 Rungnapa Sakulworakan1,2
Rungnapa Sakulworakan1,2 Putita Chokmangmeepisarn1,2
Putita Chokmangmeepisarn1,2 Nguyen Dinh-Hung1
Nguyen Dinh-Hung1 Elayaraja Sivaramasamy2
Elayaraja Sivaramasamy2 Ikuo Hirono3Rungthip Chuanchuen4
Ikuo Hirono3Rungthip Chuanchuen4 Pattanapon Kayansamruaj5*
Pattanapon Kayansamruaj5* Channarong Rodkhum2*
Channarong Rodkhum2*- 1The International Graduate Program of Veterinary Science and Technology (VST), Faculty of Veterinary Science, Chulalongkorn University, Bangkok, Thailand
- 2Center of Excellent in Fish Infectious Diseases (CE FID), Department of Veterinary Microbiology, Faculty of Veterinary Science, Chulalongkorn University, Bangkok, Thailand
- 3Tokyo University of Marine Science and Technology, Minato-ku, Japan
- 4Department of Veterinary Public Health, Faculty of Veterinary Science, Chulalongkorn University, Bangkok, Thailand
- 5Department of Aquaculture, Faculty of Fisheries, Kasetsart University, Bangkok, Thailand
Aeromonas veronii outbreaks in tilapia farming caused relatively high mortalities, and the bacteria was resistant to many kinds of antimicrobials used in Thailand aquaculture. According to the CLSI standard, the determination of antimicrobials efficacy has been limited to phenotypic analyses, and a genomics study is required. This research aimed to analyze the resistome of A. veronii isolated from diseased tilapia in Chainat, Nong Khai, and Uttaradit provinces in Thailand. A total of 12 isolates of A. veronii were identified based on the gyrB sequencing and then, the MIC values to eight antimicrobials (AMP, AML, GEN, ENR, OXO, OTC, SXT, and FFC) were determined. According to the MIC patterns, whole genome sequencing (WGS) of five representatives and resistome analysis were performed, including 15 genomes of A. veronii isolated from freshwater fish available in the NCBI. All tilapia isolates were susceptible to FFC but resistant to AML and AMP while OTC resistance was the most dominant. In addition to the WGS analysis, 4.5 Mbp of A. veronii was characterized. A total of 20 ARGs were detected by resistome analysis and 16 genes were shared among the A. veronii population. In conclusion, A. veronii strains isolated from tilapia exhibited a resistance to several antimicrobials and multidrug resistance (MDR) which was related to the presence of multiple ARGs. Aeromonas veronii shared the ARGs in their population worldwide with a possibility of a plasmid-mediated acquisition due to the presence of resistance islands.
Introduction
Thailand is an important aquaculture producer and one of the top world fish exporting countries (Food and Agriculture Organization (FAO), 2018). The total production of Nile tilapia (Oreochromis niloticus) reached 217.9 metric tons in 2017 and was regarded the highest among other economically important freshwater aquaculture (52.7% of the total production yield) (Department of Fisheries (DOF), 2019). Nowadays, inland intensive farming system has become the standard practice for Nile tilapia production which corresponds to the continual increase of consumer demand. However, the intensive farming can lead to more stressful conditions and eventually bring the risk of infectious diseases which threatens the tilapia industry as a whole (Dong et al., 2015). Aeromonas veronii, an etiological organism of Motile Aeromonas Septicemia (MAS), contributed to major losses all-year-round with the cumulative mortality varying from 10 to 100% (Dong et al., 2017). More importantly, these bacteria can also cause disease in a wide range of hosts such as human, amphibian, and other aquatic animals (Martino et al., 2016). Antimicrobial therapy is a standard practice for controlling bacterial diseases occurring in tilapia farms. However, several antimicrobial groups that are allowed for use in Thailand aquaculture have also been widely used in human medication such as Beta-lactam, Quinolone, Tetracycline, or Sulfonamides (FCSTD, 2012). The situation of antimicrobial resistance (AMR) and multi drug resistance (MDR; resistant to at least one drug in three or more group of antimicrobials) (Magiorakos et al., 2012) has not only been found in human medicine but also in aquaculture as well, with the report of resistant isolates from different countries (Caruso, 2016; Deng et al., 2016; Baron et al., 2017; Yang et al., 2017; Roh et al., 2019). Wastewater from aquaculture may contain drug residues and resistant bacteria which subsequently pass on to the environment and, ultimately, the consumers. The AMR property can be transferred horizontally to other terrestrial bacteria through the mobile genetic elements (MGEs) (Subramani and Michael, 2017). Aeromonas spp. has been well-known as potential bacteria harboring several AMR determinants including plasmid, integron, and genomic island (Piotrowska and Popowska, 2015). However, concerning the current situation of A. veronii outbreaks in Thailand, antimicrobial susceptibility is very limited with little available AMR information. Therefore, this research aimed to gain the comprehensive understanding of AMR, as well as the potentially correlated genetic determinants in the recent isolates of A. veronii isolated from tilapia using a genome-wide investigation of the AMR genetic determinants (resistome analysis). Due to the potency of whole genome sequencing, WGS can simultaneously investigate the collection of resistance-associated genes which can lead to the in silico prediction of AMR traits (Zankari et al., 2012). Nevertheless, the information of intrinsic and acquired resistance genes can be beneficial to the public health concern by revealing the potential transferable AMR determinants in A. veronii and increases the awareness of the possible distribution of AMR traits in the future.
Materials and Methods
Bacterial Isolates
A total of 12 isolates of Aeromonas veronii were obtained from the bacterial collection of FID-RU (Fish Infectious Disease Research Unit, Chulalongkorn University, Thailand), which were isolated from Nile tilapia (Oreochromis niloticus) and hybrid red tilapia (O. mossambicus × O. niloticus) during the disease outbreaks from 2015 and 2018 in Thailand. The details of each isolate are presented in Table 1. The bacterial isolation method and biosecurity were approved by Chulalongkorn University, Faculty of Veterinary Science Institutional Biosafety Committee (CU-VET-IBC) under the permit ID: IBC 183104.
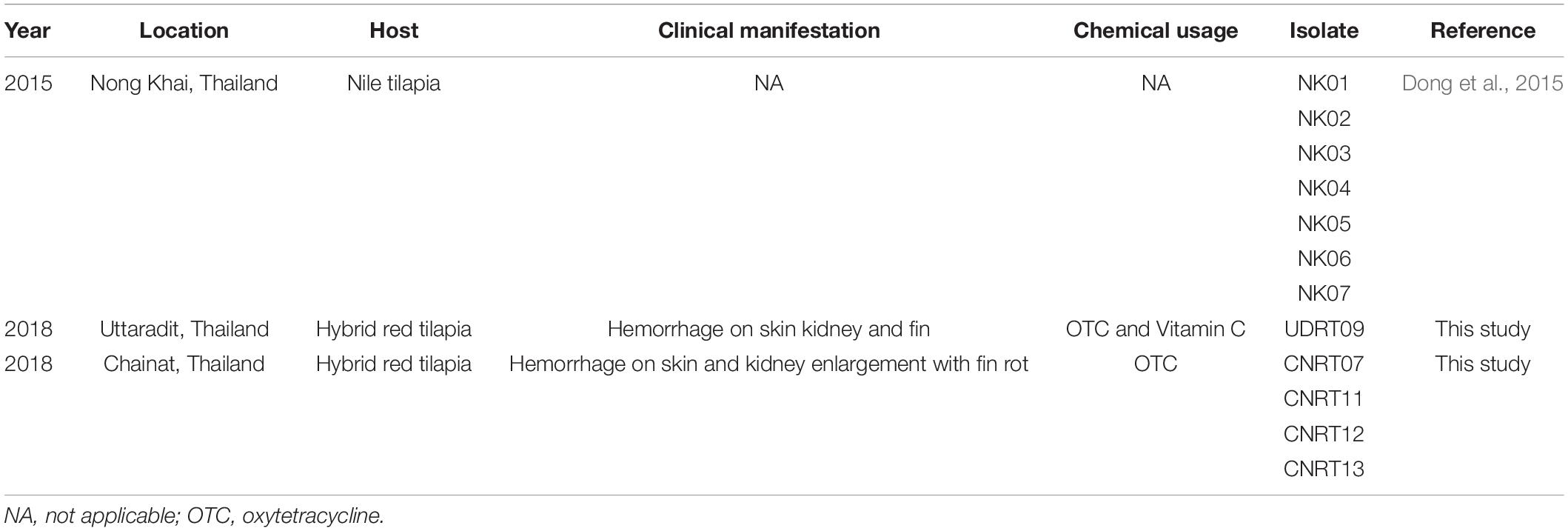
Table 1. Details of Aeromonas veronii isolates used in this study.
