscopy. Proc. Natl. Acad. Sci. USA 115, 3563–3568. doi: 10.1073/pnas.1721927115
Noguchi, T., Fukami, Y., Oh-oka, H., and Inoue, Y. (1997). Fourier transform infrared study on the primary donor P798 of Heliobacterium modesticaldum: cysteine S-H coupled to P798 and molecular interactions of carbonyl groups. Biochemistry 36, 12329–12336. doi: 10.1021/bi970853c
Norris, J. R., Uphaus, R. A., Crespi, H. L., and Katz, J. J. (1971). Electron spin resonance of chlorophyll and the origin of signal I in photosynthesis. Proc. Natl. Acad. Sci. USA 68, 625–628. doi: 10.1073/pnas.68.3.625
Novoderezhkin, V. I., Dekker, J. P., and van Grondelle, R. (2007). Mixing of exciton and charge-transfer states in photosystem II reaction centers: modeling of Stark Spectra with modified Redfield theory. Biophys. J. 93, 1293–1311. doi: 10.1529/biophysj.106.096867
Nuijs, A. M., Dorssen, R. J. V., Duysens, L. N. M., and Amesz, J. (1985a). Excited states and primary photochemical reactions in the photosynthetic bacterium Heliobacterium chlorum. Proc. Natl. Acad. Sci. USA 82, 6865–6868. doi: 10.1073/pnas.82.20.6865
Nuijs, A. M., Vasmel, H., Joppe, H. L. P., Duysens, L. N. M., and Amesz, J. (1985b). Excited states and primary charge separation in the pigment system of the green photosynthetic bacterium Prosthecochloris aestuarii as studied by picosecond absorbance difference spectroscopy. Biochim. Biophys. Acta Bioenerg. 807, 24–34.
Oh-oka, H., Kamei, S., Matsubara, H., Lin, S., van Noort, P. I., and Blankenship, R. E. (1998). Transient sbsorption spectroscopy of energy-transfer and trapping processes in the reaction center complex of Chlorobium tepidum. J. Phys. Chem. B 102, 8190–8195. doi: 10.1021/jp981478g
Olson, J. M. (1998). Chlorophyll organization and function in green photosynthetic bacteria. Photochem. Photobiol. 67, 61–75. doi: 10.1111/j.1751-1097.1998.tb05166.x
Olson, J. M. (2004). The FMO Protein. Photosynth. Res. 80, 181–187. doi: 10.1023/B:PRES.0000030428.36950.43
Olson, J. M. (2013). “Green bacteria: the light-harvesting chlorosome,” in Encyclopedia of Biological Chemistry, Second Edn, eds W. J. Lennarz and M. D. Lane (Waltham: Academic Press), 513–516.
Olson, T. L., Williams, J. C., and Allen, J. P. (2013). Influence of protein interactions on oxidation/reduction midpoint potentials of cofactors in natural and de novo metalloproteins. Biochim. Biophys. Acta Bioenerg. 1827, 914–922. doi: 10.1016/j.bbabio.2013.02.014
Oppenheimer, J. R., and Schwinger, J. (1941). On the interaction of mesotrons and nuclei. Phys. Rev. 60, 150–152. doi: 10.1103/PhysRev.60.150
Oren, A. (2011). “Characterization of pigments of prokaryotes and their use in taxonomy and classification,” in Methods in Microbiology, eds F. Rainey and A. Oren (Cambridge: Academic Press), 261–282.