Taxonomic Identification
Aeromonas veronii stocks (TSB with 20% of glycerol) were recovered by streaking on a tryptic soy agar (TSA; Difco, United States) with an overnight incubation at 28°C. A single colony was then inoculated into 3 mL of tryptic soy broth (TSB; Bacto, United States) and incubated at 28°C for 24 h with shaking at 160 rpm. Bacterial suspension was used for DNA extraction using the Wizard Genomic DNA purification kit (Promega Corporation, United States) following the instructions of the manufacturer. To confirm the bacterial taxonomy, partial gyrB (∼1.4-kb-long) was amplified using the universal primers (gyrB3F: TCC GGC GGT CTG CAC GGC GT and gyrB14R: TTG TCC GGG TTG TAC TCG TC) with PCR conditions described in the original publication (Hoel et al., 2017). PCR product was separated on 1% agarose Tris-borate-EDTA gel using NucleoSpin® Gel and PCR cleanup (Macherey-Nagel, United States). The purified amplicon was submitted to FirstBASE laboratory service (Malaysia) for Sanger sequencing. The reads were assembled into a contiguous sequence using the ContigExpress software. The identity of the contig was searched against the NCBI nucleotide database using Megablast. Species level demarcation cut-off was set to a sequence coverage of ≥99% (Hoel et al., 2017).
Determination of the Minimum Inhibitory Concentration
A total of 12 isolates from the glycerol stock were recovered on the TSA supplemented with 5% sheep blood and incubated at 28°C for 24 h (Hassan et al., 2017). Each single colony was sub-cultured on the Mueller-Hinton agar (MHA) for the MIC determination by broth microdilution assay with eight antimicrobials including Amoxicillin, AMC [256–0.5 mg/L]; Ampicillin, AMP [256–0.5 mg/L]; Enrofloxacin, ENR [16–0.03 mg/L]; Florfenicol, FFC [64–0.125 mg/L]; Gentamicin, GEN [256–0.5 mg/L]; Oxolinic acid, OXO [64–0.125 mg/L]; Oxytetracycline, OTC [256–0.5 mg/L]; and Sulfamethoxazole + trimethoprim, SXT [256–0.5 mg/L] (Oxoid, United Kingdom) according to the CLSI guideline VET04 (Clinical and Laboratory Standards Institute (CLSI), 2014). The concentration of the bacterial suspension was adjusted by aliquoting 0.85% normal saline to obtain 0.5 McFarland turbidity. Subsequently, the adjusted bacterial suspension was mixed with the cation-adjusted Mueller-Hinton broth (CAMHB) containing an antimicrobial agent at a ratio of 1:1 (V/V) (Baron et al., 2017). Incubation was carried out at 28°C for 24 h. The MIC value was interpreted from a visible growth of bacteria in the medium solution. The resistance trait of each bacterial isolate was categorized into either resistant, sensitive, or multidrug resistant (at least one drug from three or more antimicrobial class) based on the epidemiological cut-off value of the Aeromonas species published previously (Baron et al., 2017). Herein, Escherichia coli ATCC 25922 was used as an internal control of this MIC assay.
Whole Genome Sequencing
Five representative A. veronii isolates, namely NK01 (resistant to three antimicrobial classes, 3MDR), NK02 (gentamicin resistant), NK07 (5MDR), UDRT09 (4MDR), and CNRT12 (sensitive to all tested drugs except beta-lactam antibiotics), were subjected to genome sequencing. Genomic DNA of these isolates was extracted by the Wizard Genomic DNA purification kit prior to RNaseA treatment to minimize RNA contamination (Promega Corporation, Madison, WI, United States). The integrity of the genomic DNA was determined by 1% agarose gel electrophoresis, whereas DNA purity and concentration were evaluated by the OD260/280 spectrophotometer and Qubit dsDNA BR Assay Kit Fluorometric Quantitation (Invitrogen, Carlsbad, CA, United States), respectively. Paired-end libraries were prepared using the NEBNext® UltraTM DNA Library Prep Kit for Illumina® and genome sequencing was performed using an Illumina HiSeq instrument with the read length of 150 bp.
Genome Assembly and Annotation
Adaptor sequences and low-quality bases (Q score < 25) were trimmed from raw reads using the Trimmomatic ver. 0.32 (Bolger et al., 2014). The improvement of the read quality was determined using the FastQC ver. 0.11.8.1 Trimmed reads were assembled into contigs by the SPAdes ver. 3.13.0 software and assembly quality was verified by the QUAST web service2 (Bankevich et al., 2012). To construct the scaffold, contigs were aligned to the reference genome (A. veronii B565, GCA_000204115.1) using the Medusa server.3 To improve the genome assembly quality, trimmed reads were mapped to the obtained scaffolds by the BWA software4 and gaps were filled using Pilon and GMcloser, sequentially (Li and Durbin, 2010; Walker et al., 2014; Kosugi et al., 2015). The assembled genomes were annotated automatically by the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) when the assembled genomes were submitted to the NCBI whole genome shotgun (WGS) web portal (Tatusova et al., 2016). The genomes were assigned the accession numbers GCA_012029595.1 (UDRT09), GCA_012029535.1 (CNRT12), GCA_012029545.1 (NK01), GCA_012029575.1 (NK02), and GCA_012029585.1 (NK07).
Genome-Based Phylogenetic Analysis
The genome-level taxonomic identification was conducted by submitting the assembled genomes to the Type Strain Genome Server5 in which the closest species was determined via the MASH algorithm (Meier-Kolthoff and Goker, 2019). Additionally, the average nucleotide identity (ANI) of the newly assembled genomes in comparison with A. veronii biovar sobria [strain 312M (GCA_003859745.1) and LMG13067 (GCA_000820385.1)] and biovar veronii [strain CECT4257 (GCA_000820225.1), CCM4359 (GCA_001908535.1), and C198 (GCA_013697145.1)] was calculated using the ANI calculator6 with the ANI cut-off value ≥ 95% (Jain et al., 2018).
Phylogenetic analysis of the newly assembled genomes and other reference strains (n = 15, Table 2) was carried out. The sequence type (ST) of the A. veronii genomes was classified in comparison with the PubMLST database using the Sequence Query tool provided in the PubMLST website (Jolley et al., 2018). The MLST scheme for Aeromonas spp. included the gltA, groL, gyrB, metG, ppsA, and redA genes. The DNA sequence of each locus was exported from the PubMLST: Sequence Query output page. Multilocus sequence analysis (MLSA) was conducted in the PhyloSuite v1.2.1 program based on the DNA sequence variation among these loci (Zhang et al., 2020). Briefly, a multiple sequence alignment of each locus was performed using MAFFT (Katoh and Standley, 2013). All aligned sequences were then concatenated into a contig, and the maximum likelihood tree was constructed using IQ-tree (Nguyen et al., 2015) under the TIM2 + R3 + F model for 5,000 ultrafast bootstraps (Minh et al., 2013). Herein, Aeromonas schubertii WL1483 (accession no. NZ_CP013067) was used as the outgroup for MLSA. The consensus tree was visualized using the MEGA X software (Kumar et al., 2018). A total of 14 gyrB sequences were retrieved from the A. veronii genomes and were used to generate the phylogenetic tree separately from the MLST tree. Aeromonas schubertii was used as an outgroup. Phylogenetic analysis was performed using the Molecular Evolutionary Genetic Analysis X (MEGA X) software (Kumar et al., 2018). The Maximum likelihood tree was constructed under the Tamura 3-parameter + GI model with 1,000 replicates. Moreover, a genome-level phylogenetic analysis was also performed based on the sequence variation in the core genome, also called genome-wide SNPs. Core genome among the distinct A. veronii genomes was detected and SNPs were called/concatenated/aligned using the automated web-tool CSI Phylogeny v1.4 using the default setting (Kaas et al., 2014). Aligned SNP sequence was used for the maximum likelihood tree reconstruction via the PhyloSuite program as described previously.
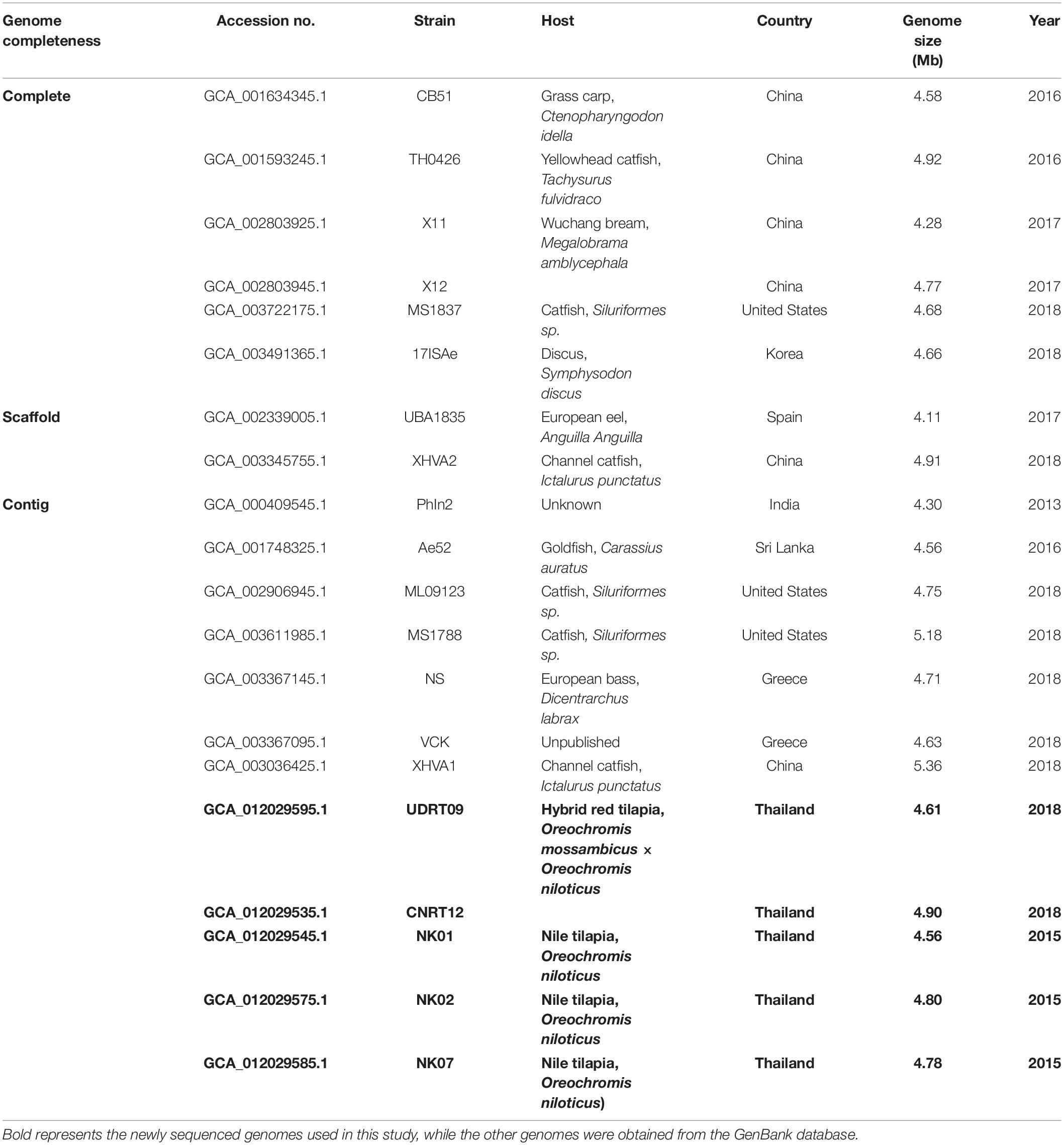
Table 2. List of Aeromonas veronii genome included in the resistome analysis.
Resistome Analysis
The A. veronii genomes listed in Table 2 were also used for the resistome analysis. The genomes were queried against the Comprehensive Antibiotic Resistance Database (CARD7) (Alcock et al., 2019) for the in silico prediction of the possible antimicrobial resistance gene (ARGs) contained in the genome. Resistance Gene Identifier (RGI) automate tool was used for the gene analysis in CARD. The search criteria were set as “perfect and strict hits only” with a “high quality/coverage” sequence quality. Nucleotide identity to query sequences lower than 96% were excluded (Lomonaco et al., 2018). In addition, the presence of acquired antimicrobial resistance genes in the A. veronii genomes were also analyzed through ResFinder V3.18 (Bortolaia et al., 2020). Genome (fasta file) was uploaded to the web resource with a 95% identity of sequence threshold and 80% minimum length of alignment setting (Lomonaco et al., 2018). The amino acid sequences of the predicted ARGs were downloaded from CARD and ResFinder and were used to construct the local blast database on the Blast2GO software in order to perform a reciprocal BLAST. The deduced amino acid sequences (proteome) from each A. veronii isolate of this study were queried against the local blast database using blastp implemented in the Blast2GO program (Conesa et al., 2005).
Genomic Islands Analysis
The presence of genomic island (GI) and resistance island (RI) within the newly sequenced A. veronii genomes (NK01, NK02, NK07, CNRT12, and UDRT09) were predicted using IslandViewer49 (Bertelli et al., 2017). The GenBank file was uploaded to the IslandViewer4 web interface in which A. veronii 17ISAe was selected as a reference genome for the alignment. GI was predicted using the IslandPick, SIGI-HMM, and IslandPath-DIMOB methods (Bertelli et al., 2017) implemented in IslandViewer. The output was manually compared to the complete A. veronii genomes (strain CB51, TH0426, X11, X12, MS1837, and 17ISAe).
Results
Taxonomic Identification
According to the partial gyrB sequence (1.4-kb-long), all bacterial isolates were mostly similar to A. veronii with a 98%–99% sequence identity (100% query coverage). Likewise, the TYGS web tool also indicated that the representative genomes of this study (NK01, NK02, NK07, CNRT12, and UDRT09) are genotypically closest to A. veronii CECT 4257. The digital DNA-DNA hybridization (dDDH) value of the current genomes in comparison with A. veronii CECT 4257 is 69.18% (d4 formula) and the GC% difference is 0.28. The dDDH cut-off for the bacterial species demarcation is ≥70%. Thus, the Aeromonad isolates used in this study should be classified as A. veronii. Herein, the type strain A. veronii CECT 4257 belongs to biovar veronii. This study also attempted to identify the biovar of the current isolates based on a pairwise genome comparison against both biovars of A. veronii, namely biovar sobria and biovar veronii. The ANI calculator revealed that the genomes of this study were similar to both biovar sobria (ANI = 96.05%–96.45%) and biovar veronii (ANI = 96.16%–96.48%). The ANI values in comparison with both biovars are demonstrated in the supplementary information (Supplementary Table 1). Since the ANI cut-off for species demarcation is ≥95%, we cannot differentiate the biovar of the current A. veronii isolates based on their genomes alone. However, there was evidence that biovar veronii could be differentiated from biovar sobria by the exhibition of ornithine decarboxylase negative and arginine dihydrolase positive activities (Abbott et al., 2003). According to the biochemical profiles shown in Supplementary Table 2, the recent isolates shared a similar characteristic with the isolates determined in our previous study (Dong et al., 2015). Thus, with reference to the biochemical profile, A. veronii in the present study can be distinguished as biovar veronii.
Determination of the Minimum Inhibitory Concentration
The broth microdilution method was used to evaluate the MIC and the results are demonstrated in Table 3. The results suggested that the isolates of this study were completely resistant to beta-lactams (ampicillin and amoxicillin; MIC > 256 mg/L). On contrary, all isolates were susceptible to florfenicol with the MIC lower than 1 mg/L. The percentage of resistance to other antimicrobials ranged from oxytetracycline (67%) to oxolinic acid (33%) and enrofloxacin (17%). More importantly, this research found the first resistance of A. veronii isolated from freshwater fish to gentamicin and Trimethoprim/sulfamethoxazole (MIC > 256 mg/L) in the NK02 and NK07 isolates, respectively. Furthermore, the isolates with a diverse degree of multidrug resistance (MDR), namely NK07 (six drugs from five antimicrobial classes, 5MDR), UDRT09 (four antimicrobial classes, 4MDR), NK01 (three antimicrobial classes, 3MDR), NK02 (gentamicin resistant), and CNRT12 (resistant to only beta-lactam, Sense), were included in the subsequent genome investigation.
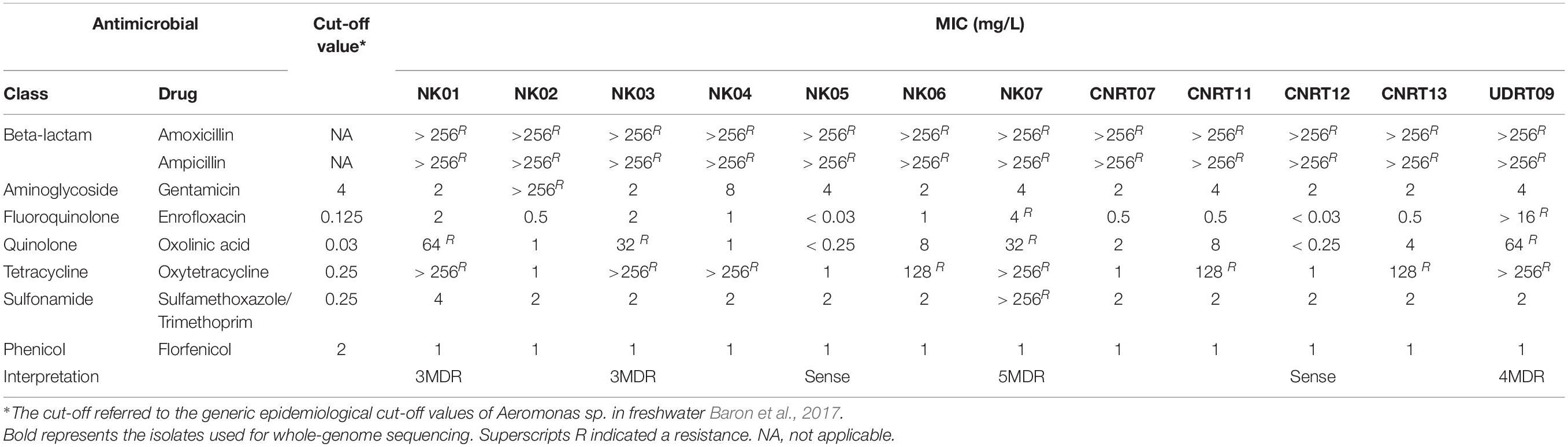
Table 3. MIC values Aeromonas veronii of this study.