Orf, G. S., Gisriel, C., and Redding, K. E. (2018). Evolution of photosynthetic reaction centers: insights from the structure of the heliobacterial reaction center. Photosynth. Res. 138, 11–37. doi: 10.1007/s11120-018-0503-2
Papiz, M. Z., Prince, S. M., Howard, T., Cogdell, R. J., and Isaacs, N. W. (2003). The structure and thermal motion of the B800-850 LH2 complex from Rps. acidophila at 2.0 Å resolution and 100 K: new structural features and functionally relevant motions. J. Mol. Biol. 326, 1523–1538. doi: 10.1016/s0022-2836(03)00024-x
Partensky, F., Hess, W. R., and Vaulot, D. (1999). Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microbiol. Mol. Biol. Rev. 63, 106–127. doi: 10.1128/MMBR.63.1.106-127.1999
Pawlowicz, N. P., van Grondelle, R., van Stokkum, I. H. M., Breton, J., Jones, M. R., and Groot, M. L. (2008). Identification of the first steps in charge separation in bacterial photosynthetic reaction centers of Rhodobacter sphaeroides by ultrafast mid-infrared spectroscopy: electron transfer and protein dynamics. Biophys. J. 95, 1268–1284. doi: 10.1529/biophysj.108.130880
Pearson, R. G. (1963). Hard and soft acids and bases. J. Am. Chem. Soc. 85, 3533–3539. doi: 10.1021/ja00905a001
Peloquin, J. M., Williams, J. C., Lin, X., Alden, R. G., Taguchi, A. K. W., Allen, J. P., et al. (1994). Time-dependent thermodynamics during early electron transfer in reaction centers from Rhodobacter sphaeroides. Biochemistry 33, 8089–8100. doi: 10.1021/bi00192a014
Peterman, E. J. G., van Amerongen, H., van Grondelle, R., and Dekker, J. P. (1998). The nature of the excited state of the reaction center of photosystem II of green plants: a high-resolution fluorescence spectroscopy study. Proc. Natl. Acad. Sci. USA 95, 6128–6133. doi: 10.1073/pnas.95.11.6128
Petrenko, A., Maniero, A. L., van Tol, J., MacMillan, F., Li, Y., Brunel, L.-C., et al. (2004). A high-field EPR study of P700+⋅ in wild-type and mutant photosystem I from Chlamydomonas reinhardtii. Biochemistry 43, 1781–1786. doi: 10.1021/bi035466j
Pierella Karlusich, J. J., and Carrillo, N. (2017). Evolution of the acceptor side of photosystem I: ferredoxin, flavodoxin, and ferredoxin-NADP+ oxidoreductase. Photosynth. Res. 134, 235–250. doi: 10.1007/s11120-017-0338-2
Plato, M., Krauß, N., Fromme, P., and Lubitz, W. (2003). Molecular orbital study of the primary electron donor P700 of photosystem I based on a recent X-ray single crystal structure analysis. Chem. Phys. 294, 483–499. doi: 10.1016/S0301-0104(03)00378-1
Poluektov, O. G., Paschenko, S. V., Utschig, L. M., Lakshmi, K. V., and Thurnauer, M. C. (2005). Bidirectional electron transfer in photosystem I: direct evidence from high-frequency time-resolved EPR spectroscopy. J. Am. Chem. Soc. 127, 11910–11911. doi: 10.1021/ja053315t
Poluektov, O. G., Utschig, L. M., Schlesselman, S. L., Lakshmi, K. V., Brudvig, G. W., Kothe, G., et al. (2002). Electronic structure of the P700 special pair from high-frequency electron paramagnetic resonance spectroscopy. J. Phys. Chem. B 106, 8911–8916. doi: 10.1021/jp021465+
Prince, R. C., Gest, H., and Blankenship, R. E. (1985). Thermodynamic properties of the photochemical reaction center of Heliobacterium chlorum. Biochim. Biophys. Acta Bioenerg. 810, 377–384. doi: 10.1016/0005-2728(85)90224-5
Prince, R. C., and Olson, J. M. (1976). Some thermodynamic and kinetic properties of the primary photochemical reactants in a complex from a green photosynthetic bacterium. Biochim. Biophys. Acta Bioenerg. 423, 357–362. doi: 10.1016/0005-2728(76)90191-2