Phylogenetic Analysis
Among the 20 A. veronii genomes used for the genome analysis, ST could be assigned to only 4 genomes, i.e., MS1837 (ST 254), 17ISAe (ST485), NS, and VCK (ST 23). On the other hand, the other isolates were defined as an unknown ST due to the presence of new allele variants (Supplementary Table 2). In this study, only four MLST loci (gltA, groA, metG, and recA), with a total length of 2,070 bp, were included for the phylogenetic tree reconstruction since gyrB and ppsA were absent in some isolates due to the incompleteness of the genomes. However, since gyrB is a key genetic marker for Aeromonas species identification, a phylogenetic analysis of gyrB was separately performed to compare with the MLST loci. The results from both the gyrB-based tree and MLSA-based tree suggested a high genetic diversity among the A. veronii isolates (Figure 1A and Supplementary Figure 2). The observed phylogeny was likely unrelated to the geographical origin, host species, and resistance phenotype of the analyzed taxa. For instance, the isolates from Thailand are clustered in distinct clade disperse, whereas the isolates NK02 and CNRT12 were grouped into the same subclade regardless of the difference in the resistance phenotype (aminoglycoside resistant vs. sensitive). Similarly, the phylogenetic tree based on the genome-wide SNPs also suggested a high genetic variability (Figure 1B). The numbers of core genome SNPs (shown in the Supplementary Figure 1) of the pairwise comparison across the A. veronii genomes were diverse with between 11 and 45,536 SNPs. It is worth mentioning that two out of five newly assembled genomes (NK01 and UDRT09) were almost genetically identical (only 27 SNPs) and have similar resistance phenotypes (resistant to quinolone, tetracycline, and beta-lactam). This indicated that NK01 and UDRT09 were derived from the same clone.
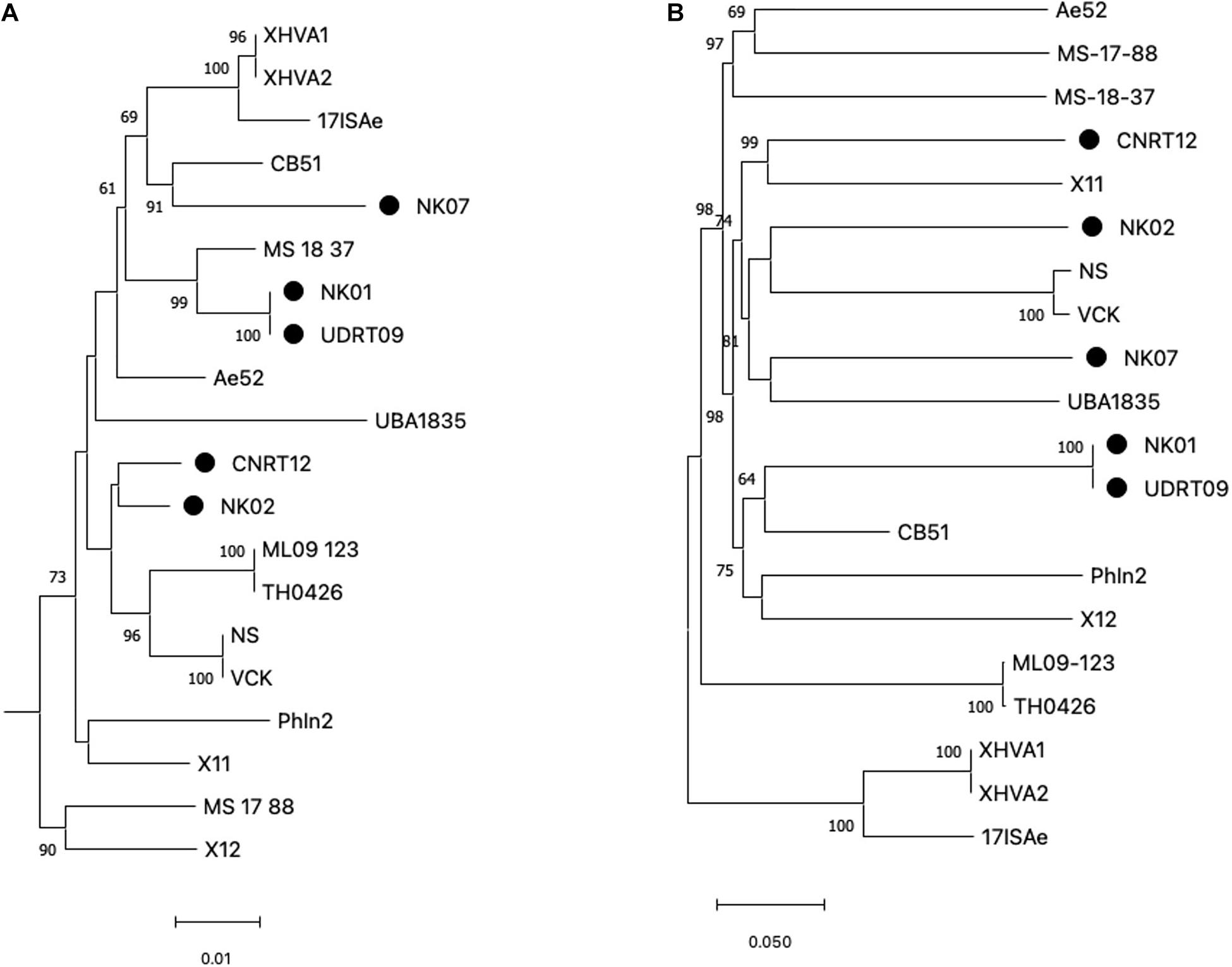
Figure 1. Maximum likelihood tree generated from concatenated MLST loci (A) and genome-wide SNPs (B). MLST loci included gltA, groA, metG, and recA with the total length of 2,070 bp and Aeromonas schubertii WL1483 as an outgroup (omitted from the tree). The genome-wide SNPs was concatenated into contig with a total length of 175,597 bp. Trees were constructed using IQ-tree with 5,000 ultrabootstrap. Numbers at the tree node represent the bootstrap value in percentage (only ≥ 60 value is shown). The newly assembled genomes (Thai isolates) are indicated by the filled circle adjacent to the taxa. Scale bar represents the nucleotide substitution per site.
Resistome Analysis
Aeromonas veronii genomes were queried against two AMR databases, namely CARD and ResFinder, to identify the ARG repertoire, also known as resistome. The bacteria used in this analysis comprised of (i) the strains isolated from tilapia and (ii) other freshwater fishes whose genomes were retrieved from the NCBI (Table 2). The potential ARGs observed in each group of A. veronii strains were described as follows:
Resistome of Aeromonas veronii Isolated From Tilapia
There were 17 ARGs identified from the newly assembled genomes derived from tilapia (n = 5; NK01, NK02, NK07, UDRT09, and CNRT12). These ARGs can be categorized into nine groups according to the antimicrobial classes, i.e., aminoglycoside, beta-lactam, chloramphenicol, macrolide, organic compound, quinolone, sulfonamide, tetracycline, and multidrug resistance-associated (Figure 2). Among the five isolates, UDRT09 and NK07 carried the greatest number of ARGs (14 genes), while NK02, NK01, and CNRT12 carried 13, 12, and 12 genes, respectively. Of these 17 ARGs, 11 genes [ceph-A3, qnr32, dfrA12, mcr-3, sul1, tetA, tetC, tetD, tetE, aac(6’)-Ib-cr, and ade-F] were shared by all A. veronii tilapia isolates. Herein, two ARGs associated with macrolide resistant (mphA), aminoglycoside resistant (aac(3)-IIb), and chloramphenicol (catA1) were specifically found in NK07, NK02, and UDRT09, respectively.