Prisner, T., Rohrer, M., and MacMillan, F. (2001). Pulsed EPR spectroscopy: biological applications. Annu. Rev. Phys. Chem. 52, 279–313. doi: 10.1146/annurev.physchem.52.1.279
Ptushenko, V. V., Cherepanov, D. A., Krishtalik, L. I., and Semenov, A. Y. (2008). Semi-continuum electrostatic calculations of redox potentials in photosystem I. Photosynth. Res. 97:55. doi: 10.1007/s11120-008-9309-y
Ralf, G., and Repeta, D. J. (1992). The pigments of Prochlorococcus marinus: the presence of divinylchlorophyll a and b in a marine procaryote. Limnol. Oceanogr. 37, 425–433. doi: 10.4319/lo.1992.37.2.0425
Rappaport, F., Guergova-Kuras, M., Nixon, P. J., Diner, B. A., and Lavergne, J. (2002). Kinetics and pathways of charge recombination in photosystem II. Biochemistry 41, 8518–8527. doi: 10.1021/bi025725p
Rasmussen, B., Fletcher, I. R., Brocks, J. J., and Kilburn, M. R. (2008). Reassessing the first appearance of eukaryotes and cyanobacteria. Nature 455, 1101–1104. doi: 10.1038/nature07381
Rigby, S. E. J., Nugent, J. H. A., and O’Malley, P. J. (1994a). ENDOR and special triple resonance studies of chlorophyll cation radicals in photosystem 2. Biochemistry 33, 10043–10050. doi: 10.1021/bi00199a031
Rigby, S. E. J., Thapar, R., Evans, M. C. W., and Heathcote, P. (1994b). The electronic structure of P840+⋅ the primary donor of the Chlorobium limicola f. sp. thiosulphatophilum photosynthetic reaction centre. FEBS Lett. 350, 24–28. doi: 10.1016/0014-5793(94)00724-1
Roberts, J. A., Holten, D., and Kirmaier, C. (2001). Primary events in photosynthetic reaction centers with multiple mutations near the photoactive electron carriers. J. Phys. Chem. B 105, 5575–5584. doi: 10.1021/jp010280k
Robinson, H. H., and Crofts, A. R. (1983). Kinetics of the oxidation—reduction reactions of the photosystem II quinone acceptor complex, and the pathway for deactivation. FEBS Lett. 153, 221–226. doi: 10.1016/0014-5793(83)80152-5
Romberger, S. P., and Golbeck, J. H. (2012). The FX iron-sulfur cluster serves as the terminal bound electron acceptor in heliobacterial reaction centers. Photosynth. Res. 111, 285–290. doi: 10.1007/s11120-012-9723-z
Romero, E., van Stokkum, I. H. M., Novoderezhkin, V. I., Dekker, J. P., and van Grondelle, R. (2010). Two different charge separation pathways in photosystem II. Biochemistry 49, 4300–4307. doi: 10.1021/bi1003926
Roszak, A. W., Howard, T. D., Southall, J., Gardiner, A. T., Law, C. J., Isaacs, N. W., et al. (2003). Crystal structure of the RC-LH1 core complex from Rhodopseudomonas palustris. Science 302, 1969–1972. doi: 10.1126/science.1088892
Saito, K., and Ishikita, H. (2011). Cationic state distribution over the P700 chlorophyll pair in photosystem I. Biophys. J. 101, 2018–2025. doi: 10.1016/j.bpj.2011.09.010
Santabarbara, S., Kuprov, I., Poluektov, O., Casal, A., Russell, C. A., Purton, S., et al. (2010). Directionality of electron-transfer reactions in photosystem I of prokaryotes: universality of the bidirectional electron-transfer model. J. Phys. Chem. B 114, 15158–15171. doi: 10.1021/jp1044018
Sauer, K., and Yachandra, V. K. (2004). The water-oxidation complex in photosynthesis. Biochim. Biophys. Acta 1655, 140–148. doi: 10.1016/j.bbabio.2003.07.004
Savikhin, S., and Jankowiak, R. (2014). “Mechanism of primary charge separation in photosynthetic reaction centers,” in The Biophysics of Photosynthesis, eds J. H. Golbeck and A. van der Est (New York, NY: Springer), 193–240.
Savikhin, S., Xu, W., Chitnis, P. R., and Struve, W. S. (2000). Ultrafast primary processes in PS I from Synechocystis sp. PCC 6803: roles of P700 and A0. Biophys. J. 79, 1573–1586. doi: 10.1016/S0006-3495(00)76408-3
Schäfer, A., Horn, H., and Ahlrichs, R. (1992). Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 97, 2571–2577. doi: 10.1063/1.463096
Schatz, G. H., Brock, H., and Holzwarth, A. R. (1987). Picosecond kinetics of fluorescence and absorbance changes in photosystem II particles excited at low photon density. Proc. Natl. Acad. Sci. USA 84, 8414–8418. doi: 10.1073/pnas.84.23.8414
Schlau-Cohen, G. S., Dawlaty, J. M., and Fleming, G. R. (2012). Ultrafast multidimensional spectroscopy: principles and applications to photosynthetic systems. IEEE J. Sel. Top Quantum Electron. 18, 283–295. doi: 10.1109/JSTQE.2011.2112640
Schmidt, K. A., Neerken, S., Permentier, H. P., Hager-Braun, C., and Amesz, J. (2000). Electron transfer in reaction center core complexes from the green sulfur bacteria Prosthecochloris aestuarii and Chlorobium tepidum. Biochemistry 39, 7212–7220. doi: 10.1021/bi992861u
Şener, M., Strümpfer, J., Hsin, J., Chandler, D., Scheuring, S., Hunter, C. N., et al. (2011). Förster energy transfer theory as reflected in the structures of photosynthetic light-harvesting systems. Chem. Phys. Chem. 12, 518–531. doi: 10.1002/cphc.201000944
Seo, D., Tomioka, A., Kusumoto, N., Kamo, M., Enami, I., and Sakurai, H. (2001). Purification of ferredoxins and their reaction with purified reaction center complex from the green sulfur bacterium Chlorobium tepidum. Biochim. Biophys. Acta Bioenerg. 1503, 377–384. doi: 10.1016/s0005-2728(00)00245-0
Shelaev, I. V., Gostev, F. E., Mamedov, M. D., Sarkisov, O. M., Nadtochenko, V. A., Shuvalov, V. A., et al. (2010). Femtosecond primary charge separation in Synechocystis sp. PCC 6803 photosystem I. Biochim. Biophys. Acta 1797, 1410–1420. doi: 10.1016/j.bbabio.2010.02.026
Shkuropatov, A. Y., and Shuvalov, V. A. (1993). Electron transfer in pheophytin a-modified reaction centers from Rhodobacter sphaeroides (R-26). FEBS Lett. 322, 168–172. doi: 10.1016/0014-5793(93)81561-d
Shuvalov, V. A., Amesz, J., and Duysens, L. N. M. (1986). Picosecond spectroscopy of isolated membranes of the photosynthetic green sulfur bacterium Prosthecochloris aestuarii upon selective excitation of the primary electron donor. Biochim. Biophys. Acta Bioenerg. 851, 1–5. doi: 10.1016/0005-2728(86)90242-2
Sirohiwal, A., Neese, F., and Pantazis, D. A. (2020). Protein matrix control of reaction center excitation in photosystem II. J. Am. Chem. Soc. 142, 18174–18190. doi: 10.1021/jacs.0c08526
Song, Y., Sechrist, R., Nguyen, H. H., Johnson, W., Abramavicius, D., Redding, K. E., et al. (2021). Excitonic structure and charge separation in the heliobacterial reaction center probed by multispectral multidimensional spectroscopy. Nat. Commun. 12:2801. doi: 10.1038/s41467-021-23060-9
Srinivasan, N., and Golbeck, J. H. (2009). Protein-cofactor interactions in bioenergetic complexes: the role of the A1A and A1B phylloquinones in photosystem I. Biochim. Biophys. Acta 1787, 1057–1088. doi: 10.1016/j.bbabio.2009.04.010
Staroverov, V. N., Scuseria, G. E., Tao, J., and Perdew, J. P. (2003). Comparative assessment of a new nonempirical density functional: molecules and hydrogen-bonded complexes. J. Chem. Phys. 119, 12129–12137. doi: 10.1063/1.1626543
Steglich, C., Mullineaux, C. W., Teuchner, K., Hess, W. R., and Lokstein, H. (2003). Photophysical properties of Prochlorococcus marinus SS120 divinyl chlorophylls and phycoerythrin in vitro and in vivo. FEBS Lett. 553, 79–84. doi: 10.1016/s0014-5793(03)00971-2
Stowell, M. H. B., McPhillips, T. M., Rees, D. C., Soltis, S. M., Abresch, E., and Feher, G. (1997). Light-induced structural changes in photosynthetic reaction center: implications for mechanism of electron-proton transfer. Science 276, 812–816. doi: 10.1126/science.276.5313.812
Strain, H. H., and Svec, W. A. (1966). “Extraction, separation, estimation, and isolation of the chlorophylls,” in The Chlorophylls, eds L. P. Vernon and G. R. Seely (Cambridge: Academic Press), 21–66.