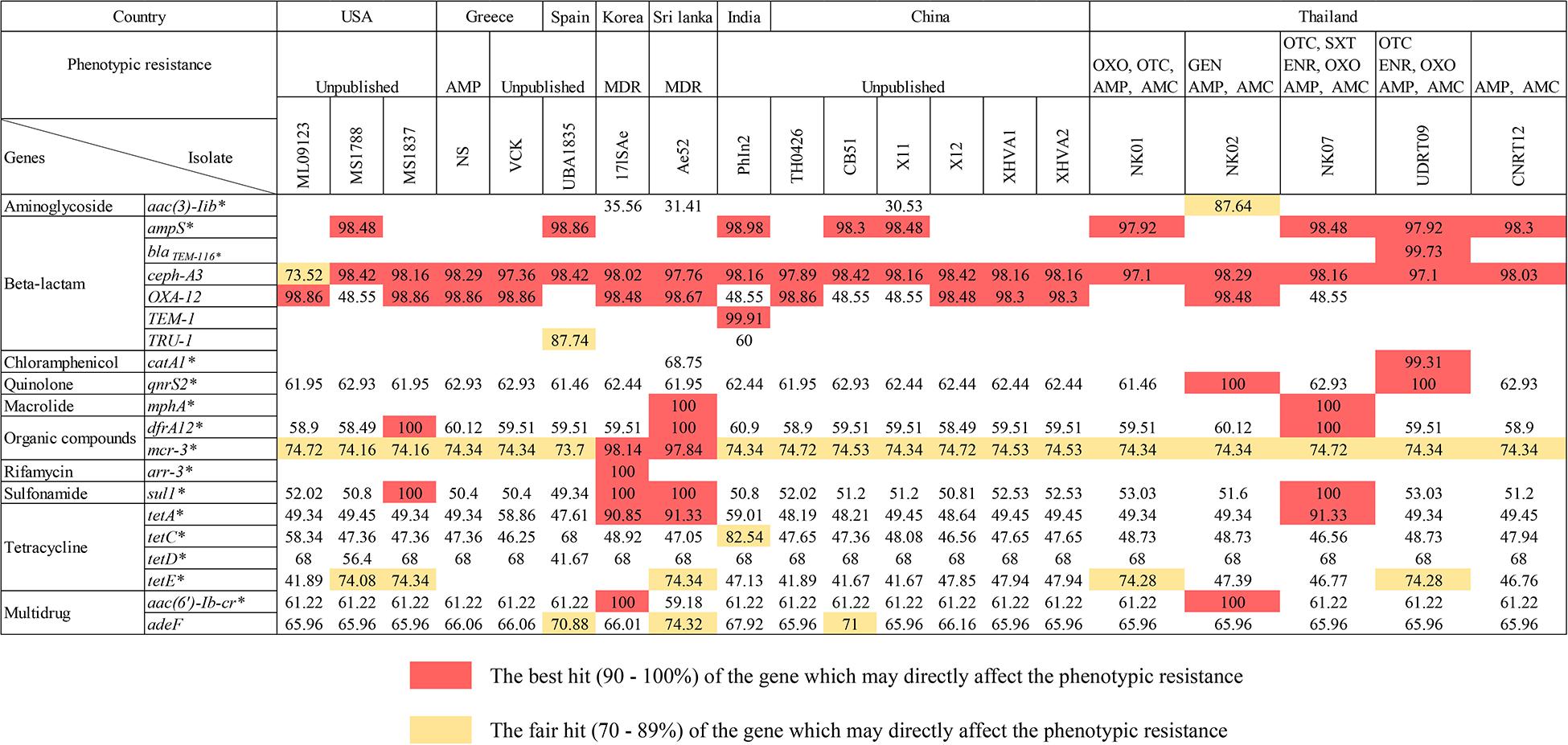
Figure 2. The presence of ARGs within the genomes of A. veronii isolated from freshwater fishes (n = 20). The numbers represent the pairwise percent identity (obtained from protein BLAST) using the deduced amino acid sequence and its homolog as query and subject sequence, respectively. High percent identity is suggested by a darker color, whereas an empty box indicated that no homologous gene is observed in either the CARD or ResFinder database. Strains isolated from tilapia (newly sequenced genomes) are defined by an underlined name. Acquired antimicrobial resistant genes are labeled with an asterisk.
Resistome of Aeromonas veronii Isolated From Other Freshwater Fishes
A total of 19 ARGs (ten drug classes) were identified from 15 genomes of A. veronii associated with an invasive disease in various freshwater fish species (Figure 2). The genomes from PhIn2 and Ae52 (isolated from India and Sri Lanka) harbored the highest number of ARGs (15 genes), followed by X11 (China), CB51 (China), 17ISAe (Korea), and MS1788 (United States) which carried 14, 13, 13, and 13 ARGs, respectively. The strains UBA1835 (Spain); ML09123 and MS1837 from United States; and TH0426, X12, XHVA1, and XHVA2 from China each carried 12 ARGs. Lastly, 11 ARGs were detected in VCK and NS from Greece. There was one strain, namely 17ISAe, that carried the rifamycin resistance gene (arr-3) which was absent in other A. veronii isolates, including the tilapia isolates.
Concerning the sharing of ARGs among the Aeromonad population, there were 16 ARGs detected in both tilapia and freshwater fish isolates (n = 20). The following genes: aac (6′)-lb-cr, adeF, cphA3, qnrS2, drf A12, mcr-3, sul-1, tetA, tetC, and tetD were detected in all isolates. The OXA-12 gene was observed in only two out of five tilapia isolates (NK02 and NK09), but abundant in the strains isolated from other fish species (14 out of 15). This was similar to the absence of tetE in only four isolates from freshwater fish. In addition, catA1 and mphA were identified in one strain, i.e., Ae52, similar to the gene observed in UDRT09 and NK07 from the tilapia group.
Genomic Islands Analysis
Since the newly sequenced genomes used for GI detection are incomplete genomes, any predicted GI located near the boundary of contigs (< 1,000 bp from 5′ and 3′ termini) were omitted from this analysis to avoid a false positive detection. Herein, IslandViewer 4 predicted the presence of potential GIs in every tilapia A. veronii isolate genome. A manual investigation of the gene contents resided within these potential GIs showed that the NK07 isolate harbored a large resistance island (RI) with a size of 22,323 bp (Figure 3). This RI contained several AMRs and heavy metal resistance genes (MRGs) [mercury resistance (mer) operon and chromate (chrA) efflux pump]. There were at least six AMRs potentially associated with the resistant to diverse drug classes including sulfonamide (sul1), trimethoprim (drfA12), tetracycline (tetR/acrR), aminoglycoside (aadA2), macrolide (mphA), and multidrug efflux pump (MFS transporter). Additionally, this NK07-derived RI contained one antiseptic resistance gene (qacE). Interestingly, some resistance elements, i.e., sul1, qacE, and trimethropim-resistant dihydrofolate reductase (dfrA), were also observed in an important 19-kb-long RI presented in the reference strain 17ISAe (Figure 3), although other genetic contents were dissimilar to the NK07-derived RI. Regarding the other A. veronii tilapia isolates, the 12-kb-long RI containing tetracycline resistance genes (tetE and tetR) was identified in the genome of the NK01 and UDRT09 isolates (Supplementary Figure 3), while no GI carrying AMRs was observed in the NK02 and CNRT12 isolates.
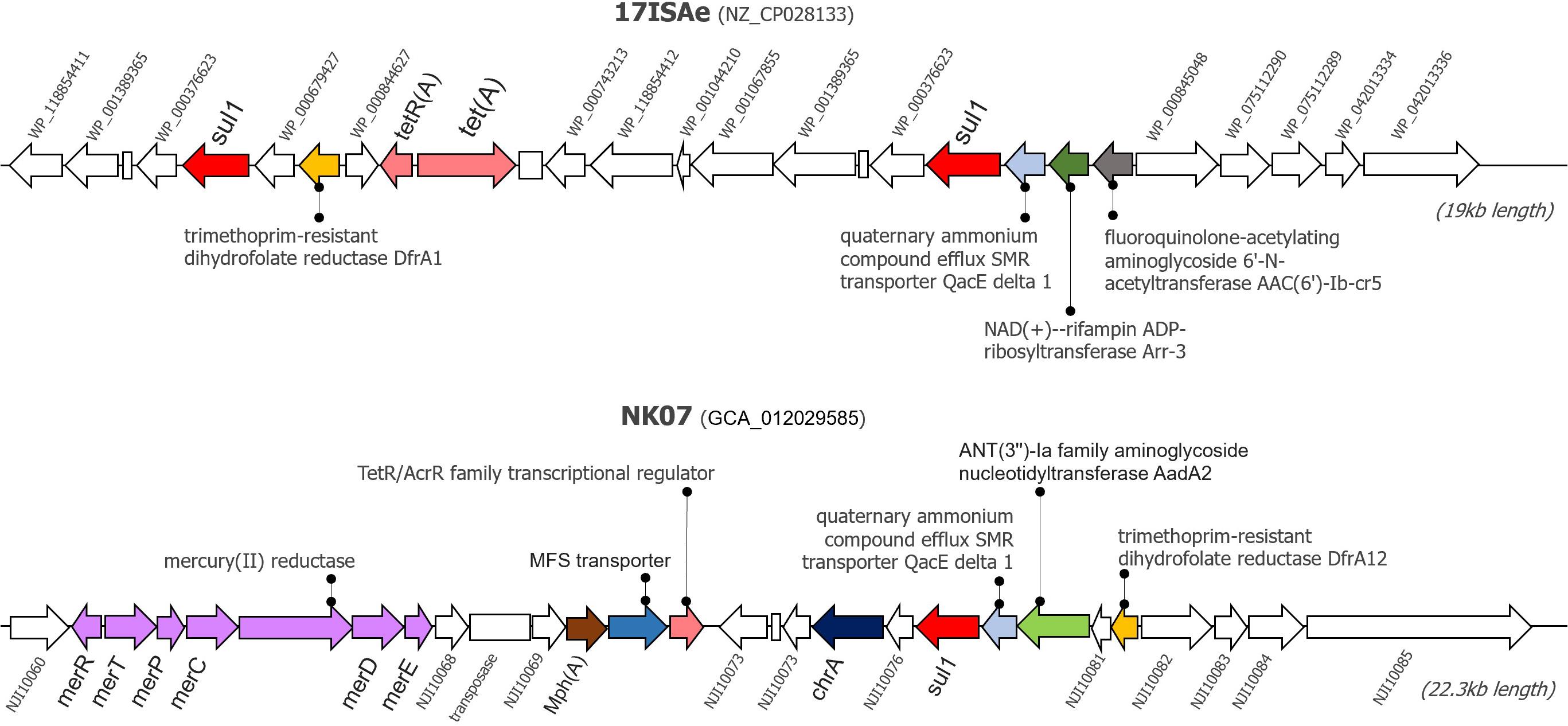
Figure 3. Graphical representation of resistance islands presented in the genome of A. veronii 17ISAe and NK07. These resistance islands were predicted using the IslandPath-DEMOB and SIGI-HMM methods on the IslandViewer 4 web server. Arrows represents the coding sequences, whereas an empty box indicates a pseudogene. CDS accession numbers or gene name are described adjacent to the gene. Antimicrobial and heavy metal resistance genes are labeled by color.