Sugiura, M., Boussac, A., Noguchi, T., and Rappaport, F. (2008). Influence of Histidine-198 of the D1 subunit on the properties of the primary electron donor, P680, of photosystem II in Thermosynechococcus elongatus. Biochim. Biophys. Acta Bioenerg. 1777, 331–342. doi: 10.1016/j.bbabio.2008.01.007
Sun, J., Hao, S., Radle, M., Xu, W., Shelaev, I., Nadtochenko, V., et al. (2014). Evidence that histidine forms a coordination bond to the A0A and A0B chlorophylls and a second H-bond to the A1A and A1B phylloquinones in M688HPsaA and M668HPsaB variants of Synechocystis sp. PCC 6803. Biochim. Biophys. Acta Bioenerg. 1837, 1362–1375. doi: 10.1016/j.bbabio.2014.04.004
Sun, Y., Wang, H., Zhao, F., and Sun, J. (2004). The effect of axial Mg2+ ligation and peripheral hydrogen bonding on chlorophyll a. Chem. Phys. Lett. 387, 12–16. doi: 10.1016/j.cplett.2004.01.103
Taguchi, A. T., Mattis, A. J., O’Malley, P. J., Dikanov, S. A., and Wraight, C. A. (2013). Tuning cofactor redox potentials: the 2-methoxy dihedral angle generates a redox potential difference of >160 mV between the primary (QA) and secondary (QB) quinones of the bacterial photosynthetic reaction center. Biochemistry 52, 7164–7166. doi: 10.1021/bi4011896
Takahashi, R., Hasegawa, K., and Noguchi, T. (2008). Effect of charge distribution over a chlorophyll dimer on the redox potential of P680 in photosystem II as studied by density functional theory calculations. Biochemistry 47, 6289–6291. doi: 10.1021/bi8007998
Tamura, H., Saito, K., and Ishikita, H. (2020). Acquirement of water-splitting ability and alteration of the charge-separation mechanism in photosynthetic reaction centers. Proc. Natl. Acad. Sci. USA 117, 16373–16382. doi: 10.1073/pnas.2000895117
Tao, J., Perdew, J. P., Staroverov, V. N., and Scuseria, G. E. (2003). Climbing the density functional ladder: nonempirical meta–generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 91:146401. doi: 10.1103/PhysRevLett.91.146401
Tice, M. M., and Lowe, D. R. (2004). Photosynthetic microbial mats in the 3,416-Myr-old ocean. Nature 431, 549–552. doi: 10.1038/nature02888
Tomi, T., Shibata, Y., Ikeda, Y., Taniguchi, S., Haik, C., Mataga, N., et al. (2007). Energy and electron transfer in the photosynthetic reaction center complex of Acidiphilium rubrum containing Zn-bacteriochlorophyll a studied by femtosecond up-conversion spectroscopy. Biochim. Biophys. Acta 1767, 22–30. doi: 10.1016/j.bbabio.2006.10.008
Tomo, T., Okubo, T., Akimoto, S., Yokono, M., Miyashita, H., Tsuchiya, T., et al. (2007). Identification of the special pair of photosystem II in a chlorophyll d-dominated cyanobacterium. Proc. Natl. Acad. Sci. USA 104, 7283–7288. doi: 10.1073/pnas.0701847104
Umena, Y., Kawakami, K., Shen, J. R., and Kamiya, N. (2011). Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 A. Nature 473, 55–60. doi: 10.1038/nature09913
van Amerongen, H., and Croce, R. (2013). Light harvesting in photosystem II. Photosynth. Res. 116, 251–263. doi: 10.1007/s11120-013-9824-3
Van Brederode, M. E., Jones, M. R., Van Mourik, F., Van Stokkum, I. H. M., and Van Grondelle, R. (1997). A new pathway for transmembrane electron transfer in photosynthetic reaction centers of Rhodobacter sphaeroides not involving the excited special pair. Biochemistry 36, 6855–6861. doi: 10.1021/bi9703756
van Brederode, M. E., Ridge, J. P., van Stokkum, I. H. M., van Mourik, F., Jones, M. R., and van Grondelle, R. (1998). On the efficiency of energy transfer and the different pathways of electron transfer in mutant reaction centers of Rhodobacter sphaeroides. Photosynth. Res. 55, 141–146. doi: 10.1023/A:1005925917867