Discussion
Aeromonad infection is one of the global concerns to the loss of aquaculture production. Due to the rise of antimicrobial resistance (AMR), the solution and choice of antimicrobial use is a challenge to the farmer and researcher. In this study, Aeromonas spp. were isolated from tilapia and according to our findings, these were classified as A. veronii biovar veronii based on their biochemical characterization (Abbott et al., 2003).
Resistome analysis is capable of investigating the comprehensive data of AMR to improve the efficacy of antimicrobial use (Zankari et al., 2012). As a result, in the MIC assay, florfenicol showed the best efficacy against A. veronii. However, the isolates in this study were altogether resistant to beta-lactams (amoxicillin and ampicillin) with a high MIC value, similar to that reported in the previous publication (Janda and Abbott, 2010; Yang et al., 2017). Referring to the epidemiological cut-off values from the study of Aeromonas diversity in France (Baron et al., 2017), the MIC values of A. veronii in this study were seriously higher than the previous publication. Likewise, this is the first discovery of gentamicin and sulfamethoxazole/trimethoprim resistant A. veronii isolated from tilapia. These rare resistances were reported previously in the clinical and environmental samples from Australia where Aeromonas spp. was susceptible to gentamicin and sulfamethoxazole/trimethoprim, with more than 98% and 99%, respectively (Aravena-Roman et al., 2012).
According to the licensed antimicrobials allowed for use in Thailand (FCSTD, 2012), oxytetracyclines are commonly used in tilapia farming which promotes a high resistance as evidenced by the MIC values that were higher than those in the previous report (Skwor et al., 2014). In addition, oxolinic acid and enrofloxacin were both less effective against A. veronii, in contrast to the study from China whereby A. veronii isolated from Chinese long-snout catfish was susceptible to these antimicrobials (Cai et al., 2012). Besides, A. veronii isolated from tilapia revealed a resistance to multiple antimicrobials similar to the study in channel catfish (Yang et al., 2017). MDR was observed mainly in NK07 (5MDR) followed by UDRT09 and NK01, which are 4MDR and 3MDR, respectively.
Regarding the ARGs, their presence and transmission were implicated in the efficacy of human and animal diseases treatment caused by the resistant A. veronii (Yang et al., 2017). The resistome analysis of the recent A. veronii isolates from tilapia and other freshwater fish isolates was performed by the consideration of the ARGs associated with a phenotypic resistance expression. It revealed 20 ARGs, which belong to nine groups of antimicrobial classes as shown in Figure 2. Overall, the ARGs were shared among the isolates from both tilapia and other fish species. The reciprocal protein BLAST against CARD-derived ARGs sequences indicated a relatively high percent identity (over 80%) suggesting a potentially complete or functional genes in this study.
Aminoglycoside Resistome
The resistant phenotype observed in NK02 was also in accordance with the presence of the potentially completed aminoglycoside resistance genes [aac (3)-IIb and aac (6′)-Ib-cr] exclusively found in its genome. Notably, there were few reports of aminoglycoside resistance in Aeromonas sp. from channel catfish and discus (Yang et al., 2017; Roh et al., 2019). In any case without evidence of drug use before, ARGs may then be acquired from the other pathogens or surrounding the environment (Heuer et al., 2002).
Beta-Lactam Resistome
Beta-lactams are broadly used worldwide; however, the recent A. veronii isolates were resistant to both amoxicillin and ampicillin categorized as broad-spectrum beta-lactams. The set of beta-lactam resistance genes were detected in all isolates which are typically found in the Aeromonas species (Chen et al., 2019). Previously, A. hydrophila and other species of Aeromonas have been reported as intrinsically ampicillin-resistant (Joseph et al., 1979; Yucel and Aslim, 2005). However, there is no official report of intrinsic resistance in A. veronii. Referring to the MIC values that were supported by the presence of beta-lactams resistance-associated genes (ampS, ceph-A3 and OXA-12) with over a 97% sequence identity, A. veronii should be noted as an intrinsically broad-spectrum beta-lactam resistant as mentioned in the previous publications (Baron et al., 2017). Extended spectrum beta-lactamases (ESBLs) are enzymes which enable bacteria to hydrolyze an extended spectrum cephalosporin (Ghafourian et al., 2015). So far, there are no reports about ESBL drug group use in Thai aquaculture. However, the ESBL-related gene detected in this study was probably acquired from agricultural, human medication, or other sources as reported from a previous publication (Piotrowska et al., 2017). Further investigation of the phenotypic susceptibility to ESBL is needed.
Quinolone and Fluoroquinolone Resistome
The presence of the qnrS2 gene refers to the plasmid-mediated quinolone resistance protein which was originally found in Salmonella enterica and plays a role on horizontal gene transfer (Jia et al., 2017). This gene was detected in all isolates but only showed a perfect identity in NK02 and UDRT09. However, there were few publications which reported that A. veronii encoded the qnrS2 gene on a plasmid since the first report in 2008 (Sanchez-Cespedes et al., 2008). Generally, missense mutations in DNA gyrase (gyrA or gyrB) and topisomerase IV (parC or parE) encoded genes are common mechanisms that confer a resistance to fluoroquinolones while qnrS2 plays a role as a supportive resistance gene (Sanchez-Cespedes et al., 2008; Redgrave et al., 2014).
Tetracycline Resistome
Five ARGs were blasted against the A. veronii isolates, and these included the tetA, tetC, tetD, tetE, and adeF genes. The adeF works as a secondary resistance gene and enhances tetracycline and fluoroquinolone resistance (Mobasseri et al., 2018). In addition, a set of tet genes are located on a plasmid and functionally work for tetracycline resistance (Jia et al., 2017). Similar to the previous study, A. veronii resistance to tetracycline and their associated genes have been reported worldwide (Skwor et al., 2014; Baron et al., 2017; Yang et al., 2017). The set of tetA, tetC, tetD, tetE, and adeF genes were found in most of the isolates. As seen in the NK01, NK07, and UDRT09 isolates, the involvement of tet genes (high percent identity) coincide with higher MIC values as mentioned in the previous publication (Balassiano et al., 2007).
Sulfonamide Resistome
A sulfonamide resistance gene (sul1) is a gene encoding dihydropteroate synthase and it was reported as multidrug resistance mediated by class 1 integrons in Aeromonas (Deng et al., 2016). The gene was presented in all strains used in this analysis, although only four strains (MS1788, 17ISAe, Ae52, and NK07) presented a 100% sequence identity. Interestingly, the presence of a complete sul1 in NK07 consistent to its Sulfamethoxazole/Trimethoprim resistance phenotype was indicated by the MIC assay.
Other Resistance Genes
The group of class 1 integron resistance association consists of genes catA1 and dfrA12. These genes are acquired differently from other pathogens. Generally, catA1 is a gene encoding chloramphenicol acetyltransferase from Shigella flexneri 2a and dfrA12 is a gene encoding dihydrofolate reductase from Vibrio cholera (Jia et al., 2017). Similar to this study, catA1 was detected in A. salmonicida and recently in A. veronii (Tanaka et al., 2016; Syrova et al., 2018). In addition, dfrA12 was reported as MDR mediated by class 1 integrons in Aeromonas isolates (Deng et al., 2016). However, the effect of the high identity of catA1 and dfrA12 gene to the phenotypic resistance was not evaluated in UDRT09 and NK07. Lastly, mcr-3, a transferable colistin resistance gene which was firstly isolated from the pWJ1 plasmid of E. coli, showed a high amino acid identity to the Aeromonas species in this study (Yin et al., 2017). However, an evaluation of the colistin MIC value to confirm the potential contribution of the gene to phenotypic resistance was not performed in the current study.
Genomic Islands Analysis
Genomic Islands analysis enables the prediction of the potential resistance elements known as the Horizontal Gene Transfer (HGT) foundation (Soares et al., 2016). This study analyzed the potential of resistance and GIs association as clearly seen on the NK07 resistance island (NK07-RI). First of all, NK07-GIs are larger than 17ISAe (MDR isolate from discus) and much larger when compared to the less resistant isolates (NK01 and UDRT09). ARGs (sul1, drfA12, tetR/acrR, aadA2, and mphA) of NK07-RI had perfect sequence identity hits which are related to high MIC values. These genes have been reported as a mobile genetic element associated, as described before. Multidrug efflux pump (MFS transporter) plays an important role together with the mobile element protein to promote the MDR isolate (Pasqua et al., 2019). Moreover, antiseptic resistance gene (qacE) detected in NK07-RI has been studied in many pathogens with the effect of reducing the susceptibility of biocide (antiseptics and disinfectants) and antimicrobials (Vijayakumar and Sandle, 2019). Although, there is no study about this effect in A. veronii, the presence of qacE and resistance potential should be of concern. As seen in Figure 3, several MRGs were detected in NK07-RI. The chromate efflux pump associated gene (chrA) and mercury resistance (mer) operon are both generally encoded on a plasmid which mainly works on cell detoxify mechanism (Boyd and Barkay, 2012; Baaziz et al., 2017). Previously, Mer operon was detected in MDR A. veronii strain MS-18-37 isolated from United States catfish but no study about its function was demonstrated (Abdelhamed et al., 2019). Due to the ability of heavy metal uptake mechanism, Aeromonas spp. was tested as a bioremediation property for wastewater treatment (Ogugbue and Sawidis, 2011). This finding represents an adaptation ability to selective pressure in a microorganism induced by heavy metal residues contaminated in a water source. Although, there are several studies of MRGs with ARGs, their functions remain unclear (Chen et al., 2019).