van Brederode, M. E., and van Grondelle, R. (1999). New and unexpected routes for ultrafast electron transfer in photosynthetic reaction centers. FEBS Lett. 455, 1–7. doi: 10.1016/s0014-5793(99)00810-8
van Brederode, M. E., van Mourik, F., van Stokkum, I. H. M., Jones, M. R., and van Grondelle, R. (1999). Multiple pathways for ultrafast transduction of light energy in the photosynthetic reaction center of Rhodobacter sphaeroides. Proc. Natl. Acad. Sci. USA 96, 2054–2059. doi: 10.1073/pnas.96.5.2054
van der Est, A., Chirico, S., Karyagina, I., Cohen, R., Shen, G., and Golbeck, J. H. (2009). Alteration of the axial Met ligand to electron acceptor A0 in photosystem I: an investigation of electron transfer at different temperatures by multifrequency time-resolved and CW EPR. Appl. Magn. Reson. 37:103. doi: 10.1007/s00723-009-0043-1
van Gorkom, H. J., Tamminga, J. J., and Haveman, J. (1974). Primary reactions, plastoquinone and fluorescence yield in subchloroplast fragments prepared with deoxycholate. Biochim. Biophys. Acta 347, 417–438. doi: 10.1016/0005-2728(74)90080-2
van Stokkum, I. H. M., Beekman, L. M. P., Jones, M. R., van Brederode, M. E., and van Grondelle, R. (1997). Primary electron transfer kinetics in membrane-bound Rhodobacter sphaeroides reaction centers: A global and target analysis. Biochemistry 36, 11360–11368. doi: 10.1021/bi9707943
Vermaas, J. V., Taguchi, A. T., Dikanov, S. A., Wraight, C. A., and Tajkhorshid, E. (2015). Redox potential tuning through differential quinone binding in the photosynthetic reaction center of Rhodobacter sphaeroides. Biochemistry 54, 2104–2116. doi: 10.1021/acs.biochem.5b00033
Vinyard, D. J., Ananyev, G. M., and Dismukes, C. (2013). Photosystem II: the reaction center of oxygenic photosynthesis. Annu. Rev. Biochem. 82, 577–606. doi: 10.1146/annurev-biochem-070511-100425
Vos, M. H., Breton, J., and Martin, J.-L. (1997). Electronic energy transfer within the hexamer cofactor system of bacterial reaction centers. J. Phys. Chem. B 101, 9820–9832. doi: 10.1021/jp971486h
Wachtveitl, J., Farchaus, J. W., Das, R., Lutz, M., Robert, B., and Mattioli, T. A. (1993a). Structure, spectroscopic, and redox properties of Rhodobacter sphaeroides reaction centers bearing point mutations near the primary electron donor. Biochemistry 32, 12875–12886. doi: 10.1021/bi00210a041
Wachtveitl, J., Farchaus, J. W., Mathis, P., and Oesterhelt, D. (1993b). Tyrosine 162 of the photosynthetic reaction center L-subunit plays a critical role in the cytochrome c2 mediated rereduction of the photooxidized bacteriochlorophyll dimer in Rhodobacter sphaeroides. 2. Quantitative kinetic analysis. Biochemistry 32, 10894–10904. doi: 10.1021/bi00091a045
Wakeham, M. C., and Jones, M. R. (2005). Rewiring photosynthesis: engineering wrong-way electron transfer in the purple bacterial reaction centre. Biochem. Soc. Trans. 33, 851–857. doi: 10.1042/BST0330851
Wasielewski, M. R., Fenton, J. M., and Govindjee. (1987). The rate of formation of P700+–A0– in photosystem I particles from spinach as measured by picosecond transient absorption spectroscopy. Photosynth. Res. 12, 181–189. doi: 10.1007/BF00047947
Wasielewski, M. R., and Tiede, D. M. (1986). Sub-picosecond measurements of primary electron transfer in Rhodopseudomonas viridis reaction centers using near-infrared excitation. FEBS Lett. 204, 368–372. doi: 10.1016/0014-5793(86)80845-6