Conclusion
Resistome analysis of A. veronii isolated from tilapia in Thailand provided evidence that conventional antimicrobials used in aquaculture are going to lose their effectiveness. According to the licensed antimicrobials allowed for use in Thailand, amoxicillin, oxytetracycline, and oxolinic acid may not be recommended for a longer use, likewise, enrofloxacin requires a high dosage for its usage (more than 16 mg/L) but there should be concern about the effect of resistant A. veronii in human medication. The last choice of the recommended antimicrobial use is sulfamethoxazole/trimethoprim and florfenicol (after license announcement by the FDA). In this study, A. veronii isolates were adapted into a multidrug-resistance related to the presence of multiple ARGs, and several genes were shared in the aquatic system among the A. veronii population worldwide. The prevalence of resistance against ESBL, beta-lactam, and colistin in A. veronii is highlighted and requires more insight study. Moreover, the possibility of plasmid-mediated resistance gene acquisition especially in gentamicin and sulfamethoxacin should be of concern as these can affect human and other animal health care. It should be noted that A. veronii has a broad-spectrum beta-lactam intrinsic resistance as revealed by the current and previous studies. At last, the outcomes of this study can be applied for AMR prediction and further treatment plans for effective antimicrobial use.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA612772.
Author Contributions
RS, PC, ND-H, and PK: data curation. RS and PK: investigation and writing of the original draft. IH and CR: resources. PK and CR: supervision and validation. RS, PC, ND-H, ES, PK, RC, and CR: writing—review and editing. CR: funding acquisition. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Funding
This study was financially supported by the Oversea Research Experience Scholarship for Graduate Student, Chulalongkorn University, the 90th year Anniversary of Chulalongkorn University Scholarship (Ratchadaphiseksomphot Endowment Fund), and the Thailand Science Research and Innovation (TSRI) Fund [Grant number Chulalongkorn University_FRB64001_01_31_6].
Acknowledgments
We would like to thank the Laboratory of Genome Science and Technology, Graduate School of Marine Science and Technology, Tokyo University of Marine Science and Technology for providing the whole genome sequence facilities.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.733668/full#supplementary-material
Footnotes
- ^ http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- ^ http://quast.bioinf.spbau.ru/
- ^ http://combo.dbe.unifi.it/medusa
- ^ http://bio-bwa.sourceforge.net/
- ^ https://tygs.dsmz.de/
- ^ https://www.ezbiocloud.net/tools/ani
- ^ http://arpcard.mcmaster.ca/
- ^ http://cge.cbs.dtu.dk/services/ResFinder
- ^ https://www.pathogenomics.sfu.ca/islandviewer
References
Abbott, S. L., Cheung, W. K., and Janda, J. M. (2003). The genus Aeromonas: biochemical characteristics, atypical reactions, and phenotypic identification schemes. J. Clin. Microbiol. 41, 2348–2357. doi: 10.1128/jcm.41.6.2348-2357.2003
Abdelhamed, H., Lawrence, M. L., and Waldbieser, G. (2019). Complete genome sequence data of multidrug-resistant Aeromonas veronii strain MS-18-37. Data Brief 23:103689. doi: 10.1016/j.dib.2019.01.037
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2019). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525. doi: 10.1093/nar/gkz935
Aravena-Roman, M., Inglis, T. J., Henderson, B., Riley, T. V., and Chang, B. J. (2012). Antimicrobial susceptibilities of Aeromonas strains isolated from clinical and environmental sources to 26 antimicrobial agents. Antimicrob. Agents Chemother. 56, 1110–1112. doi: 10.1128/AAC.05387-11
Baaziz, H., Gambari, C., Boyeldieu, A., Ali Chaouche, A., Alatou, R., Méjean, V., et al. (2017). ChrASO, the chromate efflux pump of Shewanella oneidensis, improves chromate survival and reduction. PLoS One 12:e0188516. doi: 10.1371/journal.pone.0188516
Balassiano, I. T., Bastos, M. D. C. D. F., Madureira, D. J., Da Silva, I. G., De Freitas-Almeida, A. C., and De Oliveira, S. S. (2007). The involvement of tetA and tetE tetracycline resistance genes in plasmid and chromosomal resistance of Aeromonas in Brazilian strains. Mem. Inst. Oswaldo Cruz 102, 861–866. doi: 10.1590/s0074-02762007005000121
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Baron, S., Granier, S. A., Larvor, E., Jouy, E., Cineux, M., Wilhelm, A., et al. (2017). Aeromonas Diversity and Antimicrobial Susceptibility in Freshwater-An Attempt to Set Generic Epidemiological Cut-Off Values. Front. Microbiol. 8:503. doi: 10.3389/fmicb.2017.00503
Bertelli, C., Laird, M. R., Williams, K. P., Simon Fraser University Research Computing, G, Lau, B. Y., Hoad, G., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bortolaia, V., Kaas, R. S., Ruppe, E., Roberts, M. C., Schwarz, S., Cattoir, V., et al. (2020). ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 75, 3491–3500. doi: 10.1093/jac/dkaa345
Boyd, E., and Barkay, T. (2012). The Mercury Resistance Operon: from an Origin in a Geothermal Environment to an Efficient Detoxification Machine. Front. Microbiol. 3:349. doi: 10.3389/fmicb.2012.00349
Cai, S. H., Wu, Z. H., Jian, J. C., Lu, Y. S., and Tang, J. F. (2012). Characterization of pathogenic Aeromonas veronii bv. veronii associated with ulcerative syndrome from chinese longsnout catfish (Leiocassis longirostris günther). Br. J. Microbiol. 43, 382–388. doi: 10.1590/s1517-83822012000100046
Caruso, G. (2016). Antibiotic Resistance in Fish Farming Environments: a Global Concern [Online]. J. FisheriesSciences.com 10, 9–13.
Chen, J., Li, J., Zhang, H., Shi, W., and Liu, Y. (2019). Bacterial Heavy-Metal and Antibiotic Resistance Genes in a Copper Tailing Dam Area in Northern China. Front. Microbiol. 10:1916. doi: 10.3389/fmicb.2019.01916
Clinical and Laboratory Standards Institute (CLSI) (2014). Performance Standards For Antimicrobial Susceptibility Testing Of Bacteria Isoloated From Aquatic Animals; Second Information Supplement. Annapolis Junction: Clinical and Laboratory Standards Institute.
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Deng, Y., Wu, Y., Jiang, L., Tan, A., Zhang, R., and Luo, L. (2016). Multi-Drug Resistance Mediated by Class 1 Integrons in Aeromonas Isolated from Farmed Freshwater Animals. Front. Microbiol. 7:935. doi: 10.3389/fmicb.2016.00935
Department of Fisheries (DOF) (2019). Fisheries Statistics of Thailand. Bangkok: Department of Fisheries, Ministry of Agriculture.
Dong, H. T., Nguyen, V. V., Le, H. D., Sangsuriya, P., Jitrakorn, S., Saksmerprome, V., et al. (2015). Naturally concurrent infections of bacterial and viral pathogens in disease outbreaks in cultured Nile tilapia (Oreochromis niloticus) farms. Aquaculture 448, 427–435. doi: 10.1016/j.aquaculture.2015.06.027
Dong, H. T., Techatanakitarnan, C., Jindakittikul, P., Thaiprayoon, A., Taengphu, S., Charoensapsri, W., et al. (2017). Aeromonas jandaei and Aeromonas veronii caused disease and mortality in Nile tilapia, Oreochromis niloticus (L.). J. Fish Dis. 40, 1395–1403. doi: 10.1111/jfd.12617
Food and Agriculture Organization (FAO) (2018). THE STATE OF THE WORLD series of the Food and Agriculture Organization of the United Nations. Rome: Food and Agriculture Organization.
FCSTD (2012). Veterinary Drugs, Chemicals And Hazardous Substances With Standard Aquaculture [Online]. Department of fisheries. Available online at: https://www.fisheries.go.th/thacert/index.php/knowledge/76-drug-animal (Accessed September 1, 2018)
Ghafourian, S., Sadeghifard, N., Soheili, S., and Sekawi, Z. (2015). Extended Spectrum Beta-lactamases: definition, Classification and Epidemiology. Curr. Issues Mol. Biol. 17, 11–21.