Webber, A. N., and Lubitz, W. (2001). P700: the primary electron donor of photosystem I. Biochim. Biophys. Acta Bioenerg. 1507, 61–79. doi: 10.1016/s0005-2728(01)00198-0
Webber, A. N., Su, H., Bingham, S. E., Käβ, H., Krabben, L., Kuhn, M., et al. (1996). Site-directed mutations affecting the spectroscopic characteristics and midpoint potential of the primary donor in photosystem I. Biochemistry 35, 12857–12863. doi: 10.1021/bi961198w
Weigend, F. (2006). Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 8, 1057–1065. doi: 10.1039/b515623h
Weigend, F., and Ahlrichs, R. (2005). Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305. doi: 10.1039/b508541a
White, N. T. H., Beddard, G. S., Thorne, J. R. G., Feehan, T. M., Keyes, T. E., and Heathcote, P. (1996). Primary charge separation and energy transfer in the photosystem I reaction center of higher plants. J. Phys. Chem. 100, 12086–12099. doi: 10.1021/jp9604709
Widdel, F., Schnell, S., Heising, S., Ehrenreich, A., Assmus, B., and Schink, B. (1993). Ferrous iron oxidation by anoxygenic phototrophic bacteria. Nature 362, 834–836. doi: 10.1038/362834a0
Williams, J. C., Alden, R. G., Murchison, H. A., Peloquin, J. M., Woodbury, N. W., and Allen, J. P. (1992). Effects of mutations near the bacteriochlorophylls in reaction centers from Rhodobacter sphaeroides. Biochemistry 31, 11029–11037. doi: 10.1021/bi00160a012
Williams, J. C., and Allen, J. P. (2009). “Directed modification of reaction centers from purple bacteria,” in The Purple Phototrophic Bacteria, eds C. N. Hunter, F. Daldal, M. C. Thurnauer, and J. T. Beatty (Netherlands: Springer), 337–353.
Williams, J. C., Haffa, A. L. M., McCulley, J. L., Woodbury, N. W., and Allen, J. P. (2001). Electrostatic interactions between charged amino acid residues and the bacteriochlorophyll dimer in reaction centers from Rhodobacter sphaeroides. Biochemistry 40, 15403–15407. doi: 10.1021/bi011574z
Wraight, C. (2004). Proton and electron transfer in the acceptor quinone complex of photosynthetic reaction centers from Rhodobacter sphaeroides. Front. Biosci. 9, 309–337. doi: 10.2741/1236
Wraight, C. A. (1979). Electron acceptors of bacterial photosynthetic reaction centers II. H+ binding coupled to secondary electron transfer in the quinone acceptor complex. Biochim. Biophys. Acta Bioenerg. 548, 309–327. doi: 10.1016/0005-2728(79)90138-5
Xu, H., Zhang, R.-B., Ma, S.-H., Qu, Z.-W., Zhang, X.-K., and Zhang, Q.-Y. (2002). Theoretical studies on the mechanism of primary electron transfer in the photosynthetic reaction center of Rhodobacter sphaeroides. Photosynth. Res. 74, 11–36. doi: 10.1023/A:1020811911956
Keywords: primary donor, reaction center, heterodimer, homodimer, chlorophyll, electron paramagnetic resonance, density functional theory
Citation: Gorka M, Baldansuren A, Malnati A, Gruszecki E, Golbeck JH and Lakshmi KV (2021) Shedding Light on Primary Donors in Photosynthetic Reaction Centers. Front. Microbiol. 12:735666. doi: 10.3389/fmicb.2021.735666
Received: 03 July 2021; Accepted: 30 August 2021;
Published: 01 October 2021.
Edited by:
Catarina M. Paquete, Universidade Nova de Lisboa, PortugalReviewed by:
Heiko Lokstein, Charles University, CzechiaDong-Woo Lee, Yonsei University, South Korea
Copyright © 2021 Gorka, Baldansuren, Malnati, Gruszecki, Golbeck and Lakshmi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John H. Golbeck, amhnNUBwc3UuZWR1; K. V. Lakshmi, bGFrc2hrQHJwaS5lZHU=