Hassan, M. A., Noureldin, E. A., Mahmoud, M. A., and Fita, N. A. (2017). Molecular identification and epizootiology of Aeromonas veronii infection among farmed Oreochromis niloticus in Eastern Province, KSA. Egypt. J. Aquat. Res. 43, 161–167. doi: 10.1016/j.ejar.2017.06.001
Heuer, H., Krögerrecklenfort, E., Wellington, E. M., Egan, S., van Elsas, J. D., van Overbeek, L., et al. (2002). Gentamicin resistance genes in environmental bacteria: prevalence and transfer. FEMS Microbiol. Ecol. 42, 289–302. doi: 10.1111/j.1574-6941.2002.tb01019.x
Hoel, S., Vadstein, O., and Jakobsen, A. N. (2017). Species Distribution and Prevalence of Putative Virulence Factors in Mesophilic Aeromonas spp. Isolated from Fresh Retail Sushi. Front. Microbiol. 8:931. doi: 10.3389/fmicb.2017.00931
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Janda, J. M., and Abbott, S. L. (2010). The genus Aeromonas: taxonomy, pathogenicity, and infection. Clin. Microbiol. Rev. 23, 35–73. doi: 10.1128/CMR.00039-09
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Jolley, K. A., Bray, J. E., and Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 3:124. doi: 10.12688/wellcomeopenres.14826.1
Joseph, S. W., Daily, O. P., Hunt, W. S., Seidler, R. J., Allen, D. A., and Colwell, R. R. (1979). Aeromonas Primary Wound Infection of a Diver in Polluted Waters. J. Clin. Microbiol. 10, 46–49. doi: 10.1128/jcm.10.1.46-49.1979
Kaas, R. S., Leekitcharoenphon, P., Aarestrup, F. M., and Lund, O. (2014). Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS One 9:e104984. doi: 10.1371/journal.pone.0104984
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kosugi, S., Hirakawa, H., and Tabata, S. (2015). GMcloser: closing gaps in assemblies accurately with a likelihood-based selection of contig or long-read alignments. Bioinformatics 31, 3733–3741. doi: 10.1093/bioinformatics/btv465
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Lomonaco, S., Crawford, M. A., Lascols, C., Timme, R. E., Anderson, K., Hodge, D. R., et al. (2018). Resistome of carbapenem- and colistin-resistant Klebsiella pneumoniae clinical isolates. PLoS One 13:e0198526. doi: 10.1371/journal.pone.0198526
Magiorakos, A. P., Srinivasan, A., Carey, R. B., Carmeli, Y., Falagas, M. E., Giske, C. G., et al. (2012). Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 18, 268–281. doi: 10.1111/j.1469-0691.2011.03570.x
Meier-Kolthoff, J. P., and Goker, M. (2019). TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 10:2182. doi: 10.1038/s41467-019-10210-3
Minh, B. Q., Nguyen, M. A., and von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi: 10.1093/molbev/mst024
Mobasseri, P., Azimi, L., Salehi, M., Hosseini, F., and Fallah, F. (2018). Distribution and Expression of Efflux Pump Gene and Antibiotic Resistance in Acinetobacter baumannii. Arch. Clin. Infect. Dis. 13:e67143. doi: 10.5812/archcid.67143
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Ogugbue, C. J., and Sawidis, T. (2011). Bioremediation and Detoxification of Synthetic Wastewater Containing Triarylmethane Dyes by Aeromonas hydrophila Isolated from Industrial Effluent. Biotech. Res. Int. 2011:967925. doi: 10.4061/2011/967925
Pasqua, M., Grossi, M., Zennaro, A., Fanelli, G., Micheli, G., Barras, F., et al. (2019). The Varied Role of Efflux Pumps of the MFS Family in the Interplay of Bacteria with Animal and Plant Cells. Microorgan 7:285. doi: 10.3390/microorganisms7090285
Piotrowska, M., and Popowska, M. (2015). Insight into the mobilome of Aeromonas strains. Front. Microbiol. 6:494. doi: 10.3389/fmicb.2015.00494
Piotrowska, M., Przygodzinska, D., Matyjewicz, K., and Popowska, M. (2017). Occurrence and Variety of beta-Lactamase Genes among Aeromonas spp. Isolated from Urban Wastewater Treatment Plant. Front. Microbiol. 8:863. doi: 10.3389/fmicb.2017.00863
Redgrave, L. S., Sutton, S. B., Webber, M. A., and Piddock, L. J. (2014). Fluoroquinolone resistance: mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol. 22, 438–445. doi: 10.1016/j.tim.2014.04.007
Roh, H. J., Kim, B. S., Kim, A., Kim, N. E., Lee, Y., Chun, W. K., et al. (2019). Whole-genome analysis of multi-drug-resistant Aeromonas veronii isolated from diseased discus (Symphysodon discus) imported to Korea. J. Fish Dis. 42, 147–153. doi: 10.1111/jfd.12908
Sanchez-Cespedes, J., Blasco, M. D., Marti, S., Alba, V., Alcalde, E., Esteve, C., et al. (2008). Plasmid-mediated QnrS2 determinant from a clinical Aeromonas veronii isolate. Antimicrob. Agents Chemother. 52, 2990–2991. doi: 10.1128/AAC.00287-08
Skwor, T., Shinko, J., Augustyniak, A., Gee, C., and Andrasoc, G. (2014). Aeromonas hydrophila and Aeromonas veronii Predominate among Potentially Pathogenic Ciprofloxacin- and Tetracycline-Resistant Aeromonas Isolates from Lake Erie. Appl. Environ. Microbiol. 80, 841–848. doi: 10.1128/AEM.03645-13
Soares, S. C., Geyik, H., Ramos, R. T., de Sa, P. H., Barbosa, E. G., Baumbach, J., et al. (2016). GIPSy: genomic island prediction software. J. Biotechnol. 232, 2–11. doi: 10.1016/j.jbiotec.2015.09.008
Subramani, P. A., and Michael, R. D. (2017). “Prophylactic and Prevention Methods Against Diseases in Aquaculture,” in Fish Diseases, ed. G. Jeney (Amsterdam: Elsevier), 81–117. doi: 10.1016/b978-0-12-804564-0.00004-1
Syrova, E., Kohoutova, L., Dolejska, M., Papezikova, I., Kutilova, I., Cizek, A., et al. (2018). Antibiotic resistance and virulence factors in mesophilic Aeromonas spp. from Czech carp fisheries. J. Appl. Microbiol. doi: 10.1111/jam.14075 [Online ahead of print]
Tanaka, K. H., Vincent, A. T., Trudel, M. V., Paquet, V. E., Frenette, M., and Charette, S. J. (2016). The mosaic architecture of Aeromonas salmonicida subsp. salmonicida pAsa4 plasmid and its consequences on antibiotic resistance. PeerJ. 4:e2595. doi: 10.7717/peerj.2595
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Vijayakumar, R., and Sandle, T. (2019). A review on biocide reduced susceptibility due to plasmid-borne antiseptic-resistant genes-special notes on pharmaceutical environmental isolates. J. Appl. Microbiol. 126, 1011–1022. doi: 10.1111/jam.14118
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Yang, Q., Zhao, M., Wang, K. Y., Wang, J., He, Y., Wang, E. L., et al. (2017). Multidrug-Resistant Aeromonas veronii Recovered from Channel Catfish (Ictalurus punctatus) in China: prevalence and Mechanisms of Fluoroquinolone Resistance. Microb. Drug Resist. 23, 473–479. doi: 10.1089/mdr.2015.0296
Yin, H. L., Shen, Y., Liu, Z., Wang, S., Shen, Z., Zhang, R., et al. (2017). Novel Plasmid-Mediated Colistin Resistance Gene mcr-3 in Escherichia coli. Am. Soc. Microbiol. 8, e00543–17. doi: 10.1128/mBio
Yucel, N., and Aslim, B. (2005). Prevalence and resistance to antibiotics for aeromonas species isolated from retail fish in Turkey. J. Food Qual. 28, 313–324. doi: 10.1111/j.1745-4557.2005.00037.x
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Keywords: Aeromonas veronii, antimicrobial resistance, genomic islands, resistome analysis, tilapia, whole genome sequencing
Citation: Sakulworakan R, Chokmangmeepisarn P, Dinh-Hung N, Sivaramasamy E, Hirono I, Chuanchuen R, Kayansamruaj P and Rodkhum C (2021) Insight Into Whole Genome of Aeromonas veronii Isolated From Freshwater Fish by Resistome Analysis Reveal Extensively Antibiotic Resistant Traits. Front. Microbiol. 12:733668. doi: 10.3389/fmicb.2021.733668
Received: 30 June 2021; Accepted: 19 August 2021;
Published: 17 September 2021.
Edited by:
Bo Peng, Sun Yat-sen University, ChinaReviewed by:
Guilherme Campos Tavares, Federal University of Minas Gerais, BrazilSoojin Jang, Korea Pasteur Institute, South Korea
Copyright © 2021 Sakulworakan, Chokmangmeepisarn, Dinh-Hung, Sivaramasamy, Hirono, Chuanchuen, Kayansamruaj and Rodkhum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pattanapon Kayansamruaj, cGF0dGFuYXBvbi5rQGt1LnRo; Channarong Rodkhum, Y2hhbm5hcm9uZy5SQGNodWxhLmFjLnRo